
Submitted:
26 June 2024
Posted:
26 June 2024
You are already at the latest version
Abstract
Arrhythmogenic cardiomyopathy (AC) is a hereditary cardiac disorder characterized by the gradual replacement of cardiomyocytes with fibrous and adipose tissue, leading to ventricular wall thinning, chamber dilation, arrhythmias, and sudden cardiac death. Despite advances in treatment, disease management remains challenging. Animal models, particularly mice and zebrafish, have become invaluable tools for understanding AC's pathophysiology and testing potential therapies. Mice models, although useful for scientific research, cannot fully replicate the complexity of the human AC. However, they have provided valuable insights into gene involvement, signalling pathways, and disease progression. Zebrafish offer a promising alternative to mammalian models, despite the phylogenetic distance, due to their economic and genetic advantages. By combining animal models with in vitro studies, researchers can comprehensively understand AC, paving the way for more effective treatments and interventions for patients and improving their quality of life and prognosis.
Keywords:
Arrhythmogenic Cardiomyopathy
; animal models
; mouse
; zebrafish
; heart
Introduction
Arrhythmogenic cardiomyopathy (AC) is an inherited cardiac disorder characterized by fibrous and adipose tissue that gradually replaces cardiomyocytes, beginning at the epicardium and progressing to the endocardium [1,2]. This transmural damage results in myocardial atrophy, with possible wall thinning and aneurysms usually in the right ventricle, and accounts for the clinical presentation with syncope, palpitations, ventricular arrhythmias, and impaired ventricular systolic function. In many cases, it culminates in sudden cardiac death (SCD) in young individuals, including athletes [1,2,3,4]. Notably, physical exercise and competitive sports can trigger life-threatening ventricular arrhythmias, accelerating disease progression and the risk of SCD in AC patients [5]. The prevalence of AC in the general population is approximately 1: 2000-5000 [3,4,6,7], although this frequency might be underestimated due to diagnostic challenges [8]. Males are affected up to three times more frequently than females, a disparity linked to sex-related differences in exercise volume and intensity or the physiological effects of sex hormones [9,10,11,12]. The fibro-fatty substitution of the myocardial tissue, the main hallmark of AC, appears to be linked to the instability and disruption, driven by genetic factors, of desmosomes' structure [1,2,13]. Desmosomes, symmetrical multi-protein structures within intercalated disks (ID), play a critical role in cell adhesion, mechanical anchorage and communication between cardiomyocytes, facilitating force transmission and maintaining electrical continuity [14,15,16,17]. These complexes also contribute to apoptosis, electrochemical coupling, tissue differentiation, and cell-to-cell communication [18]. The desmosome structure consists of cytoplasmic proteins (plakin and armadillo families) and transmembrane sticky glycoproteins (cadherin superfamily). Desmoplakin (DSP), Plakoglobin (JUP) and Plakophilin-2 (PKP2) form the intracellular portion, separated into an outer and inner dense plaque, while the intercellular portion is characterized by the interaction between cadherin proteins, Desmoglein-2 (DSG2) and Desmocolin-2 (DSC2) [19,20] (Figure 2). Ventricular arrhythmias in AC result from pathological changes in myocardial tissue, causing inhomogeneity, altered conduction and abnormal repolarization and depolarization of the action potential [2,13,21]. AC patients often experience periodic inflammatory cells infiltrates in the myocardium, which resemble acute responses like those seen in myocarditis or myocardial infarction and could accelerate the progression of the disease [2,8,22,23,24]. Necrotic and/or apoptotic myocyte death is implicated in the cascade of events leading to fibro-fatty replacement, possibly connected to AC pathogenesis [25,26,27]. In end-stage cardiomyopathy, myocyte loss occurs due to both apoptosis and necrosis, contributing to the progression of cardiac dysfunction and degeneration [25,27,28,29] (Figure 1). Treatments are mainly focused on SCD by using antiarrhythmic drugs, beta-blockers, implantable cardiac defibrillators (ICD), and catheter ablation; heart transplantation is applied either for refractory life-threatening arrhythmias or more rarely for end-stage heart failure [30]. However, these cures are palliative, thus ongoing research aims to develop preventive therapies based on newly identified biomarkers [31], addressing the genetic nature of AC.
Genetics of AC
AC is primarily considered a heterogeneous inherited cardiomyopathy transmitted as an autosomal dominant trait with incomplete penetrance and variable expressivity. Compound/digenic heterozygotes and homozygous recessive forms have also been described as severe forms of the disease, associated with almost complete penetrance and cutaneous abnormalities [32,33,34,35]. About 40% of affected individuals carry mutations in genes encoding desmosomal proteins. However, for more than a half of AC patients, a definitive genetic cause has not been identified, suggesting an oligogenic inheritance or complex genomic rearrangement. Currently, mostly desmosomal genes have been definitively proven to be associated to AC; all other genes either have strong or moderate evidence or are recently published with limited data, identified in few families with AC phenotype [36].
Signalling Pathways Related to AC
The disruption and instability of desmosomes in AC contribute to the dysregulation of signalling pathways involved in myocardial replacement by fibro-adipose tissue, such as Wnt/β-catenin, Hippo/YAP-TAZ and TGFβ [37,38,39]. The Wnt/β-catenin and Hippo/YAP-TAZ pathways, in particular, play a crucial role in cardiac development, controlling proliferation, differentiation, tissue remodelling, and apoptosis [40,41,42,43,44,45,46]. Recent studies have linked the dysregulation of Wnt/β-catenin signalling to AC through desmosome destabilization and subsequent JUP protein release, leading to adipogenesis and fibrogenesis [37,45,47]. Dysregulated activation of Hippo/YAP-TAZ in AC, instead, results in the remodelling and instability of IDs, leading to elevated levels of phosphorylated YAP (p-YAP) and altered gene expression associated with adipogenesis and apoptosis [48,49]. Furthermore, p-YAP has been identified to suppress the Wnt/β-catenin pathway by binding and sequestering β-catenin, reducing transcriptional activities [45]. The TGF-β signalling pathway, upregulated in AC patients, contributes to cardiac repair, remodelling and fibrosis [50,51]. Excessive or prolonged signalling alteration exacerbates unfavourable remodelling, promoting myofibroblast trans-differentiation and facilitating the transition to scar formation [42,52,53].
Search Strategies and Selection Criteria
Search engines in medical literature included PubMed/Medline and Scopus, using the following keyword search strings: “AC” OR “ACM” OR “Arrhythmogenic cardiomyopathy” AND “animal models” OR “Mice” OR “Zebrafish”. We carefully reviewed reference lists of original publications and review articles for missing studies. Duplicates were eliminated. All studies were filtered independently by 3 reviewers (RG, RC and MC), and occasional disagreements were settled by additional authors (NT, KP and CB). Only original peer-reviewed and review articles providing accurate animal models phenotypic characterization were considered. ZFIN database was used to check the availability of not yet characterized zebrafish lines.
Animal Models for AC
Animal models have significantly enhanced our comprehension of human diseases at the cellular and molecular levels, offering insights into pathophysiology, disease progression, and the impact of environmental factors, while also aiding in the development and testing of novel therapeutic approaches. Over the past decade, scientists have established a range of in vivo models incorporating targeted genetic mutations and/or transgenic overexpression of AC-related genes [19]. Notably, instances of spontaneous AC manifestation have been observed in domestic animals, including a case in a primate [54]. Moving to other animals, particularly Boxer dogs and related English bulldogs [55], instances of cardiomyopathy were commonly associated with the disease [56]. Mutations causing AC in Boxer dogs were found triggering significant alterations in the mechanical and electrical connections between cardiomyocytes. AC also affects other dog breeds, including Fila Brasileiro dog [57], Springer Spaniel dog [58], Labrador retriever [59], Weimaraner dog [60], Shetland sheepdog, Dalmatian [61] and Siberian husky [62]. In feline models, clinically significant cardiomyopathies closely resembling the human condition have been discovered, with marked myocardial injury, fibrous and/or fatty replacement, myocarditis, and apoptosis [63,64,65]. While these feline models exhibit all the common clinical characteristics of AC, their entirely spontaneous nature poses limitations for examining the pathophysiological process from the onset of AC. To address this limitation, laboratory animal models, such as mice or zebrafish with genetically induced disease have been developed, overcoming the constraints of spontaneous models. The mouse (Mus musculus) is a widely-utilized animal model for studying human diseases due to the substantial similarities between the human and murine genomes. Approximately 99% of human genes have direct orthologues in mice, and there are notable parallels in their morphology, cell biology and physiology. Over the last 15 years, the mouse model has particularly emerged as an excellent organism for researching human cardiac diseases. The zebrafish (Danio rerio) has emerged as a valuable alternative to mice due to its ease of manipulation and biological relevance. Rodent studies, often marked by cost, time and ethical constraints, can be addressed by leveraging zebrafish, which share orthologs for approximately 71% of human proteins, with 82% linked to human diseases [66,67]. The structural and electrical similarities between zebrafish and human hearts make Danio rerio an effective model for understanding cardiac genes associated with various cardiovascular diseases, potentially facilitating the discovery of new treatments [68,69,70,71]. Despite these advantages, zebrafish, like mice, struggle to exhibit adipogenic substitution in cardiac tissue. Notably, adult zebrafish cardiomyocytes display proliferative and regenerative capacities after injuries, resembling features seen in the early postnatal mammalian ventricle [72]. Mice and zebrafish models with mutations in AC-related genes have been developed in the past years, employing various strategies such as gene knock-down (KD) and knock-out (KO), transgenic overexpression (Tg), or knock-in (KI) of human AC-gene variants. Among these methods, KD/KO-mediated gene inactivation is widely utilized to model loss-of-function mutations.
Gene Definitively Associated to AC
Plakoglobin
The N- and C-terminal domains of JUP are connected by a central region that displays highly conserved armadillo repeats (12 arm repeats), which are typical of this protein family [73]. JUP demonstrates two distinct locations: adherent junctions, where it can be replaced for the closely related armadillo protein β-catenin (CTNNB1), and desmosomes, where it interacts strongly with the cytoplasmic domains of DSG2 and DSC2 cadherins in the outer dense plaque. By interacting with DSP, JUP connects the inner dense plaque to the extracellular portion of the desmosome, forming a bridge [74]. On the Greek island of Naxos in 1986, Protonotarios and colleagues for the first time linked JUP to a recessive form of AC [32]. According to a 2007 study by Asimaki et al., an in-frame insertion of a serine residue p.(Ser39_Lys40insSer) at the N-terminus of the JUP protein has been associated to the dominant form of AC in a small German family [75]. Less than 1% of AC patients carry JUP pathogenic variants [41,75,76] but seems significantly higher in specific geographic regions [77].
Mouse. In 1996, the central role of JUP in the stability of desmosomal structure in both heart and skin tissues was demonstrated in vivo for the first time. Specifically, a Jup homozygous null-mutant mouse model showed significant embryonic lethality due to severe heart defects. The surviving embryos exhibited a reduced number of desmosomes and developed cardiac dysfunction, with larger right ventricle, higher frequency of spontaneous ventricular arrhythmias, and slower right ventricular conduction. No abnormalities in CX43 distribution and localization were observed, and there was no replacement fibrosis or remodelling of the junctions. Heterozygous animals appeared quite healthy, showing no cardiac structural defects [78,79]. However, right ventricular dilation, decreased function, and ventricular arrhythmia was detected. Jup mutated mice mimicked the cutaneous phenotype in humans, with skin blistering and subcorneal acantholysis. Swimming endurance training hastened the onset of ventricular failure and arrhythmias in these animals, while reduction of the ventricular load and training prevented the development of AC [80,81]. Cardiac specific (CS)-Jup ablation in mice resulted in a human AC phenotype condition with increased dilatation, fibrosis, cell death events and SCD, without evidence of myocardial fat [82]. To better understand the role of β-catenin (CTNNB1), a double CS-Jup/Ctnnb1 KO mouse model was generated, demonstrating the crucial role of both proteins in preserving IDs structures and mechano-electrical coupling. Indeed, this model showed clear conduction problems, fibrous tissue replacement and spontaneous ventricular arrhythmia, all leading to SCD. The IDs structures were collapsed with a reduce presence of CXC43 in gap junction plaques, as in AC human hearts [83]. A Tg mouse line with cardiac overexpression of human Naxos-associated truncated JUP protein presented premature mortality, heart dysfunction and fibro-adiposis. This truncated form had less membrane localization and decreased binding with DSP and DSG2 proteins [84]. The normalization of the levels of JUP in both these models alleviated the condition, providing a possible novel therapeutic approach for AC [85]. Epidermal growth factor receptor (EGFR) suppression was assessed under both healthy and pathological conditions as a possible therapeutic approach, surprisingly improving cardiomyocyte cohesion. This revealed a direct interaction between EGFR and DSG2 [86].
Zebrafish. The initial strategy to analyse AC-related genes in zebrafish employed KD techniques. A jup KD using antisense morpholino oligomers induced an immediate cardiac phenotype characterized by oedema, reduced heart size, blood reflux and a twisted tail. This pathological manifestation correlated with dysregulated Wnt/β-catenin signalling, reduced localization of desmosomes and adherens junctions in the IDs [87]. Subsequently, Asimaki and colleagues (2014) established a Tg zebrafish line expressing the human JUP (2057del2) mutation responsible for Naxos syndrome, recapitulating a similar phenotype. The jup-mutant zebrafish model exhibited abnormal cardiac physiology, penetrant cardiomyopathy marked by atrial and ventricular wall thinning, pericardial effusion, and mortality due to arrhythmias or heart failure. A high-throughput pharmacological screen on this mutant line identified SB216763 as a suppressor of the disease phenotype, reducing mortality and preventing heart failure [38]. This study demonstrated the utility of zebrafish models in uncovering novel AC mechanisms and identifying mechanism-based drugs to alleviate AC characteristics.
Desmoplakin
DSP, the most prevalent protein in desmosomes, mediates the interaction between the intermediate filaments (IFs) and the plasma membrane and is necessary for continuous adhesion [88]. DSP is a member of the plakin family and consists of three domains: a central ɑ-helical coiled-coil domain (rod domain) involved in protein dimerization, a globular N-terminal domain important for localization and heterophilic protein-protein interactions including with PKP2, a C-terminal domain made up of three plakin repeat domains (A-B-C), and a glycine-serine-arginine rich domain (GSR) that directly interacts with IFs [89]. Two isoforms of DSP are produced by alternative mRNA splicing, differing in the length of the central rod domain: DSP I, primarily expressed in the heart with 2871 amino acids, and DSP II, with only 2271 amino acids. Recently, DSP II was found in both ventricular and atrial tissues, challenging fundamental assumptions about AC once again [88]. Carvajal syndrome was the first autosomal recessive form of cardiomyopathy found in South America and associated to mutations in DSP, characterized by keratoderma, woolly hair, and AC [33,90]. The first autosomal dominant pathogenic variants found in the DSP gene, located on the short arm of chromosome 6 (6p24.3), were a heterozygous nonsense variant, p.(Gly331Ter), and a splicing site variant, c.939+1G>A [91]. Later, an autosomal dominant heterozygous missense variant p.(Ser299Arg) in the DSP gene was shown to be present in six AC-affected individuals from an Italian family living in the Veneto Region [92].
