
Submitted:
28 June 2024
Posted:
29 June 2024
You are already at the latest version
Abstract
Keywords: spinal muscular atrophy (SMA); survival of motor neuron 1 (SMN1); SMN2; SMN protein; antisense oligonucleotide (ASO); nusinersen; gene therapy; onasemnogene; risdiplam; small molecule, combination therapy
Keywords:
spinal muscular atrophy (SMA)
; survival of motor neuron 1 (SMN1)
; SMN2
; SMN protein
; antisense oligonucleotide (ASO)
; nusinersen
; gene therapy
; onasemnogene
; risdiplam
; small molecule
; combination therapy
1. Introduction
Spinal Muscular Atrophy (SMA) is one of the most prevalent autosomal recessive genetic disorders, affecting approximately 1 in 10,000 births[1]. SMA is mainly characterized by the degeneration of alpha motor neurons located in the anterior horn of the spinal cord and motor nuclei of the lower brainstem resulting in muscle wasting, weakness, hypotonia and difficulties with feeding and respiration[2,3,4,5].
The underlying cause of classic spinal muscular atrophy (SMA) is typically the homozygous deletion or, less commonly, smaller mutations within the SMN1 gene located on chromosome 5. Additionally, due to intrachromosomal duplication, humans have another paralogous gene SMN2[6]. However, SMN2 cannot compensate for the loss of SMN1 due to a single-nucleotide polymorphism in exon 7 (C—to—T transition) which creates an intronic splicer silencer N1 (ISS-N1). Splicing modulators can bind to ISS-N1, leading to the exclusion of exon 7 in approximately 90% of SMN2 transcripts. This results in the production of a truncated and unstable protein, SMNΔ7, which is rapidly degraded[7]. The genetic alterations in SMN1 result in decreased expression of the survival motor neuron protein (SMN). SMN protein is expressed ubiquitously in almost every cell, both in the nucleus and cytoplasm, and it plays a crucial role in various cellular mechanisms. These include the assembly of the spliceosomal machinery, endocytosis, protein translation, and maintenance of cellular homeostasis. Due to its diverse functions and widespread expression, the loss of SMN can result in systemic pathology extending beyond the motor neuron[8,9].
Several therapeutic strategies, such as nusinersen, onasemnogene abeparvovec, and risdiplam, have gained regulatory approval by the FDA and EMA for the treatment of SMA. These approaches aim to enhance SMN production either by modifying SMN2 splicing or by replacing the defective SMN1 gene [10]. Additionally, various other strategies have been explored, including elevating SMN transcript levels, stabilizing SMN protein, implementing neuroprotective measures, employing muscle activators, and more. While these therapies don't directly address the primary deficiency of SMN, they could be used in conjunction with treatments that boost SMN production to offer additional benefits to patients[11].
This review aims to comprehensively discuss the molecular characteristics, pathophysiology, and currently approved treatments for SMA. Furthermore, we will explore alternative approaches focused on increasing SMN levels.
1.1. Genetic Background of SMA & Diverse Role of SMN
Spinal muscular atrophy (SMA) is caused by deletions or mutations in the survival of motor neuron gene (SMN1), which was originally cloned and characterized by Melki and colleagues[2]. SMN1, also referred to as SMNT (with T standing for telomere), spans 20 kb and is located in the telomeric region of a 500 kb inverted duplication on chromosome 5q13. This genomic locus is characterized by a high number of repetitions, resulting in instability and frequent deletions in most SMA patients. Unlike other species, such as mice, which have only one copy of the SMN gene, humans have a variable number of centromeric SMN2 genes[12]. Notably, SMN2 appears to be unique to humans, as chimpanzees have multiple SMN1 copies but no SMN2. SMN2, also referred to as SMNC (with C indicating centromere), shares over 99% nucleotide identity with SMN1. Each SMN gene comprises nine exons, including exons 1, 2a, 2b, 3, 4, 5, 6, 7, and 8 (which encode the 3′ untranslated region (UTR))) responsible for encoding the survival of motor neuron (SMN) protein. However, SMN1 and SMN2 differ by 8 nucleotides, 5 of which are intronic and 3 of which occur in the last 3 exons[13]. Among these differences, a single functional, coding variant, c.840C>T in exon 7 of SMN2, disrupts an exonic splicing enhancer and simultaneously creates an exonic splicing silencer[14,15] (Figure 1). Normally, SMN1 produces full-length mRNA, which translates into functional SMN protein (294 amino acids, 38 kDa). The SMN1 gene is essential in diverse organisms, in which null mutations are lethal during early development[16]. In contrast to SMN1, alternative splicing due to the presence of an exonic splicing silencer results in the exclusion of exon 7 in SMN2. This leads to a shortened mRNA that encodes a truncated and unstable SMN protein (SMNΔ7; 282 amino acids, 30.5 kDa), which is rapidly degraded. However, a minority of SMN2 pre-mRNA transcripts retain exon 7 after splicing, leading to the production of approximately 10% of functional protein from SMN2, thereby making it a disease-modifying gene[17] (Figure 1).
The SMN protein, translated from the SMN gene, is ubiquitously expressed in all cells and tissues, with particularly high levels in the nervous system, especially the spinal cord[18]. During gestational and neonatal stages, SMN protein expression is high beyond the neuromuscular system and declines with age, but motor neurons of the spinal cord maintain high SMN levels throughout life[19,20]. SMN forms a complex with Gemins2-8 in the cytoplasm and in nuclear bodies called gems, similar to Cajal bodies[21,22]. In addition to gemins, other proteins also interact with SMN. These include Sm, Sm-like proteins, RNA helicase A, fibrillarin, GAR1, ribonucleoproteins (RNPs)- heterogenous nuclear RNP U (hnRNP U), hnRNP Q, hnRNP R, and p80-coilin-the marker for Cajal bodies[23,24]. The primary role of SMN is in the biogenesis and maintenance of spliceosomal small nuclear ribonucleoprotein (snRNP) assembly, crucial for pre-mRNA splicing. Additionally, SMN is involved in transcriptional regulation, telomerase regeneration, autophagy, cellular trafficking and homeostasis, signal transduction, DNA repair, and recombination[24].
Structurally, the 294-amino-acid-long SMN protein comprises multiple domains: the N-terminal Gemin2 and nucleic acids binding domain, the central Tudor domain, and the C-terminal proline- and YG box-rich domain. The Tudor domain interacts with coilin protein, a marker of Cajal bodies. The domain also binds to the C-terminal rich arginine- and glycine-rich tails of Sm core proteins to facilitate spliceosome assembly. The YG box is a tyrosine/glycine-rich region in the C-terminus of SMN protein, enabling SMN oligomerization through a glycine zipper structure. Mutations in these domains are linked to SMA, highlighting the importance of SMN's structural integrity for its function[25].
Reduced expression of SMN protein in SMA leads to impaired α-motor neuron development and degeneration, manifesting as skeletal muscle weakness, the most evident clinical manifestation of SMA[26]. Early stages of SMA involve impaired motor neuron development, affecting the cell body, axon, and myofibers. This results in delayed synaptic input acquisition, immature firing patterns[27,28,29], reduced motor axon growth and myelination[30], and deficient NMJ synapse function[31,32,33]. As the disease progresses, terminal motor axons withdraw from NMJ postsynaptic terminals, proximal motor axons degenerate, and motor neuron cell bodies lose synaptic inputs and undergo cell death[24]. Prolonged denervation of myofibers leads to their replacement by fibro-adipose tissue[34]. Later stages of SMA involve slower neurodegeneration, primarily affecting distal motor components. Key molecular mediators of neurodegeneration in SMA include the p53 pathway, which is activated due to altered splicing of Mdm2 and Mdm4, as well as JNK signaling, ER stress, and DNA damage[24].
Ninety-five percent of affected individuals have homozygous deletions of both exons 7 and 8 or only exon 7 of SMN1, regardless of the disease phenotype. Most of the remaining 5% are typically compound heterozygotes with an SMN1 exon 7 deletion and an SMN1 point mutation[35]. In patients with less severe forms of SMA (types 2 and 3), gene conversion (where SMN1 exon 7 is replaced by SMN2 exon 7) often occurs instead of genuine deletions of SMN1, which are more common in SMA type 1[36]. The protein domain encoded by exon 7 (amino acids 280–294) is essential for SMN's function[7,37]. The SMNΔ7 protein, which lacks this exon, is unstable, rapidly degraded, and deficient in oligomerization[14,38] and binding to Sm core proteins[39], and gem formation[40]. SMNΔ7 has a significantly shorter half-life compared to full-length SMN due to degradation via the ubiquitin-proteasome system[41]. However, its stability can be enhanced by coexpressing full-length SMN, which recruits SMNΔ7 into oligomeric complexes[42]. Additionally, exon 7 includes a cytoplasmic targeting signal vital for transporting SMN into neuronal processes[43].
Additional intragenic mutations, including missense, nonsense, splice site mutations, insertions, deletions, and duplications, are particularly common in exons 3 and 6 [44,45]. These missense mutations are of significant interest as they may alter specific properties of the SMN protein without causing a complete loss of function. Most SMA mutations are clustered in the Tudor domain encoded by exon 3 (e.g., W92S, V94G, G95R, A111G, I116F, Y130C, E134K, and Q136E) and in exon 6, within or near the Y/G box (e.g., L260S, S262G, S262I, M263R, M263T, S266P, Y272C, H273R, T274I, G275S, G279C, and G279V). This mutation distribution indicates that both the Tudor domain and the Y/G box, along with its surrounding regions, are critical for SMN function[46].
1.2. Clinical Manifestation of SMA
Spinal muscular atrophy (SMA) was first described by Guido Werdnig in 1891 in a case involving muscle weakness in two infant brothers, followed by seven additional cases reported by Johan Hoffmann from 1893 to 1900[47,48]. SMA leads to the progressive loss of alpha motor neurons in the ventral spinal cord and motor nuclei of the lower brainstem. Clinically, SMA presents as hypotonia, muscle weakness, and atrophy, with severity varying by genotype. The muscle weakness is predominantly proximal, with greater involvement of the lower extremities, and is usually symmetric, accompanied by diffuse areflexia[1]. In severe cases, bulbar and respiratory muscle weakness can occur, although facial and ocular muscles are generally spared[49]. The various phenotypes of spinal muscular atrophy (SMA) were formalized into a classification scheme at a 1991 International Consortium on Spinal Muscular Atrophy, sponsored by the Muscular Dystrophy Association (MDA). This initial classification identified three SMA types based on the highest level of motor function achieved (e.g., sitting or standing) and age of onset. Later modifications further divided the type 3 category by introducing type 4 for adult-onset cases and type 0 for prenatal onset cases resulting in death within weeks[35,47] (Table 1).

| SMA type | Age of Onset | Life Expectancy | SMN2 Copy Number | Clinical Manifestation | Alternative Name | Estimated SMA Portion |
|---|---|---|---|---|---|---|
| 0 | Prenatal | < 1month | 1 | Require assisted respiration at birth; Fetus dysplay reduced mobement | - | Unclear, maybe < 1% |
| 1 | 0-6 months | <2 years | 2 | Unable to sit independently, respiratory and feeding support required | Werdnig–Hoffman disease | ~60% |
| 2 | <18 months | >2 years | 3, 4 | Ability to independently sit, inability to walk, respiratory (often non-invasive) and feeding support required | Dubowitz disease | ~27% |
| 3a | 18 months – 3 years | Adult | 3, 4 | Full ambulation, but slowly progressive muscle atrophy and weakness. | Kugelberg–Welander disease | ~12% |
| 3b | >3 years | Adult | 4 | |||
| 4 | >21 years | Adult | ≥4 | Usually preserved walking ability | Adult- onset SMA | ~1% |
2. FDA Approved Gene-targeting SMN Replacement Therapies
2.1. Nusinersen
Outline
Nusinersen (Spinraza®; Biogen, Cambridge, MA, USA) is an antisense oligonucleotide (ASO) that was the first treatment approved for spinal muscular atrophy (SMA). The FDA granted approval in December 2016, followed by EMA approval in May 2017[54]. Nusinersen works by promoting the inclusion of SMN2 exon 7 via ISS-N1 inhibition, thereby increasing the production of full-length SMN2 mRNA and subsequently full-length SMN protein. For effective delivery to motor neurons in the central nervous system, nusinersen must be administered intrathecally because ASOs do not cross the intact blood-brain barrier. The dosing regimen includes a loading phase of four intrathecal injections over a two-month period (on days 0, 15, 30, and 60) with a dose of 12 milligrams (4-5 milliliters, depending on age). After the loading phase, a maintenance dose is administered every four months to sustain therapeutic levels[55] (Table 2).
