
Submitted:
27 June 2024
Posted:
01 July 2024
You are already at the latest version
Abstract
Mesenchymal stem cells (MSCs) are one of the main residents in the bone marrow (BM), having an essential role in the regulation of hematopoietic stem cell (HSCs) differentiation and proliferation. Myelodysplastic syndromes (MDS) are a group of myeloid disorders impacting hematopoietic stem and progenitor cells (HSCPs) and characterised by BM failure, ineffective haematopoiesis, cytopenia, a high risk of transformation through the expansion of MDS clones together with additional genetic defects. It has been indicated that MSCs play anti-tumorigenic roles such as cell cycle arrest and pro-tumorigenic roles including induction of metastasis in MDS and leukaemia. Growing evidence has shown that MSCs have impaired functions in MDS, such as decreased proliferation capacity, differentiation ability, haematopoiesis support, immunomodulation function, and increased inflammatory alterations within the BM through some intracellular pathways such as Notch and Wnt and extracellular modulators secreted by MSCs abnormally, including increased expressions of inflammatory factors and decreased expressions of hematopoietic factors, contributing to the development and progression of MDS. Therefore, MSCs can be targeted for the treatment of MDS and leukaemia. However, it remains unclear what drives MSCs to behave abnormally. In this review, dysregulations in MSCs and their contributions to myeloid haematological malignancies will be discussed.
Keywords:
mesenchymal stem cells
; leukaemia
; bone marrow microenvironment
; myelodysplastic syndromes
1. Introduction
The International Society for Cell & Gene Therapy Mesenchymal Stromal Cell (ISCT MSC) committee defines mesenchymal stem cells (MSCs) as multipotent progenitor cells that possess the ability to self-renew and differentiate into various cell types of the mesodermal lineage including osteocytes, chondrocytes, adipocytes, myoblasts, and as well as of embryonic lineages [1]. Due to their capacity for multipotent differentiation, MSCs have immense value in regenerative medicine and tissue engineering applications. MSCs are also characterised by the positive expression of CD105, CD90, and CD73 as well as negative expression of endothelial and hematopoietic markers, such as CD14, CD34, CD45, human leukocyte antigen-DR isotype (HLA-DR), and CD19 in humans [2] (Figure 1). The adherence to culture surfaces is another characteristic of in vitro expanded MSCs. MSCs can be collected from various body sources, including bone marrow (BM), placenta, adipose tissue, muscles, umbilical cord, amniotic fluid, menstrual blood, cord blood, dental pulp, dermal tissue, and urine [3].
They also play a vital role in regulating and maintaining the properties of hematopoietic stem cells (HSCs) [5]. The interaction between HSCs and MSCs prevents the differentiation of HSC into B-cells and dendritic cells (DCs) and protects them from apoptosis, promoting HSC stemness and self-renewal [6]. Furthermore, MSCs have an immunosuppressive effect that can suppress various immune cells like macrophages, natural killer (NK) cells, B cells, T cells, and dendritic cells, which can be useful in treating autoimmune diseases and conditions involving excessive inflammation [7].
MSCs can produce several bioactive molecules [8], including growth factors (transforming growth factor beta, (TGF-β); epidermal growth factor, (EGF); granulocyte-macrophage colony-stimulating factor, (GM-CSF), adhesion molecules (activated leukocyte cell adhesion molecule, ALCAM; intercellular adhesion molecule-1, ICAM-1), cytokines (e.g. interleukin-IL-1α, IL-1β, IL-6, TNF-α), immunomodulatory molecules (prostaglandin E2, PGE2; human leukocyte antigen, HLA-G), [9] and angiogenic factors (vascular endothelial factor, VEGF; platelet-derived growth factor, PDGF [10] that are responsible for the paracrine effect of MSCs on neighbouring cells. Due to the paracrine effect, MSCs can influence the behaviour of surrounding cells, making them valuable for modulating tissue regeneration and immune and inflammatory responses [11].
MSCs facilitate the proliferation of regulatory T/B cells (T/B regs) and inhibit immune cell proliferation and functions of almost all immune cells. MSCs also regulate haematopoiesis within the BM via interactions with HSCs and migrate to injured tissues for regenerative purposes. Consequently, MSCs have become an important contributing factor in various diseases, such as dermatitis [12], asthma [13], and arthritis [14].
BM was the primary course of MSCs for many years. However, studies have shown that the BM is made up of different cell types, including CXCL12-expressing adventitial cells (CAR cells), endothelial cells, MSCs, pericytes, adipocytes, osteoblasts, HSCs, and hematopoietic cells, such as macrophages, osteoclasts, neutrophils, and regulatory T cells [15]. BM-resident cells like HSCs, endothelial cells, and cells derived from MSCs, in conjunction with the extracellular matrix (ECM), create a highly specialised microenvironment responsible for regulating the formation of mature hematopoietic cells and their proper function [16]. Moreover, BM plays a crucial role in maintaining the blood cell system circulating throughout the body, while a limited number of HSCs residing in the BM are responsible for producing blood cells. Thus, it can be said that BM regulates the activity of HSCs and their progeny [17]. The balance modulated by hematopoietic and non-hematopoietic cells between HSC quiescence and activation and the subsequent processes of cell fate determination, proliferation, self-renewal, and differentiation are orchestrated by a complex interplay between chemical and physical factors within the BM. For this reason, dysregulation of BM niche cells interferes with stem cell behaviour, resulting in malignant transformation [18].
2. Role of MSCs in Pre-Leukaemia Myelodysplastic Pathogenesis
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of clonal hematopoietic disorders that affect hematopoietic stem and progenitor cells. These disorders are characterised by inefficient haematopoiesis, BM failure, and peripheral blood cytopenia with a high risk of progression to acute leukaemia [19]. Studies have shown that various mutations in somatic genes [20], particularly those involved in RNA-splicing (SBDS and DICER1) [21], signal transduction, DNA methylation, chromatin modification, cohesion regulation, transcription factors (p53), cytogenetic abnormalities in HSCs [22].
MDS has historically been subdivided into various subclasses by classification schemes over time [23]. The first classification was made by the French-American-British (FAB) group in 1982 based on morphologic criteria, such as BM blast percentage, ring sideroblasts, Auer rods, and monocytic proliferation in BM and blood. FAB divided MDS into five categories, such as refractory anaemia (RA), refractory anaemia with ringed sideroblasts (RARS), refractory anaemia with excess blasts (RAEB), refractory anaemia with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukaemia (CMML) (Table 1).
Then, in 2002, World Health Organisation (WHO) modified the FAB classification and divided MDS subtypes by unilineage or multilineage dysplasia and increased blasts (Table 2).
In 2016, WHO changed MDS terminology and revised the MDS subtypes based on the number of dysplastic lineages, the presence of cytopenia, the presence of ring sideroblasts, the percentage of blasts, and chromosome abnormalities specific to MDS (Table 3) [24].
More recently, in 2022, WHO divided MDS subtypes based on BM cellularity, the presence of mutations, ring sideroblasts, concomitant pathologies such as lymphoma, infection, and metastasis, dysplasia, the level of BM fibrosis, and BM karyotype by adopting hematologic, morphologic, cytogenetic, and molecular genetic approaches (Table 4) [25].
Finally, The International Prognostic Scoring System (IPSS), a system used for the assessment of prognosis in MDS patients, classifies MDS based on the percentage of BM blasts, karyotype, and cytopenia(s) and scores patients as below (Table 5 and 6).
Scientific research about MDS could be hindered due to the lack of existing utilisable models. Also, MDS cells could be challenging to study due to a high apoptosis rate. Therefore, BM support is essential to maintain some MDS cells ex vivo, even though the mechanism of this support is not fully understood yet. For this reason, bone marrow mesenchymal stem cells (BMSCs) derived from MDS patients allow the maintenance of MDS clones.
Some in vivo models have been developed for MDS studies using mice, rats, and zebrafish (Table 7). For instance, some laboratories have established xenotransplant mice by engrafting immortalised human cell lines derived from MDS patients. Moreover, genetically engineered mouse models have been established by reverse transcription BM transduction/transplantation, including harvesting murine BM nucleated cells with a retroviral construct that can express the gene of interest. Another type of genetically engineered mouse model is to knock out the genes associated with MDS by gene targeting to attain mouse embryonic stem cells that feature genes engineered through homologous recombination via DNA fragment modification and intricate gene positioning to form hematopoietic mouse cells that have human MDS properties. The last mouse model is induced mouse model which is artificially generated by biological or chemical agents [26]. Similarly, MDS can be developed in rat exposed to chemical agents like DMBA. Moreover, genetically engineered model of zebrafish can be utilised for MDS studies. For example, a zebrafish model was established through the disruption of Tet2 catalytic domain whose loss-of-function mutations is common in MDS [27].
To study MDS in vitro, some cell lines derived from MDS patients such as MDS92, MDS-L, M-TAT, and TER-3 and cell lines obtained from leukaemia developed after MDS including MDS-KZ and SKM-I can be utilised. Moreover, induced pluripotent stem cells obtained from somatic cells of MDS patients are able to differentiate into different hematopoietic lineages, iPSC clones can be processed along the spectrum from CHIP to HR-MDS through additional mutations. Also, they can be reverted to modelling lower-stage disease via gene corrections [28]. Lastly, co-culture systems can be created with MDS cells and stromal and/or hematopoietic cells to investigate cellular crosstalk. For instance, MDS cells and MSCs co-culture would be a good way to study the dysfunctionalities of MSCs in MDS as it has been known that MSCs with impaired characteristics, such as inflammatory alterations, defective differentiation potential, decreased growth and proliferation, and dysregulation of immune modulation, also contribute to the disease [29].
2.1. Impaired Morphology and Immunophenotype of MSCs in MDS
Cultured MDS BMSCs have been found to exhibit an irregular morphology. For example, one study found that ex vivo expanded patient-derived BMSCs exhibited thicker and granular morphology [30]. However, several studies suggest that variations in experimental methodologies and patient heterogeneity may contribute to conflicting findings on this topic. Specifically, some studies [31] have failed to detect morphological abnormalities in BMSCs in MDS patients in comparison to their healthy counterparts [32]. Despite these discrepancies, most studies [33] agree that patient BMSCs do not differ significantly from normal MSCs in terms of immunophenotype [34]. However, some studies report reduced expression of several key markers, including CD34, CD90, and CD45, on the surface of patient BMSCs [35].
2.2. Cytogenetic and Genetic Abnormalities of MSCs in MDS
Patients with de novo MDS frequently have clonal cytogenetic abnormalities found in their hematopoietic cells, with a frequency range of about 40–70%. On the other hand, there is inconsistent information on genetic abnormalities in MDS-BMSCs. While some research [36] has demonstrated that patient ex-vivo grown BMSCs display chromosomal abnormalities such as 5q deletion, other publications suggest that cytogenetic analysis of these cells is normal. It is noteworthy that the clonal chromosomal aberrations seen in MDS-BMSCs consistently differ from those found in the patient's hematopoietic cells, indicating that the two cell types are not produced from the same clone. This idea is further supported by the discovery that the mesenchymal compartment of BM-mononuclear cells from MDS patients did not include any mutations of epi-genetic or spliceosomal genes, including TET2, DNMT3A, SRSF2, and SF3B1 [29]. Notably, research on the detection of chromosomal abnormalities in BMSCs from MDS patients is currently ongoing. It was noted that chromosomal abnormalities were also present in BMSCs obtained from healthy donors, and that some of these aberrations were only found in later passages. MDS-BMSCs displayed a higher mutational burden and different mutational signatures compared to healthy BMSCs, according to a recent study that used exome sequencing to examine the prevalence of clonal mutations in ex vivo grown BMSCs from both patients and healthy donors. However, primary stroma cells from the same patients did not exhibit the extremely recurrent mutations found during culture. These results imply that the stroma compartment of MDS patients does not contain any evidence of clonal mutations [37].
Patients with MDS exhibit different genetic aberrations in their MSCs than in their HSCs [38]. Comparing MDS-associated MSCs to their healthy counterparts, the former show a higher susceptibility to mutations during culture. Studies have demonstrated that, in comparison to healthy MSCs, MDS-associated MSCs exhibit increased levels of genotoxic stress indicators, such as the frequency of phosphorylation of yH2AX foci [39] and replication protein A (RPA) [40]. The staining results of yH2AX in MDS patients were found to be correlated with the frequency of mutations. Mutations in MSCs activated by β-catenin are linked to the emergence of myelodysplastic alterations and common genetic changes in AML in MDS [41]. When MSCs' β-catenin is activated, Jagged1 is expressed, which in turn causes hematopoietic stem and progenitor cells (HSPCs) to activate Notch signalling, which in turn accelerates the development of leukaemia. Fully activating the inflammatory response, which involves nuclear factor kappa B (NF-κB), TGF-β, tumour necrosis factor (TNF) signalling, and EGF, is possible with highly purified CD271+ MSCs obtained from low-risk MDS patients [42]. Overall, because of these anomalies, BMSCs may accelerate the course of the disease [43].
2.3. Abnormal Hematopoietic Microenvironment Induced by MSCs in MDS
MDS-BMSCs have demonstrated a capability of contributing to the abnormal hematopoietic microenvironment. The presence of BMSCs within the tumour microenvironment can result in immune dysregulation and immune disorders through the regulation of hematopoietic-related factors [44]. In this context, there is no consistent answer as to whether MDS-BMSCs can effectively support haematopoiesis due to differences in experimental procedures and patient heterogeneity. Some studies suggest that patient BMSCs can sustain the growth of both leukaemic/autologous HSPCs and HSPCs derived from healthy individuals [45]. Conversely, other studies indicate that MDS-BMSCs have a diminished capacity to support normal HSCPs [46]. These observations are attributed to the impaired expression of niche-derived molecules that are known to influence haematopoiesis, such as angiopoietin (ANGPT), osteopontin, Jagged-1, hepatocyte growth factor (HGF), kit ligand, C-X-C motif chemokine ligand 12 (CXCL12), thrombopoietin (TPO), and insulin growth factor binding protein 2 (IGFBP2), as well as TGFβ1 [47].
Notably, Jagged-1, involved in the Notch signalling pathway, is crucial for the differentiation of HSCs. The impaired Jagged 1 expression might lead to the disruption of this process [48]. Furthermore, CD73+ MDS-BMSCs have been shown to negatively affect the clonogenic potential of autologous hematopoietic cells, compared to CD73+ BMSCs derived from healthy donors on normal hematopoietic cells. This is associated with an increased expression of focal adhesion kinase, a protein responsible for different processes, including differentiation, adhesion, survival, and proliferation, in patient-derived BMSCs [49]. Additionally, the upregulation of inflammatory factors and inhibitors of haematopoiesis due to the activation of NF- kB, such as IL-8, IL-6, and C-C motif chemokine ligand (CCL3), has been demonstrated to attenuate HSPC numbers and function ex vivo in CD271+ BMSCs from lower risk-MDS (LR-MDS) [50]. Moreover, Sbds deletion observed in osterix+ MDS-MSCs promotes genotoxic stress in HSPCs through the inflammatory p53-S100A8/9-TLR signalling, leading to MDS progression [51]. Another study has indicated that when healthy haematopoietic cells are exposed to MDS-MSCs, they will have a toxic effect on CD34+ HSPCs. These abnormal MDS-MSCs might lose their capacity to support normal haematopoiesis and impact the clonal proliferation of HSCs [52]. Furthermore, MSCs derived from patients with both early-stage and late-stage MDS exhibit decreased expression of angiopoietin/angiopoietin-like, a protein that regulates HSPC quiescence [43].
Exosomes secreted by ex-vivo expanded BMSCs from LR-MDS patients have been shown to have a different miRNA cargo than their normal counterparts. Patient BMSCs-derived exosomes are incorporated into CD34+ cells from healthy donors, change their gene expression through miRNA transfer (such as miR-15a and miR-10a), and increase their clonogenic potential and viability [53]. Figure 2 illustrates the relationship between MSCs and HSPCs in MDS. Notably, MSCs can regulate the differentiation and proliferation of HSCs via the secretion of chemokines, E-selectin, and crosstalk molecules like Jagged-1 and CXCL12. Aberrant secretion of these molecules might lead to abnormal differentiation and proliferation of HSCs. Also, it was found that the co-culturing of megakaryocytes with MSCs has been shown to induce alterations in LAT and Rap1b gene expression, resulting in the generation of platelets that exhibit low basal activation levels, suggesting its contribution to the MDS development [54].
2.4. Immunomodulatory Dysfunction by MSCs in MDS
MSCs possess immunomodulatory properties exerted through cell-to-cell contact and in a paracrine pattern. MDS is characterised by significant deregulation of the immune system, which may contribute to the progression of early MDS to advanced MDS [55].
Recent studies have suggested that BMSCs may play a significant role in the development of MDS via the immune abnormalities they induce. For example, BMSCs in MDS patients have shown a defect in their ability to inhibit T-cell proliferation and activation [56]. Some studies have also demonstrated that MDS-BMSCs have an impaired capacity to inhibit T-cell proliferation when stimulated in mixed-lymphocyte reactions [34]. Furthermore, one study found that MDS-BMSCs were less effective in inhibiting T-cell proliferation but had a similar capacity to induce regulatory T-cells compared to normal BMSCs. They showed that high-risk MDS MSCs (HR-MDS) showed lower inhibition effect on T cell apoptosis compared to low-risk MDS MSCs (LR-MDS MSCs). Even though both HR-MDS MSCs and LR-MDS MSCs suppressed T cell proliferation, the immunosuppressive impact of LR-MDS MSCs on the proliferation of T cells was less than that of HR-MDS MSCs. Moreover, they did not find any significant differences in the formation of CD4+CD25+Foxp3+Tregs in the absence of TGF-B1 in MDS-MSCs or control MSCs, suggesting the role of TGF-β1 produced by MDS-MSCs or control MSCs in the generation of Tregs [57]. Interestingly, it has been reported that BMSCs obtained from MDS patients can induce normal monocytes to acquire the properties of myeloid-derived suppressor cells, which eventually leads to the downregulation of NK and T cell function. However, this phenomenon was not observed in BMSCs obtained from healthy donors [58]. The present findings indicate a disturbance in the balance of the bone marrow macroenvironment (BMME), leading to altered immune regulation. The impaired ability of BMSCs to inhibit the activation and proliferation of T cells may contribute to immune dysregulation, potentially allowing for heightened T cell responses and chronic inflammation. Such alterations in the immune milieu could have an impact on the progression of MDS, as they may permit the survival and expansion of abnormal hematopoietic clones, thereby potentially contributing to the evolution of MDS to AML [59].