Mouse. The constitutive homozygous deletion of Dsp in mouse provoked the premature embryonic death even before the evaluation of a possible cardiac phenotype [93]. To address this issue, a cardiac specific (CS-Dsp KO) mouse line was generated by using αMHCcre induction. This model in heterozygosity showed early ultrastructural defects in desmosomes, cardiac dysfunction, cell death, inflammation, ventricular arrhythmias, an excess of adipocytes and fibrosis in the myocardium, SCD and Hippo/YAP-TAZ and Wnt/β-catenin signalling pathways dysregulation. Homozygous CS-Dsp-KO mice died at embryonic stages with only a partially formed heart and no chamber specification [37,94,95] In a cardiac conduction system-specific Dsp ablated mouse model, a clear connection between this protein and the regulatory electrical activity of the heart was observed [96]. Moreover, a new autosomal recessive Dsp mutation called Ruffled (rul) was recently found causing abnormal hair coat along with the classic AC condition, strictly resembling the human Carvajal-Huerta Syndrome [33,97,98]. In 2019, Malireddi et al. proposed a novel form of cell death called PANoptosis, characterized by pyroptosis, apoptosis and necroptosis, but that cannot be explained by any of them on its own [99,100]. Mutant CS Dsp-KO mice presented this new cell death phenomenon as a prominent phenotypic feature, associated with all pathological events described before [101]. In these mice with Dsp deficiency exclusively confined to cardiomyocytes, the severity of the pathological phenotype is intensified by exercise, and cardiomyocytes showed electrical problems linked to CX43 expression in both homozygous and heterozygous background [102,103]. Although not lethal, the cardiac overexpression of a mutant DSP cDNA with a C-terminal mutation p.(Arg2834His) led to impaired ventricular function, dilated ventricles, and elevated apoptosis with fibrosis. Altered connection between DSP and JUP was also observed [104]. Endurance exercise protocols and the related cardiovascular stress accelerate AC pathogenesis in both overexpressing and KI/KO mouse models, revealing electrical and structural abnormalities, as well as perturbed GSK3-β signalling [102,103,105,106].
Zebrafish. Morpholino injections targeting the DSP orthologs dspa and dspb in zebrafish caused desmosome structure destabilization, mirroring human patients’ phenotypes [39]. Altered signalling pathways associated with AC, such as Wnt/β-catenin, TGFβ/Smad3, and Hippo/YAP-TAZ, were identified. The drug SB216763, previously effective in a jup-KI zebrafish model [38], was later tested in the dsp double KD model, confirming its rescuing effects [39]. Recently, our group investigated the relationship between DSP and AC phenotypes in zebrafish, generating a stable dsp-KO zebrafish model. They followed the progression of the disease from the larval stage to adulthood and, for the first time, confirmed that AC can be faithfully mimicked in all its phases in fish as in mice [107]. Due to the duplication of the dsp gene, the study focused on the double heterozygous dspa/dspb combination, being both members expressed in the heart [39], in this way mimicking the heterozygosity commonly found in humans. At larval stage, this model presented an early AC-related phenotype, with cardiac alterations, oedema, and accumulation of inflammatory cells in the cardiac region. The ventricle ejection fraction, contractility and heart rhythm were altered, with a significant detection of arrhythmic events. The histological examination of mutated adult hearts revealed diminished contractile structures, an irregular ventricle shape, myocardial layer thinning, dilated vessels, and the presence of adipocytes in the myocardium. Additionally, "pale", disordered, and delocalized desmosomes were found by Transmission Electron Microscopy (TEM) examination. An intense physical training program accelerated the disease progression by exacerbating the cardiac phenotype at both early and late stages of the disease. Dysregulation of several signalling pathways, among which Wnt/β-catenin, prompted the use of the Wnt/β-catenin agonist SB216763 to mitigate the phenotype. This treatment rescued the pathway expression but also the cardiac abnormalities, stabilizing the heart rhythm and reducing the frequency of the arrhythmic episodes, confirming Wnt/β-catenin as a potential therapeutic target for this disease [107].
Plakophilin-2
PKP occurs in many forms (PKP1-3), with PKP2 being expressed mostly in heart tissue [108]. It is a structural protein interacting with desmosomal cadherins and DSP [109]. The insertion of 44 amino acids between arm repeats 2 and 3 characterized PKP2 isoforms, transcript 2b (881 amino acids) and 2a, exclusively expressed in the heart (837 amino acids) [110,111]. The core region of the protein contains nine arm repeats with a flexible loop in the middle, between the fifth and sixth arm repeats. PKP2 pathogenic variants were first identified in a Dutch AC cohort by Cox and colleagues [76].
Mouse. The homozygous deletion of Pkp2 gene caused embryonic fatal changes in heart morphogenesis, characterised by decreased trabeculation, unorganized cytoskeleton, ruptures of the cardiac walls and blood release into the pericardial cavity. DSP was observed to be separated from the desmosome structure, and instead aggregating in the cytoplasm; no defects were detected in the skin tissue. Therefore, a heterozygous mouse strain was used, although only electrical but not histological signs of the disease were detected [112]. In the hearts of heterozygous CS-Pkp2 KO mice a reduced number of sodium channels and altered IDCs were associated with sodium dysfunction and arrhythmias [113,114]. In addition, Camors et al. observed in a KI mouse model, expressing a human truncating variant of PKP2, p.(Lys405Ter), characterized by the insertion of thymidine in exon 5, which mimics a familial case of AC in humans, a reduced actin expression and a related lack of ventricle contraction [115]. A training protocol can create a pro-arrhythmogenic state in a CS-Pkp2-induced conditional KO [116,117] and, specifically, the Tg overexpression of the human PKP2 truncated mutation p.(Arg735Ter) resulted in an exercise-dependent AC phenotype with a clear right ventricle dysfunction [118]. A cardiac overexpression of human PKP2 variant p.(Ser329Ter), instead, resulted in a phenotype like the one described in the CS-Pkp2 mutant mouse model, with a CX43 delocalization but no fibro-fatty replacement [119].
Zebrafish. Morpholino-mediated KD of PKP2 in zebrafish embryos induced cardiac oedema, incomplete heart looping, reduced heart rate and a twisted tail, akin to the jup-KD model [87,120]. The cardiac desmosome structure was altered, and co-injection of wild type (WT) pkp2 mRNA rescued the morpholino-induced ("morphant") phenotype [120].
Desmoglein-2
Four different DSGs with a highly similar structure (DSG 1-4) are expressed in different tissues and facilitate calcium-dependent cell-cell adhesion [121]. DSG2 is present in all tissues harbouring desmosomes, even though it appears to be the only isoform expressed in cardiac tissue [122,123]. DSG2 is composed of four extracellular cadherin domains: a transmembrane domain, an intracellular anchoring domain, a tiny signal domain known as RUD (repeated-unit domains), whose function is still unknown, and a calcium-binding site that stabilizes each domain. DSG2 was first connected with AC in 2006 [124], with the discovery of 9 missense variants affecting highly conserved amino acids in 8 Italian families presenting a clear pathological phenotype [41,76,124].
Mouse. Dsg2-KO mutant mice display cardiomyocyte loss and fibrotic and hypertrophic cardiac remodelling. Only 30% of Dsg2-KO homozygous mice survived the embryonic stage [125,126,127,128,129]. Moreover, the Pkp2 KI mutant mice exhibited premature death during swimming activities and environmental stresses, displaying myocardial dysfunction, necrosis, inflammation, calcium overload and mitochondrial apoptosis [130,131,132,133]. Additionally, pharmacological inhibition of GSK3β (a Wnt/β-catenin modulator) improved cardiac function [130,134]. In CS-Dsg2 KO the heart function was significantly compromised in response to rising mechanical demands, with prominent morphological abnormalities [135]. Cardiomyocyte mechanical stress sets off an early immune response and tissue remodelling in the heart at a later stage in a CS-KO of Dsg2 [136]. Lin et al. demonstrated lipid accumulation and heart failure in their CS-Dsg2 KO mouse model, caused by impaired mTOR-4EBP1-PPARα-dependent fatty acid β-oxidation. Adjusting PPARα activity alleviated the pathological phenotype, suggesting it as a potential target for AC [137]. Tg mice with cardiac overexpression of p.(Asn271Ser) mutation were generated to investigate its role in the disease, directly paralleling the human AC mutation p.(Asn266Ser). Notably, a reduction in desmosomal structures with “pale” and non-compact IDs was associated with SCD events. Additional phenotypes included ventricular dilatation, aneurysms, conduction slowness, spontaneous ventricular arrhythmias, myocyte necrosis, inflammation, and fibrous tissue generation [27,124]. A reduced action potential (AP) velocity was also observed, demonstrating a direct interaction between DSG2 and the sodium channel protein Na(V)1.5 [138]. The results mirrored those observed in Dsg2-KO mice [126,128,135]. A Dsg2-KI mouse model, eliminating the tryptophan exchange (W2A) crucial for DSG2 interactions, displayed a severe cardiac profile with arrhythmia, cardiac fibrosis and decreased systolic function. Altered integrin-αVβ6 and TGFβ signalling, along with decreased fibrosis and pro-fibrotic marker expression upon integrin-αVβ6 blockade, were observed [139]. A treatment with extracellular vesicles (EVs) in a homozygous KI mutant Dsg2 mouse decreased cardiac inflammation and arrhythmias episodes, with a global enhancement of cardiac function [24,130,131].
Desmocollin-2
There are three known isoforms of DSC (DSC 1- 3), although only DSC2 is expressed throughout the body and identified in cardiac tissue [121]. The protein structure consists of four extracellular domains that are extremely conserved, the first of which is called CAR and is in charge of the heterophilic contact with the DSGs in neighbouring cells. A transmembrane domain connects them to an intracellular anchor domain at the N-terminus, which binds the intracellular portion of the desmosome in the inner dense plaque, making up the structure of DSC2. Four probands affected by AC were discovered to carry heterozygous DSC2 frameshift variants [140]. Since then, less than fifty DSC2 nucleotide variants, which account for 1–3% of AC cases, have been identified [41,76,140,141].
Mouse. KI mice p.(Gly790del) did not show any sign of AC [142], whereas cardiac overexpression of WT DSC2 in mice was shown to induce necrosis, acute inflammation and cardiac fibrotic remodelling leading to cardiomyopathy [143].
Zebrafish. Heuser et al. utilized morpholino oligomer injections to KD dsc2 expression in zebrafish, resulting in cardiac oedema, decreased fractional shortening and altered desmosomal structure. The model exhibited reduced desmosomal plaque area, loss of extracellular electron-dense midlines and associated myocardial contractility abnormalities. This underscores the essential role of the dsc2 gene in normal myocardium development in both zebrafish and humans [141].
Transmembrane Protein-43
Transmembrane Protein-43 (TMEM43) is essential to the inner nuclear membrane. It is composed of a broad hydrophilic domain that is exposed to the endoplasmic reticulum (ER) and four transmembrane domains (TMD). It has been demonstrated that TMEM43 interacts with emerin and lamins, two elements of the linker of nucleoskeleton and cytoskeleton (LINC) complex, and is therefore implicated in the organization of the nuclear membrane [144,145,146]. TMEM43 was added to the pool of genes strongly correlated with AC in the last years [36]. The TMEM43 founder mutation p.(Ser358Leu) was discovered in a well-characterized AC community in Newfoundland and later confirmed in populations in the UK, Denmark, Germany, and Spain. The TMEM43 founder variant causes a severe sex-influenced fatal AC that results in left ventricular dilatation, fibro-fatty replacement, heart failure and SCD [147,148,149,150].
Mouse. A Tmem43 CS-KO mouse line showed an age-dependent phenotype characterized by an increased mortality, cardiac dilatation and dysfunction, myocardial fibrosis, adipogenesis, and apoptosis [151]. In contrast, the KI mouse model carrying the p.(Ser358Leu) presented with gender-specific cardiac dysfunction and dysregulation of signalling pathways, such as Wnt/β-catenin and PPARG signalling, as in humans. The Wnt/β-catenin alteration was validated by translocation of JUP into the nuclei of mutant cardiomyocytes. Although the systolic dysfunction appeared earlier in homozygous mutant mice, stress test intolerance was observed in both genetic combinations, accompanied by arrhythmias and fibro-fatty infiltration. In the cardiac tissues of these mice, NF-κB activation was present, boosting downstream signalling and the expression of pro-fibrotic TGFβ1 [152,153,154]. Moreover, Padrón-Barthe and colleagues demonstrated that administering a GSK3β inhibitor to Tg mice overexpressing TMEM43 in either its WT or p.(Ser358Leu) mutant form, enhanced heart function and activity [155]. The same model treated with Enalapril, an ACE inhibitor, showed reduced fibrosis, improved ECG and echocardiographic parameters, and increased survival [155,156].
Zebrafish. In a tmem43-KO zebrafish model, significant ventricular enlargement was observed only in adulthood, with no overt cardiac abnormalities or contractile dysfunction during early embryogenesis stages. To further explore this aspect, Tg CS zebrafish lines overexpressing human WT TMEM43 and two genetic variants, p.(Ser358Leu) and p.(Pro111Leu), were created. Heterozygosity in this line resulted in mTOR pathway activation, ribosome biogenesis, and enlarged hearts with cardiomyocyte hypertrophy, cardiac morphological defects at juvenile stages, and ultrastructural changes within the myocardium, accompanied by dysregulated gene expression profiles in adulthood [146].
Genes Strongly or Moderately Associated to AC
Phospholamban
The PLN gene codes for the 52-amino acid protein Phospholamban, located in the sarcoplasmic reticulum (SR) membrane. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), which moves calcium from the cytosol into the SR, is primarily regulated by PLN, which is a key player in cardiomyocyte calcium management. In its dephosphorylated form, PLN inhibits calcium absorption by reducing SERCA's affinity for Ca2+. When PLN is phosphorylated at serine 16 by protein kinase A (PKA) or at threonine 17 by Ca2+/calmodulin-dependent protein kinase II (CaMKII), PLN-mediated inhibition of SERCA is relieved, leading to an increase in SERCA activity and calcium uptake. Heart contraction and relaxation depend on the PLN-SERCA interaction, which is controlled by the β-adrenergic receptor pathway to adjust cardiac output to physiological demands [157,158]. A Greek family affected by hereditary heart failure was found to be carrier of the c.40_42delAGA variant, a heterozygous deletion of arginine 14 p.(Arg14del) [159]. Remarkably, this pathogenic variant was identified in about 14% of Dutch patients with AC and DCM, being classified as a founder mutation in the Netherlands [160,161,162,163,164,165,166].