Discovery and Mechanism of action
The discovery of nusinersen is rooted in modifying the splicing pattern of the SMN2 gene to produce full-length, functional SMN protein. This process involves targeting splicing regulatory elements to enhance exon 7 inclusion, which is crucial to produce the functional protein. The exclusion of exon 7 from the SMN2 transcript is regulated by the intronic splicing silencer N1 (ISS-N1), located downstream of the 5′ splice site of exon 7. Deleting or masking the ISS-N1 region can promote the inclusion of exon 7 in most SMN2 transcripts[56]. The use of antisense oligonucleotides (ASOs) to manipulate such regulatory regions has emerged as an effective therapeutic approach. ASOs are single-stranded, DNA-like molecules designed to hybridize to mRNA splicing factors, thereby modifying gene expression[57]. Nusinersen is a 2′-O-methoxyethyl-modified ASO which works by binding to ISS-N1 and inhibiting the binding of other splicing factors, thereby promoting the inclusion of exon 7 into the mRNA. Studies suggested that ISS-N1 interacts with various splicing factors, such as hnRNP A1/A2, which contribute to its inhibitory effect[58]. However, another proposed mechanism is that ISS-N1-targeting ASO alters the secondary structure of pre-mRNA, facilitating the recruitment of U1 snRNP at the 5′ splice site of exon 7. U1 snRNP binds to the 5′ exon-intron junction of pre-mRNA, playing a crucial role in the early stages of splicing[59].
The development of nusinersen involved testing multiple ASOs targeting the ISS-N1 region in mouse models and SMA fibroblast, with the sequence shifting one base at a time to determine the most effective target sequence for exon 7 inclusion[58]. The ASO targeting intron 7 position +10-27 was identified as the strongest sequence. This 18-mer 2′-O-methoxyethyl-modified ASO significantly increased SMN protein levels and improved the SMA phenotype in mice, particularly with early treatment[60]. The successful delivery of ASOs to motor neurons in the central nervous system was a significant challenge due to the blood-brain barrier. Williams J et al. overcame this challenge by demonstrating that repeated intracerebroventricular (ICV) administration of a 2′-O-methyl (2′OMe) antisense oligonucleotide increased SMN levels in the central nervous system tissues of SMNΔ7 SMA mice (SMNΔ7+/+; SMN2 +/+; Smn−/−)[61]. Subsequent studies confirmed the efficacy of ICV injections in delivering MOE AONs targeting ISS-N1 to the brain and spinal cord. Preclinical studies in mouse models and non-human primates showed good ASO distribution throughout the spinal cord and dose-dependent effects on SMN expression following a single intrathecal infusion[62,63]. However, subsequent studies underscored the critical role of peripheral SMN restoration using MOE antisense oligonucleotides, which had the strongest effect on both survival and vascular-related clinical signs in a severe mouse model of SMA[64,65].
Clinical trials
The ENDEAR trial (NCT02193074) was a 13-month, international, randomized, multicenter, sham-controlled, phase III study designed to evaluate the clinical efficacy and safety of nusinersen in infants diagnosed with spinal muscular atrophy (SMA). These infants had two copies of the SMN2 gene and exhibited symptoms at or before six months of age. The trial enrolled 121 infants, with two-thirds (81 infants) receiving nusinersen and one-third (41 infants) receiving a sham treatment. The primary endpoints were the proportion of motor milestone responders, assessed by the Hammersmith Infant Neurological Examination Section 2 (HINE2), and event-free survival, evaluated by the time to death or the need for permanent ventilatory support. Secondary endpoints included subgroup analyses of overall survival and event-free survival rates based on the duration of the disease at screening. Results showed that a significantly higher percentage of infants in the nusinersen group achieved motor milestone responses compared to the control group (51% vs. 0%). Additionally, the likelihood of event-free survival was higher in the nusinersen group, though only 8% of treated infants achieved independent sitting. The trial also highlighted the importance of early intervention, showing that infants treated earlier (under three months of age) demonstrated quicker and more pronounced motor improvements. The incidence of adverse events was similar between the nusinersen group (96%) and the control group (98%)[66].
The CHERISH trial (NCT02292537) was a phase III study designed to assess the efficacy and safety of nusinersen in children with SMA type II and III who presented symptoms after six months of age. The trial enrolled 126 children, with two-thirds (84 children) receiving nusinersen and one-third (42 children) receiving a sham treatment. The primary endpoint of the study was the least-squares mean change from baseline in the Hammersmith Expanded Functional Motor Scale (HFMSE) score at 15 months post-treatment initiation. The secondary endpoint was the percentage of children who showed a clinically meaningful increase in the HFMSE score, defined as an improvement of at least 3 points, indicating better performance in at least two motor skills. Interim analysis revealed a significant improvement in motor function for the treated group, with the HFMSE score increasing by a mean of 4 points after 15 months of treatment. In contrast, the sham group showed a mean decrease of 1.9 points. Final analysis confirmed these findings, with 57% of children in the nusinersen group showing an increase of at least 3 points in the HFMSE score compared to 26% in the control group (P<0.001). The most frequent adverse events in the nusinersen group were related to the lumbar puncture procedure, such as headaches, back pain, and post-lumbar puncture syndrome. Following the interim analysis, both trials were terminated, and all patients were enrolled into the SHINE (NCT02594124) open-label extension study to continue evaluating long-term safety and efficacy[67].
The NURTURE (NCT02386553) study is a Phase 2, multisite, open-label, single-arm trial focusing on pre-symptomatic SMA patients likely to develop SMA Type I or II. The goal was to evaluate the long-term safety and efficacy of intrathecal nusinersen when initiated early, before the onset of clinical SMA signs. The trial enrolled 25 infants with genetically diagnosed SMA, including 15 with two copies of the SMN2 gene and 10 with three copies. The primary endpoint was the time to death or the need for respiratory intervention (either invasive or non-invasive for at least 6 hours per day for a minimum of 7 days, or tracheostomy). Secondary endpoints included survival rates, motor milestone achievement, and maintenance of weight. Motor milestones were assessed using the World Health Organization (WHO) criteria, Hammersmith Infant Neurological Examination Section 2 (HINE-2), and the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scale. Participants began nusinersen treatment in infancy while still pre-symptomatic. By the time the report was published, all 25 children were past the expected age of symptom onset for SMA Type I or II. At the time of analysis, all participants were alive, and none required permanent ventilation, though some needed temporary respiratory support during acute illnesses. Motor function improvements were significant: every participant achieved the ability to sit without support, 92% could walk with assistance, and 88% could walk independently. The results underscore the effectiveness of early treatment. The treatment was well-tolerated, with no new safety concerns, and no participants withdrew due to treatment-related adverse events[68].
Nusinersen has emerged as a notable success in treating various types of Spinal Muscular Atrophy (SMA). It has been effective in improving muscle strength, re-establishing motor neuron connections, and addressing some respiratory issues. Currently, over 11,000 pediatric and adult patients worldwide with all types of SMA are receiving this treatment[69]. However, patients with SMA types 1 and 2 often still require assisted ventilation (non-invasive ventilation, NIV) due to ongoing respiratory challenges[70]. Despite its effectiveness, nusinersen therapy is associated with several adverse effects. Notably, in the clinical studies, 16% of patients with normal or elevated baseline platelet levels developed thrombocytopenia, and 58% of treated patients exhibited elevated urine protein levels, indicating renal toxicity, including potentially fatal glomerulonephritis. These adverse effects have led the FDA to issue warnings and recommend baseline and pre-dose monitoring of platelet counts, coagulation tests, and urine protein levels[71]. These findings underscore the importance of monitoring and optimizing ASO distribution to enhance treatment outcomes.
2.2. Onasemnogene Abeparvovec
Outline
Onasemnogene Abeparvovec (ZOLGENSMA), initially known as AVXS-101, is a gene therapy aimed at treating SMA by delivering a functional SMN1 gene to produce the vital SMN protein. Developed by AveXis, the therapy became part of Novartis’ portfolio following their acquisition of AveXis in May 2018, subsequently rebranded as Novartis Gene Therapies[69]. Zolgensma received FDA approval in May 2019, followed by EMA approval in May 2020, for use in patients under two years of age with various types of SMA. This treatment is administered one-time intravenously at a dose of 1.1 × 10^14 vector genomes per kilogram of body weight, using a non-replicating, self-complementary adeno-associated virus (AAV) 9 vector to deliver the therapeutic gene[72] (Table 2).
Discovery and Mechanism of action
Onasemnogene abeparvovec is a gene therapy that uses an scAAV9 vector to cross the blood-brain barrier and deliver therapeutic genes[73]. Adeno-associated viruses (AAVs) are non-enveloped, single-stranded DNA viruses that require a helper virus to complete their life cycle and can transduce neurons effectively. Different AAV serotypes exhibit varying capsid properties, which influence their receptor affinity and tissue tropism[74]. The scAAV9 vectors are unique because they contain double-stranded, self-complementary DNA, which allows for faster protein synthesis after entering host cells[75]. In onasemnogene abeparvovec AAV9 vector contains human SMN1 cDNA under the control of a hybrid cytomegalovirus (CMV) enhancer/chicken-β-actin (CB) promoter. The hybrid CMV enhancer and CB promoter ensure continuous and sustained production of SMN protein[11]. Overcoming the challenge of gene transduction into the CNS posed by the blood-brain barrier, the AAV9 vector has shown superior CNS transduction in various animal models, including mice, cats, rats, and non-human primates[73,76].
Clinical trials
The phase I START trial (NCT02122952) evaluated the safety and efficacy of onasemnogene abeparvovec. Conducted between December 2014 and August 2017, the study enrolled 15 patients with homozygous SMN1 exon 7 deletion and 2 copies of the SMN2 gene (mean age: 3.4 months, range: 0.9–7.9 months). The patients were divided into two cohorts: the first cohort of three patients received a low dose of gene therapy (6.7 × 10^13 vector genomes per kilogram [vg/kg]) over the initial four months, while the second cohort of twelve patients received a high dose (2.0 × 10^14 vg/kg). The therapy was administered intravenously through a venous catheter inserted into a peripheral vein[77]. The primary outcome focused on determining safety by monitoring any treatment-related adverse events of grade 3 or higher. Secondary outcomes included measuring the time until death or the need for permanent ventilatory assistance (defined as at least 16 hours of respiratory support per day for at least 14 days, in the absence of any acute, reversible illness or perioperative state). Additionally, motor milestones and CHOP INTEND scores were assessed as part of the exploratory outcomes. At the 18-month first data cut-off, all 15 patients in the study had a 100% survival rate at 20 months of age, with only one patient from the low-dose cohort requiring permanent ventilation at 29 months. All patients showed an increase in CHOP INTEND scores, with a mean increase of 24.6 points in the high-dose cohort and 16.3 points in the low-dose cohort. In the high-dose cohort, 11 out of 12 patients achieved sitting unassisted for at least 5 seconds, full head control, and the ability to speak; nine of these patients could roll over, and two could crawl, stand, and walk independently. Seven patients in this cohort could feed orally without an enteral tube. The treatment also reduced hospitalizations per year (2.1 vs. 7.6), the median length of stay (6.7 vs. 13 days), and hospitalization duration (4.4% vs. 18.5%) compared to the natural history cohort studies, respectively[77,78]. A subgroup analysis of these 12 infants revealed that early dosing (<3 months) led to quicker achievement of motor milestones, highlighting the importance of early diagnosis and treatment[79]. During the clinical trial, one patient in the low-dose cohort experienced serious liver injury. This led to the administration of oral prednisolone (1 mg/kg/day) to the remaining 14 patients for 30 days, starting 24 hours before gene vector administration. In the high-dose cohort, one patient had significantly elevated AST and ALT levels, which were managed with additional prednisolone. Two other patients had asymptomatic AST and ALT elevations that resolved without intervention[80,81].
An interventional phase III, open-label trial (STR1VE-US, NCT03306277) was conducted from 2018 to 2020, involving 22 SMA type 1 patients under 6 months of age who received an intravenous dose of onasemnogene. Similar phase III trials, STR1VE-EU (NCT03461289) and STR1VE-AP (NCT03837184), aimed to assess the efficacy and safety of the treatment in SMA infants of similar age and SMN2 copy number. The STR1VE-US trial has been completed, demonstrating that 91% of the enrolled patients had event-free survival at 14 months of follow-up, and 59% were able to sit without support for at least 30 seconds. Additionally, 81.8% of patients were free of ventilation support at 18 months old, 95% achieved a CHOP-INTEND score of 40 or higher, and 64% attained a score of 50 or higher[69,80,81].
Another clinical trial investigated the intrathecal administration of onasemnogene for patients with milder SMA (three copies of SMN2). The STRONG trial (NCT03381729), a phase I, open-label study, evaluated different intrathecal doses of the gene therapy (6.7 × 10^13, 1.2 × 10^14, or 2.4 × 10^14 vg/kg) in 51 patients. However, A partial clinical hold was placed by the FDA due to preclinical safety concerns about dorsal root ganglia (DRG) inflammation and neuronal cell body degeneration or loss. This hold was lifted by the FDA in August 2021, and the study was completed in November 2021, demonstrating significant improvements in the HFMSE. These positive results have led to the commencement of the STEER trial (NCT05089656) in February 2022[69,81].
Finally, the Phase III SPR1NT trial (NCT03505099) evaluated the efficacy and safety of onasemnogene abeparvovec in presymptomatic infants with spinal muscular atrophy (SMA) type 1 who have two copies of the SMN2 gene. Conducted as a multicenter, single-arm study, the trial included 14 infants treated at six weeks or younger. Compared to a matched natural-history cohort, all infants treated with onasemnogene abeparvovec showed significant improvements. Notably, all 14 infants were able to sit independently for at least 30 seconds by 18 months, and 13 of them maintained a healthy body weight. None of the children required permanent ventilation or nutritional support, and no serious adverse events related to the treatment were reported. These results highlight the potential of onasemnogene abeparvovec to significantly alter the course of SMA when administered early, emphasizing the importance of universal newborn screening for SMA[82].
