
Submitted:
29 June 2024
Posted:
02 July 2024
You are already at the latest version
Abstract
Caveolin is a structural protein within caveolae, which may be involved in transmembrane molecular transport and/or various intercellular interactions within cells. Specific mutations of caveolin-3 in muscle cells are well-known to cause a limb-girdle muscular dystrophy. Altered expression of the caveolin-3 has been also detected in Duchenne's muscular dystrophy, which may be a part of the pathological process leading to muscle weakness. Interestingly, it has been shown that the renovation of nitric oxide synthase (NOS) to the sarcolemma with muscular dystrophy could improve the muscle health, suggesting that NOS may be involved in the pathology of muscular dystrophy. Here, we summarize the notable function of caveolin and/or NOS in skeletal muscle cells, and would like to discuss about their involvement in the pathology as well as possible tactics for the innovative treatment of muscular dystrophies.
Keywords:
caveolae
; caveolin
; NOS
; Duchenne muscular dystrophy
; limb-girdle muscular dystrophy
; gut microbiota
; probiotics
1. Introduction
Muscular dystrophies represent a group of disorders characterized by the primary skeletal muscle deterioration and/or the subsequent emergence of co-morbidities such as inflammation, mitochondrial dysfunction, and metabolic irregularities. These conditions may predominantly result from mutations in certain gene products that connect the cytoskeleton to the basal lamina of muscle cells [1]. (Figure 1 and Figure 2) The Duchenne muscular dystrophy (DMD) is the most common inherited X-linked form of muscular dystrophy, causing a severe and progressive neuromuscular disorder characterized by insufficient production of dystrophin due to specific mutations in the dystrophin gene [2]. In patients with Duchenne muscular dystrophy, the X-linked mutation can interfere with the ability to produce functional dystrophin in the muscles [3]. Duchenne muscular dystrophy may end in respiratory failure and premature death [4]. Although this disorder is primarily known to affect skeletal muscle, cardiac complications such as left ventricular dysfunction are another common appearance of Duchenne muscular dystrophy. The functional deficiency of dystrophin may lead to plasma-membrane instability, causing myofiber necrosis and/or muscle weakness [5], which also results in muscle fiber inflammation, impaired regeneration of muscle fibers, and replacement of muscle by fibrotic and adipose tissue, directing to advanced deterioration of muscle function [6,7]. Therefore, this disease shows through compromised membrane integrity, chronic inflammation and fibrosis, as well as impaired tissue remodeling [8]. A previous study has shown that lipid metabolism might be a critical metabolic disturbance in mdx mice (a genetic model of Duchenne muscular dystrophy), which also uncovered dysregulation of cholesterol and fatty acid metabolism transcription factors and accumulation of cholesterol in dystrophic muscles [9,10]. In addition, xanthine oxidase activity has been as a contributing factor in mdx mouse [11]. Most of the dysregulated metabolite have a connection to energy and phospholipid metabolism, revealing the intricate metabolic remodeling of phospholipids and energy metabolism in Duchenne muscular dystrophy. Further, another study has revealed the muscle metabolic remodeling patterns of Duchenne muscular dystrophy by using high resolution mass spectrometry [12].
Although a progress has been made, fundamental pathological mechanisms are still worthy of further study in order to discover beneficial therapeutic targets for Duchenne muscular dystrophy. There is no cure for Duchenne muscular dystrophy up to the present time. Metabolic insufficiencies may have further promotional effect in the disease progression [13]. Calcium channel blockers to restore calcium homeostasis and/or anabolic steroids to restore muscle mass may not have any clinical benefits associated with the overall development of the disease [6,14]. Glucocorticoids are currently the only medications recognized to benefit for Duchenne muscular dystrophy with significant side effects [15,16], which may prolong survival [17]. In addition, this treatment may be associated with various complications [18]. Compared with monotherapy, a reduction in motor function decline could be detected among the stable subgroup of patients treated with combination therapy with combination of citrulline and/or metformin treatment [19]. Interestingly, some studies have correlated the intestinal microbiota with lifetime cardiovascular risk of muscular dystrophy [20]. These findings underline the urgent need to identify strategies targeting the gut microbial system and conduct in-depth analyses of the functional relationship between food and microbiota composition to modulate various human diseases including Duchenne muscular dystrophy, which may contribute to the elucidation of potential therapeutic targets and/or biomarkers.
2. Inflammation and Redox System May Be Involved in the Regulation of Muscular Dystrophy
Inflammation and oxidative stress may be involved in the development of muscle dystrophy. For instance, some anti-oxidants have shown to successfully reduce muscle damage in an animal model for Duchenne muscular dystrophy [21]. In addition, inflammatory markers and specific lymphocyte subsets have been identified in the blood/muscles of DMD patients. Additionally, T lymphocytes from the murine model of DMD (mdx mice) could induce muscular damage when injected into healthy murine muscle [22]. Following the muscular damage, the inflammatory phenotype may be started by damage-associated molecular arrangements that activate neutrophils by specific membrane markers such as Toll-like receptor. Recruited pro-inflammatory macrophages may induce muscle lysis, producing inducible nitric oxide synthase (iNOS). In dystrophic mdx mice, excess population of this macrophage could lead to further tissue destruction with the employment of CD4/CD8+ T-cells [23]. In these ways, the pathological manifestation of DMD is complicatedly linked to inflammation and/or oxidative stress involvement [24]. Loss of dystrophin leads to membrane fragility of muscle cells, resulting in chronic inflammation, myofiber death, and/or regeneration. Eventually, functional muscle cells might be replaced by fibrous features and/or fat [25,26]. Interestingly, restoration of nitric oxide synthase (NOS) to the sarcolemma would be supposed to improve muscle health in DMD. In addition, mis-located nNOS could produce dysregulated ROS, which might contribute to the pathology of DMD [25,26]. Generally, NOS is transcriptionally upregulated by cytokines as well as by hypoxia. Dystrophin-deficient muscles may exhibit reductions in expression of NOS, suggesting that NO deficiency could affect the pathology of DMD [27]. NO is a kind of signaling molecule with complexed and/or controversial effects. Occasionally, NOS could facilitate several cellular processes imperative for cellular functions [28]. Remarkably, NO is usually considered to be cardioprotective [29]. Therefore, altered NOS function may contribute to different cardiovascular diseases including high blood pressure and/or heart failure [30]. In the pathology of DMD, the increasing of oxidative stress may depend on over-expression of inflammatory cells, leading to upregulation of NOS [31]. The accumulation of inflammatory molecules could also affect the function of proteins and lipids [32,33]. Likewise, the neutrophils causes damages to muscular tissues [34]. Additionally, the oxidation of thiol (-SH) groups of cysteine residues is associated with the development of necrosis and fatty tissue [35]. Dystrophin can connect to intricate transmembrane proteins including dystroglycan as well as key signaling molecules including NOS in sarcolemma [36]. In particular, NOS activities might be coupled to oxygen concentration. Therefore, mislocalized NOS could trigger abnormal protein nitrosylation and ROS, which are believed to contribute to the pathology of DMD. In skeletal muscle, NOS may be associate with caveolin-3. It has been suggested that muscle regeneration may be mediated by the upregulation of endothelial NOS activity and increased expression of VEGF and/or adhesion molecules [37]. Interestingly, female-specific factors including sex hormones may play a role in those alterations for crucial aspects of DMD pathology [38]. Additionally, the neuronal isoform of NOS homeostasis is tightly associated with dystrophin [39]. In addition, the NOS is almost absent in the skeletal muscle of DMD patients [40]. Deficiency in NOS-derived NO might exacerbate the phenotype of DMD by impairing skeletal muscle function.
