
Submitted:
27 June 2024
Posted:
28 June 2024
You are already at the latest version
Abstract
Studies on animal virome have mainly concentrated on chordates and medically significant invertebrates, often overlooking sylvatic mosquitoes, constituting the major part of mosquito species diversity. Despite their potential role in arbovirus transmission, the viromes of sylvatic mosquitoes remain largely unexplored. These mosquitoes may also harbor insect-specific viruses (ISVs), affecting arboviral transmission dynamics. The Cerrado biome, known for rapid deforestation and its status as a biodiversity hotspot, offers an ideal setting for investigating mosquito viromes due to potential zoonotic spillover risks from land use changes. This study aimed to characterize the viromes of sylvatic mosquitoes collected from various locations within Minas Gerais state, Brazil. Total RNA was extracted from mosquito pools of Psorophora albipes, Sabethes albiprivus, Sa. chloropterus, Psorophora ferox, and Coquillettidia venezuelensis species, followed by high-throughput sequencing (HTS). Bioinformatic analysis included quality control, contig assembly, and viral detection. Sequencing data analysis revealed 11 near-complete viral genomes (new viruses are indicated with asterisks) across seven viral families and one unassigned genus. These included: Xinmoviridae (Ferox mosquito mononega-like virus* and Albipes mosquito Gordis-like virus*), Phasmaviridae (Sabethes albiprivus phasmavirus*), Lispiviridae (Pedras lispivirus isolate MG), Iflaviridae (Sabethes albiprivus iflavivirus*), Virgaviridae (Buriti virga-like virus isolate MG and Sabethes albiprivus virgavirus 1*), Flaviviridae (Psorophora ferox flavivirus*), Mesoniviridae (Alphamesonivirus cavallyense isolate MG), and the genus Negevirus (Biggie virus isolate MG virus and Coquillettidia venezuelensis negevirus*). Phylogenetic analyses revealed evolutionary relationships with varying degrees of similarity to known sequences in distinct host species. In some cases, new viruses and potential new genera were discovered, emphasizing the importance of studying sylvatic mosquito virome to understand viral diversity and evolution in ecologically sensitive regions. Moreover, the presence of ISVs and potential novel arboviruses underscores the need for ongoing surveillance and control strategies to mitigate the risk of emerging infectious diseases.
Keywords:
Sylvatic mosquitoes
; RNA virome
; Brazilian Cerrado
; Novel viral species
; Arbovirus transmission
; Biodiversity hotspot
1. Introduction
Studies on animal virome have predominantly focused on chordates and medically significant invertebrates such as ticks and mosquitoes [1]. Within mosquitoes, urban species like Aedes aegypti, Ae. albopictus and Culex quinquefasciatus, known for their role in arbovirus transmission, have been the primary subjects of virome investigations [2,3]. However, despite an estimated global mosquito species count exceeding 3,700 [4], sylvatic mosquitoes remain underexplored regarding their viromes. This gap is significant given the critical role of certain species, such as Sabethes spp in Yellow Fever Virus transmission [5] and Coquillettidia spp. in Eastern Equine Encephalitis virus [6] and Oropouche virus transmission [7,8].
In addition to arboviruses, mosquitoes may harbor insect-specific viruses (ISVs) which, although incapable of infecting vertebrates, offer valuable perspectives into viral evolution due to their close phylogenetic relationship with arboviruses. Some ISVs have shown the ability to modulate arboviral transmission from vectors to mammalian hosts, suggesting their potential role in developing innovative strategies for controlling arboviral outbreaks [9,10].
In parallel, high-throughput sequencing (HTS) has emerged as a critical tool for discovering new viruses, including ISVs, and determining virome in mosquitoes [11,12,13]. HTS enables the comprehensive and unbiased analysis of viral genetic material present in mosquito samples, facilitating the identification of both known and novel viruses [14]. This technology is particularly valuable for exploring the viromes of sylvatic mosquito species, which are often understudied compared to their urban counterparts [15]. By offering intricate perspectives into the diversity and composition of mosquito viromes, HTS enhances our understanding of viral ecology and evolution, supports the development of more effective vector control and surveillance strategies, and can be utilized to discover new viruses in the largely unexplored Brazilian biomes.
The Cerrado, a biodiversity hotspot in Brazil [16], faces alarming rates of deforestation, driven in part by expanding monoculture activities [17]. Given that anthropogenic alterations in land use are major contributors to zoonotic spillover events [18], understanding the viromes associated with arthropod vectors in this biome is crucial for enhancing genomic surveillance of emerging arboviruses. Therefore, the present study aims to deepen our understanding of RNA viromes within sylvatic mosquitoes inhabiting the Cerrado biome.
2. Materials and Methods
2.1. Sample Collection
Sylvatic mosquitoes were collected across five municipalities within the Minas Gerais state (Figure 1). Additionally, a mixed pool comprising diverse mosquito species was included in the study, including Sabethes spp and Ps. ferox (Table 1).
Adult mosquitoes were captured employing the protected human attraction method [19] and the Shannon (1939) [20] trap, with assistance from entomological nets and oral aspirators [21]. Following capture, the mosquitoes were sorted by genera and cryopreserved in liquid nitrogen (-196°C) before being transported to the laboratory. Taxonomic classification was conducted on a chilled table at -20◦C utilizing a stereoscopic microscope and dichotomous keys [22,23].
2.2. RNA Extraction and High-Throughput Sequencing
Total RNA was extracted from mosquito pools of varying sizes (20 to 200 individuals - Table 1) using the QIAmp Viral RNA (Qiagen) kit. The extracted RNA was quantified using a Quantifluor® RNA system (Promega) kit, following the manufacturer’s instructions. For samples containing at least 1 µg of RNA, 0.1 v/v of 3M sodium acetate and 2 v/v of absolute ethanol were added. These samples were then dispatched to Macrogen (South Korea), where Illumina TruSeq stranded Total RNA library + Ribo-zero Gold libraries were generated for NovaSeq 6000, 100bp paired-end sequencing, resulting in 20M to 30M reads per library.
2.3. Bioinformatic Quality Control, Assembly of Contigs, and Viral Detection
We conducted quality control analysis of the raw reads using fastQC [24], Trim-galore for adapter removal v0.6.10 [25] and Trimmomatic v 0.39 [26] to trim 10 bp from each end of the reads. The assembly of trimmed reads into contigs was performed using Spades v3.15.5, with the -rnaviral flag [27]. Viral contigs were identified through local diamond blastx searches [28] of RNA-dependent RNA-polymerases compiled in RdRP-scan [29]. To identify M and S segments of Phasmaviridae, local diamond blastx searches were conducted using Refseq sequences deposited in Genbank [30] (assessed on 08/07/2023). Viral contigs were verified with BLASTx searches against the non-redundant protein sequences in NCBI, and viral completeness was assessed using CheckV [31]. Viral genomes were annotated utilizing Geneious v. 11.1.5 (Biomatters) and ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). The obtained viral sequences were deposited in Genbank with accession numbers PP946236 - PP946246.
2.4. Phylogenetic Analyses
Global multiple alignments were created for each viral family and protein marker using Mafft v7.520 [32]. The resulting alignments were subsequently trimmed by employing TrimAI v1.4.15 [33] with the -automated1 flag. The amino acid substitution models for each alignment were determined using modeltest-NG v0.1.7 [34]. Maximum Likelihood trees were then constructed utilizing RAxML-NG v1.2.0 [35] with 1000 bootstraps. All trees, otherwise stated, were mid-point rooted.
3. Results
Each sequencing library comprised approximately 20.5 to 30.4 million reads. Post-assembly, each library produced around 2,014 to 37,238 contigs (Table 2). The higher contig count in the mixed mosquito pool is attributed to its diverse species composition, compared to the single-species composition of the other insect pools.
The HTS data analysis uncovered 11 near-complete viral genomes spanning seven distinct viral families and one genus of an unassigned family. Among these were the ssRNA- families Xinmoviridae, Phasmaviridae, and Lispiviridae, and the ssRNA+ families Iflaviridae, Virgaviridae, Flaviviridae, Mesoniviridae along with the genus Negevirus (Table 2). Of the 11 viral genomes described below, 7 are new, while the 4 previously known genomes were identified in new host species.
3.1. Xinmoviridae
We identified two consensus putative viral sequences, measuring 13 kb and 12 kb, in Ps. ferox and Ps. albipes, respectively. Their encoded RdRps exhibited 59.02% and 51.10% similarity with their closest matches in Genbank (Guadeloupe mosquito mononega-like virus, MN053735, and Gordis virus, MW435014) within the Xinmoviridae family (Figure 2A,B). According to ICTV criteria [36], RdRp sequences showing less than 60% similarity within this family may indicate the presence of novel genera. Both of these present genes for a putative nucleoprotein, an envelope glycoprotein, and an RdRp. Despite these viruses being discovered [37,38] in similar hosts (same Culicidae family, but different genera), their positions within the RdRp tree are notably distinct (Figure 2C). Hence, we tentatively named these viruses Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus, respectively.
3.2 Phasmaviridae
Our investigation revealed three distinct putative viral segments - small (S), medium (M), and large (L) segments - in the Sa. albiprivus pool, with lengths of 6.5 kb, 2.1 kb, and 1.7 kb, coding for its RdRp, envelope glycoprotein and capsid protein, respectively (Figure 3A). Comparison with Genbank sequences identified an RdRp sequence showing 45.50% similarity to Miglotas virus (QRW41774.1) [38] within the Phasmaviridae family. As this RdRp shares less than 95% identity, as per ICTV criteria, we suggest the classification of a novel virus species, tentatively named Sabethes albiprivus phasmavirus. Phylogenetic analysis supports the distinction of this new virus, forming a separate monophyletic branch (Figure 3B).
3.3. Lispiviridae
We identified a consensus putative viral genome of 12 kb for the Sa. albiprivus pool, containing five open reading frames (ORF) (Figure 4A). Its genes include a nucleoprotein, a phosphoprotein, an envelope glycoprotein, and an RdRp. Notably, its RdRp sequence shares more than 85% similarity with that of Pedras lispivirus [39], indicating classification within the same species within the Lispiviridae family, according to ICTV criteria [40]. Hence, we designate this virus as Pedras lispivirus isolate MG. In their study [39], previously identified this virus in Sa. quasicyaneus, inhabiting a transitional zone between the Cerrado and Amazon biomes. Despite geographical separation, the hosts of this virus share a close evolutionarily relationship (same subgenus - Sabethes Sabethes) (Figure 4B), suggesting potential occurrence in other species within this mosquito genus, a hypothesis warranting further investigation.
3.4. Iflaviridae
We identified a consensus putative viral genome of approximately 9 kb for the Sa. albiprivus pool (Figure 5A). This genome encodes a single polyprotein comprising both structural and non-structural components, showing homology to viruses from the Iflaviridae family. The structural component, responsible for coding the capsid proteins, has its closely related sequence in Genbank with 62% coverage and 71.65% similarity to Aedes Iflavi-like virus 1 (QQD36915.1), a virus isolated from Ae. aegypti in the Brazilian Amazon [41]. According to the criteria set by the ICTV [42], the structural component’s similarity is lower than 90%, and its occurrence in Sa. albiprivus, suggests that this is a novel species. We tentatively name this species Sabethes albiprivus iflavivirus (Figure 5B).
3.5. Virgaviridae
We identified two putative viral consensus genomes of approximately 8.8 kb and 9.1 kb in the mixed pool (Sa. albiprivus, Sa. chloropterus, and Ps.ferox) and Sa. albiprivus pool, respectively (Figure 6A), showing homology to viruses from the Virgaviridae family, each with a polyprotein coding gene and a putative capsid coding gene, albeit in differing synthesis. We tentatively name these viruses, respectively, Buriti virga-like virus isolate MG and Sabethes albiprivus virga virus 1. In the phylogenetic analysis, Buriti virga-like virus isolate MG clusterswith Buriti virga-like virus found in Sa. chloropterus [39] (Figure 6B). Considering the ICTV criteria for taxa classification within this family (https://ictv.global/report_9th/RNApos/Virgaviridae), metadata such as the mode of transmission is required. Therefore, we are currently unable to propose a taxon for Sabethes albiprivus virgavirus 1.