Onasemnogene abeparvovec has demonstrated significant benefits, including substantial improvements in motor milestone achievements and ventilator-free survival in SMA patients. These improvements offer a promising outlook for better quality of life. Additionally, despite its high initial cost, this one-time intravenous gene therapy may prove to be more cost-effective in the long run compared to the repeated intrathecal administrations required by treatments such as nusinersen. However, the therapy presents several challenges. The clinical data supporting its benefits is limited to about five years, restricting the understanding of its long-term efficacy and safety. The therapy carries common adverse effects, including elevated aminotransferases and vomiting, with a significant risk of acute liver injury. This necessitates a boxed warning and rigorous liver function monitoring before and frequently after the infusion for at least three months. Baseline assessments should include liver function tests and anti-AAV9 antibodies, with systemic corticosteroids (1 mg/kg/day oral prednisolone) administered starting one day before the infusion for 30 days. Additionally, monitoring platelet counts, and cardiac troponin-I levels is crucial due to transient decreases observed post-infusion. Animal studies have shown dose-dependent cardiac and hepatic toxicities, including mortality at higher doses. After the 30-day corticosteroid regimen, liver function must be reassessed. If normal, corticosteroids should be tapered off over 28 days; if abnormal, they should continue until normalization[83]. These challenges underscore the need for careful patient management and long-term monitoring to ensure the therapy's safety and efficacy.
2.3. Risdiplam
Outline
Risdiplam (EVRYSDI®), developed by Roche (RG7916, RO7034067), is the only approved oral medication for spinal muscular atrophy (SMA). It received FDA approval in August 2020 and EMA approval in March 2021 for patients two months and older with SMA types 1, 2, and 3, or with four copies of the SMN2 gene. In May 2022, the FDA expanded its approval to include patients of all ages, supported by promising results in treating pre-symptomatic individuals[72]. The approved dosage regimen for risdiplam is based on patient age and body weight: 0.2 mg/kg/day for patients aged 2 months to 2 years, 0.25 mg/kg/day for patients aged 2 years and older weighing less than 20 kg, and 0.5 mg/kg/day for those aged 2 years and older weighing more than 20 kg[84] (Table 2).
Discovery and Mechanism of action
The discovery of risdiplam was initiated through a high-throughput screening (HTS) designed to find small molecules that promote exon 7 inclusion in SMN2 pre-mRNA splicing. This process identified a promising coumarin derivative[85]. After multiple optimization efforts to address issues such as in vitro Ames flag and phototoxicity, this led to compound 2 (RG7800), a pyridopyrimidinone derivative[86]. However, the development of RG7800 was halted due to retinal toxicity observed in non-human primates[87]. Further improvements were then implemented to compound 2 to develop risdiplam, the compound 1 /molecule 3[87]. These included enhanced selectivity against off-target genes and increased on-target potency, allowing for reduced efficacious doses and a better therapeutic window. Additionally, the incorporation of a basic amine moiety with a low pKa value-maintained potency while preventing hERG inhibition or phospholipidosis. The resulting compound 1, risdiplam, exhibited an excellent pharmacokinetic profile with the optimal volume of distribution and half-life, along with favorable systemic tissue distribution[88].
Mechanistically, risdiplam functions as an SMN2 splice modulator by promoting the inclusion of exon 7 in SMN2 pre-mRNA splicing, leading to the production of functional SMN protein[89]. During splicing, the U1 small nuclear ribonucleoprotein (U1 snRNP) must bind to the 5′ splice site (5′ss) at the exon-intron border. While some deviations from the 5′ss consensus sequence are tolerated, certain substitutions can weaken U1 snRNP binding, causing exon skipping[90]. SMN2 exon 7 exclusion is an example of such splice-skipping. Risdiplam-like compounds stabilize the transient double-strand RNA structure formed between the 5′ss of SMN2 exon 7 and the U1 snRNP complex. This stabilization strengthens the weak 5′ splice site of SMN2 exon 7, compensating for the sequence mismatch and increasing U1 snRNP's binding affinity. This enhanced binding ensures proper splicing and the production of functional SMN protein [91].
In preclinical studies, risdiplam demonstrated significant efficacy in increasing SMN protein levels and improving motor function in animal models of spinal muscular atrophy (SMA). Adult C/C allele mice treated daily with risdiplam at doses of 1, 3, or 10 mg/kg for 10 days showed elevated SMN levels, increased motor neuron counts, and enhanced neuromuscular junction (NMJ) innervation. Similarly, SMNΔ7 mice received intraperitoneal injections of risdiplam from postnatal day 3 to 9, resulting in comparable improvements[87]. In non-human primates, oral administration of risdiplam at 3 mg/kg/day for 7 days achieved good biodistribution in relevant tissues[88]. Despite initial concerns about retinal toxicity observed in monkeys, subsequent studies in pigmented and albino rats showed no retinal changes[87]. These findings supported the continued development of risdiplam, which proved more efficacious than its predecessor, compound 2, in clinical trials.
Clinical trials
FIREFISH (NCT02913482), a phase 2/3 open-label trial in SMA type I patients for risdiplam, began in December 2016 and consisted of two parts. The main focus of part 1 of the trial was to establish the safety, tolerability, pharmacokinetics, and pharmacodynamics of varying doses of risdiplam. It enrolled 21 infants aged 1 to 7 months with type 1 SMA[92]. The dosing strategy started with daily oral administration at 0.04 mg/kg, 0.08 mg/kg, or 0.2 mg/kg, depending on the participant's age, and was adjusted to 0.2 mg/kg within a few months of starting the treatment This was ultimately adjusted to 0.25. mg/kg when the participant reached 3 years of age. After eight months of daily treatment, 93% of participants showed significant motor improvement, with an average CHOP-INTEND score increase of 16 points and several motor milestones achieved, such as rolling, kicking, sitting with or without support, and full head control, as measured by HINE-2[92]. A 16-month follow-up confirmed continued motor development, with 82% of the high-dose cohort reaching a CHOP-INTEND score of 40[92,93]. None of the 90.5% (19/21) survived infants required permanent ventilation or tracheostomies[94]. No drug-related adverse events led to withdrawal from the study after 19 months of treatment[95]. Part 2 of the trial was confirmatory, assessing the efficacy and safety of the selected dose from Part 1. After 12 months of treatment, 29% of infants could sit unsupported for at least 5 seconds, a milestone typically not achieved by untreated Type 1 SMA patients[95]. Overall, 78% responded positively according to the HINE-2 tool, with 76% gaining head control and 61% achieving some form of sitting. Additionally, for 90% treated infants a significant increase in CHOP-INTEND scores was observed, with 56% achieving scores of 40 or more. By the end of the 23 months, 95% maintained the ability to swallow, with 89% able to feed orally [96,97]. Part 2 demonstrated the clinical benefit of risdiplam with a 93% survival rate. Furthermore, during the 12-month treatment course, 49% of the infants did not require hospitalizations. The most common serious adverse events included respiratory issues like pneumonia and bronchiolitis, while non-life-threatening events included upper respiratory infections, pyrexia, and gastrointestinal issues such as constipation and diarrhea[97]. No retinal toxic effects as seen in cynomolgus monkeys were observed in this human study, and the treatment was generally well-tolerated[92].
Another study, SUNFISH (NCT02908685), is a phase III, randomized, double-blind, placebo-controlled study that began in October 2016. This study enrolled children and adults aged 2–25 years with a clinical diagnosis of SMA Type 2 or Type 3. Similar to the FIREFISH study, the aim of this study was to test the safety, tolerability, PK, PD, and the efficacy of risdiplam and consisted of two parts. In Part 1, 51 participants were enrolled to investigate an exploratory dose over 12 weeks. Adolescents and adults aged 12 to 25 years received either 3 mg or 5 mg of risdiplam for at least 12 weeks[84]. Once the appropriate dose for Part 2 was determined, participants transitioned to the open-label study in Part 2. Part 2 involved 180 participants and was a randomized, placebo-controlled, double-blind study assessing the safety and efficacy of the dose established in Part 1. The primary endpoint was the change in MFM32 scores after 12 months of treatment. Patients treated with risdiplam showed significantly greater improvements in MFM32 scores compared to those receiving placebo. Secondary endpoints, including the Revised Upper Limb Module (RULM) and Hammersmith Functional Motor Score-Expanded (HFMSE), also improved significantly. Patients aged 12 and older reported increased independence using the SMA Independence Scale (SMAIS). There were no major drug-related adverse events leading to withdrawal, with most events related to the underlying SMA. Common adverse events included upper respiratory infections, nasopharyngitis, pyrexia, and headache, while serious adverse events were primarily respiratory-related but not linked to risdiplam. Overall, risdiplam was well tolerated and demonstrated significant efficacy in improving motor function and independence in patients with Type 2 and Type 3 SMA[84,98].
JEWELFISH ((NCT03032172), a trial investigating the effects of risdiplam in patients previously treated with other SMA therapies, began in March 2017. The trial aimed to evaluate the pharmacokinetics, pharmacodynamics, safety, and efficacy of risdiplam in non-naïve SMA patients aged 6 months to 60 years who had a clinical diagnosis of SMA Type 1, 2 or 3. This multicenter, open-label study included participants who had previously received treatments such as RG7800, nusinersen, or onasemnogene abeparvovec (ZOLGENSMA)[99]. Among the 12 participants, nine were non-ambulatory, and three were ambulatory, with an average MFM32 score of 48.44 at the start of the study. Over 12 months of risdiplam treatment, there was a consistent increase in SMN protein levels, averaging more than twice the baseline levels, similar to those observed in the SUNFISH Part 1 trial[100].
Finally, RAINBOWFISH (NCT03779334), studying risdiplam in pre-symptomatic SMA infants 0–6 weeks of age who were genetically diagnosed with SMA, began in August 2019. The primary endpoint of this study is the ability to sit unsupported for five seconds, assessed after 12 months of treatment in 10 infants with at least two SMN2 copies and a compound muscle action potential (CMAP) of ≥ 1.5 mV. Additional endpoints include survival, need for permanent ventilation, ability to swallow independently, CHOP-INTEND motor function score, development of SMA symptoms, and SMN protein levels in the blood. The estimated completion date of the study is early January 2029[101].
Risdiplam has demonstrated substantial therapeutic benefits through systemic administration, as evidenced by preclinical rodent studies and clinical trials. It effectively crosses the blood-brain barrier without restricted permeability, thereby targeting the central nervous system as well as peripheral tissues, including various organs and muscles. However, administration of risdiplam is associated with some adverse effects. Commonly reported side effects include constipation, diarrhea, rash, fever, pneumonia, and vomiting. Less frequently, patients may experience urinary tract infections, joint pain, and ulcers[102]. Additionally, risdiplam may have harmful effects on the fetus when administered to pregnant women and may also compromise male fertility. Consequently, the use of risdiplam in pregnant individuals is currently advised against. Although there is a lack of comprehensive data from pregnant women undergoing risdiplam treatment, animal studies have shown risks such as embryofetal mortality, congenital malformations, and reduced fetal weight. Therefore, the prescribing information for risdiplam recommends that females undergo pregnancy testing prior to the initiation of treatment [102]. To enhance the quality of risdiplam treatment, further research is needed to better understand and mitigate these adverse effects. More studies should also focus on the drug's safety profile during pregnancy and compare its efficacy and safety against other available SMA therapies.
Table 2.
Summary of FDA approved therapies for Spinal Muscular Atrophy (SMA)[2,35,47,50,51,103,104].
| Drug/company | Type | Mechanism of action | Status | Route of administration | Protocol | Targeted population | Cost |
|---|---|---|---|---|---|---|---|
| Nusinersen (Spinraza)/Biogen | Antisense oligonucleotide | splicing modifier of SMN2 (MOE chemistry targeting SMN2 ISS-N1 that promotes exon 7 inclusion) | FDA approval- December 2016; EMA approval- May 2017 | intrathecal | 3 loading doses at 14-day interval, 4th loading dose 30 days after the 3rd dose, and maintenance dose every 4 months thereafter | all ages and all types of SMA | up to $125,000 per dose; drug cost for the first year: $750,000 and then $375,000 annually |
| Onasemnogene abeparvovec-xioi (Zolgensma)/Novartis | Gene therapy | replacement of SMN1 gene (scAAV9-SMN under the control of a CBA promoter) | FDA approval- May 2019; EMA approval- conditional approval on May 2020 | intravenous | Single dose | FDA: SMA patient less than 2 years of age with bi-allelic mutations in the SMN1 gene. EMA: patients with 5q SMA with a bi-allelic mutation in the SMN1 gene and a clinical diagnosis of SMA type 1, or patients with 5q SMA with a bi-allelic mutation in the SMN1 gene and up to 3 copies of the SMN2 gene | $2,125,000 (single injection) |
| Risdiplam (Evrysdi)/Roche | Small molecule | splicing modifier of SMN2 | FDA approval- August 2020; EMA approval March 2021 | orally | Once daily | patients 2 months of age and older | up to $340,000 a year (cheaper in younger patient as dosing dependes on weight) |
3. Broader Therapeutic Strategies: SMN-Dependent and Independent Approaches
Research continues to develop drugs for SMA by targeting and increasing SMN expression via splice modulation, leading to the development of additional drugs targeting SMN2.