Mouse. Mice harbouring the human mutation p.(Arg14del) exhibited increased arrhythmia, ventricular action potential prolongation, unresponsive to β-adrenergic stimulation, and electric remodelling with affected calcium homeostasis and suppression of Sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activity [158,161,167,168,169]. Specifically, the mutant PLN protein is localised at the plasma membrane and modifies the activity of the Na/K ATPase (NKA), failing to co-localize with SERCA2 [159,161]. Interestingly, in Tg mice, the disruption of the human allele by AAV9-CRISPR/Cas9 approach strongly improved the cardiac function, providing preclinical evidence for therapeutically suppressing the AC phenotype in these AC patients [170].
Zebrafish. Adult zebrafish carrying the p.(Arg14del) variant manifested tissue remodelling with sub-epicardial inflammation, fibrosis and adipogenic substitution. Echocardiography revealed contractile variations correlating with action potential duration alternance at the cellular level. Calcium level alterations were detected at both embryonic and adult stages. Treatment with Istaroxime, a calcium regulator, improved cardiac relaxation, restored cellular action potential duration and mitigated calcium dysregulation [171].
Desmin
Desmin is connected to many cellular structures such as desmosomes, Z-bands, mitochondria, and nuclei, and is located in the cytoskeleton of cardiomyocytes. Its role is connected to preserving the structural integrity of cardiomyocytes [172]. Mutations in this protein can disrupt subcellular organelle organization, lead to the development of inclusion bodies, weaken the DES cytoskeleton, and ultimately cause myofibril disintegration. Heterozygous missense or in-frame deletion mutations account for most pathogenic DES variants, causing abnormal filament assembly and aggregation [173,174]. A case report by Otten et al. in 2010 described two Dutch families with characteristics resembling AC. These patients experienced muscular weakness and cardiac arrhythmias both in early childhood and later in life [175].
Mouse. Both KO and KI p.(Arg349Pro) mice developed a cardiac phenotype with fibrosis, arrhythmias, protein aggregates, mitochondrial and conductions defects, partially mimicking AC [176,177,178,179]. In the homozygous Des-KO mouse model, coagulation and complement system activation interacted, exacerbating myocardial injury and impairing sinoatrial pacemaker function. This alteration was mitigated by using the thrombin inhibitor Lepirudin [180,181,182,183].
Zebrafish. Morpholino antisense oligomers were used to KD both orthologs desma and desmb in zebrafish. These morphant lines developed cardiac oedema, arrhythmia, and dysfunction, resulting in decreased viability [184]. Furthermore, Ramspacher and colleagues characterized two KI zebrafish models (ct122Gta and ct122RGt) in 2015, revealing that the loss of Desmin function promotes skeletal muscle defects, alters heart biomechanics and affects contraction [185].
Non-Desmosomal Genes with Disputed, Limited or no- Curated Evidence of Disease Association
Conclusions and Limitations
Progressive fibro-fatty substitution of the myocardium, leading to life-threatening electrical instability and eventually ventricular dysfunction, are characteristic features of AC. Despite ongoing research using various techniques, fully replicating AC's clinic-pathological characteristics in laboratory settings remains challenging. Developing more sophisticated AC models is crucial for comprehensively understanding its pathophysiology, as such human clinical evidence should drive outcomes and derivable searched in these models. In vitro cell models, while valuable, have limitations related to origin, genetic factors, maturity, cell type, and environmental interactions. In vivo animal models play a fundamental role in validating and delving deeper into observed in vitro results. Adult cardiomyocytes directly derived from AC patients, while close to human physiology, have limited accessibility and maintenance challenges. Various alternative models have been explored, but their main drawback is the absence of the complex microenvironment found in the heart tissue, including interactions among different cell types like fibroblasts, immune cells, vascular cells, cardiac progenitors and cardiac myocytes. These interactions are essential for the mechanical and electrical properties of cardiac tissue, contributing to tissue morphogenesis, differentiation and homeostasis. Moreover, addressing the progressive nature of AC is crucial when discussing its limitations in cell models. Over the past two decades, efforts have been made to overcome these limitations by generating and characterizing numerous in vivo models. Various genetic approaches have been employed to investigate gene involvement, test drug molecules and study disease progression in animals, particularly mice and zebrafish, which are commonly used for cardiac research. Mouse models, due to their cardiac similarity to humans, have been immensely valuable in studying all aspects of AC, including gene involvement, specific human variants, signalling pathway interactions and disease progression. While whole-body or cardiac-specific KO mouse models have been useful, they often mimic conditions resembling non-sense mutations and truncating protein formation. However, homozygous and whole-body application of this approach can lead to severe phenotypes and foetal mortality, and heterozygous models may not consistently exhibit a phenotype. The use of Tg-CS models, which overexpress human mutated proteins in cardiac tissue, has been employed to more faithfully reproduce phenotypes related to human mutations. However, this approach does not always mirror the effects observed in human conditions, especially due to the persistence of the functional ortholog in the transgenic animal. Studying human missense mutations in KI animal models, where the mutation is inserted into the endogenous gene, proves to be more effective, functioning through its own promoter. Notably, despite similarities, the human heart differs from mouse heart, and characteristic features like adipogenic substitution found in human ventricles are rarely observed in mouse models [113,126,228]. Conversely, the potential of the zebrafish model is evident, considering its economic, spatial, ethical, and genetic advantages, even though the development of zebrafish models for AC study was initially delayed, resulting in fewer stable mutant lines. Despite the challenge of duplicate gene copies, zebrafish's heart structure and protein organization allow the reproduction of a clear AC phenotype in generated models. While AC research in zebrafish is still in its early stages, with most models being transient KD, the few stable models so far established have validated the reproducibility of the disease at both molecular and morphological levels. Additionally, drug testing is simpler, faster and more extensive due to the rapid development and the abundance of embryos obtained from each mating. These promising results suggest that zebrafish, in conjunction with mouse and in vitro models, could contribute significantly to a better understanding of AC, aiming ultimately to find a cure or alleviate severe phenotypic conditions observed in human patients.
Acknowledgments
GR is a Post-Doc fellow supported by the Italian Ministry of University and Research (grant PRIN 2022WZCXRZ to SV). RBC is a student of the UniPD Biosciences PhD School (CARIPARO fellowship). CB, GT, KP were supported by the ARCA association, the Regional Registry for Cardio-cerebro-vascular Pathology, Veneto Region, Venice, Italy; Ministry of Health Grants RF-2019-12370183, Rome, Italy; Veneto Region Target Research, Venice 933/2015; PRIN Ministry of Education, University and Research 20173ZWACS and 20229FE439 Rome, Italy; University Research Grants BIRD212725, Padua, Italy; PNRR Next-Generation EU PNRR-MR1-2022-12376614, Rome, Italy. The University-Hospital of Padua is a member of the European Reference Network for rare, low-prevalence and complex diseases of the heart (ERN GUARD-Heart). NT was supported by the Italian Telethon Foundation (grant GGP19287) and the Italian Ministry of University and Research (grant PNRR M4C2 CN00000041).
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N Engl J Med 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic Right Ventricular Cardiomyopathy. Dysplasia, Dystrophy, or Myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of Arrhythmogenic Cardiomyopathy. Nat Rev Cardiol 2011, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Cipriani, A.; Bariani, R.; Pilichou, K.; Corrado, D.; Bauce, B. Role of Exercise as a Modulating Factor in Arrhythmogenic Cardiomyopathy. Curr Cardiol Rep 2021, 23, 57. [Google Scholar] [CrossRef] [PubMed]
- Nava, A.; Bauce, B.; Basso, C.; Muriago, M.; Rampazzo, A.; Villanova, C.; Daliento, L.; Buja, G.; Corrado, D.; Danieli, G.A.; et al. Clinical Profile and Long-Term Follow-up of 37 Families with Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol 2000, 36, 2226–2233. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: Clinical Impact of Molecular Genetic Studies. Circulation 2006, 113, 1634–1637. [Google Scholar] [CrossRef]
- Thiene, G.; Corrado, D.; Basso, C. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Orphanet J Rare Dis 2007, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise Increases Age-Related Penetrance and Arrhythmic Risk in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy-Associated Desmosomal Mutation Carriers. J Am Coll Cardiol 2013, 62, 1290–1297. [Google Scholar] [CrossRef]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and Digenic Heterozygosity Predicts Lifetime Arrhythmic Outcome and Sudden Cardiac Death in Desmosomal Gene-Related Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Genet 2013, 6, 533–542. [Google Scholar] [CrossRef]
- Choudhary, N.; Tompkins, C.; Polonsky, B.; McNitt, S.; Calkins, H.; Mark Estes, N.A.; Krahn, A.D.; Link, M.S.; Marcus, F.I.; Towbin, J.A.; et al. Clinical Presentation and Outcomes by Sex in Arrhythmogenic Right Ventricular Cardiomyopathy: Findings from the North American ARVC Registry. J Cardiovasc Electrophysiol 2016, 27, 555–562. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex Hormones Affect Outcome in Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: From a Stem Cell Derived Cardiomyocyte-Based Model to Clinical Biomarkers of Disease Outcome. Eur Heart J 2017, 38, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right Ventricular Dysplasia: A Report of 24 Adult Cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- North, A.J.; Bardsley, W.G.; Hyam, J.; Bornslaeger, E.A.; Cordingley, H.C.; Trinnaman, B.; Hatzfeld, M.; Green, K.J.; Magee, A.I.; Garrod, D.R. Molecular Map of the Desmosomal Plaque. J Cell Sci 1999, 112 ( Pt 23) Pt 23, 4325–4336. [Google Scholar] [CrossRef]
- Awad, M.M.; Calkins, H.; Judge, D.P. Mechanisms of Disease: Molecular Genetics of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Nat Clin Pract Cardiovasc Med 2008, 5, 258–267. [Google Scholar] [CrossRef]
- Estigoy, C.B.; Pontén, F.; Odeberg, J.; Herbert, B.; Guilhaus, M.; Charleston, M.; Ho, J.W.K.; Cameron, D.; Dos Remedios, C.G. Intercalated Discs: Multiple Proteins Perform Multiple Functions in Non-Failing and Failing Human Hearts. Biophys Rev 2009, 1, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Qiu, Y.; Zhang, H.M.; Yang, D. Intercalated Discs: Cellular Adhesion and Signaling in Heart Health and Diseases. Heart Fail Rev 2019, 24, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Merritt, A.J.; Berika, M.Y.; Zhai, W.; Kirk, S.E.; Ji, B.; Hardman, M.J.; Garrod, D.R. Suprabasal Desmoglein 3 Expression in the Epidermis of Transgenic Mice Results in Hyperproliferation and Abnormal Differentiation. Mol Cell Biol 2002, 22, 5846–5858. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Bezzina, C.R.; Thiene, G.; Basso, C. Arrhythmogenic Cardiomyopathy: Transgenic Animal Models Provide Novel Insights Into Disease Pathobiology. Circ Cardiovasc Genet 2011, 4, 318–326. [Google Scholar] [CrossRef]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Perazzolo Marra, M.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic Cardiomyopathy. Orphanet J Rare Dis 2016, 11, 33. [Google Scholar] [CrossRef]