3. Relationship between Caveolin and Muscular Dystrophy
In general, NOS-derived NO could regulate a wide range of cell functions including inflammation, apoptosis, permeability and cell growth. The NOS may be localized mainly near specific intracellular membrane domains including the cytoplasmic face of the Golgi apparatus and plasma membrane caveolae [41]. Caveolae are specialized lipid domains of the plasma membrane containing caveolins, which are key structural components and serve as a scaffold for signaling proteins [42]. In addition, caveolae can classify signal transduction molecules which regulate multiple functions including the production of nitric oxide (NO) by the caveolae resident enzyme NOS [43]. Flask-shaped invaginations of the plasma membrane known as caveolae could work as critical regulators for human health and/or disease [44]. (Figure 2) The integral membrane protein caveolin-1 (CAV1) is a major structural component of caveolae and is required in place of caveolae formation in non-muscle cells [45]. Association of CAV1 into oligomeric protein complexes is an indispensable step in caveolae biogenesis, and faults in the oligomerization could cause several diseases [46]. In mammals, CAV1 and/or caveolae are widely distributed in many tissues where they operate for lipid homeostasis, protection, endocytosis, and signal transduction [47]. Conversely, dysregulation of caveolae may contribute to the development/progression of several diseases including cancers, inflammatory respiratory diseases, hypertension, asthma, and lipodystrophy [48]. Interestingly, the P132L mutation of wild type CAV1 has been identified in several different patient samples including breast cancer and/or lung adenocarcinomas [49]. In addition to the P132L mutation, a variety of other pathogenic mutations in caveolin homologs have been identified in humans [50]. The P132L mutation has also been reported to disturb the ability of CAV1 to correctly move to the plasma membrane, suggesting it might function as a dominant negative with the wild type CAV1 [51]. These findings emphasize the importance of P132 mutation in caveolin also function in the development/progression of health and disease. Therefore, the P132L mutation is also a powerful tool to investigate how defects in trafficking and/or oligomerization of caveolins could impede the caveolae formation [52]. Some protein mutations that could lead to muscular dystrophy may often generate deficiencies in cytoskeletal support of the muscle sarcolemma. In particular, caveolae are cholesterol-rich microdomains that could construct mechanically deformable invaginations of the sarcolemma. For example, it is well-known that mutations to caveolin-3, the main scaffolding protein of caveolae in muscle, can trigger limb-girdle muscular dystrophy (LGMD). An equivalent mutation in caveolin-3 (CAV3), P105L (curiously denoted as P104L in many studies), could show the phenotype of muscular dystrophies both in humans and/or animal models [53]. Therefore, the most equivalent to P132L may be the P105L mutation of CAV3 frequently associated with LGMD [54,55]. As for dominant negative function, muscle biopsies from patients harboring the P105L mutation of CAV3 with an autosomal form of limb-girdle muscular dystrophy may detect noticeably decreased CAV3 levels compared to normal controls [55]. In addition, oligomerization and/or trafficking defects have been also observed for the P105L mutants of CAV3 [56]. Resembling the P132L mutation of CAV1, the P105L mutant protein can intracellularly entrap the wild type CAV3 in mammalian heterologous expression systems [57]. However, the fine character of the oligomerization defects appears to be slightly different, as P105L mutation of CAV3 may incline to form larger oligomers rather than wild type CAV3 [56,58]. It seems possible that the P105L mutation of CAV3 may play a comparable role in destabilizing the structure of CAV3 complexes, as is the situation for CAV1 [59]. Standard expression levels of CAV3 may be protective for the sarcolemma, while the upregulation of CAV3 may be detected in several muscular dystrophies owing to the probable compensation for additional functional deficiencies [60].
4. Gut-Brain Axis and microRNAs Might Be Involved in the Pathogenesis of Muscular Dystrophies
It has been described that up-regulation of several microRNAs in the atrial myocardium of patients may lead to translational suppression of dystrophin and enhance the decay of the mRNA of NOS, which may contribute to the atrial remodeling that can promote the exacerbation of cardiac atrial arrhythmia [61]. Consistent with this finding, NOS1 knock out mice may exhibit atrial characteristic features of the remodeling as well as an increased tendency to develop the arrhythmia [61]. However, the occurrence of cardiac arrhythmia in DMD patients may be comparatively low after the development of cardiomyopathy [62]. Probably because, deficiency of dystrophin might not result in the increase of the relevant microRNA in the atrial myocardium of DMD [62]. Several miRNAs associated with the skeletal muscle development and/or regeneration in the context of DMD have been intensely investigated, which have revealed several elevated levels of microRNAs including miR-1, miR-133, and/or miR-206 in the serum of DMD patients [63,64]. Interestingly, dystrophin depletion in the mdx mouse may also be associated with a decrease in atrial NOS protein content and/or such microRNAs expression. Compensatory changes in caveolin-1 and NOS may explain the biochemical phenotype of mdx atria, and could elucidate why DMD patients might not show an advanced prevalence of the atrial arrhythmias in spite of a decrease in myocardial NOS protein level [65]. The NOS reduction in mdx mice might come from failure of the dystrophin complex, which may induce the ubiquitin-mediated degradation of the complex including NOS by the proteasome. In other words, the dystrophin could influence the stability of NOS protein as well as its subcellular localization [66]. For example, in vitro studies have described that NOS1 may undertake the proteasomal degradation via the ubiquitination of the calmodulin-association site [67]. As the ubiquitination-proteasomal degradation may specially take place on the inactive and/or damaged form of protein, this autophagy may organize an imperative mechanism for removing nonfunctional NOS proteins [67]. Amplified caveolae domain and NOS expression level may also be linked to the downregulation of miRNA-124a and/or miRNA-155 with a greater antioxidant mechanism [68].
The target genes of the downregulated miRNAs might be related with the construction of caveolae, which are supposed to be also indispensable for exosome generation and/or internalization [69]. For example, the relationship among the content of caveolin-1, caveolin-3, and hnRNPA2B1, which may play a related role in discerning transport of miRNAs into some exosomal vesicles, has been recognized [70]. In addition, the decrease of caveolin-3 expression with several miRNAs could also arise the mechanosensitive channel current in myotubes [60,71]. These miRNAs are a class of noncoding RNAs, which are single-stranded RNAs that work as gene regulators on the post-transcriptional level [72]. Interaction of miRNAs with their target RNAs may result in either translational inhibition or degradation of the target mRNAs, which can contribute to the suppression of their resultant protein. Interestingly, muscle-specific ablation of dicer, a key enzyme for the maturation of precursor miRNAs, has shown that miRNAs are also necessary for muscle development [73]. Furthermore, miRNAs have emerged as powerful regulators of skeletal muscle regeneration that might influence many transcriptional pathways [74]. Profiling during myogenic differentiation has shown multiple miRNAs with distinction of expression patterns in the development of skeletal muscle, which might operate as important myogenic regulators. In particular, some specific miRNAs including miR-1, miR-133 and miR-206, could play imperative roles during muscle cell proliferation, differentiation and/or regeneration [75]. Additionally, miR-3074 can regulate myogenic differentiation by targeting caveolin-1 gene, indicating its potential role to progress muscle regeneration [76]. It has been found that exosomes from fibroblasts within muscular dystrophies may exhibit higher levels of miR-199a-5p compared to control exosomes [77]. Exosomes, natural carriers of mRNAs, non-coding RNAs and/or several proteins may vigorously contribute to the cell-cell communication. Interestingly, injection of fibroblast-derived exosomes from the patients of muscular dystrophies into mouse muscle has shown higher fibrosis rather than those of control [77]. Therefore, exosomes created from fibroblasts in the muscle of muscular dystrophies could bring a phenotypic alteration of normal fibroblasts to myofibroblasts thereby enhancing the fibrotic reaction. This transformation may be associated with conveying of miR-199a-5p and/or decreasing of its target caveolin-1, which might suggest potential therapeutic strategy. The mechanisms of caveolae-mediated cellular uptake might encompass other forms of endocytosis including phagocytosis and/or pinocytosis [78]. Extracellular vesicles may contribute to the pathology of muscular dystrophies, which may become a good biomarker for identifying the status of specific pathological processes that occur in dystrophic muscle [79]. In addition again, transfer of muscle progenitor cell-derived exosomes might be a favorable approach for treating muscle diseases such as DMD [80].