Branaplam: Branaplam (also known as LMI070), similar to risdiplam, is an orally available, small molecule developed by Novartis as a potential therapy for spinal muscular atrophy (SMA). It was identified through a high-throughput screen designed to find molecules that promote the inclusion of exon 7 in SMN2 pre-mRNA, stabilizing it with splicing factor complexes. In SMA mice, daily administration of Branaplam resulted in a dose-dependent increase in exon 7 inclusion and SMN protein expression, improving body weight and lifespan. The first human phase I/II trial (NCT02268552) for Branaplam began in 2015, targeting SMA patients under six months old with two copies of SMN2. This trial aimed to evaluate the safety, tolerability, and early effectiveness of Branaplam [11]. However, recruitment was halted in 2016 due to adverse events observed in animal studies while the trial was underway, which included damage to nerves, the spinal cord, testes, and kidney blood vessels. Enrollment resumed in late 2017 with modifications to the study design, including the allowance of both feeding tube and oral administration, and the addition of nerve tests as safety measures. Recruitment was completed in May 2019, encompassing 13 infants in part 1 and 25 infants in part 2. Later that year, the company announced positive progress, with some infants receiving therapy for more than four years, though full results were not disclosed, leaving the complete safety profile unknown (branaplam (LMI070) (smanewstoday.com)). Additionally, clinical deterioration was observed after reducing the subsequent target dose to one-tenth. Due to rapid advancements in SMA treatments, Novartis discontinued Branaplam's development for SMA in mid-2021[69]. However, Branaplam has shown potential in reducing huntingtin mRNA, the mutated protein in Huntington’s disease, earning FDA Orphan Drug Designation for Huntington's, with a phase IIb trial planned for 2021[11].
Histone deacetylase inhibitors (HDAC): Histone deacetylase (HDAC) inhibitors have been extensively investigated for their role in treating spinal muscular atrophy (SMA) due to their ability to activate SMN2 transcription by inhibiting deacetylation of chromatin histones, thereby promoting gene expression[105,106,107,108]. Notably, several HDAC inhibitors, including valproic acid, trichostatin A, and sodium phenylbutyrate (Figure 2), have demonstrated promising results in increasing SMN levels both in vitro and in vivo[109]. Valproic acid, a classic class I HDAC inhibitor, has shown beneficial effects in SMA mouse models and patient fibroblasts, leading to its rapid progression to clinical trials[110]. However, a systematic review of these trials indicated an improvement in motor function but little effect on survival[111]. Similarly, another class I HDAC inhibitor, phenylbutyrate, was shown to increase SMN protein levels and the number of Gems in SMA fibroblast culture[108], however a randomized, placebo-controlled trial showed no significant improvement in motor function, leading to the premature termination of its clinical trial (NCT00439569)[112]. Other small molecules with HDAC inhibitor properties, such as suberoylanilide hydroxamic acid (SAHA/ Vorinostat) [113], trichostatin A [114], and resveratrol[115], also demonstrated success in laboratory models of SMA but have not advanced to clinical trials. While HDAC inhibitors alone do not provide therapeutic benefits equivalent to SMN replacement, they may offer additional neuroprotective support when used in combination with other SMN-targeting therapies[1,111]. This concept is supported by recent research demonstrating the advantages of combining the HDAC inhibitor LBH589 (panobinostat) with low doses of Spinraza-like antisense oligonucleotides (ASOs), suggesting a potential synergistic approach for enhancing SMA treatment efficacy[116].
Neuromuscular Junction targeting therapies: SMA is associated with an impairment of neuromuscular junction dysfunction (NMJ) development, maturation, and function, contributing to muscle weakness and fatigue. Targeting NMJ pathology presents a potential complementary therapy for SMA. Therapeutic strategies targeting NMJ under development include Salbutamol, Amifampridine, NT-1654, and Pyridostigmine [11,69] (Figure 2).
Salbutamol/albuterol, beta- adrenoreceptors agonists, primarily act on Beta2-adrenoreceptors in the smooth muscle of blood vessels, lungs, and intestines. They have shown promise in improving muscle strength by increasing full-length SMN mRNA, SMN protein, and Gem numbers by promoting exon 7 inclusion in SMN2 in vitro and possibly stabilizing acetylcholine receptor clusters at the NMJ[117,118]. Although small clinical studies suggest benefits in maintaining motor function or improving respiratory function in SMA type 2 patients, large-scale, placebo-controlled studies are lacking [69].
Amifampridine, a voltage-dependent K+ channel blocker, enhances neuromuscular transmission and muscle function. Approved for Lambert-Eaton myasthenic syndrome[11], it was also tested in a randomized, double-blind, placebo-controlled crossover study (NCT03781479) for SMA involving 13 type 3 SMA patients who could walk unaided. The primary outcome was the HFMSE score change, with secondary outcomes including timed tests and quality-of-life assessments. The study indicated that patients receiving amifampridine showed a mean HFMSE improvement of 0.792 compared to the placebo group, with no serious adverse events reported. This suggests that the NMJ could be a potential SMN-independent therapeutic target, highlighting the need for larger studies to confirm amifampridine's role as an adjunctive therapy in SMA treatment[119].
Pyridostigmine, an acetylcholinesterase inhibitor, slows the degradation of acetylcholine within the synaptic cleft, enhancing cholinergic transmission efficiency. Used to improve muscle strength in myasthenia gravis, an autoimmune disorder that also causes muscle weakness. The SPACE trial (NCT02941328) is a phase 2, monocentre, double-blind, placebo-controlled cross-over trial to assess the efficacy of the pyridostigmine in SMA type II-IV involving 37 participants aged 12 and older. The trial assessed motor fatigue and function using the repeated nine-hole peg test (R9HPT) and the motor function measure (MFM). Results showed no significant difference in R9HPT scores between treatments, and while MFM scores were slightly better with pyridostigmine (average 42.4% vs. 41.6%), the difference was not statistically significant. However, 74.4% of pyridostigmine-treated patients reported less fatigue compared to 29.7% on placebo, indicating a significant reduction in fatigue[120].
Additionally, subcutaneous administration of NT-1654, the active portion of agrin, has been shown to delay disease progression in SMA mouse models[121]. The agrin/MuSK signaling pathway, crucial for NMJ formation and maturation, is dysregulated in SMA. Overexpressing agrin or administering agrin-like molecules or downstream mediators like DOK7 can enhance NMJ structure and reduce disease severity in SMA[122,123].
Neuroprotective and muscle-enhancing therapies: Recent studies have shown that selectively depleting SMN in the skeletal muscle of mice leads to muscle and neuromuscular junction (NMJ) pathology. It is hypothesized that improving muscle pathology may help preserve proprioceptive synapses on motor neurons, which are typically lost in SMA. Consequently, targeting muscle is seen as a promising therapeutic approach through various strategies (Figure 2).
Myostatin Inhibitors: Myostatin acts as a negative regulator of muscle growth. Inhibition of the myostatin signaling pathway has shown promising results, especially in less severe models of SMA or in addition to SMN-restoring therapies. Myostatin-deficient animals and humans demonstrate significantly increased musculature[123,124].
Apitegromab (SRK-015) is an investigational fully human monoclonal antibody which inhibits the activation of myostatin, thereby preserving muscle mass. In SMA mice, apitegromab improved muscle mass and function[125]. The Phase 2 TOPAZ clinical trial evaluated the safety and efficacy of apitegromab in nonambulatory patients with SMA types 2 and 3, including 58 participants aged 2 to 21 years. The trial demonstrated sustained improvements in motor function over 24 months, particularly in younger patients given higher doses. The trial's extension phase up to three years confirmed these positive outcomes, with patients showing continued improvement in motor abilities and reduced fatigue. Apitegromab was well-tolerated, with minor adverse effects such as headache, upper respiratory tract infections, pyrexia, nasopharyngitis, cough, and vomiting[126].
Follistatin is an endogenous antagonist of myostatin; over-expression of recombinant follistatin in SMA mouse muscle leads to increased skeletal muscle mass and survival[127].
A hybrid activin II receptor (ACTIIR) ligand trap, BIIB110 (ALG 801) inhibits activin receptor type IIB (ActRIIB) ligands, promoting muscle growth, which in turn enhances muscle mass and function. It is currently in phase 1 of clinical development[104].
Fast Skeletal Muscle Troponin Activators: Reldesemtiv (formally CK-2127107): This drug enhances muscle contractility by prolonging calcium binding to the troponin complex in fast skeletal muscles, reducing the energetic cost of muscle contraction[128]. In a phase 2 trial (NCT02644668) for SMA type II-IV patients, participants received either 150 mg or 450 mg twice daily for 8 weeks. The higher dose group showed better performance in the 6-minute walk test (6MWT) and the Timed Up and Go (TUG) test, as well as increased maximal expiratory pressure (MEP). Some patients reported serious adverse events such as elevated blood creatine phosphokinase and aspartate aminotransferase levels, along with gastrointestinal infections[129]. Further studies are required to establish its efficacy, including in type 1 SMA infants.
Neuroprotection: In SMA, neurons and muscles—the primary tissues affected—have high energy demands, making energy pathway targeting potentially neuroprotective and therapeutic[130]. Olesoxime (TRO19622), a member of the trophos cholesterol-oxime compound family, functions as a mitochondrial pore modulator with neuroprotective properties. Pre-clinical studies indicated its potential to enhance neuron function and survival[131]. However, a phase 2 placebo-controlled trial in patients with types 2 and 3 SMA revealed only a modest but consistent benefit compared to placebo. A subsequent 18-month follow-up study (OLEOS, NCT02628743) failed to demonstrate significant clinical benefits, leading the pharmaceutical company to halt its development for SMA in June 2018[132]. Patients from this study were subsequently enrolled in the JEWELFISH (RO7034067) clinical trial with risdiplam[69].
Other potentially neuroprotective agents, such as riluzole and gabapentin, have been explored for their effects on treating SMA by targeting excitotoxicity. Riluzole, known for its modest effects in ALS, was tested in a small preliminary phase I trial involving seven participants, which suggested some potential benefit[133]. Subsequently, a phase II/III multicenter, randomized, double-blind study was conducted to assess riluzole's efficacy and safety in young adults with types 2 and 3 SMA (NCT00774423). However, the majority of results were not encouraging, and the studies failed to demonstrate significant efficacy[109,134]. Similarly, gabapentin was tested in type 2/3 SMA patients due to its effects in ALS. While one study showed some improvement in motor function, another did not demonstrate any significant benefit[135,136].
Targeting cell death mechanism: In SMA, neurodegeneration is linked to disruptions in core pathways of cell homeostasis, including autophagy, ubiquitin homeostasis, and apoptotic pathways[137,138]. Therapeutic strategies targeting molecules involved in these pathways are under investigation. Celecoxib (Figure 2), a selective cyclooxygenase-2 (COX-2) inhibitor that can cross the blood-brain barrier, has shown potential in increasing SMN protein levels through the activation of the p38 pathway in SMA cell and rodent models[139]. However, despite these promising preclinical findings, a phase II clinical trial of celecoxib for SMA was prematurely terminated, and the results have not yet been published (NCT02876094)[11].
Targeting cytoskeleton: In SMA, the upregulation of the RhoA/Rho kinase (ROCK) pathway disrupts actin dynamics, which disrupts cytoskeleton structure and hinders neuronal growth and regeneration[140]. Therefore, targeting this pathway can be a possible therapeutic option. Y-27632[141] and the FDA-approved fasudil[142], pharmacological inhibitors of ROCK pathway (Figure 2), have demonstrated improvements in survival, neuromuscular junction (NMJ) maturation, and muscle development in SMA mouse models.
4. Challenges in the treatment era: Are We There Yet in the Battle Against SMA?
The therapeutic landscape for Spinal Muscular Atrophy (SMA) has seen remarkable advancements with significant progress in extending patient lives and improving motor function. Despite the success of FDA-approved treatments like nusinersen, onasemnogene abeparvovec, and risdiplam in improving patient outcomes, these therapies come with several challenges.
Nusinersen, for instance, must be injected directly into the cerebrospinal fluid (CSF) intrathecally to ensure it reaches the CNS to treat lower motor neurons, although this method is often associated with thrombocytopenia and injection-site adverse events[55,143,144]. Adult SMA patients face additional challenges such as scoliosis, spine or thorax deformities, and prior spinal surgeries, which complicate the safe administration of nusinersen[145]. Furthermore, intrathecal administration limits nusinersen's efficacy to the central nervous system, failing to address SMA's multi-organ impact effectively[146]. Recent studies in animal models and humans indicate that SMA affects multiple organs, including the heart, peripheral nervous system, skeletal muscle, liver, and vasculature[5,147]. For example, muscle-specific SMN loss in SMA mouse models results in compromised motor performance, premature death, muscle fiber defects, and neuromuscular junction abnormalities. In human fetuses with SMA, SMN deficiency causes delayed growth and maturation of myotubules[148]. Untreated severe SMA patients with only one copy of SMN2 have shown thrombotic occlusions of small blood vessels, leading to digital necrosis, suggesting that severe SMN deficiency may also manifest as a vascular disease[149]. The increasing evidence of multi-organ involvement in SMA suggests that ASO therapies targeting motor neurons alone may be insufficient for long-term management of SMA pathology. Additionally, a recent study[150] investigating nusinersen's biodistribution in post-mortem infant spinal cord samples found variability in ASO distribution, with notably lower concentrations in the cranial portion of the spinal cord and in the brain suggesting uneven distribution within the CNS and potentially affecting treatment efficacy. Despite positive outcomes from pre-symptomatic intrathecal treatment in the NURTURE trial, the long-term effects and efficacy of nusinersen remain uncertain[66,67]. Furthermore, gaps remain in understanding whether nusinersen helps achieve significant milestones, regain function, or avoid serious side effects and ventilation support. More research and long-term data are needed to justify its use in specific SMA patient subgroups.