2.5. Cytokine Dysregulation Mediated by MSCs in MDS
The pathogenesis of MDS might be caused by various defects in the BMME, including improper secretion of angiogenic cytokines by MSCs. Studies have shown that MDS patients have elevated levels of cytokine production in their BM, such as TNF-α, IL-6, and interferon-gamma (IFN-γ) [60]. Further research has provided a disease-progression model in which the overproduction of IFN-γ and TNF-α by MSCs in the BM is the primary event. This results in the expression of the B7-H1 molecule on MDS blasts, which creates a bifunctional signal causing T-cell apoptosis and enhancing the proliferation of blasts [61].
MDS-MSCs demonstrate signs of ongoing stromal stimulation and response to an inflammatory environment, which is accompanied by increased expression of genes associated with fibrosis (ADAMTS4, SPARC, and LOXL2), a feature of MDS [43]. A study has also highlighted that a gene set, including cytokine-cytokine receptor interaction, is significantly enriched in MDS MSCs. The CXCR4/CXCL12 signalling pathway sustains haematopoiesis in the BM through MSCs, as well as a reduction in expression of some haematopoietic factors, such as stem cell factor (SCF), insulin-like growth factor 1 (IGF1), and TPO [62], leading to MDS by an impairment in the normal process of blood cell formation, growth and differentiation of HSCs and progenitor cells, contributing to the ineffective haematopoiesis characteristic of MDS and the production of dysfunctional and abnormal blood cells. More detailed, SCF is known to be involved in maintaining the HSC niche within BM to provide a supportive environment for haematopoiesis; for this reason, a decrease in SCF expression might disrupt the integrity of the niche, affecting the interactions between HSCs and the BMME and the self-renewal/differentiation of HSCs, contributing to MDS pathogenesis. TPO is a key regulator of platelet production, stimulating the differentiation of megakaryocytes and subsequent platelet release. A reduction in TPO expression might lead to thrombocytopenia characterised by low platelet counts, which is a common feature in MDS and can contribute to bleeding tendencies [63].
A decrease in IGF1 playing a role in promoting cell survival and growth might affect growth signalling pathways, potentially affecting the survival and proliferation of haematopoietic cells. Dysregulation of growth signalling is implicated in the abnormal expansion of malignant clones seen in MDS [64].
2.6. Altered Growth Kinetics and Elevated Cellular Senescence of MSCs in MDS
Studies have shown that MDS-derived BMSCs have impaired growth potential, associated with a decrease in the expression of CD44 and CD49e [65]. CD49e is a marker expressed on the surface of 97.5% of BMSCs [66]. Its presence is crucial for facilitating cellular attachment to the ECM and for the formation of adhesion complexes with cytoskeleton proteins and the activation of kinases regulating the signalling pathways related to cell division, survival, and differentiation [67]. CD44 expressed on the surface of BMSCs with a rate of 99% is essential for homing and adhesion of haematopoietic progenitor cells (HPCs) to MSCs, facilitating the establishment of a supportive microenvironment for haematopoiesis [68].
It has been observed that in some cases, the reduced growth potential is linked to an increase in cellular senescence [37]. The lower proliferative capacity of MDS-BMSCs could also be attributed to the downregulation of the canonical Wnt signalling pathway and the up-regulation of the non-canonical pathway. A recent study confirmed the downregulation of the canonical WNT signalling pathway in BMSCs derived from MDS patients [69]. Additionally, another study that used the samples of patients with different MDS subtypes such as RCMD, RAEB I, RAEB II, del5q, CMML, and MDS-U has shown that MSCs from MDS patients are more susceptible to cellular senescence. This is evident by a significantly higher number of SA-β-galactosidase-positive cells compared to healthy controls. Moreover, the same study also showed reduced clonogenic potential of all MDS subtypes, and it has been indicated that only osteogenic differentiation ability of all MDS subtypes was reduced, which is reflected by a reduction in expressions of Osterix and osteocalcin (OC) and is linked to specific methylation signatures of some genes such as T-Box transcription factor 15 (TBX15), paired-like homeodomain transcription factor 2 (PITX2), and homeobox B1 (HOXB1). Specifically, HOX genes are responsible for stem cell differentiation towards osteogenesis, chondrogenesis, and adipogenesis and are involved in joint and bone formation. HOXB1 hypermethylation at its promoter region led to a decrease in messenger ribonucleic acid (mRNA) expression. TBX15 expression in mesenchymal precursor cells is essential for bone development and TBX15 hypermethylation in MDS MSCs led to a reduction in mRNA expression of TBX15, suggesting that TBX15 silencing due to hypermethylation might be reason for decreased osteogenic differentiation potential. Finally, PITX2 hypermethylation at its gene body caused an overexpression of PIT2X mRNA in MDS MSCs [52].
The increased senescence and impaired growth kinetics may affect the ability of MSCs to maintain a supportive microenvironment for HSCs and consequently hinder normal haematopoiesis. Moreover, one study found that MDS-MSCs are dysfunctional, causing abnormalities in healthy HSPCs. These transformed self-replicative MDS-MSCs that can transmit biological abnormalities to their progeny can result in impaired haematopoietic regeneration in healthy CD34+ HSPCs following exposure to these cells. Notably, the ensuing impairment can be sustained autonomously in HSPCs for significant periods of time by an MDS stroma. Findings have demonstrated a decrease in the haematopoietic expansion of CD34+ cells and colony-forming cell (CFC) potential following the co-culture of healthy HSPCs and HR-MDS-MSCs [50]. On the other hand, a study conducted by Rathnayake et al analysed the patients with different de novo MDS subtypes, such as RCMD, RCUD, RAEB I, RAEB II, and Del(5q). They found that culture-expanded MDS MSCs across all MDS types and healthy controls exhibited thin spindle and fibroblast-like cellular morphology. Both all MDS MSCs and healthy controls showed positive expression of CD90, CD105, and CD73 and negative expression of CD45 and CD34, however, the percentage of positive cells in RCMD was lower in comparison with other subtypes. 33% of patients with RAEB, 60% of patients with RCMD, and 16.7% of patients with RCUD had abnormalities in karyotype including random loss of chromosomes in their MSCs and these abnormalities were different than that in BM karyotypes. Unlike other studies indicating that MDS MSCs show disrupted hematopoietic support because of their reduced growth characteristics and increased cellular senescence, doubling time of MDS subtypes were not remarkably different from that of controls or among the subtypes, indicating that there is no significant change in growth kinetics between controls and MDS subtypes. Moreover, unlike other studies, all MDS subtypes had adipogenic and osteogenic differentiation capacity. These discrepancies between this study and existing literature might be due to differences in culture conditions or experimental methodologies, different pathophysiology of MDS classes, and heterogeneity of the MDS biology [70].
2.7. Reduced Osteogenic Differentiation Caused by MSCs in MDS
Studies have shown that MDS-MSCs do not exhibit significant differences in adipogenic or chondrogenic differentiation, but their potential for osteogenic differentiation is remarkably reduced. This reduction in osteogenic differentiation is due to a decrease in the levels of microRNAs (miRNAs) involved in the early differentiation process toward osteoblasts, such as Osterix, a transcription factor, and OC, a marker of mature osteoblasts. A deletion of Dicer1, a protein involved in the processing of miRNAs, has been linked to impaired osteogenic differentiation of MSCs related to MDS evolution in mice. In detail, Dicer1 reduction is already linked to an onset of cellular senescence in different types of cancer cells. In the case of MDS MSCs, a study showed that the ablation of Dicer1 caused MSCs to undergo senescence until the seventh day. Also, Dicer deletion has been shown to inhibit adipogenic and osteogenic differentiation and decrease the support of healthy CD34+ cells. The same study also showed that all MDS subtypes exhibited osteogenic differentiation capacity, which is evident by decreased Runx2, essential for skeletal morphogenesis and osteoblastic differentiation, and alkaline phosphatases (ALP), an indicator of the new bone formation and the presence of osteoblast cells. In contrast, all MDS subtypes showed 2-fold higher adipogenic differentiation potential, indicated by increased expression of fatty acid-binding protein 4 (FABP4), important in maintaining adipocyte homeostasis and regulating adipogenesis. Interestingly, when Dicer1 is overexpressed, these impaired functions are reversed [71].
Numerous studies have demonstrated that MDS-MSCs exhibit impaired PI3K/AKT and Wnt/β-catenin signalling pathways. Dysregulation of genes within the PI3K/AKT pathway in MSCs may impede their capacity to interact with normal hematopoietic cells, ultimately resulting in bone marrow failure and the development of myelodysplasia Falconi et al., 2016). Aberrant Wnt/B-catenin signalling pathways have been reported to stimulate MSC proliferation while inhibiting osteogenic proliferation, thereby contributing to the pathogenesis of MDS [72]. Consistent with the impaired osteogenic differentiation potential of BMSCs derived from patients, an early study on transiliac bone biopsies obtained from MDS patients demonstrated abnormalities in bone remodelling, characterised by reduced numbers of osteoclasts and osteoblasts, as well as decreased bone formation, as evidenced by the diminished mineral apposition rate [73]. A report consistent with these findings has also demonstrated bone loss in MDS patients [74]. Altogether, the reduced osteogenic differentiation capacity of MSCs can significantly impair their ability to support HSPCs, thereby leading to ineffective haematopoiesis in MDS.
MDS-MSCs have been found to exhibit irregular morphologies, form disorganised colonies, and demonstrate significantly reduced osteogenic differentiation capacity, suggesting their contribution to the ineffective haematopoiesis of the MDS and their insufficient stromal support in MDS. An imbalance between osteoblastic cells and adipocytes, which are differentiated from MSCs, may also be the reason for this. Osteoblastic cells usually support haematopoiesis, while adipocytes suppress it. If the process of osteogenic differentiation is disturbed, adipocytes can suppress haematopoiesis, contributing to MDS [75]. However, despite the proven low proliferative capacity of MSCs in MDS, the causes are unclear.
The origin of MSC abnormalities in relation to MDS remains a subject of inquiry. One possibility is that the BM may trigger oncogenesis. When the integrity of haematopoiesis is damaged, it creates an environment for hematopoietic progenitor cells to initiate clonal transformation, leading to the pathophysiology of MDS. Specifically, BM abnormalities contribute to the development and expansion of MDS clones, which in turn disrupt the BM environment by producing abnormal levels of cytokines, promoting clonal expansion and disease progression (Figure 3). It is also possible that the agent responsible for causing genetic damage in myelodysplastic hematopoietic cells, such as aromatic compounds, viruses, or radiation exposure, might have also affected MSCs. Another possibility is that an abnormal microenvironment could have created a favourable condition for the clonal expansion of hematopoietic stem cells. Reciprocal heterotypic signalling between MSC and disease-propagating hematopoietic cells within the BM might be required for the disease initiation/progression. The first step could be the "reprogramming" of MSCs by hematopoietic cells, followed by a series of events that have yet to be defined, which would promote the progression of the disease [43].
3. Role of MSCs in Leukaemia Pathogenesis
Leukaemia is a group of malignant tumours characterised by the abnormal and uncontrolled growth of cells that originate from hematopoietic precursor cells in the BM. Therefore, the BMME plays a crucial role in the development of leukaemia.
The classification of leukaemia is an integral component of the World Health Organization (WHO) classification of tumours, based on evidence used for different purposes, such as research and diagnosis, monitoring public health, and registries of cancer on a global scale. The WHO Classification of haematolymphoid Tumour's forthcoming 5th edition is grounded in molecular biology and emphasises the utilisation of biomarkers that can be therapeutically and/or prognostically [77]. The classification places a priority on categorising tumour types based on the identification of defining genetic abnormalities, whenever possible, and includes emerging entities as subtypes of the disease under a framework of other specified genetic alterations. The classification integrates data from various sources, such as morphology, cytogenetics, and molecular biology, to provide a comprehensive and precise classification of leukaemia [78]. Different categories of leukaemia based on their origin are as follows.
Myeloid leukaemia is a form of leukaemia that results from the abnormal proliferation of myeloid cells, which are responsible for producing blood components (platelets, white blood cells, and red blood cells). Myeloid leukaemia encompasses subtypes: AML and chronic myeloid leukaemia (CML). CML is divided into chronic and blast phases, focusing on risk factors in the chronic phase. Lymphoid leukaemia arises from the abnormal growth of lymphoid cells, which play a role in producing immune cells known as lymphocytes. It includes subtypes such as acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL). Histiocytic/dendritic cell leukaemia is a type of leukaemia which originates from abnormal growth of histiocytic or dendritic cells and is part of the immune system. It is considered a very uncommon distinction of leukaemia. MDS-related leukaemia refers to a group of disorders characterised by abnormal production of blood cells. In certain cases, MDS can progress to leukaemia, known as MDS-related leukaemia or AML myelodysplasia-related (AML-MR).
Leukaemia is also associated with specific genetic conditions; for example, this condition can occur in individuals with certain genetic conditions, such as Down syndrome, RASopathies and Fanconi anaemia. These cases are classified based on the specific genetic condition and the associated myeloid disease phenotype [79].
MSCs located in the BMME are believed to be important in leukaemia development [80]. It is worth mentioning that MSCs exhibit both anti-tumorigenic and pro-tumorigenic effects in leukaemia. On one hand, MSCs, irrespective of their origin, can impede tumour growth by suppressing tumour cell proliferation, which is achieved by arresting the tumour cell cycle, inhibiting angiogenesis, decreasing vascular density, and their paracrine action, including their ability to secrete extracellular vesicles that can exert an anti-proliferative impact on leukaemic cells. On the other hand, they can facilitate tumour growth by inhibiting tumour cell apoptosis [81]. The biphasic nature of MSCs could conceivably have implications for leukaemia, as depicted in Figure 4.
3.1. Pro-tumorigenic Effects of MSCs in Leukaemia
Numerous studies demonstrated that MSCs can protect leukaemic cells and enhance their survival through multiple mechanisms. One of these mechanisms is their capacity to impede apoptosis of leukaemic cells by utilising signalling pathways and soluble factors present in the co-culture medium, which in turn enhances their viability in experimental settings. Additionally, the interplay between leukaemic cells and MSCs plays a crucial role in the survival of leukaemic cells, indicating that both cell types can impact each other.
In a study conducted by Manabe et al., it was discovered that MSCs possess anti-apoptotic activity in acute lymphoblastic leukaemia via the secretion of cytokines and soluble factors [83]. Another study found that MSCs were able to reduce apoptosis in BV173 CML cells. Upon contact with MSCs, leukemic cells were found to be in a resting state (G0/G1), and a downregulation of cyclin D2 was observed. This downregulation is believed to preserve the proliferative capacity of the leukemic cells, ultimately improving their chances of survival and self-renewal ability. The same study also demonstrated that the characteristics of MSCs vary based on the type of study conducted, whether in vitro or in vivo [84]. Another study revealed that MSCs can facilitate the growth of ALL cells in an in vivo model by increasing luciferase activity. This suggests that MSCs may have a negative effect on the recurrence of ALL cells [85]. In addition, a study conducted by Naderi et al. using BMSC demonstrated that MSCs can protect BCP-ALL cells from p53 accumulation and apoptosis. This is achieved through the production of PGE2 and activation of the cAMP-PKA signalling pathway, ultimately inducing leukaemogenesis [86].
Leukaemic cells need to progress through vascular support, which is facilitated by MSCs. MSCs directly contribute to the density of tumour vasculature by differentiating into endothelial cells [87] or pericytes involved in tumour stabilisation [88]. Additionally, MSCs indirectly support tumour vasculature by secreting pro-angiogenic factors, including VEGF and other pro-angiogenic cytokines, such as fibroblast growth factor (FGF), ANGPT-1, and IL-6, which are necessary for the angiogenic activity of VEGF [89,90,91]. As a supporting study, AML-MSCs have been found to have a high level of VEGFA and IL-6, which contribute to tumour-supportive angiogenesis [92,93]. Similarly, CLL patients have an increased amount of PDGF and VEGF. In MSCs, PDGF selectively activates PDGF receptor (PDGFR), which in turn induces MSC VEGF production through a PI3K-dependent mechanism, leading to neovascularisation and disease progression [94].
MSCs can lead to tumour growth by their immunomodulation effect on leukaemia. For instance, a study demonstrated that granulocytic-myeloid-derived suppressor cells primed with CML-MSCs exhibited an increase in immunomodulatory factors, including TNF-α, IL-1β, Arginase-1, IL-6, and cyclooxygenase 2 (COX-2), thereby supporting the contribution of MSCs to tumorigenesis [95]. Another study revealed that MSC stimulation in ALL led to the upregulation of C-C motif chemokine ligand 2 (CCL2) and IL-8 inflammatory cytokines, which increased the adhesion support of MSCs for ALL cells, ultimately enhancing the proliferation and survival of ALL cells [96]. Additionally, MSCs were found to cause the dysfunction of T cells and the proliferation of Tregs in HR-MDS in CML patients, and they were strongly associated with changes in MSCs [97,98]. Furthermore, while MSCs are known to inhibit DC maturation under normal physiological conditions, one study demonstrated that CML-MSCs could activate regulatory DCs, leading to the inhibition of T cell function or the promotion of Tregs proliferation, hence indirectly promoting immune escape in CML [99].
MSCs can create an environment that promotes tumour growth by increasing the stemness of tumour cells. MSCs can also differentiate into various types of cells associated with the tumour microenvironment, including cancer-associated fibroblasts (CAFs) [100]. In addition, MSCs have been found to enhance the stemness of cells in blood cancers through the activation of the Bruton tyrosine kinase signalling pathway [101]. However, further research is needed to fully understand this mechanism.
MSCs can exert their pro-tumorigenic effects by inducing metastasis. Leukaemic cells can exploit the BMME and modify the chemokine axes to create a malignant niche. In line with this, the chemokines produced by MSCs can affect the migration of leukaemic cells and increase their metastatic potential [102]. Moreover, Activin A, which is a protein that can elevate the migratory capacity of leukaemic cells in response to chemoattractant CXCL12 while suppressing the migration of healthy CD34+ cells, was found to be highly expressed by MSCs in the BM of paediatric B-ALL patients, compared to healthy counterparts, giving B-ALL cells a greater chance to metastasise, as observed in a xenograft mouse model and in vitro [103].
