
Submitted:
25 June 2024
Posted:
25 June 2024
You are already at the latest version
Abstract
Superior fiber quality is one of the most important objectives in cotton breeding. To detect the genetic basis underlying fiber quality, an F2 population containing 413 plants was constructed by crossing Jifeng 914 and Jifeng 173, both of which have superior fiber quality, with Jifeng 173 being better. Five fiber quality traits were investigated in the F2, F2:3, F2:4, and F2:5 populations. Quantitative trait loci (QTL) mapping was conducted based on a high-density genetic map containing 11,488 single nucleotide polymorphisms (SNPs) and spanning 4,202.12 cM in length. Transgressive segregation patterns and complex correlations in the five tested traits were observed. A total of 108 QTLs were found, including 27 for fiber length (FL), 16 for fiber strength (FS), 24 for micronaire (MC), 17 for fiber uniformity (FU), and 24 for fiber elongation rate (FE). Chromosome A7 contained 12 QTL, ranking the first. No QTL was found on chromosome D1 and D11. Each QTL contributed 1.98%–21.45% to the phenotypic variation (PV), including 13 major effect QTLs that contributed more than 10% toward PV. Two QTLs could be repeatedly detected in three populations, including qFL-D3-2 in F2, F2:4, and F2:5 with 9.18%–21.45% of PV, qFS-A11-1 in F2:3, F2:4 and F2:5 with 6.05%–10.41% of PV. Another seven stable QTLs could be detected in two populations, including four major effect QTLs: qFL-A12-3, qFS-D10-2, qMC-D6-2, and qMC-D8-1. Fourteen QTL-overlapping regions were found, which might explain the complex correlations among the five phenotypic traits. Four regions on chromosome A11, D3, D6, and D10 covered by both stable and major effect QTLs are promising for further fine mapping. The genomic regions of the two QTLs detected in three populations and the four major effect QTLs contain 810 genes. Gene functional analysis revealed that the annotated genes are mainly involved in protein binding and metabolic pathways. Fifteen candidate genes in the qFL-D3-2 region are highly expressed in fiber or ovules during fiber initiation, elongation, secondary cell wall thickening, or maturation stages. qRT-PCR revealed that Ghir_D03G005440.1 and Ghir_D03G011310.1 may play a role in promoting fiber initiation, Ghir_D03G006470.1 may be beneficial for promoting fiber elongation. This study provides more information for revealing the molecular genetic basis underlying cotton fiber quality.
Keywords:
upland cotton
; fiber quality
; QTL mapping
; candidate gene
; qRT-PCR
1. Introduction
There are four cultivated cotton species, two diploids, Gossypium arboretum L. and G. herbaceum L., and two tetraploids G. hirsutum L. and G. barbadense L. Gossypium hirsutum L., also known as upland cotton, is the most widely planted species in the world and contributes to more than 95% of the world’s cotton production [1,2]. Lint fiber is the most important economic cotton product, and superior quality fiber is an important cotton breeding target. In recent years, textile industries have put forward more stringent requirements on fiber quality, and higher quality fiber can attain higher prices. It is a great challenge to improve fiber quality further under the premise of ensuring cotton yield and a negative correlation between fiber quality and yield has dragged down the breeding efficiency.
Molecular markers are powerful tools to dissect complex traits such as yield and fiber quality using quantitative trait loci (QTL) mapping as well as genome-wide association analysis (GWAS). Marker-assisted selection is a new approach to increasing breeding efficiency and improving cotton yield and fiber quality simultaneously. Many QTL mapping studies about cotton yield and fiber quality have been reported both in intra- and inter-specific populations, and numerous QTLs have been mapped on the 26 chromosomes [3,4,5,6].
The development of high-throughput sequencing technology has made it convenient to detect the genetic basis underlying a phenotype. The reference genome sequence quality of the standard upland cotton line TM-1 has been improved since its first release [7,8,9,10,11]. The release of the NDM8 high-quality genome sequence will be a great source for modern cultivar genetics research [12]. QTL mapping studies for various traits based on reference genome sequences have been carried out, such as cotton earliness [13,14,15], plant architecture [16,17,18,19], okra leaf [20], yield and fiber quality [6,21,22,23,24]. Wang et al. adopted restriction site-associated DNA sequencing (RAD-seq) for high-density genetic map construction and detected 33 yield and fiber quality-related QTLs [21]. Islam et al. used genotyping by sequencing (GBS) for single nucleotide polymorphism (SNP) detection in upland cotton and validated its utility [25]. Qi et al. constructed a high-density genetic map using GBS and mapped 17 QTLs for plant architecture traits [16]. Su et al. used specific-locus amplified fragment sequencing (SLAF-seq) for GWAS analysis and found two genomic regions associated with fiber quality [26]. Sun et al. conducted GWAS analysis using 719 upland cotton accessions and detected 46 significant SNPs associated with five fiber quality traits [27]. Ma et al. resequenced 419 upland cotton accessions and found 7,383 unique SNPs associated with 13 fiber-related traits [6]. Wang et al. constructed 239 RILs with chromosome segrements introgressed from Gossypium barbadense into G. hirsutum and used to detect 67 QTL for fiber quality and 37 QTL for yield, and 3 putative candidate genes were identified for fiber length QTL [1]. Due to the high yield and wide adaptability of G. hirsutum and the super fiber quality of G. barbadense, this research had important reference value for synchronously improving cotton yield and fiber quality. Although more than 1000 QTLs for fiber quality have been published, few QTLs have been mapped repeatedly in different populations mainly due to the differences in materials or environments [1,3,4,28,29]. The genetic mechanism of fiber quality in high-yield cotton cultivars is still unclear, and effective molecular markers are rare.
Jifeng 914 is a nationally certified variety with a high and stable yield as well as relatively better fiber length (FL) and fiber strength (FS). Jifeng 173 is a super quality fiber cultivar especially its FL, FS, and micronaire (MC). To detect the genetic basis underlying fiber quality, an F2 population was constructed using Jifeng 914 as the maternal material and Jifeng 173 as the paternal material. The GBS technique was applied to detect SNP markers and construct a high-density genetic map. Five fiber quality traits were observed in the F2, F2:3, F2:4, and F2:5 populations and used for QTL mapping. Genes in the stable and major QTL regions were annotated, and gene functions were first analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. Online RNA-seq data and qRT-PCR were used for gene expression patterns analyses and candidate genes selection.