Onasemnogene abeparvovec (Zolgensma) is a one-time gene therapy that offers significant survival benefits for SMA patients. However, it raises concerns about liver toxicity and reduced efficacy over time[151,152]. This reduction happens because cell division dilutes the episomal SMN cDNA within the AAV vector, limiting expression to non-dividing cells like neurons[11]. The body's immune response to the viral vector complicates repeated treatments, while systemic delivery and maintenance of the treatment is crucial for long-term benefits due to the involvement of peripheral organs in the disease. Onasemnogene has to be administered within the first two years of life, making newborn screening for SMA crucial for early diagnosis and timely treatment, given the limited window for effective intervention. Serious side effects, such as liver damage, thrombotic microangiopathy, thrombocytopenia, and cardiac toxicity has also been observed for onasemnogene and therefore require careful patient monitoring. Furthermore, recent studies have highlighted a toxic gain of function of SMN, especially in the sensorimotor circuit, including the loss of proprioceptive neurons. Similar results were also found when intrathecal injection was explored as an alternative delivery method (NCT03381729). Pre-clinical trials involving intrathecal injection have shown concerns about neurotoxicity, including ataxia, loss of proprioceptive synapses, neuroinflammation, and neurodegeneration[153,154,155]. These issues highlight the need for more long-term data to fully understand the therapy's effectiveness and safety.
Risdiplam, an oral medication, provides a more accessible treatment option, especially for older patients after receiving its approval in 2020 for all SMA types in patients 2 years of age. Although it overcomes the necessity for invasive intrathecal injections, its long-term effects are still unknown. Potential risks include male fertility impairment, growth issues, retinal toxicity, and off-target effect although these have not yet been observed in humans[102,156,157]. The drug is contraindicated for patients with hepatic abnormalities, which could limit its use in many older SMA patients who often present with liver issues.
Apart from these, the extremely high cost of these treatments poses a significant barrier to widespread accessibility. While early treatment is critical for optimal outcomes[158], there remains a subset of patients who do not respond to these therapies[66]. The variability in response can be attributed to factors beyond SMN1 and SMN2 genes, including other genetic factors, environmental influences, and disparities in medical care quality and accessibility.
5. Future directions: Advancing the battle against SMA
The field of Spinal Muscular Atrophy treatment has significantly evolved with the introduction of nusinersen, onasemnogene abeparvovec, and risdiplam, which have become the standard of care. Despite their ground-breaking nature, it has become clear through long-term follow-ups that SMN2 gene modulation or SMN1 gene replacement alone does not constitute a cure. Future directions in SMA treatment involve incorporating SMN-independent therapies alongside SMN-dependent ones to provide broader benefits, especially for patients diagnosed later or with milder forms of SMA. A cross-disease approach, repurposing drugs with established safety profiles, could also expedite the development of effective therapies. Additionally, combining currently approved therapies has garnered interest due to their different approaches to treating SMA. For example, Lee et al. provided insights on patients first treated with nusinersen followed by onasemnogene[159], while another small study involving five patients treated with both therapies showed phenotype improvements but highlighted potential liver toxicity risks[160]. More research is needed to determine the therapeutic benefits and cost-benefit analysis of combination treatments.
In addition to drug-based therapies, a multidisciplinary approach is essential for effective SMA treatment, incorporating physiotherapy, rehabilitation, respiratory management, orthopedic care, and nutritional support[161]. Further research is needed to determine the most effective combination of these treatments.
Recent evidence strongly supports that early intervention provides greater clinical benefits than treating symptomatic patients, with a high potential to achieve age-appropriate motor milestones[158]. Therefore, neonatal genetic screening is crucial for early diagnosis and treatment. In Belgium, scientists completed an observational newborn screening study titled "Sun May Arise on SMA"[162]. Similarly, Prof. Servais launched a newborn screening study in Oxford, UK, in March 2022, expected to conclude in August 2025[163].
Finally, researching to improve existing treatment approaches for SMA is essential to offer more effective, safer, and accessible options. One promising area of research focuses on enhancing the efficacy and minimizing the toxicity of antisense oligonucleotides (ASOs) as nusinersen's success highlights the promise and efficiency of ASO-based therapies, yet there is room for enhancement. A notable advancement is the development of phosphorodiamidate morpholino oligomers (PMOs), a different chemistry of ASO, which are neutrally charged due to their phosphorodiamidate linkage, offer enhanced stability and minimal toxicity[164]. These properties have made PMOs effective therapeutic options, as evidenced by the approval of four PMO-based ASOs for Duchenne Muscular Dystrophy (DMD)[165]. In the context of SMA, studies on mouse models have demonstrated that PMOs can significantly increase SMN2 levels and improve survival rates through various delivery routes, including intrathecal and intravenous injections. Although the superiority of these delivery routes remains debated. Most notably, PMOs have shown lower toxicity, prolonged survival, and most importantly, effective SMN restoration in central nervous system (CNS) tissues. Additionally, PMOs' neutral charge allows them to be conjugated with cell-penetrating peptides (CPPs) for improved delivery and uptake in target tissues[166,167,168]. CPPs, short sequences that facilitate the uptake of cargo into target cells, were discovered in the late 1980s[165]. In the context of SMA, CPPs such as Pip6a, RXR peptides, and DG9 have shown promising results by enhancing cellular uptake and crossing the blood-brain barrier, leading to better restoration of SMN levels, prolonged survival, and improved phenotypes in mouse models[169,170,171]. Overall, it is crucial to continue researching ways to enhance current treatment approaches while exploring new options to ensure the best possible outcomes for SMA patients.
6. Conclusions
The field of Spinal Muscular Atrophy (SMA) treatment is witnessing unprecedented advancements, bringing life-changing options to patients and their families. The approval of three SMA therapies—nusinersen, onasemnogene abeparvovec, and risdiplam—has opened new possibilities and transformed the standard of care. However, several challenges persist. Addressing these challenges in the coming years will be critical for the future development of SMA therapies. Continued research is essential, particularly for chronic forms of SMA and to establish optimal treatment regimens. Monitoring the evolution of new phenotypes and complications that have not been previously observed is also necessary. A combination of therapies and a multidisciplinary approach may play a significant role in future developments, though testing these approaches will be complex given the evolving phenotypes. Moreover, as more therapies become available, it will be crucial to consider the ethical and practical implications for all stakeholders, including the healthcare system, industry, patients, and caregivers. Overall, advancing SMA treatment will require a holistic and collaborative effort to ensure the best outcomes for patients.
Author Contributions
Conception and design, US.H. and T.Y.; Literature review and writing-original draft preparation, US.H; Writing-review and editing, US.H, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
No specific grant support was received for this study.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the Canadian Institutes of Health Research, the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, and the Women and Children’s Health Research Institute for their support.
Conflicts of Interest
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Groen, E.J.N.; Talbot, K.; Gillingwater, T.H. Advances in Therapy for Spinal Muscular Atrophy: Promises and Challenges. Nat. Rev. Neurol. 2018, 14, 214–224. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the Survival Motor Neuron Gene, a Candidate Gene for Human Spinal Muscular Atrophy, Leads to Massive Cell Death in Early Mouse Embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef]
- Burghes, A. H.; Beattie, C. E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat Rev Neurosci 2009, 10, 597–609. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal Muscular Atrophy: Going Beyond the Motor Neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Rochette, C.; Gilbert, N.; Simard, L. Smn Gene Duplication and the Emergence of the Smn2 Gene Occurred in Distinct Hominids: Smn2 Is Unique to Homo Sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef]
- Lorson, C.L.; Androphy, E.J. An Exonic Enhancer Is Required for Inclusion of an Essential Exon in the Sma-Determining Gene Smn. Hum. Mol. Genet. 2000, 9, 259–265. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (Smn) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The Spinal Muscular Atrophy Disease Gene Product, Smn, and Its Associated Protein Sip1 Are in a Complex with Spliceosomal Snrnp Proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef]
- Mercuri, E.; Pera, M.C.; Scoto, M.; Finkel, R.; Muntoni, F. Spinal Muscular Atrophy - Insights and Challenges in the Treatment Era. Nat. Rev. Neurol. 2020, 16, 706–715. [Google Scholar] [CrossRef]
- Chaytow, H.; Faller, K.M.; Huang, Y.-T.; Gillingwater, T.H. Spinal Muscular Atrophy: From Approved Therapies to Future Therapeutic Targets for Personalized Medicine. Cell Rep. Med. 2021, 2, 100346. [Google Scholar] [CrossRef]
- Kolb, S.J.; Battle, D.J.; Dreyfuss, G. Molecular Functions of the Smn Complex. J. Child Neurol. 2007, 22, 990–994. [Google Scholar] [CrossRef]
- Keinath, M. C.; Prior, D. E.; Prior, T. W. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl Clin Genet 2021, 14, 11–25. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the Smn Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the Sma Gene Smn1 from the Copy Gene Smn2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Schmid, A.; DiDonato, C.J. Animal Models of Spinal Muscular Atrophy. J. Child Neurol. 2007, 22, 1004–1012. [Google Scholar] [CrossRef]
- Han, K.-J.; Foster, D.G.; Zhang, N.-Y.; Kanisha, K.; Dzieciatkowska, M.; Sclafani, R.A.; Hansen, K.C.; Peng, J.; Liu, C.-W. Ubiquitin-Specific Protease 9x Deubiquitinates and Stabilizes the Spinal Muscular Atrophy Protein-Survival Motor Neuron. J. Biol. Chem. 2012, 287, 43741–43752. [Google Scholar] [CrossRef]
- Battaglia, G.; Princivalle, A.; Forti, F.; Lizier, C.; Zeviani, M. Expression of the Smn Gene, the Spinal Muscular Atrophy Determining Gene, in the Mammalian Central Nervous System. Hum. Mol. Genet. 1997, 6, 1961–1971. [Google Scholar] [CrossRef]
- Lefebvre, S.; Sarret, C. Pathogenesis and Therapeutic Targets in Spinal Muscular Atrophy (Sma). Arch. De Pediatr. 2020, 27, 7S3–7S8. [Google Scholar] [CrossRef]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H.M. The Survival Motor Neuron Protein in Spinal Muscular Atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. A Novel Nuclear Structure Containing the Survival of Motor Neurons Protein. Embo J. 1996, 15, 3555–65. [Google Scholar] [CrossRef]
- Carvalho, T.; Almeida, F.; Calapez, A.; Lafarga, M.; Berciano, M. T.; Carmo-Fonseca, M. The Spinal Muscular Atrophy Disease Gene Product, Smn: A Link between Snrnp Biogenesis and the Cajal (Coiled) Body. J Cell Biol 1999, 147, 715–728. [Google Scholar] [CrossRef]
- Feng, W.; Gubitz, A.K.; Wan, L.; Battle, D.J.; Dostie, J.; Golembe, T.J.; Dreyfuss, G. Gemins Modulate the Expression and Activity of the Smn Complex. Hum. Mol. Genet. 2005, 14, 1605–1611. [Google Scholar] [CrossRef]