3.6. Flaviviridae
In the Ps. ferox pool, we identified a consensus putative viral genome of approximately 11 kb (Figure 7A) showing homology to viruses from the Flaviviridae family. The genome includes the ORFs characteristic of flaviviruses, encoding the polyprotein that is cleaved into structural (C, prM, E) and non-structural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) proteins. NS5 encoded RdRp that presented 55.73% identity with its most closely related sequence in GenBank, Mansonia flavivirus (LC567153) [43]. We have tentatively named this virus Psorophora freaks flavivirus. Considering the ICTV criteria for the family (https://ictv.global/report_9th/RNApos/Flaviviridae), classification requires metadata beyond sequence information, including antigenicity and ecological characteristics, which are beyond the scope of this study. Therefore, we lack sufficient data to propose a new taxon for Psorophora ferox flavivirus. Given its placement within the phylogenetic tree, it is most likely an ISV (Figure 7B).
3.7. Mesoniviridae
We identified a consensus putative viral genome consisting of 20,198 bases in Cq. venezuelensis (Figure 8A), showing homology to viruses within the Mesoniviridae family. Its gene synteny is the same as observed in the species, including genes encoding for two polyproteins for its non-structural proteins and smaller structural coding genes near its 3’ end. The encoded putative RdRp has over 99% similarity to that of Alphamesonivirus cavallyense (AXL48235.1) found in Culex pipiens [44]. Notably, Cq. venezuelensis and Cx. pipiens differ significantly in terms of phylogeny, behavior, and geographic distribution. These species belong to different tribes, Mansonini and Culicini, respectively. Cq. venezuelensis exhibits wild habits and is restricted to the Americas [45], whereas Cx. pipiens is urban and invasive across multiple continents [46]. In the phylogenetic analysis, this virus found in Cq. venezuelensis formed a monophyletic branch (Figure 8B). We tentatively name this Alphamesonivirus cavallyense isolate MG.
3.8. Negevirus
We identified two putative consensus viral genomes of 9.2 Kb in Cq. velezuelensis (Figure 9A,B), showing homology to viruses within the Negevirus genus. Both of these present a gene synteny typical of the genus, including a non-structural polyprotein, an envelope glycoprotein, and a capsid protein. One of these genomes encodes a replicase with 74% coverage and over 99% similarity to its closest sequence in Genbank, Biggie virus Mos11 (KX924639.1) found in Culex pipiens collected in the USA [47]. We have tentatively named this Biggie virus isolate MG. As previously mentioned, these hosts differ significantly, which may suggest considerable plasticity of this virus. The second genome encodes a replicase with 73% coverage and 66% similarity to its closely related sequence, Aqua Salud Negevirus (MT197494.1) detected in Culex declarator in Panama [48], also a quite different host. We tentatively named this virus Coquillettidia velezuelensis negevirus (Figure 9C).
4. Discussion
In this study, we identified seven novel viruses and four previously known genomes identified in new host species, all of which are underrepresented in virological research. The discovery of new ISVs and potential arboviruses is particularly significant as it broadens our understanding of viral diversity within mosquito populations and reveals the potential ecological roles these viruses may play. Given the capacity of ISVs to interact with or impede arbovirus transmission [49], our findings underscore the necessity of further exploring these interactions. Moreover, HTS once again demonstrates its remarkable ability to discover new viruses, even in pools of mosquitoes from different species, thereby enhancing our understanding of comprehensive virome surveillance in regions undergoing rapid environmental changes, such as the Cerrado Brazilian biome. Compared to the work of Silva et al. (2023) [13], the number of full RdRp sequences detected was similar to our study. However, the viruses’ characterization by authors was partial. Interestingly, only one mosquito species, Cq. venezuelensis, was the same host searched, but the viruses identified were different. This highlights the diversity of sylvatic mosquitoes and underscores the need for further research. Below we discuss in detail the viruses detected and characterized here.
4.1. Xinmoviridae
Infections with anpheviruses (family Xinmoviridae; order Mononegavirales) seem to be relatively prevalent, evidenced by their presence in laboratory colonies and wild populations of Aedes albopictus from various geographic locations over several years [50,51]. Notably, Wolbachia bacteria enhance Aedes anphevirus replication, while this virus marginally inhibits DENV replication in mosquito cell lines [50]. Anpheviruses have also been identified in populations of Culex spp. [52] and anophevirus-like species have been found in Amazonian anophelines, including Anopheles marajoara and An. darlingi [53]. Here, we described two putative new Xinmoviridae species in two Psorophora sp. mosquitoes. The widespread distribution of most Psorophora species in the Americas [23] poses a potential threat to public health due to their capacity to transmit several arboviruses such as Yellow Fever Virus, Rocio, West Nile Virus, Eastern Equine Encephalitis and Ilheus Virus [54,55,56,57,58,59]. The capability of arboviruses to infect Psorophora species suggests a potential for disease transmission. The discovery of Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus in Ps. ferox and Ps. albipes, respectively, presents new avenues for future studies aiming to understand the interactions between ISVs and other arboviruses in the context of pathogen transmission. The identification of these novel viruses characterized by their distinct positions within the RdRp phylogenetic tree and less than 60% similarity to known viruses, hints at the potential presence of new genera within the Xinmoviridae family, emphasizing the necessity for further research into the diversity and ecological roles of ISVs in mosquito populations.
4.2. Phasmaviridae
Phasmaviruses remain relatively enigmatic, with our understanding largely shaped by HTS endeavors. In our investigation, we introduce novel viral entities identified within Sa. albiprivus, tentatively classified as Sabethes albiprivus phasmavirus. These findings contribute to the expanding spectrum of phasmaviral diversity. Across arthropod hosts spanning Diptera, Hymenoptera, and Coleoptera orders, phasmaviruses have been sporadically detected [60]. Presently, the ICTV acknowledges seven genera within the Phasmaviridae family [61]. Nevertheless, our grasp of the physicochemical attributes of these entities remains rudimentary, with detailed characterizations limited to merely two of these genera [61]. The dearth of comprehensive in vitro analyses underscores the pressing need for elucidating the fundamental properties of phasmaviruses, including their constituent subunits. Additionally, exploring the socio-virological dynamics associated with these viruses represents a fertile ground for future inquiry, promising perspectives into their ecological roles and potential impacts on host populations. Thus, bridging these knowledge gaps stands as imperative for unraveling the enigmatic realm of phasmaviral biology.
4.3. Lispiviridae
Members of the Lispiviridae family have been detected in various hosts across different regions worldwide, including arthropods such as hemipterans, odonatans, hymenopterans, and orthopterans [60,62,63,64,65,66]. Infection by viruses representing the Lispiviridae family appears rare in mosquitoes. In this study, we identified a lispivirus (Pedras lispivirus isolate MG) in the host species Sa. albiprivus, previously identified in Sa. quasicyaneus [39], suggesting a close relationship between virus species and their hosts within the genus Sabethes, subgenus Sabethes (Sabethes). Members of the genus Sabethes are significant vectors of the wild yellow fever virus in the Americas [67]. Notably, Sa. albiprivus has already been found naturally infected by the amaryllic virus [5,68]. Future studies are warranted to investigate the occurrence of this virus in other species within this mosquito genus to enhance our understanding of its epidemiology and potential impact on public health.
4.4. Iflaviridae
Iflaviruses have been described in insects across several orders, including Lepidoptera, Hymenoptera, and Hemiptera, as well as in bee parasitic mites [69]. In bees, iflaviruses are major pathogens and are primarily associated with wing deformation [70,71]. The iflavivirus found in Sa. albiprivus (named Sabethes albiprivus iflavivirus) in this study shares similarities with Aedes Iflavi-like virus 1, isolated from Ae. aegypti in the Brazilian Amazon [41]. Despite the phylogenetic distinction and geographical / behavioral separation between the host mosquitoes Ae. aegypti and Sa. albiprivus, they harbor similar Iflaviruses. This discovery suggests a potential broader host range and adaptability of iflaviruses across different mosquito species and regions. Further investigations are necessary to comprehend the ecological and epidemiological implications of these viruses, particularly regarding their impact on mosquito populations and their potential role in disease transmission.
4.5. Virgaviridae
The Virgaviridae family has emerged prominently across diverse mosquito virome studies, underscoring its prevalence and potential significance in mosquito ecology [72,73]. In our investigation, we introduce two novel viral entities, tentatively designated as Buriti virga-like virus isolate MG and Sabethes albiprivus virgavirus 1, expanding the repertoire of known Virgaviridae members. Traditionally associated with plant hosts, the detection of virgavirus in mosquito viromes raises intriguing questions regarding their ecological roles and transmission dynamics within insect populations. While it is conceivable that certain virgavirus species have adapted to exploit mosquito hosts for replication, an alternative explanation implicates dietary factors in their presence within mosquito viromes. The multifaceted nature of mosquito viromes encompasses a broad spectrum of viruses, including ISVs, arboviruses, and even plant viruses [74]. The phylogenetic positioning of Buriti virga-like virus isolate MG alongside its counterpart in Sa. chloropterus suggests a shared evolutionary ancestry, indicative of potential host-specific adaptations. Conversely, the distinct monophyletic branch formed by Sabethes albiprivus virgavirus 1 underscores its unique evolutionary trajectory within the Virgaviridae family. Further exploration into the ecological interactions between virgaviruses and their mosquito hosts promises valuable perspectives into their adaptive strategies and ecological impacts within vector populations.
4.6. Flaviviridae
The Flaviviridae family harbors a notable array of ISVs with intriguing implications for arboviral dynamics. Key among these are ISVs like Nhumirim virus [75], Cell fusion agent virus [76], and Culex flavivirus [77], which have been documented to exhibit inhibitory effects on their arboviral counterparts, albeit with variable outcomes across studies. While the majority of these findings stem from cell-line investigations, insights gleaned from mosquito-based studies are also noteworthy. In our study, we introduce Psorophora ferox flavivirus as a novel member of the Flaviviridae family. This discovery adds to the growing catalog of ISVs within the flavivirus lineage and underscores the diverse virome composition within mosquito populations. The inhibitory mechanisms attributed to ISV flaviviruses are postulated to stem from their close genetic relatedness to arboviruses within the family (reviewed in Carvalho and Long, 2021) [78]. However, the nuanced nature of these interactions is underscored by the variability in outcomes observed across different experimental settings. Despite the discordance among studies, the intricate interplay between flaviviruses holds promise for arboviral control strategies and enhances our comprehension of vector competence across mosquito species. Continued exploration of these viral dynamics, both in laboratory settings and within natural vector populations, is crucial for elucidating the full spectrum of ISV effects on arboviral transmission and for informing targeted interventions aimed at mitigating vector-borne disease burdens.
4.7. Mesoniviridae
Alphamesonivirus stands out as a ubiquitous presence across diverse mosquito species (over 34) and geographic regions, encompassing Aedes spp., Culex spp., Anopheles spp., Armigeres subalbatus, and Cq. xanthogaster [3]. Our study adds a significant dimension to this narrative by documenting the presence of Alphamesonivirus cavallyense isolate MG for the first time in Cq. venezuelensis, a notable inclusion given the species' unique ecological and geographical context. The robust adaptability of Alphamesonivirus across different host species and environments underscores its remarkable success in host colonization, warranting comprehensive investigation into the factors driving its epidemiological dynamics. Notably, the Yichang virus, a close relative within the Mesoniviridae family, has demonstrated both horizontal and vertical transmission among Ae. albopictus and Cx. quinquefasciatus, alongside the intriguing ability to hinder flavivirus replication in mosquito cell lines [79]. However, elucidating similar relationships between other mesonivirids and arboviruses remains an open avenue for research. The discovery of Alphamesonivirus cavallyense isolate MG in Cq. venezuelensis, a species with distinct phylogenetic, behavioral, and ecological attributes compared to its typical hosts like Cx. pipiens, offers a unique lens into the dynamics of mesonivirus-host interactions. By forming a distinct monophyletic branch within the phylogenetic tree, this novel isolate underscores the genetic diversity and evolutionary adaptability of mesoniviruses across divergent mosquito taxa. Further exploration into the ecological drivers shaping the distribution and dynamics of mesoniviruses, alongside their potential implications for arboviral transmission dynamics, remains imperative. Unraveling the intricate interplay between mesonivirids and arboviruses holds promise for advancing our understanding of vector-borne disease ecology and informing targeted interventions aimed at mitigating mosquito-borne disease burdens.