5. Possible Treatment Tactics with the Alteration of Gut Microbiota against Muscular Dystrophies
The human gut may emerge to be a key organ of bacterium-host communication for the homeostasis of physical health. In addition to its function as a digestive organ, the human gastrointestinal tract is a home of an extremely complexed gut microbiota consisted by more than 100 trillion participants [81]. It is well-known that gut microbiota may employ various metabolic and/or immunogenic interactions, which could influence on many biological activities including enteric protection, immune barrier function, and/or brain function. These energetic cross talk between gut microbiota and host organs may be an imperative feature of physical homeostasis, which could occasionally provoke several diseases. Several studies have emphasized the robust relationship between gut microbiota and skeletal muscle of muscle dystrophy [82]. In addition, the gut microbiota may play a crucial role in regulating several immune responses. Therefore, the modulation of gut microbiota could contribute to the improvement of immunological disorder by influencing the expression of genes involved in immune cells-dependent inflammatory reactions [83]. Therefore, dysbiosis, known as disruptions of gut microbial composition, may be connected to various immune-mediated and/or autoimmune diseases [84]. In other words, the intricate interactions between the gut microbiota and immune cells are definitely regulated, and dysfunctions in this system could lead to several inflammatory conditions [85]. The commensal population comprising the microbiota may extensively diverge among individuals intensely affected by immune responses and/or host genotypes [86]. Additionally, the microbiota is implicated in the development of obesity and diabetes, which could link the gut to systemic insufficient inflammation and/or different muscular adipogenic pathways [87]. The quality of bacterial species in the gut microbiota may precisely influence on the development of inflammation and/or adiposity in individuals [88]. Interestingly, some studies have correlated the gut microbiota with lifetime cardiovascular risk [20,89]. Short-chain fatty acids (SCFAs) are metabolites made by the gut microbiota with a key role in immune regulation. For example, acetate can enhance the killing activity of macrophages to Streptococcus pneumoniae in an NO-dependent manner [90]. In addition, acetate could commonly improve the bactericidal activity of macrophages. Acetate-induced NOS expression and increased NO production has been revealed in endothelial cells, which is also linked to inflammatory cytokine production [91]. These findings may emphasize the requirement to identify strategies targeting the gut microbial system for the treatment of DMD. In-depth analyses of the functional relationship between food and microbiota composition may contribute to the strategy against various human diseases. Possibly, these diseases may be associated with conditions that indorse intestinal dysbiosis with other pathologic circumstances. For instance, an inactive lifestyle has malicious effects on microbial composition, which may also influence to treatments of patients with DMD [92]. Similar to human patients with DMD, mdx mice may also exhibit amplified gut peristalsis and relatively decreased fecal excretion [93,94,95,96]. Gut microbiota dysbiosis, characterized by reduced microbial diversity and/or increased maleficent bacteria, has been observed in mdx mice. Dysbiosis may cause gut inflammation and immune dysregulation, which exacerbates the muscle damage of DMD [97]. Therefore, an improved strategy targeting gut dysbiosis could help to reduce inflammation and/or could rescue muscle strength [98]. Studies have confirmed the probiotic and anti-gut inflammatory properties in mice fed with high fat diet [99]. Come to think of this, gut microbiota could play a crucial role in connecting food intervention with disease improvement as gut could absolutely be influenced by some diets, which should be worth exploring. The potential clinical utility of these diet modifiers would apply into distinct phenotypes of DMD in the future. Interestingly, it has been directed the clinical heterogeneity of DMD by stratifying based on the seriousness of muscle and brain dysfunction [100].
6. Future Perspectives
Lipid metabolism disorders and fatty infiltration are typical pathological changes in DMD muscle [101]. In particular, diseased muscle exhibits excessive accumulation of fibro-adipogenic progenitors, which cause fibrosis and fatty replacement [102]. In addition, mitochondrial dysfunction, caused by loss of dystrophin, further contributes to lipid deposition in DMD muscle by impairing glycolipid utilization and enhancing oxidative stress [103]. Abnormal lipid accumulation in muscle may lead to the production of detrimental lipid intermediates [104], which in turn exacerbates mitochondrial damage [105]. This creates a vicious cycle of lipid metabolism in DMD muscle. Lipid metabolism disorder means an abnormal alteration in the lipid profile including hypertriglyceridemia, low-density lipoprotein hypercholesterolemia, and lowered high-density lipoprotein cholesterol in the blood, liver and/or other tissues [106]. Unhealthy lifestyles with high-fat diet, too little exercise, and/or alcohol consumption may be more prone to muscular dystrophy [107]. It also modulates gut–liver interactions to improve lipid metabolism by regulating gut microbiota and their metabolites including SCFAs [108]. Skeletal muscle, Adipose tissue, along with the liver are the central metabolic organs that are involved in obesity-associated metabolic disorders via the secretion of adipokines, myokines, and/or hepatokines. It is worth mentioning that these metabolic organs may also discharge several exosomes to communicate with peripheral cells along with distant cells/organs and might control body metabolism and/or homeostasis. Again, it has been shown that probiotics and/or fecal microbiota transplantation (FMT) could improve the gut microbiota structure and decrease the amount of harmful bacteria in patients with hyperlipidemia [109]. Therefore, it is recommended to use probiotics combined with medication to treat patients with muscular dystrophy [109]. (Figure 3) To expedite the translation of fundamental knowledge into clinical applications, the search for innovative biomarkers and disease targets gains fruitful consequence. For example, numerous studies spanning various domains have aimed to pinpoint potential biomarkers for assessment in DMD patients [110]. This endeavor holds the promise of enhancing our understanding and designing innovative therapeutic strategies, ultimately contributing to improved care for individuals grappling with these debilitating diseases.
7. Conclusions
Several microRNAs, NO, NOS, and/or caveolins could be involved in the development of certain muscular dystrophies, in which the association between gut and immune/liver/brain/muscle might play an important role. In addition, the correlation between caveolae and exosome might also influence the development of muscular dystrophies. An in-depth knowledge of the role of microRNAs in exosomes may be valuable for advancing new clinical diagnosis and treatment.
Author Contributions
Conceptualization, MN, SY, NS, and SM; original draft preparation and editing, MN, and SM; visualization, MN and SM; supervision, SM. Each author (MN, NS, SY, and SM) has participated sufficiently in this work of drafting the article and/or revising the article for the important rational content. Then, all authors gave final approval of the version to be submitted. Finally, all authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no competing financial interests.
