
Submitted:
30 June 2024
Posted:
01 July 2024
You are already at the latest version
Abstract
Glycogen storage disorders (GSDs) are a group of inherited metabolic disorders characterized by defects in enzymes involved in glycogen metabolism. Deficiencies in enzymes responsible for glycogen breakdown and synthesis can impair mitochondrial function. For instance, in GSD type II (Pompe disease), acid alpha-glucosidase deficiency leads to lysosomal glycogen accumulation, which secondarily impacts mitochondrial function through dysfunctional mitophagy, which disrupts mitochondrial quality control generating oxidative stress. In GSD type III (Cori disease), the lack of the debranching enzyme causes glycogen accumulation and affects mitochondrial dynamics and biogenesis by disrupting the integrity of muscle fibers. Malfunctional glycogen metabolism can disrupt various cascades thus causing mitochondrial and cell metabolic dysfunction through various mechanisms. These dysfunctions include altered mitochondrial morphology, impaired oxidative phosphorylation, increased production of reactive oxygen species (ROS), and defective mitophagy. The oxidative burden typical of GSDs compromises mitochondrial integrity and exacerbates the metabolic derangements observed in GSDs. The intertwining of mitochondrial dysfunction and GSDs underscores the complexity of these disorders and has significant clinical implications. GSD patients often present with multisystem manifestations, including hepatomegaly, hypoglycemia, and muscle weakness, which can be exacerbated by mitochondrial impairment. Moreover, mitochondrial dysfunction may contribute to the progression of GSD-related complications, such as cardiomyopathy and neurocognitive deficits. Targeting mitochondrial dysfunction thus represents a promising therapeutic avenue in GSDs. Potential strategies include antioxidants to mitigate oxidative stress, compounds that enhance mitochondrial biogenesis, and gene therapy to correct the underlying mitochondrial enzyme deficiencies. Mitochondrial dysfunction plays a critical role in the pathophysiology of GSDs. Recognizing and addressing this aspect can lead to more comprehensive and effective treatments, improving the quality of life of GSD patients.
This review aims to elaborate on the intricate relationship between mitochondrial dysfunction and various types of GSDs. The review presents challenges and treatment options for several GSD
Keywords:
mitochondrial dysfunction
; glycogen storage disorders
; reactive oxygen species
; oxidative stress
; autophagy and mitophagy
; myopathy
1. Background
Glycogen storage disorders (GSDs) are a group of inherited glycogen metabolic disorders caused by deficiency of enzymes involved in the synthesis or degradation of glycogen. Most GSDs are congenital and can present at any age from newborn to adulthood [1]. GSDs primarily affect the liver and skeletal muscles, where glycogen is predominantly stored, however, depending on the specific enzyme deficiency and its distribution across various tissues, other organs, including heart, kidneys, and brain can also be affected [2,3,4]. GSDs classified according to the specific enzyme deficiency and the affected organ like GSD 0a, GSD 0b, GSD I, GSD II (Pompe disease), GSD III, GSD IV, GSD V, GSD VI, GSD VII, GSD IX, GSD X, GSD XI (lactate dehydrogenase subunit A deficiency), GSD XII, GSD XIII, phosphogluco-mutase 1 deficiency–congenital disorder of glycosylation (PGM1-CDG; previously called GSD XIV), GSD XV, Fanconi–Bickel syndrome (FBS) and phosphoglycerate kinase (PGK) deficiency [1,4]. Mitochondria are essential organelles known for their role in ATP production through OXPHOS, regulation of cellular metabolism, and involvement in apoptosis. Impairment in mitochondrial function can have widespread effects on cellular energy status and metabolic homeostasis [2,5,6]. In GSDs, mitochondrial dysfunction can exacerbate the metabolic derangements caused by impaired glycogen metabolism, leading to a more complex pathological conditions and influencing disease outcomes [7,8,9]. Mitochondrial dysfunction has emerged as a critical component in the pathology of various GSDs. The abnormal accumulation or deficient production of glycogen, primarily in the liver and muscles, resulting in a spectrum of clinical manifestations. Although traditionally the focus has been on the direct consequences of enzyme deficiencies on glycogen metabolism, recent studies have shed light on the significant role of mitochondrial dysfunction in the progression and severity of GSDs. One of the most extensively studied GSDs with respect to mitochondrial dysfunction is GSD type II (Pompe disease) [10]. Pompe disease results from mutations in the acid alpha-glucosidase (GAA) gene, leading to the accumulation of glycogen within lysosomes. This lysosomal storage disorder primarily affects muscle cells, including cardiac and skeletal muscles. Recent research has indicated that secondary mitochondrial dysfunction plays a pivotal role in the disease’s pathophysiology. The accumulated glycogen disrupts normal cellular processes, leading to mitochondrial swelling, reduced mitochondrial membrane potential, and impaired OXPHOS. These mitochondrial abnormalities contribute to the muscle weakness and respiratory complications characteristic of Pompe disease. Similarly, GSD III (Cori disease) is caused by a deficiency in the debranching enzyme amylo-1,6-glucosidase, accumulating abnormal glycogen with shorter outer branches. This condition not only affects liver and muscle tissues but also shows a significant impact on mitochondrial function. Studies have demonstrated that patients with GSD III exhibit reduced activities of mitochondrial respiratory chain complexes, increased oxidative stress, and altered mitochondrial morphology. These mitochondrial impairments are believed to contribute to the muscle weakness, cardiomyopathy, and hepatic dysfunction observed in GSD III patients [11,12]. The interplay between glycogen storage and mitochondrial dysfunction is also evident in GSD V, (McArdle disease) caused by deficiency in myophosphorylase, an enzyme crucial for glycogen breakdown in muscle cells [13]. The inability to mobilize glycogen during exercise leads to muscle cramps, fatigue, and, in severe cases, rhabdomyolysis. Recent studies have highlighted that mitochondrial dysfunction, characterized by decreased mitochondrial biogenesis and impaired oxidative metabolism, exacerbates the exercise intolerance seen in McArdle disease. Mitochondrial dysfunction has emerged as a critical component in the pathophysiology of several GSDs, influencing the clinical manifestations and therapeutic approaches.
Based on an extensive literature search using keywords like mitochondrial dysfunction in GSDs, mitochondrial impairment and GSDs, metabolic disorders and GSDs, mitochondria-related myopathy, and autophagy in GSDs, we went through the literature and following major abnormalities related to mitochondrial dysfunction across different GSDs have been reported. This review provides an extensive overview of mitochondrial dysfunction in GSDs, discussing the underlying mechanisms, clinical implications, and potential therapeutic strategies.
2. Glycogen Storage Disorders (GSDs) an Overview
GSDs primarily affect the liver and skeletal muscles due to the high abundance of glycogen in these tissues. Glucose and glycogen convert into one another via synthesis or degradation through various steps in the glycogen metabolism pathways chiefly in the liver and skeletal muscles, catalyzed by different metabolic enzymes. Mutations in genes encoding individual enzymes in the glycogen metabolism pathway led to GSDs, depending on the enzyme defect and its relative expression in the liver, skeletal muscle as well as in the kidney, heart, and brain also, clinical manifestations of GSDs vary from one disorder to the other. Liver GSDs are commonly caused by fasting hypoglycemia and/or hepatomegaly, muscle GSDs are present in one of two different ways: exercise intolerance with rhabdomyolysis or fixed muscle weakness without rhabdomyolysis. GSDs are categorized based on the specific enzyme deficiency and the resultant metabolic abnormalities, details are summarized in (Table 1).