4.8. Negevirus
Several viruses in this genus have been first isolated from mosquito cell lines exposed to filtered mosquito and phlebotomine specimens, particularly using C6/36 cell lines [80]. However, recent reports of new negeviruses have expanded to agricultural pests, including Hemiptera [81,82]. The detection of mosquito-associated negeviruses, using either cell cultures or HTS, has occurred across the globe [3,80,83], indicating wide host ranges and genetic variability. Their closest relatives include plant arboviruses, such as kitavirids [82,83]. Furthermore, diverse viruses in this genus also negatively modulate the multiplication of flaviviruses and alphaviruses in co-cultures in mosquito cell lines, possibly through superinfection exclusion [84,85]. This modulator capacity may be explored in the future for arbovirus control, which could help explain the lack of vectorial capacity of diverse mosquito genera in transmitting arboviruses. The negeviruses from the current study (named Biggie virus isolate MG and Coquillettidia velezuelensis negevirus) represent a promising avenue for further exploration, particularly in isolating viruses from sylvatic mosquitoes using mosquito cell lines for further characterization.
Our study shares several limitations common to RNA virome studies. First, host attribution of the identified viruses is because these viruses might replicate in other organisms, such as plants consumed by mosquitos. This limitation is exacerbated by using whole-body mosquitoes for pools, rather than dissected parts such as salivary glands. To better determine the hosts of the viruses identified in our study, complementary approaches are necessary, such as virus culturing in mosquito cell lines and small RNA sequencing. The latter can provide sequences derived from the RNAi machinery of the cell, indicating active replication. Second, although none of the virus genomes we identified possess genes similar to host genes, we cannot exclude the possibility that these viruses might be endogenous viral elements (EVEs) [9]. Third, associating different segments into a single genome for multipartite viral genomes is very challenging without isolated viruses. We were able to associate the phasmavirus segments into a single genome because they were the only segments found for these viral taxa in their pool. However, this is only sometimes the case for different samples. Lastly, we used sequence-similarity approaches to identify RdRps in contigs. While RdRps are hallmarks enzymes of RNA viruses, this strategy cannot identify highly divergent RdRp sequences, leaving them in the realm of viral dark matter. Small RNA sequencing may aid in properly characterizing viral dark matter by associating sequence cleavage patterns with otherwise unrelated contigs to form complete viral genomes.
5. Conclusions
This study expands our understanding of the RNA virome of sylvatic mosquitoes from the Brazilian Cerrado biome, uncovering eleven near-complete viral genomes in seven distinct viral families and one genus of an unassigned family, including Ferox mosquito mononega-like virus* and Albipes mosquito Gordis-like virus* (Xinmoviridae), Sabethes albiprivus phasmavirus* (Phasmaviridae), Pedras lispivirus isolate MG (Lispiviridae), Sabethes albiprivus iflavivirus* (Iflaviridae), Buriti virga-like virus isolate MG and Sabethes albiprivus virga virus 1* (Virgaviridae), Psorophora ferox flavivirus* (Flaviviridae), Alphamesonivirus cavallyense isolate MG (Mesoniviridae), and Biggie virus isolate MG and Coquillettidia velezuelensis negevirus* (Negevirus), with seven being new and four previously known genomes identified in new host species. The discovery of two xinmovirids in Ps. ferox and Ps. albipes presents new avenues for future studies aimed at understanding the interactions between ISVs and other arboviruses in the context of pathogen transmission. Additionally, HTS once again demonstrates its remarkable capacity to discover new viruses, even in pools of mosquitoes from different species, thereby enhancing our understanding of comprehensive virome surveillance in regions undergoing rapid environmental changes, as such the Brazilian Cerrado biome. Moreover, the presence of ISVs and potential novel arboviruses underscores the need for ongoing surveillance and control strategies to mitigate the risk of emerging infectious diseases. Further research is needed to elucidate the ecological roles of these viruses, their potential impacts on vector competence, and their implications for human and animal health in the context of emerging infectious diseases. Continuing to explore the viromic landscape of sylvatic mosquitoes can better prepare us for and mitigate the threats posed by future viral outbreaks.
Author Contributions
Conceived and designed the experiments: L.J.M. and A.B.S.; Performed the experiments: L.J.M, A.B.S., C.H.O., and F.V.S.A; Collected and identified mosquitoes: C.H.O and F.V.S.A. Analyzed the data: L.J.M., A.B.S., and F.S.C.; Contributed to the writing of the manuscript: L.J.M., A.B.S., C.H.O., F.S.C., L.A.S., F.V.S.A., and B.M.R.; Reviewed the manuscript: L.J.M., A.B.S., C.H.O., F.S.C., L.A.S., F.V.S.A, and B.M.R.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported with grants from the National Council for Scientific and Technological Development (CNPq award number: 304223/2021-2 to BMR) and Fundação de Apoio à Pesquisa do Distrito Federal, FAPDF (award numbers: 193.00001749/2022-31 and 193-00002148/2023-27 to BMR). UnB, UFT and UFRGS helped to pay publishing charges for Open Access articles.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper. The viruses' genome sequences have been deposited in GenBank under the accession numbers PP946236 to PP946246. HTS raw data has been deposited in the SRA platform under BioProject accession number PRJNA1126168, with BioSample accession numbers SAMN41926027 to SAMN41926031 and SRA (SRR) accession numbers SRR29474085 to SRR29474089.
Acknowledgments
We thank the entire team at Insect Behavior Laboratory, Instituto Federal do Norte de Minas Gerais, Salinas for collecting the mosquitoes. L.J. was granted a post-doctoral scholarship from CAPES. C.H.O was granted a PhD scholarship from Fapemig. B.M.R and F.S.C. are CNPq research fellows. F.V.S.A is grateful to Fapemig and the Centro Colaborador de Entomologia (SES-MG) for their funding. We would like to thank Ronaldo Medeiros dos Santos for creating the map of the state of Minas Gerais with the main biomes and municipalities collected.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Harvey, E.; Holmes, E.C. Diversity and evolution of the animal virome. Nat Rev Microbiol. 2022, 20, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Atoni, E.; Zhao, L.; Karungu, S.; Obanda, V.; Agwanda, B.; Xia, H.; Yuan, Z. The discovery and global distribution of novel mosquito-associated viruses in the last decade (2007-2017). Rev Med Virol. 2019, 29, e2079. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.P.; Schinkel, M.; van der Most, T.; Miesen, P.; van Rij, R.P. Composition and global distribution of the mosquito virome - A comprehensive database of insect-specific viruses. One Health 2023, 16, 100490. [Google Scholar] [CrossRef]
- Harbach, R.E. Mosquito Taxonomic Inventory. Available online: https://mosquito-taxonomic-inventory.myspecies.info/ (accessed on 18 January 2024).
- de Oliveira, C.H.; Andrade, M.S.; Campos, F.S.; da, C. Cardoso, J.; Gonçalves-Dos-Santos, M.E.; Oliveira, R.S.; Aquino-Teixeira, S.M.; Campos, A.A.; Almeida, M.A.; Simonini-Teixeira, D.; da P. Sevá, A.; Temponi, A.O.D.; Magalhães, F.M.; da Silva Menezes, A.S.; Lo-pes, B.T.; Almeida, H.P.; Pedroso, A.L.; Gonçalves, G.P.; Chaves, D.C.C.; de Menezes, G.G.; Bernal-Valle, S.; Müller, N.F.; Janssen, L.; Dos Santos, E.; Mares-Guia, M.A.; Albuquerque, G.R.; Romano, A.P.; Franco, A.C.; Ribeiro, B.M.; Roehe, P.M.; Lourenço-de-Oliveira, R.; de Abreu, F.V.S. Yellow Fever Virus Maintained by Sabethes Mosquitoes during the Dry Season in Cerrado, a Semiarid Region of Brazil, in 2021. Viruses 2023, 15, 757. [Google Scholar] [CrossRef]
- Sherwood, J.A.; Stehman, S.V.; Howard, J.J.; Oliver, J. Cases of Eastern equine encephalitis in humans associated with Aedes canadensis, Coquillettidia perturbans and Culiseta melanura mosquitoes with the virus in New York State from 1971 to 2012 by analysis of aggregated published data. Epidemiol Infect. 2020, 148, e72. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.R.; Spence, L.; Downs, W.G.; Aitken, T.H. Oropouche virus: a new human disease agent from Trinidad, West Indies. Am J Trop Med Hyg. 1961, 10, 574–578. [Google Scholar] [CrossRef]
- Romero-Alvarez, D.; Escobar, L.E. Oropouche fever, an emergent disease from the Americas. Microbes Infect. 2018, 20, 135–146. [Google Scholar] [CrossRef]
- de Almeida, J.P.; Aguiar, E.R.; Armache, J.N.; Olmo, R.P.; Marques, J.T. The virome of vector mosquitoes. Curr Opin Virol. 2021, 49, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Olmo, R.P.; Todjro, Y.M.H.; Aguiar, E.R.G.R.; de Almeida, J.P.P.; Ferreira, F.V.; Armache, J.N., et al. Mosquito vector competence for dengue is modulated by insect-specific viruses. Nat Microbiol. 2023, 8(1), 135–49.
- Maia, L.J.; Oliveira, C.H.; Silva, A.B.; Souza, P.A.A.; Müller, N.F.D.; Cardoso, J.D.C.; Ribeiro, B.M.; Abreu, F.V.S.; Campos, F.S. Arbovirus surveillance in mosquitoes: Historical methods, emerging technologies, and challenges ahead. Exp Biol Med. 2023, 248, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-throughput sequencing (HTS) for the analysis of viral populations. Infect Genet Evol. 2020, 80, 104208. [Google Scholar] [CrossRef]
- Silva, A.F.; Machado, L.C.; Silva, L.M.I.; Dezordi, F.Z.; Wallau, G.L. RNA virome of sylvatic mosquitoes from northeast Brazil reveals a divergent and diverse insect-specific viral community. bioRxiv 2023, 2023.06. 27.546706.
