
Preprint
Article
An In-silico Designing of Vaccine against All Serotypes of Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes
Altmetrics
Downloads
215
Views
96
Comments
0
A peer-reviewed article of this preprint also exists.
supplementary.rar (9.50MB )


This version is not peer-reviewed
Submitted:
28 June 2024
Posted:
01 July 2024
You are already at the latest version
Alerts
Abstract
Dengue virus is a global health problem most prevalent in tropical and subtropical regions. However, after the approval of two vaccines, Dengvaxia, TV003/TV005 of Takeda, for its prevention, problems were reported, like enhanced risks of infection or less efficiency of protection. Since then, more struggles have been in demand for the development of better vaccines. Here, we conducted a new design through an in-silico strategy. Initially, epitopes were chosen based on their antigenicity, immunogenicity, and binding affinity with MHC molecule, while excluded with allergenicity, toxicity and potential risk of antibody dependent enhancement. Subsequently, a core antigen was constructed with selected epitopes and linked with distinct adjuvant proteins to achieve three candidate vaccines, PSDV-1~3. PSDV-2 was chosen for further validation based on their advantage in physicochemical and structural properties. With a panel of simulations, this artificial protein showed tight binding with pattern recognition receptors, good stability, robust immune induction, and ultimately was confirmed as a high quality vaccine candidate. The plasmid for its recombinant expression was then designed accordingly. With our new design, one more choice was obtained for the effective protection from Dengue virus. Further experimental validations are still require to confirm it protection capacity and safety.
Keywords:
Subject: Biology and Life Sciences - Immunology and Microbiology
1. Introduction
Dengue Virus (DENV) infection and consequent diseases, including dengue fever (DF), dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS), have emerged as a significant challenge to global health. Over 50 million people are under threat of DENV globally, with around 500,000 cases of hospitalization and approximately 25,000 deaths every year. The prevalence of these diseases imposes considerable socioeconomic and health-related stresses, especially in tropical and subtropical areas [1,2]. Hence, Dengue Virus has been regarded as one of the most dangerous pathogens, as announced by WHO.
Dengue Virus, a member of the Flaviviridae family, is characterized as a virus based on a single-strand, positive-sense RNA genome encapsulated with icosahedral Capsid protein and in the surrounding Envelope protein. With its RNA genome of ~11kb, a single polyprotein is encoded and then hydrolyzed to three structural proteins (C, prM, and E) and seven nonstructural ones (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). These highly immunogenic viral proteins induce strong reactions in the host immune system upon infection. Due to the rapid evolution of its genome and the consequent genetic variations in viral proteins, distinct antigenic properties were widely observed among DENV strains. Accordingly, four unique serotypes DENV-1, 2, 3, and 4, were defined, among which similarity in genomic sequences was revealed as approximately 65% [3]. Due to the complexity of antigenic determinants of dengue serotypes, vaccination against a single serotype failed to confer immune protection but even induced some certain risks in the host upon secondary infection [4], which means the more robust balance of immune across serotypes is necessary for the safety and effectiveness in immunity.
Along with developing the vaccine against DENV, 6 candidates based on distinct technical platforms have been pushed out in different preclinical and clinical evaluation stages, encompassing live attenuated viruses, viral vectors, whole-inactivated viruses, subunit vaccines, and DNA vaccines. Two live-attenuated virus vaccines have been approved recently by the World Health Organization (WHO); one is Dengvaxia, manufactured by Sanofi Pasteur, and the other is TV003/TV005 of Takeda [5]. Both vaccines are tetravalent, trying to induce balanced sera immune against all serotypes. In Dengvaxia, yellow fever vaccine backbone 17D was utilized and integrated with dengue prM and E proteins from serotypes 1~4 and TV003/TV005 vaccine was attenuated primarily with a 30-nt deletion (rΔ30) introduced within the 3’ untranslated region of viral genomes. Besides, the chimera vaccine virus against DENV-2 was further generated through a replacement in DENV-4 rΔ30 backbone of with prM and E from DENV-2 New Guinea C strain. In 2015, Dengvaxia was authorized for clinical application in 20 countries, including the Philippines, Thailand, and Singapore and etc. Disappointedly, full protection hadn’t been achieved and the efficacy was varied among serotypes: 51% for DENV1, 34% for DENV2, 75% for DENV3, and 77% for DENV4 [6]. Even worse, thousands of deaths and other severe hospitalizations were reported among children <9 years old and innocent of dengue infection history. Later, TV003/TV005 was approved in November 2019 [7], but less efficiency than DENV-2 and 3 had also been exhibited [8]. In other words, further studies are still seriously demanded to construct a highly protective and safe vaccine.
The major obstacle to DENV vaccine development was considered as the immune-complexities lying behind four serotypes. Due to the genomic similarity among serotypes, cross-reactive antibodies against the E protein are dominantly called up in secondary infections [9]. Unfortunately, these antibodies are highly potent in binding with E antigen and consequently facilitating viral entry, rather than protective in a cross-serotype manner. This effect was defined as Antibody-Dependent Enhancement (ADE) and frequently leads to fatal symptoms. In clinics, serious conditions such as dengue hemorrhagic fever or dengue shock syndrome were constantly observed in patients who experienced sequential infection with distinct serotypes of DENV [10,11]. For a safe and fully protective vaccine against DENV, risky epitopes associated with ADE effects should be carefully identified and eliminated. Meanwhile, the induction of antibodies should be kept at a similar level among the four serotypes to reach the immune balance.
Based on the rapid progression of immunoinformatics approaches, the computational designing of vaccines become highly feasible and efficient. In-silico prediction of epitopes will highlight the activator of B cells, cytotoxic and helper T lymphocytes (CTLs and HTLs), and clarify the inducer of allergy and other side-effects, which will significantly benefit the efficiency and safety of vaccination. Some new proposals have been published for vaccine construction of DENV in-silico. However, there are still some hidden problems in these designs. First of all, the worldwide accumulation of the Dengue viral genome contains intensive complexities and variations, which is always a tough job to cover within the limited number of epitopes and restrain the molecular size at a reasonable level for the designed antigen. Secondly, to balance the antibody induction for four serotypes and avoid ADE effects, tetravalent composition was still employed, even in the designs state-of-art. However, due to this formulation, difficulties in manufacturing and validation will increase, while efficacy will be decreased in lateral applications. The third point, which should be considered seriously, is that no special attention was paid to exclude the epitopes related to ADE risks in the published designs.
Therefore, we carried out the current study with the following measures: 1) obtaining the conserved fragments in viral polyprotein as the bases of epitope selection. 2) Exclusion of potential ADE-associated epitopes based on literature collection. 3) Combining serotype-specific B-cell epitopes from E protein and the pan-serotype T-cell epitopes from other proteins NS1, NS3, NS5, and Capsid. 4) Integrating all epitopes and adjuvant proteins into a single molecule of reasonable size rather than four. Prospectively, a strategy based on these measures will provide advantages to the new design of the dengue vaccine, including effectiveness, safety, and feasibility.
2. Materials and Methods
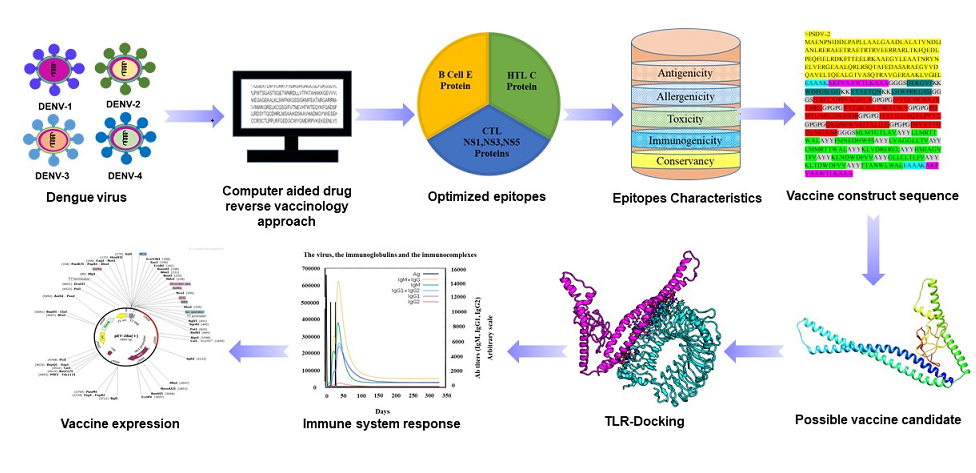
The entire procedure of designing was presented in Figure 1.Web addresses of databases and platforms utilized in this study were listed as Table Supplementary (S) 1. All supplementary(S) tables were included in Supplementary File 1 as well as all supplementary figures in Supplementary File 2.
2.1. Retrieval and Analysis of Full-Length Dengue Polyprotein Sequences
To initiate designing, full-length sequences of DENV-1~4 polyproteins isolated from 10 representative countries and four reference strains (Table S2, S3, S4) were retrieved from NCBI (National Center for Biotechnology Information) and VIPR (Virus Pathogen Database and Analysis Resource). Conserved fragments were generated within serotypes based on an alignment of multiple sequences (MSA). Primary alignment was performed with MUSCLE [12] and verified with CLUSTALW version 2.0 [13] and MAFFT [14]. Conserved fragments were then harvested with the assistance of BioEdit 7.2 [15] and listed in Table S5. Phylogenetic trees were constructed one by one within the same serotypes. The molecular Evolutionary Genetics Analysis (MEGA) 4 software package elucidated the evolutionary distance among DENV strains [16]. The phylogenetic tree was then constructed with the neighbor-joining method [17] and visualized in the platform Interactive Tree Of Life (iTOL) [18].
2.2. Step-by-Step Selection of Epitopes and Estimation of Coverage in Population
Continuous B cell epitopes were predicted in conserved sequences of DENV-1~4Polyproteins with the IEDB database through the Bepipred2.0 method [19]. A hidden Markov model was used in the prediction, with 0.5 as the threshold. Prediction for CTLs (MHC I) and HTLs (MHC II) was carried out respectively in platform NetMHC 4.0 [20] and NetMHC MHC class II 2.3 [21]. The length of epitopes was defined as 6-12aa for B cells, 9aa for CTLs and 15aa for HTLs.
Stepwise selections were conducted for all three types of epitopes with immunoinformatic platforms, including VaxiJen [22], Allertop v.2.0 [23], Toxinpred [24], and IEDB [25]. Inclusion was conducted based on antigenicity and intra-serotype conservancy to collect B cell epitopes, while exclusion was conducted based on allergenicity, toxicity, and cross-serotype conservancy. In a further step, clearance of potentially ADE-related B cell epitopes was conducted based on the collection of literature (Supplementary Table 11). For CTLs and HTLs, epitopes were primarily selected with binding affinity with MHC I/II receptors. Thresholds for strong binders (SB) and weak binders (WB) of CTL epitopes were within the top-ranked <0.5% and <2% and within the top <1% and < 5% for HTL epitopes. The following selection was performed according to their antigenicity, immunogenicity, intra, and cross-conservancy, without toxicity or allergenicity.