3. Mitochondrial Metabolic Dysfunction
Mitochondria are essential organelles responsible for energy production through oxidative phosphorylation (OXPHOS), regulation of apoptosis, and maintenance of cellular metabolism. Mitochondria also play a role in calcium homeostasis, reactive oxygen species (ROS) production, and biosynthesis of certain macromolecules. Mitochondria provide most of the cell’s oxidative metabolism and energy production and are the major sites of the tri-carboxylic cycle (TCA cycle), OXPHOS, and fatty acid β-oxidation [49]. Primary mitochondrial impairment leads to cardiomyopathy, progressive muscular and neuro-degeneration. OXPHOS involves a series of oxidation-reduction reactions performed by five protein complexes present in the inner mitochondrial membrane: complex I (CI) also known as NADH: ubiquinone oxidoreductase accepts electrons from NADH and reduces ubiquinone (CoQ), which serves as the substrate for complex III; complex II (CII) or succinate: ubiquinone oxidoreductase accepts electrons from FADH2 also reduces CoQ; complex III (CIII) called ubiquinol: ferricytochrome c oxidoreductase, it transfers electrons from reduced ubiquinone (ubiquinol) to cytochrome c; complex IV (CIV) also called cytochrome c oxidase passes electrons from cytochrome c to molecular oxygen (O2), reducing it to form water (H2O) and complex V (CV) known as FoF1-ATP synthetase utilizes the proton gradient generated by the other complexes to synthesize ATP from ADP and inorganic phosphate [6,50]. Defective OXPHOS can lead to an overproduction of reactive oxygen species (ROS) that damage DNA, proteins, and lipid membranes [51]. Lactate dehydrogenase (LDHD) convert Pyruvate into Lacate and vice versa during glycogen metabolism, its deficiency causes GSD XI. A case study reported elevated D-lactate levels due to LDHD cause complex IV deficiency [34]. Mitochondrial disorders may stem from pathogenic mutations in either mitochondrial DNA (mtDNA) or nuclear DNA (nDNA), giving rise to diverse inheritance patterns such as maternal, autosomal dominant, autosomal recessive, and X-linked. These mutations disrupt normal mitochondrial function, leading to a spectrum of clinical presentations and inheritance modes that vary in severity and manifestation [52]. Mutations in OXPHOS complexes proteins can cause severe pathological manifestations from the neonatal period to adult-onset, including fatal infantile lactic acidosis, infancy/early childhood onset neuropathological disorders [53], cardiomyopathy, liver disease, and myopathy [54]. Mitochondrial impairment was found in patients with GSD I, GSD III, GSD VI, and GSD IX by examining enzymatic activities in lymphocytes. The results revealed significant alterations in enzyme activity profiles across all GSD types. Notably, reduced activity of succinate dehydrogenase (a component of complex II of the respiratory chain) and glycerol-3-phosphate dehydrogenase (involved in glycolysis) was observed. Conversely, increased enzymatic activity of NADH dehydrogenase and lactate dehydrogenase (LDH) was noted in all GSD types, with the most pronounced changes found in GSD I [55]. These findings provide evidence of mitochondrial dysfunction in various forms of GSDs, highlighting the complex interplay between metabolic pathways and mitochondrial function in these disorders (Figure 1). GSDs disrupt the normal metabolism of glycogen, leading to impaired synthesis, breakdown, and transport of this essential carbohydrate. Since carbohydrates serve as major energy sources for cells, researchers have investigated the involvement of mitochondria and other energy-producing pathways in GSD pathogenesis. A reduction in the use of carbohydrates as fuel and reduced regeneration of phosphorylated molecules in mitochondria during exercise has been reported [56,57,58]. GSD Ia patient fibroblast exhibited a lower concentration of malate, responsible for translocating electrons produced during glycolysis across the semipermeable inner membrane of the mitochondrion for OXPHOS [59]. Peripheral lymphocytes from GSD I patients have shown decreased activity of succinate dehydrogenase and NADH dehydrogenase [55]. A previous study has indicated the presence of indirect markers of TCA cycle and fatty acid oxidation (FAO) overload in the urine and plasma of GSD Ia patients. [60]. Accumulation of free carnitine and long-chain acylcarnitines, a transporter of fatty acids into the mitochondria for subsequent β-oxidation was found and also increased excretion of various TCA cycle metabolites was observed in urine of GSD I patients [61,62]. The cause of mitochondrial dysfunction is not fully known, decrease in mitochondrial oxidation could result solely from reduced mitochondrial content. In GSD Ia animal model of G6pc−/− mice, several mitochondrial dysfunctions have been identified in the liver, including reduced mitochondrial content, abnormal morphology, impaired respiration, and disturbed TCA cycle function. Significant abnormalities in liver mitochondria were observed included reduced mitochondrial content, irregular ultrastructure of cristae (the internal membranes of mitochondria), and overall morphological alterations. Additionally, there are indications of disrupted mitophagy, the process by which cells remove damaged or dysfunctional mitochondria. The ultrastructural analyses of knockout (KO) mice and knockdown (KD) AML-12 cells show a significant number of damaged mitochondria. The changes in membrane potential also cause defects in membrane structure, possibly due to altered or imbalanced incorporation of fatty acids into the mitochondrial membrane [12]. Glucotoxicity or lipotoxicity due to lipid species such as saturated free fatty acids and ceramides might also mediate mitochondrial damage, it is possible that the excessive intracellular carbohydrates or lipids could lead to mitochondrial dysfunction [63,64]. Mitochondrial dynamics, including fusion and fission, are crucial for maintaining mitochondrial function and integrity [65]. In several GSDs, abnormal glycogen accumulation disrupts these processes, in GSD III, the accumulation of structurally abnormal glycogen affects mitochondrial morphology, leading to impaired fusion and fission. This results in dysfunctional mitochondria that are less efficient in energy production and more prone to oxidative damage., [12]. Progressive skeletal muscle hypotonia and cardiac hypertrophy are classical manifestations of infantile-onset Pompe disease [66]. Mitochondria play a crucial role in cellular metabolism, patients with hypertrophic cardiomyopathy, disrupted metabolic signaling and mitochondrial dysfunction have been reported [67]. Damaged mitochondria impair ATP synthase activity and ATP production by reducing the inner membrane potential, resulting in energy deficiency and contractile failure in muscle tissues [68].Huang et al. carried out a study in Pompe disease-specific induced pluripotent stem cell (Pompe disease-iCMs), exhibited a significant reduction in OXPHOS compared to control- iCMs which was partially ameliorated by treatment with recombinant alpha-glucosidase (rhGAA), An increased basal respiration, maximal respiration, and spare respiratory capacity, along with a subsequent rise in proton leakage and ATP production was also observed. Pompe disease-iCMs had a depolarized mitochondrial membrane potential and elevated reactive oxygen species (ROS) levels relative to Ctrl-iCMs, both of which were also mitigated by rhGAA treatment. These findings indicate that, in addition to reducing lysosomal glycogen accumulation, rhGAA can partially restore mitochondrial biogenesis and function, potentially addressing the cardiac pathology observed in Pompe disease [8] another study reported markedly swollen cristae and mitochondrial dysfunction, including decreased glycolysis and OXPHOS in Pompe disease -iCMs [69]. Lim et al. identified mitochondrial dysfunction associated with abnormal energy metabolism in skeletal muscle biopsies of patients and in animal models of Pompe disease [70].
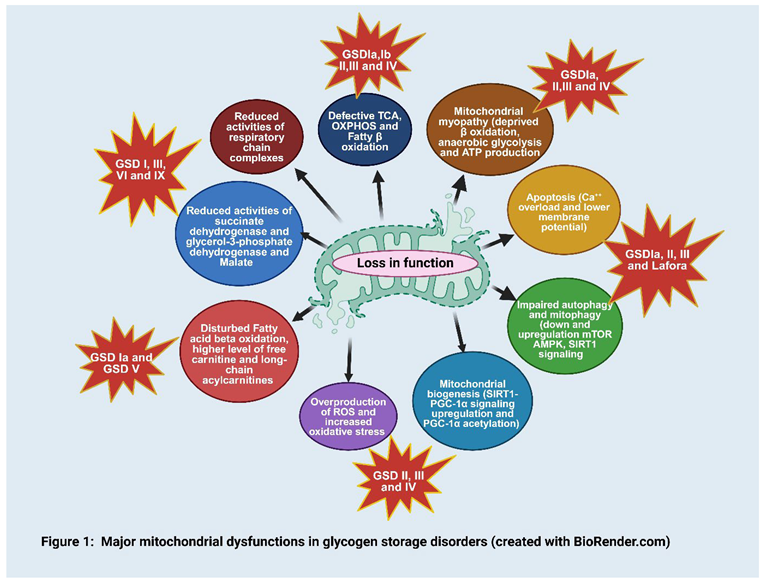
3.1. Overproduction of ROS and Oxidative Stress
Besides ATP production, mitochondria are also a key source of reactive oxygen species (ROS) [71]. ATP is generated through the electron transport chain (ETC), where high-energy electrons are transferred to oxygen in a controlled manner. Occasionally, some electrons deviate from this path and directly react with oxygen, forming superoxide radicals. These superoxide radicals can then be converted, either enzymatically or spontaneously, into hydrogen peroxide [72]. Hydrogen peroxide can participate in the Fenton reaction to produce hydroxyl radicals, which are highly reactive and capable of causing significant damage to cellular structures such as membranes, proteins, enzymes, and DNA, potentially leading to cell death [73,74]. To mitigate ROS damage, mitochondria possess a sophisticated antioxidant defense system. Superoxide dismutase (SOD) converts superoxide radicals into hydrogen peroxide, which is then reduced to water by glutathione peroxidase in the presence of reduced glutathione. This process effectively reduces the formation of harmful hydroxyl radicals, thereby scavenging most ROS generated within the mitochondria. Additionally, mitochondria assist in scavenging ROS originating from other cellular organelles, like peroxisomes [75]. However, mitochondrial dysfunction can result in excessive ROS production, causing cellular damage. When ROS production exceeds the capacity of the cellular antioxidant defenses, oxidative stress ensues, leading to damage of cellular macromolecules and impairment of cellular functions and viability [76]. Oxidative stress is increasingly recognized as a pivotal mechanism in the development of various diseases, including metabolic syndrome (MetS). Mitochondria are crucial in maintaining appropriate ROS levels within cells. An increased oxidative stress exacerbates mitochondrial dysfunction by damaging components of the electron transport chain (ETC) and other mitochondrial constituents, it enhances mitochondrial fragmentation and further decline in OXPHOS. [77]. Elevated superoxide and hydrogen peroxide levels can disrupt intracellular signaling pathways, leading to metabolic changes that promote increased fat synthesis and storage. Therefore, heightened ROS production may contribute to the pathogenesis of MetS through mechanisms involving oxidative stress and metabolic reprogramming [78]. All GSD types are groups of inherited metabolic disorders and these mitochondrial dysfunctions may lead to disease development and progression.