- Moutailler, S.; Yousfi, L.; Mousson, L.; Devillers, E.; Vazeille, M.; Vega-Rúa, A.; Perrin, Y.; Jourdain, F.; Chandre, F.; Cannet, A.; Chantilly, S.; Restrepo, J.; Guidez, A.; Dusfour, I.; Vieira Santos de Abreu, F.; Pereira Dos Santos, T.; Jiolle, D.; Visser, T.M.; Koenraadt, C.J.M.; Wongsokarijo, M.; Diallo, M.; Diallo, D.; Gaye, A.; Boyer, S.; Duong, V.; Piorkowski, G.; Paupy, C.; Lourenco de Oliveira, R.; de Lamballerie, X.; Failloux, A.B. A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas. Viruses 2019, 11, 904. [Google Scholar] [CrossRef]
- Gómez, M.; Martínez, D.; Páez-Triana, L.; Luna, N.; Ramírez, A.; Medina, J.; Cruz-Saavedra, L.; Hernández, C.; Castañeda, S.; Bohórquez Melo, R.; Suarez, L.A.; Palma-Cuero, M.; Murcia, L.M.; González Páez, L.; Estrada Bustos, L.; Medina, M.A.; Ariza Campo, K.; Padilla, H.D.; Zamora Flórez, A.; De Las Salas, J.L.; Muñoz, M.; Ramírez, J.D. Influence of dengue virus serotypes on the abundance of Aedes aegypti insect-specific viruses (ISVs). J Virol. 2024, 98, e0150723. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.M.C.; Bates, J.M. Biogeographic Patterns and Conservation in the South American Cerrado: A Tropical Savanna Hotspot: The Cerrado, which includes both forest and savanna habitats, is the second largest South American biome, and among the most threatened on the continent. BioScience 2002, 52, 225–234. [Google Scholar] [CrossRef]
- Plowright, R.K.; Reaser, J.K.; Locke, H.; Woodley, S.J.; Patz, J.A.; Becker, D.J.; Oppler, G.; Hudson, P.J.; Tabor, G.M. Land use-induced spillover: a call to action to safeguard environmental, animal, and human health. Lancet Planet Health 2021, 5, e237–e245. [Google Scholar] [CrossRef] [PubMed]
- Brazil. Guia para o Planejamento das Acoes de Captura de Anofelinos pela Tecnica de Atracao por Humano Protegido (TAHP) e Acompanhamento dos Riscos a Saude do Profissional Capturador. Ministerio da Saude, Secretaria de Vigilancia em Saude, Departamento de Imunizacao e Doencas Transmissiveis. Brasilia Ministerio da Saude, Brasil, 27 p. 2019. Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/svsa/malaria/vigilancia-entomologica-e-controle-vetorial-da-malaria/guia-para-o-planejamento-das-acoes-de-captura-de-anofelinos-pela-tecnica-de-atracao-por-humano-protegido-tahp-e-acompanhamento-dos-riscos-a-saude-do-profissional-capturador.pdf. (accessed on 17 May 2024).
- Shannon, R.C. Methods for collecting and feeding mosquitoes in jungle yellow fever studies. Am. J. Trop. Med. Hyg. 1939, 19, 131–140. [Google Scholar] [CrossRef]
- Abreu, F.V.S.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Santos, A.A.C.D.; Miranda, R.M.; Bonelly, I.S.; Neves, M.S.A.S.; Bersot, M.I.; Santos, T.P.D.; Gomes, M.Q.; et al. Haemagogus leucocelaenus and Haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in Brazil, 2016–2018. Emerg. Microbes Infect. 2019, 8, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Consoli, R.A.G.B.; Oliveira, R.L. Principais mosquitos de importância sanitária no Brasil [online]. Rio de Janeiro: Editora Fiocruz, 1994. 228 p. ISBN 85-85676-03-5. Available online: https://static.scielo.org/scielobooks/th/pdf/consoli-9788575412909.pdf (accessed on 17 May 2024).
- Forattini, O.P. Culicidologia Médica: Identificação, Biologia, Epidemiologia. Editora da Universidade de São Paulo: São Paulo, Brasil, 2002; Volume 2, p. 1924.
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (accessed on 17 May 2024).
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Boeckler, B.S. FelixKrueger/TrimGalore: v0.6.7 - 2021. [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Meleshko, D.; Hajirasouliha, I.; Korobeynikov, A. coronaSPAdes: from biosynthetic gene clusters to RNA viral assemblies. Bioinformatics 2021, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Charon, J.; Buchmann, J.P.; Sadiq, S.; Holmes, E.C. RdRp-scan: A bioinformatic resource to identify and annotate divergent RNA viruses in metagenomic sequence data. Virus Evol. 2022, 8, veac082. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US); 1988. Available online: https://www.ncbi.nlm.nih.gov/. (accessed on 17 May 2024).
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–3. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol Biol Evol. 2020, 37(1), 291–294. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.; Paraskevopoulou, S. ICTV Virus Taxonomy Profile: Xinmoviridae 2023. J Gen Virol. 2023, 104. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.B.; Matthijnssens, J. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Dudas, G.; Haas-Stapleton, E.; Kistler, A.L.; Li, L.M.; Logan, P.; Ratnasiri, K.; Retallack, H. Single mosquito metatranscriptomics identifies vectors, emerging pathogens and reservoirs in one assay. Elife 2021, 10, e68353. [Google Scholar] [CrossRef] [PubMed]
- Aragão, C.F.; da Silva, S.P.; do Nascimento, B.L.S.; da Silva, F.S.; Nunes Neto, J.P.; Pinheiro, V.C.S.; Cruz, A.C.R. Shotgun Metagenomic Sequencing Reveals Virome Composition of Mosquitoes from a Transition Ecosystem of North-Northeast Brazil. Genes 2023, 14, 1443. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Wang, F.; Ye, G.; Paraskevopoulou, S. ICTV Virus Taxonomy Profile: Lispiviridae 2023. J Gen Virol. 2023, 104. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, G.O.; Morais, V.S.; Monteiro, F.J.C.; Ribeiro, E.S.D.; Rego, M.O.D.S.; Souto, R.N.P.; Villanova, F.; Tahmasebi, R.; Hefford, P.M.; Deng, X.; Delwart, E.; Cerdeira Sabino, E.; Fernandes, L.N.; da Costa, A.C.; Leal, É. Aedes aegypti from Amazon Basin Harbor High Diversity of Novel Viral Species. Viruses 2020, 12, 866. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; Ictv Report Consortium. ICTV Virus Taxonomy Profile: Iflaviridae. J Gen Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Orba, Y.; Matsuno, K.; Nakao, R.; Kryukov, K.; Saito, Y.; Kawamori, F.; Loza Vega, A.; Watanabe, T.; Maemura, T.; Sasaki, M.; Hall, W.W.; Hall, R.A.; Pereira, J.A.; Nakagawa, S.; Sawa, H. Diverse mosquito-specific flaviviruses in the Bolivian Amazon basin. J Gen Virol. 2021, 102. [Google Scholar] [CrossRef]
- Hang, J.; Klein, T.A.; Kim, H.C.; Yang, Y.; Jima, D.D.; Richardson, J.H.; Jarman, R.G. Genome Sequences of Five Arboviruses in Field-Captured Mosquitoes in a Unique Rural Environment of South Korea. Genome Announc. 2016, 4, e01644-15. [Google Scholar] [CrossRef]
- Alencar, J.; Pacheco, J.B.; Correa, F.F.; Silva, J. dos S.; Guimarães, A.É. New report on the bionomics of Coquillettidia venezuelensis in temporary breeding sites (Diptera: Culicidae). Rev Soc Bras Med Trop. 2011, 44, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Farajollahi, A.; Fonseca, D.M.; Kramer, L.D.; Marm Kilpatrick, A. "Bird biting" mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology. Infect Genet Evol. 2011, 11, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 thousand Culex mosquitoes from throughout California. Journal of Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, K.; Marklewitz, M.; Zirkel, F.; Overheul, G.J.; Page, R.A.; Loaiza, J.R.; Drosten, C.; van Rij, R.P.; Junglen, S. Agua Salud alphavirus defines a novel lineage of insect-specific alphaviruses discovered in the New World. J Gen Virol. 2020, 101, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Villinger, J.; Muthoni, J.N.; Dobel-Ober, L.; Hughes, G.L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr Opin Insect Sci. 2020, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.; Asgari, S. Aedes Anphevirus: an Insect-Specific Virus Distributed Worldwide in Aedes aegypti Mosquitoes That Has Complex Interplays with Wolbachia and Dengue Virus Infection in Cells. Journal of Virology 2018, 92, e00224-18. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Zdobnov, E.M. A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway. Viruses 2020, 12, 1264. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. Journal of Virology 2017, 91, e00680-17. [Google Scholar] [CrossRef] [PubMed]
- Scarpassa, V.M.; Debat, H.J.; Alencar, R.B.; Saraiva, J.F.; Calvo, E.; Arcà, B.; Ribeiro, J.M.C. An insight into the sialotranscriptome and virome of Amazonian anophelines. BMC Genomics 2019, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.S.; Rocco, I.M.; Bergo, E.S.; Brasil, R.A.; Siciliano, M.M.; Suzuki, A.; Silveira, V.R.; Bisordi, I.; Souza, R.P. Yellow Fever Working Group. Reemergence of yellow fever: detection of transmission in the State of São Paulo, Brazil, 2008. Revista da Sociedade Brasileira de Medicina Tropical 2008, 44, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Lopes, O.S.; Sacchetta, L.A.; Francy, D.B.; Jakob, W.L.; Calisher, C.H. Emergence of a new arbovirus disease in Brazil III. Isolation of Rocio virus from Psorophora ferox (Humboldt, 1819). American Journal of Epidemiology 1981, 113, 122–125. [Google Scholar]
- Kulasekera, V.L.; Kramer, L.; Nasci, R.S.; Mostashari, F.; Cherry, B.; Trock, S.C.; Glaser, C.; Miller, J.R. West Nile virus infection in mosquitoes, birds, horses, and humans, Staten Island, New York, 2000. Emerging infectious diseases 2017, 4, 722–725. [Google Scholar] [CrossRef]
- Cupp, E.W.; Zhang, D.; Yue, X.; Cupp, M.S.; Guyer, C.; Sprenger, T.R.; Unnasch, T.R. Identification of reptilian and amphibian blood meals from mosquitoes in an eastern equine encephalomyelitis virus focus in central Alabama. The American Journal of Tropical Medicine and Hygiene, 2004, 71, 272–276. [Google Scholar] [CrossRef]
- Turell, M.; O'Guinn, M.; Jones, J.; Sardelis, M.; Dohm, D.; Watts, D.; Fernandez, R.; Travassos Da Rosa, A.; Guzman, H.; Tesh, R.; Rossi, C.; Ludwig, G.; Mangiafico, J.; Kondig, J.; Wasieloski, L.; Pecor, J.; Zyzak, M.; Schoeler, G. Isolation of viruses from mosquitoes (Diptera: Culicidae) collected in the Amazon Basin region of Peru. Journal of Medical Entomology 2005, 42, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.F. Febre amarela [Yellow Fever]. Revista da Sociedade Brasileira de Medicina Tropical 2003, 36, 275–293. [Google Scholar] [CrossRef]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; Junglen, S.; Misof, B.; Drosten, C. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS pathogens 2019, 15, e1008224. [Google Scholar] [CrossRef] [PubMed]
- ICTV, 2024. Phasmaviridae family. Available on: https://ictv.global/report/chapter/phasmaviridae/phasmaviridae. (accessed on 11 june 2024).
- Viljakainen, L.; Holmberg, I.; Abril, S.; Jurvansuu, J. Viruses of invasive Argentine ants from the European Main supercolony: characterization, interactions and evolution. The Journal of General Virology, 2018, 99, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Lay, C.L.; Shi, M.; Buček, A.; Bourguignon, T.; Lo, N.; Holmes, E.C. Unmapped RNA Virus Diversity in Termites and their Symbionts. Viruses 2020, 12, 1145. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Ye, Z.X.; Wang, X.; Yan, X.T.; Zhang, Y.; He, Y.J.; Qi, Y.H.; Zhang, X.D.; Zhuo, J.C.; Lu, G.; Lu, J.B.; Mao, Q.Z.; Sun, Z.T.; Yan, F.; Chen, J.P.; Zhang, C.X.; Li, J.M. Diversity and infectivity of the RNA virome among different cryptic species of an agriculturally important insect vector: whitefly Bemisia tabaci. NPJ Biofilms and Microbiomes 2021, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yuan, B.; Xiao, S.; Zhang, J.; Jia, W.; Fang, Q.; Wang, F.; Song, Q.; Ye, G. Diverse RNA Viruses Discovered in Three Parasitoid Wasps of the Rice Weevil Sitophilus oryzae. mSphere 2021, 6, e00331-21. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.X.; Wang, S.M.; Lu, G.; Chen, J.P.; Zhang, C.X.; Xu, L.Y.; Li, J.M. Complete genome sequence of a novel arlivirus from a yellow spotted stink bug (Erthesina fullo (Thunberg, 1783). Archives of Virology 2022, 167, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Hervé, J.P.; Dégallier, N.; Travassos-da-Rosa, A.P.A.; Pinheiro, F.P.; Sá Filho, G.C. Arboviroses: aspectos ecológicos. In: Instituto Evandro Chagas: 50 anos. Belém: Instituto Evandro Chagas 1986, 408-437.