- Nava, A.; Canciani, B.; Buja, G.; Martini, B.; Daliento, L.; Scognamiglio, R.; Thiene, G. Electrovectorcardiographic Study of Negative T Waves on Precordial Leads in Arrhythmogenic Right Ventricular Dysplasia: Relationship with Right Ventricular Volumes. J Electrocardiol 1988, 21, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of Clinicopathologic Manifestations of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Multicenter Study. J Am Coll Cardiol 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Lubos, N.; van der Gaag, S.; Gerçek, M.; Kant, S.; Leube, R.E.; Krusche, C.A. Inflammation Shapes Pathogenesis of Murine Arrhythmogenic Cardiomyopathy. Basic Res Cardiol 2020, 115, 42. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-N.; Ibrahim, A.; Marbán, E.; Cingolani, E. Pathogenesis of Arrhythmogenic Cardiomyopathy: Role of Inflammation. Basic Res Cardiol 2021, 116, 39. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Tedgui, A.; Fontaliran, F.; Frank, R.; Durigon, M.; Fontaine, G. Evidence of Apoptosis in Arrhythmogenic Right Ventricular Dysplasia. N Engl J Med 1996, 335, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.; Calabrese, F.; Thiene, G.; Angelini, A.; Basso, C.; Nava, A.; Rossi, L. In Vivo Evidence of Apoptosis in Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Pathol 1998, 152, 479–484. [Google Scholar]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.B.; de Bakker, J.M.T.; et al. Myocyte Necrosis Underlies Progressive Myocardial Dystrophy in Mouse Dsg2-Related Arrhythmogenic Right Ventricular Cardiomyopathy. J Exp Med 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Virmani, R.; DiSalvo, T.G.; Kolodgie, F.D.; Hajjar, R.J.; Schmidt, U.; Semigran, M.J.; Dec, G.W.; Khaw, B.A. Apoptosis in Myocytes in End-Stage Heart Failure. N Engl J Med 1996, 335, 1182–1189. [Google Scholar] [CrossRef]
- Rayment, N.B.; Haven, A.J.; Madden, B.; Murday, A.; Trickey, R.; Shipley, M.; Davies, M.J.; Katz, D.R. Myocyte Loss in Chronic Heart Failure. J Pathol 1999, 188, 213–219. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Bueno Marinas, M.; Celeghin, R.; Cason, M.; Bariani, R.; Frigo, A.C.; Jager, J.; Syrris, P.; Elliott, P.M.; Bauce, B.; Thiene, G.; et al. A microRNA Expression Profile as Non-Invasive Biomarker in a Large Arrhythmogenic Cardiomyopathy Cohort. IJMS 2020, 21, 1536. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac Abnormalities in Familial Palmoplantar Keratosis. Br Heart J 1986, 56, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Huerta, L. Epidermolytic Palmoplantar Keratoderma with Woolly Hair and Dilated Cardiomyopathy. J Am Acad Dermatol 1998, 39, 418–421. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a Deletion in Plakoglobin in Arrhythmogenic Right Ventricular Cardiomyopathy with Palmoplantar Keratoderma and Woolly Hair (Naxos Disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive Mutation in Desmoplakin Disrupts Desmoplakin-Intermediate Filament Interactions and Causes Dilated Cardiomyopathy, Woolly Hair and Keratoderma. Hum Mol Genet 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ Genom Precis Med 2021, 14, e003273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of Canonical Wnt/Beta-Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J Clin Invest 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Karin Arndt, A.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci Transl Med 2014, 6, 240ra74. [Google Scholar] [CrossRef] [PubMed]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of Cardiac Wnt/β-Catenin Signalling in Desmoplakin-Deficient AC8 Zebrafish Models Is Rescuable by Genetic and Pharmacological Intervention. Cardiovasc Res 2018, 114, 1082–1097. [Google Scholar] [CrossRef]
- Brade, T.; Männer, J.; Kühl, M. The Role of Wnt Signalling in Cardiac Development and Tissue Remodelling in the Mature Heart. Cardiovasc Res 2006, 72, 198–209. [Google Scholar] [CrossRef]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.-C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal Gene Analysis in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Spectrum of Mutations and Clinical Impact in Practice. Europace 2010, 12, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming Growth Factor (TGF)-β Signaling in Cardiac Remodeling. J Mol Cell Cardiol 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.-L. The Hippo Pathway in Heart Development, Regeneration, and Diseases. Circ Res 2015, 116, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, A.; Calore, M.; Poloni, G.; De Windt, L.J.; Braghetta, P.; Rampazzo, A. Wnt/β-Catenin Pathway in Arrhythmogenic Cardiomyopathy. Oncotarget 2017, 8, 60640–60655. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front Cell Dev Biol 2019, 7, 211. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; De Bortoli, M.; Poloni, G.; Rampazzo, A. Arrhythmogenic Cardiomyopathy: A Disease of Intercalated Discs. Cell Tissue Res 2015, 360, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The Hippo Pathway Is Activated and Is a Causal Mechanism for Adipogenesis in Arrhythmogenic Cardiomyopathy. Circ Res 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat Rev Cardiol 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb Perspect Biol 2016, 8, a022053. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Maione, A.S.; Stadiotti, I.; Pilato, C.A.; Perrucci, G.L.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-Β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int J Mol Sci 2021, 22, 2673. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming Growth Factor-β in Myocardial Disease. Nat Rev Cardiol 2022, 19, 435–455. [Google Scholar] [CrossRef]
- Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic RV Cardiomyopathy - Dysplasia: Recent Advances; Springer-Verlag: Milan, 2007; ISBN 978-88-470-0489-4. [Google Scholar]
- Holdt, S.L.; Peckens, N.K.; Rosenthal, S.; Cober, R. Arrhythmogenic Right Ventricular Cardiomyopathy in Bulldogs: Evaluation of Clinical and Histopathologic Features, Progression, and Outcome in 71 Dogs (2004-2016). J Vet Cardiol 2022, 40, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Palermo, V.; Stafford Johnson, M.J.; Sala, E.; Brambilla, P.G.; Martin, M.W.S. Cardiomyopathy in Boxer Dogs: A Retrospective Study of the Clinical Presentation, Diagnostic Findings and Survival. Journal of Veterinary Cardiology 2011, 13, 45–55. [Google Scholar] [CrossRef]
- Belerenian, G.; Donati, P.A.; Rodríguez, C.D.; Castillo, V.; Guevara, J.M.; Olivares, R.W.I. Left-Dominant Arrhythmogenic Cardiomyopathy in a Fila Brasileiro Dog. Open Vet J 2022, 12, 495–501. [Google Scholar] [CrossRef]
- Bertola, L.; Cappelleri, A.; Tomba, R.M.; Dotti, E.; Caniatti, M.; Dall’Ara, P.; Recordati, C. Vaccine-Associated Anaphylactic Shock in a Springer Spaniel Dog with Arrhythmogenic Right Ventricular Cardiomyopathy. J Comp Pathol 2022, 194, 34–38. [Google Scholar] [CrossRef]
- Möhr, A.J.; Kirberger, R.M. Arrhythmogenic Right Ventricular Cardiomyopathy in a Dog. J S Afr Vet Assoc 2000, 71, 125–130. [Google Scholar] [CrossRef]
- Eason, B.D.; Leach, S.B.; Kuroki, K. Arrhythmogenic Right Ventricular Cardiomyopathy in a Weimaraner. Can Vet J 2015, 56, 1035–1039. [Google Scholar]
- Nakao, S.; Hirakawa, A.; Yamamoto, S.; Kobayashi, M.; Machida, N. Pathological Features of Arrhythmogenic Right Ventricular Cardiomyopathy in Middle-Aged Dogs. J Vet Med Sci 2011, 73, 1031–1036. [Google Scholar] [CrossRef]
- Fernández del Palacio, M.J.; Bernal, L.J.; Bayón, A.; Bernabé, A.; Montes de Oca, R.; Seva, J. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in a Siberian Husky. J Small Anim Pract 2001, 42, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.R.; Maron, B.J.; Basso, C.; Liu, S.-K.; Thiene, G. Spontaneously Occurring Arrhythmogenic Right Ventricular Cardiomyopathy in the Domestic Cat: A New Animal Model Similar to the Human Disease. Circulation 2000, 102, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.M.; Battersby, I.A.; Faena, M.; Fews, D.; Darke, P.G.G.; Ferasin, L. Arrhythmogenic Right Ventricular Cardiomyopathy in Two Cats. J Small Anim Pract 2005, 46, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ciaramella, P.; Basso, C.; Di Loria, A.; Piantedosi, D. Arrhythmogenic Right Ventricular Cardiomyopathy Associated with Severe Left Ventricular Involvement in a Cat. J Vet Cardiol 2009, 11, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Dahm, R.; Geisler, R. Learning from Small Fry: The Zebrafish as a Genetic Model Organism for Aquaculture Fish Species. Mar Biotechnol (NY) 2006, 8, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Genge, C.E.; Lin, E.; Lee, L.; Sheng, X.; Rayani, K.; Gunawan, M.; Stevens, C.M.; Li, A.Y.; Talab, S.S.; Claydon, T.W.; et al. The Zebrafish Heart as a Model of Mammalian Cardiac Function. Rev Physiol Biochem Pharmacol 2016, 171, 99–136. [Google Scholar] [CrossRef]
- Vornanen, M.; Hassinen, M. Zebrafish Heart as a Model for Human Cardiac Electrophysiology. Channels (Austin) 2016, 10, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Giardoglou, P.; Beis, D. On Zebrafish Disease Models and Matters of the Heart. Biomedicines 2019, 7, E15. [Google Scholar] [CrossRef] [PubMed]
- Bowley, G.; Kugler, E.; Wilkinson, R.; Lawrie, A.; van Eeden, F.; Chico, T.J.A.; Evans, P.C.; Noël, E.S.; Serbanovic-Canic, J. Zebrafish as a Tractable Model of Human Cardiovascular Disease. Br J Pharmacol 2022, 179, 900–917. [Google Scholar] [CrossRef]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart Regeneration in Zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Garrod, D.; Chidgey, M. Desmosome Structure, Composition and Function. Biochim Biophys Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Chitaev, N.A.; Leube, R.E.; Troyanovsky, R.B.; Eshkind, L.G.; Franke, W.W.; Troyanovsky, S.M. The Binding of Plakoglobin to Desmosomal Cadherins: Patterns of Binding Sites and Topogenic Potential. J Cell Biol 1996, 133, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Hum Genet 2007, 81, 964–973. [Google Scholar] [CrossRef]
- Cox, M.G.P.J.; van der Zwaag, P.A.; van der Werf, C.; van der Smagt, J.J.; Noorman, M.; Bhuiyan, Z.A.; Wiesfeld, A.C.P.; Volders, P.G.A.; van Langen, I.M.; Atsma, D.E.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Pathogenic Desmosome Mutations in Index-Patients Predict Outcome of Family Screening: Dutch Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Genotype-Phenotype Follow-up Study. Circulation 2011, 123, 2690–2700. [Google Scholar] [CrossRef]
- Celeghin, R.; Pilichou, K. The Complex Molecular Genetics of Arrhythmogenic Cardiomyopathy. Int J Cardiol 2019, 284, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Bierkamp, C.; Mclaughlin, K.J.; Schwarz, H.; Huber, O.; Kemler, R. Embryonic Heart and Skin Defects in Mice Lacking Plakoglobin. Dev Biol 1996, 180, 780–785. [Google Scholar] [CrossRef]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Günthert, U.; Franke, W.W.; et al. Targeted Mutation of Plakoglobin in Mice Reveals Essential Functions of Desmosomes in the Embryonic Heart. The Journal of cell biology 1996, 135, 215–225. [Google Scholar] [CrossRef]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schäfers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.-H.; Baba, H.A.; et al. Age- and Training-Dependent Development of Arrhythmogenic Right Ventricular Cardiomyopathy in Heterozygous Plakoglobin-Deficient Mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Hoogendijk, M.G.; Scicluna, B.P.; van Amersfoorth, S.C.M.; Fortmueller, L.; Wolf, S.; Laakmann, S.; Kreienkamp, N.; Piccini, I.; Breithardt, G.; et al. Load-Reducing Therapy Prevents Development of Arrhythmogenic Right Ventricular Cardiomyopathy in Plakoglobin-Deficient Mice. J Am Coll Cardiol 2011, 57, 740–750. [Google Scholar] [CrossRef]
- Li, J.; Swope, D.; Raess, N.; Cheng, L.; Muller, E.J.; Radice, G.L. Cardiac Tissue-Restricted Deletion of Plakoglobin Results in Progressive Cardiomyopathy and Activation of {beta}-Catenin Signaling. Mol Cell Biol 2011, 31, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.L. Loss of Cadherin-Binding Proteins β-Catenin and Plakoglobin in the Heart Leads to Gap Junction Remodeling and Arrhythmogenesis. Mol Cell Biol 2012, 32, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear Plakoglobin Is Essential for Differentiation of Cardiac Progenitor Cells to Adipocytes in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Res 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Stroud, M.J.; Zhang, J.; Fang, X.; Ouyang, K.; Kimura, K.; Mu, Y.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; et al. Normalization of Naxos Plakoglobin Levels Restores Cardiac Function in Mice. J Clin Invest 2015, 125, 1708–1712. [Google Scholar] [CrossRef] [PubMed]
- Shoykhet, M.; Dervishi, O.; Menauer, P.; Hiermaier, M.; Moztarzadeh, S.; Osterloh, C.; Ludwig, R.J.; Williams, T.; Gerull, B.; Kääb, S.; et al. EGFR Inhibition Leads to Enhanced Desmosome Assembly and Cardiomyocyte Cohesion via ROCK Activation. JCI Insight 2023, 8, e163763. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Moriarty, M.A.; Byrnes, L.; Grealy, M. Plakoglobin Has Both Structural and Signalling Roles in Zebrafish Development. Dev Biol 2009, 327, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-Y.; Cheng, L.-T.; Wang, Z.-F.; Wu, Y.-Q. Desmoplakin and Clinical Manifestations of Desmoplakin Cardiomyopathy. Chin Med J (Engl) 2021, 134, 1771–1779. [Google Scholar] [CrossRef]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The Desmosome. Cold Spring Harb Perspect Biol 2009, 1, a002543. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Carvajal-Huerta, L.; Ruiz-Cabezas, J.C.; Thiene, G.; Saffitz, J.E. Structural and Molecular Pathology of the Heart in Carvajal Syndrome. Cardiovasc Pathol 2004, 13, 26–32. [Google Scholar] [CrossRef]