- Mercuri, E.; Sumner, C. J.; Muntoni, F.; Darras, B. T.; Finkel, R. S. Spinal Muscular Atrophy. Nat Rev Dis Primers 2022, 8, 52. [Google Scholar] [CrossRef]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse Role of Survival Motor Neuron Protein. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between Severity and Smn Protein Level in Spinal Muscular Atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Ling, K.K.Y.; Lin, M.-Y.; Zingg, B.; Feng, Z.; Ko, C.-P. Synaptic Defects in the Spinal and Neuromuscular Circuitry in a Mouse Model of Spinal Muscular Atrophy. PLOS ONE 2010, 5, e15457. [Google Scholar] [CrossRef]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O'Donovan, M.J. Early Functional Impairment of Sensory-Motor Connectivity in a Mouse Model of Spinal Muscular Atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef]
- Fletcher, E. V.; Simon, C.M.; Pagiazitis, J.G.; Chalif, J.I.; Vukojicic, A.; Drobac, E.; Wang, X.; Mentis, G.Z. Reduced Sensory Synaptic Excitation Impairs Motor Neuron Function Via Kv2.1 in Spinal Muscular Atrophy. Nat Neurosci 2017, 20, 905–916. [Google Scholar] [CrossRef]
- Kong, L.; Valdivia, D.O.; Simon, C.M.; Hassinan, C.W.; Delestrée, N.; Ramos, D.M.; Park, J.H.; Pilato, C.M.; Xu, X.; Crowder, M.; et al. Impaired Prenatal Motor Axon Development Necessitates Early Therapeutic Intervention in Severe Sma. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Kariya, S.; Park, G.-H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced Smn Protein Impairs Maturation of the Neuromuscular Junctions in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective Vulnerability of Motor Neurons and Dissociation of Pre- and Post-Synaptic Pathology at the Neuromuscular Junction in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2007, 17, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marcé, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired Synaptic Vesicle Release and Immaturity of Neuromuscular Junctions in Spinal Muscular Atrophy Mice. J. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a Potential Biomarker for Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S. J.; Kissel, J. T. Spinal Muscular Atrophy. Neurol Clin 2015, 33, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021, 44, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Coovert, D.D.; Monani, U.R.; Morris, G.E.; Burghes, A.H.M. The Survival Motor Neuron (Smn) Protein: Effect of Exon Loss and Mutation on Protein Localization. neurogenetics 2000, 3, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Young, P. J.; Man, N.T.; Lorson, C.L.; Le, T.T.; Androphy, E.J.; Burghes, A.H.; Morris, G.E. The Exon 2b Region of the Spinal Muscular Atrophy Protein, Smn, Is Involved in Self-Association and Sip1 Binding. Hum Mol Genet 2000, 9, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. Smn Mutants of Spinal Muscular Atrophy Patients Are Defective in Binding to Snrnp Proteins. Proc. Natl. Acad. Sci. 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [PubMed]
- Frugier, T.; Tiziano, F.D.; Cifuentes-Diaz, C.; Miniou, P.; Roblot, N.; Dierich, A.; Le Meur, M.; Melki, J. Nuclear Targeting Defect of Smn Lacking the C-Terminus in a Mouse Model of Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 849–858. [Google Scholar] [CrossRef]
- Burnett, B.G.; Muñoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of Smn Protein Stability. Mol. Cell. Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. Smndelta7, the Major Product of the Centromeric Survival Motor Neuron (Smn2) Gene, Extends Survival in Mice with Spinal Muscular Atrophy and Associates with Full-Length Smn. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Pan, F.; Hong, D.; Shenoy, S.M.; Singer, R.H.; Bassell, G.J. Active Transport of the Survival Motor Neuron Protein and the Role of Exon-7 in Cytoplasmic Localization. J. Neurosci. 2003, 23, 6627–6637. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Schönling, J.; Rudnik-Schöneborn, S.; Raschke, H.; Zerres, K.; Wirth, B. Missense Mutations in Exon 6 of the Survival Motor Neuron Gene in Patients with Spinal Muscular Atrophy (Sma). Hum. Mol. Genet. 1997, 6, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, R.d.H.; Matsui, C.; Polido, G.J.; Silva, A.M.S.; Kulikowski, L.; Dias, A.T.; Zanardo, E.A.; Solla, D.J.F.; Gurgel-Giannetti, J.; de Moura, A.C.M.L.; et al. Intragenic Variants in the Smn1 Gene Determine the Clinical Phenotype in 5q Spinal Muscular Atrophy. Neurol. Genet. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Beattie, C.E.; Carrel, T.L.; McWhorter, M.L. Fishing for a Mechanism: Using Zebrafish to Understand Spinal Muscular Atrophy. J. Child Neurol. 2007, 22, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S. J.; Kissel, J. T. Spinal Muscular Atrophy: A Timely Review. Arch Neurol 2011, 68, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V. Ramblings in the History of Spinal Muscular Atrophy. Neuromuscul. Disord. 2009, 19, 69–73. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era. Muscle Nerve 2014, 51, 157–167. [Google Scholar] [CrossRef]
- Finkel, R.; Bertini, E.; Muntoni, F.; Mercuri, E.; Enmc Sma Workshop Study Group. 209th Enmc International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9 November 2014, Heemskerk, the Netherlands. Neuromuscul Disord 2015, 25, 593–602. [Google Scholar] [CrossRef]
- Awano, T.; Kim, J.-K.; Monani, U.R. Spinal Muscular Atrophy: Journeying from Bench to Bedside. Neurotherapeutics 2014, 11, 786–795. [Google Scholar] [CrossRef]
- D'Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet J Rare Dis 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Chen, T. H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. Fda Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid. Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef] [PubMed]
- FDA. Spinraza™ Safely and Effectively. Food and Drug Administration, 2016.
- Singh, N. K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a Critical Exon of Human Survival Motor Neuron Is Regulated by a Unique Silencer Element Located in the Last Intron. Mol Cell Biol 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W. Iss-N1 Makes the First Fda-Approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense Masking of an Hnrnp A1/A2 Intronic Splicing Silencer Corrects Smn2 Splicing in Transgenic Mice. Am J Hum Genet 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the Discovery of Iss-N1 Led to the First Medical Therapy for Spinal Muscular Atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense Correction of Smn2 Splicing in the Cns Rescues Necrosis in a Type Iii Sma Mouse Model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Williams, J.H.; Schray, R.C.; Patterson, C.A.; Ayitey, S.O.; Tallent, M.K.; Lutz, G.J. Oligonucleotide-Mediated Survival of Motor Neuron Protein Expression in Cns Improves Phenotype in a Mouse Model of Spinal Muscular Atrophy. J. Neurosci. 2009, 29, 7633–7638. [Google Scholar] [CrossRef]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense Oligonucleotides Delivered to the Mouse Cns Ameliorate Symptoms of Severe Spinal Muscular Atrophy. Sci. Transl. Med. 2011, 3, 72ra18–72ra18. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a Central Nervous System Delivered 2'-O-Methoxyethyl-Modified Survival of Motor Neuron Splicing Oligonucleotide in Mice and Nonhuman Primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral Smn Restoration Is Essential for Long-Term Rescue of a Severe Spinal Muscular Atrophy Mouse Model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Miller, M.R.; Robbins, K.L.; Lombardi, A.M.; Atkinson, A.K.; Brehm, A.J.; Lorson, C.L. Morpholino Antisense Oligonucleotides Targeting Intronic Repressor Element1 Improve Phenotype in Sma Mouse Models. Hum. Mol. Genet. 2014, 23, 4832–4845. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen Versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen Versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.-L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen Initiated in Infants During the Presymptomatic Stage of Spinal Muscular Atrophy: Interim Efficacy and Safety Results from the Phase 2 Nurture Study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Willis, T.A. Therapeutic Advances in Spinal Muscular Atrophy. Paediatr. Child Heal. 2023, 33, 23–28. [Google Scholar] [CrossRef]
- Sansone, V.A.; Pirola, A.; Albamonte, E.; Pane, M.; Lizio, A.; D'Amico, A.; Catteruccia, M.; Cutrera, R.; Bruno, C.; Pedemonte, M.; et al. Respiratory Needs in Patients with Type 1 Spinal Muscular Atrophy Treated with Nusinersen. J. Pediatr. 2020, 219, 223–228. [Google Scholar] [CrossRef]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.-B.; Manautou, J.E. Adverse Drug Reactions and Toxicity of the Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. 2022, 50, 879–887. [Google Scholar] [CrossRef]
- Nakevska, Z.; Yokota, T. Challenges and Future Perspective of Antisense Therapy for Spinal Muscular Atrophy: A Review. Eur. J. Cell Biol. 2023, 102, 151326. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular Aav9 Preferentially Targets Neonatal Neurons and Adult Astrocytes. Nat. Biotechnol. 2008, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, J.; Nobre, R.J.; de Almeida, L.P. Gene Therapy for the Cns Using Aavs: The Impact of Systemic Delivery by Aav9. J. Control. Release 2016, 241, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic Delivery of Scaav9 Expressing Smn Prolongs Survival in a Model of Spinal Muscular Atrophy. Sci. Transl. Med. 2010, 2, 35ra42. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Ferraiuolo, L.; Schmelzer, L.; Braun, L.; McGovern, V.; Likhite, S.; Michels, O.; Govoni, A.; Fitzgerald, J.; Morales, P.; et al. Improving Single Injection Csf Delivery of Aav9-Mediated Gene Therapy for Sma: A Dose-Response Study in Mice and Nonhuman Primates. Mol. Ther. 2015, 23, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. New Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health Outcomes in Spinal Muscular Atrophy Type 1 Following Avxs-101 Gene Replacement Therapy. Pediatr. Pulmonol. 2018, 54, 179–185. [Google Scholar] [CrossRef]
- Lowes, L. P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; Menier, M.; Feltner, D.E.; Wells, C.; Kissel, J.T.; Al-Zaidy, S.; Mendell, J. Impact of Age and Motor Function in a Phase 1/2a Study of Infants with Sma Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr Neurol 2019, 98, 39–45. [Google Scholar] [CrossRef]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef]
- Naveed, A.; Calderon, H. Onasemnogene Abeparvovec (AVXS-101) for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2021, 26, 437–444. [Google Scholar] [CrossRef]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene Abeparvovec for Presymptomatic Infants with Two Copies of Smn2 at Risk for Spinal Muscular Atrophy Type 1: The Phase Iii Spr1nt Trial. Nat. Med. 2022, 28, 1381–1389. [Google Scholar] [CrossRef]
- FDA. Zolgensma Safely and Effectively. Food and Drug Administration, 2019.
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor Neuron Disease. Smn2 Splicing Modifiers Improve Motor Function and Longevity in Mice with Spinal Muscular Atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine to Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( Smn2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (Sma). J Med Chem, 2018; 61, 6501–6517. [Google Scholar]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam Distributes and Increases Smn Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to Smn2 Pre-Mrna-Protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat Commun 2017, 8, 1476. [Google Scholar] [CrossRef]
- Wan, R.; Bai, R.; Shi, Y. Molecular Choreography of Pre-Mrna Splicing by the Spliceosome. Curr Opin Struct Biol 2019, 59, 124–133. [Google Scholar] [CrossRef]
- Ratni, H.; Scalco, R.S.; Stephan, A.H. Risdiplam, the First Approved Small Molecule Splicing Modifier Drug as a Blueprint for Future Transformative Medicines. ACS Med. Chem. Lett. 2021, 12, 874–877. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. New Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. Firefish Part 1: 1-Year Results on Motor Function in Babies with Type 1 Sma (S25.003). Neurology 2019, 92. [Google Scholar] [CrossRef]
- Servais, L.; Baranello, G.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. Firefish Part 1: Survival, Ventilation and Swallowing Ability in Infants with Type 1 Sma Receiving Risdiplam (Rg7916) (S25.008). Neurology 2019, 92. [Google Scholar] [CrossRef]
- Servais, L.; Baranello, G.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Rose, K.; Vlodavets, D.; Xiong, H.; Zanoteli, E.; El-Khairi, M.; Fuerst-Recktenwald, S.; et al. Firefish Part 2: Efficacy and Safety of Risdiplam (Rg7916) in Infants with Type 1 Spinal Muscular Atrophy (Sma) (1302). Neurology 2020, 94. [Google Scholar] [CrossRef]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Foley, A.R.; Yang, M.L.; Martens, W.B.; et al. Observational Study of Spinal Muscular Atrophy Type I and Implications for Clinical Trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef]
- Analyst R, Webcast A. Roche Ir - Aan 2020 Highlights.