2. Materials and Methods
2.1. Mapping population and trait evaluation
Two upland cotton varieties, Jifeng 914 and Jifeng 173, bred by the Institute of Cereal and Oil Crops, Hebei Academy of Agriculture and Forestry Sciences, were selected as parents. Jifeng 914 is a nationally certified variety with outstanding high and stable yield and super fiber quality. And the most outstanding character of Jifeng 173 is the super fiber quality with longer FL, stronger FS and better MC. Before hybridization, both Jifeng 914 and Jifeng 173 underwent multiple generations of self pollination to obtain homozygous lines. In the summer of 2018, Jifeng 914 was used as a maternal cultivar and crossed with Jifeng 173 at Shijiazhuang. All of the F1 seeds were planted and self-pollinated in the following winter at Sanya, Hainan province. The F2 seeds were harvested from the bulked self-pollinated seeds of all F1 plants and planted in 15 rows (7.0 m long and 0.76 cm apart) at Shijiazhuang on April 30, 2019, with 413 plants reserved after final singling. Two hundred normal plants from the F2 population were randomly selected and continuously self-pollinated. The derived F2:3, F2:4, and F2:5 populations were planted in a completely randomized block design with two replicates (5.0 m long and 0.76 cm apart) at Shijiazhuang on April 27, 2020, 2021, and 2022. Field management was performed under local practices.
Thirty naturally opened bolls from each row were hand-harvested. After ginning, five fiber quality traits, including FL, FS, fiber uniformity (FU), MC, and fiber elongation rate (FE), were evaluated by a high-volume instrument (HFT9000) at the Cotton Quality Testing Center in Institute of Cotton Research, Chinese Academy of Agricultural Science (CRI CAAS). Fiber quality data were analyzed using Excel 2010 and SPSS (17.0) software.
2.2. QTL mapping
Previously, a high-density genetic map has been constructed [30]. A total of 416 G sequence data was obtained by genotyping by GBS, with an average of 25.91 G and 9× depth in the parents, 1.82 G and 0.7× depth in the F2 plants. The Q30 score reached 95.68%. The genetic map contained 11,488 SNPs and spanned 4,202.12 cM in length, ranging from 150.74 cM on A3 to 178.90 cM on A9. The SNPs were unevenly distributed on the 26 linkage groups, with only 30 SNPs on A2 and 1,318 SNPs on D5. Based on this genetic map, additive effect QTLs were analyzed using inclusive composite interval mapping (ICIM) with QTL IciMapping 4.0 software. The parameters were set as Step = 1cM and PIN = 0.001. LOD scores were determined using a 1000 permutation test. QTLs contributing more than 10% of the phenotypic variation (PV) are denoted as major effect QTLs. QTLs mapped in at least two populations are denoted as stable QTLs.
2.3. Gene annotation and candidate gene selection
Annotated genes in the major or stable QTLs were obtained using the reference genome sequence information [9] using the CottonFGD database (https://cottonfgd.net/) [31]. Gene expression levels were analyzed using the online software CottonMD (http://yanglab.hzau.edu.cn/CottonMD) [32]. Candidate genes were first screened based on the highly mapped GO and KEGG terms. qRT-PCR was conducted to analyze gene expression patterns, using ovule samples (-1DPA, 0DPA, 1DPA) from Jifeng 914 and Jifeng 173 and fiber samples (5DPA, 10DPA, 15DPA) from Jifeng 4 (JF4) and Jifeng 4xuan (JF4x) as materials. The difference in lint percentage between Jifeng 914 and Jifeng 173 is greater. And JF4 and JF4x are sister lines. JF4 has better fiber quality than JF4x, especially fiber length and fiber strength. Total RNA was extracted using a Plant RNA Purification Kit (Tiangen, Beijing, China). First-strand cDNA was reverse transcribed from 1 μg total RNA using a FastKing gDNA Dispelling RT SuperMix Kit (Tiangen, Beijing, China). qRT-PCR was carried out with the SYBR Premix Ex Taq (TAKARA, Japan) on a LightCycler480 instrument (Rotkreuz, Switzerland).
3. Results
3.1. Phenotypic variation and trait correlation analysis
Five fiber quality traits, including FL, FS, FU, MC, and FE, were investigated. Significant differences were found between the parents, especially FL, FS, and MC (Table 1). Jifeng 173 showed longer FL, stronger FS, and smaller MC in all four years. Both the maximum and minimum values in the offspring populations exceeded the parents, indicating the transgressive segregation of the five observed traits, and minor effect alleles controlling these traits existed in both parents (Additional file 1). The absolute values of skew and kurt in the tested populations were smaller than one except for FE in 2022, which indicates that most of the observed traits presented nearly normal distributions. MC has the largest coefficient variations (CVs) over four years, followed by FS, FL, FU, and FE. Thus, this population is appropriate for fiber quality-related QTLs mapping.
Complex correlations were observed among FL, FS, FU, MC, and FE over four years (Additional file 2). Significant positive correlations were observed within the trait pairs, such as FL-FS, FL-FE, FS-FE, MC-FU, and FE-FU. Significant negative correlations were observed between FL and MC, between FS and MC, between FE and MC. No consistent correlation was observed between FL and FU, between FS and FU. The complex phenotypic correlations reflected the complex genetic interactions underlying these fiber quality traits.
3.2. QTL mapping for fiber quality traits
In this study, a total of 108 QTLs were found, including 27 for FL, 16 for FS, 24 for MC, 17 for FU, and 24 for FE (Additional file 3). Chromosome A7 contained the highest number of QTLs with 12. No QTL was found on chromosomes D1 and D11. Each QTL contributed 1.98%–21.45% to the PV, 13 QTLs contributed more than 10% to the PV and 9 QTLs could be mapped repeatedly (Figure 1). Jifeng 914 conferred favorable alleles for 49 QTLs, and Jifeng 173 conferred another 59 favorable alleles.