- Armstrong, D.K.; McKenna, K.E.; Purkis, P.E.; Green, K.J.; Eady, R.A.; Leigh, I.M.; Hughes, A.E. Haploinsufficiency of Desmoplakin Causes a Striate Subtype of Palmoplantar Keratoderma. Hum Mol Genet 1999, 8, 143–148. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in Human Desmoplakin Domain Binding to Plakoglobin Causes a Dominant Form of Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Hum Genet 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is Required Early in Development for Assembly of Desmosomes and Cytoskeletal Linkage. J Cell Biol 1998, 143, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise Restores Dysregulated Gene Expression in a Mouse Model of Arrhythmogenic Cardiomyopathy. Cardiovasc Res 2020, 116, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Olcum, M.; Fan, S.; Rouhi, L.; Cheedipudi, S.; Cathcart, B.; Jeong, H.-H.; Zhao, Z.; Gurha, P.; Marian, A.J. Genetic Inactivation of β-Catenin Is Salubrious, Whereas Its Activation Is Deleterious in Desmoplakin Cardiomyopathy. Cardiovasc Res 2023, 119, 2712–2728. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, V.; Liang, Y.; Wright, A.T.; Lyon, R.C.; Pfeiffer, E.; Song, M.Y.; Gu, Y.; Dalton, N.D.; Scheinman, M.; Peterson, K.L.; et al. Desmosomal Junctions Are Necessary for Adult Sinus Node Function. Cardiovasc Res 2016, 111, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A. Naxos Disease and Carvajal Syndrome: Cardiocutaneous Disorders That Highlight the Pathogenesis and Broaden the Spectrum of Arrhythmogenic Right Ventricular Cardiomyopathy. Cardiovasc Pathol 2004, 13, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Herbert Pratt, C.; Potter, C.S.; Fairfield, H.; Reinholdt, L.G.; Bergstrom, D.E.; Harris, B.S.; Greenstein, I.; Dadras, S.S.; Liang, B.T.; Schofield, P.N.; et al. Dsp Rul: A Spontaneous Mouse Mutation in Desmoplakin as a Model of Carvajal-Huerta Syndrome. Exp Mol Pathol 2015, 98, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-Optosis). Front Cell Infect Microbiol 2019, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Cao, P.; Wang, Y.; Zhang, Q.; Zhang, D.; Wang, Y.; Wang, L.; Gong, Z. PANoptosis: A Cell Death Characterized by Pyroptosis, Apoptosis, and Necroptosis. J Inflamm Res 2023, 16, 1523–1532. [Google Scholar] [CrossRef]
- Olcum, M.; Rouhi, L.; Fan, S.; Gonzales, M.M.; Jeong, H.-H.; Zhao, Z.; Gurha, P.; Marian, A.J. PANoptosis Is a Prominent Feature of Desmoplakin Cardiomyopathy. J Cardiovasc Aging 2023, 3, 3. [Google Scholar] [CrossRef]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological Abnormalities Precede Overt Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy Due to Mutations in Desmoplakin-A Combined Murine and Human Study. Eur Heart J 2012, 33, 1942–1953. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin Defects Underlie Arrhythmogenic Right Ventricular Cardiomyopathy in a Novel Mouse Model. Hum Mol Genet 2014, 23, 1134–1150. [Google Scholar] [CrossRef]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal Dysfunction Due to Mutations in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ Res 2006, 99, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Martherus, R.; Jain, R.; Takagi, K.; Mendsaikhan, U.; Turdi, S.; Osinska, H.; James, J.F.; Kramer, K.; Purevjav, E.; Towbin, J.A. Accelerated Cardiac Remodeling in Desmoplakin Transgenic Mice in Response to Endurance Exercise Is Associated with Perturbed Wnt/β-Catenin Signaling. Am J Physiol Heart Circ Physiol 2016, 310, H174–187. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.L.; Manring, H.R.; Wallace, M.J.; Argall, A.; Dew, T.; Papaioannou, P.; Antwi-Boasiako, S.; Xu, X.; Campbell, S.G.; Akar, F.G.; et al. Humanized Dsp ACM Mouse Model Displays Stress-Induced Cardiac Electrical and Structural Phenotypes. Cells 2022, 11, 3049. [Google Scholar] [CrossRef] [PubMed]
- Celeghin, R.; Risato, G.; Beffagna, G.; Cason, M.; Bueno Marinas, M.; Della Barbera, M.; Facchinello, N.; Giuliodori, A.; Brañas Casas, R.; Caichiolo, M.; et al. A Novel DSP Zebrafish Model Reveals Training- and Drug-Induced Modulation of Arrhythmogenic Cardiomyopathy Phenotypes. Cell Death Discov 2023, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Hatzfeld, M. Plakophilins: Multifunctional Proteins or Just Regulators of Desmosomal Adhesion? Biochim Biophys Acta 2007, 1773, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bonne, S.; Hatzfeld, M.; van Roy, F.; Green, K.J. Protein Binding and Functional Characterization of Plakophilin 2. Evidence for Its Diverse Roles in Desmosomes and Beta -Catenin Signaling. J Biol Chem 2002, 277, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Kuhn, C.; Franke, W.W. Plakophilins 2a and 2b: Constitutive Proteins of Dual Location in the Karyoplasm and the Desmosomal Plaque. J Cell Biol 1996, 135, 1009–1025. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Charron, P.; Fressart, V.; Lorin de la Grandmaison, G.; Simon, F.; Gary, F.; Vite, A.; Hainque, B.; Hidden-Lucet, F.; Komajda, M.; et al. Plakophilin 2A Is the Dominant Isoform in Human Heart Tissue: Consequences for the Genetic Screening of Arrhythmogenic Right Ventricular Cardiomyopathy. Heart 2011, 97, 844–849. [Google Scholar] [CrossRef]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of Plakophilin 2 for Heart Morphogenesis and Cardiac Junction Formation. J Cell Biol 2004, 167, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Noorman, M.; Lin, X.; Chkourko, H.; Liang, F.-X.; van der Nagel, R.; Hund, T.; Birchmeier, W.; Mohler, P.; van Veen, T.A.; et al. Sodium Current Deficit and Arrhythmogenesis in a Murine Model of Plakophilin-2 Haploinsufficiency. Cardiovasc Res 2012, 95, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Leo-Macías, A.; Liang, F.-X.; Delmar, M. Ultrastructure of the Intercellular Space in Adult Murine Ventricle Revealed by Quantitative Tomographic Electron Microscopy. Cardiovasc Res 2015, 107, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Camors, E.M.; Roth, A.H.; Alef, J.R.; Sullivan, R.D.; Johnson, J.N.; Purevjav, E.; Towbin, J.A. Progressive Reduction in Right Ventricular Contractile Function Attributable to Altered Actin Expression in an Aging Mouse Model of Arrhythmogenic Cardiomyopathy. Circulation 2022, 145, 1609–1624. [Google Scholar] [CrossRef]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 Is Required for Transcription of Genes That Control Calcium Cycling and Cardiac Rhythm. Nat Commun 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- van Opbergen, C.J.M.; Bagwan, N.; Maurya, S.R.; Kim, J.-C.; Smith, A.N.; Blackwell, D.J.; Johnston, J.N.; Knollmann, B.C.; Cerrone, M.; Lundby, A.; et al. Exercise Causes Arrhythmogenic Remodeling of Intracellular Calcium Dynamics in Plakophilin-2-Deficient Hearts. Circulation 2022, 145, 1480–1496. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; García-Prieto, J.; García-Ruiz, J.M.; Pizarro, G.; Jiménez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruíz-Cabello, J.; et al. Exercise Triggers ARVC Phenotype in Mice Expressing a Disease-Causing Mutated Version of Human Plakophilin-2. J Am Coll Cardiol 2015, 65, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Moncayo-Arlandi, J.; Guasch, E.; Sanz-de la Garza, M.; Casado, M.; Garcia, N.A.; Mont, L.; Sitges, M.; Knöll, R.; Buyandelger, B.; Campuzano, O.; et al. Molecular Disturbance Underlies to Arrhythmogenic Cardiomyopathy Induced by Transgene Content, Age and Exercise in a Truncated PKP2 Mouse Model. Hum Mol Genet 2016, 25, 3676–3688. [Google Scholar] [CrossRef]
- Moriarty, M.A.; Ryan, R.; Lalor, P.; Dockery, P.; Byrnes, L.; Grealy, M. Loss of Plakophilin 2 Disrupts Heart Development in Zebrafish. Int J Dev Biol 2012, 56, 711–718. [Google Scholar] [CrossRef]
- Green, K.J.; Simpson, C.L. Desmosomes: New Perspectives on a Classic. J Invest Dermatol 2007, 127, 2499–2515. [Google Scholar] [CrossRef]
- Schäfer, S.; Koch, P.J.; Franke, W.W. Identification of the Ubiquitous Human Desmoglein, Dsg2, and the Expression Catalogue of the Desmoglein Subfamily of Desmosomal Cadherins. Exp Cell Res 1994, 211, 391–399. [Google Scholar] [CrossRef]
- Nuber, U.A.; Schäfer, S.; Schmidt, A.; Koch, P.J.; Franke, W.W. The Widespread Human Desmocollin Dsc2 and Tissue-Specific Patterns of Synthesis of Various Desmocollin Subtypes. Eur J Cell Biol 1995, 66, 69–74. [Google Scholar] [PubMed]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in Desmoglein-2 Gene Are Associated with Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.E. Loss of Desmoglein 2 Suggests Essential Functions for Early Embryonic Development and Proliferation of Embryonal Stem Cells. Eur J Cell Biol 2002, 81, 592–598. [Google Scholar] [CrossRef]
- Krusche, C.A.; Holthöfer, B.; Hofe, V.; van de Sandt, A.M.; Eshkind, L.; Bockamp, E.; Merx, M.W.; Kant, S.; Windoffer, R.; Leube, R.E. Desmoglein 2 Mutant Mice Develop Cardiac Fibrosis and Dilation. Basic Res Cardiol 2011, 106, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Krull, P.; Eisner, S.; Leube, R.E.; Krusche, C.A. Histological and Ultrastructural Abnormalities in Murine Desmoglein 2-Mutant Hearts. Cell Tissue Res 2012, 348, 249–259. [Google Scholar] [CrossRef]
- Gerçek, M.; Gerçek, M.; Kant, S.; Simsekyilmaz, S.; Kassner, A.; Milting, H.; Liehn, E.A.; Leube, R.E.; Krusche, C.A. Cardiomyocyte Hypertrophy in Arrhythmogenic Cardiomyopathy. Am J Pathol 2017, 187, 752–766. [Google Scholar] [CrossRef]
- Buck, V.U.; Hodecker, M.; Eisner, S.; Leube, R.E.; Krusche, C.A.; Classen-Linke, I. Ultrastructural Changes in Endometrial Desmosomes of Desmoglein 2 Mutant Mice. Cell Tissue Res 2018, 374, 317–327. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central Role for GSK3β in the Pathogenesis of Arrhythmogenic Cardiomyopathy. JCI Insight 2016, 1, 85923. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti, C.B.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise Triggers CAPN1-Mediated AIF Truncation, Inducing Myocyte Cell Death in Arrhythmogenic Cardiomyopathy. Sci Transl Med 2021, 13, eabf0891. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, J.; Scalco, A.; Agafonova, J.; Williams Iii, L.; Pansari, N.; Keceli, G.; Jun, S.; Wang, N.; Mastorci, F.; Tichnell, C.; et al. Psychosocial Stress Hastens Disease Progression and Sudden Death in Mice with Arrhythmogenic Cardiomyopathy. J Clin Med 2020, 9, 3804. [Google Scholar] [CrossRef]
- Hamstra, S.I.; Braun, J.L.; Chelko, S.P.; Fajardo, V.A. GSK3-Inhibition Improves Maximal SERCA Activity in a Murine Model of Arrhythmogenic Cardiomyopathy. Biochim Biophys Acta Mol Basis Dis 2022, 1868, 166536. [Google Scholar] [CrossRef]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ Cardiovasc Genet 2015, 8, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.E.; Delaney, P.J.; Thenet, D.; Murtough, S.; Webb, C.M.; Zaman, N.; Tsisanova, E.; Mastroianni, G.; Walker, S.L.M.; Westaby, J.D.; et al. Early Inflammation Precedes Cardiac Fibrosis and Heart Failure in Desmoglein 2 Murine Model of Arrhythmogenic Cardiomyopathy. Cell Tissue Res 2021, 386, 79–98. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, R.; Huang, Y.; Yang, Z.; Xian, J.; Huang, J.; Qiu, Z.; Lin, X.; Zhang, M.; Chen, H.; et al. Reactivation of PPARα Alleviates Myocardial Lipid Accumulation and Cardiac Dysfunction by Improving Fatty Acid β-Oxidation in Dsg2-Deficient Arrhythmogenic Cardiomyopathy. Acta Pharm Sin B 2023, 13, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated Disc Abnormalities, Reduced Na(+) Current Density, and Conduction Slowing in Desmoglein-2 Mutant Mice Prior to Cardiomyopathic Changes. Cardiovasc Res 2012, 95, 409–418. [Google Scholar] [CrossRef]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.-T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated with Mutations in the Desmosomal Gene Desmocollin-2. Am J Hum Genet 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant Desmocollin-2 Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Hum Genet 2006, 79, 1081–1088. [Google Scholar] [CrossRef]