Leukaemia drug resistance triggered by MSCs is a prevalent occurrence. A study conducted by Vianello et al revealed that BMSCs offer non-selective protection to CML cells from imatinib-induced apoptosis through the CXCR4/CXCL12 axis [104]. A study determined that MSCs have an anti-apoptotic effect on ALL cells treated with Adriamycin, leading to an increase in the expression of the Bcl-2 gene and an enhanced Bcl-2/Bax ratio [105]. A different study indicated that cell-to-cell contact between AML cells and MSCs promotes resistance against mitoxantrone in AML through c-Myc upregulation, preventing cancer cells from undergoing apoptosis [106]. Lastly, Carter et al found that the interaction between AML cells and MSCs regulated by the ARC protein induces the expression of IL-1β in AML cells, which leads to resistance against cytarabine [107].
3.2. Anti-tumorigenic Effects of MSCs in leukaemia
MSCs can also have anti-tumorigenic effects through various mechanisms. One of the widely accepted mechanisms is their ability to cause tumour cell cycle arrest. For instance, a study conducted by Fonseka et al showed that human umbilical cord-derived MSCs (hUC-MSCs) inhibited the proliferation of K562 cells by inducing an arrest in the G0/G1 phase through the secretion of IL-6 and IL-8 [108]. Similarly, Zhu et al. investigated the impact of MSCs derived from human adipose tissue on the K562 CML cell line and found that these MSCs impeded tumour cell proliferation by inducing cell cycle arrest through dickkopf-related protein 1 (DKK1) secretion [109]. In another study, Wei et al. co-cultured K562 CML cells with MSCs derived from BM of leukaemia patients and observed a decrease in the number of leukaemia cells in the S phase and an increase in the number of cells in the G0-G1 phase. Meanwhile, MSCs were observed to decrease apoptosis through the PI3K-Akt-Bad signalling pathway, as evidenced by the phosphorylation of Akt and Bad proteins, indicating a protective effect against apoptosis [110]. Another study by Tian et al. revealed that UC-MSCs inhibited the growth of HL60 (AML) and K562 (CML) cells by arresting them in the G0-G1 phase through increasing the phosphorylation of p38 MAPK in these cells [111]. The Wnt signalling pathway has also been shown to be involved in the anti-tumorigenic effects of MSCs, in addition to the p38 MAPK signalling pathway. Han et al found that BMSCs from both healthy individuals and those with CML increased the anti-apoptotic ability of cancer cells. This was accomplished through regulation of apoptosis-related protein expression, such as Bcl-2, Bax, caspase-3, and activation of the Wnt signalling pathway in CML patients [112]. Furthermore, a study conducted with human UC-MSCs from T-Acute lymphoblastic leukaemia (Jurkat cell line) revealed that MSCs inhibit cell proliferation by arresting the cells in the G0/G1 phase and protect the cells from apoptosis by activating the Notch signalling pathway [113].
In a study by Song et al the effects of MSCs on the growth of various types of leukaemia and lymphoma cells were examined. The results revealed that the MSCs inhibited the growth of these cells, arrested cell cycle progression at the G0/G1 phase and promoted early apoptosis. These effects depended on factors such as the length of co-culture, the number of MSCs used, and the duration of exposure, as the anti-proliferative effects were no longer observed when the MSCs and cancer cells were separated by a permeable membrane. The study also demonstrated that the inhibition of cell proliferation by mouse MSCs may not involve the inhibition of Akt, a protein that promotes cell survival and growth. Furthermore, the study found that intravenous administration of MSCs could reduce graft-versus-host disease (GVHD) without increasing tumour growth or incidence in a model of allogeneic BMT [114]. Similarly, Sarmadi et al. revealed that MSCs can effectively hinder the growth of a specific number of CML cells, surpassing the effect on Jurkat cells (T-ALL). The study also highlighted that the dose-dependent inhibition of cancerous cell proliferation is mainly attributed to cell-to-cell contact. Furthermore, MSCs demonstrated the ability to arrest the cell cycle of leukaemia cells in the G0/G1 phase, which prevented further division in the S phase [115]. Furthermore, a study has shown that the secretome of human Wharton's jelly derived-MSC has anti-tumour and cytotoxic effects on K562 CML cells. This suggests that specific substances or paracrine signals are involved, indicating that MSCs could be used to reduce the dose of cytotoxic drugs and their side effects [116]. Zhu et al. conducted a study which demonstrated that MSCs can impede the proliferation of K562 cells in the humoral microenvironment through the secretion of DKK-1, a negative regulator of the Wnt signalling pathway [109]. According to a study, MSCs have been found to inhibit the growth of CML cells, likely through the production of IFN-α. This mechanism could provide a promising approach for immunotherapy of CML [117]. Additionally, another study has shown that BMSCs can inhibit the proliferation of K562 CML and arrest the cell cycle at the G0/G1 phase. This inhibition occurs via caspase3, while co-culture media of K562 cells and BMSCs contained cytokines such as cytokine-induced neutrophil chemoattractant-1 (CINC-1) and tissue inhibitor of metalloproteinases-1 (TIMP-1) [82].
Notably, it has been suggested that MSCs play a dual role in leukaemia due to the heterogeneity of both leukaemia and MSCs [118]. Also, it is important to consider the number of leukaemic cells and MSCs cultured together as it might affect their dual role. The density of MSCs in the culture has a significant impact on their proliferation rate, secreted factors, and morphology. Studies have shown that MSCs have multiple functions, such as immunoregulation, which can alter their impact on tumour suppression or promotion [119,120]. Moreover, the anti-tumorigenic effects of MSCs on solid cancers have been associated with a lower number of MSCs, whereas the pro-tumorigenic effect is observed with a higher number of MSCs [121]. However, this association has not been studied for leukaemia owing to a lack of data. Therefore, further research is necessary to understand the mechanism of their anti-tumorigenic effect and standardise the concentration of leukaemia cells and MSCs.
3.3. Changes in MSCs in Leukaemia
MSCs are believed to contribute to leukaemogenesis through genetic defects in MSCs from leukaemia patients. For example, MLL-AF4 is a fusion gene commonly found in infant B-acute lymphoblastic leukaemia (B-ALL). a study showed that MLL-ENL, MLL-AF10, or MLL-AF9 fusion genes were absent in BMSCs of childhood leukaemia, but MLL-AF4 was detected and expressed in BMSCs from all cases of MLL-AF4+ B-ALL, indicating its possible tumour-associated role [122]. Additionally, MSCs from patients with childhood B cell precursor ALL have been found to exhibit various chromosomal translocations, including E2A-PBX1, TEL-AML1, or MLL rearrangement [123]. Another study found that BMSCs were cytogenetically abnormal in 54% of AML patients [124].
MSCs isolated from leukaemia patients have been found to exhibit functional abnormalities as well as genetic abnormalities. For example, a comparative study of the growth kinetics and integrin mRNA expression levels of BMSCs obtained from patients with CML revealed that the growth characteristics and cell proliferation of MSCs from CML patients were indistinguishable from those of the control group. However, the expression of integrin mRNA was found to be significantly higher in the former group. Elevated levels of integrin mRNA expression might enhance adhesion, supporting CML cells and shielding them from the effects of immune surveillance [125].
MSCs in leukaemia are known to have immunomodulatory defects. In several studies, MSCs from patients with Hodgkin disease (HD), non-Hodgkin lymphoma (NHL), MDS, ALL, and AML were tested for their immunomodulatory effects on T lymphocytes. It was found that MSCs from patients with ALL, HD, and NHL could inhibit T-lymphocyte proliferation stimulated in a mixed-lymphocyte reaction in a dose-dependent manner through TGFβ1 and HGF. In contrast, the MSCs from AML patients failed to suppress T cell proliferation [126]. A separate study showed that MSCs from CML patients exhibited a reduced inhibitory effect on T cell activation and proliferation. The MSCs' ability to silence T cells in the G0/G1 phase of the cell cycle was reduced in MSCs from CML patients.
The immunomodulatory effects of MSCs in various types of leukaemia have been found to exhibit changes. The immune response type and severity vary according to the pathophysiological features of different kinds of leukaemia, which might explain why the immunomodulatory effects of MSCs could be abnormal in one type of leukaemia and normal in another. Factors and cytokines secreted by MSCs in leukaemic conditions differ from those secreted in normal conditions. For instance, BMSCs in AML patients produce much lower levels of SCF, TNF-α, monocyte chemoattractant protein-1 (MCP-1), GM-CSF, and IL-6 compared to normal BMSCs from healthy controls. Lower levels of MCP-1 preclude the anti-tumour activity of MSCs, and lower levels of GM-CSF suggest that the leukaemic BMME results in a diminished ability to support normal haematopoiesis [44]. It has also been suggested that the heightened level of IL-8 produced by MSCs in AML leads to increased vessel density of AML BM, contributing to leukaemogenesis [127]. Furthermore, the elevated level of indoleamine 2,3-dioxygenase (IDO) secreted by MSCs in leukaemia leads to apoptosis of T cells, thereby reducing the immune system's ability to cope with leukaemia [128].
4. Role of MSCs in Lymphoma and Multiple Myeloma Pathogenesis
Since the discovery of MSCs, a plethora of investigations have been conducted globally to comprehend their characteristics and functions. They demonstrate the ability to migrate to damaged tissue, undergo self-renewal, and exert immunomodulatory and antitumor effects [129]. Indeed, MSCs have been extensively employed in regenerative medicine for the purposes of bone and cardiovascular repair [130]. Despite the extensive research conducted over the past decade, the question of whether MSCs exhibit tumour-suppressing or tumour-promoting effects remains unresolved. There are around 42 clinical trials that have been registered, all of which are focused on examining the impact of MSCs on tumours. However, it is worth noting that only 13 of these trials specifically concentrate on investigating the effect of MSCs on hematologic malignancies, encompassing myelodysplastic syndrome, leukaemia, lymphoma, and multiple myeloma [81].
MSCs have both antitumorigenic and pro-tumorigenic effects in leukaemia by either inhibiting tumour growth by suppressing tumour cell proliferation or by promoting it through suppressing tumour cell apoptosis [131]. This multifaceted function of MSCs can be characterized as a "paradoxical phenomenon".
Although MSCs have the capability to hinder and exacerbate hematologic malignancies, they also possess the potential to decrease the proliferation of tumour cells under laboratory conditions. Furthermore, these cells have been observed to exhibit comparable immunosuppressive properties, surface antigens and phenotypes [132].
Song et al conducted a study where they cultivated MSCs derived from the BM of C57BL/6 mice together with A20 murine B-lymphoma. They assessed various aspects such as cell proliferation, progressive cell cycle stages, apoptosis, and secretion of cytokines. As a result, MSCs exhibited the ability to inhibit the growth of lymphoma cells in vitro by inducing cell cycle arrest and reducing the production IL-10 [114]. This study also illustrated the antitumor properties of MSCs through the suppression of tumour development in an in vivo setting. After intravenous administration of MSCs into BALB/c mice implanted with BALB/c-derived B-lymphoma A20 cells, Song et al. observed a decrease in the frequency of lymphoma and an enhancement in survival rates. Following co-culture with MSCs, the concentration of IL-10 obtained from the supernatant of A20 cell cultures exhibited a significant reduction in an immoderate manner. This phenomenon could contribute to the evasion of immune responses. Additionally, in the presence of MSCs, the proportion of A20 cells expressing intracellular IL-10 markedly increased, indicating that MSCs hinder the IL-10 secretion by A20 cells. The researchers concluded that the unforeseen tumorigenic impact of MSCs observed in diabetic/non-obese and immunodeficient mice with lymphoma could be attributed to the specific animal model used. They noted that immunodeficient mice fail to mirror the environment in which autologous tumour growth occurs, thus emphasising the superior reliability of their in vivo data obtained using BALB/c mice.
Extracellular vesicles (EVs) derived from MSCs, particularly endosome-derived exosomes, have shown potential anti-tumour effects [133]. MSCs can inhibit vascular growth by inducing endothelial cell apoptosis and reducing vascular density, leading to tumour growth inhibition. The modulation of a vascular endothelial cadherin signalling pathway is involved in this process [81]. In a study by Roccaro et al, in the context of multiple myeloma, exosomes derived from BMSCs associated with the tumour were found to transfer miR-15a, a tumour-suppressive miRNA, at low or undetectable levels [134]. This resulted in an increase in tumour burden and heightened dissemination of cancer cells to distant bone marrow niches. Conversely, exosomes secreted by normal BMSCs, which carry normal levels of miR-15a, contribute to the inhibition of multiple myeloma tumour growth by reducing the spread of cancer cells. The mechanisms by which anti-tumour agents induce cancer cell arrest and the effects of MSCs on lymphoma and other hematologic malignancies are not well understood, and further research is needed to study the molecular processes involved in tumour cell cycle arrest and the antitumor effects of MSCs on hematologic malignancies.
MSCs have pro-tumorigenic effects in hematologic malignancies in vivo and in vitro by promoting tumour growth and suppressing apoptosis of tumour cells. Mechanisms favouring tumour growth involve signalling pathways and a wide range of factors, which further require cellular communication to be set in motion. A study emphasized the importance of cellular communication and an interplay of soluble factors by demonstrating MSCs to be anti-apoptotic in B-ALL cells [83]. The Notch signalling and the Akt-Bad signalling pathways are recognized as mechanisms leading to antiapoptotic activity and tumour development, in addition to tumour cell cycle arrest, as a version explained for MSCs possessing antitumor effects.
MSCs play a crucial role in supporting hematologic malignancies by promoting vascular support. MSCs secrete various soluble factors such as VEGF, IL-6, ANGP-1, etc., which contribute to tumour angiogenesis and neovascularisation [135]. In an experiment MSCs with multiple myeloma MSCs’ tumorigenicity (MM-MSC) were shown to influence granulocytic-myeloid-derived suppressor cells (G-MDSCs), which led to an increase in the levels of certain immunomodulatory factors, such as TNF-α, COX-2, arginase-1 and IL-1β. These factors have been associated with promoting tumour development and progression [95].
MSCs also contribute to the tumour-promoting microenvironment and increase tumour cell stemness through various mechanisms. As in the case of hematologic malignancy like MM, the MSCs advance the tumour cell stemness by activating a unique signalling pathway of Bruton tyrosine kinase. Several studies have investigated the drug resistance of hematologic malignancies induced by MSCs and showed tumour cells to acquire chemoresistance. IL-6 is a crucial cytokine secreted by MSCs in the tumour microenvironment (TME). This cytokine has a significant role in modulating the immune response, as well as regulating tumour cell proliferation, apoptosis, and tumorigenesis. Research indicates that IL-6 activates the JAK2/STAT3 and Akt signalling pathways to facilitate tumour growth and induce drug resistance in various tumours, including Mantle Cell Lymphoma (MCL) [136]. Elevated levels of IL-6 in the TME of MCL contribute to tumour progression and drug resistance. The growth of Diffuse Large B-cell lymphoma (DLBCL) is facilitated by human BMSCs not only through the secretion of IL-6 into the TME, both in vitro and in vivo but also by inducing the differentiation of Th17 cells, thus leading to increased IL-17A levels in vitro. The combined action of IL-6 and IL-17A synergistically promotes the growth and drug resistance of DLBCL cells by protecting them from spontaneous or drug-induced apoptosis in vitro. IL-6 promotes DLBCL cell growth via the JAK2/STAT3 pathway, while IL-17A enhances the growth of DLBCL cells by upregulating cyclin D2 through the activation of the PI3K/Akt signalling pathway [137]. In a study conducted, they investigated the drug-resistant ability of MSCs and found that the upregulation of two specific proteins, CXCR4 and CXCL12, contributed to their ability to resist the effect of drugs used in the treatment of chronic myeloid leukaemia. This upregulation suggests that these proteins play a role in protecting these MSCs from the effect of drugs, making them resistant to treatment [104].
5. Potential Use of MSCs in Therapies for Blood Cancers
MDS is treated with hypomethylating agents (HA), such as decitabine and azacitidine (AZA), immunomodulatory drugs, including lenalidomide, allogeneic stem cell transplantation, chemotherapy, targeted therapy combined with HA, such as venetoclax (BLC-2 inhibitor), ivosidenib (IDH1(2 inhibitors), midostaurin (FLT3 inhibitor) and immune therapy, depending on the subtype of MDS and the severity of symptoms [138].
MSCs are a promising treatment option for MDS, with the potential to improve their functions in this context. Ex vivo expanded MDS-BMSCs exhibit significantly different DNA methylation patterns than their normal counterparts, characterised by aberrant hypermethylation. To combat this abnormality, AZA treatment was found to restore the aberrant hypermethylation pattern of MDS-BMSCs, leading to increased proliferation potential and osteogenic capacity of the cells and improved ability to support HSPCs for in vivo engraftment. In other words, the therapeutic effects of AZA in MDS may be attributed to the reversal of the specific epigenetic signature of MDS-MSCs [52]. Another study has reported that AZA regulates various genes responsible for the support of haematopoiesis, which is associated with IFN-γ and ECM receptor interaction pathways, inpatient BMSCs, improving the adverse effect of MSCs on haematopoiesis [139]. Additionally, MSCs treated with AZA cause a remarkable reduction in the increased level of IL-6 in MDS-MSCs, ameliorating the inflammatory environment induced by MSCs [140]. Another study has revealed that AZA-treated MDS-MSCs improved haematopoiesis in HR-MDS and reversed MDS-MSCs’ proliferation and osteogenic differentiation capacities by normalising the BMME disrupted by MSCs [50].
Treatment with lenalidomide that can regulate the expression of chemokines in MSCs has been demonstrated to decrease C-X-C motif chemokine ligand 12 (CXCL12) secretion by MDS-BMSCs. MDS-BMSCs combined with Lenalidomide have been reported to improve BMSCs' capacity to support normal HSPCs [46].
Decitabine treatment reduces the ability of MDS-MSCs to induce the differentiation of T cells into Treg cells. This reduction is related to decreased programmed death-ligand 1 (PD-L1) expression in decitabine-treated MDS-MSCs. The same study also found that decitabine treatment altered the cell cycle of MDS-MSCs, leading to a decrease in the number of cells in the G1 phase and an increase in the number of cells in the G2/M phase. This change was related to a decrease in cyclin-dependent kinase inhibitor 1A (CDKN1A) expression, which suggests that the senescence of MDS-MSCs was improved [141]. These findings imply that administering AZA, decitabine, and lenalidomide treatments to MDS-MSCs can improve their functionality, which could be significant in treating MDS.