Fiber length. Twenty-seven QTLs were mapped on 18 chromosomes, including three major effect QTLs and four stable QTLs. The favorable alleles for 13 and 14 QTLs were conferred by Jifeng 914 and Jifeng 173, respectively. The most outstanding QTL was qFL-D3-1 conferred by Jifeng 914, which was repeatedly mapped in the F2, F2:4, and F2:5 populations and contributed 9.18%–21.45% to the PV. qFL-A12-3 was mapped in the F2:3 and F2:4 populations and contributed 10.18%–13.80% to the PV, and was conferred by Jifeng 173. In addition, qFL-A6-1, qFL-A12-1, and qFL-D9-1 were mapped in two populations with favorable alleles conferred by Jifeng 914, qFL-A11-1 contributed 10.00% to the PV with a favorable allele conferred by Jifeng 173.
Fiber strength. Sixteen QTLs were mapped on ten chromosomes, including only two stable and major effect QTLs. Jifeng 914 and Jifeng 173 conferred favorable alleles for nine and seven QTLs, respectively. qFS-A11-1 was mapped in the F2:3, F2:4 and F2:5 populations and contributed 6.05%–10.41% to the PV, and was conferred by Jifeng 173. qFS-D10-2 was mapped in the F2 and F2:4 populations and contributed 10.54%–17.51% to the PV, and was conferred by Jifeng 914.
Micronaire. Twenty-four QTLs were mapped on seventeen chromosomes, and five major effect and two stable QTLs were found. Jifeng 914 and Jifeng 173 conferred favorable alleles for nine and fifteen QTLs, respectively. qMC-D6-2 was mapped in the F2 and F2:3 populations and contributed 10.15%–10.91% to the PV, and was conferred by Jifeng 173. qMC-D8-1 was mapped in the F2:3 and F2:4 populations and contributed 6.39%–10.97% to the PV, and was conferred by Jifeng 914. In addition, qMC-D6-1, qMC-D7-1, and qMC-D10-1 contributed 10.81%, 13.16%, and 12.90% to the PV, respectively.
Fiber uniformity. Seventeen QTLs were mapped on 13 chromosomes. Jifeng 914 and Jifeng 173 conferred favorable alleles for eight and nine QTLs, respectively. While only two major QTLs were found, including qFU-D7-1 and qFU-D10-1, which contributed 14.77% and 10.19% to the PV, respectively. No stable QTL was found over four years, indicating a significant environmental effect on FU.
Fiber elongation. Twenty-four QTLs were mapped on 13 chromosomes. Jifeng 914 and Jifeng 173 conferred favorable alleles for 10 and 14 QTL, respectively. In addition, six QTLs were mapped on chromosome D5 and Jifeng 173 conferred favorable alleles, which might reflect the importance of D5 from Jifeng 173 in regulating FE. Only one major effect QTL was found. qFE-D6-1 contributed 10.72% to the PV and was conferred by Jifeng 914. No stable QTL was found over the four years, which indicated a significant environmental effect on FE.
QTL-overlapping regions. A total of 14 QTL-overlapping regions were found on 12 chromosomes (Figure 1). The additive effect directions of the QTL in 10 out of these 14 regions are different, which might explain the complex correlations among the five phenotypic traits. Four regions on chromosome A11, D3, D6, and D10 covered by both stable and major effect QTLs are promising for further fine mapping. The 0-4.5 cM region on A11 was covered by 3 QTLs (qMC-A11-1, qFL-A11-1 and qFS-A11-1). qFL-A11-1 is a major effect QTL, and qFS-A11-1 is a stable QTL with 10.41% contribution ratio to the PV. The 49.5-57.5 cM region on D3 was covered by 2 QTLs (qFL-D3-2 and qFU-D3-1). qFL-D3-2 is a stable QTL with 9.18-21.45% of contribution ratios to the PV. The 165.5-170 cM region on D6 was covered by 2 major QTLs (qFE-D6-1 and qMC-D6-2) and qMC-D6-2 could be repeatedly mapped. The 99.5-100.5 cM on D10 was covered by 4 QTLs (qFL-D10-2, qFS-D10-2, qFU-D10-1 and qMC-D10-1), of which, 3 are major effect QTLs and 1 is stable QTL.
3.3. Candidate gene analysis
There are six major effect QTLs that could be repeatedly mapped in at least two populations, including qFL-D3-2 and qFS-A11-1 mapped in three populations, qFL-A12-3, qFS-D10-2, qMC-D6-2, and qMC-D8-1 mapped in two populations. There are no genes in qFS-D10-2 (16596338 bp-16630630 bp), qMC-D6-2 (1054134 bp-1066524 bp), and qMC-D8-1 (9129475 bp-9137306 bp), while 810 genes were annotated in qFS-A11-1, qFL-A12-3, and qFL-D3-2 including 6 genes in qFS-A11-1 (119649722 bp-119686364 bp), 6 genes in qFL-A12-3 (10414004 bp-10555066 bp), and 799 genes in qFL-D3-2 (6090201 bp-41836768 bp) (Additional file 4). Upon using GO and KEGG analysis, 392 GO terms and 62 KEGG pathways were mapped under the corrected p value < 0.05 (Additional file 5). The most significant GO term is protein binding, and the most significant KEGG pathway is metabolic pathways. Among the 265 genes in the mapped protein binding term and metabolic pathways, 15 genes located in the qFL-D3-2 region are highly expressed at different fiber development stages ( Additional file 6). Thirteen genes are highly expressed during the fiber elongation stage (5-25DPA fiber), which determines FL. Five genes Ghir_D03G010890, Ghir_D03G010910, Ghir_D03G006470, Ghir_D03G010430, and Ghir_D03G010470, highly expressed during the lint fiber initiation stage (−3-3DPA ovule). As FU-related QTL qFU-D3-1 overlapped with qFL-D3-2, these five genes may affect FL and FU by regulating fiber initiation. Based on the present sequencing depth, a total of 116 SNPs and 40 InDels markers were found in the qFL-D3-2 region, and none of these markers located in protein coding regions of the above mentioned genes (Additional file 7). And based on the 8 mapped SNP markers in the qFL-D3-2 region, it is difficult to narrow down the interval length to fine map qFL-D3-2 (Additional file 8).