- Mercuri, E.; Barisic, N.; Boespflug-Tanguy, O.; Deconinck, N.; Kostera-Pruszczyk, A.; Masson, R.; Mazzone, E.; Nascimento, A.; Saito, K.; Vlodavets, D.; et al. Sunfish Part 2: Efficacy and Safety of Risdiplam (Rg7916) in Patients with Type 2 or Non-Ambulant Type 3 Spinal Muscular Atrophy (Sma) (1260). Neurology 2020, 94. [Google Scholar] [CrossRef]
- Chiriboga, C.; Bruno, C.; Day, J.W.; Duong, T.; Fischer, D.; Kirschner, J.; Muntoni, F.; Fuerst-Recktenwald, S.; Gerber, M.; Gorni, K.; et al. Jewelfish: Safety and Pharmacodynamic Data in Non-Naïve Patients with Spinal Muscular Atrophy (Sma) Receiving Treatment with Risdiplam (Rg7916) (772). Neurology 2020, 94. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Mercuri, E.; Fischer, D.; Kraus, D.; Yeung, W.Y.; Kletzl, H.; Gerber, M.; Cleary, Y.; Gorni, K.; Khwaja, O. Jewelfish: Risdiplam (Rg7916) Increases Smn Protein in Non-Naïve Patients with Sma. In 23rd International Annual Congress of the World Muscle Society; Mendoza, Argentina: Roche, 2018. [Google Scholar]
- Bertini, E.; Day, J.; Muhaizea, M.; Xiong, H.; Servais, L.; Prufer, A.; Tichy, M.; Yeung, W.; Gorni, K. P.362rainbowfish: A Study of Risdiplam (Rg7916) in Newborns with Pre-Symptomatic Spinal Muscular Atrophy (Sma). Neuromuscul. Disord. 2019, 29, S187–S187. [Google Scholar] [CrossRef]
- Kakazu, J.; Walker, N.L.; Babin, K.C.; Trettin, K.A.; Lee, C.; Sutker, P.B.; Kaye, A.M.; Kaye, A.D. Risdiplam for the Use of Spinal Muscular Atrophy. Orthop. Rev. 2021, 13, 25579. [Google Scholar] [CrossRef]
- D'Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet Journal of Rare Diseases 2011, 6, 71. [Google Scholar] [CrossRef]
- Chen, T.-H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? International Journal of Molecular Sciences 2020. [Google Scholar] [CrossRef]
- Calder, A.N.; Androphy, E.J.; Hodgetts, K.J. Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 10067–10083. [Google Scholar] [CrossRef]
- Brichta, L.; Hofmann, Y.; Hahnen, E.; Siebzehnrubl, F.A.; Raschke, H.; Blumcke, I.; Eyupoglu, I.Y.; Wirth, B. Valproic Acid Increases the Smn2 Protein Level: A Well-Known Drug as a Potential Therapy for Spinal Muscular Atrophy. Hum Mol Genet 2003, 12, 2481–2489. [Google Scholar] [CrossRef]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic Acid Increases Smn Levels in Spinal Muscular Atrophy Patient Cells. Ann. Neurol. 2003, 54, 647–654. [Google Scholar] [CrossRef]
- Andreassi, C.; Angelozzi, C.; Tiziano, F.D.; Vitali, T.; De Vincenzi, E.; Boninsegna, A.; Villanova, M.; Bertini, E.; Pini, A.; Neri, G.; et al. Phenylbutyrate Increases Smn Expression in Vitro: Relevance for Treatment of Spinal Muscular Atrophy. Eur. J. Hum. Genet. 2003, 12, 59–65. [Google Scholar] [CrossRef]
- Wadman, R. I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug Treatment for Spinal Muscular Atrophy Types Ii and Iii. Cochrane Database Syst Rev 2020, 1, CD006282. [Google Scholar] [CrossRef]
- Tsai, L.-K.; Tsai, M.-S.; Ting, C.-H.; Li, H. Multiple Therapeutic Effects of Valproic Acid in Spinal Muscular Atrophy Model Mice. J. Mol. Med. 2008, 86, 1243–1254. [Google Scholar] [CrossRef]
- Elshafay, A.; Hieu, T.H.; Doheim, M.F.; Kassem, M.A.M.; Eldoadoa, M.F.; Holloway, S.K.; Abo-Elghar, H.; Hirayama, K.; Huy, N.T. Efficacy and Safety of Valproic Acid for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis. CNS Drugs 2019, 33, 239–250. [Google Scholar] [CrossRef]
- Brahe, C.; Vitali, T.; Tiziano, F.D.; Angelozzi, C.; Pinto, A.M.; Borgo, F.; Moscato, U.; Bertini, E.; Mercuri, E.; Neri, G. Phenylbutyrate Increases Smn Gene Expression in Spinal Muscular Atrophy Patients. Eur. J. Hum. Genet. 2004, 13, 256–259. [Google Scholar] [CrossRef]
- Hauke, J.; Riessland, M.; Lunke, S.; Eyüpoglu, I.Y.; Blümcke, I.; El-Osta, A.; Wirth, B.; Hahnen, E. Survival Motor Neuron Gene 2 Silencing by DNA Methylation Correlates with Spinal Muscular Atrophy Disease Severity and Can Be Bypassed by Histone Deacetylase Inhibition. Hum. Mol. Genet. 2008, 18, 304–317. [Google Scholar] [CrossRef]
- Narver, H.L.; Kong, L.; Burnett, B.G.; Choe, D.W.; Bosch-Marcé, M.; Taye, A.A.; Eckhaus, M.A.; Sumner, C.J. Sustained Improvement of Spinal Muscular Atrophy Mice Treated with Trichostatin a Plus Nutrition. Ann. Neurol. 2008, 64, 465–470. [Google Scholar] [CrossRef]
- Dayangaç-Erden, D.; Bora, G.; Ayhan, P.; Kocaefe, C.; Dalkara, S.; Yelekçi, K.; Demir, A.S.; Erdem-Yurter, H. Histone Deacetylase Inhibition Activity and Molecular Docking of (E )-Resveratrol: Its Therapeutic Potential in Spinal Muscular Atrophy. Chem. Biol. Drug Des. 2009, 73, 355–364. [Google Scholar] [CrossRef]
- Pagliarini, V.; Guerra, M.; Di Rosa, V.; Compagnucci, C.; Sette, C. Combined Treatment with the Histone Deacetylase Inhibitor Lbh589 and a Splice-Switch Antisense Oligonucleotide Enhances Smn2 Splicing and Smn Expression in Spinal Muscular Atrophy Cells. J. Neurochem. 2019, 153, 264–275. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Lomastro, R.; Pinto, A.M.; Messina, S.; D'Amico, A.; Fiori, S.; Angelozzi, C.; Pane, M.; Mercuri, E.; Bertini, E.; et al. Salbutamol Increases Survival Motor Neuron (Smn) Transcript Levels in Leucocytes of Spinal Muscular Atrophy (Sma) Patients: Relevance for Clinical Trial Design. J. Med Genet. 2010, 47, 856–858. [Google Scholar] [CrossRef]
- Clausen, L.; Cossins, J.; Beeson, D. Beta-2 Adrenergic Receptor Agonists Enhance Achr Clustering in C2c12 Myotubes: Implications for Therapy of Myasthenic Disorders. J. Neuromuscul. Dis. 2018, 5, 231–240. [Google Scholar] [CrossRef]
- Bonanno, S.; Giossi, R.; Zanin, R.; Porcelli, V.; Iannacone, C.; Baranello, G.; Ingenito, G.; Iyadurai, S.; Stevic, Z.; Peric, S.; Maggi, L. Amifampridine Safety and Efficacy in Spinal Muscular Atrophy Ambulatory Patients: A Randomized, Placebo-Controlled, Crossover Phase 2 Trial. J Neurol 2022, 269, 5858–5867. [Google Scholar] [CrossRef]
- Stam, M.; A Wijngaarde, C.; Bartels, B.; Asselman, F.-L.; Otto, L.A.M.; E Habets, L.; A van Eijk, R.P.; Middelkoop, B.M.; Goedee, H.S.; de Groot, J.F.; et al. Randomized Double-Blind Placebo-Controlled Crossover Trial with Pyridostigmine in Spinal Muscular Atrophy Types 2-4. Brain Commun. 2022, 5, fcac324. [Google Scholar] [CrossRef]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell. Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef]
- Kim, J.-K.; Caine, C.; Awano, T.; Herbst, R.; Monani, U.R. Motor Neuronal Repletion of the Nmj Organizer, Agrin, Modulates the Severity of the Spinal Muscular Atrophy Disease Phenotype in Model Mice. Hum. Mol. Genet. 2017, 26, 2377–2385. [Google Scholar] [CrossRef]
- Kaifer, K. A.; Villalon, E.; Smith, C.E.; Simon, M.E.; Marquez, J.; Hopkins, A.E.; Morcos, T.I.; Lorson, C.L. Aav9-Dok7 Gene Therapy Reduces Disease Severity in Smn(2b/-) Sma Model Mice. Biochem Biophys Res Commun 2020, 530, 107–114. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin Inhibition in Combination with Antisense Oligonucleotide Therapy Improves Outcomes in Spinal Muscular Atrophy. J. Cachex- Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef]
- Long, K.K.; O’shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific Inhibition of Myostatin Activation Is Beneficial in Mouse Models of Sma Therapy. Hum. Mol. Genet. 2018, 28, 1076–1089. [Google Scholar] [CrossRef]
- Crawford, T. O.; Darras, B.T.; Day, J.W.; Young, S.D.; Duong, T.; Nelson, L.L.; Barrett, D.; Song, G.; Bilic, S.; Cote, S.; et al. Safety and Efficacy of Apitegromab in Patients with Spinal Muscular Atrophy Types 2 and 3: The Phase 2 Topaz Study. Neurology 2024, 102, e209151. [Google Scholar] [CrossRef]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically Induced Mouse Model of Adult Spinal Muscular Atrophy to Evaluate Effectiveness of Therapeutics after Disease Onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Hwee, D.T.; Kim, L.H.; Durham, N.; Yang, H.T.; Hinken, A.C.; Kennedy, A.R.; Terjung, R.L.; Jasper, J.R.; Malik, F.I.; et al. Fast Skeletal Muscle Troponin Activator Ck-2066260 Increases Fatigue Resistance by Reducing the Energetic Cost of Muscle Contraction. J. Physiol. 2019, 597, 4615–4625. [Google Scholar] [CrossRef]
- Marisa Wexler, MS. Reldesemtiv (Formerly Ck-2127107) for Sma. Nov. 15, 2022 2022.
- Boyd, P.J.; Tu, W.-Y.; Shorrock, H.K.; Groen, E.J.N.; Carter, R.N.; Powis, R.A.; Thomson, S.R.; Thomson, D.; Graham, L.C.; Motyl, A.A.L.; et al. Bioenergetic Status Modulates Motor Neuron Vulnerability and Pathogenesis in a Zebrafish Model of Spinal Muscular Atrophy. PLOS Genet. 2017, 13, e1006744. [Google Scholar] [CrossRef]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and Characterization of Cholest-4-En-3-One, Oxime (Tro19622), a Novel Drug Candidate for Amyotrophic Lateral Sclerosis. J Pharmacol Exp Ther 2007, 322, 709–720. [Google Scholar] [CrossRef]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet 2020, 21, 231–261. [Google Scholar] [CrossRef] [PubMed]
- Russman, B.S.; Iannaccone, S.T.; Samaha, F.J. A Phase 1 Trial of Riluzole in Spinal Muscular Atrophy. Arch. Neurol. 2003, 60, 1601–1603. [Google Scholar] [CrossRef]
- Darras, B. T. Spinal Muscular Atrophies. Pediatr Clin North Am 2015, 62, 743–766. [Google Scholar] [CrossRef]
- Merlini, L.; Solari, A.; Vita, G.; Bertini, E.; Minetti, C.; Mongini, T.; Mazzoni, E.; Angelini, C.; Morandi, L. Role of Gabapentin in Spinal Muscular Atrophy: Results of a Multicenter, Randomized Italian Study. J Child Neurol 2003, 18, 537–541. [Google Scholar] [CrossRef]
- Miller, R.; Moore, D.; Dronsky, V.; Bradley, W.; Barohn, R.; Bryan, W.; Prior, T.; Gelinas, D.; Iannaccone, S.; Kissel, J.; et al. A Placebo-Controlled Trial of Gabapentin in Spinal Muscular Atrophy. J. Neurol. Sci. 2001, 191, 127–131. [Google Scholar] [CrossRef]
- Custer, S.K.; Androphy, E.J. Autophagy Dysregulation in Cell Culture and Animals Models of Spinal Muscular Atrophy. Mol. Cell. Neurosci. 2014, 61, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.; Schiaffino, L.; Boido, M.; Valsecchi, V.; Guglielmotto, M.; De Amicis, E.; Puyal, J.; Garcera, A.; Tamagno, E.; Soler, R.M.; et al. Inhibition of Autophagy Delays Motoneuron Degeneration and Extends Lifespan in a Mouse Model of Spinal Muscular Atrophy. Cell Death Dis. 2017, 8, 3223. [Google Scholar] [CrossRef] [PubMed]
- Farooq, F.; Abadía-Molina, F.; MacKenzie, D.; Hadwen, J.; Shamim, F.; O'Reilly, S.; Holcik, M.; MacKenzie, A. Celecoxib Increases Smn and Survival in a Severe Spinal Muscular Atrophy Mouse Model Via P38 Pathway Activation. Hum. Mol. Genet. 2013, 22, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tönges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The Spinal Muscular Atrophy Disease Protein Smn Is Linked to the Rho-Kinase Pathway Via Profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-Kinase Inactivation Prolongs Survival of an Intermediate Sma Mouse Model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef]
- Bowerman, M.; Murray, L.M.; Boyer, J.G.; Anderson, C.L.; Kothary, R. Fasudil Improves Survival and Promotes Skeletal Muscle Development in a Mouse Model of Spinal Muscular Atrophy. BMC Med. 2012, 10, 24–24. [Google Scholar] [CrossRef] [PubMed]
- Lowery, S.; Oliver, A. Incidence of Postdural Puncture Headache and Backache Following Diagnostic/Therapeutic Lumbar Puncture Using a 22g Cutting Spinal Needle, and after Introduction of a 25g Pencil Point Spinal Needle. Paediatr Anaesth 2008, 18, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Hache, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children with Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J Child Neurol 2016, 31, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.; Chehade, L.; Kothary, R. Curing Sma: Are We There Yet? Gene Ther 2023, 30, 8–17. [Google Scholar] [CrossRef]
- Heitschmidt, L.; Pichlmaier, L.; Eckerland, M.; Steindor, M.; Olivier, M.; Fuge, I.; Kölbel, H.; Hirtz, R.; Stehling, F. Nusinersen Does Not Improve Lung Function in a Cohort of Children with Spinal Muscular Atrophy - a Single-Center Retrospective Study. Eur. J. Paediatr. Neurol. 2021, 31, 88–91. [Google Scholar] [CrossRef]
- Kim, J.-K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.-P.; Monani, U.R. Muscle-Specific Smn Reduction Reveals Motor Neuron-Independent Disease in Spinal Muscular Atrophy Models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hernández, R.; Soler-Botija, C.; Also, E.; Alias, L.; Caselles, L.; Gich, I.; Bernal, S.; Tizzano, E.F. The Developmental Pattern of Myotubes in Spinal Muscular Atrophy Indicates Prenatal Delay of Muscle Maturation. J. Neuropathol. Exp. Neurol. 2009, 68, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Vogelgesang, S.; Armbrust, S.; Graul-Neumann, L.; Fusch, C.; Zerres, K. Digital Necroses and Vascular Thrombosis in Severe Spinal Muscular Atrophy. Muscle Nerve 2010, 42, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.M.; D’ydewalle, C.; Gabbeta, V.; Dakka, A.; Klein, S.K.; Norris, D.A.; Matson, J.; Taylor, S.J.; Zaworski, P.G.; Prior, T.W.; et al. Age-Dependent Smn Expression in Disease-Relevant Tissue and Implications for Sma Treatment. J. Clin. Investig. 2019, 129, 4817–4831. [Google Scholar] [CrossRef] [PubMed]
- Deguise, M.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal Fatty Acid Metabolism Is a Core Component of Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal Fatty Acid Metabolism in Childhood Spinal Muscular Atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Katz, N.; Vite, C.H.; Louboutin, J.-P.; Bote, E.; Yu, H.; Zhu, Y.; Casal, M.L.; Bagel, J.; et al. Evaluation of Intrathecal Routes of Administration for Adeno-Associated Viral Vectors in Large Animals. Hum. Gene Ther. 2018, 29, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Dyer, C.; Goode, T.; Johansson, J.; Bell, P.; Richman, L.; Buza, E.; Wilson, J.M. Translational Feasibility of Lumbar Puncture for Intrathecal Aav Administration. Mol. Ther. - Methods Clin. Dev. 2020, 17, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Van Alstyne, M.; Tattoli, I.; Delestrée, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of Toxic Function by Long-Term Aav9-Mediated Smn Overexpression in the Sensorimotor Circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef]
- Chand, D.; Mohr, F.; McMillan, H.; Tukov, F.F.; Montgomery, K.; Kleyn, A.; Sun, R.; Tauscher-Wisniewski, S.; Kaufmann, P.; Kullak-Ublick, G. Hepatotoxicity Following Administration of Onasemnogene Abeparvovec (Avxs-101) for the Treatment of Spinal Muscular Atrophy. J. Hepatol. 2020, 74, 560–566. [Google Scholar] [CrossRef]
- FDA. Evrysdi Safely and Effectively. Food and Drug Administration, 2020.