3.2. Mitochondrial Biogenesis
Mitochondria contain their own DNA, which encodes a few components of the ETC and 22 mitochondrial tRNAs [79]. The majority of ETC components and other mitochondrial machinery are derived from nuclear genes. Therefore, mitochondrial biogenesis requires coordinated expression of both nuclear and mitochondrial genes [79,80]. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) is a crucial regulator of this process, activating various transcription factors like Nuclear respiratory factors (NRF-1 and NRF-2), and estrogen-related receptors (ERRs), which induce mitochondrial transcription factor A (TFAM) to promote mitochondrial gene transcription [81,82]. PGC-1α also enhances mitochondrial fatty acid oxidation by co-activating PPARα and PPARδ, leading to increased mitochondrial mass and substrate oxidation [83]. PGC-1α activation, triggered by elevated AMP (via AMPK) and increased NAD+ (via Sirtuin-1), also reduces cellular oxidative stress by boosting mitochondrial antioxidant enzyme expression [84,85]. Thus, PGC-1α is a key target for therapeutic strategies against metabolic syndrome (MetS), focusing on enhancing mitochondrial biogenesis to improve mitochondrial efficiency and reduce oxidative stress, providing multifactorial benefits. G6Pase-α deficiency causes glycogen storage disease type 1a (GSD Ia). In a study using L-G6pc−/− mice, G6Pase-α deficiency in the liver resulted in elevated levels of inactive, acetylated PGC-1α and mitochondrial dysfunction, as evidenced by reduced basal oxygen consumption, fewer functional and total mitochondria per hepatocyte, and decreased levels of mitochondrially encoded cytochrome C oxidase I (MTCO1) in complex IV and mitochondrially encoded ATP Synthase membrane subunit 6 (MTATP6) in complex V. There was also an increase in ATP citrate lyase (ACLY) activity and acetyl-CoA levels, which would further promote the acetylation-based inactivation of PGC-1α. Crucially, hepatic overexpression of Sirtuin-1 (SIRT1), a NAD+-dependent deacetylase that can deacetylate and activate PGC-1α, normalized acetyl-PGC-1α levels, restored the expression of ETC components, and increased mitochondrial complex IV activity, which shows downregulation of SIRT1-PGC-1α signaling is a key factor in the mitochondrial dysfunction observed in GSD Ia [86].
3.3. Autophagy and Mitophagy
Cells possess a quality control system to restore mitochondrial function, but when repair is not possible, the entire organelle is removed via the macroautophagic pathway. Macroautophagy, commonly known as autophagy, is an evolutionarily conserved process that supplies amino acids and energy during periods of starvation. In the absence of nutrients, cells utilize their internal resources by enclosing portions of the cytoplasm in autophagic vesicles (autophagosomes) [87,88]. These vesicles then transport their contents to lysosomes for degradation and recycling. Mitophagy, an autophagic degradation of damaged mitochondria), is crucial for removing damaged mitochondria and maintaining cellular homeostasis. Mitophagy plays a crucial role in post-mitotic cells like neurons, which cannot increase glycolytic ATP production. Eliminating damaged mitochondria is essential to prevent the buildup of ROS, which inevitably result from dysfunctional OXPHOS. Accumulation of ROS can further impair mitochondrial function, thereby compromising the cell’s energy homeostasis [89,90]. Mitophagy begins with the accumulation of PINK1, a serine-threonine protein kinase, on the outer mitochondrial membrane (OMM) when membrane potential (ΔΨm) is lost. PINK1 then signals mitochondrial dysfunction to PARK2, a cytosolic E3 ubiquitin ligase, which facilitates the autophagosomal engulfment of damaged mitochondria, leading to their degradation in lysosomes. In GSDs, impaired autophagy can lead to the accumulation of dysfunctional mitochondria, exacerbating oxidative stress and leads to cellular damage [91,92]. Downregulation of the pro-autophagic AMPK signaling and up-regulation of anti-autophagic mTOR signaling was found to be associated with impaired autophagy. Several signaling pathways, including the AMPK-mTOR pathway regulate mitophagy and plays a cardioprotective role by clearing out abnormal mitochondria, thereby preventing oxidative stress and reducing apoptosis in cardiomyocytes [3,93,94]. Autophagy was induced using either ULK1 overexpression or by rapamycin treatment to inhibit mTOR led to decreased hepatocyte glycogen and lipid stores in neonatal G6pc knock-out mice, as well as G6PC-deficient dog of GSD la [95]. In contrast, subsequent research by Cho et al., in adult liver-specific G6pc knockout mice, indicated that the down-regulation of SIRT1 and its target gene, FOXO1, played a crucial role in the suppression of autophagy. Restoration of SIRT1 signaling re-established autophagy but failed to correct other metabolic anomalies, suggesting that pathways beyond SIRT1 regulation of autophagy are likely disrupted in GSD Ia [96]. Additionally, this model exhibited minimal dysfunction in mTOR signaling, and rapamycin treatment did not reinstate autophagy [96]; discrepancies in model type and the age of mice used might explain the variations in mTOR signaling observed across these studies. Further investigation in the adult liver-specific knockout mice suggested that an increased flux of metabolites through the hexose-monophosphate shunt could influence SIRT1 signaling and reduce hepatic autophagy [96]. Impaired autophagy may also result in diminished mitochondrial function, as mitochondria damaged by oxidative stress are unable to undergo removal via mitophagy [97]. Lafora disease is a glycogen storage disorder caused by mutations in either laforin or malin, in which cells accumulate polyglucosan bodies (branched polymers of glucose) called Lafora bodies in tissues such as brain, heart, liver, muscle, and skin [98]. Cell culture studies have demonstrated impaired mitophagy in fibroblasts lacking laforin suggesting mitochondria could still be targeted for degradation but could not progress further to mitophagy [99]. Mitochondrial dysfunction due to excessive ROS activity and oxidative stress and flawed autophagy are common features of many GSDs. A study of mouse and human models of Pompe disease identified several mitochondrial defects in cardiac and skeletal muscle myopathy, including profound dysregulation of Ca2+ homeostasis, mitochondrial Ca2+ overload, increased reactive oxygen species, decreased mitochondrial membrane potential, elevated caspase-independent apoptosis, as well as reduced oxygen consumption and ATP production in mitochondria and concluded that disturbances in Ca2+ homeostasis and mitochondrial abnormalities are early pathogenic changes in Pompe disease [70].
3.4. Apoptosis and Mitochondrial Dysfunction
Apoptosis is a highly regulated form of cell death closely associated with mitochondrial function which plays a critical role in mitochondrial-associated neurodegenerative diseases [100]. Mitochondria are not limited to energy production through ATP synthesis but also to integrating essential cellular signaling pathways. In apoptosis, damaged mitochondria can no longer maintain the proper electrical potential across mitochondrial membranes, thus releasing cytochrome c into the cytosol, where it binds to proteins that form the apoptosome, activating caspases that execute the cell program death [101,102]. Mitochondrial Ca2+ overload can lead to necrosis or apoptosis [70]. An increased Ca2+ level causes mitochondrial abnormalities, which in turn contribute to muscle damage and wasting through mitochondria-mediated apoptosis. In G6PC KD cells found a striking decrease in membrane potential. It was associated with the translocation of cytochrome c into the cytoplasm and cleavage and activation of caspase 9, a specific marker of the mitochondrial apoptosis pathway [12] that can cause hepatocellular apoptosis one of the important clinical features of GSD la [103]. Autophagy and apoptosis are two distinct yet interconnected pathways that significantly influence cell fate. Autophagy is essential for cell survival during periods of starvation and acts as a cellular cleanup mechanism under various stress conditions. In contrast, apoptosis is a form of programmed cell death crucial for maintaining cellular homeostasis and eliminating damaged or unnecessary cells. Decreased mitochondrial membrane potential, reactive oxygen species (ROS) production, calcium overload, and apoptosis indeed precede the development of autophagic buildup and lipofuscin accumulation in Pompe disease [70].