The utility of IEDB population coverage was employed to ensure the population coverage of selected epitopes. MHC I and II epitopes were selected and submitted to the analysis, and all global regions were incorporated [26].
2.3. Formulation and Evaluation of Vaccine Candidates
To formulate candidate vaccine molecules, selected epitopes were grouped for B cells, CTLs, and HTLs, respectively, and assembled with linker sequences and adjuvant protein domains [27]. Human beta-defensin 3 and Heparin-binding hemagglutinin (HBHA) of Mycobacterium sp were used as adjuvants to enhance vaccination [28,29]. The pan-HLA DR binding epitopes sequence (PADRE, 13aa) was incorporated into the vaccine to reinforce immunogenicity further and activate CD4+helper T cells [27,30]. A panel of linker peptides was employed to achieve the integral vaccine candidates from selected immune elements. Organization of B cell epitopes was mediated with a flexible linker KK [31], CTL epitopes with another flexible linker AAY, HTL epitopes with a glycine-proline-rich linker GPGPG, while GGGS linker [27] was utilized for the connection between distinct groups of epitopes. Besides, the N- or C-terminus of grouped epitopes was conjugated with adjuvant proteins through the rigid linker EAAAK to obtain the full antigen molecule.
2.4. Predictions and Validation of Molecular Structure for Vaccine Candidates
A PSIPRED online server was hired to predict the secondary structure of vaccine protein [37]. The tertiary structure was predicted with a protein sequence submitted to the PHYRE-2 protein folding recognition server [38]. The best model of 3D structures were superimposed using UCSF Chimera to present conformational changes after prediction [39]. Ramachandran plot was generated through the online server PROCHECK [40], while ProSA-web [41] was used to validate the 3D structure of the modeled vaccine.
2.5. Molecular Docking and Dynamics of the Vaccine-Immune Receptor Complexes
To evaluate the binding of vaccine protein to receptors of pattern recognition and antigen presentation, Hawk Dock was used for the molecular docking and binding energy calculation of antigen-immune receptor complexes [26]. The resulted models were visualized and evaluated using the UCSF Chimera tool [42]. Prevalent panel of HLA alleles and TLRs were examined, with their protein structures downloaded from Protein Databank (PDB) with the PDB IDs listed in Table S12 [26,27]. With PDBSum, amino acid residues were generated, non-covalent interactions (Hydrogen bonds, Salt bridge interactions) and interface areas were then analyzed [43].
Molecular dynamics simulations were then conducted for top qualified complexes of vaccine ensemble with TLR2 and TLR4 on the AMBER18 platform [44] with protein and water box margins maintained as 12-angstrom padding. TLR settings were configured with the ff14SB force field [45,46], while vaccine molecules were placed in a TIP3P water box using the GAFF force field [47]. Simulation steps included 500 for hydrogen atoms, 300 for non-heavy atoms, 1000 for carbon alpha atoms, and 1000 for the solvent box, with the system neutralized by counter ions. The minimization process employed the steepest descent or conjugate gradient approach, with transitions every 1000 cycles up to a maximum of 5000 cycles, aiming to eliminate collisions in the complexes' structures. The systems were heated over 250 ps to reach thermal equilibrium at 300 K, followed by a 100ns production cycle. The SHAKE algorithm [48] ensured the rigidity of covalently bound hydrogen atoms with periodic boundary conditions in the canonical ensemble. Temperature was maintained at 300 K using the Langevin thermostat, with a 10 Å threshold for non-bounded interactions and Ewald simulations for long-range interactions. The CPPTRAJ module [49] analyzed statistical parameters, and Pymol [50] visualized the results. Studies focused on Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Radius of Gyration (Rg), and hydrogen bonds of the vaccine-receptor complex.
2.6. Immune Simulation of Selected Candidate Vaccine
The immune simulations were conducted in the C-ImmSim server to reflect the vaccine administration and the consequent immune responses [51]. Three doses with 2-week intervals were set for simulation. Since 1 time step was equivalent to 8 hours in real life, time points of dosing were set to steps 1, 84, and 168 [52]. Other parameters were set as default.
2.7. Codon Optimization and In-Silico Cloning of Selected Candidate Vaccine
The peptide sequence of the candidate vaccine was submitted to the JCat server for codon optimization [53], choosing E. coli K12 as the preferred host organism. This optimization process adhered to specific guidelines that avoided: (1) restriction enzyme cleavage sites, (2) prokaryotic ribosome binding sites, and (3) transcription rho-independent termination sites. The optimized sequence was assessed for a codon adaptation index (CAI) of over 0.8 and a GC content of 30 to 70% [53]. Subsequently, the modified nucleotide sequence underwent an in-silico cloning step to the pET28a (+) expression vector. This procedure was facilitated by SnapGene version 4.2 [54].
3. Results
3.1. Conserved Fragments Were Extracted from DENV Polyprotein Sequences Collected Worldwide
Based on the epidemiology, geographical location, and data availability, 10 representative countries were selected as the source of viral sequences, including Bangladesh, Brazil, China, India, Pakistan, Philippines, Singapore, Thailand, USA, and Vietnam. A total of 2929 full-length polyprotein sequences of DENV deposited from these source countries from the years 1960 to 2023 were downloaded and placed in Table S4. In brief, the Number of sequences for DENV-1/2/3/4 was 1355, 1031, 400, and 143, respectively. Detail numbers of each serotype in individual source countries were listed in Table S2.
After aligning the sequences, phylogenetic trees were constructed for each serotype with an overall mean distance of 0.02 (Figure S1). As visualized in the trees, sequences were tightly clustered within a serotype, while the sequences isolated from the same region/country resembled more closely in a corresponding clad, which suggested certain common ancestries of them. Based on the similarity of DENV polyprotein sequences, conserved fragments were then generated for each serotype and subjected to epitope screening (Table S5).
3.2. Effective and Non-Risky Epitopes Were Selected Stepwise from Conserved Fragments of DENV Polyprotein for B-Cells, CTLs, and HTLs
For B cell epitopes, the initial list was obtained with 75%sensitivity and 50% specificity, containing 113 epitopes for DENV-1 and 92, 92, and 85 epitopes for DENV-2, 3, 4, respectively (Table S6 and Figure S2). After being shortlisted with antigenicity score ≥ 0.4, non-allergenic, non-toxic, intra and cross conservancy, the list of epitopes was shortened significantly, as 4 for DENV-1and 3 each for DENV-2, 3, and 4 (Tables S7-10). Further exclusion of ADE-associated epitopes was executed according to the previous reports [55,56] about the ADE-corresponding regions in protein E (literature listed in Table S11), which mainly referred to Domains I and II. Finally, one epitope was selected for four serotypes (Table 1). As mapped, selected B cell epitopes were all located on the surface of the Envelope Protein (Figure S2), among which the ones for DENV-1~3 were contained in Domain III and the one for DENV-4 was in Domain II.
For either CTLs or HTLs, long lists containing more than 2000 epitopes were primarily generated for each serotype. The predicted epitopes were then filtered with strong affinity (IC50≤ 50 nM), and 30~60 epitopes were then actually collected for both classes in each serotype (Tables S13-17 for MHC-I, 18-23 for MHC-II) and Figures S3, 4). The aggregative ranking was then performed, adding up the ranks of binding affinity, antigenicity, and immunogenicity for MHC I, while binding affinity plus antigenicity for MHC II. After that, exclusion was carried out based on allergenicity and toxicity. Ultimately, 11 epitopes for MHC I were selected in nonstructural proteins NS1, 3, and 5, while 7 epitopes for MHC II were obtained from structural protein C (Table 2 and Table 3 and Tables S17, 23). Among the epitopes selected for T cells, the cross-serotype conservancy was seen very little in the MHC I group but dominant in the MHC II group (Table 2 and Table 3). Coverage in population was estimated to be 62.26% with selected HLA alleles (Figure S5).
3.3. Three Candidate Vaccines Composed of Selected Epitopes Were Predicted as Stable, Water Soluble, and Antigenic
As the core antigen of candidate vaccines, the selected epitopes (4 for B cells, 11 for CTLs and 7 for HTLs) were firstly organized with peptide linkers and flanked with two copies of PADRE peptide. Three candidate vaccine molecules, termed PSDV1~3, were further constructed through conjugation of two adjuvant proteins, HBHA and/or Beta-defensin, with core antigen as follows (Figure 2):
PSDV1: HBHA at N-terminus and Beta-Defensin at C-terminus.
PSDV2: HBHA at N-terminus.
PSDV3: Beta-Defensin at N-terminus.
For the size of PSDV1~3, peptide length was calculated as 577aa, 508aa and 394aa (Line 1 in Table 4), while molecular weight was 63.4kDa, 55.3kDa and 42.7kDa (Line 2 in Table 4).
Their stability was estimated using the Instability Index, Aliphatic Index, and half-life integrally. All values of the Instability Index fell under threshold 40, listed as 25.25, 24.28, and 20.52 for PSDV1~3 (Line 6 in Table 4). Their Aliphatic Index was calculated as 86.55, 88.33, and 85.056 (Line 7 in Table 4). The intracellular half-life was estimated to be more than 30 hours in mammalian cells for all three molecules (Line 5 in Table 4). A good stability for PSDV1~3 molecules was implied in vitro and in vivo.
Good solubility in water was suggested to PSDV1~3 with the Solubility Index about 0.79, 0.76, and 0.56 (Line 12 in Table 4). With GRAVY (Grand average of hydropathicity), PSDV1 was predicted hydrophilic due to a negative value of -0.027. In contrast, PSDV3 is hydrophobic with a positive value of 0.151, and PSDV2 scored 0.018 nearly neutrally (Line 9-11 in Table 4). Based on theoretical pI, PSDV1 and 3 were weakly alkaline proteins, and PSDV2 was weakly acidic (Line 3 in Table 4).
Ultimately, all three molecules were assessed as antigenic, non-allergenic, and non-toxic (Line 6 in Table 4) and primarily suggested as qualified for further evaluation.