- Hamada, Y.; Yamamoto, T.; Nakamura, Y.; Sufu-Shimizu, Y.; Nanno, T.; Fukuda, M.; Ono, M.; Oda, T.; Okuda, S.; Ueyama, T.; et al. G790del Mutation in DSC2 Alone Is Insufficient to Develop the Pathogenesis of ARVC in a Mouse Model. Biochemistry and Biophysics Reports 2020, 21, 100711. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS One 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, L.; Otto, H. LUMA Interacts with Emerin and Influences Its Distribution at the Inner Nuclear Membrane. Journal of Cell Science 2008, 121, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Franke, W.W.; Dörflinger, Y.; Kuhn, C.; Zimbelmann, R.; Winter-Simanowski, S.; Frey, N.; Heid, H. Protein LUMA Is a Cytoplasmic Plaque Constituent of Various Epithelial Adherens Junctions and Composite Junctions of Myocardial Intercalated Disks: A Unifying Finding for Cell Biology and Cardiology. Cell Tissue Res 2014, 357, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Zink, M.; Seewald, A.; Rohrbach, M.; Brodehl, A.; Liedtke, D.; Williams, T.; Childs, S.J.; Gerull, B. Altered Expression of TMEM43 Causes Abnormal Cardiac Structure and Function in Zebrafish. IJMS 2022, 23, 9530. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. Am J Hum Genet 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.F.M.; Merner, N.D.; Hodgkinson, K.A.; Houston, J.; Syrris, P.; Booth, V.; Connors, S.; Pantazis, A.; Quarta, G.; Elliott, P.; et al. Recurrent Missense Mutations in TMEM43 (ARVD5) Due to Founder Effects Cause Arrhythmogenic Cardiomyopathies in the UK and Canada. Eur Heart J 2013, 34, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Klauke, B.; Christensen, A.H.; Müsebeck, J.; Walhorn, V.; Grannemann, S.; Münnich, T.; Šarić, T.; Rasmussen, T.B.; Jensen, H.K.; et al. The TMEM43 Newfoundland Mutation p.S358L Causing ARVC-5 Was Imported from Europe and Increases the Stiffness of the Cell Nucleus. Eur Heart J 2015, 36, 872–881. [Google Scholar] [CrossRef]
- Dominguez, F.; Zorio, E.; Jimenez-Jaimez, J.; Salguero-Bodes, R.; Zwart, R.; Gonzalez-Lopez, E.; Molina, P.; Bermúdez-Jiménez, F.; Delgado, J.F.; Braza-Boïls, A.; et al. Clinical Characteristics and Determinants of the Phenotype in TMEM43 Arrhythmogenic Right Ventricular Cardiomyopathy Type 5. Heart Rhythm 2020, 17, 945–954. [Google Scholar] [CrossRef]
- Rouhi, L.; Cheedipudi, S.M.; Chen, S.N.; Fan, S.; Lombardi, R.; Chen, X.; Coarfa, C.; Robertson, M.J.; Gurha, P.; Marian, A.J. Haploinsufficiency of Tmem43 in Cardiac Myocytes Activates the DNA Damage Response Pathway Leading to a Late-Onset Senescence-Associated pro-Fibrotic Cardiomyopathy. Cardiovasc Res 2021, 117, 2377–2394. [Google Scholar] [CrossRef]
- Zheng, G.; Jiang, C.; Li, Y.; Yang, D.; Ma, Y.; Zhang, B.; Li, X.; Zhang, P.; Hu, X.; Zhao, X.; et al. TMEM43-S358L Mutation Enhances NF-κB-TGFβ Signal Cascade in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Protein Cell 2019, 10, 104–119. [Google Scholar] [CrossRef]
- Gu, Q.; Xu, F.; Orgil, B.-O.; Khuchua, Z.; Munkhsaikhan, U.; Johnson, J.N.; Alberson, N.R.; Pierre, J.F.; Black, D.D.; Dong, D.; et al. Systems Genetics Analysis Defines Importance of TMEM43/LUMA for Cardiac- and Metabolic-Related Pathways. Physiol Genomics 2022, 54, 22–35. [Google Scholar] [CrossRef]
- Orgil, B.-O.; Munkhsaikhan, U.; Pierre, J.F.; Li, N.; Xu, F.; Alberson, N.R.; Johnson, J.N.; Wetzel, G.T.; Boukens, B.J.D.; Lu, L.; et al. The TMEM43 S358L Mutation Affects Cardiac, Small Intestine, and Metabolic Homeostasis in a Knock-in Mouse Model. Am J Physiol Heart Circ Physiol 2023, 324, H866–H880. [Google Scholar] [CrossRef] [PubMed]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- Domínguez, F.; Lalaguna, L.; López-Olañeta, M.; Villalba-Orero, M.; Padrón-Barthe, L.; Román, M.; Bello-Arroyo, E.; Briceño, A.; Gonzalez-Lopez, E.; Segovia-Cubero, J.; et al. Early Preventive Treatment With Enalapril Improves Cardiac Function and Delays Mortality in Mice With Arrhythmogenic Right Ventricular Cardiomyopathy Type 5. Circ Heart Fail 2021, 14, e007616. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A Crucial Regulator of Cardiac Contractility. Nat Rev Mol Cell Biol 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; van de Kolk, C.W.A.; Stege, N.M.; Te Rijdt, W.P.; Hoorntje, E.T.; van der Zwaag, P.A.; van Rooij, E.; et al. The Phospholamban p.(Arg14del) Pathogenic Variant Leads to Cardiomyopathy with Heart Failure and Is Unreponsive to Standard Heart Failure Therapy. Sci Rep 2020, 10, 9819. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Pritchard, T.; Bossuyt, J.; Waggoner, J.R.; Yuan, Q.; Fan, G.-C.; Osinska, H.; Anjak, A.; Rubinstein, J.; Robbins, J.; et al. The Human Phospholamban Arg14-Deletion Mutant Localizes to Plasma Membrane and Interacts with the Na/K-ATPase. J Mol Cell Cardiol 2012, 52, 773–782. [Google Scholar] [CrossRef]
- DeWitt, M.M.; MacLeod, H.M.; Soliven, B.; McNally, E.M. Phospholamban R14 Deletion Results in Late-Onset, Mild, Hereditary Dilated Cardiomyopathy. Journal of the American College of Cardiology 2006, 48, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.-C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A Mutation in the Human Phospholamban Gene, Deleting Arginine 14, Results in Lethal, Hereditary Cardiomyopathy. Proc Natl Acad Sci U S A 2006, 103, 1388–1393. [Google Scholar] [CrossRef]
- Posch, M.G.; Perrot, A.; Geier, C.; Boldt, L.-H.; Schmidt, G.; Lehmkuhl, H.B.; Hetzer, R.; Dietz, R.; Gutberlet, M.; Haverkamp, W.; et al. Genetic Deletion of Arginine 14 in Phospholamban Causes Dilated Cardiomyopathy with Attenuated Electrocardiographic R Amplitudes. Heart Rhythm 2009, 6, 480–486. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del Mutation in Patients Diagnosed with Dilated Cardiomyopathy or Arrhythmogenic Right Ventricular Cardiomyopathy: Evidence Supporting the Concept of Arrhythmogenic Cardiomyopathy. Eur J Heart Fail 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- López-Ayala, J.M.; Boven, L.; Van Den Wijngaard, A.; Peñafiel-Verdú, P.; Van Tintelen, J.P.; Gimeno, J.R. Phospholamban p.Arg14del Mutation in a Spanish Family With Arrhythmogenic Cardiomyopathy: Evidence for a European Founder Mutation. Revista Española de Cardiología (English Edition) 2015, 68, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Rao, M.; Hu, Z.; Zhang, Y.; Gu, M.; et al. A Novel Genotype-Based Clinicopathology Classification of Arrhythmogenic Cardiomyopathy Provides Novel Insights into Disease Progression. European Heart Journal 2019, 40, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.C.; Healey, J.S.; Hamilton, R.; Spears, D.; Gollob, M.H.; Mellor, G.; Steinberg, C.; Sanatani, S.; Laksman, Z.W.; Krahn, A.D. Phospholamban Cardiomyopathy: A Canadian Perspective on a Unique Population. Neth Heart J 2019, 27, 208–213. [Google Scholar] [CrossRef]
- Raad, N.; Bittihn, P.; Cacheux, M.; Jeong, D.; Ilkan, Z.; Ceholski, D.; Kohlbrenner, E.; Zhang, L.; Cai, C.-L.; Kranias, E.G.; et al. Arrhythmia Mechanism and Dynamics in a Humanized Mouse Model of Inherited Cardiomyopathy Caused by Phospholamban R14del Mutation. Circulation 2021, 144, 441–454. [Google Scholar] [CrossRef]
- Rogalska, M.E.; Vafiadaki, E.; Erpapazoglou, Z.; Haghighi, K.; Green, L.; Mantzoros, C.S.; Hajjar, R.J.; Tranter, M.; Karakikes, I.; Kranias, E.G.; et al. Isoform Changes of Action Potential Regulators in the Ventricles of Arrhythmogenic Phospholamban-R14del Humanized Mouse Hearts. Metabolism 2023, 138, 155344. [Google Scholar] [CrossRef] [PubMed]
- Maniezzi, C.; Eskandr, M.; Florindi, C.; Ferrandi, M.; Barassi, P.; Sacco, E.; Pasquale, V.; Maione, A.S.; Pompilio, G.; Teixeira, V.O.N.; et al. Early Consequences of the Phospholamban Mutation PLN-R14del+/- in a Transgenic Mouse Model. Acta Physiol (Oxf) 2024, e14082. [Google Scholar] [CrossRef]
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene Editing Reverses Arrhythmia Susceptibility in Humanized PLN-R14del Mice: Modelling a European Cardiomyopathy with Global Impact. Cardiovasc Res 2022, 118, 3140–3150. [Google Scholar] [CrossRef]
- Kamel, S.M.; van Opbergen, C.J.M.; Koopman, C.D.; Verkerk, A.O.; Boukens, B.J.D.; de Jonge, B.; Onderwater, Y.L.; van Alebeek, E.; Chocron, S.; Polidoro Pontalti, C.; et al. Istaroxime Treatment Ameliorates Calcium Dysregulation in a Zebrafish Model of Phospholamban R14del Cardiomyopathy. Nat Commun 2021, 12, 7151. [Google Scholar] [CrossRef]
- Sarantitis, I.; Papanastasopoulos, P.; Manousi, M.; Baikoussis, N.G.; Apostolakis, E. The Cytoskeleton of the Cardiac Muscle Cell. Hellenic J Cardiol 2012, 53, 367–379. [Google Scholar] [PubMed]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Šarić, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual Color Photoactivation Localization Microscopy of Cardiomyopathy-Associated Desmin Mutants. Journal of Biological Chemistry 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular Insights into Cardiomyopathies Associated with Desmin (DES) Mutations. Biophys Rev 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Otten, E.; Asimaki, A.; Maass, A.; Van Langen, I.M.; Van Der Wal, A.; De Jonge, N.; Van Den Berg, M.P.; Saffitz, J.E.; Wilde, A.A.M.; Jongbloed, J.D.H.; et al. Desmin Mutations as a Cause of Right Ventricular Heart Failure Affect the Intercalated Disks. Heart Rhythm 2010, 7, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Colucci-Guyon, E.; Pinçon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular Lesions and Skeletal Myopathy in Mice Lacking Desmin. Developmental Biology 1996, 175, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Stöckigt, F.; Strucksberg, K.-H.; Chevessier, F.; Winter, L.; Schütz, J.; Bauer, R.; Thorweihe, J.-M.; Wenzel, D.; Schlötzer-Schrehardt, U.; et al. The Toxic Effect of R350P Mutant Desmin in Striated Muscle of Man and Mouse. Acta Neuropathol 2015, 129, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Wittig, I.; Peeva, V.; Eggers, B.; Heidler, J.; Chevessier, F.; Kley, R.A.; Barkovits, K.; Strecker, V.; Berwanger, C.; et al. Mutant Desmin Substantially Perturbs Mitochondrial Morphology, Function and Maintenance in Skeletal Muscle Tissue. Acta Neuropathol 2016, 132, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Stöckigt, F.; Eichhorn, L.; Beiert, T.; Knappe, V.; Radecke, T.; Steinmetz, M.; Nickenig, G.; Peeva, V.; Kudin, A.P.; Kunz, W.S.; et al. Heart Failure after Pressure Overload in Autosomal-Dominant Desminopathies: Lessons from Heterozygous DES-p.R349P Knock-in Mice. PLoS ONE 2020, 15, e0228913. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of Muscle Architecture and Myocardial Degeneration in Mice Lacking Desmin. The Journal of cell biology 1996, 134, 1255–1270. [Google Scholar] [CrossRef]
- Mavroidis, M.; Davos, C.H.; Psarras, S.; Varela, A.; C Athanasiadis, N.; Katsimpoulas, M.; Kostavasili, I.; Maasch, C.; Vater, A.; van Tintelen, J.P.; et al. Complement System Modulation as a Target for Treatment of Arrhythmogenic Cardiomyopathy. Basic Res Cardiol 2015, 110, 27. [Google Scholar] [CrossRef]
- Mavroidis, M.; Athanasiadis, N.C.; Rigas, P.; Kostavasili, I.; Kloukina, I.; Te Rijdt, W.P.; Kavantzas, N.; Chaniotis, D.; van Tintelen, J.P.; Skaliora, I.; et al. Desmin Is Essential for the Structure and Function of the Sinoatrial Node: Implications for Increased Arrhythmogenesis. Am J Physiol Heart Circ Physiol 2020, 319, H557–H570. [Google Scholar] [CrossRef]
- Ren, J.; Tsilafakis, K.; Chen, L.; Lekkos, K.; Kostavasili, I.; Varela, A.; Cokkinos, D.V.; Davos, C.H.; Sun, X.; Song, J.; et al. Crosstalk between Coagulation and Complement Activation Promotes Cardiac Dysfunction in Arrhythmogenic Right Ventricular Cardiomyopathy. Theranostics 2021, 11, 5939–5954. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Andersson-Lendahl, M.; Sejersen, T.; Arner, A. Knockdown of Desmin in Zebrafish Larvae Affects Interfilament Spacing and Mechanical Properties of Skeletal Muscle. J Gen Physiol 2013, 141, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ramspacher, C.; Steed, E.; Boselli, F.; Ferreira, R.; Faggianelli, N.; Roth, S.; Spiegelhalter, C.; Messaddeq, N.; Trinh, L.; Liebling, M.; et al. Developmental Alterations in Heart Biomechanics and Skeletal Muscle Function in Desmin Mutants Suggest an Early Pathological Root for Desminopathies. Cell Rep 2015, 11, 1564–1576. [Google Scholar] [CrossRef]
- Yu, J.; Hu, J.; Dai, X.; Cao, Q.; Xiong, Q.; Liu, X.; Liu, X.; Shen, Y.; Chen, Q.; Hua, W.; et al. SCN5A Mutation in Chinese Patients with Arrhythmogenic Right Ventricular Dysplasia. Herz 2014, 39, 271–275. [Google Scholar] [CrossRef]
- Papadatos, G.A.; Wallerstein, P.M.R.; Head, C.E.G.; Ratcliff, R.; Brady, P.A.; Benndorf, K.; Saumarez, R.C.; Trezise, A.E.O.; Huang, C.L.-H.; Vandenberg, J.I.; et al. Slowed Conduction and Ventricular Tachycardia after Targeted Disruption of the Cardiac Sodium Channel Gene Scn5a. Proc Natl Acad Sci U S A 2002, 99, 6210–6215. [Google Scholar] [CrossRef]
- Hesse, M.; Kondo, C.S.; Clark, R.B.; Su, L.; Allen, F.L.; Geary-Joo, C.T.M.; Kunnathu, S.; Severson, D.L.; Nygren, A.; Giles, W.R.; et al. Dilated Cardiomyopathy Is Associated with Reduced Expression of the Cardiac Sodium Channel Scn5a. Cardiovasc Res 2007, 75, 498–509. [Google Scholar] [CrossRef]
- Watanabe, H.; Yang, T.; Stroud, D.M.; Lowe, J.S.; Harris, L.; Atack, T.C.; Wang, D.W.; Hipkens, S.B.; Leake, B.; Hall, L.; et al. Striking In Vivo Phenotype of a Disease-Associated Human SCN5A Mutation Producing Minimal Changes in Vitro. Circulation 2011, 124, 1001–1011. [Google Scholar] [CrossRef]
- Daniel, L.L.; Yang, T.; Kroncke, B.; Hall, L.; Stroud, D.; Roden, D.M. SCN5A Variant R222Q Generated Abnormal Changes in Cardiac Sodium Current and Action Potentials in Murine Myocytes and Purkinje Cells. Heart Rhythm 2019, 16, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Huttner, I.G.; Trivedi, G.; Jacoby, A.; Mann, S.A.; Vandenberg, J.I.; Fatkin, D. A Transgenic Zebrafish Model of a Human Cardiac Sodium Channel Mutation Exhibits Bradycardia, Conduction-System Abnormalities and Early Death. J Mol Cell Cardiol 2013, 61, 123–132. [Google Scholar] [CrossRef]
- Chopra, S.S.; Stroud, D.M.; Watanabe, H.; Bennett, J.S.; Burns, C.G.; Wells, K.S.; Yang, T.; Zhong, T.P.; Roden, D.M. Voltage-Gated Sodium Channels Are Required for Heart Development in Zebrafish. Circ Res 2010, 106, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.A.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Genet 2017, 10, e001605. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole Exome Sequencing with Genomic Triangulation Implicates CDH2-Encoded N-Cadherin as a Novel Pathogenic Substrate for Arrhythmogenic Cardiomyopathy. Congenit Heart Dis 2017, 12, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced Deletion of the N-Cadherin Gene in the Heart Leads to Dissolution of the Intercalated Disc Structure. Circ Res 2005, 96, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic Variation in Titin in Arrhythmogenic Right Ventricular Cardiomyopathy-Overlap Syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, M.; Michely, B.; Krohne, C.; Heuser, A.; Erdmann, B.; Klaassen, S.; Hudson, B.; Magarin, M.; Kirchner, F.; Todiras, M.; et al. Stress-Induced Dilated Cardiomyopathy in a Knock-in Mouse Model Mimicking Human Titin-Based Disease. J Mol Cell Cardiol 2009, 47, 352–358. [Google Scholar] [CrossRef]
- Xu, X.; Meiler, S.E.; Zhong, T.P.; Mohideen, M.; Crossley, D.A.; Burggren, W.W.; Fishman, M.C. Cardiomyopathy in Zebrafish Due to Mutation in an Alternatively Spliced Exon of Titin. Nat Genet 2002, 30, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Tran, D.; Baalbaki, M.; Tang, L.F.; Poon, A.; Pelonero, A.; Titus, E.W.; Yuan, C.; Shi, C.; Patchava, S.; et al. An Internal Promoter Underlies the Difference in Disease Severity between N- and C-Terminal Truncation Mutations of Titin in Zebrafish. Elife 2015, 4, e09406. [Google Scholar] [CrossRef]
- Shih, Y.-H.; Dvornikov, A.V.; Zhu, P.; Ma, X.; Kim, M.J.; Ding, Y.; Xu, X. Exon- and Contraction-Dependent Functions of Titin in Sarcomere Assembly. Development, 1392. [Google Scholar] [CrossRef]
- Santiago, C.F.; Huttner, I.G.; Fatkin, D. Mechanisms of TTNtv-Related Dilated Cardiomyopathy: Insights from Zebrafish Models. J Cardiovasc Dev Dis 2021, 8, 10. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C Gene Mimic Arrhythmogenic Right Ventricular Cardiomyopathy. Eur Heart J 2012, 33, 1128–1136. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in Nuclear Structure and Function Promote Dilated Cardiomyopathy in Lamin A/C-Deficient Mice. J Clin Invest 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.M.; Wang, L.; Alcalai, R.; Pizard, A.; Burgon, P.G.; Ahmad, F.; Sherwood, M.; Branco, D.M.; Wakimoto, H.; Fishman, G.I.; et al. Lamin A/C Haploinsufficiency Causes Dilated Cardiomyopathy and Apoptosis-Triggered Cardiac Conduction System Disease. J Mol Cell Cardiol 2008, 44, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-J.; Lee, Y.-K.; Lau, Y.-M.; Ho, J.C.-Y.; Lai, W.-H.; Wong, N.L.-Y.; Huang, D.; Hai, J.-J.; Ng, K.-M.; Tse, H.-F.; et al. Expression of Lmna-R225X Nonsense Mutation Results in Dilated Cardiomyopathy and Conduction Disorders (DCM-CD) in Mice: Impact of Exercise Training. Int J Cardiol 2020, 298, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sun, J.; Chen, Z.; Liu, L.; Sun, Y.; Lin, J.; Hu, X.; Zhao, M.; Ma, Y.; Lu, D.; et al. The LMNA p.R541C Mutation Causes Dilated Cardiomyopathy in Human and Mice. Int J Cardiol 2022, 363, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory Mutations in Transforming Growth Factor-Beta3 Gene Cause Arrhythmogenic Right Ventricular Cardiomyopathy Type 1. Cardiovasc Res 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of Mutations in the Cardiac Ryanodine Receptor Gene in Families Affected with Arrhythmogenic Right Ventricular Cardiomyopathy Type 2 (ARVD2). Hum Mol Genet 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Mitchell, B.M.; Goonasekera, S.A.; Chelu, M.G.; Zhang, W.; Sood, S.; Kearney, D.L.; Danila, C.I.; De Biasi, M.; Wehrens, X.H.T.; et al. Mice with the R176Q Cardiac Ryanodine Receptor Mutation Exhibit Catecholamine-Induced Ventricular Tachycardia and Cardiomyopathy. Proc Natl Acad Sci U S A 2006, 103, 12179–12184. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.-X.; Wronska, A.; Marks, A.R. Calcium Leak through Ryanodine Receptors Leads to Atrial Fibrillation in 3 Mouse Models of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, N.; Kuwahara, M.; Hirata, Y.; Oku, Y.; Nishio, H. A Knock-in Mouse Model of N-Terminal R420W Mutation of Cardiac Ryanodine Receptor Exhibits Arrhythmogenesis with Abnormal Calcium Dynamics in Cardiomyocytes. Biochem Biophys Res Commun 2014, 452, 665–668. [Google Scholar] [CrossRef]
- Bround, M.J.; Asghari, P.; Wambolt, R.B.; Bohunek, L.; Smits, C.; Philit, M.; Kieffer, T.J.; Lakatta, E.G.; Boheler, K.R.; Moore, E.D.W.; et al. Cardiac Ryanodine Receptors Control Heart Rate and Rhythmicity in Adult Mice. Cardiovasc Res 2012, 96, 372–380. [Google Scholar] [CrossRef]
- Takeshima, H. Embryonic Lethality and Abnormal Cardiac Myocytes in Mice Lacking Ryanodine Receptor Type 2. The EMBO Journal 1998, 17, 3309–3316. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of FilaminC (FLNc) Results in Severe Defects in Myogenesis and Myotube Structure. Mol Cell Biol 2006, 26, 6522–6534. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Z.; Zhang, L.; Zhu, M.; Tan, C.; Zhou, X.; Evans, S.M.; Fang, X.; Feng, W.; Chen, J. Loss of Filamin C Is Catastrophic for Heart Function. Circulation 2020, 141, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.D.; Kirkland, N.J.; Liu, C.; Razu, S.S.; Fang, X.; Engler, A.J.; Chen, J.; McCulloch, A.D. Subcellular Remodeling in Filamin C Deficient Mouse Hearts Impairs Myocyte Tension Development during Progression of Dilated Cardiomyopathy. Int J Mol Sci 2022, 23, 871. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; Zhao, M.; Currie, P.D.; Bryson-Richardson, R.J. Characterization and Investigation of Zebrafish Models of Filamin-Related Myofibrillar Myopathy. Human Molecular Genetics 2012, 21, 4073–4083. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin Electrophysiol 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, Encoding Integrin-Linked Kinase, Are Associated with Arrhythmogenic Cardiomyopathy. Transl Res 2019, 208, 15–29. [Google Scholar] [CrossRef]
- Quang, K.L.; Maguy, A.; Qi, X.-Y.; Naud, P.; Xiong, F.; Tadevosyan, A.; Shi, Y.-F.; Chartier, D.; Tardif, J.-C.; Dobrev, D.; et al. Loss of Cardiomyocyte Integrin-Linked Kinase Produces an Arrhythmogenic Cardiomyopathy in Mice. Circ Arrhythm Electrophysiol 2015, 8, 921–932. [Google Scholar] [CrossRef]
- Bendig, G.; Grimmler, M.; Huttner, I.G.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Katus, H.A.; Fishman, M.C.; Rottbauer, W. Integrin-Linked Kinase, a Novel Component of the Cardiac Mechanical Stretch Sensor, Controls Contractility in the Zebrafish Heart. Genes Dev 2006, 20, 2361–2372. [Google Scholar] [CrossRef]
- Pott, A.; Shahid, M.; Köhler, D.; Pylatiuk, C.; Weinmann, K.; Just, S.; Rottbauer, W. Therapeutic Chemical Screen Identifies Phosphatase Inhibitors to Reconstitute PKB Phosphorylation and Cardiac Contractility in ILK-Deficient Zebrafish. Biomolecules 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Cason, M.; Celeghin, R.; Marinas, M.B.; Beffagna, G.; Della Barbera, M.; Rizzo, S.; Remme, C.A.; Bezzina, C.R.; Tiso, N.; Bauce, B.; et al. Novel Pathogenic Role for Galectin-3 in Early Disease Stages of Arrhythmogenic Cardiomyopathy. Heart Rhythm 2021, 18, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Fan, Y.; Gu, J.; Han, Z.; Wang, Y.; Gao, L.; Zeng, H.; Mao, C.; Wang, C. Effect of Lgals3a on Embryo Development of Zebrafish. Transgenic Res 2021, 30, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Guo, G.; Li, M.; Chen, S.; Chen, K.; Chen, X.; Song, J.; Hu, S. The Homozygous Variant c.245G > A/p.G82D in PNPLA2 Is Associated with Arrhythmogenic Cardiomyopathy Phenotypic Manifestations. Clin Genet 2019, 96, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.-L.; Sun, X.; Yang, J.; Wang, X.-L.; Ackerman, M.J.; Wang, R.-X.; Xu, X.; Lee, H.-C.; Lu, T. Changes in Ion Channel Expression and Function Associated with Cardiac Arrhythmogenic Remodeling by Sorbs2. Biochim Biophys Acta Mol Basis Dis 2021, 1867, 166247. [Google Scholar] [CrossRef]
- Sommariva, E.; Stadiotti, I.; Perrucci, G.L.; Tondo, C.; Pompilio, G. Cell Models of Arrhythmogenic Cardiomyopathy: Advances and Opportunities. Dis Model Mech 2017, 10, 823–835. [Google Scholar] [CrossRef]
Figure 1.
Hallmarks of AC disease pathogenesis and progression detected in human patients. The central hallmark of AC is the fibro-fatty substitution of the myocardial tissue, being linked to the instability/disruption of intercalated disks (IDs) and desmosomes, activation of FAPs (fibroblast activation proteins), dysregulation of signalling pathways such as Wnt/β-catenin, and inflammation. Inflammatory infiltrates are associated with necrotic and/or apoptotic myocyte death, leading to electrical instability, arrhythmias and SCD. Created with BioRender.
Figure 1.
Hallmarks of AC disease pathogenesis and progression detected in human patients. The central hallmark of AC is the fibro-fatty substitution of the myocardial tissue, being linked to the instability/disruption of intercalated disks (IDs) and desmosomes, activation of FAPs (fibroblast activation proteins), dysregulation of signalling pathways such as Wnt/β-catenin, and inflammation. Inflammatory infiltrates are associated with necrotic and/or apoptotic myocyte death, leading to electrical instability, arrhythmias and SCD. Created with BioRender.
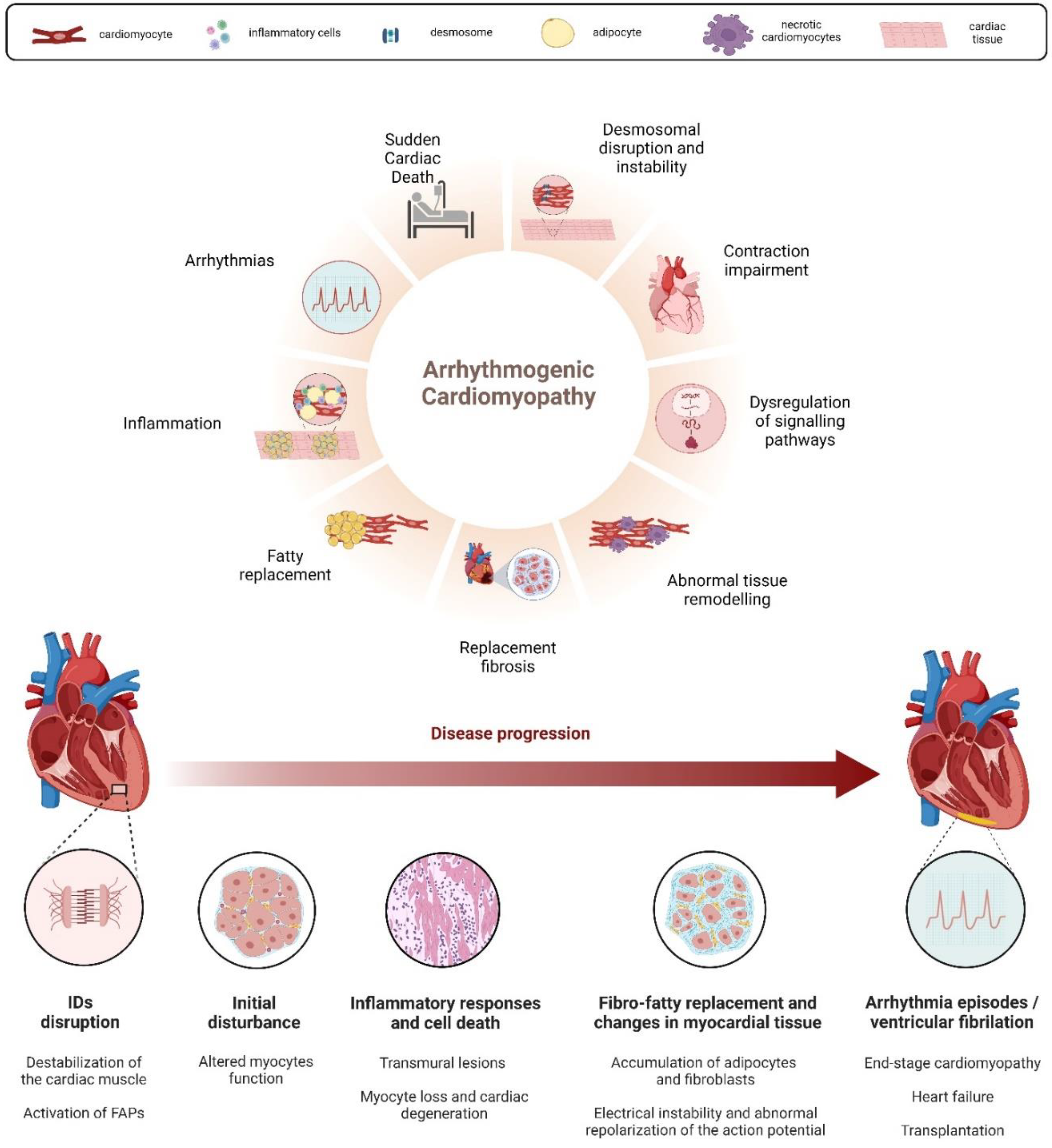
Figure 2.
Genetic and Signalling pathway dysregulation in AC. A: Desmosomes play a crucial role in cell adhesion, mechanical anchorage, and communication between cardiomyocytes. B: The desmosome structure consists of several proteins: Desmoplakin (DSP), Plakoglobin (JUP) and Plakophilin-2 (PKP2) form the intracellular portion, separated into an outer and inner dense plaque, while the intercellular portion is characterized by the interaction between cadherin proteins, Desmoglein-2 (DSG2), and Desmocolin-2 (DSC2). C: The dysregulation of signalling pathways fundamental in the progression of the disease. Wnt/β-catenin dysregulation occurs through desmosome destabilization and subsequent JUP protein release, leading to adipogenesis and fibrogenesis. The activation of Hippo/YAP-TAZ pathway leads to accumulation p-YAP in the cytosol, its degradation and the subsequent altered gene expression associated with adipogenesis and apoptosis. p-YAP also binds and sequesters β-catenin, reducing its transcriptional activities. The upregulated TGF-β signalling pathway contributes to cardiac repair, remodelling, and fibrosis. Created with Biorender.
Figure 2.
Genetic and Signalling pathway dysregulation in AC. A: Desmosomes play a crucial role in cell adhesion, mechanical anchorage, and communication between cardiomyocytes. B: The desmosome structure consists of several proteins: Desmoplakin (DSP), Plakoglobin (JUP) and Plakophilin-2 (PKP2) form the intracellular portion, separated into an outer and inner dense plaque, while the intercellular portion is characterized by the interaction between cadherin proteins, Desmoglein-2 (DSG2), and Desmocolin-2 (DSC2). C: The dysregulation of signalling pathways fundamental in the progression of the disease. Wnt/β-catenin dysregulation occurs through desmosome destabilization and subsequent JUP protein release, leading to adipogenesis and fibrogenesis. The activation of Hippo/YAP-TAZ pathway leads to accumulation p-YAP in the cytosol, its degradation and the subsequent altered gene expression associated with adipogenesis and apoptosis. p-YAP also binds and sequesters β-catenin, reducing its transcriptional activities. The upregulated TGF-β signalling pathway contributes to cardiac repair, remodelling, and fibrosis. Created with Biorender.
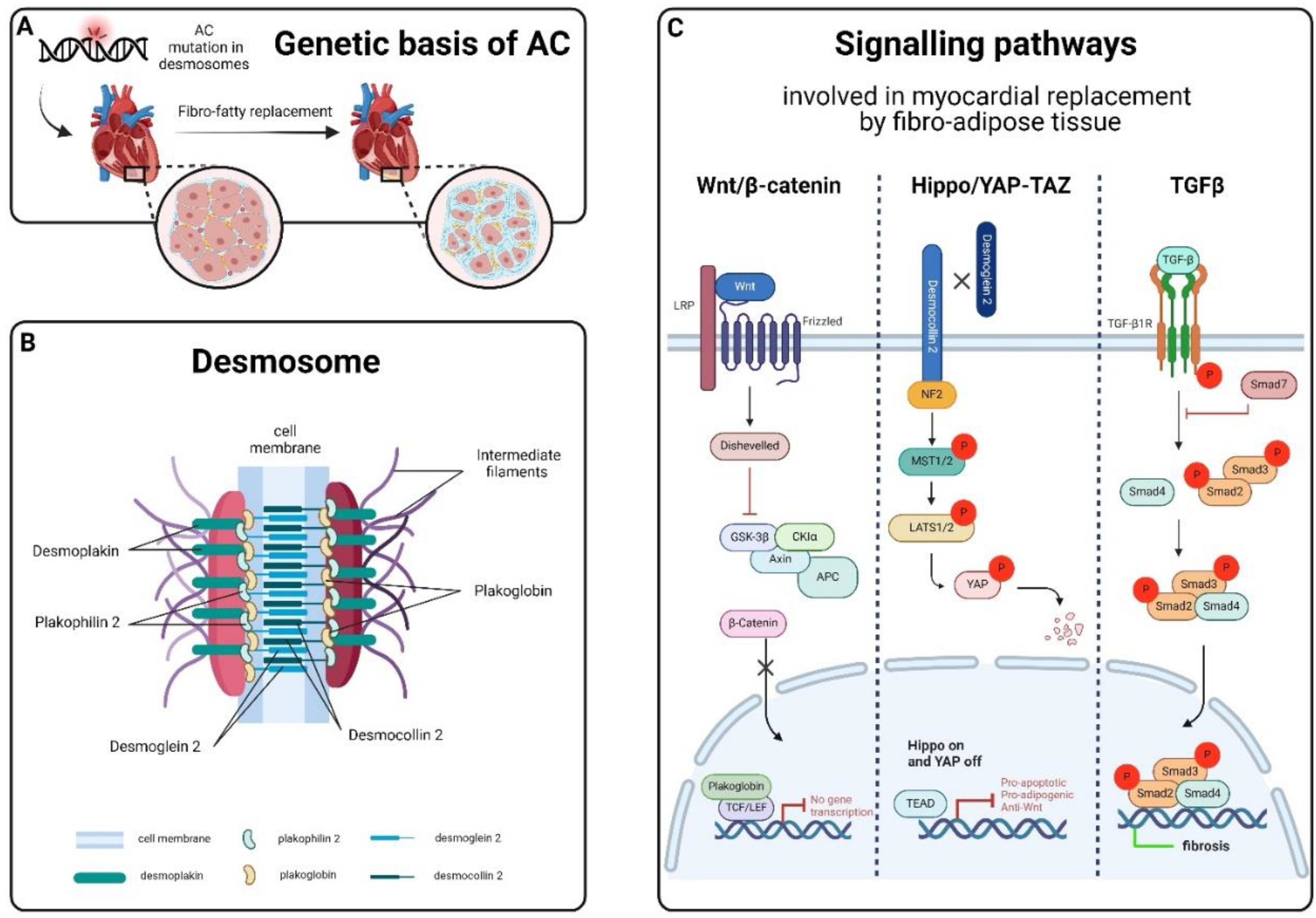
Figure 3.
Timeline overview of in vivo animal models for AC-related genes, organized by the year of publication. Abbreviations: CS = cardiac specific; Tg = transgenic; Hs = Homo sapiens; KD = knock down; KO = knock out; KI = knock in. Updated from Gerull and Brodehl, 2020. Created with Biorender.
Figure 3.
Timeline overview of in vivo animal models for AC-related genes, organized by the year of publication. Abbreviations: CS = cardiac specific; Tg = transgenic; Hs = Homo sapiens; KD = knock down; KO = knock out; KI = knock in. Updated from Gerull and Brodehl, 2020. Created with Biorender.
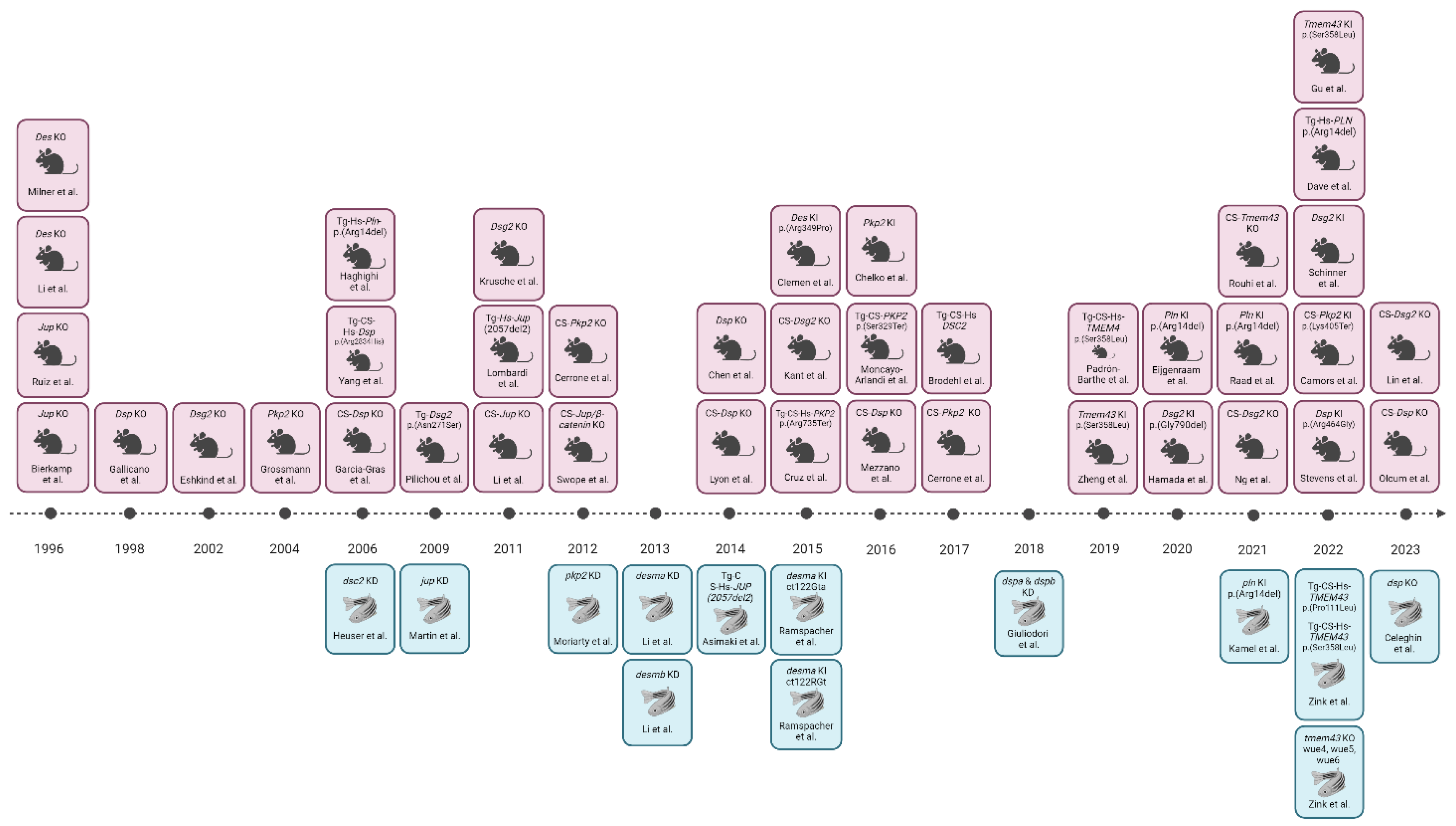

Table 1.
Overview on non-desmosomal models.
| GENE | MOUSE MODEL | ZEBRAFISH MODEL |
|---|---|---|
| LIMITED | ||
| Sodium voltage-gated channel alpha subunit 5 (SCN5A) [186] |
CS-Scn5A-KO mouse models showed severe biventricular cardiomyopathy and conduction defects in heterozygosity, whereas homozygotes can also present foetal lethality [187,188]. Scn5A-KI mouse models presenting human p.(Asp1275Asn) or p.Asp222Gln mutations showed a progressive cardiac dysfunction but not a clear AC morphological phenotype [189,190]. | A CS-Tg zebrafish expressing the human Asp1275Asn mutation displayed bradycardia, conduction-system abnormalities and premature death, highlighting zebrafish as a valuable model for understanding the gene’s role in the disease [191]. Similarly, KD of scn5a in zebrafish delayed early chamber development and disrupted the ventricle’s patterned growth, suggesting that cardiac sodium channels influence heart development through a non-electrophysiological mechanism [192]. |
| N-cadherin (CDH2) [193,194] |
Ventricular arrhythmia and aberrant conduction in mice with CS-Cdh2 deletion were observed, resulting in early death after two months. However, more studies are needed to understand CDH2 role in the pathogenesis of the disease [195]. | According to the ZFIN database, insertional, ENU- and CRISPR-induced mutants are available but not characterized as putative AC models. |
| Titin (TTN) [196] |
Only after hemodynamic stress, the Ttn KI mouse model, mimicking the human p.(Met14544*) mutation, developed left ventricular dilatation, impaired fractional shortening and diffuse myocardial fibrosis [197]. | Homozygous N-terminal and C-terminal titin truncations led to severe cardiac contractility defects and premature death in zebrafish. C-terminal truncations caused severe skeletal muscle myopathies, while heterozygous truncations resulted in spontaneous DCM with decreased baseline ventricular systolic function, mirroring human conditions [198,199,200,201]. |
| Lamin A/C (LMNA) [202] |
Homozygous Lmna-KO mice died in few weeks due to delayed postnatal development and the onset of DCM [203]. Heterozygous KO mutant mice, instead, survived showing conduction defects, arrhythmia events and cardiac contractility problems [204]. KI mouse models expressing human mutations p(Arg225Ter) and p.(Arg541Cis) confirmed the conductions alterations and in general the phenotype observed in KO ones [205,206]. | According to the ZFIN database, morphants, insertional, ENU- and CRISPR-induced mutants are available but not characterized as putative AC models. |
| Transforming growth factor β-3 (TGFB3) [207] |
Tgfβ3-KO mice failed to display cardiac defects or AC phenotypes, therefore this protein is nowadays more recognized for its indirect role in the disease, being found differently expressed in several other AC models [39,42,52,53,152]. | According to the ZFIN database, morphants, ENU- and CRISPR-induced mutants are available but not characterized as putative AC models. |
| DISPUTED | ||
| Cardiac Ryanodine receptor 2 (RYR2) [208] |
RYR2 primarily impacts cardiac electrical activity rather than tissue remodelling. The KI mouse model with the human disease-associated RYR2 mutation p.Arg176Gln exhibited calcium-dependent ventricular arrhythmias without histological changes [209], like other KI models [210,211] and a CS-KO one [212]. Whole body KO homozygosity provoked embryonic lethality [213]. | According to the ZFIN database, ENU- and CRISPR-induced mutants are available but so far not characterized. |
| NOT CURATED | ||
| Filamin C (FLNC) [214] |
CS-Flnc deficient mice exhibited cardiac fibrosis, dysfunction, elevated expression of cardiac stress markers and early mortality. These findings connected FLNC with the regulation of cardiomyocyte structure [215,216,217]. | Zebrafish flnc models (morphants and Tg) display cardiac alterations both functionally and structurally, with reduced survival. The observed effects included pericardial oedema, dysmorphic or dilated cardiac chambers with protein aggregation, abnormal heart tube looping, reduced blood circulation and overall weaker contractility. TEM analysis revealed irregular or seemingly absent Z-discs [218,219]. |
| Integrin-linked kinase (ILK) [220] |
In mouse cardiomyocytes, Ilk deletion produced a lethal AC phenotype with relevant ion channel and structural remodelling, connecting this protein to the disease [221]. | CS expression of human wild-type and mutated variants (H77Y and P70L) of ILK resulted in cardiac malfunction, decreased fractional shorting and premature mortality by the time the fish were two to three weeks old [220]. Likewise, a spontaneous homozygous ilk mutation (p.Leu308Pro) in zebrafish resulted in cardiac oedema, reduced heart function, and early mortality [222,223]. |
| Galectin-3 (LGALS-3/GAL-3) [224] |
Gal-3 emerged as one of the differentially expressed genes (DEGs) in the myocardium of Tg mice with the early AC phenotype. Dysregulation in the myocardium was found in Tg mice overexpressing the DSG2 p.Asn271Ser mutation. This finding was further validated in three AC patients who experienced SCD without structural remodelling [224]. | A pharmacological KD zebrafish lgals3a line exhibited developmental defects, decreased macrophages, apoptotic cardiomyocytes, and dysregulation of the Wnt/β-catenin pathway, suggesting a potential role of lgals3a in the aetiology of AC and other cardiac diseases [224,225]. |
| Patatin Like Phospholipase Domain Containing 2 (PNPLA2) [226] |
A KI mouse model carrying the human Pnpla2 mutation presented with arrhythmias and significant cardiac dysfunction. Moreover, those mice suffered SCD with extensive lipogenesis in cardiomyocytes and cardiac fibrosis in the myocardium [226]. | According to the ZFIN database, morphants and CRISPR-induced mutants are available but not characterized as putative AC models. |
| SH3 domain-containing 2b (SORBS2) [227] |
KO mice with a complete SORBS2 depletion exhibited a phenotype resembling that of AC patients, featuring right ventricular dilatation, dysfunction, spontaneous ventricular tachycardia, and sudden cardiac death. The absence of SORBS2 caused significant cardiac electrical remodelling, affecting action potentials, impulse conduction, and inducing life-threatening arrhythmias [227]. | According to the ZFIN database, morphants and mutants are available but not characterized as putative AC models. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