Abbreviations
| CAV1: caveolin-1 |
| CAV3: caveolin-3 |
| CNS: central nervous system |
| DMD: Duchenne muscular dystrophy |
| FMT: fecal microbiota transplantation |
| iNOS: inducible nitric oxide synthase |
| LGMD: limb-girdle muscular dystrophy |
| mRNA: messenger RNA |
| mdx mice: genetic model of Duchenne muscular dystrophy |
| miRNA: microRNA |
| NO: nitric oxide |
| NOS: nitric oxide synthase |
| QOL: quality of life |
| ROS: reactive oxygen species |
| SCFAs: short-chain fatty acids |
| siRNA: short interference RNA |
| UTR: untranslated region |
References
- Cossu, G.; Sampaolesi, M. New therapies for Duchenne muscular dystrophy: Challenges, prospects and clinical trials. Trends Mol. Med. 2007, 13, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Cho, D.S.; Doles, J.D. Metabolomic analyses reveal extensive progenitor cell deficiencies in a mouse model of duchenne muscular dystrophy. Metabolites. 2018, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Starosta, A.; Konieczny, P. Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell Mol Life Sci. 2021, 78, 4867–4891. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Razini, P.; Erratico, S.; Torrente, Y.; Meregalli, M. Cell based therapy for Duchenne muscular dystrophy. J. Cell Physiol. 2009, 221, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Scripture-Adams, D.D.; Chesmore, K.N.; Barthélémy, F.; Wang, R.T.; Nieves-Rodriguez, S.; Wang, D.W.; Mokhonova, E.I.; Douine, E.D.; Wan, J.; Little, I.; et al. Single nuclei transcriptomics of muscle reveals intra-muscular cell dynamics linked to dystrophin loss and rescue. Commun Biol. 2022, 5, 989. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and nutriceutical approaches to address energy dysregulation and skeletal muscle wasting in Duchenne muscular dystrophy. Nutrients. 2015, 7, 9734–9767. [Google Scholar] [CrossRef]
- Xu, H.; Cai, X.; Xu, K.; Wu, Q.; Xu, B. The metabolomic plasma profile of patients with Duchenne muscular dystrophy: providing new evidence for its pathogenesis. Orphanet J Rare Dis. 2023, 18, 273. [Google Scholar] [CrossRef]
- Lindsay, A.; McCourt, P.M.; Karachunski, P.; Lowe, D.A.; Ervasti, J.M. Xanthine oxidase is hyper-active in Duchenne muscular dystrophy. Free Radic Biol Med. 2018, 129, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Dabaj, I.; Ferey, J.; Marguet, F.; Gilard, V.; Basset, C.; Bahri, Y.; Brehin, A.C.; Vanhulle, C.; Leturcq, F.; Marret, S.; et al. Muscle metabolic remodelling patterns in Duchenne muscular dystrophy revealed by ultra-high-resolution mass spectrometry imaging. Sci Rep. 2021, 11, 1906. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses. 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Cozzoli, A.; Capogrosso, R.F.; Sblendorio, V.T.; Dinardo, M.M.; Jagerschmidt, C.; Namour, F.; Camerino, G.M.; De Luca, A. GLPG0492, a novel selective androgen receptor modulator, improves muscle performance in the exercised-mdx mouse model of muscular dystrophy. Pharmacol Res. 2013, 72, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, Z.; Yu, Y.; Zhang, N.; Jiang, H.; Zhang, G.; Zhang, Z.; Zhang, B. Current pharmacological strategies for Duchenne muscular dystrophy. Front Cell Dev Biol. 2021, 9, 689533. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, D.; Wu, L.; Li, M.; Song, L.; Wei, C.; Lu, C.; Zhang, X.; Hong, S.; Ma, M.; et al. Genotype characterization and delayed loss of ambulation by glucocorticoids in a large cohort of patients with Duchenne muscular dystrophy. Orphanet J. Rare Dis. 2021, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Klein, A.; Rubino, D.; Gocheva, V.; Schmidt, S.; Schroeder, J.; Bernert, G.; Laugel, V.; Steinlin, M.; et al. Effect of combination l-Citrulline and metformin treatment on motor function in patients with Duchenne muscular dystrophy: a Randomized Clinical Trial. JAMA Netw Open. 2019, 2, e1914171. [Google Scholar] [CrossRef]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Montecino-Rodriguez, E.; Dorshkind, K.; Tidball, J.G. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 2001, 98, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.C.; Desjardins, Y.; Furtos, A.; Marcil, V.; Dudonne, S.; Montoudis, A.; Garofalo, C.; Delvin, E.; Marette, A.; Levy, E. Prevention of oxidative stress, inflammation and mitochondrial dysfunction in the intestine by different cranberry phenolic fractions. Clin. Sci. 2015, 128, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.H.; McNally, E.M. Modifier genes and their effect on Duchenne muscular dystrophy. Curr. Opin. Neurol. 2015, 28, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Uryash, A.; Mijares, A.; Esteve, E.; Adams, J.A.; Lopez, J.R. Cardioprotective Effect of Whole Body Periodic Acceleration in Dystrophic Phenotype mdx Rodent. Front Physiol. 2021, 12, 658042. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.R.; McNally, E.M. Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics. Matrix Biol. 2018, 68-69, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Sessa, WC. eNOS at a glance. J Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Beqiri, D.; Shen, J.B.; Redden, J.M.; Dodge-Kafka, K.; Jacobson, K.A.; Liang, B.T. P2X4 receptor-eNOS signaling pathway in cardiac myocytes as a novel protective mechanism in heart failure. Comput. Struct. Biotechnol. J. 2015, 13, 1–7. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Kim, D.Y.; Lim, S.G.; Suk, K.; Lee, W.H. Mitochondrial dysfunction regulates the JAK-STAT pathway via LKB1-mediated AMPK activation ER-stress-independent manner. Biochem. Cell Biol. 2020, 98, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef]
- Terrill, J.R.; Duong, M.N.; Turner, R.; Le Guiner, C.; Boyatzis, A.; Kettle, A.J.; Grounds, M.D.; Arthur, P.G. Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy. Redox Biol. 2016, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Pinniger, G.J.; Graves, J.A.; Grounds, M.D.; Arthur, P.G. Increasing taurine intake and taurine synthesis improves skeletal muscle function in the mdx mouse model for Duchenne muscular dystrophy. J. Physiol. 2016, 594, 3095–3110. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiological reviews. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, P.J.; Sobrino, A.; Laguna-Fernandez, A.; Novella, S.; Tarín, J.J.; García-Pérez, M.A.; Sanchís, J.; Cano, A.; Hermenegildo, C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol Cell Endocrinol. 2011, 335, 96–103. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.; Del Gatto, A.; De Rosa, L.; Romanelli, A.; Pedone, C. Peptides targeting angiogenesis related growth factor receptors. Curr Pharm Des. 2009, 15, 2414–2429. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013, 110, 525–530. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Iwakiri, Y. S-nitrosylation of proteins: a new insight into endothelial cell function regulated by eNOS-derived NO. Nitric Oxide. 2011, 25, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Schwencke, C.; Braun-Dullaeus, R.C.; Wunderlich, C.; Strasser, R.H. Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc. Res. 2006, 70, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012, 729, 51–62. [Google Scholar] [PubMed]