- Dangouloff, T.; Servais, L. Clinical Evidence Supporting Early Treatment of Patients with Spinal Muscular Atrophy: Current Perspectives. Ther. Clin. Risk Manag. 2019, ume 15, 1153–1161. [Google Scholar] [CrossRef]
- Lee, B.H.; Collins, E.; Lewis, L.; Guntrum, D.; Eichinger, K.; Voter, K.; Abdel-Hamid, H.Z.; Ciafaloni, E. Combination Therapy with Nusinersen and Avxs-101 in Sma Type 1. Neurology 2019, 93, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Rao, V.K.; Arya, K.; Kuntz, N.L.; DiDonato, C.J.; Napchan-Pomerantz, G.; Agarwal, A.; Stefans, V.; Katsuno, M.; Veerapandiyan, A. Combination Molecular Therapies for Type 1 Spinal Muscular Atrophy. Muscle Nerve 2020, 62, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 1: Recommendations for Diagnosis, Rehabilitation, Orthopedic and Nutritional Care. Neuromuscul Disord 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Caberg, J.-H.; Beckers, P.; Dideberg, V.; di Fiore, S.; Bours, V.; Marie, S.; Dewulf, J.; Marcelis, L.; Deconinck, N.; et al. Three Years Pilot of Spinal Muscular Atrophy Newborn Screening Turned into Official Program in Southern Belgium. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laurent Servais, Manu Vatish "First Uk Pilot Study of Newborn Screening for Spinal Muscular Atrophy Launched in Oxford." University of Oxford, 2022.
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Haque, U. S.; Yokota, T. Enhancing Antisense Oligonucleotide-Based Therapeutic Delivery with Dg9, a Versatile Cell-Penetrating Peptide. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.; Burghes, A.H. A Single Administration of Morpholino Antisense Oligomer Rescues Spinal Muscular Atrophy in Mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A Novel Morpholino Oligomer Targeting Iss-N1 Improves Rescue of Severe Spinal Muscular Atrophy Transgenic Mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Salani, S.; Ruepp, M.D.; Rizzo, F.; Ruggieri, M.; Zanetta, C.; Brajkovic, S.; Moulton, H.M.; Muehlemann, O.; et al. Effect of Combined Systemic and Local Morpholino Treatment on the Spinal Muscular Atrophy Delta7 Mouse Model Phenotype. Clin Ther 2014, 36, 340–356.e5. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic Peptide-Mediated Oligonucleotide Therapy Improves Long-Term Survival in Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed]
- Bersani, M.; Rizzuti, M.; Pagliari, E.; Garbellini, M.; Saccomanno, D.; Moulton, H.M.; Bresolin, N.; Comi, G.P.; Corti, S.; Nizzardo, M. Cell-Penetrating Peptide-Conjugated Morpholino Rescues Sma in a Symptomatic Preclinical Model. Mol. Ther. 2021, 30, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Erkut, E.; Ren, J.; Lim, K.R.Q.; Woo, S.; Hatlevig, S.; Moulton, H.M.; Gosgnach, S.; Greer, J.; Maruyama, R.; et al. Dg9-Conjugated Morpholino Rescues Phenotype in Sma Mice by Reaching the Cns Via a Subcutaneous Administration. J. Clin. Investig. 2023, 8. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genetic basis of SMA; (A) Healthy individuals produce 100% functional SMN protein from their SMN1 genes and only 10% from their SMN2 genes. (B) Patients with SMA do not have SMN1 and rely on the 10% SMN protein produced by SMN2 due to a C to T transition mutation in exon 7. Most of the SMN protein encoded by SMN2 is rapidly degraded.
Figure 1.
Genetic basis of SMA; (A) Healthy individuals produce 100% functional SMN protein from their SMN1 genes and only 10% from their SMN2 genes. (B) Patients with SMA do not have SMN1 and rely on the 10% SMN protein produced by SMN2 due to a C to T transition mutation in exon 7. Most of the SMN protein encoded by SMN2 is rapidly degraded.
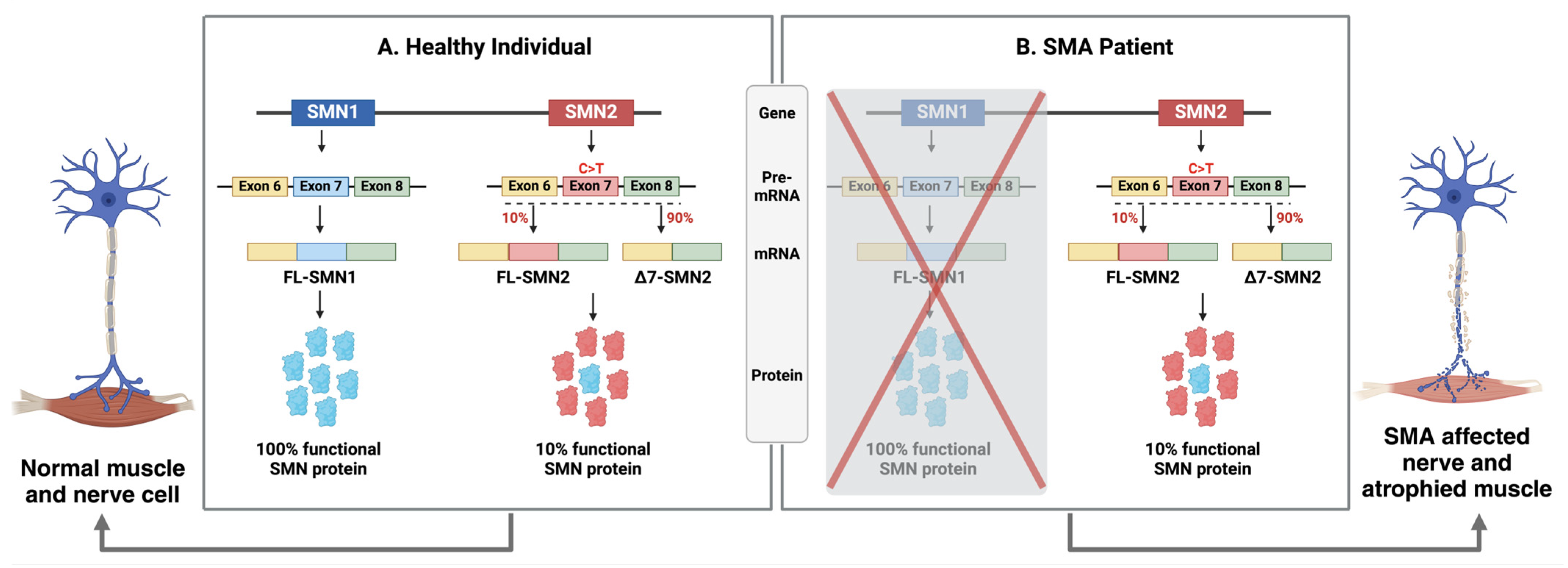
Figure 2.
Schematic diagram of SMN-dependent (FDA approved) and SMN-independent therapies (in development).
Figure 2.
Schematic diagram of SMN-dependent (FDA approved) and SMN-independent therapies (in development).
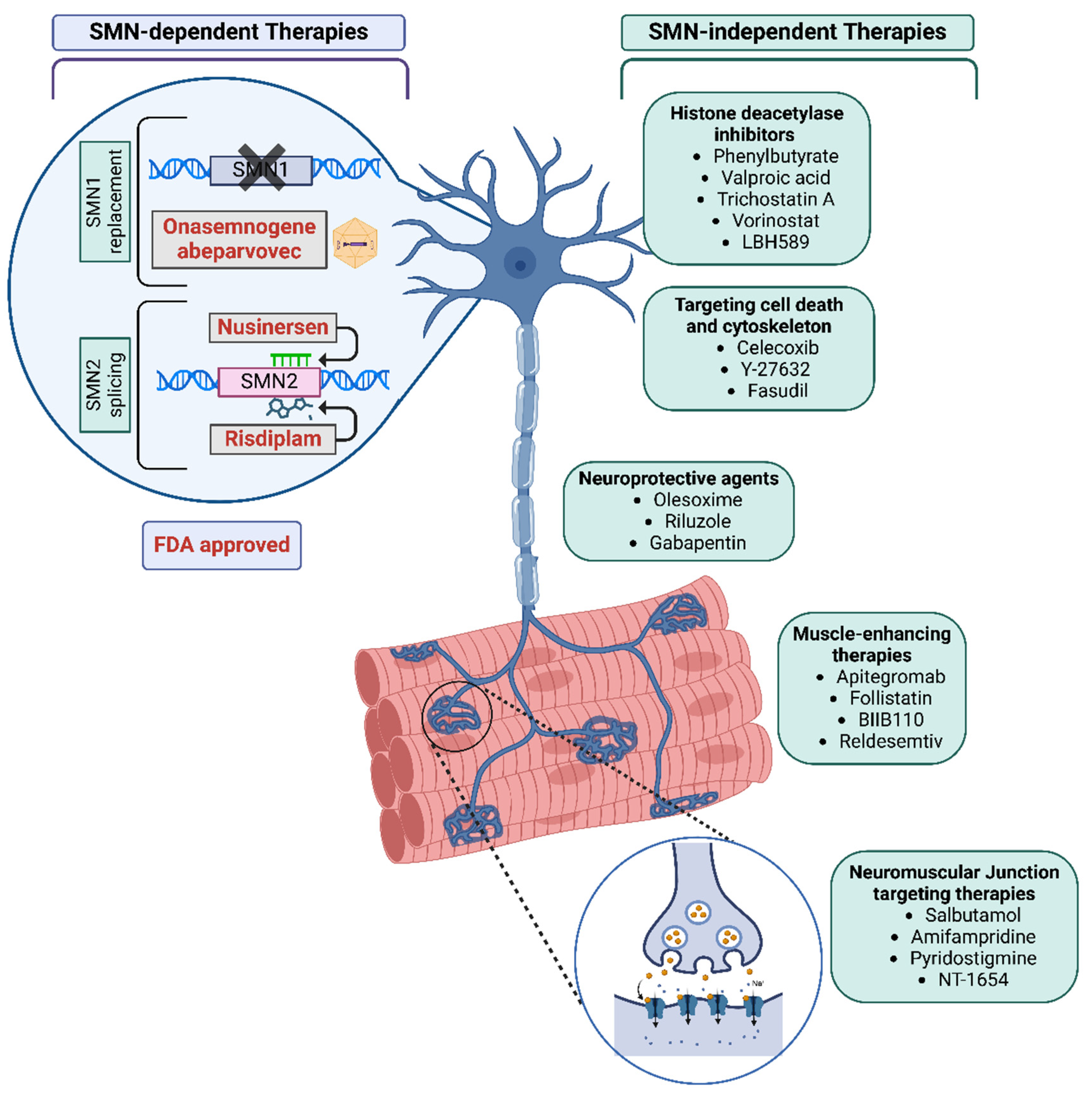
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