- Goenaga, S.; Fabbri, C.; Dueñas, J.C.; Gardenal, C.N.; Rossi, G.C.; Calderon, G.; Morales, M.A.; Garcia, J.B.; Enria, D.A.; Levis, S. Isolation of yellow fever virus from mosquitoes in Misiones province, Argentina. Vector Borne and Zoonotic Diseases 2012, 12, 986–993. [Google Scholar] [CrossRef]
- van Oers, M.M. Genomics and Biology of Iflaviruses. In K. Johnson, & S. Agari (Eds.), Caister Academic Press. Insect Virology 2010, 231-250.
- Bowen-Walker, P.L.; Martin, S.J.; Gunn, A. The transmission of deformed wing virus between honeybees (Apis melliferaL.) by the ectoparasitic mite Varroa jacobsoni Oud. J. Invertebr. Pathol 1999, 73, 101–106. [Google Scholar] [CrossRef]
- Posada-Florez, F.; Childers, A.K.; Heerman, M.C.; Egekwu, N.I.; Cook, S.C.; Chen, Y.; Evans, J.D.; Ryabov, E.V. Deformed wing virus type A, a major honey bee pathogen, is vectored by the mite Varroa destructor in a non-propagative manner. Sci Rep 2019, 9, 12445. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Dovrolis, N.; Kouvela, A.; Kassela, K.; Rosa Freitas, M.G.; Nearchou, A.; de Courcy Williams, M.; Veletza, S.; Karakasiliotis, I. Defining virus-carrier networks that shape the composition of the mosquito core virome of a local ecosystem. Virus Evol. 2022, 8, veac036. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cui, F.; Liu, X.; Fu, Y.; Fang, W.; Kang, X.; Lu, H.; Li, S.; Liu, B.; Guo, W.; Xia, Q.; Kang, L.; Jiang, F. Association of virome dynamics with mosquito species and environmental factors. Microbiome 2023, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Bonning, B.C. The Insect Virome: Opportunities and Challenges. Curr Issues Mol Biol. 2020, 34, 1–12. [Google Scholar] [CrossRef]
- Pauvolid-Corrêa, A.; Solberg, O.; Couto-Lima, D.; Kenney, J.; Serra-Freire, N.; Brault, A.; Nogueira, R.; Langevin, S.; Komar, N. Nhumirim virus, a novel flavivirus isolated from mosquitoes from the Pantanal, Brazil. Arch Virol. 2015, 160, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Stollar, V.; Thomas, V.L. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology 1975, 64, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Isawa, H.; Tsuda, Y.; Yano, K.; Sasaki, T.; Yuda, M.; Takasaki, T.; Kobayashi, M.; Sawabe, K. Genetic characterization of a new insect flavivirus isolated from Culex pipiens mosquito in Japan. Virology 2007, 359, 405–414. [Google Scholar] [CrossRef]
- Carvalho, V.L.; Long, M.T. Insect-Specific Viruses: An overview and their relationship to arboviruses of concern to humans and animals. Virology 2021, 557, 34–43. [Google Scholar] [CrossRef]
- Ye, G.; Wang, Y.; Liu, X.; Dong, Q.; Cai, Q.; Yuan, Z.; Xia, H. Transmission competence of a new mesonivirus, Yichang virus, in mosquitoes and its interference with representative flaviviruses. PLoS Negl Trop Dis. 2020, 14, e0008920. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; González, A.V.; Haddow, A.D.; Watts, D.M.; da Rosa, A.P.; Weaver, S.C.; Lipkin, W.I.; Tesh, R.B. Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.H.; Xu, L.Y.; Zhai, J.; Ye, Z.X.; Lu, G.; Chen, J.P.; Zhang, C.X.; Li, J.M. Complete genome sequence of a novel nege-like virus in aphids (genus Indomegoura). Virol J. 2021, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Quito-Avila, D.F.; Reyes-Proaño, E.; Armijos-Capa, G.; Alcalá Briseño, R.I.; Alvarez, R.; Flores, F.F. Analysis of a new negevirus-like sequence from Bemisia tabaci unveils a potential new taxon linking nelorpi- and centiviruses. PLoS One 2024, 19, e0303838. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Contreras-Gutierrez, M.A.; Guzman, H.; Martins, L.C.; Barbirato, M.F.; Savit, C.; Balta, V.; Uribe, S.; Vivero, R.; Suaza, J.D.; Oliveira, H.; Nunes Neto, J.P.; Carvalho, V.L.; da Silva, S.P.; Cardoso, J.F.; de Oliveira, R.S.; da Silva Lemos, P.; Wood, T.G.; Widen, S.G.; Vasconcelos, P.F.C.; Fish, D.; Vasilakis, N.; Tesh, R.B. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 2017, 504, 152–167. [Google Scholar] [CrossRef]
- Patterson, E.I.; Kautz, T.F.; Contreras-Gutierrez, M.A.; Guzman, H.; Tesh, R.B.; Hughes, G.L.; Forrester, N.L. Negeviruses Reduce Replication of Alphaviruses during Coinfection. J Virol. 2021, 95, e0043321. [Google Scholar] [CrossRef]
- Carvalho, V.L.; Prakoso, D.; Schwarz, E.R.; Logan, T.D.; Nunes, B.T.D.; Beachboard, S.E.; Long, M.T. Negevirus Piura Suppresses Zika Virus Replication in Mosquito Cells. Viruses 2024, 16, 350. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Map of Brazil and Minas Gerais state depicting the municipalities from which mosquito samples were collected and associated biomes.
Figure 1.
Map of Brazil and Minas Gerais state depicting the municipalities from which mosquito samples were collected and associated biomes.
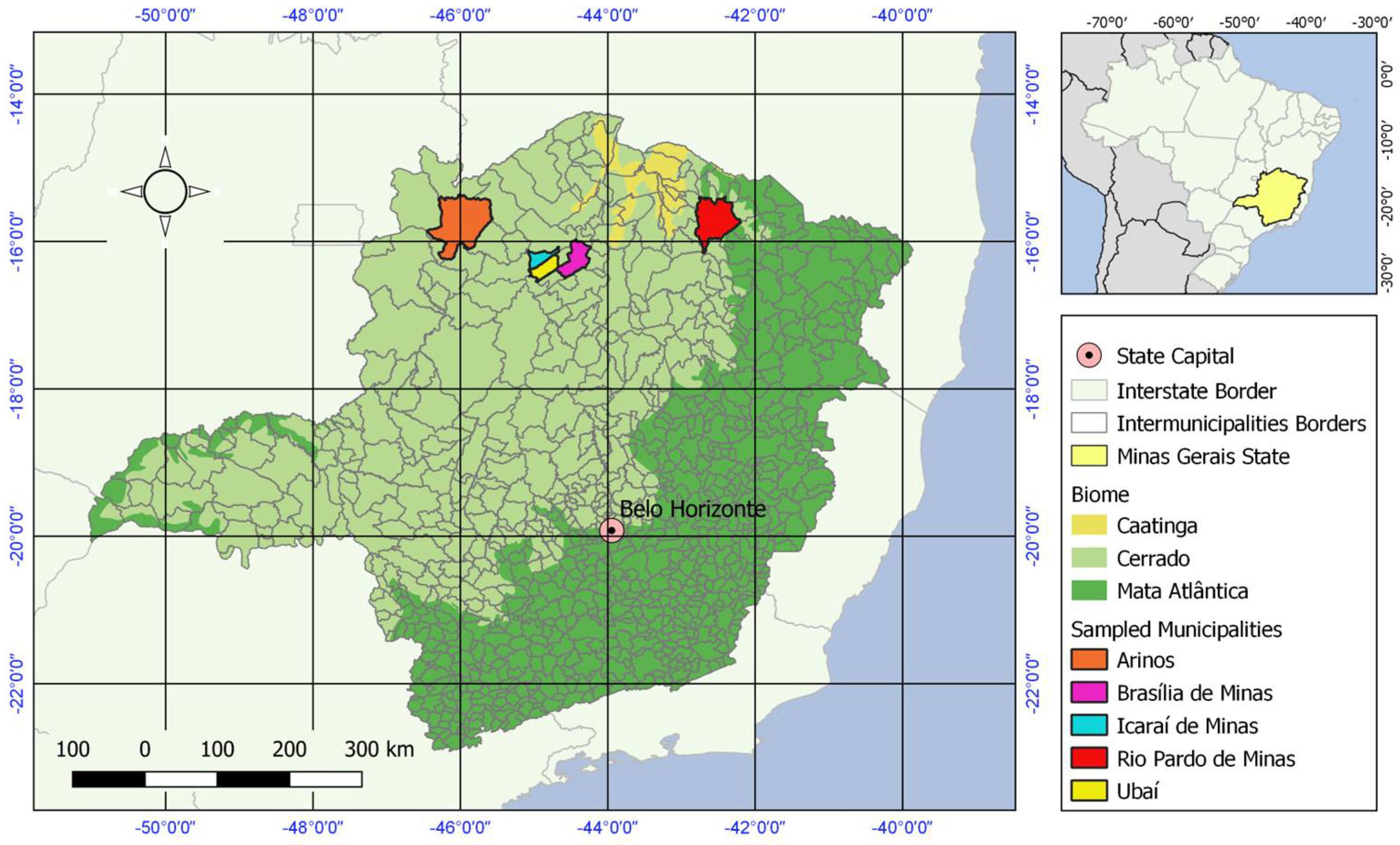
Figure 2.
Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus genomes discovered in Ps. ferox (A) and Ps. albipes (B), respectively. C. Phylogenetic analysis of the Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus discovered in Ps. ferox and Ps. albipes hosts, respectively. ML tree was constructed using the RdRp aminoacid sequences from the best hits obtained via BLASTx for each xinmovirus identified in this study against the NCBI nr-database. The tree includes currently recognized species in the family by ICTV, with Aranavirus as an outgroup.
Figure 2.
Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus genomes discovered in Ps. ferox (A) and Ps. albipes (B), respectively. C. Phylogenetic analysis of the Ferox mosquito mononega-like virus and Albipes mosquito Gordis-like virus discovered in Ps. ferox and Ps. albipes hosts, respectively. ML tree was constructed using the RdRp aminoacid sequences from the best hits obtained via BLASTx for each xinmovirus identified in this study against the NCBI nr-database. The tree includes currently recognized species in the family by ICTV, with Aranavirus as an outgroup.