- Ariotti, N.; Parton, R.G. SnapShot: caveolae, caveolins, and cavins. Cell. 2013, 154, 704–704.e1. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Lisanti, M.P. Caveolin-deficient mice: insights into caveolar function human disease. J. Clin. Invest. 2001, 108, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Copeland, C.A.; Tiwari, A.; Kenworthy, A.K. Assembly and turnover of caveolae: what do we really know? Front Cell Dev Biol. 2016, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.; Bai, J.; Gupta-Rossi, N.; Jimenez, A.J.; Delevoye, C.; Lamaze, C.; Echard, A. Caveolae promote successful abscission by controlling intercellular bridge tension during cytokinesis. Sci. Adv. 2022, 8, eabm5095. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R. Critical role of caveolin-1 loss/dysfunction in pulmonary hypertension. Med. Sci (Basel). 2021, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, M.; Nixon, S.J.; Howes, M.T.; Abi-Rached, L.; Wakeham, D.E.; Hanzal-Bayer, M.; Ferguson, C.; Hill, M.M.; Fernandez-Rojo, M.; Brown, D.A.; et al. Evolutionary analysis and molecular dissection of caveola biogenesis. J. Cell Sci. 2008, 121, 2075–2086. [Google Scholar] [CrossRef]
- Karhan, A.N.; Zammouri, J.; Auclair, M.; Capel, E.; Apaydin, F.D.; Ates, F.; Verpont, M.C.; Magré, J.; Fève, B.; Lascols, O.; et al. Biallelic CAV1 null variants induce congenital generalized lipodystrophy with achalasia. Eur. J. Endocrinol. 2021, 185, 841–854. [Google Scholar] [CrossRef]
- Lee, H.; Park, D.S.; Razani, B.; Russell, R.G.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (-/-) null mice show mammary epithelial cell hyperplasia. Am. J. Pathol. 2002, 161, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.K.; Lee, M.G.; Kim, N.H.; Lee, K.Y.; Oh, S.J.; Moon, J.R.; Kim, H.J.; Chi, S.G. Bidirectional alteration of Cav-1 expression is associated with mitogenic conversion of its function in gastric tumor progression. BMC Cancer. 2017, 17, 766. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, B.S.; Proszynski, T.J. A role for caveolin-3 in the pathogenesis of muscular dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.S.; Nisr, R.B.; Stretton, C.; Krasteva-Christ, G.; Hundal, H.S. Caveolin-3 deficiency associated with the dystrophy P104L mutation impairs skeletal muscle mitochondrial form and function. J. Cachexia Sarcopenia Muscle. 2020, 11, 838–858. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E.; Sotgia, F.; Galbiati, F.; Minetti, C.; Lisanti, M.P. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004, 62, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Volonte, D.; Minetti, C.; Chu, J.B.; Lisanti, M.P. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J. Biol. Chem. 1999, 274, 25632–25641. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Woodman, S.E.; Bonuccelli, G.; Capozza, F.; Minetti, C.; Scherer, P.E.; Lisanti, M.P. Phenotypic behavior of caveolin-3 R26Q, a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am. J. Physiol. Cell Physiol. 2003, 285, C1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Deng, Y.; Shang, L.; Yang, L.; Huang, J.; Ma, J.; Liao, X.; Zhou, H.; Xian, J.; Liang, G.; et al. Effect of type 2 diabetes mellitus caveolin-3 K15N mutation on glycometabolism. Exp Ther Med. 2019, 18, 2531–2539. [Google Scholar] [PubMed]
- Morales-Paytuví, F.; Ruiz-Mirapeix, C.; Fajardo, A.; Rae, J.; Bosch, M.; Enrich, C.; Collins, B.M.; Parton, R.G.; Pol, A. Proteostatic regulation of caveolins avoids premature oligomerisation and preserves ER homeostasis. bioRxiv. 2022. [Google Scholar] [CrossRef]
- Huang, H.; Bae, C.; Sachs, F.; Suchyna, T.M. Caveolae regulation of mechanosensitive channel function in myotubes. PLoS One 2013, 8, e72894. [Google Scholar] [CrossRef]
- Reilly, S.N.; Liu, X.; Carnicer, R.; Recalde, A.; Muszkiewicz, A.; Jayaram, R.; Carena, M.C.; Wijesurendra, R.; Stefanini, M.; Surdo, N.C.; et al. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci Transl Med. 2016, 8, 340ra374. [Google Scholar] [CrossRef] [PubMed]
- Groh, W.J.; Bhakta, D.; Tomaselli, G.F.; Aleong, R.G.; Teixeira, R.A.; Amato, A.; Asirvatham, S.J.; Cha, Y.M.; Corrado, D.; Duboc, D.; et al. 2022 HRS expert consensus statement on evaluation and management of arrhythmic risk in neuromuscular disorders. Heart Rhythm. 2022, 19, e61–e120. [Google Scholar] [CrossRef] [PubMed]
- Llano-Diez, M.; Ortez, C.I.; Gay, J.A.; Alvarez-Cabado, L.; Jou, C.; Medina, J.; Nascimento, A.; Jimenez-Mallebrera, C. Digital PCR quantification of miR-30c and miR-181a as serum biomarkers for Duchenne muscular dystrophy. Neuromuscul. Disord. NMD. 2017, 27, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Nakamura, A.; Aoki, Y.; Ito, N.; Kishi, S.; Yamamoto, K.; Sekiguchi, M.; Takeda, S.; Hashido, K. Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: Promising novel blood-based markers for muscular dystrophy. PLoS ONE. 2011, 6, e18388. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.N.; Hooper, C.; Stefanini, M.; Vrellaku, B.; Carnicer, R.; Wood, M.J.; Simon, J.N.; Casadei, B. Why is early-onset atrial fibrillation uncommon in patients with Duchenne muscular dystrophy? Insights from the mdx mouse. Cardiovasc Res. 2024, 120, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.N.; Liu, X.; Carnicer, R.; Recalde, A.; Muszkiewicz, A.; Jayaram, R.; Carena, M.C.; Wijesurendra, R.; Stefanini, M.; Surdo, N.C.; et al. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci Transl Med. 2016, 8, 340ra374. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Demady, D.R.; Osawa, Y. Ubiquitination of neuronal nitric-oxide synthase in vitro and in vivo. J Biol Chem. 2000, 275, 17407–17411. [Google Scholar] [CrossRef] [PubMed]
- Paula, S.M.; Fernandes, T.; Couto, G.K.; Jordão, M.T.; Oliveira, E.M.; Michelini, L.C.; Rossoni, L.V. Molecular Pathways Involved in Aerobic Exercise Training Enhance Vascular Relaxation. Med Sci Sports Exerc. 2020, 52, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Huang-Doran, I.; Zhang, C.Y.; Vidal-Puig, A. Extracellular vesicles: novel mediators of cell communication in metabolic disease. Trends Endocrinol Metab. 2017, 28, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Arroyo-Campuzano, M.; Flores-García, M.; Patlán, M.; Hernández-Díazcouder, A.; Alcántara, D.; Ramírez-Camacho, I.; Arana-Hidalgo, D.; Soria-Castro, E.; Sánchez, F.; et al. Citicoline Modifies the Expression of Specific miRNAs Related to Cardioprotection in Patients with ST-Segment Elevation Myocardial Infarction Subjected to Coronary Angioplasty. Pharmaceuticals (Basel). 2022, 15, 925. [Google Scholar] [CrossRef]
- Benzoni, P.; Gazzerro, E.; Fiorillo, C.; Baratto, S.; Bartolucci, C.; Severi, S.; Milanesi, R.; Lippi, M.; Langione, M.; Murano, C.; et al. Caveolin-3 and Caveolin-1 Interaction Decreases Channel Dysfunction Due to Caveolin-3 Mutations. Int J Mol Sci. 2024, 25, 980. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature. 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- O'Rourke, J.R.; Georges, S.A.; Seay, H.R.; Tapscott, S.J.; McManus, M.T.; Goldhamer, D.J.; Swanson, M.S.; Harfe, B.D. Essential role for Dicer during skeletal muscle development. Dev Biol. 2007, 311, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012, 482, 524–528. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J. The MyomiR network in skeletal muscle plasticity. Exerc Sport Sci Rev. 2011, 39, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, A.; Khorram Khorshid, H.R.; Mowla, S.J.; Mowla, S.J.; Mohebbi, H.A.; Mohammadian, A.; Yaseri, M.; Solaymani-Dodaran, M.; Sherafatian, M.; Tavallaie, M. Altered miR-223 Expression in Sputum for Diagnosis of Non-Small Cell Lung Cancer. Avicenna J Med Biotechnol. 2017, 9, 189–195. [Google Scholar] [PubMed]