Ovules from the fiber initiation stage and fibers from the fiber elongation stage were used for qRT-PCR. The expression levels of the above mentioned 15 genes in the ovules were higher in Jifeng 914 than in Jifeng 173 (Figure 2). Nine genes showed continuously increased expression trend from -1DPA to 1DPA ovules, and 2 genes (Ghir_D03G005440.1 and Ghir_D03G011310.1) showed significantly high expression levels in 1DPA ovules. There are 2 genes (Ghir_D03G005100.1 and Ghir_D03G010910.1) showed continuously decreased expression trend from -1DPA to 1DPA ovules. Gene expression patterns in fibers were more complex and most of the gene expression patterns between JF4 and JF4x were different (Figure 3). Two genes (Ghir_D03G006470.1 and Ghir_D03G007410.1) showed continuously increased expression trend from 5DPA to 15DPA fiber. The expression level of Ghir_D03G006470.1 in 15DPA fiber is fifty times higher than that in 5DPA fiber, and this gene had higher expression level in 5DPA and 10DPA fiber in JF4 and higher expression level in 15DPA fiber in JF4x. It is predicted that Ghir_D03G006470.1 has a more prominent role in promoting fiber elongation.
4. Discussion
Cotton is the leading natural fiber crop and cash crop in the world [5,33]. Yield and fiber quality traits are the most important economic traits in cotton production. Boll weight (BW), lint percentage (LP), and seed index (SI) are three vital yield components. FL, FS, FU, MC, and FE are the first five fiber quality traits. All of these traits are quantitatively genetically controlled, and significant positive or negative correlations exist among them, such as significant positive correlations between FL and FS, FL and SI, and FS and SI, and significant negative correlations between FL and MC, FS and MC, FL and LP, FS and LP, and SI and LP [28]. Similar results were found in our study, and complex correlations were observed among the five fiber quality traits. Simultaneously, improvements in cotton yield and fiber quality are more difficult to counter these unfavorable correlations through traditional breeding.
High-density genetic maps are a prerequisite for molecular genetic detection of vital traits, and reduced-representation sequencing techniques supply more convenience, which makes the molecular marker detection procedure more time efficient. Wang et al. applied RAD-seq in cotton mapping parents sequencing and found 1,323 SSR, 3,838 InDel, and 9,366 SNP markers [21]. Jia et al. constructed a genetic map containing 6,295 SNPs with only 0.63 cM between adjacent markers using RAD-seq [15]. Li et al. adopted GBS in marker detection and constructed a genetic map containing 3,978 SNPs [13]. Zhang et al. published a genetic map with 5,521 SNPs and 3,259.37 cM using SLAF-seq [34]. Ma et al. constructed a genetic map that contained 7,709 SNPs and spanned 3,433.24 cM using GBS [18]. In this study, we applied GBS in molecular marker detection, and a total of 1,305,642 polymorphic SNPs were found. A high-density genetic map containing 11,488 SNPs was constructed. The genetic map spanned 4,202.12 cM, with 0.37 cM between adjacent markers. The quality of the constructed genetic map was evaluated by marker collinearity analysis and heat map analysis. The marker density and map quality were comparable with these previously published high-density genetic maps.
Up to now, more than 1,000 fiber quality and yield-related QTLs have been published and are distributed on almost all 26 chromosomes [1,3,4,28]. By comparing with published results [1,27,28,35,36], 32 QTLs in this study have been reported, and 76 new QTLs might have been identified (Additional file 2). Only three of the sixteen major or stable QTLs overlapped with previously reported QTL regions, which reflected the parental genetic diversity used in this study. Six outstanding QTL regions were hopeful prospects for candidate gene fine mapping, including qFL-D3-2 and qFS-A11-1 mapped in three populations, qFL-A12-3, qFS-D10-2, qMC-D6-2, and qMC-D8-1 mapped in two populations.
Among the 15 genes in the qFL-D3-2 region with high expression levels at different fiber development stages, Ghir_D03G006470 is a SWEET1 gene, Ghir_D03G005100, and Ghir_D03G005720 are CYP89A9 genes. As reported, GhSWEET12 RNAi plants produced shorter fiber [37]. PAG1, one homolog gene of Arabidopsis CYP734A1, plays a crucial role in regulating FL [38]. Thus, Ghir_D03G006470, Ghir_D03G005100, and Ghir_D03G005720 may also have important functions during cotton fiber development. In addition, Ghir_D03G010890 and Ghir_D03G010910 are UDP-glycosyltransferase genes, Ghir_D03G010430 is a eukaryotic translation initiation factor 5A gene, Ghir_D03G010470 is ribosomal protein S5 family protein gene, Ghir_D03G009660 is a luminal-binding protein 5 gene, Ghir_D03G005440 is a cinnamoyl-CoA reductase-like SNL6 gene. Based on the published literature, UDP-glycosyltransferase, eukaryotic translation initiation factor 5A, ribosomal protein, luminal-binding protein 5, and cinnamoyl-CoA reductase are key factors involved in cotton fiber development [12,39,40,41,42,43]. qRT-PCR results revealed that most of the above mentioned 15 genes showed continuously increased expression from -1DPA to 1DPA ovules, Ghir_D03G005440.1 and Ghir_D03G011310.1 had significantly high expression levels in 1DPA ovules. These genes may play a role in promoting fiber initiation, which has a significant impact on fiber length and fiber uniformity. Ghir_D03G006470.1 in 15DPA fiber is fifty times higher than that in 5DPA fiber, and this gene had higher expression level in 5DPA and 10DPA fiber in JF4 and higher expression level in 15DPA fiber in JF4x, indicating that this gene may be beneficial for promoting fiber elongation.Therefore, more work needs to be performed to fine map these outstanding QTLs, and to screen out and validate the key genes regulating fiber development.
In summary, six outstanding QTLs out of 108 QTLs were screened in four populations. Fifteen genes in the qFL-D3-2 region were predicted as candidates for regulating FL as they showed high expression levels during the fiber initiation and elongation stages.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
LM and JXY conceived the project and set the scientific objectives. ZJJ, ZHX, WGY, and WSJ contributed to equipment preparation and data acquisition. JXY wrote the manuscript. LM and WGY reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (32201758), Hebei Agriculture Research System (HBCT2023070202), HAAFS Science and Technology Innovation Special Project (2022KJCXZX-LYS-14), State Key Laboratory of Cotton Bio-breeding and Integrated Utilization Open Fund (CB2023A05).