There are also some studies that aim to suppress the adhesion of leukaemia cells to the stroma by targeting the CXCR4-CXCL12 axis. CXCL12, crucial for enhancing the homing of CXCR4-expressing HSCs into BM, is secreted at higher levels in MDS-MSCs [142], possibly accounting for BM hypercellularity. Therefore, this axis might be of therapeutic relevance involving MSCs. Furthermore, the administration of α-lipoic acid (ALA) can reduce reactive oxygen species (ROS) levels in MSCs and decrease the intracellular iron content that results from ineffective erythropoiesis. This reduction in ROS and iron content can significantly decrease autophagy, which may be a useful factor against MDS [143]. Another study has revealed that impaired proliferation activity of BMSCs in MDS can be reversed by inducing CDKN2A [144]. When BMSCs are co-cultured with MDS-derived cells, menatetrenone has been observed to enhance apoptosis, indicating that it can aid BMSCs in supporting haematopoiesis. This promising finding suggests that Menatetrenone treatment could improve cytopenia in MDS [145]. When exposed to TGFβ1, healthy MSCs underwent functional deficits and exhibited a phenotype similar to that observed in stromal cells derived from patients. The inhibitory effects of TGFβ1 on stromal cell functionality were effectively nullified by SD-208, an inhibitor of TGFβ receptor signalling. The ability of patient-derived MSCs to undergo osteogenic differentiation was restored through the blockade of TGFβ signalling with SD-208 [47].
Leukaemia is traditionally treated with chemotherapy, radiation therapy, stem cell/BM transplantation, and targeted medication. However, researchers are now exploring the potential of MSCs as a viable option for treating leukaemia. This is due to their remarkable immunomodulatory properties, their ability to arrest the cell cycle, and their potential as delivery vehicles. Moreover, these cells are readily obtainable and can be extensively expanded in vitro [146]. They can be efficiently genetically modified in vitro [147]. Also, they have a natural inclination to migrate towards tumour environments and injured tissues [148].
Targeting MSCs could be another therapeutic approach for leukaemia. Researchers have identified these mechanisms as potential therapeutic targets. For instance, PI3K, which is frequently active in CLL, and CXCR4, a critical receptor for adhesion to MSCs and CLL cell migration, have both been found to play a crucial role in mediating fludarabine resistance in leukaemia caused by MSCs. Studies have shown that PI3K inhibitors, such as PIK-90 and PI-103, can reverse the MSC-induced fludarabine resistance in CLL cells [149]. Also, it has been discovered that the CXCR4 signalling pathway represents a critical communication pathway between leukaemic cells and their microenvironment. As such, other drugs targeting this pathway may prove effective. CXCR4 activation results in the retention of leukaemic cells via CXCL12 in the BM, where they find signals for drug resistance and growth. CXCR4 antagonists such as Plerixafor and T140 analogues disrupt the MSC-leukaemia crosstalk, thereby mobilising the leukaemia cells from their protective stromal environment and rendering them more susceptible to anti-cancer drugs [80]. In addition to CXCR4, the Notch signalling pathway is another therapeutic target. It is believed that the anti-apoptotic effects of MSCs on leukaemia cells might be mediated by this pathway. Studies have shown that using anti-notch molecule-neutralising antibodies can decrease B-ALL cell survival. Furthermore, blocking the Notch pathway entirely using GSI XII can cause an increase in the number of apoptotic B-ALL cells co-cultured with MSCs [150]. Finally, the use of hEGR1 blockers, such as E4031, has been proposed as a potential strategy to overcome chemotherapy resistance in ALL cells that are induced by MSCs through hEGR1 channels. A study conducted on NOD/SCID mice that were engrafted with ALL cells demonstrated that following treatment with specific hERG1 channel blockers, a decrease in leukaemic infiltration was observed, and the survival rate of the mice was significantly higher [151]. These findings suggest that the use of hEGR1 blockers and the inhibition of CXCR4 and Notch pathways exhibit therapeutic implications of MSCs. Therefore, further research is warranted to elucidate the mechanisms underlying the effects of these therapeutic strategies and to assess their clinical efficacy.
It has been discovered that transferring the hIFN-γ gene to MSCs can reduce the proliferation of leukaemia cells (identified as K562 CML). Additionally, the percentage of K562 cells in the G1 phase of the cell cycle was found to increase, while the number of cells in the S phase decreased. Co-culturing genetically modified MSCs with K562 cells led to an increase in apoptotic K562 cells compared to the negative control. This research indicates that it is possible to create hIFN-γ gene-engineered MSCs from CML donors that can inhibit proliferation and inducing apoptosis in K562 cells in vitro only through the local production of hIFN-γ [152]. Another study used MSCs genetically engineered to release an anti-CD33-anti-CD3 bispecific antibody for immunotherapy in AML [153]. Finally, it has been shown that engineered MSCs are effective delivery systems for gene therapy in treating leukaemia. When these cells were injected into mice with CML, the mice showed tumour regression and improved survival rates due to the in vivo interferon production by the engineered MSCs [154].
MSCs have been shown to have the potential to deliver chemotherapeutic drugs. A study demonstrated that when human MSCs were loaded with PTX, they were able to effectively inhibit the growth of leukaemia cells and angiogenesis in vitro. The study also observed that when MSCs were primed with PTX, it had a positive impact on the survival of leukaemia-bearing mice in vivo [155]. Moreover, it was found that MSC EVs containing RNA, miRNA, and proteins exhibit a capacity to significantly inhibit the proliferation of leukaemic cells when used in combination with doxorubicin, suggesting that MSC EVs have a potential in leukaemia [116]. Similarly, exosomes derived from BMSCs delivered miR-222-3p through the targeting of IRF2, inhibiting cell proliferation and promoting cell apoptosis in the AML cell line [156]. One study showed that MSCs carrying levamisole (sLipo leva) could produce an anti-leukaemia effect in mice affected with leukaemia through targeting macrophages [19]. These discoveries highlight the potential of MSCs as effective delivery systems in leukaemia gene therapy.
A recent study has demonstrated that intra-BM treatment that includes direct injection of donor MSCs into the tibia of a leukaemia-bearing mouse can restore the BMME. The study showed that intra-BM treatment helped in the functional recovery of host MSCs, improved thrombopoiesis, and rebalanced myelopoiesis. Additionally, intra-BM treatment with MSCs resulted in a decrease in tumour burden and prolonged the survival of leukaemia-bearing mice. The study also found that donor MSC treatment was able to restore the function of host MSCs and reprogram host macrophages into arginase1 positive phenotype with tissue-repair features. Based on these findings, the study concluded that donor MSCs could reprogram host macrophages for the restoration of the BMME and inhibition of leukaemia development [157]. Table 8 presents a compilation of all studies that have explored the impact of MSCs on leukaemia by suppressing tumour growth, and Table 9 presents the studies that targeted MSCs or used them as drug-delivery tools.
The multipotent nature of MSCs renders them promising targets for therapeutic interventions and an invaluable source of novel clinical treatments. They can be easily acquired, possess hypo-immunogenic properties, can be rapidly expanded in laboratory conditions, and can be transplanted [81]. Furthermore, MSCs are recognised for their innate capacity to migrate to tumour sites, owing to their tumour-tropism abilities [158]. These cells secrete various substances that inhibit the growth and advancement of cancer. The substances can be categorised into three main groups: therapeutic proteins, oncolytic viruses, and suicide genes (Figure 5) [159]. However, there have been several difficulties encountered in harnessing the homing capabilities of MSCs for targeted delivery. The utilisation of drug delivery systems enhances the pharmacological properties of anticancer drugs. Although there are limitations, such as the rapid elimination of nano-carriers directly from the circulating blood, the combination of the active targeting capability as well as the hypo-immunogenic capabilities of MSCs holds promise for anticancer therapies [158].
Yan et al explained a double-target therapeutic system for non-Hodgkin's lymphoma therapy has been designed using MSC homing capacity and scFvCD20 antigen-restriction to NHL. The system involves the expression of a fusion protein called scFvCD20-sTRAIL in hUC-MSCs, which demonstrated potent inhibition of cell proliferation in CD20-positive BJAB cells and caused a significant increase of cellular apoptosis. In a mouse model, the treatment with MSC.scFvCD20-sTRAIL significantly inhibited tumour growth compared to MSC.ISZ-sTRAIL treatment, with no observed toxicities [160].
Furthermore, on a cellular and molecular level, the effects of MSCs are primarily mediated through paracrine action. EVs, including exosomes and microvesicles, are vesicles enclosed by a lipid membrane secreted by MSCs. Exosomes derived from endosomes have been identified as physiologically significant and potent components of the MSC secretome [133]. A recent study demonstrated that the secretome of MSCs exerted an inhibitory effect on leukemic cells and exhibited cytotoxic effects when combined with doxorubicin, thereby indicating the potential of exosomes derived from MSCs in treating leukaemia [116]. Additionally, synthetically personalised exosome mimetics (EMs) could serve as alternative vehicles for drug delivery and show promise as effective therapeutic agents.
There are approximately 16 clinical trials registered on clinicaltrial.gov that aim to utilize MSCs in the treatment of hematologic malignancies. Out of these trials, 7 involve the use of engineered MSCs as carriers for therapeutic cytokines or oncolytic viruses, while 1 trial is specifically designed to assess the safety and efficacy of MSC-derived exosomes containing KrasG12D siRNA (iExosomes) [159]. A phase I/II randomized study registered with NCT04565665 explains that cord blood-derived MSCs, which is a form of tissue-derived MSC, have been implied for the treatment of hematopoietic and lymphoid cell neoplasms and are currently recruiting participants. Likewise, the NCT03184935 clinical phase I/II study makes use of cord blood-derived MSCs for the treatment of myelodysplastic syndrome patients. The clinical trials, currently using the tissue-derived MSCs based therapies for the treatment of hematologic malignancies have been enlisted in Table 10.
6. Conclusions and Future Perspectives
The anti-tumorigenic effects of MSCs are primarily attributed to their ability to inhibit the proliferation of leukaemia cells. While the precise mechanisms or molecules involved in this process remain unclear, it is widely acknowledged that cell cycle arrest at the G0/G1 phase contributes significantly to this phenomenon. To leverage this antitumor activity for clinical applications in the future, it is imperative to consider other factors. Other factors must be considered to utilise the anti-tumorigenic activity for clinical use in the future. MSCs possess certain beneficial properties, including the potential to be used as delivery vehicles and the ability to inhibit vascular growth and arrest the cell cycle. MSCs exhibit several valuable properties, including their potential to serve as delivery vehicles, their ability to inhibit vascular growth, and their capacity to arrest the cell cycle. However, they also possess certain unfavourable characteristics, such as their tendency to promote tumour growth by suppressing apoptosis, supporting tumour vasculature, and modulating the immune response of cancer cells. Furthermore, they contribute to the protection of cancer cells from drug-induced apoptosis, thereby causing chemo-resistance, which poses significant obstacles to their use as a therapeutic agent in leukaemia.
In addition, despite the broad acceptance of the dual role of MSCs in leukaemia, a solid principle that explains the anti-tumorigenic and pro-tumorigenic effects of MSCs is imperative. This principle will provide greater clarity on the impact of MSCs in leukaemia treatment and aid in the development of more effective therapeutic approaches.
Owing to the lack of standardised experimental methodologies, the heterogeneous characteristics of MSCs that are easily influenced by the neighbouring cells and surrounding environment, and the absence of specific cell surface markers to identify subsets of MSCs, the clinical outcomes of MSC-based therapeutic approaches have exhibited a wide range of variability. As such, further research is necessary for the development of MSC-based therapies in leukaemia treatment.
It may be feasible to develop anti-leukaemia therapies using MSCs that target individual pathways, such as CXCR4/CXCL12, Notch, and Wnt. Specifically, developing molecules that augment anti-tumorigenic effects or diminish pro-tumorigenic effects would be a promising approach for advanced leukaemia therapies. Further investigation of the anti-tumorigenic effects of MSCs is warranted to develop an effective and safe treatment for leukaemia. This could involve the development of engineered or genetically modified MSCs that are more efficacious than the heterogeneous and unstable naïve MSCs.
MSC-based clinical results have exhibited a wide range of variation, likely attributable to the absence of standardised experimental methods, specific cell surface markers to identify subsets of MSCs and the heterogeneous characteristics of MSCs that are easily influenced by the surrounding environment. Thus, further investigation is imperative to advance the development of MSCs for cancer treatment. Furthermore, there exist numerous unclear and intricate facets of cancer, particularly hematologic malignancies, such as the tumour-related impacts of MSCs. Several studies have been undertaken to examine the effects of MSCs in carcinogenesis or tumour microenvironments; however, a solitary principle fails to elucidate both the antitumorigenic and pro-tumorigenic functions of MSCs. Even though the underlying process remains elusive, the dual role of MSCs is widely recognised.
The antitumor effects of MSCs primarily result from the suppression of malignant cell proliferation. The specific mechanisms or molecules implicated remain uncertain, although arrest at the G0/G1 phase of the cell cycle is an established mechanism. To harness this antitumorigenic activity for future clinical applications, other factors must be taken into consideration. MSCs possess certain advantageous characteristics, such as their potential to serve as delivery vehicles and their capacity to inhibit vascular growth and halt the cell cycle. However, some disadvantages exist, such as immunomodulation of tumour cells, promoting tumour growth and suppressing cell apoptosis, leading to chemo-resistance in case of suppression of drug-induced apoptosis, supporting tumour vasculature, etc. Tumour-associated MSCs, integral components of the tumour microenvironment, are also implicated in a pro-tumorigenic effect as they tend to enhance tumour cell stemness.
From the various underlying mechanisms proposed and summarised herein, it may be conceivable to formulate MSC-based anticancer therapies by selectively targeting individual pathways. Specifically, creating molecules capable of augmenting the antitumorigenic effects or attenuating the pro-tumorigenic effects holds considerable promise for advancing therapeutic strategies. Elaborate studies are requisite to surmount limitations such as the heterogeneous nature of MSCs and the absence of standardised methodologies. Further investigation pertaining to the antitumor effects of MSCs ought to be conducted to devise secure and efficacious treatments for hematologic malignancies. Developing engineered or genetically modified MSCs may serve as a propitious approach, as they exhibit enhanced safety and efficiency in contrast to the unstable and heterogeneous naïve MSCs. As numerous researchers persevere in overcoming limitations and refining MSC-based cellular therapies targeting hematologic malignancies, optimism exists that successful therapeutic interventions will be realised. Until such time, it is indispensable that we adopt a cautious approach to MSC-based cellular therapies, considering the adverse outcomes delineated.
Author Contributions
I.E and R.A.L did literature review and wrote the manuscript. J. E-J instructed and checked this paper.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study.