- Zanotti, S.; Gibertini, S.; Blasevich, F.; Bragato, C.; Ruggieri, A.; Saredi, S.; Fabbri, M.; Bernasconi, P.; Maggi, L.; Mantegazza, R.; et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018, 74, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Liu, S.; Feng, X.; Luo, W.; Zhang, H.; Wu, Y.; Chen, S.; Shang, X. The Revolution of exosomes: From biological functions to therapeutic applications in skeletal muscle diseases. J Orthop Translat. 2024, 45, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yedigaryan, L.; Sampaolesi, M. Extracellular vesicles and Duchenne muscular dystrophy pathology: Modulators of disease progression. Front Physiol. 2023, 14, 1130063. [Google Scholar] [CrossRef]
- Su, X.; Shen, Y.; Kim, I.M.; Weintraub, N.L.; Hamrick, M.; Tang, Y. Extracellular Vesicles for Muscle Atrophy Treatment. Adv Exp Med Biol. 2023, 1418, 119–126. [Google Scholar]
- Shapira, M. Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Jollet, M.; Mariadassou, M.; Rué, O.; Pessemesse, L.; Ollendorff, V.; Ramdani, S.; Vernus, B.; Bonnieu, A.; Bertrand-Gaday, C.; Goustard, B.; et al. Insight into the Role of Gut Microbiota in Duchenne Muscular Dystrophy: An Age-Related Study in Mdx Mice. Am J Pathol. 2024, 194, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Kosiewicz, M.M.; Zirnheld, A.L.; Alard, P. Gut microbiota, immunity, and disease: A complex relationship. Front. Microbiol. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.M. Introduction to the special focus issue on the impact of diet on gut microbiota composition and function and future opportunities for nutritional modulation of the gut microbiome to improve human health. Gut Microbes. 2017, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013, 500, 541–546. [Google Scholar]
- Heiskanen, M.A.; Aatsinki, A.; Hakonen, P.; Kartiosuo, N.; Munukka, E.; Lahti, L.; Keskitalo, A.; Huovinen, P.; Niinikoski, H.; Viikari, J.; et al. Association of Long-Term Habitual Dietary Fiber Intake since Infancy with Gut Microbiota Composition in Young Adulthood. J Nutr. 2024, 154, 744–754. [Google Scholar] [CrossRef]
- Machado, M.G.; Patente, T.A.; Rouillé, Y.; Heumel, S.; Melo, E.M.; Deruyter, L.; Pourcet, B.; Sencio, V.; Teixeira, M.M.; Trottein, F. Acetate Improves the Killing of Streptococcus pneumoniae by Alveolar Macrophages via NLRP3 Inflammasome and Glycolysis-HIF-1α Axis. Front Immunol. 2022, 13, 773261. [Google Scholar] [CrossRef]
- Noris, M.; Todeschini, M.; Casiraghi, F.; Roccatello, D.; Martina, G.; Minetti, L.; Imberti, B.; Gaspari, F.; Atti, M.; Remuzzi, G. Effect of Acetate, Bicarbonate Dialysis, and Acetate-Free Biofiltration on Nitric Oxide Synthesis: Implications for Dialysis Hypotension. Am J Kidney Dis. 1998, 32, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Pérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; González-Soltero, R.; Larrosa, M. Gut microbiota modification: another piece in the puzzle of the benefits of physical exercise in health? Front Physiol 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Baccari, M.C.; Nistri, S.; Vannucchi, M.G.; Calamai, F.; Bani, D. Reversal by relaxin of altered ileal spontaneous contractions in dystrophic (mdx) mice through a nitric oxide-mediated mechanism. Am J Physiol Regul Integr Comp Physiol 2007, 293, R662–R668. [Google Scholar] [CrossRef] [PubMed]
- Mulè, F.; Amato, A.; Serio, R. Gastric emptying, small intestinal transit and fecal output in dystrophic (mdx) mice. J Physiol Sci. 2010, 60, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Swiderski, K.; Bindon, R.; Trieu, J.; Naim, T.; Schokman, S.; Swaminathan, M.; Leembruggen, A.J.L.; Hill-Yardin, E.L.; Koopman, R.; Bornstein, J.C.; et al. Spatiotemporal mapping reveals regional gastrointestinal dysfunction in mdx dystrophic mice ameliorated by oral L-arginine supplementation. J Neurogastroenterol Motil. 2020, 26, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Tonali, P.; Servidei, S.; Azzena, G.B. Analysis of peristaltic reflex in young mdx dystrophic mice. Neurosci Lett. 1995, 192, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Tripodi, L.; Villa, C.; Strati, F.; Facoetti, A.; Baselli, G.; Troisi, J.; Landolfi, A.; Lonati, C.; Molinaro, D.; et al. Microbiota dysbiosis influences immune system and muscle pathophysiology of dystrophin-deficient mice. EMBO Mol. Med. 2023, 15, e16244. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, H.; Pagano, E.; Paris, D.; Panza, E.; Cuozzo, M.; Moriello, C.; Piscitelli, F.; Abolghasemi, A.; Gazzerro, E.; Silvestri, C.; et al. Targeting gut dysbiosis against inflammation and impaired autophagy in Duchenne muscular dystrophy. EMBO Mol. Med. 2023, 15, e16225. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ji, Y.; Cheng, Q.; Zhu, Y.; Zhang, H.; Guo, Y.; Cao, X.; Wang, H. Dietary astaxanthin-rich extract ameliorates atherosclerosis/retinopathy and restructures gut microbiome in apolipoprotein E-deficient mice fed on a high-fat diet. Food Funct. 2022, 13, 10461–10475. [Google Scholar] [CrossRef]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE. 2009, 4, e4347. [Google Scholar] [CrossRef]
- Srivastava, N.K.; Yadav, R.; Mukherjee, S.; Pal, L.; Sinha, N. Abnormal lipid metabolism in skeletal muscle tissue of patients with muscular dystrophy: In vitro, high-resolution NMR spectroscopy based observation in early phase of the disease. Magn. Reson. Imaging. 2017, 38, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Cardone, N.; Taglietti, V.; Baratto, S.; Kefi, K.; Periou, B.; Gitiaux, C.; Barnerias, C.; Lafuste, P.; Pharm, F.L.; Pharm, J.N.; et al. Myopathologic trajectory in Duchenne muscular dystrophy (DMD) reveals lack of regeneration due to senescence in satellite cells. Acta Neuropathol. Commun. 2023, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2019, 20, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Li, K.; Peng, X.; Kan, Y.; Li, H.; Zhu, Y.; Wang, Z.; Li, Z.; Liu, H.Y.; Cai, D. Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients. 2023, 15, 4772. [Google Scholar] [CrossRef]
- Li, L.; Guo, W.L.; Zhang, W.; Xu, J.X.; Qian, M.; Bai, W.D.; Zhang, Y.Y.; Rao, P.F.; Ni, L.; Lv, X.C. Grifola Frondosa Polysaccharides Ameliorate Lipid Metabolic Disorders and Gut Microbiota Dysbiosis in High-Fat Diet Fed Rats. Food Funct. 2019, 10, 2560–2572. [Google Scholar] [CrossRef]
- Cao, S.; Liu, M.; Han, Y.; Li, S.; Zhu, X.; Li, D.; Shi, Y.; Liu, B. Effects of Saponins on Lipid Metabolism: The Gut-Liver Axis Plays a Key Role. Nutrients. 2024, 16, 1514. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wu, G.; Zhao, X.; Zhang, H.; Ren, M.; Song, X.; Chang, H.; Jing, Z. Probiotics combined with atorvastatin administration in the treatment of hyperlipidemia: A randomized, double-blind, placebo-controlled clinical trial. Medicine (Baltimore). 2024, 103, e37883. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Spitali, P. Circulating Biomarkers for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, S49–S58. [Google Scholar] [CrossRef]
Figure 1.