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Gong, J.; Song, Z.; Zhou, J.; Wang, J.; Zhao, C.; Jiao, M.; et al. Identification of candidate genes for key fibre-related QTLs and derivation of favourable alleles in Gossypium hirsutum recombinant inbred lines with G. barbadense introgressions. Plant Biotechnol. J. 2020, 18, 707–720. [Google Scholar] [CrossRef]
- Li, Y.; Mo, T.; Ran, L.; Zeng, J.; Wang, C.; Liang, A.; Dai, Y.; Wu, Y.; Zhong, Z.; Xiao, Y. Genome resequencing-based high-density genetic map and QTL detection for yield and fiber quality traits in diploid Asiatic cotton (Gossypium arboreum). Mol. Genet. Genom. 2022, 297, 199–212. [Google Scholar] [CrossRef]
- Said, J.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef]
- Said, J.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.; Zhang, J. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G.barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.; Yoo, M.; Lin, M.; Murphy, M.; Harker, D.; Byers, R.; Lipka, A.; Hu, G.; Yuan, D.; Conover, J.; et al. Genetic analysis of the transition from wild to domesticated cotton ( Gossypium hirsutum L.). G3 (Bethesda, Md.) 2020, 10, 731–754. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.; Scheffler, B.; Stelly, D.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Chang, X.; He, X.; Li, J.; Liu, Z.; Pi, R.; Luo, X.; Wang, R.; Hu, X.; Lu, S.; Zhang, X.; et al. High-quality Gossypium hirsutum and Gossypium barbadense genome assemblies reveal the landscape and evolution of centromeres. Plant Commun. 2023, 5, 100722. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, Y.; Wu, L.; Zhang, G.; Sun, Z.; Li, Z.; Jiang, Y.; Ke, H.; Chen, B.; Liu, Z.; et al. High-quality genome assembly and resequencing of modern cotton cultivars provide resources for crop improvement. Nat. Genet. 2021, 53, 1385–1391. [Google Scholar] [CrossRef]
- Li, L.; Zhao, S.; Su, J.; Fan, S.; Pang, C.; Wei, H.; Wang, H.; Gu, L.; Zhang, C.; Liu, G.; et al. High-density genetic linkage map construction by F2 populations and QTL analysis of early-maturity traits in upland cotton (Gossypium hirsutum L.). PLoS ONE 2017, 12, e182918. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Pang, C.; Wei, H.; Li, L.; Liang, B.; Wang, C.; Song, M.; Wang, H.; Zhao, S.; Jia, X.; et al. Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genom. 2016, 17, 687. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Pang, C.; Wei, H.; Wang, H.; Ma, Q.; Yang, J.; Cheng, S.; Su, J.; Fan, S.; Song, M.; et al. High-density linkage map construction and QTL analysis for earliness-related traits in Gossypium hirsutum L. BMC Genom. 2016, 17, 909. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Wang, N.; Qiao, W.; Xu, Q.; Zhou, H.; Shi, J.; Yan, G.; Huang, Q. Construction of a high-density genetic map using genotyping by sequencing (GBS) for quantitative trait loci (QTL) analysis of three plant morphological traits in upland cotton (Gossypium hirsutum L.). Euphytica 2017, 213, 83. [Google Scholar] [CrossRef]
- Su, J.; Li, L.; Zhang, C.; Wang, C.; Gu, L.; Wang, H.; Wei, H.; Liu, Q.; Huang, L.; Yu, S. Genome-wide association study identified genetic variations and candidate genes for plant architecture component traits in Chinese upland cotton. Theor. Appl. Genet. 2018, 131, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pei, W.; Ma, Q.; Geng, Y.; Liu, G.; Liu, J.; Cui, Y.; Zhang, X.; Wu, M.; Li, X.; et al. QTL analysis and candidate gene identification for plant height in cotton based on an interspecific backcross inbred line population of Gossypium hirsutum × Gossypium barbadense. Theor. Appl. Genet. 2019, 132, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xiao, X.; Gong, J.; Li, J.; Zhang, Z.; Liu, A.; Lu, Q.; Shang, H.; Shi, Y.; Ge, Q.; et al. QTL mapping for plant height and fruit branch number based on RIL population of upland cotton. J. Cotton Res. 2020, 3, 5. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, J.; Liu, D.; Stiller, W.; Liu, D.; Zhang, Z.; Llewellyn, D.; Wilson, I. Integrated mapping and characterization of the gene underlying the okra leaf trait in Gossypium hirsutum L. J. Exp. Bot. 2016, 67, 763–774. [Google Scholar] [CrossRef]
- Wang, H.; Jin, X.; Zhang, B.; Shen, C.; Lin, Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. 2015, 22, 147–160. [Google Scholar] [CrossRef]
- Jia, X.; Wang, H.; Pang, C.; Ma, Q.; Su, J.; Wei, H.; Song, M.; Fan, S.; Yu, S. QTL delineation for five fiber quality traits based on an intra-specific Gossypium hirsutum L. recombinant inbred line population. Mol. Genet. Genom. 2018, 293, 831–843. [Google Scholar] [CrossRef]