3.4. PSDV2 Was Selected Based on Structural Advantages and Showed Tight Interaction with TLRs and HLAs
As analyzed, PSDV1~3 proteins were composed of coils, helixes, and β-strands in various percentages. In detail, PSDV1 was composed of 31.71% coils, 56.67% helixes, and 11.61% β-strands; PSDV2 contained 33.46% coils, 55.70 helixes and 10.82% β-strands, while 29.08% coils, 37.81% of helixes and 23.10% of β-strands were observed in PSDV3 (Figure S6 and Figure 3). Modelling of PSDV1~3 tertiary structures was then performed in the Phyre2 server with a template-based algorithm, in which the templates 5d4w, 6tpi, and 1kj6 were employed correspondingly. The confidence levels showed 87.9%, 97.4%, and 99.9%; the coverage was 47%, 59%, and 11% for PSDV1~3 (Figure S7 and Figure 3). Based on the tertiary structure models of candidate vaccines, Ramachandran plots were generated in the PROCHECK server. They exhibited an overall quality score of 97.6% for PSDV2 (Figure S8 and Figure 3), which means 92.0% of amino acid residues were located in the most favored regions and 5.6% in allowed regions. PSDV1 and 3 were scored as 98.3% (85.2 %in favored region plus 13.1%in allowed region) and 89.5% (76.3%in favored region plus13.2%in allowed region), respectively. PSDV2 had the highest overall quality score and favored region among the 3 candidates (Figure S8 and Figure 3). A similar analysis was also carried out with ProSA, and the corresponding Z-scores for PSDV1, 2, and 3 were presented as 3.85, -5.19, and -4.75 (Figures S8 and Figure 3). Undoubtedly, PSDV2 molecule, rather than PSDV1 or 3, achieved the best qualified tertiary structure (Figure 3) and was selected for further evaluation accordingly.
Molecular Docking of PSDV2 was performed with 4 HLA alleles (HLA-DR B101:01, HLA-DRB104:01, HLA-A0201 and HLA-DRB10701) or 2 TLRs (TLR2 and TLR4) utilizing Hawk Dock (Figure S9 and Figure 4). For each complex of vaccine-receptor, ten conformations were generated in docking, and the one with the lowest binding energy was chosen for the following analysis (Table S24 and Figure 4). The binding energy for these docking complexes ranged from 5000~7000 kcal/mol, while the values for TLR2 and 4 were -7330.87 and -5386.23 kcal/mol. Figure 4 and Table S25, Table 5 show that the interaction between PSDV2 and TLR2 was mediated with 4 hydrogen bonds and 3 salt bridges. Their binding interface was formed with 20 residues in PSDV2 (1023Å2) and 21 residues in TLR2 (1066Å2). In the binding of PSDV2 with TLR4, 5 hydrogen bonds, and 3 salt bridges were formed, while 13 residues in the vaccine (893Å2) and 14 in receptor (877Å2) were involved in the shaping of the interface.
The conformational stability of PSDV2-TLR complexes was then confirmed in molecular dynamic simulation with 100ns interval (Figure 5). RMSM value always stayed within the allowed range of 4Å and reached equilibrium within 40ns. RMSF value was also kept stable in the 4Å allowed range. Rg value fell in the region close to 37Å for TLR2 or 34Å for TLR4, respectively. The number of hydrogen bonds inside TLR was also stable in the range of 20-25. Hence, the stability of the interaction between PSDV2 and TLR2/4 receptors was indicated well, which ensured the recognition of PSDV2 by the host immune system.
3.5. Robust Responses Were Induced for Both Innate and Adaptive Immunity upon Simulation of PSDV2 Candidate Vaccine
To evaluate the effectiveness of the PSDV2 candidate vaccine, immunization was simulated in the C-ImmSim platform with 3 doses and 2-week intervals.
For innate immunity, the status of DCs, macrophages, and NK cells was presented in Figure 6, and activation of macrophages was clearly observed after immunization. Production of cytokines was also robustly induced with PSDV2, with IFN-γ and IL-2 ascending rapidly as the most significant ones in panel. A remarkably response of host was indicated with the level of IFN-γ exceeding 400,000ng/ml.
Activation of the adaptive immune system is also revealed upon PSDV2 administration virtually (Figure 7). After 3 doses, B cells were strongly boosted and persisted at a high level for almost 1 year, especially for two subpopulations, IgM homotypic B cells and Memory B cells. A significant boost of memory helper T cell population was indicated and lasted over 350 days, either. The generation of antibodies was stimulated post-vaccination along with the activation of B cells. The titer of IgM+IgG reached a peak higher than 14000, a range comparable to other reports of in-silico vaccine design. The major compositions of antibodies were shown as IgM and then IgG1, with IgG2 as minority. The antibodies, total B cells, and total HTLs reached their peaks at similar time points no later than 50 days post 1st immunization and slightly earlier than the peak of CTLs.
In one word, inoculation of PSDV2 candidate vaccine will stimulate the immunity systematically, through which a powerful protection against DENVs and long-term immune memory of T and B cells will then be established in speculation.
3.6. Codon Optimization and In-Silico Cloning of the Designed Vaccines
For an efficient preparation of the PSDV2 vaccine, the E. coli system was adopted for recombinant expression. A DNA sequence of 1524bps was generated based on the designed peptide sequence. Its codon usage was then optimized in JCAT (Java Codon Adaptation Tool) with a codon adaptation index of 0.97 to meet the cellular machinery of the K12 strain. GC content was approximately 54.65% in the optimized sequence. After flanking with restriction sites SgrAI and PstI at 5’ and 3’ ends, this sequence was inserted into the pET28a (+) vector in-silico to form the expression plasmid of 6893 bps (Figure 8).
4. Discussion
Dengue virus has been investigated for over 80 years [55]; however, no specific treatment exists for this pathogen. Unfortunately, its vaccine development is also a rugged long journey. In past decades, two types of live attenuated virus vaccines were approved in clinics out of all other trials, including inactivated vaccine [55], virus-like particle-based vaccine [56], subunit vaccine [57], etc. Dengvaxia [58] and TV-003/TV005 [7] were designed as tetravalent, aiming for effective and balanced immune protection. However, disappointingly, clinical reports showed insufficient induction of humoral immunity against DENV-2 with both vaccines, and even some fatal symptoms were observed after the immunization of Dengvaxia [58]. As living vaccine, potential risks of reverse to wild type or unexpected recombination in vivo are always intrinsic and unavoidable. Obviously, pathogenic elements are always fully contained in living vaccines since a full set of intact viral proteins were employed, especially the epitopes related to ADE effects. Since then, de novo designing of a DENV vaccine has been urgently needed. It is proposed to achieve efficient, robust, and long-lasting adaptive immunity and minimize potential risks.
For this purpose, the computer-assisted design of multi-epitope vaccines has stood out as a promising pathway compared with the conventional strategies based on wet experiments. Since RinoRappuoli proposed the first reverse vaccinological immunoinformatic design in 2000 [59] and applied in Serogroup B meningococcus [60], this approach has been employed for a wide range of diseases and pathogens, including Chikungunya virus [61], Saint Louis encephalitis virus [62], SARS-CoV-2 virus [63], Dengue virus [64], Theileriaparasite [65] and so on. Several proposals for DENV-candidate vaccines have been published since 2009. The effective epitopes were selected and organized in those designs, while allergic or toxic ones were predicted and eliminated. The first in-silico vaccine of DENV was designed based on the E protein of DENV-2 and the backbone of DENV-3 but declared to target all four serotypes implausibly [66]. However, the immune simulation was not performed. After that, a panel of similar publications emerged between 2016 and 2023.
Distinguished from earlier studies, the initial step of our design is the extraction of conserved fragments from full-length sequences of DENV polyproteins collected from 10 representative countries worldwide. This action aims to simplify the following study under the dramatic diversity of DENV genomes, which corresponds to four serotypes, hundreds of genotypes, and thousands of clinical isolates. However, the former designs were based on selected representative sequences for full-length polyproteins or individual viral proteins. Starting from the conserved fragments will facilitate broad-spectrum protection with few well-chosen epitopes.
The second facet of our strategy is that all 10 viral proteins were considered for epitope screening. In two publications of the in-silico designs [26,27], a full set of DENV proteins was also employed, with which full-range induction of immunity was indicated in computational simulations. With experimental validations, the E III domain contains the critical determinants of neutralization antibodies [67]; CD8+ T cell (CTL) epitopes are preferentially located in NS1, NS3, and NS5; and the epitopes for CD4+ T cells (HTL) are dominantly found in structural protein C, as well as proteins E and NS1, which also actively contribute to B cell induction [68]. In our design, the final selection of epitopes was also conducted mostly in this range to obtain more effective elements for immune induction.
The resultant epitopes included 4 for B cells, 11 for CD8+ CTLs, and 7 for CD4+ HTLs. All these epitopes are ensured as antigenic, immunogenic, non-allergenic, and non-toxic through a stepwise screening. Further clearance of potentially risky components was performed in candidate epitopes for B-cell serotype by serotype. Once a certain epitope showed cross-serotype conservancy or was located in an ADE suspecting region, it would be eliminated. This strategy minimized ADE potential based on current knowledge, and the interference on immune balance was ultimately avoided. For T-cell epitopes, binding strength to MHC I or II receptors was considered in additional list shortening. Besides, epitopes with higher cross-serotype conservancy were preferred for cytotoxic and helper T cells, which will benefit cross-serotype immunoprotection. With these strategies, a tetravalent core antigen was constructed as a single molecule rather than four with a reasonable size and, more importantly, prospectively covering all four serotypes in the humeral and cellular immune systems.
Three candidate vaccine molecules, PSDV-1, 2, and 3, were then produced by linking core antigens with distinct adjuvant protein domains, including PADRE, HBHA, and beta-Defensin, through different strategy of assembly. The PSDV-2, which was flanked with PADRE and contained HBHA at N-terminus, was selected for further evaluation with consideration of its structural advantages. In simulated molecular docking, stable complexes were formed with pattern recognition receptors TLR2, TLR4 and HLAs. This means the designed vaccine candidate will be robustly recognized as a foreign antigen in hosts, which is so important to initiate following steps of immune reactions.
Consequently, as showed with in-silico simulation, a full-spectrum of immune protection will be achieved. On the side of innate immunity, secretion of cytokines and promotion of NK, DC, and Macrophage cells was predicted, which is important in initiating and regulating adaptive immunity. Subsequently, the generation of antibodies, and activation of B cells, HTLs, and CTLs, were also exhibited after three doses of immunization. The titer of antibody reached 1:15000, which was similar to the level achieved in former designs. Meanwhile, Long-lasting immune memories were established in both B and helper T cells. At last, population coverage of this vaccine reached 62.26% worldwide in estimation, qualified in comparison with other studies.
In brief, a candidate vaccine potentially protective against all four serotypes of DENV was designed in the current study. The vaccine molecule was reasonably sized, with good stability and other physicochemical properties. This candidate vaccine was prospectively able to provide robust immune protection. Our design strategy contained two differential points distinct from earlier publications: 1) Epitope screening was performed on the conserved fragments summarized from a worldwide collection of viral protein sequences; 2) Clearance of risky elements was conducted, especially to remove ADE-associated B-cell epitopes. With these considerations, both efficacy and safety were considered for the candidate vaccine against DENV.