Representation of the critical factors for the development of muscular dystrophies. Muscular dystrophies are a group of illnesses that are triggering progressive loss of muscle mass. Several genetic mutations are well-known for interfering with the production of molecules required to develop healthy muscle. There are various types of muscular dystrophy. Abbreviation, DMD: Duchenne muscular dystrophy, LGMD: limb-girdle muscular dystrophy.
Figure 1.
Representation of the critical factors for the development of muscular dystrophies. Muscular dystrophies are a group of illnesses that are triggering progressive loss of muscle mass. Several genetic mutations are well-known for interfering with the production of molecules required to develop healthy muscle. There are various types of muscular dystrophy. Abbreviation, DMD: Duchenne muscular dystrophy, LGMD: limb-girdle muscular dystrophy.
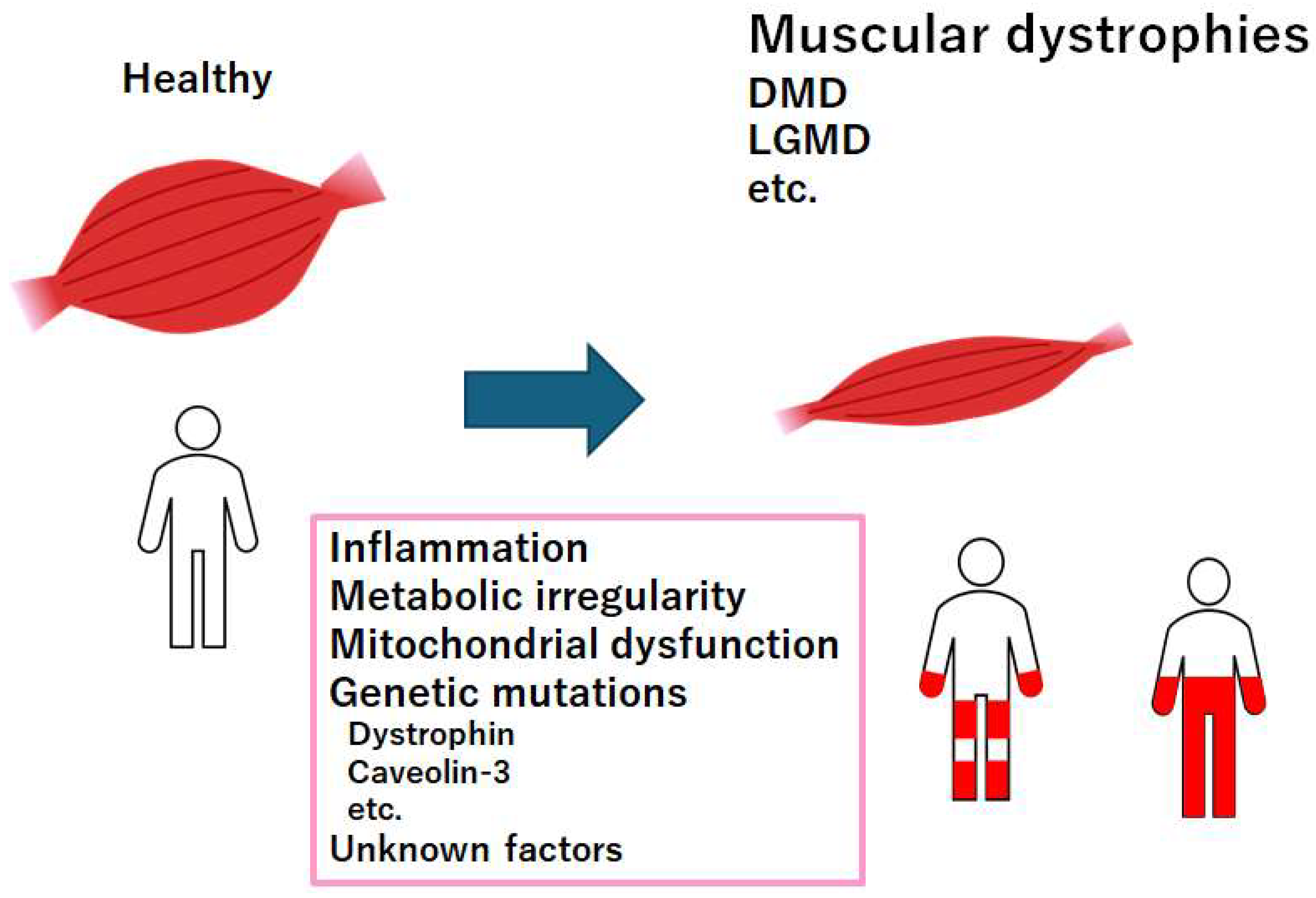
Figure 2.
A diagrammatic representation of caveolae associated with caveolin proteins. Caveolae may be a kind of platforms for various signaling and/or even exosomes, which may be related to the scaffold domain of caveolins. Note that some critical pathways for the development of various diseases have been omitted for clarity. Abbreviation, DMD: Duchenne muscular dystrophy, LGMD: limb-girdle muscular dystrophy.
Figure 2.
A diagrammatic representation of caveolae associated with caveolin proteins. Caveolae may be a kind of platforms for various signaling and/or even exosomes, which may be related to the scaffold domain of caveolins. Note that some critical pathways for the development of various diseases have been omitted for clarity. Abbreviation, DMD: Duchenne muscular dystrophy, LGMD: limb-girdle muscular dystrophy.
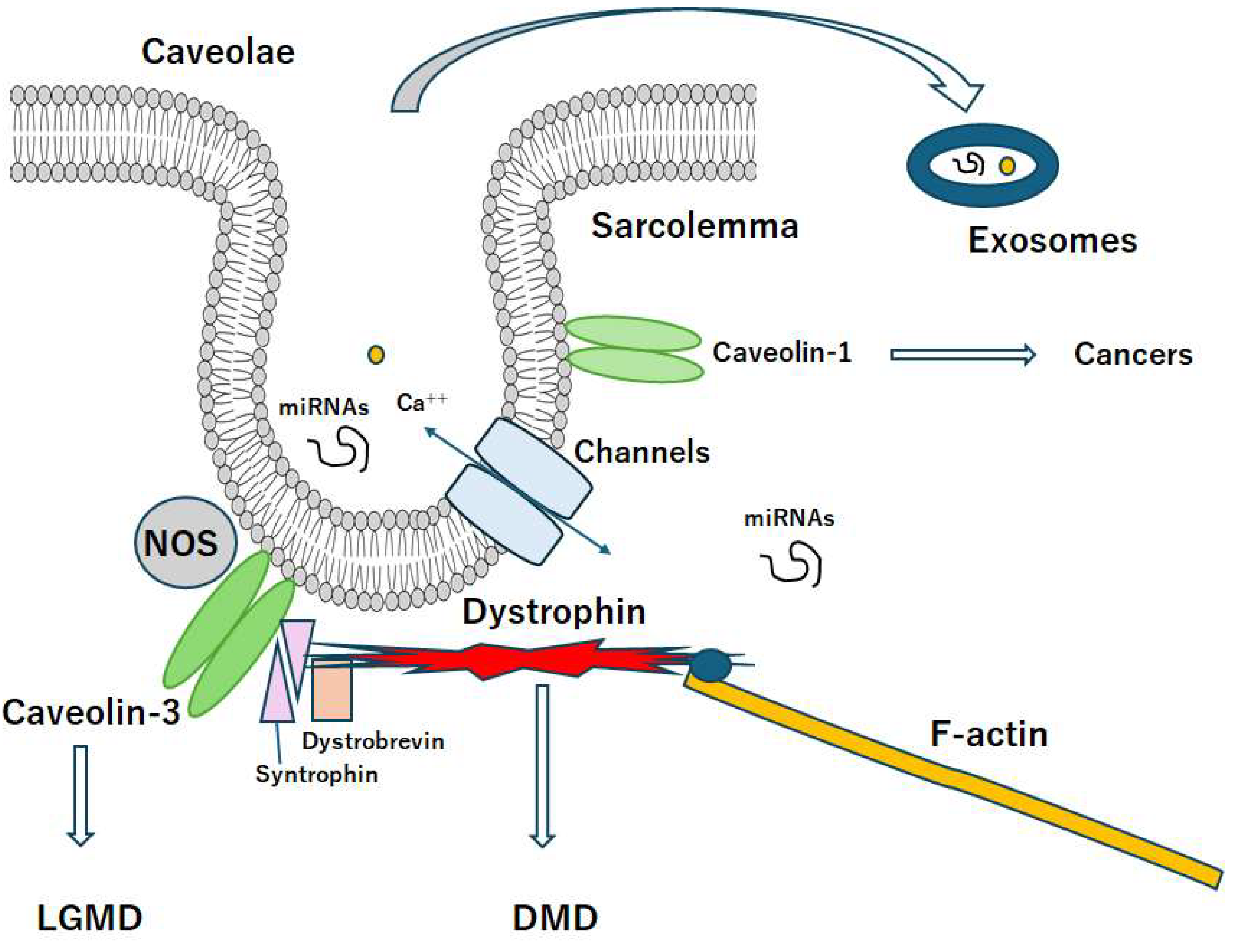
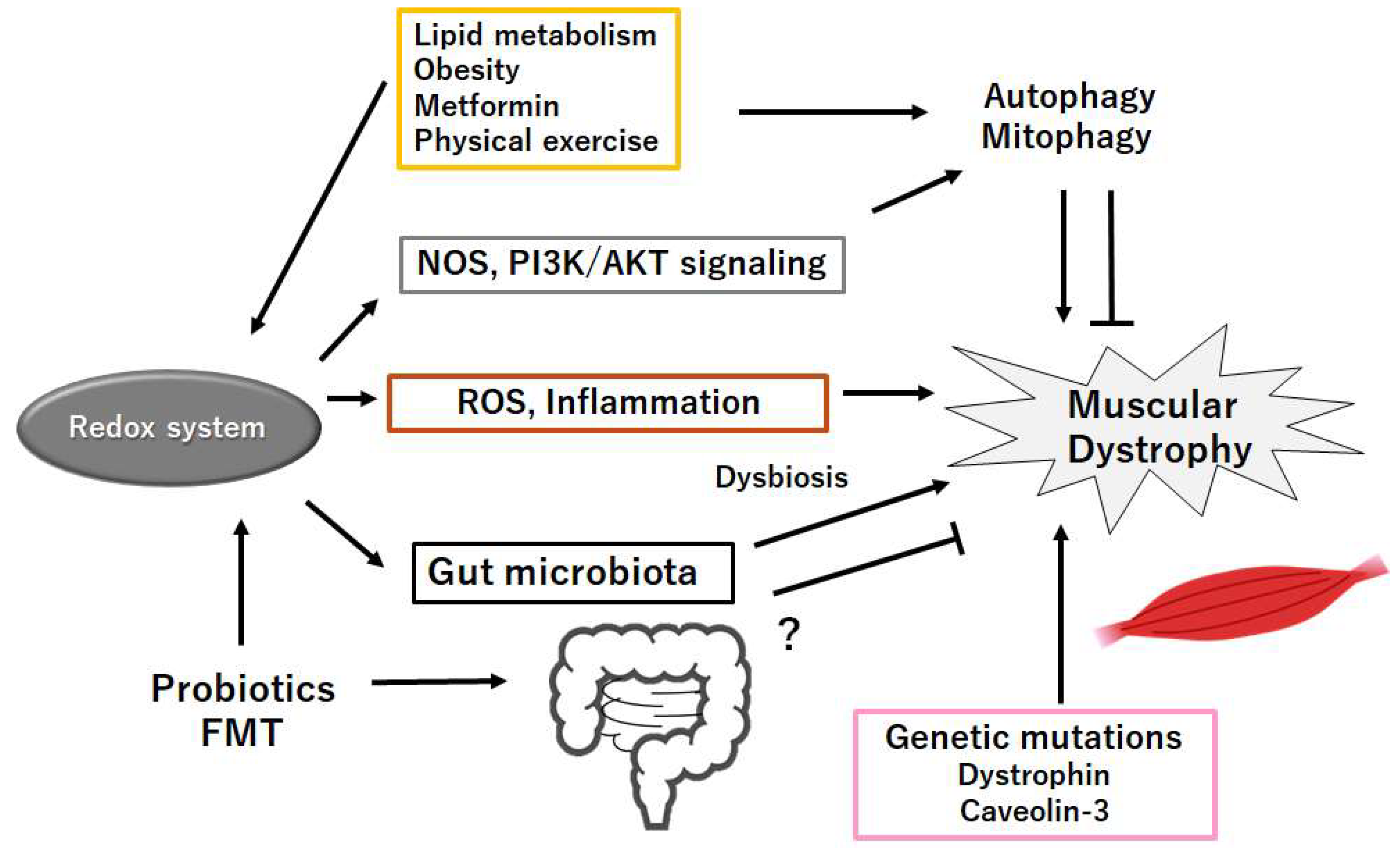
Figure 3.
Schematic representation of the probable inhibitory tactics against the pathogenesis of muscular dystrophies. Probiotics and/or fecal microbiota transplantation (FMT) might contribute to the alteration of gut microta for the production of SCFAs and/or several miRNAs, which could be beneficial for the treatment of muscular dystrophies. Example factors including metformin as well as exercise known to act on the autophagy signaling are also shown. Note that several important activities such as inflammatory reaction, autophagy initiation, and reactive oxygen species (ROS) production have been omitted for clarity. “?” means for author speculation.
Figure 3.
Schematic representation of the probable inhibitory tactics against the pathogenesis of muscular dystrophies. Probiotics and/or fecal microbiota transplantation (FMT) might contribute to the alteration of gut microta for the production of SCFAs and/or several miRNAs, which could be beneficial for the treatment of muscular dystrophies. Example factors including metformin as well as exercise known to act on the autophagy signaling are also shown. Note that several important activities such as inflammatory reaction, autophagy initiation, and reactive oxygen species (ROS) production have been omitted for clarity. “?” means for author speculation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