Acknowledgments
Figure 1, Figure 2. Figure 3, and Figure 4 were created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| MSCs Mesenchymal stem cells BM Bone marrow HSC Hematopoietic stem cells DCs Dendritic cells NKs Natural killer cells TGF-β Transforming growth factor beta EGF Epidermal growth factor GM-CSF Granulocyte-macrophage colony-stimulating factor ALCAM Activated leukocyte cell adhesion molecule ICAM-1 Intercellular adhesion molecule-1 IL Interleukin PGE2 Prostaglandin E2 HLA-G Human leukocyte antigen ECM Extracellular matrix MDS Myelodysplastic syndromes FAB French-American-British RA Refractory anaemia RARS Refractory anaemia with ringed sideroblasts RAEB Refractory anaemia with excess blasts RAEB-T Refractory anaemia with excess blasts in transformation CMML Chronic myelomonocytic leukaemia WHO World Health Organisation RCUD Refractory cytopenia with unilineage dysplasia RCMD Refractory cytopenia with multilineage dysplasia MDS-U MDS, unclassified MDS-SLD MDS with single lineage dysplasia MDS-MLD MDS with multilineage dysplasia MDS-RS MDS with ring sideroblasts MDS-RS-SLD MDS-RS with single-lineage dysplasia MDS-RS-MLD MDS-RS with multi-lineage dysplasia MDS-EB MDS with excess blasts RCC Refractory cytopenia in childhood MDS-LB MDS with low blasts MDS-h MDS hypoplastic MDS-IB MDS with increased blasts IPSS International Prognostic Scoring System BMSC Bone marrow mesenchymal stem cells AML acute myeloid leukaemia HSPC Hemaetopoietic stem and progenitor cells NF- κB Nuclear factor kappa B TNF Tumour necrosis factor ANGPT Angiopoietin HGF Hepatocyte growth factor CXCL12 C-X-C motif chemokine ligand 12 TPO Thrombopoietin IGFBP2 Insulin growth factor binding protein 2 |
CCL3 C-C motif chemokine ligand LR-MDS Low risk-MDS HR-MDS High risk-MDS BMME Bone marrow microenvironment IFN-γ Interferon-gamma SCF Stem cell factor IGF1 Insulin-like growth factor 1 HPC Haematopoietic progenitor cells TBX15 T-Box transcription factor 15 PITX2 Paired-like homeodomain transcription factor 2 HOXB1 Homeobox B1 mRNA Messenger ribonucleic acid OC Osteocalcin ALP Alkaline phosphatases FABP4 Fatty acid-binding protein 4 AML Acute myeloid leukaemia CML Chronic myeloid leukaemia ALL Acute lymphoblastic leukaemia CLL Chronic lymphocytic leukaemia VEGF Vascular endothelial factor FGF Fibroblast growth factor PDGF Platelet-derived growth factor COX-2 Cyclooxygenase 2 CCL2 C-C motif chemokine ligand 2 CAFs Cancer-associated fibroblasts hUC-MSCs Human umbilical cord-derived MSCs DKK1 Dickkopf-related protein 1 GVHD Graft-versus-host disease CINC-1 Cytokine-induced neutrophil chemoattractant-1 TIMP-1 Tissue inhibitor of metalloproteinases-1 B-ALL B-acute lymphoblastic leukaemia HD Hodgkin disease NHL Non-Hodgkin lymphoma MCP-1 Monocyte chemoattractant protein-1 IDO Indoleamine 2,3-dioxygenase EVs Extracellular vesicles MM-MSCs Multiple myeloma MSCs G-MDSCs Granulocytic-myeloid-derived suppressor cells TME Tumour microenvironment MCL Mantle Cell Lymphoma DLBCL Diffuse Large B-cell lymphoma HA Hypomethylating agents AZA Azacitidine CDKN1A Cyclin-dependent kinase inhibitor 1A ALA α-lipoic acid ROS Reactive oxygen species Ems Exosome mimetics |
References
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [CrossRef]
- Qin, L.; Liu, N.; Bao, C.-M.; Yang, D.-Z.; Ma, G.-X.; Yi, W.-H.; Xiao, G.-Z.; Cao, H.-L. Mesenchymal stem cells in fibrotic diseases-the two sides of the same coin. Acta Pharmacol. Sin. 2023, 44, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Petite, H.; Viateau, V.; Bensaïd, W.; Meunier, A.; Pollak, C. de; Bourguignon, M.; Oudina, K.; Sedel, L.; Guillemin, G. Tissue-engineered bone regeneration. Nat. Biotechnol. 2000, 18, 959–963. [Google Scholar] [CrossRef]
- Prajwal, G.S.; Jeyaraman, N.; Kanth V, K.; Jeyaraman, M.; Muthu, S.; Rajendran, S.N.S.; Rajendran, R.L.; Khanna, M.; Oh, E.J.; Choi, K.Y.; et al. Lineage Differentiation Potential of Different Sources of Mesenchymal Stem Cells for Osteoarthritis Knee. Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Cominal, J.G.; Da Costa Cacemiro, M.; Pinto-Simões, B.; Kolb, H.-J.; Malmegrim, K.C.R.; Castro, F.A. de. Emerging Role of Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Pathogenesis of Haematological Malignancies. Stem Cells Int. 2019, 2019, 6854080. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wu, Y. Paracrine molecules of mesenchymal stem cells for hematopoietic stem cell niche. Bone Marrow Res. 2011, 2011, 353878. [Google Scholar] [CrossRef]
- Asari, S.; Itakura, S.; Ferreri, K.; Liu, C.-P.; Kuroda, Y.; Kandeel, F.; Mullen, Y. Mesenchymal stem cells suppress B-cell terminal differentiation. Exp. Hematol. 2009, 37, 604–615. [Google Scholar] [CrossRef]
- Gronthos, S.; Franklin, D.M.; Leddy, H.A.; Robey, P.G.; Storms, R.W.; Gimble, J.M. Surface protein characterization of human adipose tissue-derived stromal cells. J. Cell. Physiol. 2001, 189, 54–63. [Google Scholar] [CrossRef]
- Boiret, N.; Rapatel, C.; Veyrat-Masson, R.; Guillouard, L.; Guérin, J.-J.; Pigeon, P.; Descamps, S.; Boisgard, S.; Berger, M.G. Characterization of nonexpanded mesenchymal progenitor cells from normal adult human bone marrow. Exp. Hematol. 2005, 33, 219–225. [Google Scholar] [CrossRef]
- Dazzi, F.; Ramasamy, R.; Glennie, S.; Jones, S.P.; Roberts, I. The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 2006, 20, 161–171. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine mechanisms in adult stem cell signaling and therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-R.; Zhang, Q.-Z.; Shi, S.-H.; Nguyen, A.L.; Le, A.D. Human gingiva-derived mesenchymal stromal cells attenuate contact hypersensitivity via prostaglandin E2-dependent mechanisms. Stem Cells 2011, 29, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Patel, S.A.; Kartan, S.; Axelrod, D.; Capitle, E.; Rameshwar, P. Tolerance-like mediated suppression by mesenchymal stem cells in patients with dust mite allergy-induced asthma. J. Allergy Clin. Immunol. 2012, 129, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Augello, A.; Tasso, R.; Negrini, S.M.; Cancedda, R.; Pennesi, G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum. 2007, 56, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: from embryo to adult. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Batsali, A.K.; Georgopoulou, A.; Mavroudi, I.; Matheakakis, A.; Pontikoglou, C.G.; Papadaki, H.A. The Role of Bone Marrow Mesenchymal Stem Cell Derived Extracellular Vesicles (MSC-EVs) in Normal and Abnormal Hematopoiesis and Their Therapeutic Potential. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Fröbel, J.; Landspersky, T.; Percin, G.; Schreck, C.; Rahmig, S.; Ori, A.; Nowak, D.; Essers, M.; Waskow, C.; Oostendorp, R.A.J. The Hematopoietic Bone Marrow Niche Ecosystem. Front. Cell Dev. Biol. 2021, 9, 705410. [Google Scholar] [CrossRef]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.T.; Purton, L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xie, Y.; Xu, J.-R.; Luo, Q.; Ren, Y.-X.; Chen, M.; Duan, J.-L.; Bao, C.-J.; Liu, Y.-X.; Li, P.-S.; et al. Engineered stem cell biomimetic liposomes carrying levamisole for macrophage immunity reconstruction in leukemia therapy. Chemical Engineering Journal 2022, 447, 137582. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.A.; Smith, A.E.; Kulasekararaj, A.G.; Kizilors, A.; Mohamedali, A.M.; Lea, N.C.; Mitsopoulos, K.; Ford, K.; Nasser, E.; Seidl, T.; et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica 2013, 98, 1058–1066. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Germing, U.; Malcovati, L. Diagnosis and classification of myelodysplastic syndromes. Blood 2023, 142, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; He, G. The 2016 Revision to the World Health Organization Classification of Myelodysplastic Syndromes. J. Transl. Int. Med. 2017, 5, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Attar, E.; Bennett, J.M.; Bloomfield, C.D.; Borate, U.; Castro, C.M. de; Deeg, H.J.; Frankfurt, O.; Gaensler, K.; Garcia-Manero, G.; et al. Myelodysplastic syndromes: clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 2013, 11, 838–874. [Google Scholar] [CrossRef] [PubMed]
- Beachy, S.H.; Aplan, P.D. Mouse models of myelodysplastic syndromes. Hematol. Oncol. Clin. North Am. 2010, 24, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, M.; Yang, X.; Zhang, W.; Cao, L.; Gao, R. Summary of animal models of myelodysplastic syndrome. Animal Model. Exp. Med. 2021, 4, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Modeling myeloid malignancies with patient-derived iPSCs. Exp. Hematol. 2019, 71, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Pontikoglou, C.G.; Matheakakis, A.; Papadaki, H.A. The mesenchymal compartment in myelodysplastic syndrome: Its role in the pathogenesis of the disorder and its therapeutic targeting. Front. Oncol. 2023, 13, 1102495. [Google Scholar] [CrossRef]
- Lopez-Villar, O.; Garcia, J.L.; Sanchez-Guijo, F.M.; Robledo, C.; Villaron, E.M.; Hernández-Campo, P.; Lopez-Holgado, N.; Diez-Campelo, M.; Barbado, M.V.; Perez-Simon, J.A.; et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q- syndrome. Leukemia 2009, 23, 664–672. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Arana-Trejo, R.M.; Gutiérrez-Espíndola, G.; Pérez-Cabrera, A.; Mayani, H. Mesenchymal stem cells in myelodysplastic syndromes: phenotypic and cytogenetic characterization. Leuk. Res. 2005, 29, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Kiss, J.; Várkonyi, J.; Vas, V.; Farkas, P.; Pálóczi, K.; Uher, F. Inappropriate Notch activity and limited mesenchymal stem cell plasticity in the bone marrow of patients with myelodysplastic syndromes. Pathol. Oncol. Res. 2007, 13, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Flores-Figueroa, E.; Montesinos, J.J.; Flores-Guzmán, P.; Gutiérrez-Espíndola, G.; Arana-Trejo, R.M.; Castillo-Medina, S.; Pérez-Cabrera, A.; Hernández-Estévez, E.; Arriaga, L.; Mayani, H. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk. Res. 2008, 32, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhi-Gang, Z.; Wei-Ming, L.; Zhi-Chao, C.; Yong, Y.; Ping, Z. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases. Leuk. Lymphoma 2008, 49, 2187–2195. [Google Scholar] [CrossRef]
- Campioni, D.; Rizzo, R.; Stignani, M.; Melchiorri, L.; Ferrari, L.; Moretti, S.; Russo, A.; Bagnara, G.P.; Bonsi, L.; Alviano, F.; et al. A decreased positivity for CD90 on human mesenchymal stromal cells (MSCs) is associated with a loss of immunosuppressive activity by MSCs. Cytometry B Clin. Cytom. 2009, 76, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Baldus, C.D.; Hofmann, W.-K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Jann, J.-C.; Mossner, M.; Riabov, V.; Altrock, E.; Schmitt, N.; Flach, J.; Xu, Q.; Nowak, V.; Obländer, J.; Palme, I.; et al. Bone marrow derived stromal cells from myelodysplastic syndromes are altered but not clonally mutated in vivo. Nat. Commun. 2021, 12, 6170. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Umezu, T.; Imanishi, S.; Asano, M.; Yoshizawa, S.; Katagiri, S.; Ohyashiki, K.; Ohyashiki, J.H. Genetic variations of bone marrow mesenchymal stromal cells derived from acute leukemia and myelodysplastic syndrome by targeted deep sequencing. Leuk. Res. 2017, 62, 23–28. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Chen, S.; Zambetti, N.A.; Bindels, E.M.J.; Kenswill, K.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; Sanders, M.A.; Cremers, E.M.P.; Westers, T.M.; et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia 2016, 30, 1938–1942. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef]
- Huang, J.C.; Basu, S.K.; Zhao, X.; Chien, S.; Fang, M.; Oehler, V.G.; Appelbaum, F.R.; Becker, P.S. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015, 5, e302. [Google Scholar] [CrossRef]
- Corradi, G.; Baldazzi, C.; Očadlíková, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, R.A.; Wobus, M.; List, C.; Wehner, R.; Schönefeldt, C.; Brocard, B.; Mohr, B.; Rauner, M.; Schmitz, M.; Stiehler, M.; et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica 2013, 98, 1677–1685. [Google Scholar] [CrossRef]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor β1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef]
- Zhao, Z.-G.; Xu, W.; Yu, H.-P.; Fang, B.-L.; Wu, S.-H.; Li, F.; Li, W.-M.; Li, Q.-B.; Chen, Z.-C.; Zou, P. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012, 317, 136–143. [Google Scholar] [CrossRef]
- Aanei, C.M.; Eloae, F.Z.; Flandrin-Gresta, P.; Tavernier, E.; Carasevici, E.; Guyotat, D.; Campos, L. Focal adhesion protein abnormalities in myelodysplastic mesenchymal stromal cells. Exp. Cell Res. 2011, 317, 2616–2629. [Google Scholar] [CrossRef]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1487–1500. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Oz, S.; Cadeddu, R.-P.; Fröbel, J.; Brückner, B.; Kündgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Muntión, S.; Ramos, T.L.; Diez-Campelo, M.; Rosón, B.; Sánchez-Abarca, L.I.; Misiewicz-Krzeminska, I.; Preciado, S.; Sarasquete, M.-E.; Las Rivas, J. de; González, M.; et al. Microvesicles from Mesenchymal Stromal Cells Are Involved in HPC-Microenvironment Crosstalk in Myelodysplastic Patients. PLoS One 2016, 11, e0146722. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Strat, A.N.; Bao, W.; Rosston, P.; Fallon, G.; Ohrn, S.; Zhong, H.; Lobo, C.; An, X.; Yazdanbakhsh, K. Mesenchymal stromal cells lower platelet activation and assist in platelet formation in vitro. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Epperson, D.E.; Nakamura, R.; Saunthararajah, Y.; Melenhorst, J.; Barrett, A.J. Oligoclonal T cell expansion in myelodysplastic syndrome: evidence for an autoimmune process. Leuk. Res. 2001, 25, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Sun, Z.; Liu, L.; Chen, B.; Cao, Y.; Li, K.; Zhao, R.C. Impairment in immuno-modulatory function of Flk1(+)CD31(-)CD34(-) MSCs from MDS-RA patients. Leuk. Res. 2007, 31, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Z.; Li, Q.; Li, W.; You, Y.; Zou, P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS One 2012, 7, e45675. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Wang, J.; Sunil Arvindam, U.; Hallstrom, C.; Verneris, M.R.; Grzywacz, B.; Warlick, E.; Blazar, B.R.; Miller, J.S. Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Lynch, O.F.; Calvi, L.M. Immune Dysfunction, Cytokine Disruption, and Stromal Changes in Myelodysplastic Syndrome: A Review. Cells 2022, 11. [Google Scholar] [CrossRef]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef]
- Caselli, A.; Olson, T.S.; Otsuru, S.; Chen, X.; Hofmann, T.J.; Nah, H.-D.; Grisendi, G.; Paolucci, P.; Dominici, M.; Horwitz, E.M. IGF-1-mediated osteoblastic niche expansion enhances long-term hematopoietic stem cell engraftment after murine bone marrow transplantation. Stem Cells 2013, 31, 2193–2204. [Google Scholar] [CrossRef]
- Dodillet, H.; Kreuzer, K.-A.; Monsef, I.; Skoetz, N. Thrombopoietin mimetics for patients with myelodysplastic syndromes. Cochrane Database Syst. Rev. 2017, 9, CD009883. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zheng, Q.; Xu, F.; Shi, W.; Guo, J.; Zhang, Z.; Zhao, S.; Li, X.; Chang, C. IGF-IR promotes clonal cell proliferation in myelodysplastic syndromes via inhibition of the MAPK pathway. Oncol. Rep. 2020, 44, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Aanei, C.M.; Flandrin, P.; Eloae, F.Z.; Carasevici, E.; Guyotat, D.; Wattel, E.; Campos, L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012, 21, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Rostovskaya, M.; Anastassiadis, K. Differential expression of surface markers in mouse bone marrow mesenchymal stromal cell subpopulations with distinct lineage commitment. PLoS One 2012, 7, e51221. [Google Scholar] [CrossRef]
- Roson-Burgo, B.; Sanchez-Guijo, F.; Del Cañizo, C.; Las Rivas, J. de. Insights into the human mesenchymal stromal/stem cell identity through integrative transcriptomic profiling. BMC Genomics 2016, 17, 944. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Wein, F.; Roderburg, C.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D. Adhesion of human hematopoietic progenitor cells to mesenchymal stromal cells involves CD44. Cells Tissues Organs 2008, 188, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Falconi, G.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Raffaelli, C.S.; Bellesi, S.; Hohaus, S.; Voso, M.T.; D'Alò, F.; Leone, G. Impairment of PI3K/AKT and WNT/β-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp. Hematol. 2016, 44, 75–83.e1-4. [Google Scholar] [CrossRef]
- Rathnayake, A.J.I.S.; Goonasekera, H.W.W.; Dissanayake, V.H.W. Phenotypic and Cytogenetic Characterization of Mesenchymal Stromal Cells in De Novo Myelodysplastic Syndromes. Anal. Cell. Pathol. (Amst) 2016, 2016, 8012716. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Fei, C.; Guo, J.; Gu, S.; Zhu, Y.; Xu, F.; Zhang, Z.; Wu, L.; Li, X.; et al. Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2015, 100, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, K.; Pontikoglou, C.G.; Demetriadou, A.; Batsali, A.K.; Damianaki, A.; Simantirakis, E.; Kontakis, M.; Galanopoulos, A.; Kotsianidis, I.; Kastrinaki, M.-C.; et al. Impaired proliferative potential of bone marrow mesenchymal stromal cells in patients with myelodysplastic syndromes is associated with abnormal WNT signaling pathway. Stem Cells Dev. 2014, 23, 1568–1581. [Google Scholar] [CrossRef] [PubMed]
- Mellibovsky, L.; Diez, A.; Serrano, S.; Aubia, J.; Pérez-Vila, E.; Mariñoso, M.L.; Nogués, X.; Recker, R.R. Bone remodeling alterations in myelodysplastic syndrome. Bone 1996, 19, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Weidner, H.; Rauner, M.; Trautmann, F.; Schmitt, J.; Balaian, E.; Mies, A.; Helas, S.; Baschant, U.; Khandanpour, C.; Bornhäuser, M.; et al. Myelodysplastic syndromes and bone loss in mice and men. Leukemia 2017, 31, 1003–1007. [Google Scholar] [CrossRef]
- Sugimura, R.; Li, L. Shifting in balance between osteogenesis and adipogenesis substantially influences hematopoiesis. J. Mol. Cell Biol. 2010, 2, 61–62. [Google Scholar] [CrossRef]
- Li, A.J.; Calvi, L.M. The microenvironment in myelodysplastic syndromes: Niche-mediated disease initiation and progression. Exp. Hematol. 2017, 55, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Uttley, L.; Indave, B.I.; Hyde, C.; White, V.; Lokuhetty, D.; Cree, I. Invited commentary-WHO Classification of Tumours: How should tumors be classified? Expert consensus, systematic reviews or both? Int. J. Cancer 2020, 146, 3516–3521. [Google Scholar] [CrossRef]