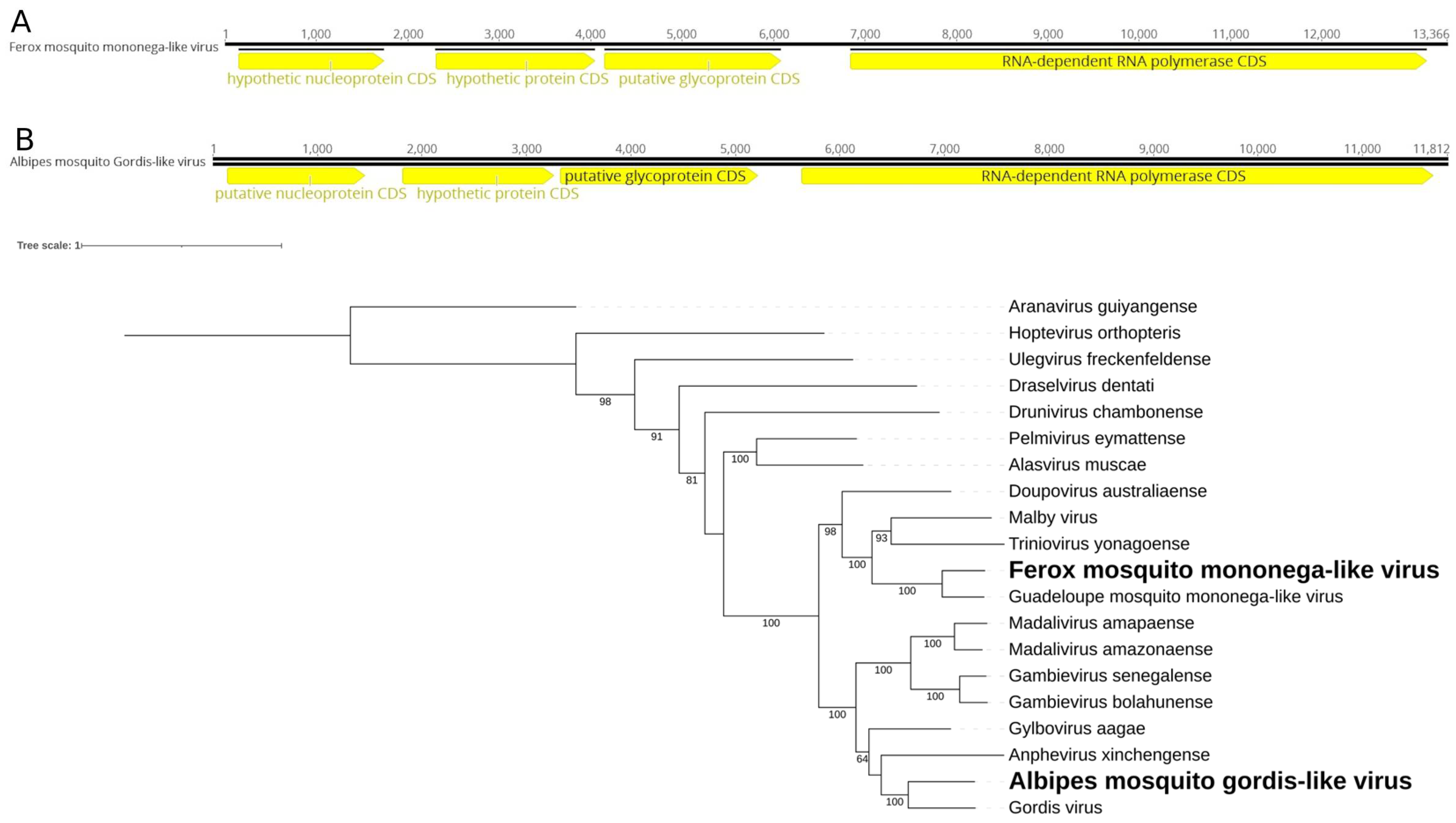
Figure 3.
A. Sabethes albiprivus phasmavirus genome exhibits L, M, and S segments identified within Sa. albiprivus. B. Phylogenetic analysis of the Sabethes albiprivus phasmavirus discovered in Sa. albiprivus host. ML tree was constructed using the RdRp aminoacid sequences of the best hits from BLASTx against the NCBI nr-database for the virus in the current study, with Sin nombre virus as an outgroup.
Figure 3.
A. Sabethes albiprivus phasmavirus genome exhibits L, M, and S segments identified within Sa. albiprivus. B. Phylogenetic analysis of the Sabethes albiprivus phasmavirus discovered in Sa. albiprivus host. ML tree was constructed using the RdRp aminoacid sequences of the best hits from BLASTx against the NCBI nr-database for the virus in the current study, with Sin nombre virus as an outgroup.
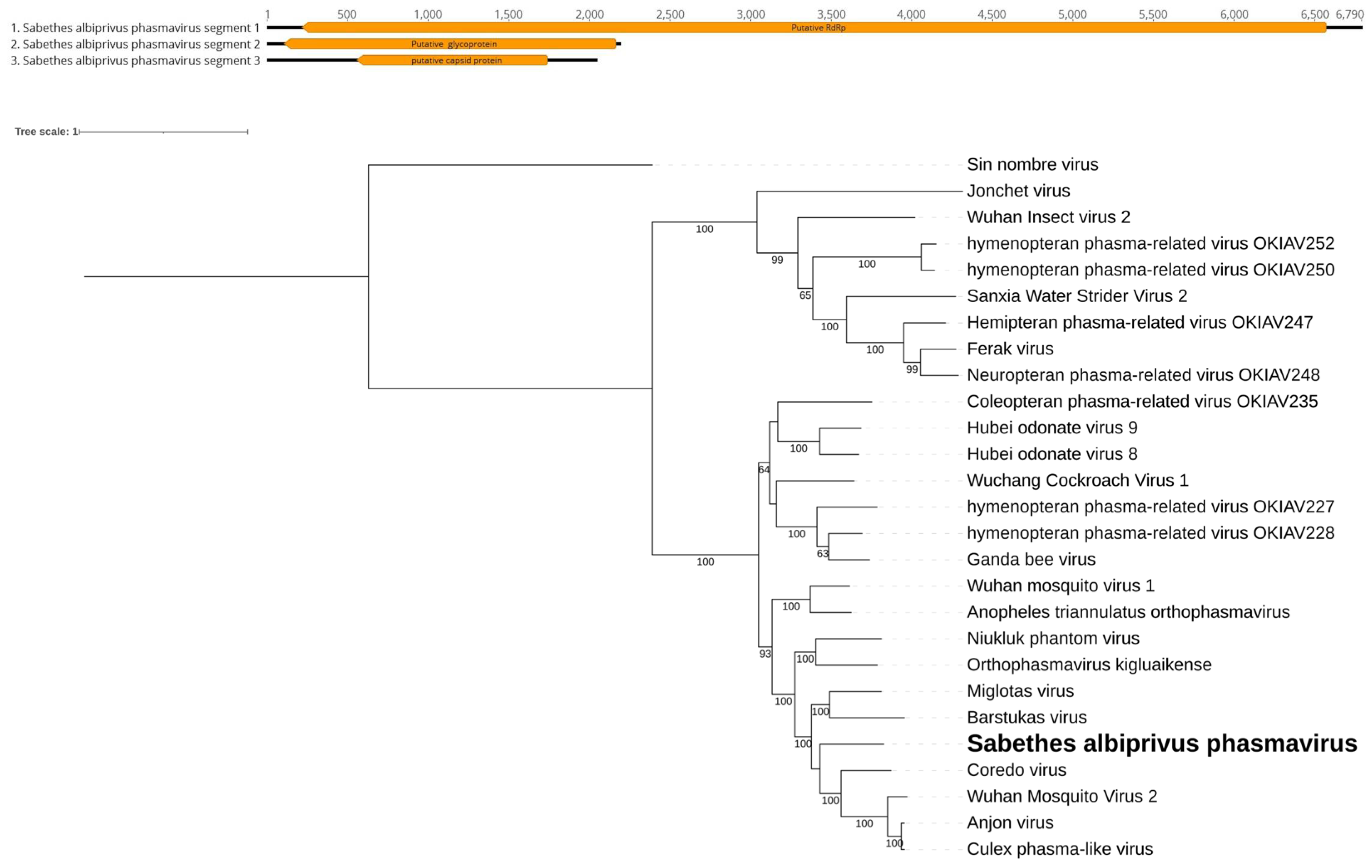
Figure 4.
A. Pedras lispivirus isolate MG genome identified within Sa. albiprivus. B. Phylogenetic analysis of the Pedras lispivirus isolate MG discovered in the Sa. albiprivus host. ML tree was constructed with the RdRp aminoacid sequences of the best hits found in the present study against the NCBI nr-database, currently recognized species in the family by ICTV, and Gambievirus as an outgroup.
Figure 4.
A. Pedras lispivirus isolate MG genome identified within Sa. albiprivus. B. Phylogenetic analysis of the Pedras lispivirus isolate MG discovered in the Sa. albiprivus host. ML tree was constructed with the RdRp aminoacid sequences of the best hits found in the present study against the NCBI nr-database, currently recognized species in the family by ICTV, and Gambievirus as an outgroup.

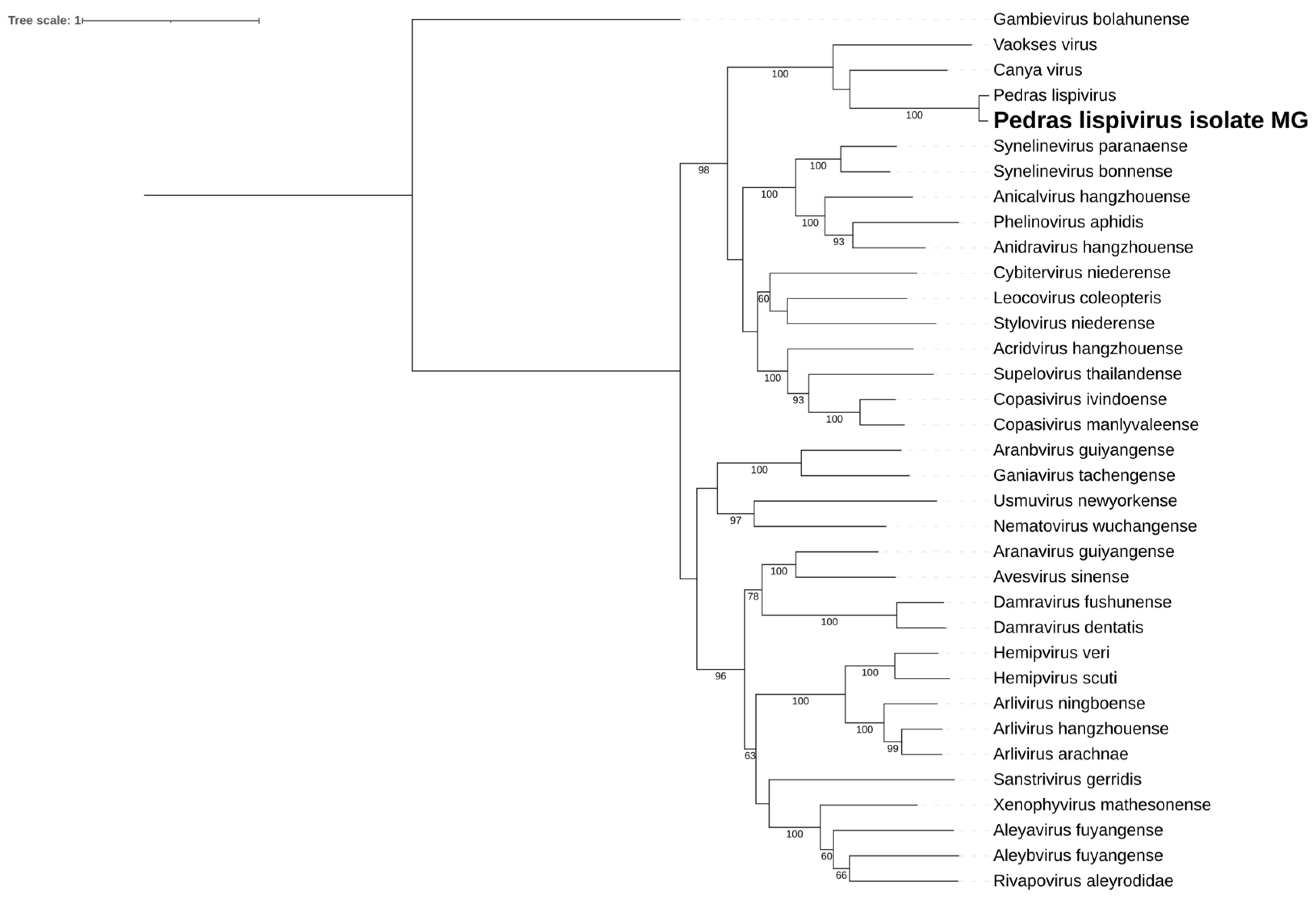
Figure 5.
A. Sabethes albiprivus iflavivirus genome identified within Sa. albiprivus. B. Phylogenetic analysis of the Sabethes albiprivus iflavivirus discovered in the Sa. albiprivus host. ML tree was constructed using the polyprotein sequences of the best BLASTx hits found in the present study against the NCBI nr-database, currently recognized species in the family by ICTV, and Cripavirus as an outgroup.
Figure 5.
A. Sabethes albiprivus iflavivirus genome identified within Sa. albiprivus. B. Phylogenetic analysis of the Sabethes albiprivus iflavivirus discovered in the Sa. albiprivus host. ML tree was constructed using the polyprotein sequences of the best BLASTx hits found in the present study against the NCBI nr-database, currently recognized species in the family by ICTV, and Cripavirus as an outgroup.
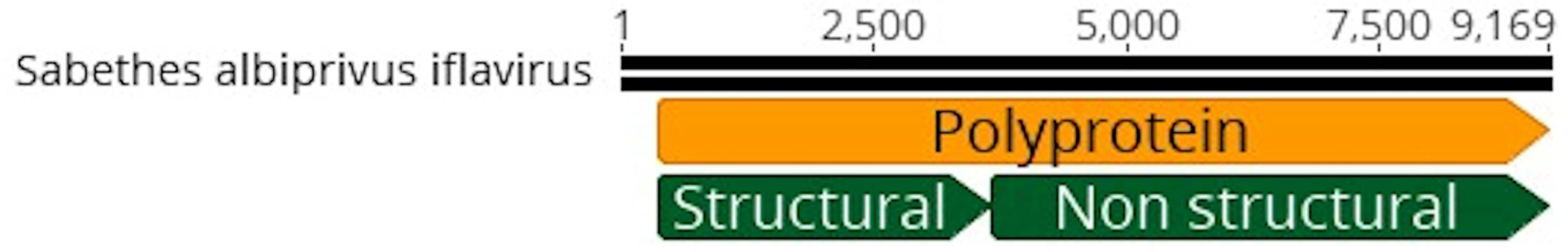
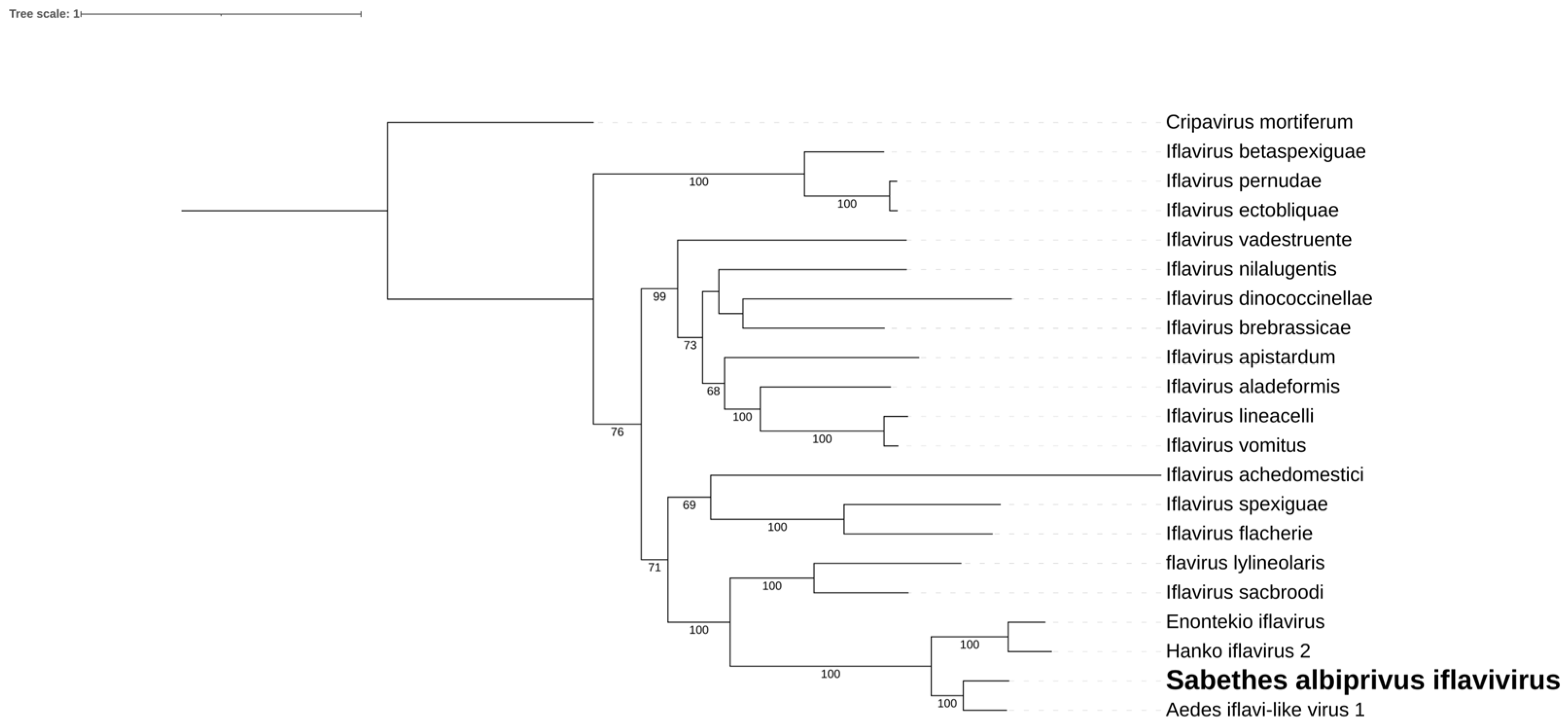
Figure 6.
A. Buriti virga-like virus isolate MG (A) and Sabethes albiprivus virgavirus 1 (B) genomes identified within Sa. albiprivus. B. Phylogenetic analysis of the Buriti virga-like virus isolate MG and Sabethes albiprivus virgavirus 1 discovered in Sa. albiprivus host. ML tree was constructed using the polyprotein aminoacid sequences of the best hits in BLASTx against the NCBI nr-database for the viruses in the current study, with Citrus leprosis virus as an outgroup.
Figure 6.
A. Buriti virga-like virus isolate MG (A) and Sabethes albiprivus virgavirus 1 (B) genomes identified within Sa. albiprivus. B. Phylogenetic analysis of the Buriti virga-like virus isolate MG and Sabethes albiprivus virgavirus 1 discovered in Sa. albiprivus host. ML tree was constructed using the polyprotein aminoacid sequences of the best hits in BLASTx against the NCBI nr-database for the viruses in the current study, with Citrus leprosis virus as an outgroup.

Figure 7.
A. Flavivirus genome identified within Ps. ferox. B. Phylogenetic analysis of the Psorophora ferox flavivirus discovered in the Ps. ferox host. ML tree was constructed using the polyprotein sequences of the best hits from BLASTx found in the present study against the NCBI nr-database.
Figure 7.
A. Flavivirus genome identified within Ps. ferox. B. Phylogenetic analysis of the Psorophora ferox flavivirus discovered in the Ps. ferox host. ML tree was constructed using the polyprotein sequences of the best hits from BLASTx found in the present study against the NCBI nr-database.
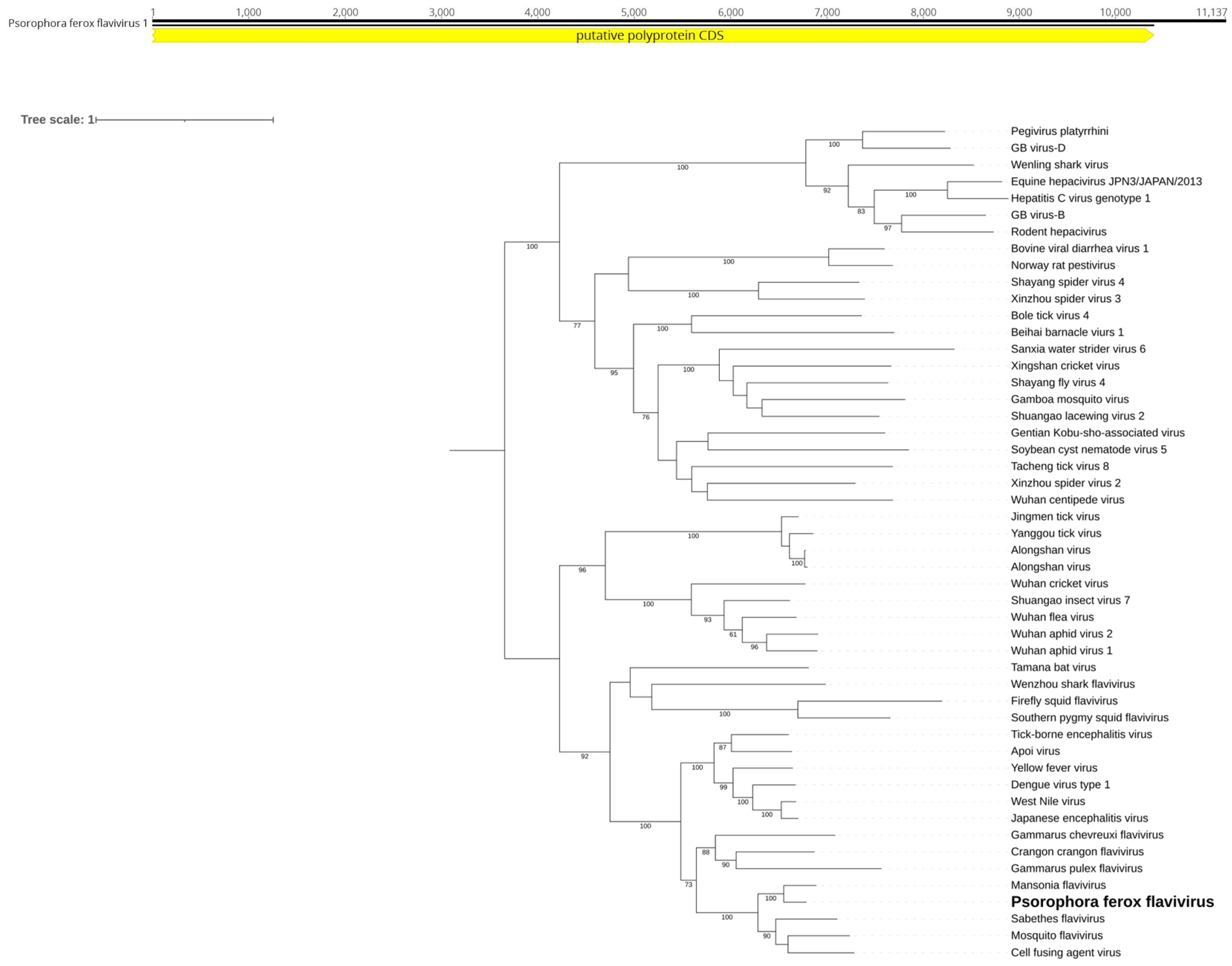
Figure 8.
A. Alphamesonivirus cavallyense isolate MG genome identifies within Cq. venezuelensis. B. Phylogenetic analysis of the Alphamesonivirus cavallyense isolate MG discovered in Cq. venezuelensis host. ML tree was constructed using the aminoacid sequences of the ORF1a replicase from representatives of the four currently recognized genera within the family, along with the two best BLASTx hits from the nr-database and Bolenivirus as an outgroup.
Figure 8.
A. Alphamesonivirus cavallyense isolate MG genome identifies within Cq. venezuelensis. B. Phylogenetic analysis of the Alphamesonivirus cavallyense isolate MG discovered in Cq. venezuelensis host. ML tree was constructed using the aminoacid sequences of the ORF1a replicase from representatives of the four currently recognized genera within the family, along with the two best BLASTx hits from the nr-database and Bolenivirus as an outgroup.
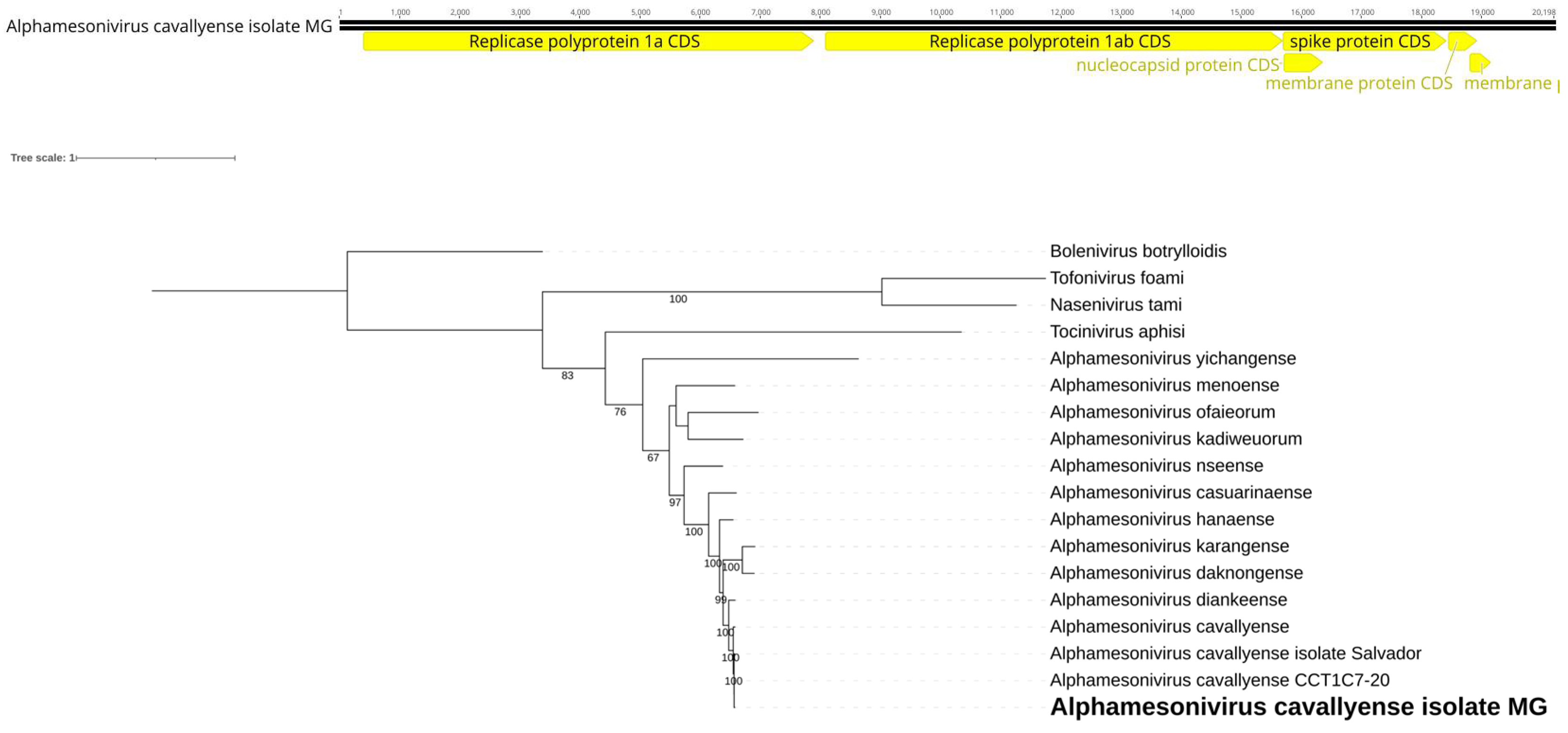
Figure 9.
Negevirus genomes identified within Cq. velezuelensis. A) Coquillettidia velezuelensis negevirus. B) Biggie virus isolate MG. C. Phylogenetic analysis of the negeviruses discovered in Cq. velezuelensis host. ML tree was constructed using the non-structural polyprotein aminoacid sequences of the best hits from BLASTx against the NCBI nr-database for the virus in the current study, with Citrus leprosis virus as an outgroup.
Figure 9.
Negevirus genomes identified within Cq. velezuelensis. A) Coquillettidia velezuelensis negevirus. B) Biggie virus isolate MG. C. Phylogenetic analysis of the negeviruses discovered in Cq. velezuelensis host. ML tree was constructed using the non-structural polyprotein aminoacid sequences of the best hits from BLASTx against the NCBI nr-database for the virus in the current study, with Citrus leprosis virus as an outgroup.

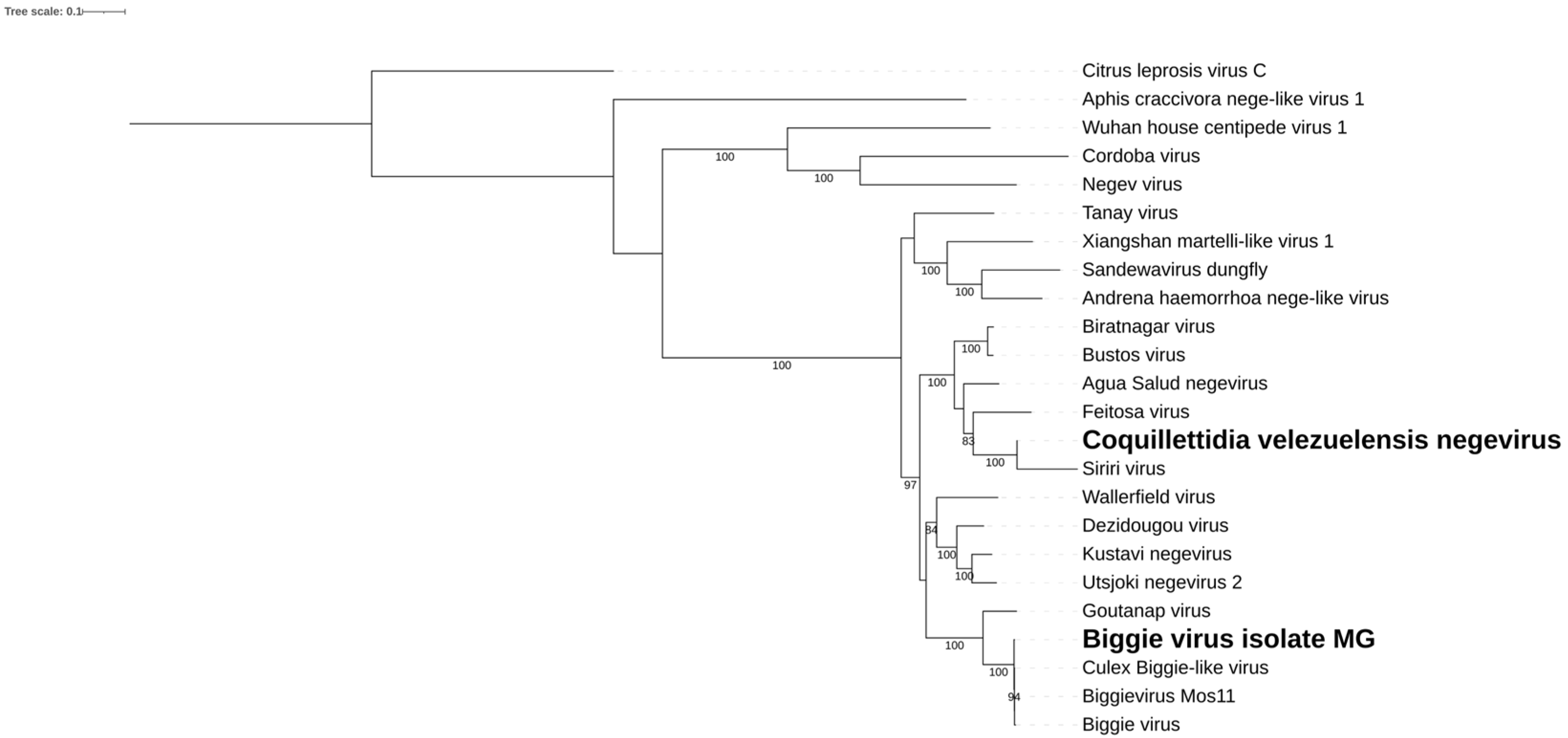

Table 1.
Description of Species, Number of Mosquitoes, and Municipality in Minas Gerais state where mosquito samples were collected.
Table 1.
Description of Species, Number of Mosquitoes, and Municipality in Minas Gerais state where mosquito samples were collected.
| Species per pool | Mosquitos per pool | Municipality |
|---|---|---|
| Psorophora (Janthinosoma) albipes (Theobald, 1907)1 | 20 | Brasília de Minas |
| Sabethes (Sabethes) albiprivus (Theobald, 1903)2 | 200 | Icaraí de Minas and Ubaí |
| Mixed pool (Sa. albiprivus, Sa. (Sabethoides) chloropterus (von Humboldt, 1819)3, and Ps. ferox) | 55 | Icaraí de Minas, Arinos, Brasília de Minas |
| Psorophora (Janthinosoma) ferox (von Humboldt, 1819)3 | 100 | Rio Pardo de Minas |
| Coquillettidia (Rhynchotaenia) venezuelensis (Theobald, 1912)4 | 100 | Arinos |
1 Theobald, F.V. 1907. A monograph of the Culicidae or mosquitoes. Volume 4. British Museum (Natural History), London. xix+ 639 pp. (1907.02.23). 2. Theobald, F.V. 1903. A monograph of the Culicidae or mosquitoes. Volume 3. British Museum (Natural History), London. xvii + 359 pp. (1903.07.25). 3. von Humboldt, F.H.A. (1819). Voyage aux regions equinoxiales du Nouveau Continent, fait en 1799, 1800, 1801, 1802, 1803 et 1804 par Al. de Humboldt et A. Bonpland; Vol. 2, 722 pp. In Humboldt, F.H.A., Ed., Voyage de MM Alexandre de Humboldt et Aime Bonpland. Pt. I: Relation historique. Paris. 4. Note sur le Culicides. Bulletin du museum national d'histoire naturelle 18 (12): 59-61. 29 Février 1912.
Table 2.
Viral diversity in insect pools included read numbers, contigs, viral families, genome type, and viruses found in this study including new viruses.
Table 2.
Viral diversity in insect pools included read numbers, contigs, viral families, genome type, and viruses found in this study including new viruses.
| Insect Pool | Read number (Millions) | Contig amount | Viral contigs | Viral families | Genome Type | Viruses found in this study (* new viruses1) |
|---|---|---|---|---|---|---|
| Ps. albipes | 20.5 | 3,882 | 1 | Xinmoviridae | ssRNA- | Albipes mosquito Gordis-like virus* |
| Sa. albiprivus | 25.1 | 13,548 | 4 | Phasmaviridae | ssRNA-2 | Sabethes albiprivus phasmavirus* |
| Lispividae | ssRNA- | Pedras lispivirus isolate MG | ||||
| Iflaviridae | ssRNA+ | Sabethes albiprivus iflavivirus* | ||||
| Virgaviridae | ssRNA+ | Sabethes albiprivus virgavirus 1* | ||||
| Mixed (Sa. albiprivus, Sa. chloropterus, and Ps.ferox) | 26.2 | 37,238 | 1 | Virgaviridae | ssRNA+ | Buriti virga-like virus isolate MG |
| Ps. ferox | 30.4 | 2,859 | 2 | Xinmoviridae | ssRNA- | Ferox mosquito mononega-like virus* |
| Flaviviridae | ssRNA+ | Psorophora ferox flavivirus* | ||||
| Cq. venezuelensis | 25.6 | 2,014 | 3 | Mesoniviridae | ssRNA+ | Alphamesonivirus cavallyense isolate MG |
| Negevirus | ssRNA+ | Biggie virus isolate MG | ||||
| Coquillettidia velezuensis negevirus* |
1 New viruses are indicated with asterisks. 2. Segmented.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