5. Conclusions
With the efficient and feasible immunoinformatics approach, our design of a multi-epitope vaccine against DENV was carried out. Through a step-by-step screening and careful removal of risky epitopes, a PSDV-2 candidate vaccine was created. The Vaccine molecule contained E protein (Domain II and III) targeted B-cell epitopes, NS1, 3, and 5 targeted CTLs epitopes, and C protein targeted HTLs epitopes, in conjugation with HBHA and PADRE adjuvant. With a panel of simulation, PSDV-2 showed good properties in molecular structure, robustly recognized with TLRs and HLAs, and would induce robust immune protection in more than 60% of populations worldwide, which strongly suggested our current design as an applicable and feasible vaccine candidate to protect from DENV. Furthermore, further lab tests in vitro and in vivo are necessary to validate its effectiveness and safety.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, L.T., and H.U.; methodology, H.U.; software, H.U.; validation, H.U., L.T. And F.Y.; formal analysis, H.U.; investigation, L.T. And F.Y.; resources, S.U and L.J.; data curation, H.U.; writing—original draft preparation, H.U.; writing—review and editing, L.T and F.Y.; visualization, Yang Fan.; supervision, L.T.; project administration, X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful for One Hundred Talented Youth Program of the Chinese Academy of Sciences to Dr. Lei Tan.
Institutional Review Board Statement
Not applicable”
Informed Consent Statement
N/A
Data Availability Statement
All the data available in the manuscript and supplementary files
Acknowledgments
We are grateful for One Hundred Talented Youth Program of the Chinese Academy of Sciences to Dr. Lei Tan.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Perera, R. and R.J. Kuhn, Structural proteomics of dengue virus. Current opinion in microbiology, 2008. 11(4): p. 369-377. [CrossRef]
- Pinheiro-Michelsen, J.R., et al., Anti-dengue vaccines: from development to clinical trials. Frontiers in Immunology, 2020. 11: p. 1252. [CrossRef]
- Kraemer, M.U., et al., The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. elife, 2015. 4: p. e08347.
- Gubler, D.J. and G.G. Clark, Dengue/dengue hemorrhagic fever: the emergence of a global health problem. Emerging infectious diseases, 1995. 1(2): p. 55.
- Thomas, R., et al., Associations of human leukocyte antigen with neutralizing antibody titers in a tetravalent dengue vaccine phase 2 efficacy trial in Thailand. Human Immunology, 2022. 83(1): p. 53-60. [CrossRef]
- Thomas, S.J. and I.-K. Yoon, A review of Dengvaxia®: development to deployment. Human vaccines & immunotherapeutics, 2019. 15(10): p. 2295-2314. [CrossRef]
- Chen, Y., et al., Progress and development of three types of live attenuated vaccines for dengue fever. Highlights in Science, Engineering and Technology, 2022. 8: p. 497-504. [CrossRef]
- Wilder-Smith, A., Dengue vaccine development by the year 2020: challenges and prospects. Current Opinion in Virology, 2020. 43: p. 71-78. [CrossRef]
- Harapan, H., et al., Dengue: a minireview. Viruses, 2020. 12(8): p. 829.
- Teo, A., et al., Understanding antibody-dependent enhancement in dengue: Are afucosylated IgG1s a concern? PLoS pathogens, 2023. 19(3): p. e1011223.
- Chan, Y., et al., Enhancement of tetravalent immune responses to highly conserved epitopes of a dengue peptide vaccine conjugated to polystyrene nanoparticles. Vaccines, 2020. 8(3): p. 417. [CrossRef]
- Edgar, R.C., MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research, 2004. 32(5): p. 1792-1797. [CrossRef]
- Thompson, J.D., T.J. Gibson, and D.G. Higgins, Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics, 2003(1): p. 2.3. 1-2.3. 22. [CrossRef]
- Edgar, R.C. and S. Batzoglou, Multiple sequence alignment. Current opinion in structural biology, 2006. 16(3): p. 368-373.
- Oliver, T., et al., Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics, 2005. 21(16): p. 3431-3432. [CrossRef]
- Kumar, S., et al., MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in bioinformatics, 2008. 9(4): p. 299-306. [CrossRef]
- Saitou, N. and M. Nei, The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution, 1987. 4(4): p. 406-425. [CrossRef]
- Letunic, I. and P. Bork, Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics, 2007. 23(1): p. 127-128. [CrossRef]
- Jespersen, M.C., et al., BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic acids research, 2017. 45(W1): p. W24-W29. [CrossRef]
- Andreatta, M. and M. Nielsen, Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics, 2016. 32(4): p. 511-517. [CrossRef]
- Nielsen, M., C. Lundegaard, and O. Lund, Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC bioinformatics, 2007. 8(1): p. 1-12. [CrossRef]
- Doytchinova, I.A. and D.R. Flower, VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics, 2007. 8: p. 1-7. [CrossRef]
- Dimitrov, I., et al., AllerTOP v. 2—a server for in silico prediction of allergens. Journal of molecular modeling, 2014. 20: p. 1-6.
- Gupta, S., et al., In silico approach for predicting toxicity of peptides and proteins. PloS one, 2013. 8(9): p. e73957. [CrossRef]
- Calis, J.J., et al., Properties of MHC class I presented peptides that enhance immunogenicity. PLoS computational biology, 2013. 9(10): p. e1003266. [CrossRef]
- Alsaiari, A.A., et al., Rational design of multi-epitope-based vaccine by exploring all dengue virus serotypes proteome: an immunoinformatic approach. Immunologic Research, 2023: p. 1-18. [CrossRef]
- Fadaka, A.O., et al., Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Scientific reports, 2021. 11(1): p. 19707. [CrossRef]
- Li, W., et al., Peptide vaccine: progress and challenges. Vaccines, 2014. 2(3): p. 515-536. [CrossRef]
- Jalal, K., et al., Pan-genome reverse vaccinology approach for the design of multi-epitope vaccine construct against Escherichia albertii. International Journal of Molecular Sciences, 2021. 22(23): p. 12814. [CrossRef]
- Ghaffari-Nazari, H., et al., Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T help in BALB/c mice. PloS one, 2015. 10(11): p. e0142563. [CrossRef]
- Dhanushkumar, T., et al., Structural immunoinformatics approach for rational design of a multi-epitope vaccine against triple negative breast cancer. International Journal of Biological Macromolecules, 2023. 243: p. 125209. [CrossRef]
- Doytchinova, I.A. and D.R. Flower, VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics, 2007. 8(1): p. 1-7. [CrossRef]
- Dimitrov, I., D.R. Flower, and I. Doytchinova. AllerTOP-a server for in silico prediction of allergens. in BMC bioinformatics. 2013. BioMed Central.
- Hebditch, M., et al., Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics, 2017. 33(19): p. 3098-3100. [CrossRef]
- Magnan, C.N., A. Randall, and P. Baldi, SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics, 2009. 25(17): p. 2200-2207. [CrossRef]
- Gasteiger, E., et al., Protein identification and analysis tools on the ExPASy server. 2005: Springer.
- Kaur, H. and G.P.S. Raghava, Prediction of β-turns in proteins from multiple alignment using neural network. Protein science, 2003. 12(3): p. 627-634.
- Kelley, L.A., et al., The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols, 2015. 10(6): p. 845-858. [CrossRef]
- Sabetian, S., et al., Exploring dengue proteome to design an effective epitope-based vaccine against dengue virus. Journal of biomolecular structure and dynamics, 2019. 37(10): p. 2546-2563. [CrossRef]
- Sasisekharan, R.G.R.C., Stereochemistry of polypeptide chain configurations J Mol Biol 79599. Ramachandran GN, Ramakrishnan C, and Sasisekharan (1963). Stereochemistry of polypeptide chain configurations. J Mol Biol, 1963. 7: p. 95-99.
- Wiederstein, M. and M.J. Sippl, ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic acids research, 2007. 35(suppl_2): p. W407-W410.
- Pettersen, E.F., et al., UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry, 2004. 25(13): p. 1605-1612. [CrossRef]
- Lovell, S.C., et al., Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics, 2003. 50(3): p. 437-450.
- Case, D., D. Pearlman, and J. Caldwell, Amber 18.(2018) University of California. San Francisco.
- Case, D., et al., The FF14SB force field. Amber, 2014. 14: p. 29-31.
- Maier, J.A., et al., ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of chemical theory and computation, 2015. 11(8): p. 3696-3713. [CrossRef]
- Wang, J., et al., Development and testing of a general amber force field. Journal of computational chemistry, 2004. 25(9): p. 1157-1174. [CrossRef]
- Kräutler, V., W.F. Van Gunsteren, and P.H. Hünenberger, A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of computational chemistry, 2001. 22(5): p. 501-508. [CrossRef]
- Roe, D.R. and T.E. Cheatham III, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. Journal of chemical theory and computation, 2013. 9(7): p. 3084-3095. [CrossRef]
- DeLano, W.L., Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr, 2002. 40(1): p. 82-92.
- Rapin, N., et al., Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PloS one, 2010. 5(4): p. e9862. [CrossRef]
- Marques, P.H., et al., Design of a Multi-Epitope Vaccine against Histoplasma capsulatum through Immunoinformatics Approaches. Journal of Fungi, 2024. 10(1): p. 43. [CrossRef]
- Grote, A., et al., JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research, 2005. 33(suppl_2): p. W526-W531. [CrossRef]
- Goldberg, M.F., et al., Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity, 2018. 49(6): p. 1090-1102. e7. [CrossRef]
- Diaz, C., et al., Phase I randomized study of a tetravalent dengue purified inactivated vaccine in healthy adults from Puerto Rico. The American journal of tropical medicine and hygiene, 2018. 98(5): p. 1435. [CrossRef]
- Boigard, H., V. Cimica, and J.M. Galarza, Dengue-2 virus-like particle (VLP) based vaccine elicits the highest titers of neutralizing antibodies when produced at reduced temperature. Vaccine, 2018. 36(50): p. 7728-7736. [CrossRef]
- Chiang, C.-Y., et al., Immunogenicity of a novel tetravalent vaccine formulation with four recombinant lipidated dengue envelope protein domain IIIs in mice. Scientific reports, 2016. 6(1): p. 30648. [CrossRef]
- Halstead, S.B., Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine, 2017. 35(47): p. 6355-6358. [CrossRef]
- Rappuoli, R., Reverse vaccinology. Current opinion in microbiology, 2000. 3(5): p. 445-450.
- Pizza, M., et al., Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science, 2000. 287(5459): p. 1816-1820. [CrossRef]
- Tahir ul Qamar, M., et al., Peptide vaccine against chikungunya virus: immuno-informatics combined with molecular docking approach. Journal of translational medicine, 2018. 16: p. 1-14. [CrossRef]
- Hasan, A., M. Hossain, and J. Alam, A computational assay to design an epitope-based Peptide vaccine against Saint Louis encephalitis virus. Bioinformatics and Biology insights, 2013. 7: p. BBI. S13402. [CrossRef]
- Sarkar, B., et al., Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology, 2020. 225(3): p. 151955.
- Chakraborty, S., et al., A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In silico biology, 2010. 10(5-6): p. 235-246. [CrossRef]
- Kar, P.P. and A. Srivastava, Immuno-informatics analysis to identify novel vaccine candidates and design of a multi-epitope based vaccine candidate against Theileria parasites. Frontiers in immunology, 2018. 9: p. 340421. [CrossRef]
- Friend, U.S., et al., In silico analysis of envelope Dengue Virus-2 and envelope Dengue Virus-3 protein as the backbone of Dengue Virus tetravalent vaccine by using homology modeling method. OnLine Journal of Biological Sciences, 2009. 9(1): p. 6-16.