- Cree, I.A. The WHO Classification of Haematolymphoid Tumours. Leukemia 2022, 36, 1701–1702. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Burger, J.A.; Peled, A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia 2009, 23, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Ryu, S.; Kim, D.S.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Mesenchymal stem cells in suppression or progression of hematologic malignancy: current status and challenges. Leukemia 2019, 33, 597–611. [Google Scholar] [CrossRef]
- Fathi, E.; Sanaat, Z.; Farahzadi, R. Mesenchymal stem cells in acute myeloid leukemia: a focus on mechanisms involved and therapeutic concepts. Blood Res. 2019, 54, 165–174. [Google Scholar] [CrossRef]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B- lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef]
- Ramasamy, R.; Lam, E.W.-F.; Soeiro, I.; Tisato, V.; Bonnet, D.; Dazzi, F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia 2007, 21, 304–310. [Google Scholar] [CrossRef]
- Lee, M.W.; Park, Y.J.; Kim, D.S.; Park, H.J.; Jung, H.L.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Human Adipose Tissue Stem Cells Promote the Growth of Acute Lymphoblastic Leukemia Cells in NOD/SCID Mice. Stem Cell Rev. Rep. 2018, 14, 451–460. [Google Scholar] [CrossRef]
- Naderi, E.H.; Skah, S.; Ugland, H.; Myklebost, O.; Sandnes, D.L.; Torgersen, M.L.; Josefsen, D.; Ruud, E.; Naderi, S.; Blomhoff, H.K. Bone marrow stroma-derived PGE2 protects BCP-ALL cells from DNA damage-induced p53 accumulation and cell death. Mol. Cancer 2015, 14, 14. [Google Scholar] [CrossRef]
- Oswald, J.; Boxberger, S.; Jørgensen, B.; Feldmann, S.; Ehninger, G.; Bornhäuser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef]
- Silva, G.V.; Litovsky, S.; Assad, J.A.R.; Sousa, A.L.S.; Martin, B.J.; Vela, D.; Coulter, S.C.; Lin, J.; Ober, J.; Vaughn, W.K.; et al. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation 2005, 111, 150–156. [Google Scholar] [CrossRef]
- Suzuki, K.; Sun, R.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef]
- Roorda, B.D.; Elst, A. ter; Kamps, W.A.; Bont, E.S.J.M. de. Bone marrow-derived cells and tumor growth: contribution of bone marrow-derived cells to tumor micro-environments with special focus on mesenchymal stem cells. Crit. Rev. Oncol. Hematol. 2009, 69, 187–198. [Google Scholar] [CrossRef]
- Kinnaird, T.; Stabile, E.; Burnett, M.S.; Lee, C.W.; Barr, S.; Fuchs, S.; Epstein, S.E. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ. Res. 2004, 94, 678–685. [Google Scholar] [CrossRef]
- Kampen, K.R.; Elst, A. ter; Bont, E.S.J.M. de. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell. Mol. Life Sci. 2013, 70, 1307–1317. [Google Scholar] [CrossRef]
- Lopes, M.R.; Pereira, J.K.N.; Melo Campos, P. de; Machado-Neto, J.A.; Traina, F.; Saad, S.T.O.; Favaro, P. De novo AML exhibits greater microenvironment dysregulation compared to AML with myelodysplasia-related changes. Sci. Rep. 2017, 7, 40707. [Google Scholar] [CrossRef]
- Ding, W.; Knox, T.R.; Tschumper, R.C.; Wu, W.; Schwager, S.M.; Boysen, J.C.; Jelinek, D.F.; Kay, N.E. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood 2010, 116, 2984–2993. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef]
- Vasconcellos, J.F. de; Laranjeira, A.B.A.; Zanchin, N.I.T.; Otubo, R.; Vaz, T.H.; Cardoso, A.A.; Brandalise, S.R.; Yunes, J.A. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 568–577. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.S.; Verma, A.; Steidl, U.; Garcia-Manero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef]
- Brück, O.; Blom, S.; Dufva, O.; Turkki, R.; Chheda, H.; Ribeiro, A.; Kovanen, P.; Aittokallio, T.; Koskenvesa, P.; Kallioniemi, O.; et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia 2018, 32, 1643–1656. [Google Scholar] [CrossRef]
- Zhao, Z.-G.; Xu, W.; Sun, L.; Li, W.-M.; Li, Q.-B.; Zou, P. The characteristics and immunoregulatory functions of regulatory dendritic cells induced by mesenchymal stem cells derived from bone marrow of patient with chronic myeloid leukaemia. Eur. J. Cancer 2012, 48, 1884–1895. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Kamga, P.T.; Tagne Simo, R.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Mesenchymal stromal cells' role in tumor microenvironment: involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129–141. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, Y.; Yue, Z.; Yuan, Y.; Wang, X. Bone marrow mesenchymal stem cells regulate stemness of multiple myeloma cell lines via BTK signaling pathway. Leuk. Res. 2017, 57, 20–26. [Google Scholar] [CrossRef]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2016, 1863, 449–463. [Google Scholar] [CrossRef]
- Fallati, A.; Di Marzo, N.; D’Amico, G.; Dander, E. Mesenchymal Stromal Cells (MSCs): An Ally of B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells in Disease Maintenance and Progression within the Bone Marrow Hematopoietic Niche. Cancers 2022, 14, 3303. [Google Scholar] [CrossRef]
- Vianello, F.; Villanova, F.; Tisato, V.; Lymperi, S.; Ho, K.-K.; Gomes, A.R.; Marin, D.; Bonnet, D.; Apperley, J.; Lam, E.W.-F.; et al. Bone marrow mesenchymal stromal cells non-selectively protect chronic myeloid leukemia cells from imatinib-induced apoptosis via the CXCR4/CXCL12 axis. Haematologica 2010, 95, 1081–1089. [Google Scholar] [CrossRef]
- Mohammadi Gorji, S.; Karimpor Malekshah, A.A.; Hashemi-Soteh, M.B.; Rafiei, A.; Parivar, K.; Aghdami, N. Effect of Mesenchymal Stem Cells on Doxorubicin-Induced Fibrosis. Cell J. 2012, 14, 142–151. [Google Scholar]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Chen, Y.; Mak, D.H.; Mu, H.; Jacamo, R.; Ruvolo, V.; Arold, S.T.; Ladbury, J.E.; Burks, J.K.; et al. Anti-apoptotic ARC protein confers chemoresistance by controlling leukemia-microenvironment interactions through a NFκB/IL1β signaling network. Oncotarget 2016, 7, 20054–20067. [Google Scholar] [CrossRef]
- Fonseka, M.; Ramasamy, R.; Tan, B.C.; Seow, H.F. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol. Int. 2012, 36, 793–801. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, Z.; Han, Q.; Liao, L.; Wang, J.; Bian, C.; Li, J.; Yan, X.; Liu, Y.; Shao, C.; et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia 2009, 23, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chen, N.; Guo, H.; Wang, X.; Xu, F.; Ren, Q.; Lu, S.; Liu, B.; Zhang, L.; Zhao, H. Bone marrow mesenchymal stem cells from leukemia patients inhibit growth and apoptosis in serum-deprived K562 cells. J. Exp. Clin. Cancer Res. 2009, 28, 141. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Yang, S.; Ren, Q.; Han, Z.; Lu, S.; Ma, F.; Zhang, L.; Han, Z. p38 MAPK contributes to the growth inhibition of leukemic tumor cells mediated by human umbilical cord mesenchymal stem cells. Cell. Physiol. Biochem. 2010, 26, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, Y.; Xu, Z.; Li, J.; Yang, J.; Li, Y.; Shang, Y.; Luo, J. Effect of bone marrow mesenchymal stem cells from blastic phase chronic myelogenous leukemia on the growth and apoptosis of leukemia cells. Oncol. Rep. 2013, 30, 1007–1013. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, D.; Chen, X.; Shao, H.; Huang, S. Human umbilical cord-derived mesenchymal stem cells inhibit proliferation but maintain survival of Jurkat leukemia cells in vitro by activating Notch signaling. Nan Fang Yi Ke Da Xue Xue Bao 2014, 34, 441–447. [Google Scholar] [PubMed]
- Song, N.; Gao, L.; Qiu, H.; Huang, C.; Cheng, H.; Zhou, H.; Lv, S.; Chen, L.; Wang, J. Mouse bone marrow-derived mesenchymal stem cells inhibit leukemia/lymphoma cell proliferation in vitro and in a mouse model of allogeneic bone marrow transplant. Int. J. Mol. Med. 2015, 36, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Sarmadi, V.H.; Tong, C.K.; Vidyadaran, S.; Abdullah, M.; Seow, H.F.; Ramasamy, R. Mesenchymal stem cells inhibit proliferation of lymphoid origin haematopoietic tumour cells by inducing cell cycle arrest. Med. J. Malaysia 2010, 65, 209–214. [Google Scholar]
- Hendijani, F.; Javanmard, S.H.; Sadeghi-aliabadi, H. Human Wharton's jelly mesenchymal stem cell secretome display antiproliferative effect on leukemia cell line and produce additive cytotoxic effect in combination with doxorubicin. Tissue Cell 2015, 47, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Zhang, L.-S. Influence of human bone marrow mesenchymal stem cells on proliferation of chronic myeloid leukemia cells. Ai Zheng 2009, 28, 29–32. [Google Scholar] [PubMed]
- Wong, R.S.Y.; Cheong, S.-K. Role of mesenchymal stem cells in leukaemia: Dr. Jekyll or Mr. Hyde? Clin. Exp. Med. 2014, 14, 235–248. [Google Scholar] [CrossRef]
- Wagner, W. Senescence is heterogeneous in mesenchymal stromal cells: kaleidoscopes for cellular aging. Cell Cycle 2010, 9, 2923–2924. [Google Scholar] [CrossRef]
- Lee, M.W.; Kim, D.S.; Ryu, S.; Jang, I.K.; Kim, H.J.; Yang, J.M.; Lee, D.-H.; Lee, S.H.; Son, M.H.; Cheuh, H.W.; et al. Effect of ex vivo culture conditions on immunosuppression by human mesenchymal stem cells. Biomed Res. Int. 2013, 2013, 154919. [Google Scholar] [CrossRef]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef]
- Menendez, P.; Catalina, P.; Rodríguez, R.; Melen, G.J.; Bueno, C.; Arriero, M.; García-Sánchez, F.; Lassaletta, A.; García-Sanz, R.; García-Castro, J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009, 206, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Eckert, C.; Seeger, K.; Pfau, M.; Prada, J.; Henze, G.; Blankenstein, T.; Kammertoens, T. Leukemia-associated genetic aberrations in mesenchymal stem cells of children with acute lymphoblastic leukemia. J. Mol. Med. (Berl) 2010, 88, 249–265. [Google Scholar] [CrossRef]
- Blau, O.; Hofmann, W.-K.; Baldus, C.D.; Thiel, G.; Serbent, V.; Schümann, E.; Thiel, E.; Blau, I.W. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp. Hematol. 2007, 35, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-L.; Yu, X.-Q.; Zhu, Y.; Ba, R.; Zhu, W.; Xu, W.-R. Expression of integrins in bone marrow mesenchymal stem cells derived from patients with chronic myeloid leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2008, 16, 755–758. [Google Scholar] [PubMed]
- Zhao, Z.-G.; Liang, Y.; Li, K.; Li, W.-M.; Li, Q.-B.; Chen, Z.-C.; Zou, P. Phenotypic and functional comparison of mesenchymal stem cells derived from the bone marrow of normal adults and patients with hematologic malignant diseases. Stem Cells Dev. 2007, 16, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Ryningen, A.; Wergeland, L.; Glenjen, N.; Gjertsen, B.T.; Bruserud, O. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk. Res. 2005, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Plumas, J.; Chaperot, L.; Richard, M.-J.; Molens, J.-P.; Bensa, J.-C.; Favrot, M.-C. Mesenchymal stem cells induce apoptosis of activated T cells. Leukemia 2005, 19, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Shao, J.; Zhang, W.; Yang, T. Using mesenchymal stem cells as a therapy for bone regeneration and repairing. Biol. Res. 2015, 48, 62. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Mesenchymal stem cells: angels or demons? J. Biomed. Biotechnol. 2011, 2011, 459510. [Google Scholar] [CrossRef]
- Klingemann, H.; Matzilevich, D.; Marchand, J. Mesenchymal Stem Cells - Sources and Clinical Applications. Transfus. Med. Hemother. 2008, 35, 272–277. [Google Scholar] [CrossRef]
- Gangadaran, P.; Rajendran, R.L.; Lee, H.W.; Kalimuthu, S.; Hong, C.M.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J. Control. Release 2017, 264, 112–126. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Invest. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, L.; Scott, P.G.; Tredget, E.E. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells 2007, 25, 2648–2659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, J.; Qian, J.; Li, H.; Romaguera, J.E.; Kwak, L.W.; Wang, M.; Yi, Q. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012, 120, 3783–3792. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhu, Z.; Xu, X.; Zhang, H.; Xiong, H.; Li, Q.; Wei, Y. Human bone marrow-derived mesenchymal stem cells promote the growth and drug-resistance of diffuse large B-cell lymphoma by secreting IL-6 and elevating IL-17A levels. J. Exp. Clin. Cancer Res. 2019, 38, 73. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Chien, K.S.; Montalban-Bravo, G. Myelodysplastic syndromes: 2021 update on diagnosis, risk stratification and management. Am. J. Hematol. 2020, 95, 1399–1420. [Google Scholar] [CrossRef]
- Wenk, C.; Garz, A.-K.; Grath, S.; Huberle, C.; Witham, D.; Weickert, M.; Malinverni, R.; Niggemeyer, J.; Kyncl, M.; Hecker, J.; et al. Direct modulation of the bone marrow mesenchymal stromal cell compartment by azacitidine enhances healthy hematopoiesis. Blood Adv. 2018, 2, 3447–3461. [Google Scholar] [CrossRef]
- Boada, M.; Echarte, L.; Guillermo, C.; Diaz, L.; Touriño, C.; Grille, S. 5-Azacytidine restores interleukin 6-increased production in mesenchymal stromal cells from myelodysplastic patients. Hematol. Transfus. Cell Ther. 2021, 43, 35–42. [Google Scholar] [CrossRef]
- Pang, Y.; Geng, S.; Zhang, H.; Lai, P.; Liao, P.; Zeng, L.; Lu, Z.; Weng, J.; Du, X. Phenotype of mesenchymal stem cells from patients with myelodyplastic syndrome maybe partly modulated by decitabine. Oncol. Lett. 2019, 18, 4457–4466. [Google Scholar] [CrossRef]
- Kastrinaki, M.-C.; Pavlaki, K.; Batsali, A.K.; Kouvidi, E.; Mavroudi, I.; Pontikoglou, C.; Papadaki, H.A. Mesenchymal stem cells in immune-mediated bone marrow failure syndromes. Clin. Dev. Immunol. 2013, 2013, 265608. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; Di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. α-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, Y.; Kang, D.; Kwon, A.; Kim, J.; Min Kim, J.; Park, S.-S.; Kim, Y.-J.; Min, C.-K.; Kim, M. Common and different alterations of bone marrow mesenchymal stromal cells in myelodysplastic syndrome and multiple myeloma. Cell Prolif. 2020, 53, e12819. [Google Scholar] [CrossRef]
- Fujishiro, A.; Iwasa, M.; Fujii, S.; Maekawa, T.; Andoh, A.; Tohyama, K.; Takaori-Kondo, A.; Miura, Y. Menatetrenone facilitates hematopoietic cell generation in a manner that is dependent on human bone marrow mesenchymal stromal/stem cells. Int. J. Hematol. 2020, 112, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Digirolamo, C.M.; Stokes, D.; Colter, D.; Phinney, D.G.; Class, R.; Prockop, D.J. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br. J. Haematol. 1999, 107, 275–281. [Google Scholar] [CrossRef]
- Hodgkinson, C.P.; Gomez, J.A.; Mirotsou, M.; Dzau, V.J. Genetic engineering of mesenchymal stem cells and its application in human disease therapy. Hum. Gene Ther. 2010, 21, 1513–1526. [Google Scholar] [CrossRef]
- Greco, S.J.; Rameshwar, P. Mesenchymal stem cells in drug/gene delivery: implications for cell therapy. Ther. Deliv. 2012, 3, 997–1004. [Google Scholar] [CrossRef]
- Niedermeier, M.; Hennessy, B.T.; Knight, Z.A.; Henneberg, M.; Hu, J.; Kurtova, A.V.; Wierda, W.G.; Keating, M.J.; Shokat, K.M.; Burger, J.A. Isoform-selective phosphoinositide 3'-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood 2009, 113, 5549–5557. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Pillozzi, S.; Masselli, M.; Lorenzo, E. de; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D'Amico, M.; Basso, G.; et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood 2011, 117, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, Y.; Huang, W.; Xu, H.; Chen, X.; Geng, Q.; Fan, H.; Tan, Y.; Xue, G.; Jiang, X. In vitro effect of adenovirus-mediated human Gamma Interferon gene transfer into human mesenchymal stem cells for chronic myelogenous leukemia. Hematol. Oncol. 2006, 24, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Aliperta, R.; Welzel, P.B.; Bergmann, R.; Freudenberg, U.; Berndt, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Stanzione, M.; Cartellieri, M.; et al. Cryogel-supported stem cell factory for customized sustained release of bispecific antibodies for cancer immunotherapy. Sci. Rep. 2017, 7, 42855. [Google Scholar] [CrossRef]
- Andreeff, M.; Studeny, M.; Dembinski, J.; Konopleva, M.; Wang, R.-Y.; Yang, H.-Y.; Fueyo, J.; Champlin, R.E.; Lang, F.; Marini, F.C. Mesenchymal stem cells as delivery systems for cancer and leukemia gene therapy. JCO 2004, 22, 3194. [Google Scholar] [CrossRef]
- Pessina, A.; Coccè, V.; Pascucci, L.; Bonomi, A.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Ciusani, E.; Crovace, A.; Falchetti, M.L.; et al. Mesenchymal stromal cells primed with Paclitaxel attract and kill leukaemia cells, inhibit angiogenesis and improve survival of leukaemia-bearing mice. Br. J. Haematol. 2013, 160, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, Y.; Wang, M.; Zhu, J.; Li, J.; Zhang, P.; Yuan, Y.; Zhu, F. Exosomes derived from human bone marrow mesenchymal stem cells transfer miR-222-3p to suppress acute myeloid leukemia cell proliferation by targeting IRF2/INPP4B. Mol. Cell. Probes 2020, 51, 101513. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Wang, T.; Cheng, H.; Dong, Y.; Weng, Q.; Sun, G.; Zhou, P.; Wang, K.; Liu, X.; Geng, Y.; et al. Mesenchymal stem cells suppress leukemia via macrophage-mediated functional restoration of bone marrow microenvironment. Leukemia 2020, 34, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, L.; Hu, J.; Sun, Y. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug, loaded nanoparticles. Nanomedicine 2013, 9, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Luo, M.; Wei, X. Mesenchymal stem/stromal cells in cancer therapy. J. Hematol. Oncol. 2021, 14, 195. [Google Scholar] [CrossRef]
- Yan, C.; Li, S.; Li, Z.; Peng, H.; Yuan, X.; Jiang, L.; Zhang, Y.; Fan, D.; Hu, X.; Yang, M.; et al. Human umbilical cord mesenchymal stem cells as vehicles of CD20-specific TRAIL fusion protein delivery: a double-target therapy against non-Hodgkin's lymphoma. Mol. Pharm. 2013, 10, 142–151. [Google Scholar] [CrossRef]
Figure 1.
Cellular markers and differentiation potential of MSCs. Human MSCs exhibit positive expression of CD73, CD105, and CD90 markers on their membrane, whereas they lack CD34, CD45, CD19, CD14, and HLA-DR expression. In addition, MSCs possess the ability to differentiate into myocytes, adipocytes, osteoblasts, and chondrocytes when subjected to culture [4].
Figure 1.
Cellular markers and differentiation potential of MSCs. Human MSCs exhibit positive expression of CD73, CD105, and CD90 markers on their membrane, whereas they lack CD34, CD45, CD19, CD14, and HLA-DR expression. In addition, MSCs possess the ability to differentiate into myocytes, adipocytes, osteoblasts, and chondrocytes when subjected to culture [4].