- Ma, J.; Geng, Y.; Pei, W.; Wu, M.; Li, X.; Liu, G.; Li, D.; Ma, Q.; Zang, X.; Yu, S.; et al. Genetic variation of dynamic fiber elongation and developmental quantitative trait locus mapping of fiber length in upland cotton (Gossypium hirsutum L.). BMC Genom. 2018, 19, 882. [Google Scholar] [CrossRef]
- Thyssen, G.; Jenkins, J.; McCarty, J.; Zeng, L.; Campbell, B.; Delhom, C.; Islam, M.; Li, P.; Jones, D.; Condon, B.; et al. Whole genome sequencing of a MAGIC population identified genomic loci and candidate genes for major fiber quality traits in upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 2019, 132, 989–999. [Google Scholar] [CrossRef]
- Islam, M.; Thyssen, G.; Jenkins, J.; Fang, D. Detection, Validation, and Application of Genotyping-by-Sequencing based single nucleotide polymorphisms in upland cotton. Plant Genome-US 2015, 8. [Google Scholar] [CrossRef]
- Su, J.; Li, L.; Pang, C.; Wei, H.; Wang, C.; Song, M.; Wang, H.; Zhao, S.; Zhang, C.; Mao, G.; et al. Two genomic regions associated with fiber quality traits in Chinese upland cotton under apparent breeding selection. Sci. Rep.-UK 2016, 6, 38496. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Jamshed, M.; Shi, Y.; Liu, A.; Gong, J.; Wang, S.; Zhang, J.; Sun, F.; Jia, F.; et al. Genome-wide quantitative trait loci reveal the genetic basis of cotton fibre quality and yield-related traits in a Gossypium hirsutum recombinant inbred line population. Plant Biotechnol. J. 2020, 18, 239–253. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Xiao, X.; Li, P.; Liu, A.; Li, J.; Gong, J.; Gong, W.; Ge, Q.; Shang, H.; et al. Genome-wide artificial introgressions of Gossypium barbadense into G. hirsutum reveal superior loci for simultaneous improvement of cotton fiber quality and yield traits. J. Advenced Res. 2023, 53, 1–16. [Google Scholar] [CrossRef]
- Jia, X.; Wang, S.; Zhao, H.; Zhu, J.; Li, M.; Wang, G. QTL mapping and BSA-seq map a major QTL for the node of the first fruiting branch in cotton. Front. Plant Sci. 2023, 14, 1113059. [Google Scholar] [CrossRef]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, J.; Huang, Y.; Wang, S.; Wei, L.; Liu, D.; Weng, Y.; Xiang, J.; Zhu, Q.; Yang, Z.; et al. CottonMD: A multi-omics database for cotton biological study. Nucleic Acids Res. 2022, 51, D1446–D1456. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, K.; Li, S.; Yu, S.; Zhai, H.; Wu, M.; Li, X.; Fan, S.; Song, M.; Yang, D.; et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 2013, 126, 275–287. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D.; et al. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative trait loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef]
- Sun, W.; Gao, Z.; Wang, J.; Huang, Y.; Chen, Y.; Li, J.; Lv, M.; Wang, J.; Luo, M.; Zuo, K. Cotton fiber elongation requires the transcription factor GhMYB212 to regulate sucrose transportation into expanding fibers. New Phytol. 2019, 222, 864–881. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, C.; Yang, X.; Liu, K.; Wu, Z.; Zhang, X.; Zheng, W.; Xun, Q.; Liu, C.; Lu, L.; et al. PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef]
- Yang, Y.; Bian, S.; Yao, Y.; Liu, J. Comparative proteomic analysis provides new insights into the fiber elongating process in cotton. J. Proteome Res. 2008, 7, 4623–4637. [Google Scholar] [CrossRef]
- Gilbert, M.; Bland, J.; Shockey, J.; Cao, H.; Hinchliffe, D.; Fang, D.; Naoumkina, M. A transcript profiling approach reveals an abscisic acid-specific glycosyltransferase (UGT73C14) induced in developing fiber of Ligon lintless-2 mutant of cotton (Gossypium hirsutum L.). PLoS ONE 2013, 8, e75268. [Google Scholar] [CrossRef]
- Hu, G.; Koh, J.; Yoo, M.; Grupp, K.; Chen, S.; Wendel, J. Proteomic profiling of developing cotton fibers from wild and domesticated Gossypium barbadense. New Phytol. 2013, 200, 570–582. [Google Scholar] [CrossRef]
- Zheng, M.; Meng, Y.; Yang, C.; Zhou, Z.; Wang, Y.; Chen, B. Protein expression changes during cotton fiber elongation in response to drought stress and recovery. Proteomics 2014, 15, 1776–1795. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhou, C.; Su, Q.; Xu, M.; Yue, H.; Zhang, S.; Zhou, B. Fine-mapping and candidate gene analysis of qFS-Chr. D02, a QTL for fibre strength introgressed from a semi-wild cotton into Gossypium hirsutum. Plant Sci. 2020, 297, 110524. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of stable QTL, major QTL and QTL-overlapping regions on the genetic map. Note: FL, fiber length; FS, fiber strength; MC, micronaire; FU, fiber uniformity; FE, fiber elongation rate.
Figure 1.
Distribution of stable QTL, major QTL and QTL-overlapping regions on the genetic map. Note: FL, fiber length; FS, fiber strength; MC, micronaire; FU, fiber uniformity; FE, fiber elongation rate.
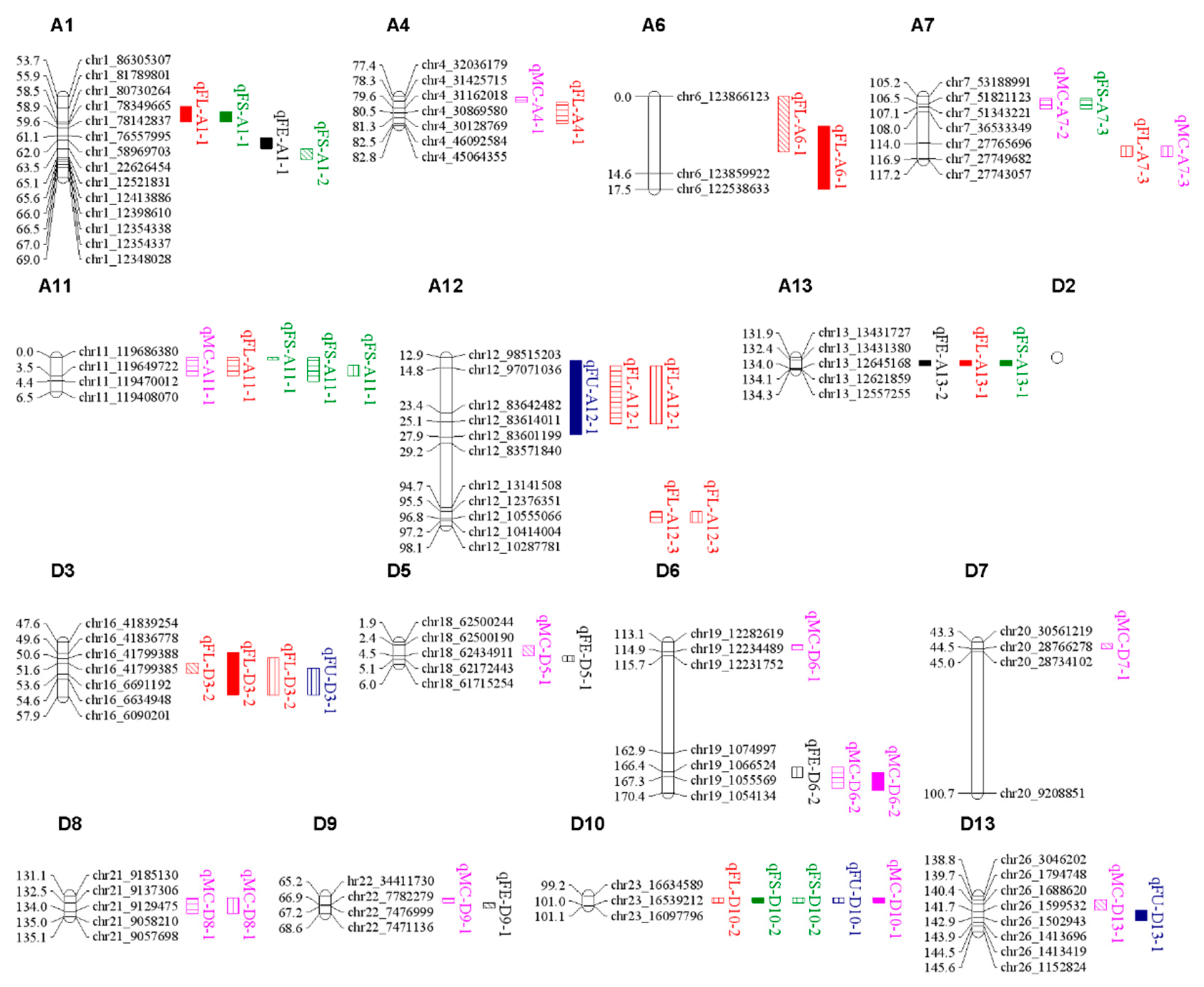
Figure 2.
Gene expression patterns in the ovules.Note: JF914, Jifeng 914; JF173, Jifeng 173.
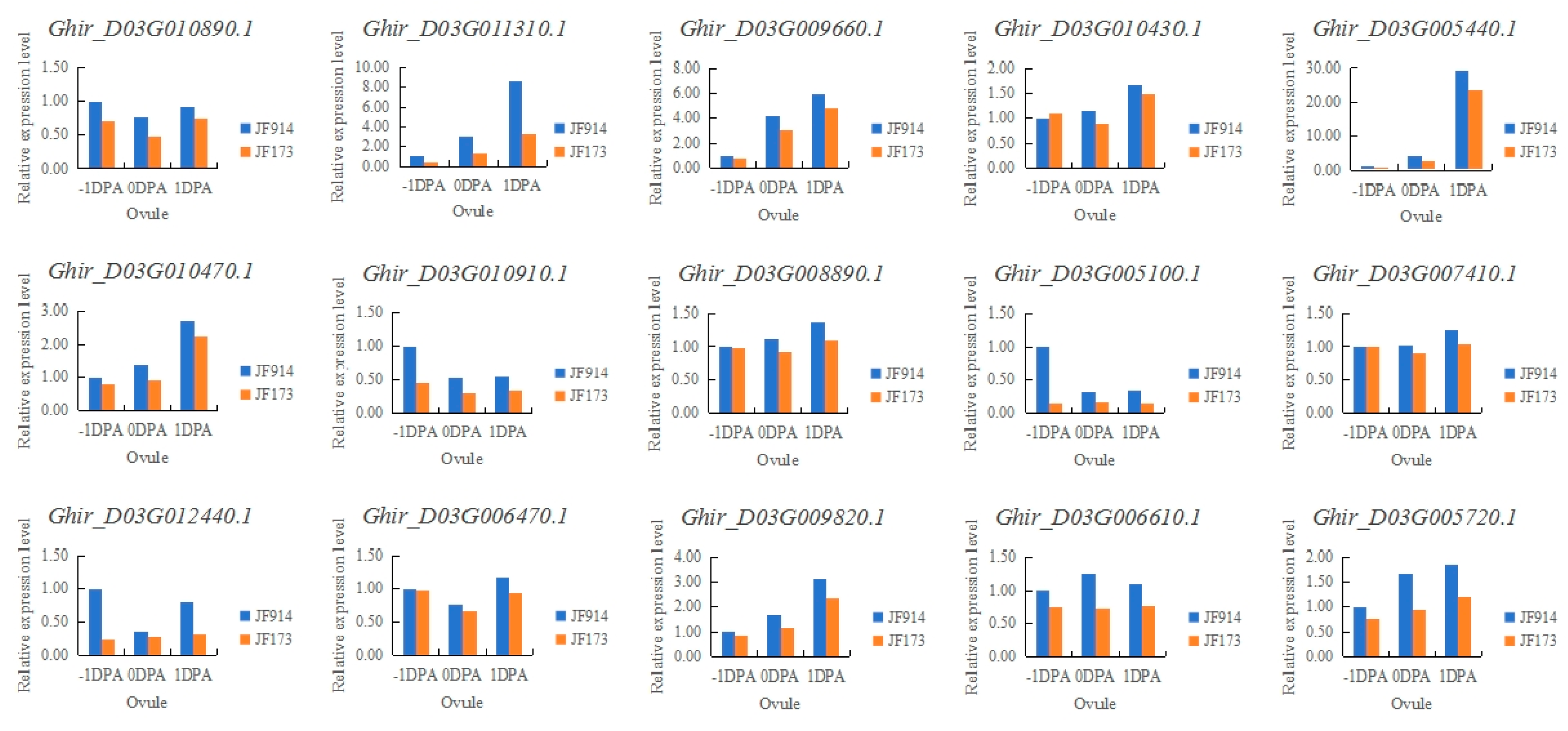
Figure 3.
Gene expression patterns in the fibers.Note: JF4, Jifeng 4; JF4x, Jifeng 4xuan.
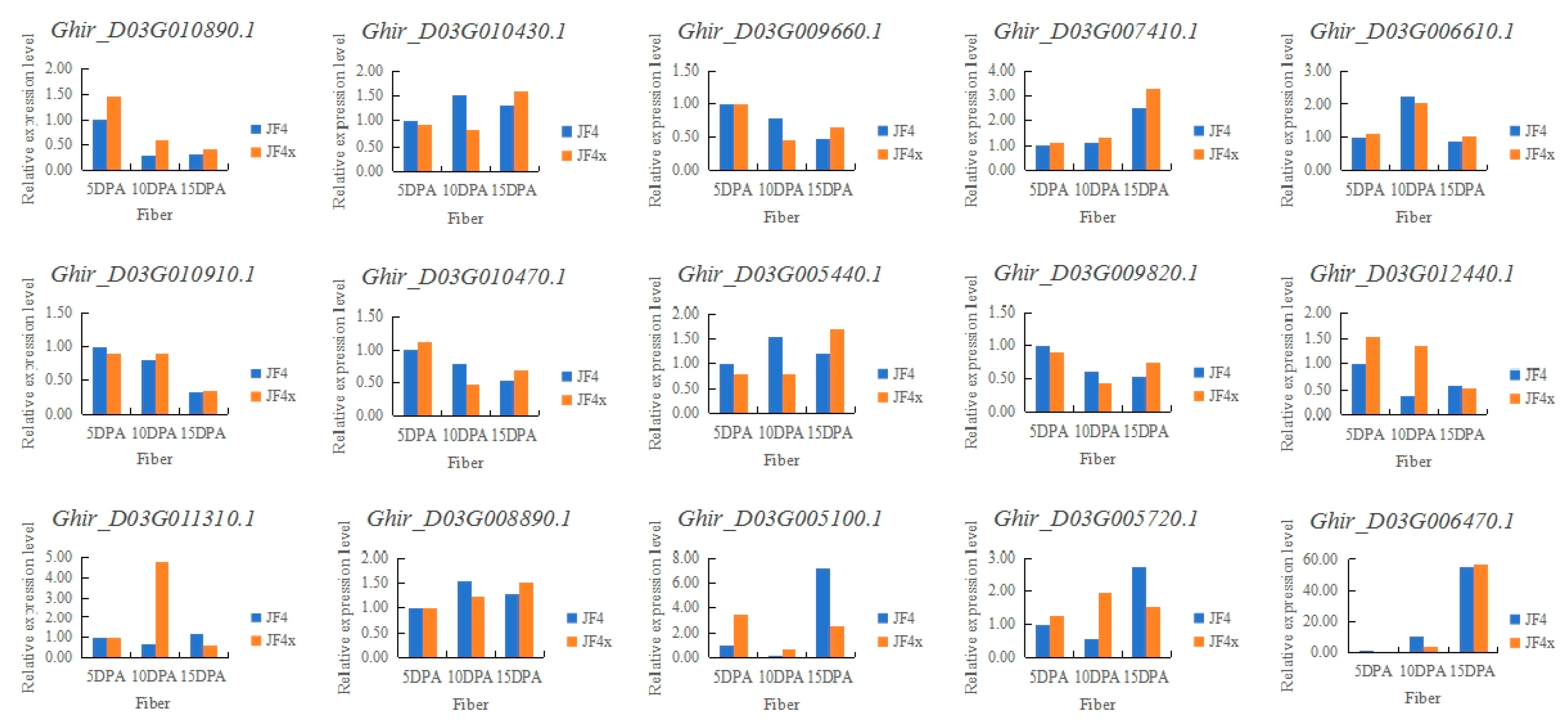

Table 1.
Basic statistics of the five tested traits in the parent and offspring populations.
| Trait | Year | Parents | Offspring populations | ||||||
| Jifeng 914 | Jifeng 173 | Max | Min | Mean | Skew | Kurt | CV(%) | ||
| FL(mm) | 2019 | 29.00 | 32.10** | 32.90 | 25.90 | 28.96 | -0.06 | -0.13 | 4.62 |
| 2020 | 29.25 | 31.65** | 35.10 | 26.70 | 30.97 | 0.00 | 0.47 | 4.37 | |
| 2021 | 30.15 | 32.65** | 35.50 | 27.60 | 31.68 | 0.18 | -0.26 | 4.61 | |
| 2022 | 29.65 | 32.40** | 33.10 | 26.80 | 30.38 | -0.16 | -0.39 | 4.14 | |
| FS(cn/tex) | 2019 | 31.10 | 34.70** | 37.50 | 24.90 | 30.82 | -0.09 | -0.56 | 6.94 |
| 2020 | 30.50 | 31.35** | 34.20 | 26.00 | 30.43 | 0.12 | 0.04 | 4.96 | |
| 2021 | 30.20 | 33.85** | 37.00 | 28.20 | 32.72 | -0.13 | -0.02 | 5.42 | |
| 2022 | 28.50 | 33.30** | 35.90 | 26.30 | 31.25 | -0.19 | 0.22 | 5.78 | |
| FU(%) | 2019 | 84.80 | 84.90 | 87.40 | 81.20 | 84.45 | -0.25 | -0.09 | 1.33 |
| 2020 | 84.60 | 84.30 | 87.30 | 80.70 | 84.48 | -0.34 | 0.34 | 1.27 | |
| 2021 | 82.65 | 83.70* | 86.30 | 81.20 | 83.79 | -0.30 | -0.03 | 1.25 | |
| 2022 | 87.80 | 85.10** | 88.00 | 82.20 | 85.53 | 0.15 | -0.28 | 1.18 | |
| MC | 2019 | 5.60 | 4.50** | 5.80 | 4.00 | 5.06 | -0.49 | -0.09 | 6.69 |
| 2020 | 5.30 | 4.80** | 5.80 | 4.00 | 4.92 | -0.14 | -0.72 | 8.25 | |
| 2021 | 5.25 | 4.45** | 5.70 | 3.70 | 4.67 | -0.42 | 0.05 | 7.96 | |
| 2022 | 5.20 | 4.50** | 5.40 | 3.50 | 4.49 | -0.24 | -0.04 | 8.35 | |
| FE(%) | 2019 | 6.80 | 6.80 | 6.90 | 6.60 | 6.77 | -0.07 | -0.22 | 0.88 |
| 2020 | 6.80 | 6.80 | 7.00 | 6.50 | 6.78 | -0.24 | 0.35 | 1.25 | |
| 2021 | 6.80 | 6.70 | 6.90 | 6.70 | 6.78 | 0.81 | -0.61 | 0.67 | |
| 2022 | 6.00 | 6.00 | 6.10 | 6.00 | 6.00 | 17.74 | 4.42 | 0.35 | |
Note: FL, fiber length; FS, fiber strength; FU, fiber uniformity; MC, micronaire; FE, fiber elongation rate; CV, coefficient variation; *, p=0.05; **, p=0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