- Gromowski, G.D., N.D. Barrett, and A.D. Barrett, Characterization of dengue virus complex-specific neutralizing epitopes on envelope protein domain III of dengue 2 virus. Journal of virology, 2008. 82(17): p. 8828-8837. [CrossRef]
- Rivino, L., et al., Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. Journal of virology, 2013. 87(5): p. 2693-2706.
Figure 1.
Flow chart of the designed Dengue vaccines study.
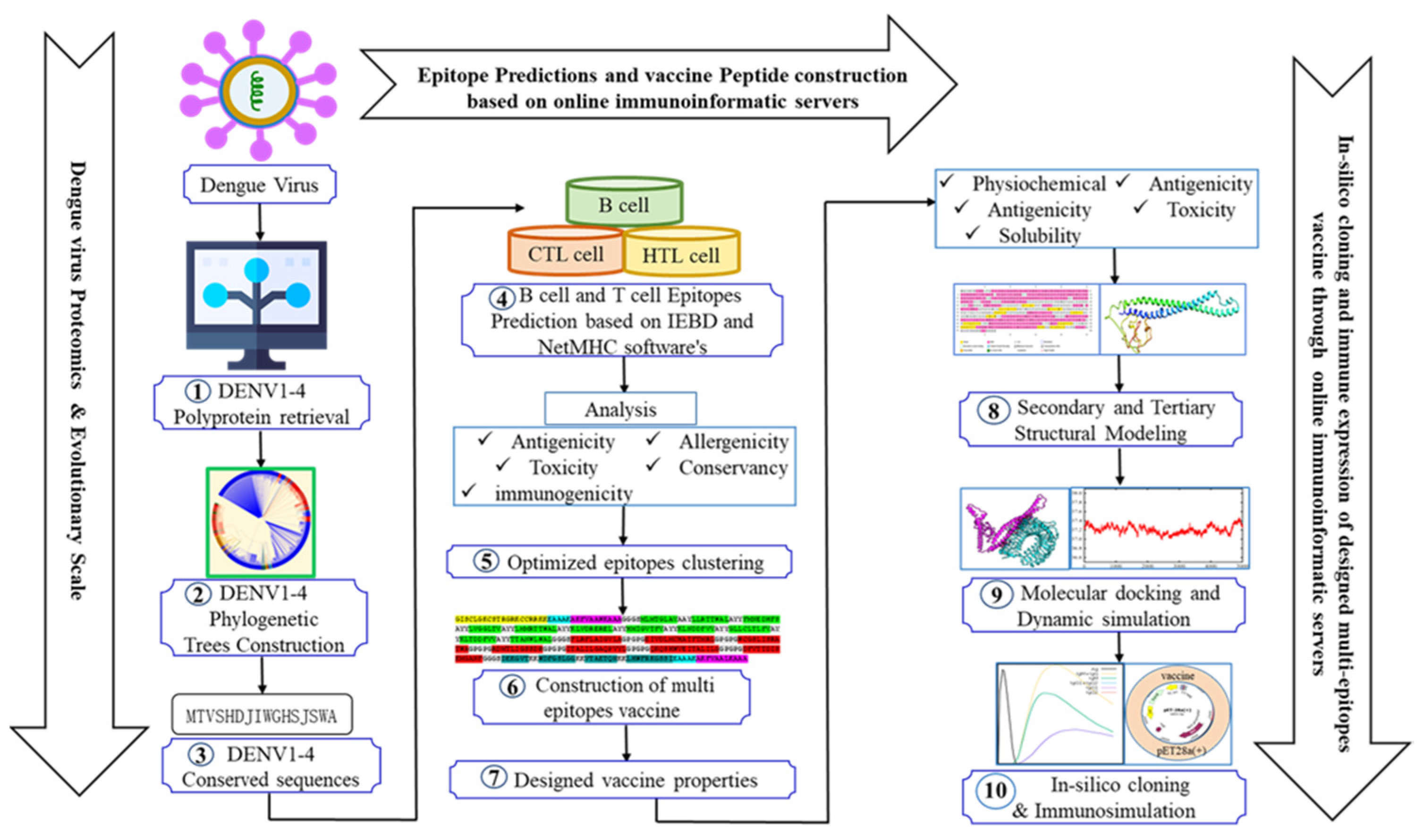
Figure 2.
Organization of multi-epitope tetravalent vaccine peptides sequence orders (A) Graphic presentation of vaccine designing strategy, (B-D) PSDV-1, 2, and 3 vaccine candidates.
Figure 2.
Organization of multi-epitope tetravalent vaccine peptides sequence orders (A) Graphic presentation of vaccine designing strategy, (B-D) PSDV-1, 2, and 3 vaccine candidates.
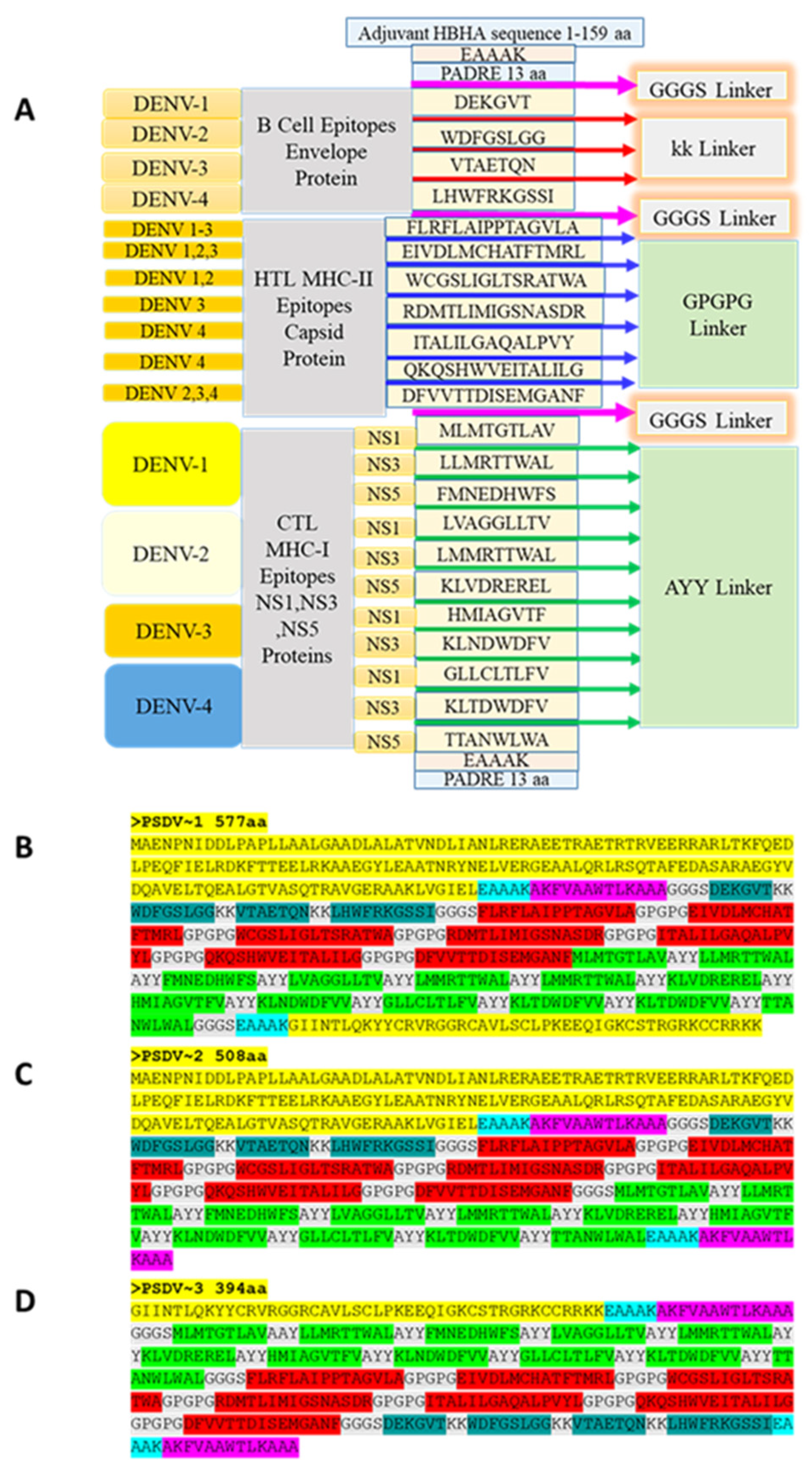
Figure 3.
Modelling and Validation of PSDV-2 Candidate Vaccine in Molecular Structure. (A) Secondary Structure, (B) Secondary Structures PSDV1~3 amino acid composition ratio, (C) Tertiary Structure, (D) Tertiary Structure Template Model c6tpiAformodeling, (E) Confidence level of PSDV 1~3, Z score of the structure model with top qualification (F) PSDV 2, (I) PSDV 1~3, (G) Coverage level of PSDV 1~3, Ramachandran plot, (H) PSDV 2, (J) PSDV 1~3,.
Figure 3.
Modelling and Validation of PSDV-2 Candidate Vaccine in Molecular Structure. (A) Secondary Structure, (B) Secondary Structures PSDV1~3 amino acid composition ratio, (C) Tertiary Structure, (D) Tertiary Structure Template Model c6tpiAformodeling, (E) Confidence level of PSDV 1~3, Z score of the structure model with top qualification (F) PSDV 2, (I) PSDV 1~3, (G) Coverage level of PSDV 1~3, Ramachandran plot, (H) PSDV 2, (J) PSDV 1~3,.