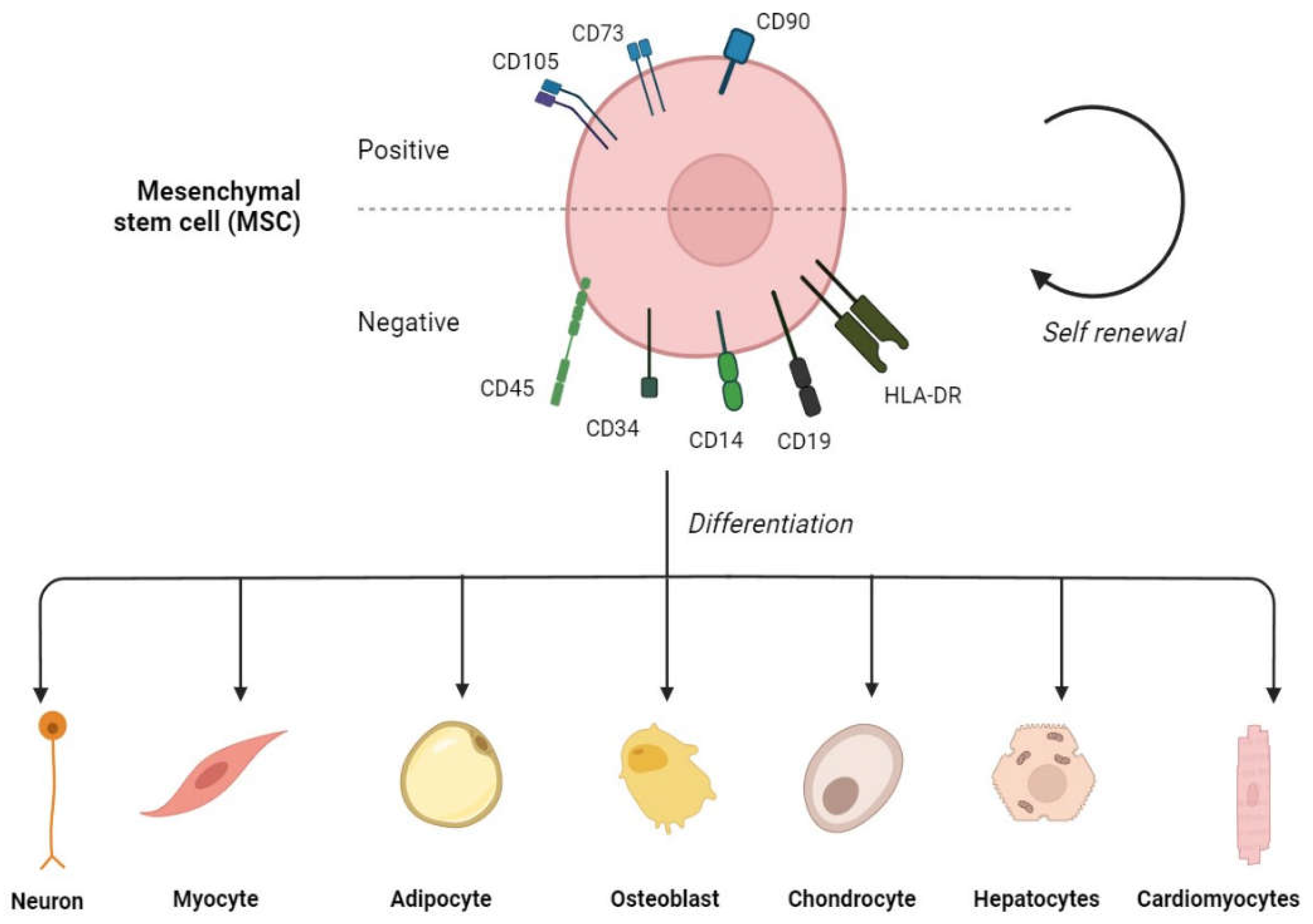
Figure 2.
Effect of impaired MSCs on HSPCs in MDS. MDS-MSCs exhibit inflammatory alterations, deregulation of immune modulation, defective differentiation potential, and a decrease in proliferation and growth, leading to an increase in apoptosis and genotoxicity, together with the expansion and clonal evolution of HSCPs in MDS and impaired support of normal HSPCs.
Figure 2.
Effect of impaired MSCs on HSPCs in MDS. MDS-MSCs exhibit inflammatory alterations, deregulation of immune modulation, defective differentiation potential, and a decrease in proliferation and growth, leading to an increase in apoptosis and genotoxicity, together with the expansion and clonal evolution of HSCPs in MDS and impaired support of normal HSPCs.
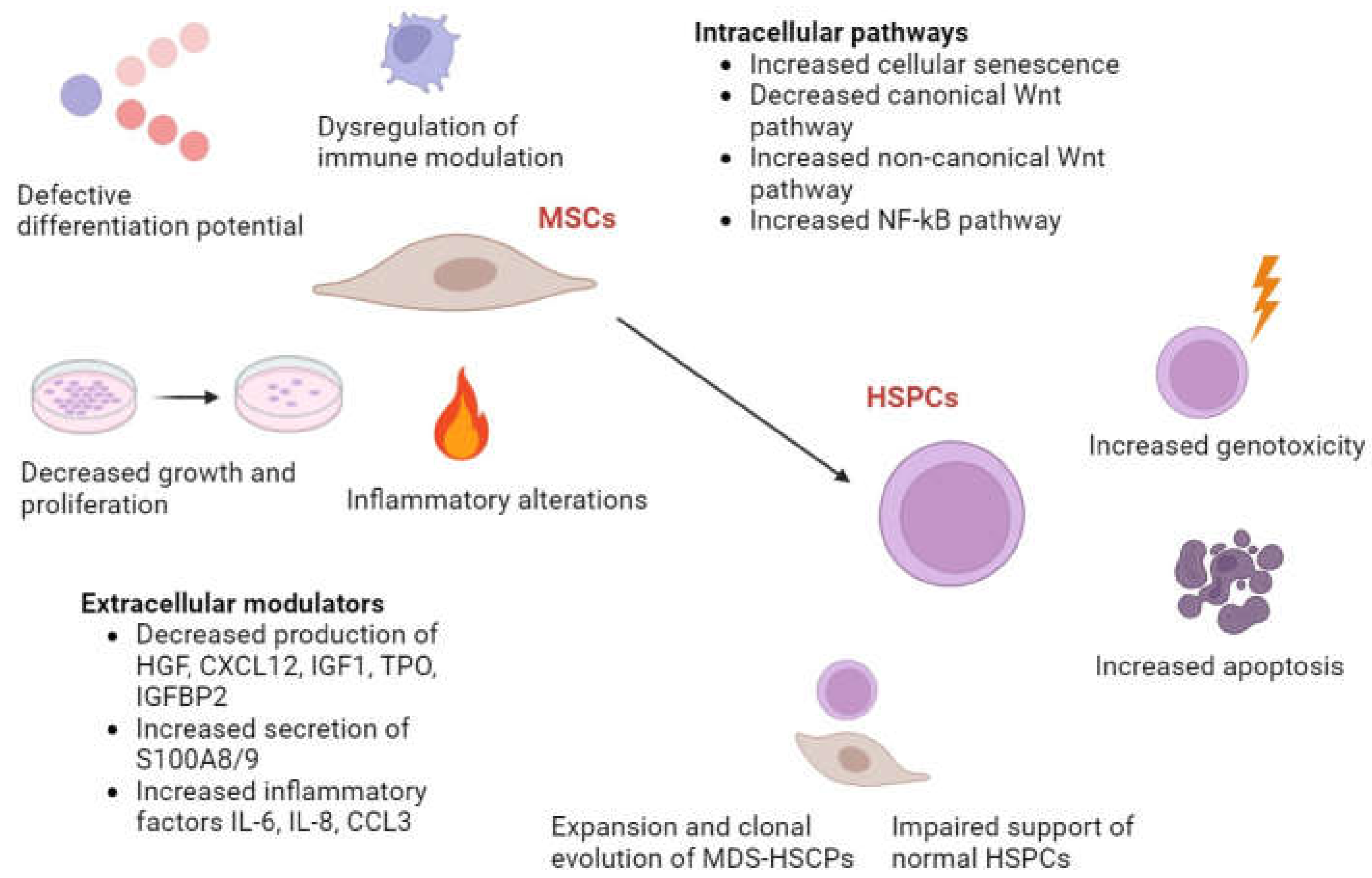
Figure 3.
Development and progression of MDS. (1) Defects in BMME may initiate or cooperate with intrinsic hematopoietic defects, causing the development of MDS clonal cells. Since MDS cells increase in number, they accumulate additional genetic defects, which may result in progression to acute leukaemia. (2) MDS cells produce cytokines that modify the mesenchymal osteolineage and vascular endothelial BMME during this process. (3) The modified BMME, together with autocrine signalling of secreted cytokines, both promote disease progression (Created with BioRender) [76].
Figure 3.
Development and progression of MDS. (1) Defects in BMME may initiate or cooperate with intrinsic hematopoietic defects, causing the development of MDS clonal cells. Since MDS cells increase in number, they accumulate additional genetic defects, which may result in progression to acute leukaemia. (2) MDS cells produce cytokines that modify the mesenchymal osteolineage and vascular endothelial BMME during this process. (3) The modified BMME, together with autocrine signalling of secreted cytokines, both promote disease progression (Created with BioRender) [76].
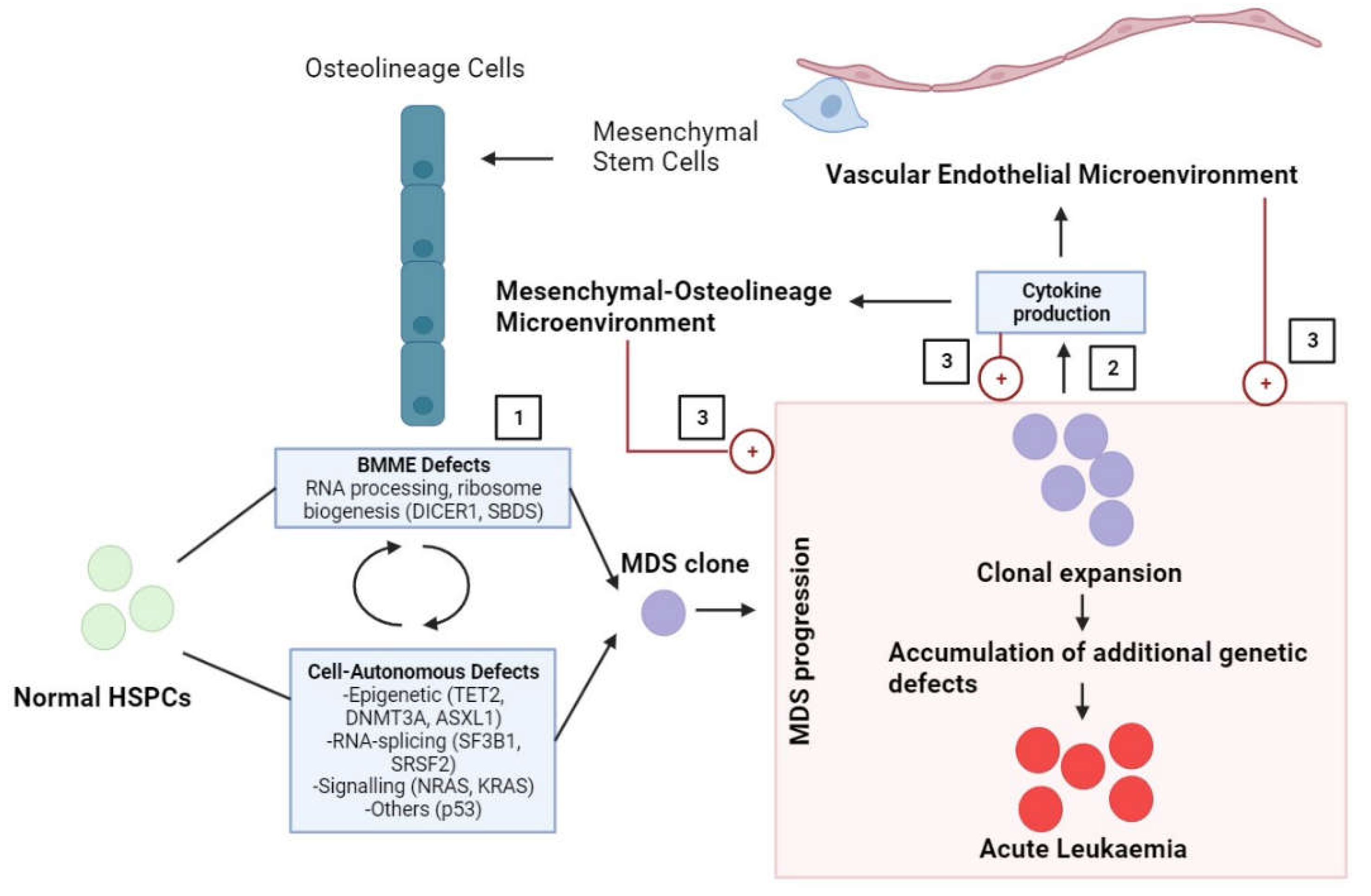
Figure 4.
Diagram for the biphasic role of MSCs in leukaemia. They have both antitumorigenic and pro-tumorigenic effects. They can suppress tumour growth by inhibiting tumour cell proliferation and induce tumour growth by inhibiting tumour cell apoptosis [82].
Figure 4.
Diagram for the biphasic role of MSCs in leukaemia. They have both antitumorigenic and pro-tumorigenic effects. They can suppress tumour growth by inhibiting tumour cell proliferation and induce tumour growth by inhibiting tumour cell apoptosis [82].
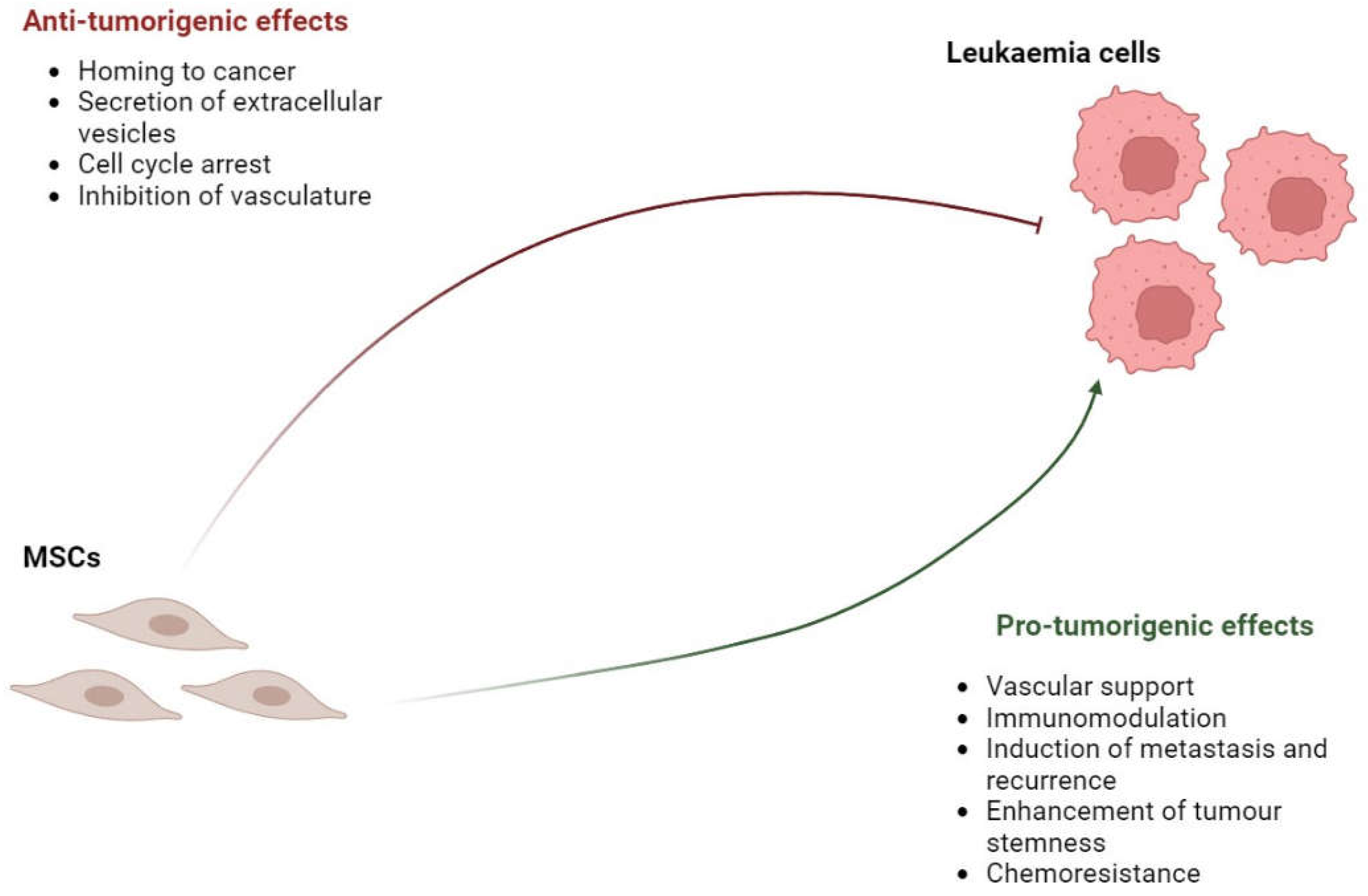
Figure 5.
Engineered MSC-based approaches as current anti-tumour therapies. Engineered MSCs can be used with therapeutic proteins, such as interferon, cytokine, growth factors, and transcription factors, oncolytic viruses including adenovirus, myxoma virus, herpes simplex virus, and measles virus, and prodrugs such as cytosine deaminase, HSV-tk, and carboxylesterase.
Figure 5.
Engineered MSC-based approaches as current anti-tumour therapies. Engineered MSCs can be used with therapeutic proteins, such as interferon, cytokine, growth factors, and transcription factors, oncolytic viruses including adenovirus, myxoma virus, herpes simplex virus, and measles virus, and prodrugs such as cytosine deaminase, HSV-tk, and carboxylesterase.