3.5. Mitochondrial Myopathy
GSDs are associated with myopathies and are well-linked to mitochondrial dysfunction. These myopathies are characterized by muscle weakness and fatigue due to impaired energy production in muscle cells. The accumulation of glycogen and defective mitochondrial function in muscle tissues contributes to these symptoms. Muscle weakness and exercise intolerance are common clinical features of several GSDs, often resulting from mitochondrial dysfunction [104]. In GSD V, muscle phosphorylase deficiency leads to a lack of glucose availability during exercise, impairing ATP production and causing muscle fatigue [13]. Similarly, in Pompe disease, the accumulation of glycogen in muscle cells disrupts mitochondrial function, leading to muscle weakness and respiratory difficulties [105,106,107]. Skeletal muscle is highly energy-dependent and therefore particularly susceptible to disorders affecting energy metabolism. ATP is the immediate and essential source of energy for both contraction and relaxation of muscle fibers. To regenerate ATP, skeletal muscle uses various substrates, including high-energy phosphate compounds, glucose, glycogen, and free fatty acids (FFAs). This regeneration occurs through multiple metabolic pathways, such as the creatine kinase (CK) reaction, anaerobic glycolysis, the β-oxidation spiral, and OXPHOS. At rest, fatty acids (FAs) are the primary source of fuel for skeletal muscle [108], High-intensity exercise relies on anaerobic glycolysis, the metabolism of glucose to produce ATP. OXPHOS is the main mechanism of producing ATP during submaximal exercise. OXPHOS metabolizes glycogen, glucose, and FAs to produce NADH and FADH2, which donate electrons in the mitochondrial electron transport chain to produce ATP. Defects in ATP production cause muscular disorders which depend on the impaired specific metabolic pathway. Mitochondrial respiratory chain complexes are essential components of the mitochondrial respiratory chain and play a major role in generating ATP. Complex V deficiency had clinical onset in the neonatal period with multiorgan failure leading to cardiomyopathy and neuromuscular disorders [109]. In a study, an impairment mitochondrial content and biogenesis, a decrease in OXPHOS complex proteins and activities, together with high muscle NADH but reduced muscle levels of glucose-6-phosphate and of the glycolytic products pyruvate and lactate were found. They suggest that the deficiency in oxidative metabolism in McArdle disease might be caused by reduced levels of OXPHOS substrates and a disruption of the mitochondrial network [13]. Exercise intolerance in McArdle disease arises from two main mechanisms: the block of anaerobic glycolysis deprives muscles of energy for isometric exercise, and the block of aerobic glycogen utilization reduces pyruvate and acetyl-CoA, impairing dynamic exercise. Impaired OXPHOS resulted in decreased oxygen extraction and maximum oxygen uptake reported in patients with myophosphorylase deficiency [110]. Dysregulation of mitochondria-mediated Ca2+ homeostasis and oxidative stress triggers muscle cell death in a variety of muscular dystrophies [111]. Muscle glycogen synthase (GYS1) deficiency, which involves in both skeletal and cardiac muscles is a characteristic feature of GSD 0b, a significant predominance of type I muscle fibers and mitochondrial proliferation suggestive of mitochondrial myopathy was reported in GYS1 deficient patient. Glycogenin-1, an enzyme involved in the biosynthesis of glycogen, encoded by the GYG1 gene, and mutations have been described in GSD XV. Muscle biopsy showed reduced glycogen, mitochondrial proliferation, and type I fiber predominance [112]; a case report in 7 patients with Glyvogenin-1 deficiency showed progressive proximal weakness, a myopathic EMG, and polyglucosan bodies in the muscle biopsy [41]. In GSD V, the enzyme myophosphorylase catalyzes the rate-limiting step in muscle glycogen metabolism by releasing glucose-1-phosphate from terminal alpha-1,4-glycosidic bonds. Due to a deficiency of this enzyme, muscle fibers cannot obtain energy from intracellular glycogen stores, leading to impaired glycolytic flux. Impaired muscle aerobic metabolism is a hallmark of GSD V characterized by very low levels of peak oxygen uptake (VO2peak) [113,114]. In, McArdle disease patients were evaluated with exercise testing, and VO2peak levels were found much lower than in healthy controls, consistent with impaired oxidative metabolism [115,116,117]. Muscle biopsy analyses from two patients revealed loss of mitochondrial and cytoskeleton integrity, particularly in type II fibers, suggesting that disruption of the mitochondrial network may contribute to the reduced muscle oxidative capacity in this disease [13]. Hypertrophic cardiomyopathy is a key feature of Pompe disease manifested by the marked thickening of the ventricular walls and associated hyperdynamic systolic function with outflow tract obstruction. Disturbed metabolic signalling and mitochondrial dysfunction are common pathogenic mechanisms of hypertrophic cardiomyopathy [67], this is obvious to speculate that mitochondria may contribute significantly to the development of cardiac hypertrophy in patients with Pompe disease. Mitochondrial dysfunction was found to be associated with aberrant energy metabolism in skeletal muscle biopsy of patients with Pompe disease and animal models [70]; Huang et al. reported the presence of swollen cristae and mitochondrial dysfunction including decreased glycolysis and OXPHOS in iPSC-derived from the Pompe disease’s patients fibroblasts [69]; deformed mitochondria were observed in muscle biopsy and exhibited a significant decrease in the number of mitochondria, consumption of oxygen, and production of ATP and a collapse in mitochondrial membrane potential with increased levels of ROS [70]. These findings support the significant role of mitochondrial dysfunction and impairment in the development of myopathy in GSDs, however, further research is needed to explore this mechanism fully.
3.6. Mitochondrial Dysfunction Targeted Diagnosis and Therapeutic Strategies in GSDs
3.6.1. Diagnosis
Based on expert’s opinions clinical practice guidelines have been developed for diagnostic and therapeutic purposes including most GSD types [11,23,31,66,118]. Newborn screening (NBS) is a good option in the diagnosis of GSDs with certain limitations requiring muscle or liver tissue for functional assays in many GSDs. It often requires metabolite detection, enzyme activity measurements, or genetic tests using dried blood specimens [119]. NBS is very common in Pompe disease using enzymatic assay [120]. The diagnosis of mitochondrial dysfunction in glycogen storage disorders requires an integrative approach combining clinical, biochemical, imaging, histological, and genetic analyses. This comprehensive evaluation helps to accurately diagnose the underlying disorder and guide appropriate management and treatment strategies. Detailed patient history, including family history of metabolic disorders, physical examination to identify symptoms such as muscle weakness, exercise intolerance, hepatomegaly, and cardiomyopathy will be useful. Laboratory tests include measuring blood glucose levels, lactate, pyruvate, amino acids, and acylcarnitines. Elevated lactate and pyruvate levels may indicate mitochondrial dysfunction. Liver function tests and creatine kinase (CK) levels can help identify liver involvement and muscle damage [66,121]. Specific enzyme activity assays in muscle or liver biopsy samples to identify deficiencies related to GSDs (e.g., glycogen synthase, debranching enzyme, branching enzyme). Imaging tests including magnetic resonance imaging (MRI) and ultrasonography (USG) for liver and abnormalities in muscle texture and composition, Echocardiography is important for individuals at risk of cardiomyopathy and arrhythmias such as those with GSD l, Pompe disease, GSD III or GSD IV. Histological examination to identify abnormal glycogen accumulation and structural changes, and Electron Microscopy can provide detailed images of mitochondrial structure to find out morphological abnormalities. The genomic analysis includes next-generation sequencing (NGS), targeted gene panels, and whole exome sequencing (WES) to identify mutations and sequencing of nuclear and mitochondrial genes. This can provide confirmatory and clear picture of the association of mitochondrial dysfunction with GSDs. Mitochondrial respiratory chain and OXPHOS studies in which measurement of the activities of respiratory chain complexes (I-IV) and oxygen consumption rates (OCR) in cells or isolated mitochondria to assess overall mitochondrial function have been recommended in studies [123,124,125].
3.7. Current Treatment Options and Therapies
Enzyme replacement therapy (ERT) has been successfully developed for some GSDs, mainly GSD II (Pompe disease). ERT aims to replace the deficient enzyme, reducing glycogen accumulation and improving mitochondrial function. Clinical studies have shown that ERT can improve muscle strength, respiratory function, and cardiac abnormalities in Pompe disease, highlighting its potential to mitigate mitochondrial dysfunction [38,126]. Gene therapy offers a promising approach to correcting the underlying genetic defects in GSDs. By delivering functional copies of the deficient gene, gene therapy can restore enzyme activity, reduce glycogen accumulation, and improve mitochondrial function. Advanced gene editing techniques, such as CRISPR-Cas9, offer the potential to precisely correct genetic mutations in GSDs. By targeting the specific mutations responsible for enzyme deficiencies, gene editing could restore normal enzyme activity and improve mitochondrial function. Preclinical studies have shown encouraging results in animal models of GSDs, and clinical trials are underway to evaluate the safety and efficacy of gene therapy in humans [127,128,129]. Given the role of oxidative stress in mitochondrial dysfunction, antioxidant therapy has been explored as a potential treatment for GSDs. Antioxidants, such as coenzyme Q10, vitamin E, and N-acetylcysteine, can reduce ROS levels and protect mitochondrial function [130,131]. Clinical studies have shown that antioxidant therapy can improve muscle function and reduce oxidative stress in patients with GSDs. A study reported that nutritional co-therapy with 1,3-butanediol and multi-ingredient antioxidants may provide an alternative to ketogenic diets for inducing ketosis and enhancing autophagic flux in Pompe disease [132]. Metabolic modulation involves altering metabolic pathways to improve energy production and reduce glycogen accumulation. For instance, ketogenic diets, which are high in fat and low in carbohydrates, can promote fatty acid oxidation and ketone body production, providing an alternative energy source for mitochondria. Clinical studies have shown that ketogenic diets can improve exercise tolerance and reduce muscle symptoms in patients with GSD V. Mitochondrial-targeted therapies aim to improve mitochondrial function directly. These therapies include compounds that enhance mitochondrial biogenesis, such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) agonists, and agents that improve mitochondrial dynamics, such as mitochondrial division inhibitor 1 (Mdivi-1). A preclinical study by our group explore the efficacy and mechanism of action of the polyglucosan-reducing compound 144DG11 (GHF 201) in GBE knockin (Gbeys/ys) mouse model and patient fibroblast of adult polyglucosan body disease (APBD). 144DG11 therapy can improve mitochondrial function and reduce disease symptoms in animal models of GSDs. Lysosomal membrane protein LAMP1 is the molecular target of 144DG11, which enhanced autolysosomal glycogen degradation by increasing autophagic flux and lysosomal acidification. 144DG11 increases carbohydrate burn at the expense of fat burn, suggesting metabolic mobilization of pathogenic polyglucosan, a key feature of APBD and increases glycolytic, mitochondrial, and total ATP production [22]. Empagliflozin, an inhibitor of the renal sodium-glucose cotransporter type 2 (SGLT2), improves redox state and oxidative stress, inhibiting reactive oxygen species (ROS) production, reducing the activity of pro-oxidant agents, and improving mitochondrial function [133]. Studies reported, that empagliflozin improved symptoms like neutropenia, hypoglycemia, and inflammatory bowel syndrome in GSD Ib patients [134,135,136]
4. Conclusions
Mitochondrial dysfunction in glycogen storage disorders (GSDs) represents a critical aspect of these metabolic diseases, underscoring the complex interplay between cellular energy management and glycogen metabolism. GSDs, characterized by deficiencies in enzymes involved in glycogen synthesis or degradation, lead to the accumulation or improper utilization of glycogen in tissues such as the liver and muscle. This metabolic dysregulation often results in impaired energy production, within mitochondria. Studies have shown that mitochondrial dysfunction in GSDs manifests through various mechanisms, including altered mitochondrial biogenesis, disturbed ROS activity, increased oxidative stress, and impaired OXPHOS. These anomalies resulted in impairing the structure and function of the mitochondria and contributed to clinical symptoms such as muscle weakness, exercise intolerance, and hepatic dysfunction, which are very common in GSDs. Furthermore, the intricate relationship between mitochondrial function and glycogen metabolism suggests that targeting mitochondrial pathways could offer therapeutic potential for managing GSDs. Advancements in molecular biology and genetics have provided deeper insights into the mitochondrial disturbances in GSDs, highlighting the need for comprehensive diagnostic and therapeutic strategies that address both glycogen metabolism and mitochondrial health. Interventions aiming to restore mitochondrial function, such as antioxidant therapy, gene therapy, and enzyme replacement therapy, hold promise but require further research and clinical validation. In conclusion, mitochondrial dysfunction plays a pivotal role in the pathophysiology of glycogen storage disorders, significantly influencing disease outcomes and patient quality of life. A multidisciplinary approach that integrates metabolic, genetic, and mitochondrial-targeted therapies is essential for developing effective treatments for GSDs, ultimately aiming to improve clinical outcomes and enhance the well-being of affected individuals.
Author Contributions
Conceptualization, Mishra K and Kakhlon O; writing and original draft preparation- Mishra K.; writing—review and editing, Kakhlon O. Authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
No new data were created or analyzed in this study. Data sharing does not apply to this article.
Acknowledgments
We would like to acknowledge the Golda Meir Foundation Hebrew University of Jerusalem, fellowship awarded to Kumudesh Mishra for his postdoctoral work.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Gümüş, E.; Özen, H. Glycogen storage diseases: An update. World J. Gastroenterol. 2023, 29, 3932–3963. [CrossRef]
- Kanungo, S.; Wells, K.; Tribett, T.; El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 2018, 6, 474–474. [CrossRef]
- Massese, M.; Tagliaferri, F.; Dionisi-Vici, C.; Maiorana, A. Glycogen storage diseases with liver involvement: a literature review of GSD type 0, IV, VI, IX and XI. Orphanet J. Rare Dis. 2022, 17, 1–12. [CrossRef]
- de Marchi, R.; Nalin, T.; Sperb-Ludwig, F.; Pinheiro, F.C.; Schwartz, I.V.D.; Steiner, C.E. Glycogen Storage Disease: Expert Opinion on Clinical Diagnosis Revisited after Molecular Testing. Genes 2023, 14, 2219. [CrossRef]
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.L.; Carelli, V. Mitochondrial diseases in adults. J. Intern. Med. 2020, 287, 592–608. [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [CrossRef]
- Gjorgjieva, M.; Oosterveer, M.H.; Mithieux, G.; Rajas, F. Mechanisms by Which Metabolic Reprogramming in GSD1 Liver Generates a Favorable Tumorigenic Environment. J. Inborn Errors Metab. Screen. 2016, 4. [CrossRef]
- Huang, W.; Zhou, R.; Jiang, C.; Wang, J.; Zhou, Y.; Xu, X.; Wang, T.; Li, A.; Zhang, Y. Mitochondrial dysfunction is associated with hypertrophic cardiomyopathy in Pompe disease-specific induced pluripotent stem cell-derived cardiomyocytes. Cell Prolif. 2023, 57, e13573. [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [CrossRef]
- Sentner, C.P.; Hoogeveen, I.J.; Weinstein, D.A.; Santer, R.; Murphy, E.; McKiernan, P.J.; Steuerwald, U.; Beauchamp, N.J.; Taybert, J.; Laforêt, P.; et al. Glycogen storage disease type III: diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 2016, 39, 697–704. [CrossRef]
- Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Lim, A.; Hirayama, M.; Landau, D.J.; Bay, B.H.; Koeberl, D.D.; Yen, P.M. Hepatic mitochondrial dysfunction is a feature of Glycogen Storage Disease Type Ia (GSDIa). Sci. Rep. 2017, 7, srep44408. [CrossRef]
- Fiuza-Luces C, Andreu A, Rodríguez-Aguilera J, Martín M, Arenas J, Vissing J, et al. Low aerobic capacity in McArdle disease: A role for mitochondrial network impairment? 2022.
- Kamenets, E.A.; Gusarova, E.A.; Milovanova, N.V.; Itkis, Y.S.; Strokova, T.V.; Melikyan, M.A.; Garyaeva, I.V.; Rybkina, I.G.; Nikitina, N.V.; Zakharova, E.Y. Hepatic glycogen synthase (GYS2) deficiency: seven novel patients and seven novel variants. JIMD Rep. 2020, 53, 39–44. [CrossRef]
- Kasapkara, .S.; Aycan, Z.; Açoğlu, E.; Senel, S.; Oguz, M.M.; Ceylaner, S. The variable clinical phenotype of three patients with hepatic glycogen synthase deficiency. J. Pediatr. Endocrinol. Metab. 2017, 30. [CrossRef]
- Musumeci, O.; Pugliese, A.; Oteri, R.; Volta, S.; Ciranni, A.; Moggio, M.; Rodolico, C.; Toscano, A. A new phenotype of muscle glycogen synthase deficiency (GSD0B) characterized by an adult onset myopathy without cardiomyopathy. Neuromuscul. Disord. 2022, 32, 582–589. [CrossRef]
- Kollberg, G.; Tulinius, M.; Gilljam, T.; Östman-Smith, I.; Forsander, G.; Jotorp, P.; Oldfors, A.; Holme, E. Cardiomyopathy and Exercise Intolerance in Muscle Glycogen Storage Disease 0. New Engl. J. Med. 2007, 357, 1507–1514. [CrossRef]
- Visser, G.; Rake, J.-P.; Fernandes, J.; Labrune, P.; Leonard, J.V.; Moses, S.; Ullrich, K.; Smit, G.A. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: Results of the European Study on Glycogen Storage Disease Type I. J. Pediatr. 2000, 137, 187–191. [CrossRef]
- Veiga-Da-Cunha, M.; Wortmann, S.B.; Grünert, S.C.; Van Schaftingen, E. Treatment of the Neutropenia Associated with GSD1b and G6PC3 Deficiency with SGLT2 Inhibitors. Diagnostics 2023, 13, 1803. [CrossRef]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [CrossRef]
- Szymańska, E.; Szymańska, S.; Truszkowska, G.; Ciara, E.; Pronicki, M.; Shin, Y.S.; Podskarbi, T.; Kępka, A.; Śpiewak, M.; Płoski, R.; et al. Variable clinical presentation of glycogen storage disease type IV: from severe hepatosplenomegaly to cardiac insufficiency. Some discrepancies in genetic and biochemical abnormalities. Arch. Med Sci. 2018, 1, 237–247. [CrossRef]
- Kakhlon, O.; Vaknin, H.; Mishra, K.; D’souza, J.; Marisat, M.; Sprecher, U.; Wald-Altman, S.; Dukhovny, A.; Raviv, Y.; Da’adoosh, B.; et al. Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism. EMBO Mol. Med. 2021, 13, e14554. [CrossRef]
- Koch, R.L.; Soler-Alfonso, C.; Kiely, B.T.; Asai, A.; Smith, A.L.; Bali, D.S.; Kang, P.B.; Landstrom, A.P.; Akman, H.O.; Burrow, T.A.; et al. Diagnosis and management of glycogen storage disease type IV, including adult polyglucosan body disease: A clinical practice resource. Mol. Genet. Metab. 2023, 138, 107525. [CrossRef]
- Da Silva, J.D.; Pereira, .; Soares, A.R.; Guimas, A.; Rocha, S.; Cardoso, M.; Garrido, C.; Soares, C.A.; Nunes, I.S.; Fortuna, A.M.; et al. Diagnostic accuracy and the first genotype–phenotype correlation in glycogen storage disease type V. Pediatr. Res. 2023, 1–7. [CrossRef]
- Labrador E, Weinstein DA. Glycogen storage disease type VI. 2019.
- Aeppli, T.R.; Rymen, D.; Allegri, G.; Bode, P.K.; Häberle, J. Glycogen storage disease type VI: clinical course and molecular background. Eur. J. Pediatr. 2020, 179, 405–413. [CrossRef]
- Musumeci, O.; Bruno, C.; Mongini, T.; Rodolico, C.; Aguennouz, M.; Barca, E.; Amati, A.; Cassandrini, D.; Serlenga, L.; Vita, G.; et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul. Disord. 2011, 22, 325–330. [CrossRef]
- DiMauro, S.; Spiegel, R. Progress and problems in muscle glycogenoses. 2011, 30, 96–102.
- Fernandes, S.A.; Cooper, G.E.; Gibson, R.A.; Kishnani, P.S. Benign or not benign? Deep phenotyping of liver Glycogen Storage Disease IX. Mol. Genet. Metab. 2020, 131, 299–305. [CrossRef]
- Roscher, A.; Patel, J.; Hewson, S.; Nagy, L.; Feigenbaum, A.; Kronick, J.; Raiman, J.; Schulze, A.; Siriwardena, K.; Mercimek-Mahmutoglu, S. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol. Genet. Metab. 2014, 113, 171–176. [CrossRef]
- Kishnani, P.S.; Goldstein, J.; Austin, S.L.; Arn, P.; Bachrach, B.; Bali, D.S.; Chung, W.K.; El-Gharbawy, A.; Brown, L.M.; Kahler, S.; et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2019, 21, 772–789. [CrossRef]
- Bali DS, Goldstein JL, Fredrickson K, Austin S, Pendyal S, Rehder C, et al. Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency. JIMD Reports, Volume 37. 2017:63-72.
- Ellingwood, S.S.; Cheng, A. Biochemical and clinical aspects of glycogen storage diseases. J. Endocrinol. 2018, 238, R131–R141. [CrossRef]
- Kwong, A.K.; Wong, S.S.; Rodenburg, R.J.T.; Smeitink, J.; Chan, G.C.F.; Fung, C. Human d-lactate dehydrogenase deficiency by LDHD mutation in a patient with neurological manifestations and mitochondrial complex IV deficiency. JIMD Rep. 2021, 60, 15–22. [CrossRef]
- Serrano-Lorenzo P, Rabasa M, Esteban J, Hidalgo Mayoral I, Domínguez-González C, Blanco-Echevarría A, et al. Clinical, biochemical, and molecular characterization of two families with novel mutations in the LDHA gene (GSD XI). Genes. 2022;13 [10]:1835.
- Jin, S.; Chen, X.; Yang, J.; Ding, J. Lactate dehydrogenase D is a general dehydrogenase for D-2-hydroxyacids and is associated with D-lactic acidosis. Nat. Commun. 2023, 14, 1–13. [CrossRef]
- Mamoune, A.; Bahuau, M.; Hamel, Y.; Serre, V.; Pelosi, M.; Habarou, F.; Morel, M.-A.N.; Boisson, B.; Vergnaud, S.; Viou, M.T.; et al. A Thermolabile Aldolase A Mutant Causes Fever-Induced Recurrent Rhabdomyolysis without Hemolytic Anemia. PLOS Genet. 2014, 10, e1004711–e1004711. [CrossRef]
- Papadopoulos, C.; Svingou, M.; Kekou, K.; Vergnaud, S.; Xirou, S.; Niotakis, G.; Papadimas, G. Aldolase A deficiency: Report of new cases and literature review. Mol. Genet. Metab. Rep. 2021, 27, 100730. [CrossRef]
- Wigley, R.; Scalco, R.S.; Gardiner, A.R.; Godfrey, R.; Booth, S.; Kirk, R.; Hilton-Jones, D.; Houlden, H.; Heales, S.; Quinlivan, R. The need for biochemical testing in beta-enolase deficiency in the genomic era. JIMD Rep. 2019, 50, 40–43. [CrossRef]
- Buch AE, Musumeci O, Wigley R, Stemmerik MPG, Eisum ASV, Madsen KL, et al. Energy metabolism during exercise in patients with β-enolase deficiency (GSDXIII). JIMD reports. 2021;61 [1]:60-6.
- Malfatti E, Nilsson J, Hedberg-Oldfors C, Hernandez-Lain A, Michel F, Dominguez-Gonzalez C, et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Annals of neurology. 2014;76 [6]:891-8.
- Visuttijai, K.; Hedberg-Oldfors, C.; Thomsen, C.; Glamuzina, E.; Kornblum, C.; Tasca, G.; Hernandez-Lain, A.; Sandstedt, J.; Dellgren, G.; Roach, P.; et al. Glycogenin is Dispensable for Glycogen Synthesis in Human Muscle, and Glycogenin Deficiency Causes Polyglucosan Storage. J. Clin. Endocrinol. Metab. 2020, 105, 557–566. [CrossRef]
- Conte, F.; Morava, E.; Abu Bakar, N.; Wortmann, S.B.; Poerink, A.J.; Grunewald, S.; Crushell, E.; Al-Gazali, L.; de Vries, M.C.; Mørkrid, L.; et al. Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots. Mol. Genet. Metab. 2020, 131, 135–146. [CrossRef]
- Altassan R, Radenkovic S, Edmondson AC, Barone R, Brasil S, Cechova A, et al. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): diagnosis, follow-up, and management. Journal of inherited metabolic disease. 2021;44 [1]:148-63.
- Grünert, S.C.; Schwab, K.O.; Pohl, M.; Sass, J.O.; Santer, R. Fanconi–Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol. Genet. Metab. 2011, 105, 433–437. [CrossRef]
- Sharari, S.; Abou-Alloul, M.; Hussain, K.; Khan, F.A. Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. [CrossRef]
- Matsumaru, S.; Oguni, H.; Ogura, H.; Shimojima, K.; Nagata, S.; Kanno, H.; Yamamoto, T. A novel PGK1 mutation associated with neurological dysfunction and the absence of episodes of hemolytic anemia or myoglobinuria. Intractable Rare Dis. Res. 2017, 6, 132–136. [CrossRef]
- Echaniz-Laguna, A.; Nadjar, Y.; Béhin, A.; Biancalana, V.; Piraud, M.; Malfatti, E.; Laforêt, P. Phosphoglycerate kinase deficiency: A nationwide multicenter retrospective study. J. Inherit. Metab. Dis. 2019, 42, 803–808. [CrossRef]
- Vega RB, Horton JL, Kelly DP. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circulation research. 2015;116 [11]:1820-34.
- Sturm, G.; Karan, K.R.; Monzel, A.S.; Santhanam, B.; Taivassalo, T.; Bris, C.; Ware, S.A.; Cross, M.; Towheed, A.; Higgins-Chen, A.; et al. OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases. Commun. Biol. 2023, 6, 1–22. [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. Am. J. Physiol. Liver Physiol. 2016, 310, G205–G214. [CrossRef]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2019, 43, 36–50. [CrossRef]
- Fassone, E.; Rahman, S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med Genet. 2012, 49, 578–590. [CrossRef]
- Rodenburg RJ. Mitochondrial complex I-linked disease. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2016;1857 [7]:938-45.
- Kurbatova, O.; Izmailova, T.; Surkov, A.; Namazova-Baranova, L.; Polyakova, S.; Miroshkina, L.; Semenova, G.; Samokhina, I.; Kapustiuna, E.; Dukhova, Z.; et al. Mitochondrial Dysfunction in Children with Hepatic Forms of Glycogen Storage Disease. Ann. Russ. Acad. Med Sci. 2014, 69, 78–84. [CrossRef]
- Wary, C.; Nadaj-Pakleza, A.; Laforêt, P.; Claeys, K.G.; Carlier, R.; Monnet, A.; Fleury, S.; Baligand, C.; Eymard, B.; Labrune, P.; et al. Investigating glycogenosis type III patients with multi-parametric functional NMR imaging and spectroscopy. Neuromuscul. Disord. 2010, 20, 548–558. [CrossRef]
- Preisler N, Pradel A, Husu E, Madsen KL, Becquemin M-H, Mollet A, et al. Exercise intolerance in glycogen storage disease type III: weakness or energy deficiency? Molecular Genetics and Metabolism. 2013;109 [1]:14-20.
- Hennis, P.J.; Murphy, E.; Meijer, R.I.; Lachmann, R.H.; Ramachandran, R.; Bordoli, C.; Rayat, G.; Tomlinson, D.J. Aerobic capacity and skeletal muscle characteristics in glycogen storage disease IIIa: an observational study. Orphanet J. Rare Dis. 2022, 17, 1–12. [CrossRef]
- Hannibal, L.; Theimer, J.; Wingert, V.; Klotz, K.; Bierschenk, I.; Nitschke, R.; Spiekerkoetter, U.; Grünert, S.C. Metabolic Profiling in Human Fibroblasts Enables Subtype Clustering in Glycogen Storage Disease. Front. Endocrinol. 2020, 11. [CrossRef]
- Rossi A, Ruoppolo M, Formisano P, Villani G, Albano L, Gallo G, et al. Insulin-resistance in glycogen storage disease type Ia: linking carbohydrates and mitochondria? Journal of inherited metabolic disease. 2018;41 [6]:985-95.
- Saavedra, H.; Yu, A.; Rodriguez-Buritica, D. The use of alanine, free carnitine and IGFBP-1 as potential biomarkers for glycogen storage disease type I. Mol. Genet. Metab. 2022, 136, S18. [CrossRef]
- Rossi, A.; Assunto, A.; Rosano, C.; Tucci, S.; Ruoppolo, M.; Caterino, M.; Pirozzi, F.; Strisciuglio, P.; Parenti, G.; Melis, D. Mitochondrial reprogramming in peripheral blood mononuclear cells of patients with glycogen storage disease type Ia. Genes Nutr. 2023, 18, 1–11. [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [CrossRef]
- Chaurasia B, Summers SA. Ceramides–lipotoxic inducers of metabolic disorders. Trends in Endocrinology & Metabolism. 2015;26 [10]:538-50.
- Milane L, Trivedi M, Singh A, Talekar M, Amiji M. Mitochondrial biology, targets, and drug delivery. Journal of controlled release. 2015;207:40-58.
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Anesthesia Analg. 2006, 8, 267–288. [CrossRef]
- Ranjbarvaziri S, Kooiker KB, Ellenberger M, Fajardo G, Zhao M, Vander Roest AS, et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation. 2021;144 [21]:1714-31. [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [CrossRef]
- Huang H-P, Chen P-H, Hwu W-L, Chuang C-Y, Chien Y-H, Stone L, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human molecular genetics. 2011;20 [24]:4851-64.
- Lim, J.-A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [CrossRef]
- Palma FR, Gantner BN, Sakiyama MJ, Kayzuka C, Shukla S, Lacchini R, et al. ROS production by mitochondria: function or dysfunction? Oncogene. 2024;43 [5]:295-303.
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial Management of Reactive Oxygen Species. Antioxidants 2021, 10, 1824. [CrossRef]
- Bou-Teen, D.; Kaludercic, N.; Weissman, D.; Turan, B.; Maack, C.; Di Lisa, F.; Ruiz-Meana, M. Mitochondrial ROS and mitochondria-targeted antioxidants in the aged heart. Free. Radic. Biol. Med. 2021, 167, 109–124. [CrossRef]
- Vujic, A.; Koo, A.N.; Prag, H.A.; Krieg, T. Mitochondrial redox and TCA cycle metabolite signaling in the heart. Free. Radic. Biol. Med. 2021, 166, 287–296. [CrossRef]
- Kim, J.; Bai, H. Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging. Antioxidants 2022, 11, 192. [CrossRef]
- Betteridge DJ. What is oxidative stress? Metabolism. 2000;49 [2]:3-8.
- Lee, B.-C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 446–462. [CrossRef]
- Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2020;1866 [10]:165838.
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [CrossRef]
- Gonçalves VF. Mitochondrial genetics. Mitochondria in Health and in Sickness. 2019:247-55.
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [CrossRef]
- Islam, H.; Edgett, B.A.; Gurd, B.J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A re-evaluation. Metabolism 2018, 79, 42–51. [CrossRef]
- Jannig, P.R.; Dumesic, P.A.; Spiegelman, B.M.; Ruas, J.L. SnapShot: Regulation and biology of PGC-1α. Cell 2022, 185, 1444–1444.e1. [CrossRef]
- Ruderman NB, Julia Xu X, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? American Journal of Physiology-Endocrinology and Metabolism. 2010;298 [4]:E751-E60.
- Carling, D.; Viollet, B. Beyond Energy Homeostasis: the Expanding Role of AMP-Activated Protein Kinase in Regulating Metabolism. Cell Metab. 2015, 21, 799–804. [CrossRef]
- Cho, J.; Kim, G.; Mansfield, B.C.; Chou, J.Y. Sirtuin signaling controls mitochondrial function in glycogen storage disease type Ia. J. Inherit. Metab. Dis. 2018, 41, 997–1006. [CrossRef]
- Majeed, S.T.; Majeed, R.; Andrabi, K.I. Expanding the view of the molecular mechanisms of autophagy pathway. J. Cell. Physiol. 2022, 237, 3257–3277. [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2017, 14, 207–215. [CrossRef]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J. Neurochem. 2018, 147, 291–309. [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLOS Biol. 2010, 8, e1000298. [CrossRef]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016, 7, e2501–e2501. [CrossRef]
- Zheng, H.; Zhu, H.; Liu, X.; Huang, X.; Huang, A.; Huang, Y. Mitophagy in Diabetic Cardiomyopathy: Roles and Mechanisms. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
- Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T.; et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3587. [CrossRef]
- Farah, B.L.; Landau, D.J.; Sinha, R.A.; Brooks, E.D.; Wu, Y.; Fung, S.Y.S.; Tanaka, T.; Hirayama, M.; Bay, B.-H.; Koeberl, D.D.; et al. Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J. Hepatol. 2015, 64, 370–379. [CrossRef]
- Cho, J.-H.; Kim, G.-Y.; Pan, C.-J.; Anduaga, J.; Choi, E.-J.; Mansfield, B.C.; Chou, J.Y. Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLOS Genet. 2017, 13, e1006819. [CrossRef]
- Farah, B.L.; Yen, P.M.; Koeberl, D.D. Links between autophagy and disorders of glycogen metabolism – Perspectives on pathogenesis and possible treatments. Mol. Genet. Metab. 2019, 129, 3–12. [CrossRef]
- Mitra, S.; Gumusgoz, E.; Minassian, B. Lafora disease: Current biology and therapeutic approaches. Rev. Neurol. 2022, 178, 315–325. [CrossRef]
- Lahuerta, M.; Aguado, C.; Sánchez-Martín, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 2018, 285, 2071–2090. [CrossRef]
- Klemmensen, M.M.; Borrowman, S.H.; Pearce, C.; Pyles, B.; Chandra, B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 2024, 21, e00292. [CrossRef]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [CrossRef]
- Sun, B.; Li, S.; Yang, L.; Damodaran, T.; Desai, D.; Diehl, A.M.; Alzate, O.; Koeberl, D.D. Activation of glycolysis and apoptosis in glycogen storage disease type Ia. Mol. Genet. Metab. 2009, 97, 267–271. [CrossRef]
- Tarnopolsky, M.A. Myopathies Related to Glycogen Metabolism Disorders. Neurotherapeutics 2018, 15, 915–927. [CrossRef]
- Tsai, L.-K.; Hwu, W.-L.; Lee, N.-C.; Huang, P.-H.; Chien, Y.-H. Clinical features of Pompe disease with motor neuronopathy. Neuromuscul. Disord. 2019, 29, 903–906. [CrossRef]
- Leslie N, Bailey L. Pompe disease. 2017.
- Leslie N, Bailey L. Pompe Disease. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle.
- Copyright © 1993-2024, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993.
- De Feo P, Di Loreto C, Lucidi P, Murdolo G, Parlanti N, De Cicco A, et al. Metabolic response to exercise. Journal of endocrinological investigation. 2003;26:851-4.
- Li X-Y, Yang Y-L. Mitochondrial disorders associated with mitochondrial respiratory chain complex V deficiency. Zhongguo Dang dai er ke za zhi= Chinese Journal of Contemporary Pediatrics. 2013;15 [7]:596-600.
- De Stefano N, Argov Z, Matthews PM, Karpati G, Arnold DL. Impairment of muscle mitochondrial oxidative metabolism in McArdle’s disease. Muscle & Nerve: Official Journal of the American Association of Electrodiagnostic Medicine. 1996;19 [6]:764-9.
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [CrossRef]
- Moslemi, A.-R.; Lindberg, C.; Nilsson, J.; Tajsharghi, H.; Andersson, B.; Oldfors, A. Glycogenin-1 Deficiency and Inactivated Priming of Glycogen Synthesis. New Engl. J. Med. 2010, 362, 1203–1210. [CrossRef]
- Santalla, A.; Valenzuela, P.L.; Rodriguez-Lopez, C.; Rodríguez-Gómez, I.; Nogales-Gadea, G.; Pinós, T.; Arenas, J.; Martín, M.A.; Santos-Lozano, A.; Morán, M.; et al. Long-Term Exercise Intervention in Patients with McArdle Disease: Clinical and Aerobic Fitness Benefits. Med. Sci. Sports Exerc. 2022, 54, 1231–1241. [CrossRef]
- Munguía-Izquierdo, D.; Santalla, A.; Lucia, A. Cardiorespiratory Fitness, Physical Activity, and Quality of Life in Patients with McArdle Disease. Med. Sci. Sports Exerc. 2015, 47, 799–808. [CrossRef]
- Scalco, R.S.; EUROMAC Consortium; Lucia, A.; Santalla, A.; Martinuzzi, A.; Vavla, M.; Reni, G.; Toscano, A.; Musumeci, O.; Voermans, N.C.; et al. Data from the European registry for patients with McArdle disease and other muscle glycogenoses (EUROMAC). Orphanet J. Rare Dis. 2020, 15, 1–8. [CrossRef]
- Santalla, A.; Nogales-Gadea, G.; Encinar, A.B.; Vieitez, I.; González-Quintana, A.; Serrano-Lorenzo, P.; Consuegra, I.G.; Asensio, S.; Ballester-Lopez, A.; Pintos-Morell, G.; et al. Genotypic and phenotypic features of all Spanish patients with McArdle disease: a 2016 update. BMC Genom. 2017, 18, 819–819. [CrossRef]
- Pizzamiglio, C.; Mahroo, O.A.; Khan, K.N.; Patasin, M.; Quinlivan, R. Phenotype and genotype of 197 British patients with McArdle disease: An observational single-centre study. J. Inherit. Metab. Dis. 2021, 44, 1409–1418. [CrossRef]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Anesthesia Analg. 2014, 16, e1–e29. [CrossRef]
- Hannah WB, Derks TG, Drumm ML, Grünert SC, Kishnani PS, Vissing J. Glycogen storage diseases. Nature Reviews Disease Primers. 2023;9 [1]:46.
- Keutzer, J.M. Establishing Pompe Disease Newborn Screening: The Role of Industry. Int. J. Neonatal Screen. 2020, 6, 55. [CrossRef]
- Mancuso M, Coppede F, Migliore L, Siciliano G, Murri L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. Journal of Alzheimer’s Disease. 2006;10 [1]:59-73.
- Rodenburg, R.J.T. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2010, 34, 283–292. [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Ermani, M.; Salviati, L.; Angelini, C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion 2011, 11, 893–904. [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; Mousson de Camaret, B.; Slama, A.; Lunardi, J.; Mazat, J.P.; Lombès, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [CrossRef]
- Winkel LP, Kamphoven JH, Van Den Hout HJ, Severijnen LA, Van Doorn PA, Reuser AJ, et al. Morphological changes in muscle tissue of patients with infantile Pompe’s disease receiving enzyme replacement therapy. Muscle & Nerve: Official Journal of the American Association of Electrodiagnostic Medicine. 2003;27 [6]:743-51.
- Koeberl, D.D.; Koch, R.L.; Lim, J.; Brooks, E.D.; Arnson, B.D.; Sun, B.; Kishnani, P.S. Gene therapy for glycogen storage diseases. J. Inherit. Metab. Dis. 2023, 47, 93–118. [CrossRef]
- Jauze, L.; Monteillet, L.; Mithieux, G.; Rajas, F.; Ronzitti, G. Challenges of Gene Therapy for the Treatment of Glycogen Storage Diseases Type I and Type III. Hum. Gene Ther. 2019, 30, 1263–1273. [CrossRef]
- Kishnani, P.S.; Sun, B.; Koeberl, D.D. Gene therapy for glycogen storage diseases. Hum. Mol. Genet. 2019, 28, R31–R41. [CrossRef]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [CrossRef]
- Nilsson, M.I.; Crozier, M.; Di Carlo, A.; Xhuti, D.; Manta, K.; Roik, L.J.; Bujak, A.L.; Nederveen, J.P.; Tarnopolsky, M.G.; Hettinga, B.; et al. Nutritional co-therapy with 1,3-butanediol and multi-ingredient antioxidants enhances autophagic clearance in Pompe disease. Mol. Genet. Metab. 2022, 137, 228–240. [CrossRef]
- Andreadi, A.; Bellia, A.; Di Daniele, N.; Meloni, M.; Lauro, R.; Della-Morte, D.; Lauro, D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: A target for new therapies against cardiovascular diseases. Curr. Opin. Pharmacol. 2021, 62, 85–96. [CrossRef]
- Grünert, S.C.; Elling, R.; Maag, B.; Wortmann, S.B.; Derks, T.G.J.; Hannibal, L.; Schumann, A.; Rosenbaum-Fabian, S.; Spiekerkoetter, U. Improved inflammatory bowel disease, wound healing and normal oxidative burst under treatment with empagliflozin in glycogen storage disease type Ib. Orphanet J. Rare Dis. 2020, 15, 1–8. [CrossRef]
- Wortmann SB, Van Hove JL, Derks TG, Chevalier N, Knight V, Koller A, et al. Treating neutropenia and neutrophil dysfunction in glycogen storage disease type Ib with an SGLT2 inhibitor. Blood, The Journal of the American Society of Hematology. 2020;136 [9]:1033-43. [CrossRef]
- Maiorana, A.; Tagliaferri, F.; Dionisi-Vici, C. Current understanding on pathogenesis and effective treatment of glycogen storage disease type Ib with empagliflozin: new insights coming from diabetes for its potential implications in other metabolic disorders. Front. Endocrinol. 2023, 14. [CrossRef]

Table 1.
Enzyme defects with clinical features and epidemiology in GSDs.
| Disorders (Common Name) | Enzyme | Gene/Chromosome/Inheritance | Clinical Manifestation | References |
|---|---|---|---|---|
| GSD 0a | Glycogen synthase in Liver | GYS2/ 12p12.1/AR | Fasting ketotic hypoglycemia, reactive hyperglycemia and lactate elevation | [14,15] |
| GSD 0b | Glycogen synthase in muscle | GYS1/ 19q13.33/AR | Hypertrophic cardiomyopathy exercise intolerance, and adult-onset myopathy without cardiomyopathy |
[16,17] |
| GSD Ia (von Gierke disease) |
Glucose-6-phosphatase | G6PC/17q21.31/ AR | Hypoglycemia, lactic acidosis, hypertriglyceridemia, hepatomegaly, renal dysfunction | [18] |
| GSD Ib |
Glucose-6-phosphate translocase |
SLC37A4/11q23.3/AR | Neutropenia, neutrophil dysfunction and inflammatory bowel disease |
[19] |
| GSD II (Pompe disease) |
alpha-1,4-glucosidase |
GAA /17q25.3/AR | Hypertrophic cardiomyopathy, hypotonia, and motor delay | [20] |
| GSD III (Cori or Forbes disease) |
Glycogen debranching (amylo-1,6 glucosidase) |
AGL/1p21.2/ AR | Hypoglycemia, elevated ketosis in hyperlipidemia, hepatomegaly, elevated liver enzyme myopathy, variable muscle and cardiac phenotype | [11] |
| GSD IV (Andersen or Adult polyglucosan body disease) |
Glycogen branching enzyme- amylo (1,4 -1,6) transglucosidase |
GBE1/3p12.2/AR | Hepatosplenomegaly, liver dysfunction, progressive cirrhosis, cardiomyopathy, hypotonia, gait difficulty, progressive neurogenic bladder, autonomic dysfunction, sensory loss and variable cognitive difficulty | [21,22,23] |
| GSD 5 (McArdle disease) |
Myophosphorylase | PYGM/11q13.1/ AR | Exercise induces fatigue, cramps, tachypnoea and tachycardia, rhabdomyolysis, myoglobinuria | [24] |
| GSD 6 (Hers disease) |
Liver glycogen phosphorylase |
PYGL/14q22.1/ AR | Hepatomegaly, hypoglycemia, with ketosis, elevated liver transaminases, hyperlipidemia, osteoporosis, liver fibrosis | [25,26] |
| GSD 7 (Tarui disease) |
Muscle phosphofructokinase |
PFKM/12q13.11/ AR | Hemolytic anemia, muscle weakness, exercise-induced muscle cramping, exertional Myopathy, gout/hyperuricemia |
[27,28] |
| GSD 9A1 (formerly GSD 8) |
Alpha-2 subunit of liver phosphorylase kinase |
PHKA2/Xp22.13 XL | Hepatomegaly, growth retardation, motor developmental delay. Hypercholesterolemia, hypertriglyceridemia, elevated liver enzymes; fasting hyperketosis |
[29] |
| GSD 9B (GSD IXb) |
Beta subunit of liver and muscle phosphorylase |
PHKB/16q12.1/ AR | Short stature, hepatomegaly, diarrhea, muscle Weakness, hypotonia |
[3,30] |
| GSD 9C (GSD IXc) |
Hepatic and testis isoform— gamma subunit of phosphorylase kinase |
PHKG2/16p11.2/AR | Growth retardation, hepatomegaly, hypotonia; cognitive delay |
[31,32] |
| GSD 9D (GSD IXd) |
Alpha subunit of muscle phosphorylase kinase |
PHKA1/Xq13.1/XL | Muscle weakness, exercise-induced muscle pain & stiffness, muscle atrophy, elevated CK |
[3,29,31] |
| GSD 10 (GSD X) | Muscle phosphoglycerate mutase |
PGAM2/7p13/AR | Exercise intolerance with cramp or pain, rhabdomyolysis, myoglobinuria, hyperuricemia, coronary arteriosclerosis | [2,33] |
| GSD 11 (GSD XI) |
Lactate dehydrogenase A | LDHA/11p15.1/AR | Exercise-induced muscle cramps & pain, uterine muscle stiffness in pregnancy, psoriatic skin lesions, Elevated serum CK during myoglobinuria, with low serum lactate dehydrogenase | [34,35,36] |
| GSD 12 (GSD XII) |
Fructose-1,6- bisphosphate aldolase |
ALDOA/16p11.2/AR | Short stature dysmorphic face, myopathy; mental retardation; delayed puberty; hemolytic anemia, hepatosplenomegaly; rhabdomyolysis with febrile illness |
[37,38] |
| GSD 13 (GSD XIII) |
Beta-enolase | ENO3/17p13.2/AR | Exercise intolerance, myalgia, rhabdomyolysis with fatty infiltration | [39,40] |
| GSD 15 (GSD XV) |
Glycogenin-1 | GYG1/3q24/AR | Weakness, arrhythmias, skeletal myopathy, cardiomyopathy | [41,42] |
| GSD 14 (Previously GSDXIV) | Phosphoglucomutase-1 | PGM1/1p31.3/AR | Hematological anomalies, hypoglycemia, growth retardation and dilated cardiomyopathy |
[43,44] |
| Fanconi–Bickel syndrome (GSD XI) |
SLC2A2 | GLUT2/3q26.2/AR | Postprandial elevations of glucose and galactose, fasting hypoglycemia, hepatomegaly, proximal tubular nephropathy, glucosuria, short stature |
[45,46] |
| PGK deficiency |
PGK1 | PGK/ Xq21.1/ XL | Nonspherocytic hemolytic anemia, myopathy with rhabdomyolysis and neurological features, myopathy, rhabdomyolysis |
[47,48] |
AR = autosomal recessive; XL = X-linked
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