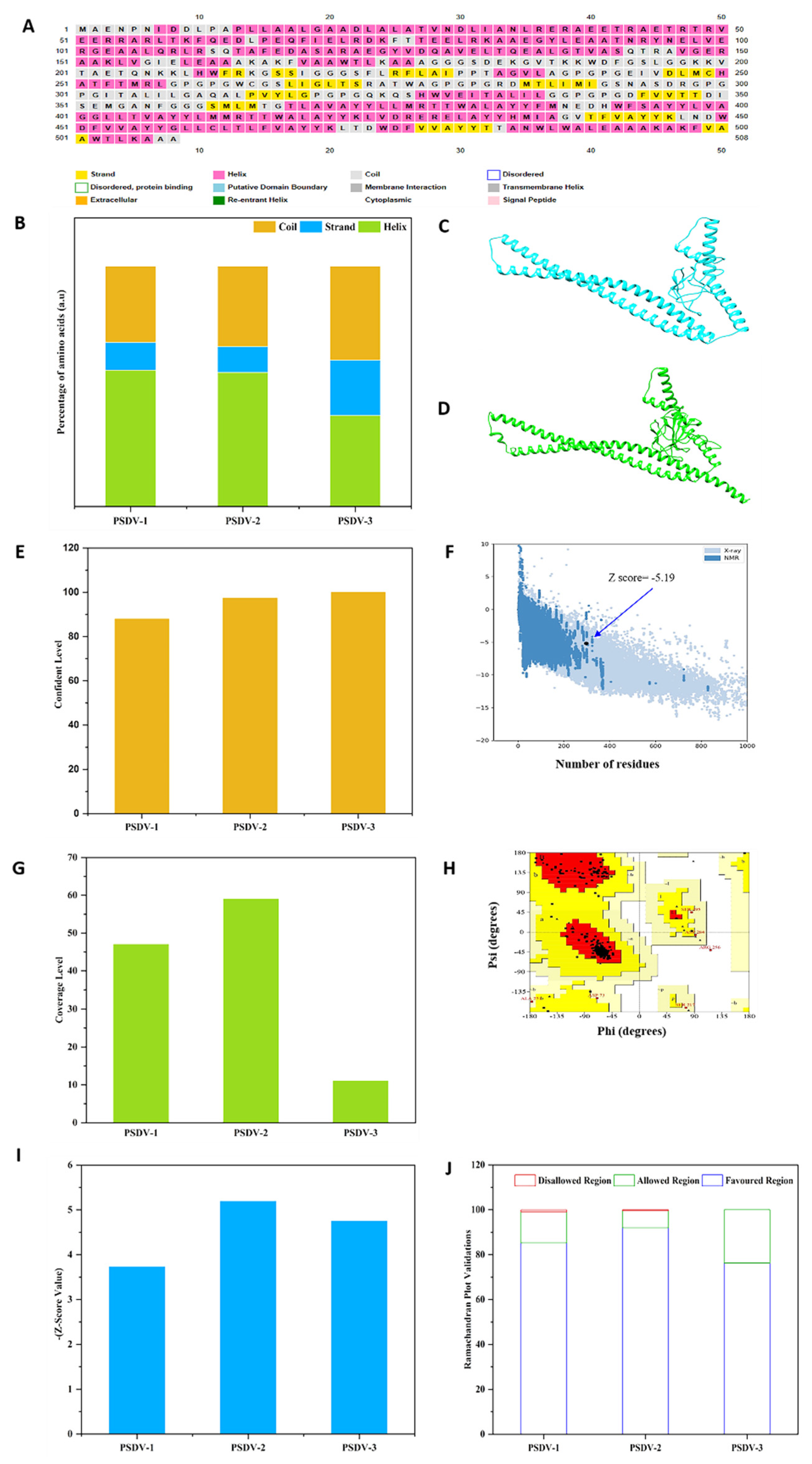
Figure 4.
Molecular Docking and Interaction Details between PSDV2 and TLR2/4 Receptors. The interactions visualized by JSmol, (A) TLR2, (E) TLR4, Schematic diagram of interactions, (C) Chain A indigo TLR2, (G) Chain A indigo TLR4, (C, G) PSDV- 2 (red), Salt bridge and Hydrogen bond interactions, (D) TLR2 (3, 4), (H) TLR4 (4, 5) TLR, Interaction sites visualized by Pymole, (B) TLR2, (F) TLR4, interface statistics (I) TLR2 and TLR4.
Figure 4.
Molecular Docking and Interaction Details between PSDV2 and TLR2/4 Receptors. The interactions visualized by JSmol, (A) TLR2, (E) TLR4, Schematic diagram of interactions, (C) Chain A indigo TLR2, (G) Chain A indigo TLR4, (C, G) PSDV- 2 (red), Salt bridge and Hydrogen bond interactions, (D) TLR2 (3, 4), (H) TLR4 (4, 5) TLR, Interaction sites visualized by Pymole, (B) TLR2, (F) TLR4, interface statistics (I) TLR2 and TLR4.
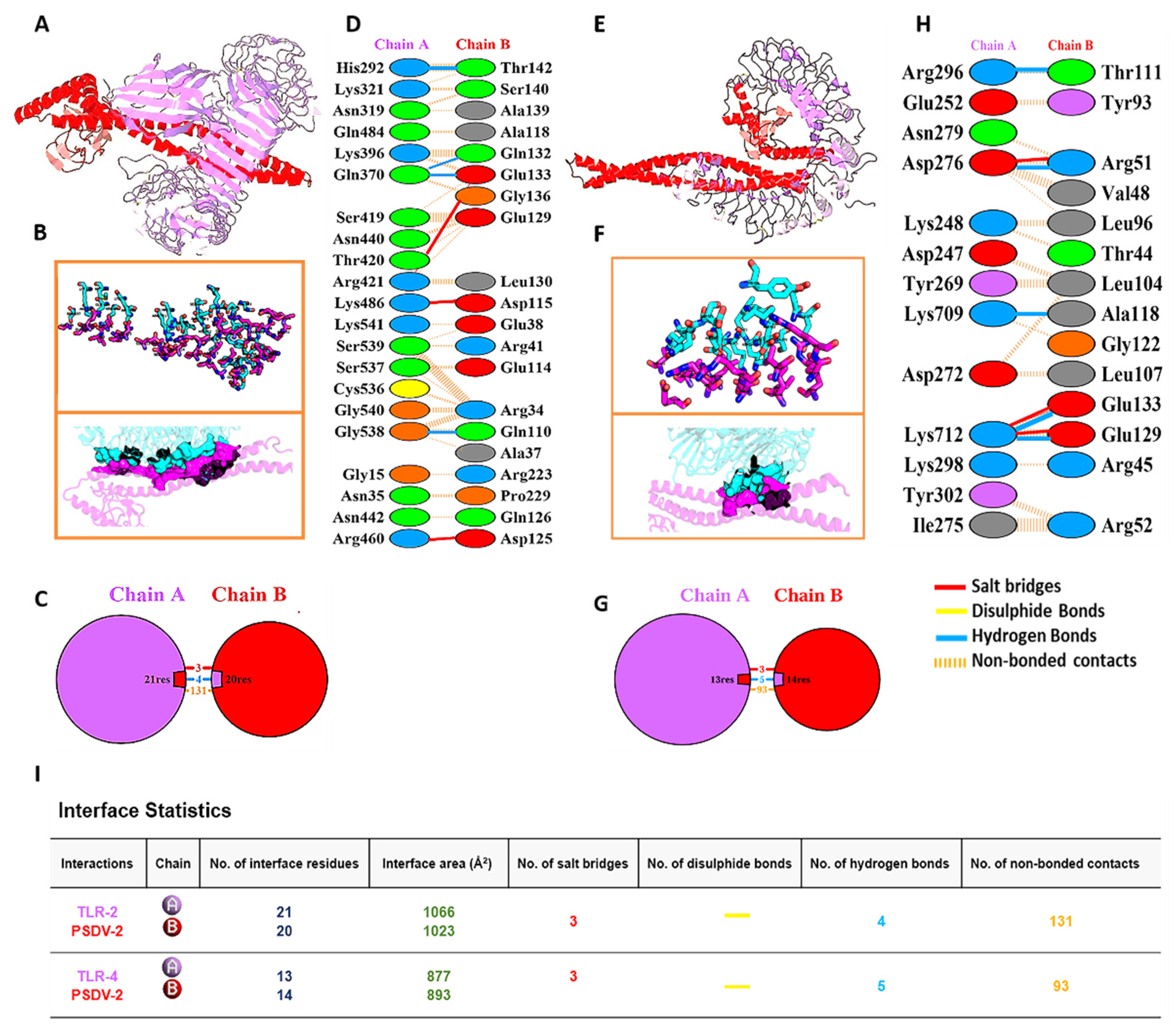
Figure 5.
Molecular Dynamics Simulation of the Complex PSDV2-TLR2/4. (A) Blue colour TLR2, (B) Red colour TLR4.
Figure 5.
Molecular Dynamics Simulation of the Complex PSDV2-TLR2/4. (A) Blue colour TLR2, (B) Red colour TLR4.
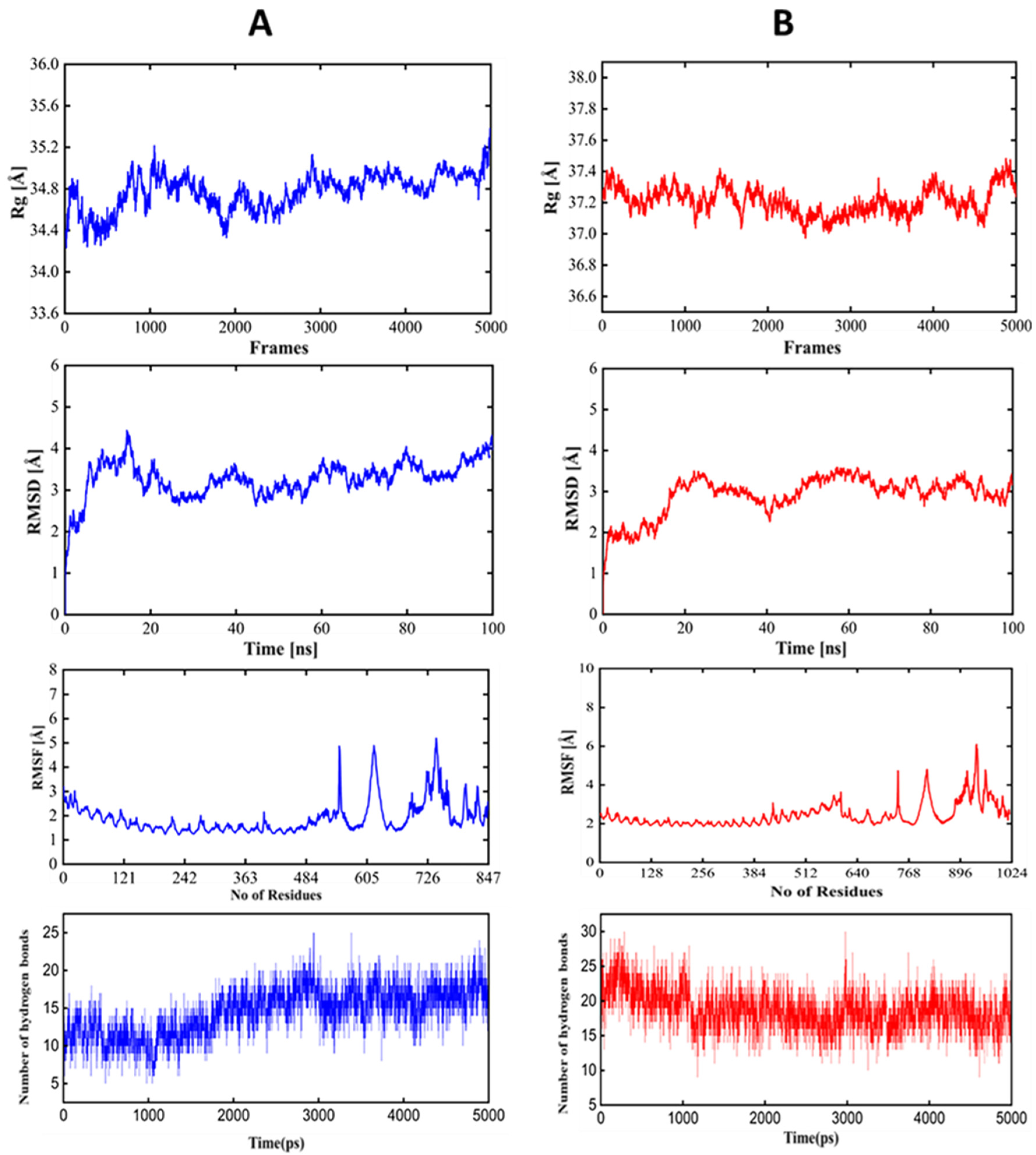
Figure 6.