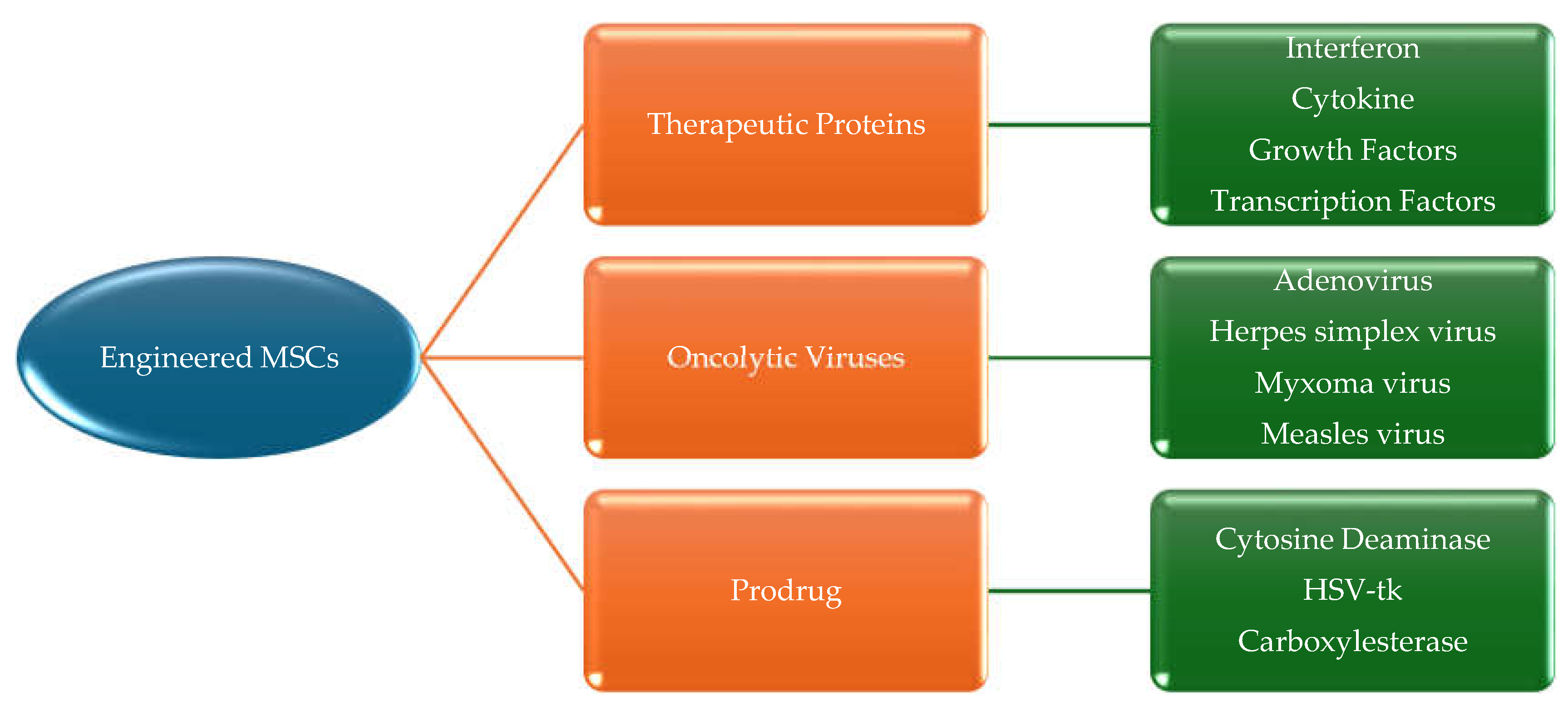

Table 1.
MDS subtypes according to FAB.
| Subtype | % of Peripheral Blasts | % of BM blasts |
|---|---|---|
| Refractory anaemia (RA) | <1 | <5 |
| Refractory anaemia with ringed sideroblasts (RARS) | <1 | <5 |
| Refractory anaemia with excess blasts (RAEB) | <5 | 5-20 |
| Refractory anaemia with excess blasts in transformation (RAEB-T) | ≥5 | 21-30 |
| Chronic myelomonocytic leukaemia (CMML) (>1000 monocytes/mcL blood) | <5 | 5-20 |
Table 2.
MDS subtypes according to WHO 2002 classification.
| Subtype | Blood | BM |
|---|---|---|
| Refractory cytopenia with unilineage dysplasia (RCUD) | Single or bicytopenia | Dysplasia in ≥ 10% of one cell line, <5% blasts |
| Refractory anaemia with ring sideroblasts (RARS) | Anaemia, no blasts | ≥ 15% of erythroid precursors with ring sideroblasts, erythroid dysplasia only, <5% blasts |
| Refractory cytopenia with multilineage dysplasia (RCMD) | Cytopenia(s), <1x109/L monocytes | Dysplasia in ≥ 10% of cells in ≥2 hematopoietic lineages, ± 15% ring sideroblasts, <5% blasts |
| Refractory anaemia with excess blasts-1 (RAEB-1) | Cytopenia(s), ≤ 2%-4% blasts, <1x109/L monocytes | Unilineage or multilineage dysplasia, no Auer rods, 5%-9% blasts |
| Refractory anaemia with excess blasts-2 (RAEB-2) | Cytopenia(s), 5%-19% blasts, <1x109/L monocytes | Unilineage or multilineage dysplasia, Auer rods, ± 10%-19% blasts |
| MDS, unclassified (MDS-U) | Cytopenia(s) | Unilineage dysplasia or no dysplasia but characteristics MDS cytogenetics, <5% blasts |
| MDS associated with isolated del(5q) | Anaemia, platelets normal or increased | Unilineage erythroid dysplasia, isolated del(5q), <5% blasts |
Table 3.
MDS subtypes according to WHO 2016 classification.
| Subtype | Dysplastic lineages | Cytopenia(s) | Ring sideroblasts in erythroid elements of BM | Blasts | Cytogenetics |
|---|---|---|---|---|---|
| MDS with single lineage dysplasia (MDS-SLD) | 1 | 1 or 2 | RS< 15% | PB<1% BM<5% No Auer rods |
Any, unless fulfils criteria for isolated del(5q) |
| MDS with multilineage dysplasia (MDS-MLD) | 2 or 3 | 1-3 | RS<15% | PB<1% BM<5% No Auer rods |
Any, unless fulfils criteria for isolated del(5q) |
| MDS with ring sideroblasts (MDS-RS) MDS-RS with single-lineage dysplasia (MDS-RS-SLD) |
1 | 1-2 | RS≥15% | PB<1% BM<5% No Auer rods |
Any, unless fulfils criteria for isolated del(5q) |
| MDS-RS with multilineage dysplasia (MDS-RS-MLD) | 2 or 3 | 1-3 | RS≥15% | PB<1% BM<5% No Auer rods |
Any, unless fulfils criteria for isolated del(5q) |
| MDS with isolated del(5q) | 1-3 | 1-2 | None or any | PB<1% BM<5% No Auer rods |
Del(5q) alone or with 1 additional abnormality except -7 or del(7q) |
| MDS with excess blasts (MDS-EB) MDS-EB1 |
0-3 | 1-3 | None or any | PB 2~4% or BM 5~9% No Aur rods |
Any |
| MDS-EB2 | 0-3 | 1-3 | None or any | PB 5~19% or BM 10%~19% or Auer | Any |
| MDS-U with 1% PB blast | 1-3 | 1-3 | None or any | PB= 1%, BM <5%, Auer rods | Any |
| With SLD and pancytopenia | 1 | 3 | None or any | PB<1% BM<5% No Auer rods |
Any |
| Defining cytogenetic abnormality | 0 | 1-3 | <15% | PB<1% BM<5% No Auer rods |
MDS defining abnormality |
| Refractory cytopenia in childhood (RCC) | 1-3 | 1-3 | None | PB<2% BM<5% No Auer rods |
Any |
Table 4.
MDS subtypes according to WHO 2022 classification.
| MDS with defining genetic abnormalities | Blasts |
| MDs with low blasts and isolated 5q deletion | <5% BM and <2% PB |
| MDS with low blasts and SF3B1 mutation | <5% BM and <2% PB |
| MDS with biallelic TP53 inactivation | <20% BM and PB |
| MDS with defining morphological abnormalities | |
| MDS with low blasts (MDS-LB) | |
| MDS, hypoplastic (MDS-h) | |
| MDS with increased blasts (MDS-IB) | |
| MDS-IB1 | 5%-9% BM or 2%-4% PB |
| MDS-IB2 | 10%-19% BM; or 2%-19% PB |
| MDS with fibrosis | 5%-19% BM; or 2%-19% PB |
Table 5.
MDS subtypes criteria according to IPSS.
| Score | |||||
|---|---|---|---|---|---|
| Prognostic variable | 0 | 0.5 | 1 | 1.5 | 2 |
| BM blasts | <5% | 5-10% | - | 11-20% | 21-30% |
| Karyotype | Good | Intermediate | Poor | - | - |
| Cytopenia(s) | 0-1 | 2-3 | - | - | - |
Table 6.
MDS subtypes` scores according to IPSS classification.
| Total score | Risk groups | Median survival (yrs) | Median time to 25% acute myeloid leukaemia (AML) evolution (yrs) |
|---|---|---|---|
| 0 | Low | 5.7 | 9.4 |
| 0.5-1 | INT-1 | 3.5 | 3.3 |
| 1.5-2 | INT-2 | 1.2 | 1.1 |
| ≥2.5 | High | 0.4 | 0.2 |
Table 7.
In vivo MDS models.
| Category | Models | Pros | Cons | Example |
|---|---|---|---|---|
| Mouse | BM transduction/transplantation | Good transplantation | No MDS-AML transformation | MLD-PLD/RUNX1-291fs BMT model |
| Gene editing/modification | Controlled gene expression MDS-AML transformation Studying mutations in particular genes |
Complex operation including embryo culture, microscope injection, and vector construction Long and expensive production cycle |
5q model Tumour suppressor model RAS mouse model Tyrosine kinase model |
|
| Xenotransplantation | Simple process Screening of targeted drugs |
Low rate of tumorigenesis | Cells derived from MDS patients were injected directly to immunodeficient mice. Subcutaneous transplantation model of human MDS cell line SKM-I |
|
| Induced animal model | Simple process MDS-AML transformation |
Unstable biological property Harmful to environment |
Benzene induced model Alkylation reagent induced model Radiation induced model |
|
| Rat | Chemical induced model | Simple process MDS-AML transformation |
Unstable biological property Harmful to environment |
Dimethylbenzanthracene (DMBA) induced model |
| Zebrafish | Genetically engineered model | Controlled gene expression Studying mutations in certain genes High throughput |
Long and expensive production cycle Non-mammalian vertebrate which is different from a patient |
c-myb-gfb model |
Table 8.
Studies reporting that MSCs impact leukaemia by suppressing tumour growth.
| MSCs type | Tumour model | Findings | Proposed mechanism | Author |
|---|---|---|---|---|
| Mouse BMSCs | ALL (P388) and B-lymphoma (A20) | Inhibit leukaemia/lymphoma cell growth | Induction of cell cycle arrest and apoptosis of tumour cells | Song et al (2015) |
| Human BMSCs | CML (BV173) and T-ALL (Jurkat) | Inhibit the proliferation of cancer cells | Induction of cell cycle arrest of leukaemic cells | Sarmadi et al (2010) |
| UC-MSCs | CML (K562) | Exert anti-proliferative effect on leukaemia cells | Paracrine signalling and secretion of substances by the secretome | Hendijani et al (2015) |
| Human AT-MSCs | AML (HL-60) and CML (K562) | Inhibit the proliferation of leukaemic cells | Secretion of DKK-1 | Zhu Y et al (2009) |
| Human BMSCs | CML (patient’s cells) | Inhibits the proliferation of CML cells | Production of IFN-a | Zhang HM et al (2009) |
| Human BMSCs | CML (K562) | Inhibit the proliferation of CML cells | Activation of caspase3 and most probably production of CINC-1 and TIMP-1 | Fathi et al (2019) |
| Human UC-MSCs | CML (K562) | Inhibit the proliferation of K562 cells | Cell cycle arrest at G0/G1 by IL-6 and IL-8 | Fonseka et al (2012) |
| Human adipose tissue-MSCs | CML (K562) and AML HL-60) | Inhibit the proliferation of cancer cells | Cell cycle arrest at G0/G1 by DKK-1 secretion | Zhu et al (2009) |
| Leukaemia patient’s BMSCs | CML (K562) | Inhibit apoptosis and growth of leukaemia cells | Cell cycle arrest at G0/G1 and induction of apoptosis via phosphorylation of Bad and Akt proteins | Wei et al (2009) |
| Human UC-MSCs | AML (HL-60) and CML (K562) | Inhibit the growth of cancer cells | Cell cycle arrest at G0/G1 and phosphorylation of p38 MAPK | Tian et al (2010) |
| Human BMSCs and CML patient’s BMSCs | CML (K562 and patient’s cells) | Increase the anti-apoptotic capacity of cancer cells | Regulation of apoptosis-associated protein expression and activation of the Wnt signalling pathway | Han et al (2013) |
| Human UC-MSCs | T-ALL (Jurkat cell line) | Inhibit the proliferation of Jurkat cells | Activation of Notch signalling pathway | Yuan et al |
Table 9.
Studies that targeted MSCs or used MSCs as drug delivery tool in MDS and leukaemia.
| MSC type | Tumour/Disease model | Findings | Proposed mechanism | Author |
|---|---|---|---|---|
| Patient derived BMSCs | MDS | Improvement of proliferation and osteogenic capacity of BMSCs with their increased support of HSPCs | Restoring aberrant DNA methylation of BMSCs | Geyh et al (2013) |
| Patient derived BMSCs | MDS | Improvement of the negative impact of MSCs on haematopoiesis by the support of MSCs on healthy HSPC expansion | Regulation of genes related to IFN-γ and ECM receptor interaction pathways by AZA for the support of haematopoiesis | Wenk et al (2018) |
| Patient derived BMSCs | MDS | Improvement of inflammatory environment through AZA | Reducing the level of IL-6 in MDS-MSCs | Boada et al (2016) |
| Patient derived BMSCs | MDS | Improvement of haematopoiesis through AZA | Normalising the BMME and reversing MSCs ability of osteogenic differentiation and proliferation | Poon et al (2019) |
| Patient derived BMSCs | MDS | Improvement of BMSCs’ capacity of supporting normal HSPCs through lenalidomide | Decreasing the secretion of CXCL12 | Ferrer et al (2013) |
| Patient derived BMSCs | MDS | Reducing the ability of MSCs to induce the differentiation of T cells into Tregs and improvement of MDS-MSC senescence through decitabine | Decreasing PD-L1 and CDKN1A expression | Pang et al (2019) |
| Patient derived BMSCs | MDS | Suppression of the adhesion of leukaemic cells to the stroma | Targeting CXCR4-CXCL12 axis | Kastrinaki et al (2013) |
| Human BMSCs | MDS | Decrease in autophagy | Decreasing ROS and iron content | Camiolo et al (2019) |
| Human BMSCs | MDS | Improvement of the proliferation activity of MSCs and BMSCs support in haematopoiesis | Inducing CDKN2A and enhancing apoptosis | Fujishiro et al (2020) |
| Patient derived BMSCs | MDS | Restoration of osteogenic differentiation of MSCs | Blocking TGFβ signalling with SD-208 | Geyh et al (2018) |
| BMSCs | CML | Regression of tumour and improvement of survival rates | Production of interferon by engineered MSCs for drug delivery | Andreeff et al (2004) |
| Mice BMSCs | T-ALL | Decrease in tumour burden and improvement of survival rate | Intra-BM treatment with MSCs | Xia et al (2020) |
| Human BMSCs | CML (K562) | Reduction in the proliferation of CML cells and induction of apoptosis | Transfer of hIFN-γ gene to MSCs and co-culture of genetically modified MSCs and K562 cells | Li et al (2006) |
| Human BMSCs | AML | Increase in the survival of MSCs | Genetically engineered MSCs to release anti-CD33-anti-CD3 bispecific antibody | Aliperta et al (2017) |
| Human BMSCs | Human T-cell ALL (MOLT-4) and mouse CLL (L1210) | Inhibition of leukaemic cells and angiogenesis | MSCs loaded with PTX | Pessina et al (2013) |
| Human UC-MSCs | CML (K562) | Suppression of leukaemic cell proliferation | MSC EVs combined with doxorubicin | Hendijani et al (2015) |
| Rat BMSCs | ALL (Ball-1) and K562 (CML) | Suppression of leukaemic cell proliferation | Targeting macrophages through MSCs-primed with sLipo leva | Liu et al (2022) |
| Human BMSCs | AML (THP-1) | Inhibition of AML cell proliferation and induction of AML apoptosis | Targeting IRF2 and delivering miR-222-3p via exosomes released from BMSCs | Zhang et al (2022) |
Table 10.
Clinical studies utilising MSC-based approaches for the treatment of hematologic malignancies, obtained from clinicaltrial.gov.
Table 10.
Clinical studies utilising MSC-based approaches for the treatment of hematologic malignancies, obtained from clinicaltrial.gov.
| NCT No. | Phase | Interventions | Treatment | Cancer Applications |
|---|---|---|---|---|
| NCT04565665 | I/II | Cord blood MSCs | MSCs IV followed by a second fusion of MSCs within 7 days of the first one. | Hematopoietic and lymphoid cell neoplasm |
| NCT03184935 | I/II | Cord blood MSCs | Allogeneic umbilical cord MSCs and decitabine (20mg/m^2) | Myelodysplastic syndromes |
| NCT02181478 | I | MSCs | Reduced-intensity conditioning with cyclophosphamide, fludarabine (with total body irradiation) or fludarabine and melphalan followed by co-transplantation of intra-osseous umbilical cord blood and MSCs. | Hematologic malignancies |
| NCT01624701 | I/II | Bone marrow MSCs | Clinically ex-vivo expanded cord blood cells are comprised of stem cell factor, Flt3 ligand, thrombopoietin, IGFBP2, and MSC co-culture. | Expanding umbilical cord blood-derived blood stem cells for treating leukaemia, lymphoma, and myeloma |
| NCT01092026 | I/II | Cord blood transplantation + MSCs | Umbilical cord blood hematopoietic stem cell transplantation co-infused with third-party MSCs | Hematologic malignancies |
| NCT01045382 | II | Hematopoietic stem cells + MSCs | 1,5-3,0 x 10E6 MSC/Kg with fludarabine and 2 Gy total body irradiation followed by HLA-is matched PBSC | Leukaemia, lymphoma, and myeloma |
| NCT01129739 | II | Cord blood MSCs | 1.0E+6 MSC/kg, Intravenous | Myelodysplastic syndromes |
| NCT05672420 | Ib/II | Umbilical cord-derived MSCs | RP2D, Intravenous | Hematologic malignancies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