Innate Immune Responses of Host after 3 Doses Vaccination of PSDV-2. (A) Exhibits the level of cytokine expression, (B) Displays the state of the DC cell population, (C) Exhibits Macrophages (MA), and (D) Shows Natural killer (NK) cells.
Figure 6.
Innate Immune Responses of Host after 3 Doses Vaccination of PSDV-2. (A) Exhibits the level of cytokine expression, (B) Displays the state of the DC cell population, (C) Exhibits Macrophages (MA), and (D) Shows Natural killer (NK) cells.
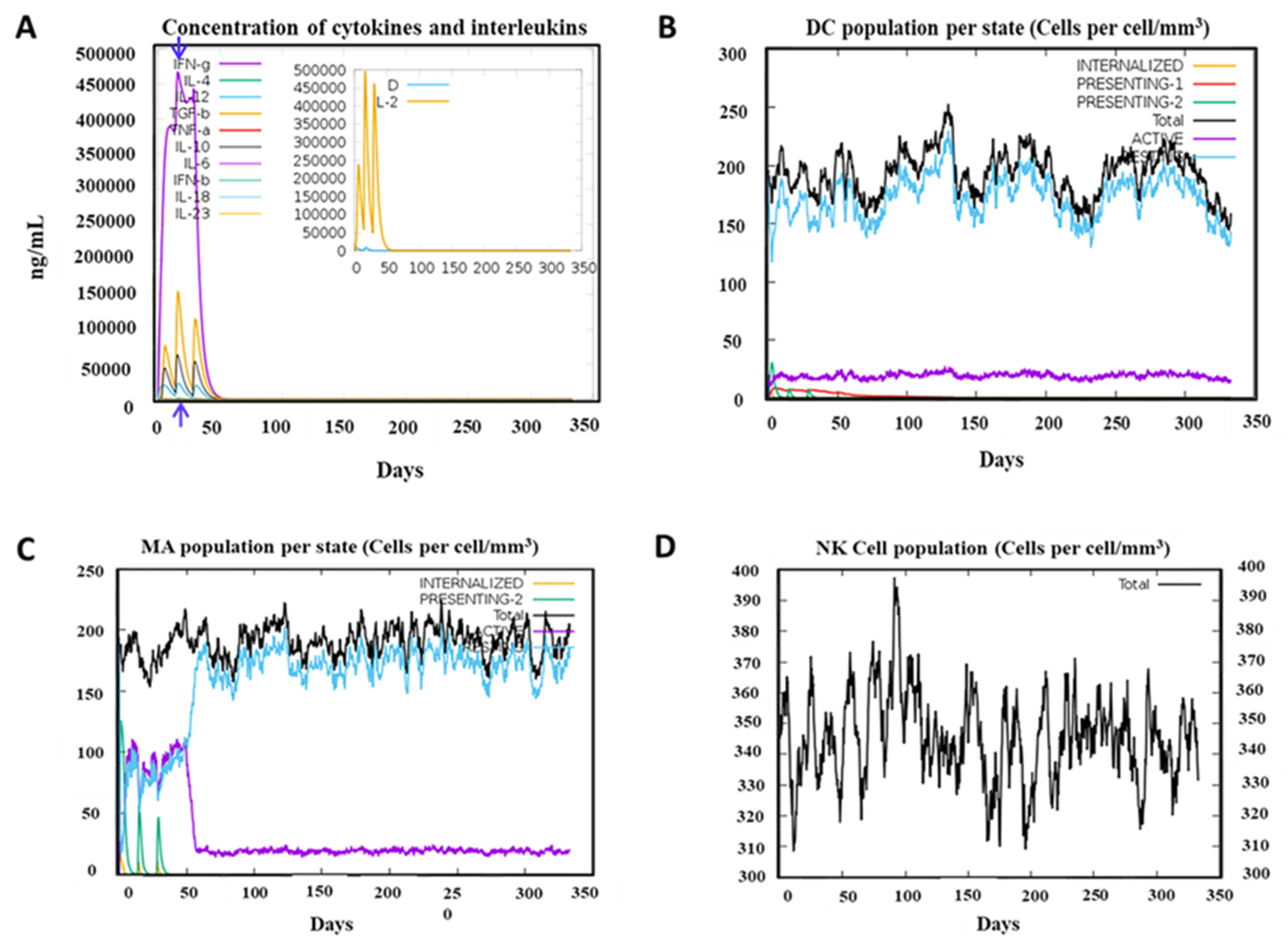
Figure 7.
Adaptive Immunity of Host in response to the exposure of PSDV-2 immunization after three injections. (A) Illustrates the state of the B cell population, (B) Displays the numbers of the B cell population, (C) Presents the state of CD8+ T-cytotoxic, (Tc) lymphocyte population, (D) Displays Tc lymphocyte, (E) Shows the state of TH cell population per, (F) Display Illustrates CD4-TH lymphocyte, (G) Production of immunoglobulins.
Figure 7.
Adaptive Immunity of Host in response to the exposure of PSDV-2 immunization after three injections. (A) Illustrates the state of the B cell population, (B) Displays the numbers of the B cell population, (C) Presents the state of CD8+ T-cytotoxic, (Tc) lymphocyte population, (D) Displays Tc lymphocyte, (E) Shows the state of TH cell population per, (F) Display Illustrates CD4-TH lymphocyte, (G) Production of immunoglobulins.
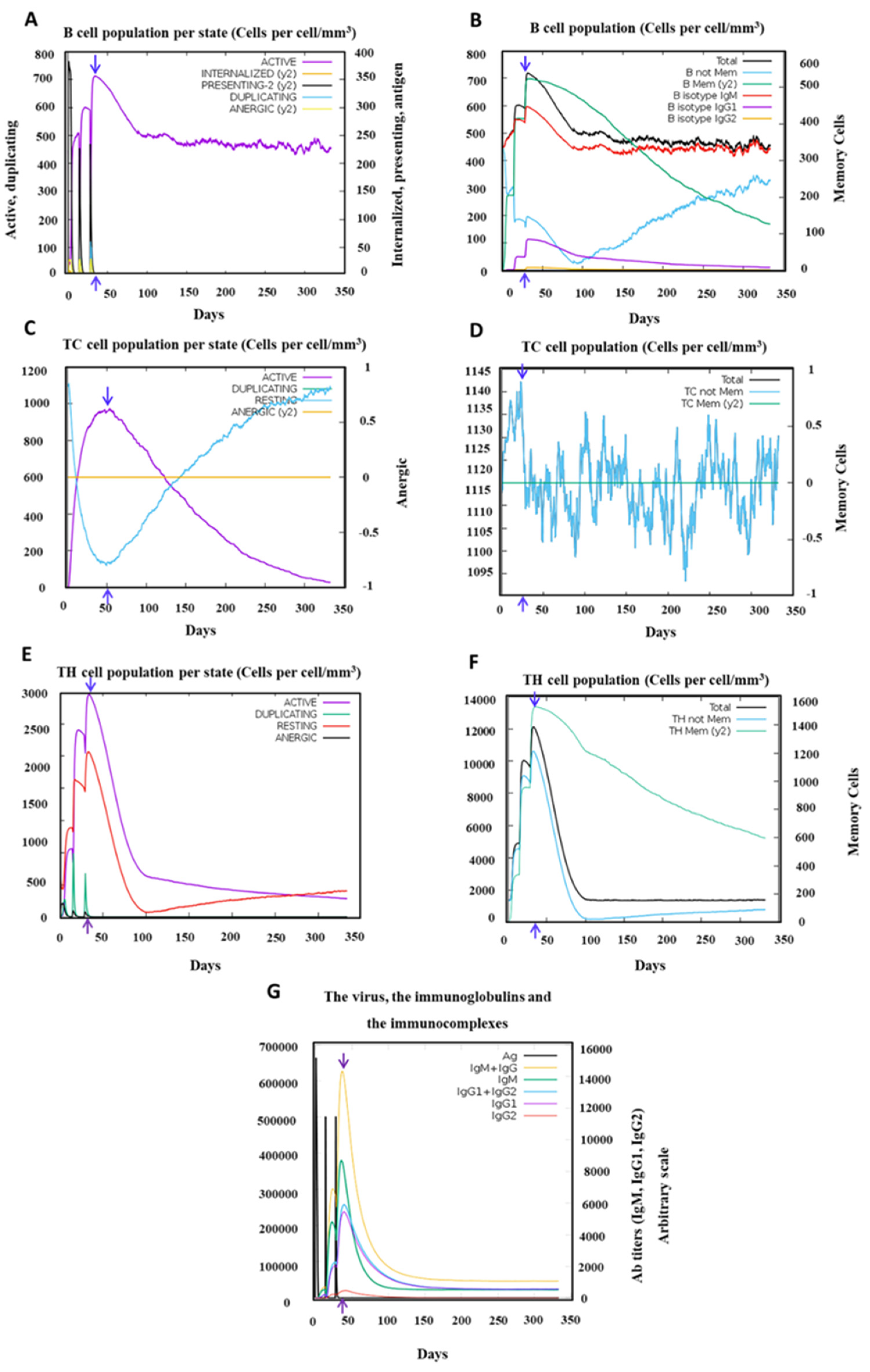
Figure 8.
The in-silico cloning of the designed vaccine into the pET-28a (þ) vector.
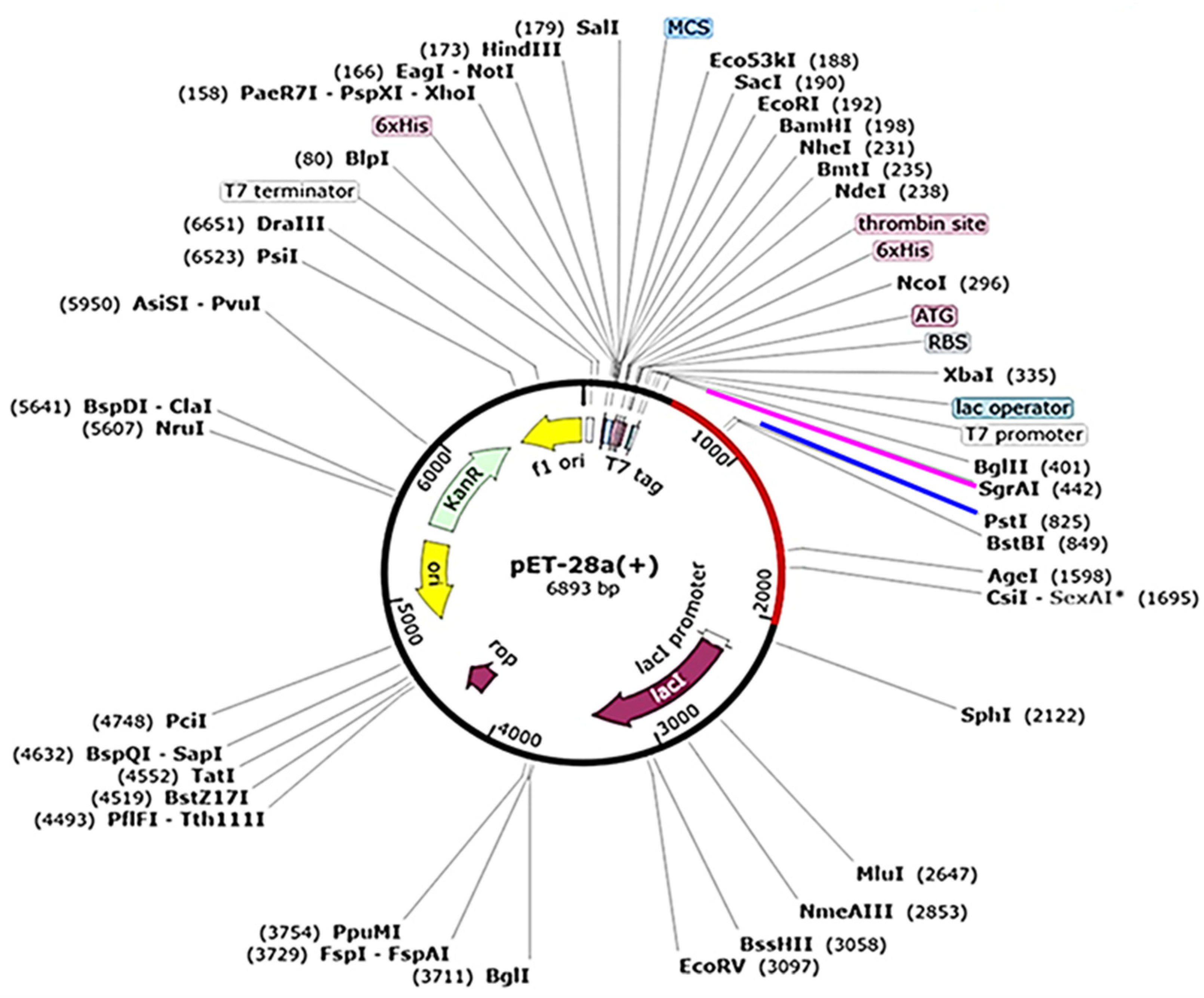

Table 1.
Final Selection of B cell Epitopes for Vaccines Designing.
| Features | DENV-1 | DENV-2 | DENV-3 | DENV-4 |
|---|---|---|---|---|
| Epitope Sequence | DEKGVT | WDFGSLGG | VTAETQN | LHWFRKGSSI |
| Start | 341 | 377 | 330 | 254 |
| End | 346 | 384 | 336 | 263 |
| Protein Region | E (ED III) | E (ED III) | E ( ED III) | E ( ED II) |
| Epitope Length | 6 | 8 | 7 | 10 |
| Allergenicity | Non-Allergen | Non-Allergen | Non-Allergen | Non-Allergen |
| Antigenicity Score | 0.7715 | 2.1175 | 1.2240 | 0.9330 |
| Toxicity | Non-Toxic | Non-Toxic | Non-Toxic | Non-Toxic |
| Intra conservancy | 100% | 100% | 100% | 100% |
| Inter DENV1-4 conservancy | 25.00% (1/4) | 25.00% (1/4) | 25.00% (1/4) | 25.00% (1/4) |
Table 2.
Final Selection of MHC-I CTLs Epitopes for Vaccines Designing.
| Serotype | Protein Region |
Epitope | Affinity (Strong Binding) |
Antigenicity, Allergenicity, Toxicity Score | immunogenicity Score |
Serotype specific, Cross Conservancy |
|---|---|---|---|---|---|---|
| DENV-1 |
NS1 | MLMTGTLAV | 3 | 0.5611, Non Allergen, Non Toxic | 0.20739 | 100%,25% |
| NS3 | LLMRTTWAL | 4.24 | 0.9556, Non Allergen, Non Toxic | 0.27922 | 100%,25% | |
| NS5 | FMNEDHWFS | 38.43 | 0.4990, Non Allergen, Non Toxic | 0.40604 | 100%,25% | |
| DENV-2 |
NS1 | LVAGGLLTV | 39.51 | 0.5463, Non-Allergen, Non Toxic | 0.08268 | 100%,25% |
| NS3 | LMMRTTWAL | 4.1 | 1.1235, Non Allergen, Non Toxic | 0.27922 | 100%,25% | |
| NS5 | KLVDREREL | 3.1 | 1.4751, Non Allergen, Non Toxic | 0.09999 | 100%,50.00% (2/4),DENV-3-2 | |
| DENV-3 |
NS1 | HMIAGVTFV | 5.19 | 0.8396, Non Allergen, Non Toxic | 0.25559 | 100%,25% |
| NS3 | KLNDWDFVV | 4.15 | 2.2249, Non Allergen, Non Toxic | 0.37972 | 100%,25% | |
| DENV-4 |
NS1 | GLLCLTLFV | 5.23 | 0.8159, Non Allergen, Non Toxic | 0.02693 | 100%,25% |
| NS3 | KLTDWDFVV | 4.15 | 2.6071, Non Allergen, Non Toxic | 0.3944 | 100%,25% | |
| NS5 | TTANWLWAL | 39.89 | 0.9911, Non Allergen, Non Toxic | 0.42125 | 100%,25% |
Table 3.
Final Selection of MHC-II HTLs Epitopes for Vaccines Designing.
| Serotype | Epitope | Allele | Protein region | affinity(Strong Binding) | Antigenicity, Allergenicity, Toxicity | , Serotype-specific Cross Conservancy |
|---|---|---|---|---|---|---|
| DENV-1 | FLRFLAIPPTAGVLA | DRB1_0101 | Capsid | 11.2 | 0.6827, Non-Allergen, Non Toxic | 100%,50.00% (2/4) DENV 1,2 |
| DENV-1 | EIVDLMCHATFTMRL | DRB1_0701 | Capsid | 6.9 | 0.9597, Non-Allergen, Non Toxic | 100%,75.00%(3/4) DENV1,2,3 |
| DENV-2 | WCGSLIGLTSRATWA | DRB1_0101 | Capsid | 7.4 | 0.8137,Non Allergen, Non Toxic | 100%,50.00% (2/4) DENV-2,3 |
| DENV-3 | RDMTLIMIGSNASDR | DRB1_0401 | Capsid | 5.9 | 0.9291, Non Allergen, Non Toxic | 100%,25%(1/4) |
| DENV-4 | ITALILGAQALPVYL | DRB1_0101 | Capsid | 4.7 | 0.6489, Non-Allergen, Non Toxic | 100%,25%(1/4) |
| DENV-4 | QKQSHWVEITALILG | DRB1_0701 | Capsid | 12.9 | 1.2944, Non-Allergen, Non Toxic | 100%,25%(1/4) |
| DENV-2 | DFVVTTDISEMGANF | DRB1_0401 | Capsid | 33.1 | 0.6533, Non Allergen, Non Toxic | 100%,75.00% (3/4) DENV-2,3,4 |
Table 4.
Properties of PSDV1~3 vaccines in physio-chemistry and immunology.
| Parameter | PSDV 1 | PSDV2 | PSDV 3 |
|---|---|---|---|
| No. of amino acids | 564 | 508 | 394 |
| Molecular weight | 62.09 kDa | 55.2733 kDa | 42.71375 kDa |
| Theoretical pI | 8.38 | 5.76 | 9.37 |
| Ext. coeff | 127200 | 112760 | 109780 |
| Half-life | 30 hours (mammalian reticulocytes, in vitro). >20 hours (yeast, in vivo). >10 hours (Escherichia coli, in vivo). |
30 hours (mammalian reticulocytes, in vitro). >20 hours (yeast, in vivo). >10 hours (Escherichia coli, in vivo). |
30 hours (mammalian reticulocytes, in vitro). >20 hours (yeast, in vivo). >10 hours (Escherichia coli, in vivo). |
| Instability Index | 26.02 | 24.28 | 20.52 |
| Aliphatic index | 86.28 | 88.33 | 85.05 |
| Hydropathicity (GRAVY) | -0.049 | 0.018 | 0.151 |
| Antigenicity | 0.6717 | 0.6727 | 0.7320 |
| Allergenicity | Non Allergenic | Non Allergenic | Non Allergenic |
| Toxicity | Non Toxic | Non Toxic | Non Toxic |
| Solubility | 0.788949 | 0.761350 | 0.569153 |
Table 5.
Receptors docked Binding Energy representations. Molecular docking and mm/gbsa analysis for HLAs and TLRs showing their binding energies and free energy for PSDV-2.Res (Residue), BFE (Binding Free Energy) CBFE (Complex binding free energy mmpbsa/kcal/mol).(Receptor A (HLAs, TLRs), (Ligand B (Vaccine).
Table 5.
Receptors docked Binding Energy representations. Molecular docking and mm/gbsa analysis for HLAs and TLRs showing their binding energies and free energy for PSDV-2.Res (Residue), BFE (Binding Free Energy) CBFE (Complex binding free energy mmpbsa/kcal/mol).(Receptor A (HLAs, TLRs), (Ligand B (Vaccine).
| Immune Receptors |
Docking Score | Res (Rec) | BFE | Res (Lig) | BFE | CBFE |
|---|---|---|---|---|---|---|
| TLR4 | -5386.23 | A-ARG-421 | -5.86 | B-ARG41 | -5.85 | -28.13 |
| A-TYR-297 | -3.68 | B-ARG45 | -4.49 | |||
| A-LYS-462 | -2.77 | B-GLU-114 | -4.15 | |||
| A-TYR-350 | -2.5 | B-LEU-104 | -3.49 | |||
| A-LEU-345 | -2.01 | B-LEU-107 | -3.1 | |||
| TLR2 | -7330.87 | A-ARG-421 | -6.24 | B-GLU-133 | -2.15 | -29.66 |
| A-ARG-460 | -4.76 | B-GLU-38 | -2.14 | |||
| A-GLY-538 | -3.68 | B-THR-142 | -1.96 | |||
| A-SER-539 | -3.42 | B-GLN-132 | -1.86 | |||
| A-HIE-292 | -2.78 | B-ASP-115 | -1.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated