
Submitted:
28 June 2024
Posted:
02 July 2024
You are already at the latest version
Abstract
Given the importance of peroxisome proliferator-activated receptor (PPAR)-gamma in epidermal inflammation and carcinogenesis, we analyzed transcriptomic changes observed in epidermal PPARγ deficient mice (Pparg-/-epi). Gene set enrichment analysis revealed a close association with epithelial malignancy, inflammatory cell chemotaxis and cell survival. Single cell sequencing of Pparg-/-epi mice verified changes to the stromal compartment, including increased inflammatory cell infiltrates, particularly neutrophils and an increase in fibroblasts expressing myofibroblast marker genes. A comparison of transcriptomic data from Pparg-/-epi and publicly available human and or mouse actinic keratoses (AK) and cutaneous squamous cell carcinomas (SCCs) revealed strong correlation between the datasets. Importantly, PPAR signaling was the top common inhibited canonical pathway in AK and SCC. Both AKs and SCCs also had significantly reduced PPARG expression and PPARγ activity z-scores. Smaller reductions in PPARA expression and PPARα activity and increased PPARD expression but reduced PPARδ activation were also observed. Reduced PPAR activity was also associated with reduced PPARα/RXRα activity while LPS/IL1-mediated inhibition of RXR activity was significantly activated in the tumor datasets. Notably. these changes are not observed in normal sun-exposed skin relative to non-exposed skin. Finally, Ppara and Pparg were heavily expressed in sebocytes while Ppard was highly expressed in myofibroblasts, suggesting a role for PPARδ in myofibroblast differentiation. In conclusion, these data indicate PPARγ and possibly PPARα represent key tumor suppressors by acting as master inhibitors of the inflammatory changes found in AKs and SCCs.
Keywords:
Peroxisome proliferators activated receptor
; inflammation
; tumor suppression
; non-melanoma skin cancer
; actinic keratoses
; cutaneous squamous cell carcinoma
; transcriptomics
; gene set enrichment analysis
1. Introduction
PPARs are members of a large family of related ligand-activated nuclear receptors that bind to a common consensus recognition sequence in the promoters of target genes, the peroxisome proliferators response element (PPRE). Ligand-induced activation of PPARs bound to the PPRE subsequently induce target gene transcription (for review, see [1]). Three different PPAR proteins have been identified (PPARα, PPARδ and PPARγ), all of which require heterodimerization with the retinoid X receptor α (RXRα) for PPRE binding and transcriptional activity [1]. Endogenous PPAR ligands include unsaturated and saturated fatty acids and fatty acid metabolites [2,3]. While the three different PPAR receptors exhibit differences in tissue distribution, all three PPARs are known to be expressed in human and mouse skin [4]. In addition, all three PPAR subtypes play key roles as homeostatic regulators of lipid metabolism, energy balance, and cellular differentiation [2,3].
In addition to their role as direct transcriptional regulators of genes involved in energy balance and lipid metabolism, PPARγ acts through mechanistically distinct transrepressive signaling pathways to suppress the activities of other transcription factors, such as NF-κB, activator protein 1 (AP-1), and nuclear factor of activated T cells (NFAT) [5]. Using whole transcriptomic analysis of differentially expressed genes in Pparg-/-epi relative to wildtype control mice, we showed that Pparg-/-epi mice exhibit a marked increase in inflammatory mediators and gene products associated with inflammasome activation, indicating a key role for PPARγ as an important immune modulator [6]. The mice also developed spontaneous inflammatory skin lesions [6]. A role for PPARγ as a suppressor of cutaneous inflammation is seen in studies of inflammatory dermatoses. Relative to normal human control skin, PPARγ transcripts in psoriatic and atopic lesions are reduced 8- and 3.3-fold, respectively [7]. Another study demonstrated that PPARG mRNA is significantly decreased in human lichen planopilaris, a form of scarring (cicatricial) alopecia [8].
PPARs have also received interest for their potential role in neoplastic development. Keratinocyte-specific loss of Pparg in mice (Pparg-/-epi mice) results in increased photocarcinogenesis and photoinflammation [9], as well as a severe defect in normal contact hypersensitivity (CHS) responses [10]. Similarly, epidermal-specific loss of either Pparg or it’s heterodimeric partner RXRα in mice resulted in an approximately 2-fold increase in DMBA/PMA-induced tumors [11]. DMBA treatment of mice with hemizygous germline loss of Pparg resulted in a cancer incidence increase of over 3-fold, while metastatic disease increased 4.6-fold [12]. The increase in cancers included a 1.7-fold increase in cutaneous papilloma multiplicity.
In addition to these loss of function models, pharmacologic gain of function studies also indicate a potential role for PPARγ agonists in cutaneous malignancy. Treatment of mice with rosiglitazone was shown to suppress chemical carcinogenesis by approximately 70% [13]. Rosiglitazone also blocks the ability of ultraviolet light to suppress both CHS responses and anti-tumor immunity [10]. In addition, rosiglitazone promotes anti-tumor immune reactions in a mouse immunogenic cutaneous SCC tumor model [14]. In yet another study, the anti-neoplastic efficacy of immune therapy consisting of CTLA4 blockade and a cancer vaccine was enhanced by the addition of rosiglitazone treatment [15]. C0llectively, these data indicate a potential tumor suppressor role for PPARγ that is mediated by its effects on inflammation and anti-tumor immune responses.
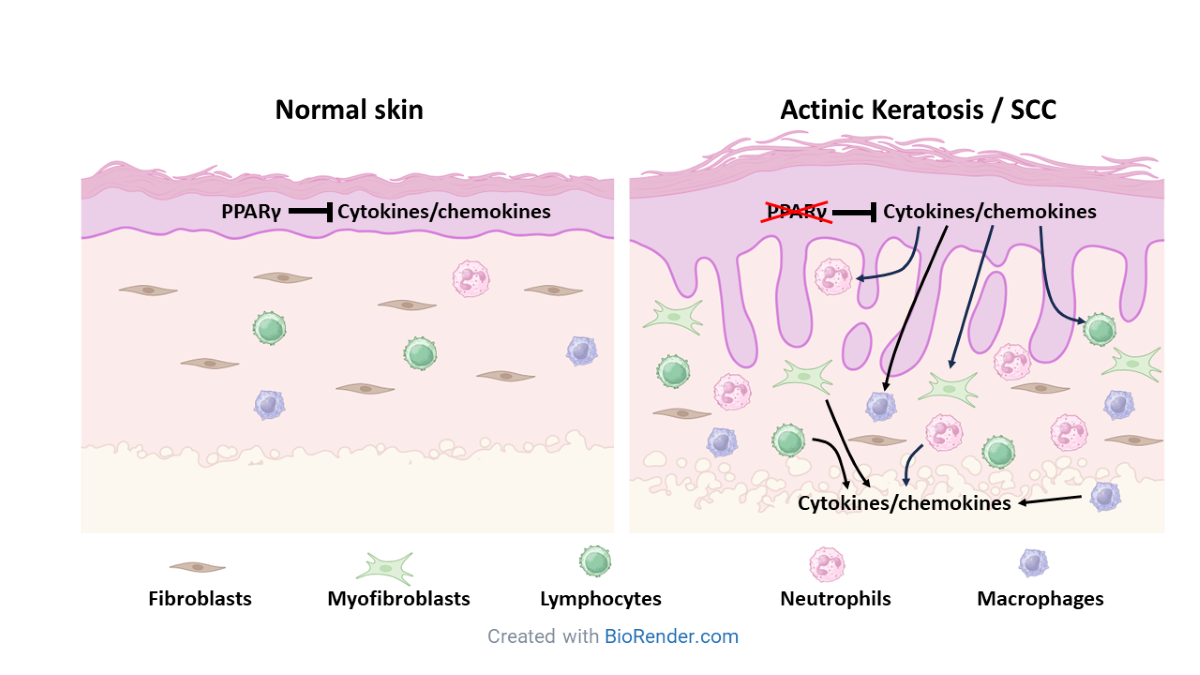
Chronic inflammation and immunosuppression are hallmarks of the tumor microenvironment [16,17]. While malignant tumors orchestrate changes to the stromal microenvironment to promote tumor growth and escape from immune surveillance, it is unclear how this process is regulated. Mice lacking epidermal Pparg (Pparg-/-epi mice) also exhibit immune suppression, chronic inflammation and increased chemical and photocarcinogenesis. We were therefore interested in determining whether the transcriptomic changes observed in Pparg-/-epi mice exhibit any overlap with non-melanoma skin cancer (NMSC). We utilized transcriptomic analysis and single cell RNA sequencing (scRNAseq) to provide evidence that PPAR signaling is the top inhibited canonical signaling pathway in NMSC and that this loss correlates with significant reductions in PPARγ expression and activity, increased cytokine & chemokine signaling. Smaller but significant reductions in PPARα mRNA expression and activity were also seen, suggesting a potential tumor suppressor role for PPARα as well. In Pparg-/-epi mice, scRNAseq further indicated that epidermal Pparg is a key epidermal regulator that modulates the recruitment of stromal myeloid, lymphoid and fibroblast populations.
2. Materials and Methods
2.1. Animal Studies
The derivation of mice lacking epidermal Pparg in the C57BL/6 background (Pparg-/-epi) and wildtype controls (lacking Cre recombinase) was previously described [10]. Mice were housed under specific pathogen-free conditions at the Indiana University School of Medicine.
2.2. Whole Transcriptomic mRNA Sequencing:
mRNA sequencing and subsequent differentially expressed gene analysis was performed on epidermal scrapings of Pparg-/-epi mice relative to wildtype controls as previously described [6]. This dataset is also publicly available at the Gene Expression Omnibus (GEO), National Center for Biotechnology Information (NCBI) depository (Accession number: GSE164024).
2.3. Single Cell Isolation
Two mice (1 male and 1 female) were euthanized for each genotype (wildtype and Pparg-/-epi). Dermal and epidermal cells were isolated from each mouse. For dermal cell preparation, the tissues were minced and digested in 2.5 mg Liberase TM, 16.7 mg DNase I, penicillin, streptomycin in RPMI media for 90 min at 37 °C. The digest was filtered through a 40 um filter. After adding 5 ml of ice cold dialyzed FBS, the samples were centrifuged at 300 x G for 10 minutes. RBC lysis (RBC Lysis buffer, eBioscienc, Cat #00-4333-57) followed by dead cell removal (Dead cell removal kit #130-090-101, Miltenyi Biotec) with centrifugation after each step was performed. The cell prep was washed twice with cell suspension buffer [PBS (calcium, magnesium, DNase and RNase free)(BioWorld # 40120706) containing 1% BSA] followed by centrifugation. RNA sequencing was then performed on a final cell prep resuspended in cell suspension buffer containing 0.5 U/µl SUPERaseIn™ RNase Inhibitor (ThermoFisher #AM2694) and 0.5 U/µl Protector RNase inhibitor (Sigma, #3335399001).
For epidermal cell preps, the dermal fat and dermis was removed using a scalpel and the epidermal sheet suspended in dispase II solution (Sigma) for 2 hrs at 37 °C. After suctioning the dispase and scraping the epidermis from the dermis, the tissue was rinsed with PBS, centrifuged and incubated in 5 ml Accutase solution (CellnTec, Cat# CnT-Accutase-100) for 20 min at room temp. After filtering through a 70 µm filter, 5 ml of dialyzed FBS was added and the cells pelleted by centrifugation. Dead cell removal and further processing was then performed as described for the dermal cell prep. The epidermal cells and dermal cells were then recombined at a 1:10 ratio (dermal vs epidermal) and the male and female cells pooled together.
2.4. Single Cells Sequencing and Library Prep Protocol
After evaluating for cell number, cell viability, and cell size, the appropriate number of cells were loaded on a multiple-channel micro-fluidics chip of the Chromium Single Cell Instrument (10x Genomics) with a targeted cell recovery of 10,000. Single cell gel beads in emulsion containing barcoded oligonucleotides and reverse transcriptase reagents were generated with the v3.1 Next GEM Single Cell 3ʹ reagent kit (10X Genomics). Following cell capture and cell lysis, cDNA was synthesized and amplified. An Illumina sequencing library was then prepared with the amplified cDNA with the Chromium™ Next GEM Single Cell 3ʹ GEM, Library & Gel Bead Kit v3.1. The resulting library was sequenced including cell barcode and UMI sequences and 100 bp RNA reads were generated with Illumina NovaSeq 6000 at the Center for Medical Genomics of Indiana University School of Medicine.
2.5. Analysis of scRNAseq data
CellRanger 6.1.1 (http://support.10xgenomics.com/) was utilized to process the raw sequence data generated. Briefly, cellranger mkfastq was implemented to demultiplex raw base sequence calls generated from the Illumina sequencer into sample-specific FASTQ files. The FASTQ files were then aligned to the mouse reference genome mm10 with RNAseq aligner STAR. The aligned reads were traced back to individual cells and the gene expression level of individual genes were quantified based on the number of UMIs (unique molecular indices) detected in each cell. The filtered feature-cell barcode matrices generated by CellRanger were used for further analysis.
2.6. SoupX Analysis
The R package SoupX version 1.5.2 [18] was used to remove the ambient RNA from the data.
2.7. Seurat Analysis
The R package Seurat version 4.0 [19,20,21,22] was used for the following analyses. This includes cell type/state discovery with graph-based clustering, cell cluster marker gene identification, and various visualization. A QC metrics of library size, number of features/genes, and mitochondrial reads (based on median-absolute-deviation (MAD), 3 MAD used here) is calculated with Scater [23]. This together with the QC analysis in Seurat are used to determine the parameters used for excluding low quality cells.
2.8. QC and Selecting Cells for Further Analysis
Low quality cells were excluded with the following criteria: cells with unique feature/gene counts over 7000 or less than 300, or >10% reads mapped to mitochondiral genome. A summary of the final scRNAseq data output is shown in Fig S1 (WT mouse cells) and Fig S2 (Pparg-/-epi mouse cells).
2.9. Unsupervised Cell Cluster Segregation and Cluster Identification
Unsupervised clustering was performed using Loupe Browser software (v6.5.0; 10x Genomics, Pleasanton, CA) by uniform manifold approximation and projection (UMAP). Using Loupe Browser, DEGs for each individual cell cluster was obtained. The cell type within each cluster were identified by uploading the top differentially expressed genes for each cluster into the Enrichr online analysis application [24,25,26], then cross-referencing with the PanglaoDB database [27].
2.10. GTEX Data Analysis
Tissue-specific (sun-exposed lower leg skin and sun protected suprapubic skin) TPM for PPARA, PPARD and PPARG were downloaded from the GTEX portal [www.gtexportal.org]. After uploading the TPM data, differential expression for the PPARs was calculated using edgeR (DEApp website: yanli.shinyapps.io/DEApp/) [28].
2.11. Gene Set Enrichment Analysis
Ingenuity Pathway Analysis (IPA) was performed to identify predicted Diseases and Biofunctions, canonical pathways and upstream regulators that are enriched in datasets relative to what would be expected by chance (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA) [29].
3. Results
3.1. Transcriptomic Changes in Pparg-/-epi Mice Shows a Strong Correlation to Cancer and Inflammatory Cell Recruitment and Mobilization
To better understand how loss of epidermal Pparg reflects disease processes, we reanalyzed our transcriptomic dataset from Pparg-/-epi mouse skin (GSE164024, [6]). After uploading the differentially expressed genes (DEGs) from the dataset into Ingenuity Pathway Analysis (IPA, Qiagen, Germantown, MD), we performed gene set enrichment (GSE) analysis for Diseases and Biofunctions (complete annotated list in Table S1). In Table 1A, the top 10 Disease and Biofunction terms that match to the Pparg-/-epi dataset are sorted by p-value. All of the Diseases and Biofunctions mapped to cancer-related biofunctions, particularly non-melanoma solid tumors and carcinoma. Thus, a strong statistical linkage to biological pathways that are over-represented in both the Pparg-/-epi mouse skin and cancer was observed.
The strong statistical linkage to cancer is somewhat surprising, as reduced PPARγ signaling has been linked to inflammatory skin diseases such as psoriasis and atopic dermatitis [7]. Thus, it was expected that these disease biofunctions would also be strongly linked to the Pparg-/-epi mouse skin dataset. However, the p-values for psoriasis and atopic dermatitis biofunctions were 3.00E-17 and 3.65E-12, respectively (Table S1). These p-values were considerably higher than those observed for the cancer-linked biofunctions seen in Table 1A.
In Table 1A, the observed activation z-scores were insufficient to predict an activating or inhibiting role for PPARγ in malignancy. This could be due to low effect size for key DEGs, inherent biological variability, or pathway complexity. It might be noted that the z-score for psoriasis (1.292) also failed to reach the 2.0 threshold that is used to predict activation while no z-score was calculated for the atopic dermatitis biofunction (Table S1).
A potential clue to how PPARγ could influence cancer is seen in Table 1B, in which the top 20 matching Disease and Biofunction terms are shown after sorting by highest positive activation z-scores. All 20 had activation z-scores of 3.942 to 5.052, well above the 2.0 cutoff that is used to predict activation. Of these 20, 15 map to biofunctions linked to inflammatory cell homing, chemotaxis or the inflammatory response. The remaining biofunctions were related to cell viability, tumor or cell invasion, and growth of lesions or tumors. Given the important role of inflammation in cancer, this data suggests that the primary interaction of PPARγ in tumor development may center on its anti-inflammatory activity. Overall, this non-biased approach found a strong linkage to cancer biofunctions in Table 1A and inflammatory biofunctions in Table 1B. In contrast, biofunctions associated with inflammatory skin diseases such as psoriasis and atopic dermatitis showed much lower p-values. This suggest that the inflammatory changes observed in Pparg-/-epi mouse skin are more relevant to neoplastic disease rather than benign causes of dermatitis.

| Table 1A: Top 10 Disease or Biofunction terms that match with the DEG dataset from Pparg-/-epi mouse skin relative to WT skin (sorted by p-value) | ||
|---|---|---|
| Diseases or Functions Annotation | p-value | z-score |
| Non-hematological solid tumor | 1.87E-66 | 0.316 |
| Epithelial neoplasm | 1.71E-65 | 0.042 |
| Non-melanoma solid tumor | 1.93E-65 | -0.047 |
| Tumorigenesis of tissue | 2.76E-65 | -0.252 |
| Nonhematologic malignant neoplasm | 1.27E-64 | 0.295 |
| Carcinoma | 3.19E-63 | 0.192 |
| Solid tumor | 1.03E-58 | 0.869 |
| Malignant solid tumor | 2.09E-58 | 0.252 |
| Cancer | 1.06E-57 | 1.516 |
| Head and neck tumor | 4.16E-57 | -0.124 |
| Table 1B: Top 20 Disease or Biofunction terms that match with the DEG dataset from Pparg-/-epi mouse skin relative to WT skin (sorted by z-score) | ||
| Diseases or Functions Annotation | p-value | Activation z-score |
| Chemotaxis of leukocytes | 1.61E-12 | 5.052 |
| Homing of leukocytes | 8.69E-14 | 4.724 |
| Cell movement of tumor cell lines | 3.36E-11 | 4.686 |
| Chemotaxis | 2.68E-18 | 4.644 |
| Migration of cells | 1.13E-30 | 4.623 |
| Homing of blood cells | 4.72E-14 | 4.574 |
| Cell survival | 2.9E-15 | 4.568 |
| Leukocyte migration | 2.98E-29 | 4.468 |
| Cell movement | 2.97E-37 | 4.464 |
| Cell movement of blood cells | 1.82E-29 | 4.36 |
| Invasion of cells | 9.29E-15 | 4.359 |
| Homing of cells | 3.91E-21 | 4.249 |
| Inflammatory response | 9.14E-20 | 4.174 |
| Cell movement of myeloid cells | 2.13E-18 | 4.171 |
| Recruitment of myeloid cells | 1.19E-13 | 4.121 |
| Cell viability | 1.77E-14 | 4.086 |
| Recruitment of blood cells | 3.44E-19 | 4.07 |
| Cell movement of phagocytes | 5.77E-19 | 4.029 |
| Growth of lesion | 5.78E-30 | 3.992 |
| Growth of tumor | 1.15E-29 | 3.942 |
3.2. Single Cell Sequencing of Pparg-/-epi Mice Reveals an Increase in Immune Cells, Particularly Neutrophils
To determine how loss of epidermal Pparg alters the stromal immune environment, we next performed single cell sequencing of skin from both WT and Pparg-/-epi mice. We found that 1560 genes were differentially expressed in the Pparg-/-epi cells relative to WT cells (Figure 1A, a full list of DEGs are found in Table S2). Of these, 94.9% of the DEGs were shared with the DEGs found in our previous whole transcriptomic RNAseq dataset [6]. After unsupervised cluster analysis, 26 different cell clusters were identified (Figure 1B). Table S3 shows the Enrichr analysis for cell types found in each cell cluster. After focusing on non-cytokeratin expressing cells, we found that 44.11% of non-keratinocytes represented Ptprc (CD45) expressing immune cells in Pparg-/-epi mouse skin (Figure 1C & Table S3). In contrast, 20.42% of non-keratinocytes were Ptprc+ in WT mouse skin (Figure 1C & Table S3). This increase in immune cells in Pparg-/-epi mouse skin relative to other stromal cell types was reflected as an increase in both myeloid and lymphoid cell populations (Figure 1C).
After further analysis of only the myeloid cell populations, we found that neutrophils represented only 0.6% of the total myeloid cell population observed in WT mice (Figure 1D & Table S3). This low neutrophil count is not particularly surprising as neutrophils are infrequent in normal mouse skin [30]. In contrast, neutrophils represented 25.93% of the total myeloid cell population in Pparg-/-epi mice, a 43-fold increase relative to WT mice (Figure 1D).
3.3. Fibroblasts Expressing Myofibroblast Markers are Increased in Pparg-/-epi Mouse Skin
In addition to differences in immune cell clusters, we also found differences in the stromal fibroblasts from Pparg-/-epi mouse skin. In Figure 2A, we show the expression of the collagen type I alpha 2 gene (Col1a2) in non-immune stromal cell clusters. Clusters 16 & 20, along with clusters 1, 3, 5, 9, 10 and 21, expressed high levels of Col1a2, consistent with the Enrichr identification of these clusters as fibroblasts. Also as expected, Col1a2 gene expression was absent in clusters representing primarily melanocytes and mast cells (cluster 4) and smooth muscle cells (cluster 18).
Enrichr analysis indicated that clusters 16 & 20 have differential gene expression overlap with pancreatic stellate cells (PSCs) (Table S3). As activated PSCs are highly fibrogenic myofibroblasts (myoFBs) [31], we verified that these fibroblast clusters expressed myofibroblast-specific genes. Figures 2B and 2C show the expression of S100 calcium binding protein A4 (S100a4) [also known as fibroblast specific protein 1] and alpha smooth muscle actin (Acta2), respectively. Both S100a4 and Acta2 are expressed in mouse dermal myofibroblasts, although Acta2 is the more specific marker [32]. Only fibroblast clusters 16 & 20 expressed both markers, with the expression particularly high in cluster 16. As an internal positive control, Acta2 was highly expressed in the smooth muscle cell cluster (cluster 18).
In Figure 2D, we show that cluster 16 myofibroblasts were infrequent in WT dermis (0.28% of non-immune stromal cells). In contrast, cluster 16 myofibroblasts were enriched in Pparg-/-epi mice, representing nearly 5% of non-immune stromal cells (relative fold change of 17.7-fold over WT mice). Similarly, cluster 20 myofibroblasts were enriched nearly 4-fold in Pparg-/-epi mouse skin (26.34% vs 6.68% of non-immune stromal cells in Pparg-/-epi and WT skin, respectively).
3.4. GSE Analysis Reveals Strong Similarity between the Pparg-/-epi Transcriptomic Data and that of Human Actinic Disease and Mouse and Human SCC
To verify that the chronic inflammatory microenvironment observed in Pparg-/-epi mice is relevant to non-melanoma skin cancer, we uploaded DEG datasets for both our whole transcriptomic RNAseq [6] and the single cell RNAseq (Table S1) for IPA analysis. We also uploaded DEGs for publicly available transcriptomic datasets of human actinic keratoses (AKs), human SCC, and mouse SCC (relative to normal skin) (Details of the publicly available datasets can be found in Table s4). To determine how closely the Pparg-/-epi datasets correlate with the different tumor datasets, we performed a comparison analysis for Diseases and Biofunctions.
Figures 3A & 3B show heatmaps that demonstrate the different Disease and Biofunction processes that are significantly associated with the datasets and are sorted by z-score. Figure 3A is limited to a comparison between Pparg-/-epi mouse skin data and human AK or SCC datasets while Figure 3B compares Pparg-/-epi data to mouse SCC. Both the whole transcriptomic and single cell RNA sequencing datasets for Pparg-/-epi mice were largely in agreement, with the observed differences generally associated with non-informative z-scores (z-score near 0) in the scRNAseq dataset due to the much smaller DEG profile for this less sensitive technique. For the tumor datasets, the activating scores varied but were largely in agreement across datasets. As with the two different Pparg-/-epi datasets, the greatest differences across different tumor datasets was largely due to non-informative terms. In addition, no consensus was seen for “Organismal death”, which was predicted to be activated, inhibited or not effected depending on the dataset. Surprisingly, the correlation between Pparg-/-epi mouse skin and human AKs and SCCs (Figure 3A) was stronger than that between Pparg-/-epi mice and mouse cutaneous SCCs (Figure 3B). When the Pparg-/-epi mouse dataset was compared to mouse SCCs, there were more non-informative functions observed in the Pparg-/-epi dataset compared to the mouse tumor datasets.
Given that Pparg-/-epi mice are highly susceptible to cutaneous carcinogenesis, it is particularly interesting that there was close agreement between the Pparg-/-epi transcriptomic datasets with the tumor datasets. In both Figures 3A & 3B, activating z-scores for functional annotations associated with cell movement, chemotaxis, and cell invasion were strongly associated with both Pparg-/-epi and tumor datasets. Other functions that were activated in most datasets included various functions associated with cell viability, tumor growth, tumor invasion and metastasis.
BCCs represent the most common NMSC observed in humans. We therefore also ran a correlation GSE analysis of our Pparg-/-epi datasets with 6 human BCC transcriptomic datasets (Figure S3). In contrast to the AK and SCC datasets, there was considerable discordance for the disease and biofunctions linked to different BCC datasets. This could indicate a greater degree of tumor heterogeneity in BCCs. Poor predictive modeling could also be due to a reduced number of gene expression changes or more modest effect size changes in the DEGs associated with indolent BCCs.
3.5. Canonical Pathway Analysis Shows Loss of PPAR Signaling is a Top Inhibited Canonical Pathway in Human AKs, Human SCCs and Mouse SCCs
To determine whether similarities in the diseases and biofunctions are associated with common signaling pathways, we utilized IPA to compare Canonical Pathways that are predicted to be activated or inhibited based on the DEGs found in Pparg-/-epi mouse skin and those from human AKs or SCCs (Figure 4A), as well as a comparison with both human and mouse SCCs (Figure 4B). As with Diseases and Biofunctions, there was a relatively high level of consistency in z-scores between the pathways mapped to Pparg-/-epi mouse skin and human tumors (Figure 4A). Canonical pathways with largely positive z-scores were heavily associated with cytokine & chemokine signaling, innate and adaptive immune responses, pyroptosis, the tumor microenvironment and fibrosis. These changes are not surprising given the known role of inflammation in tumor-stromal interactions.
The strong correlation between the predicted activation of these pathways in both malignancy and Pparg-/-epi mouse skin further supports a potential role for PPARγ as a tumor suppressing signal through its anti-inflammatory activity. It is therefore of interest that “PPAR Signaling” was the top common canonical pathway which showed largely negative z-scores in Figure 4A. Interestingly, the canonical pathway “LPS/IL-1 Mediated inhibition of RXR Function” was predicted to be activated in most of the AK and SCC datasets. This is of interest as the inhibition of RXRα activity could indirectly impact PPAR signaling as all PPARs require heterodimerization with RXRα for their transcriptional activity. In addition, as noted above, LPS and IL1β signaling also directly inhibit PPARG/Pparg transcript expression and target gene expression [33]. Thus, activation of this pathway can suppress PPARγ signaling both directly and indirectly.
In Figure 4B we show a similar heat map that illustrates common Canonical Pathways that are activated or inhibited in Pparg-/-epi mouse skin and either human or mouse SCCs. As with Fig 4A, the canonical pathway “PPAR Signaling” showed largely negative z-scores in all SCC datasets. In addition, several additional canonical pathways linked to PPAR signaling were also inhibited. “PPARα/RXRα activation” and “LXR/RXRα activation” canonical pathwasys were also predicted to be inhibited in Pparg-/-epi mouse skin and the tumor datasets. The predicted suppression of signaling by these two heterodimeric partners of RXRα is consistent with the predicted activation of “LPS/IL-mediated inhibition of RXR signaling” that we observed in human AK and SCC datasets (black arrows in Figs 4A & B).
As with Diseases and Biofunctions in Fig 3, there was more disagreement in the Canonical Pathways between the Pparg-/-epi mouse skin RNAseq data relative to mouse SCCs than there was between Pparg-/-epi mice and human SCCs (compare Figures 4A & 4B). These differences included positive z-scores for Canonical Pathways such as “Phagosome Formation”, “Role of NFAT in Regulation of the Immune Response”, “T cell receptor signaling” and “FAK Signaling” for Pparg-/-epi mice and human SCCs, but a trend to negative z-scores for these canonical pathways in mouse SCCs.
Finally, we also examined canonical signaling pathways in human BCCs (Figure S4). As with the Disease and Biofunctions analysis, canonical signaling pathways that were annotated to the Pparg-/-epi mouse or BCC datasets showed poor consensus. This suggests that the gene expression changes that occur with loss of epidermal Pparg may not be particularly relevant to the top canonical signaling pathways that are observed in BCCs.
3.6. A Shift to Reduced PPAR and RXR Activity and Reduced PPARA/Ppara & PPARG/Pparg Expression but Increased PPARD/Ppard Expression Occurs during the Progression from Sun-Exposed skin to NMSC
Given that PPAR and RXR signaling represent top inhibited canonical signaling pathways in Figures 4A & B, we further examined how PPAR signaling, PPARα/RXRα activation and LPS/IL1-mediated inhibition of RXR function were altered during tumor progression.
In Figure 5A, we plot the mean z-scores for the canonical pathway “PPAR Signaling” for the human and mouse tumor dataset as well as human sun-exposed skin (hSES). For hSES skin relative to non-sun-exposed skin (NES), “PPAR Signaling” had significantly positive mean z-scores, near the z-score activation cutoff of 2.0. In contrast, mean z-scores were significantly reduced for human AKs, human SCCs and mouse SCCs. In all three cases, the means were near the inhibitory z-score threshold of -2.0. The shift from a positive “PPAR Signaling” z-score in SES skin to a negative z-score in human SCC was also statistically significant.
In Figure 5B, the mean z-scores for canonical “PPARα/RXRα Activation” are similarly shown. In the case of SES vs NES skin, the datasets were non-informative, indicating no impact on canonical “PPARα/RXRα activation”. However, negative mean z-scores were observed for human AK, human SCC and mouse SCC. Again, the mean z-score for human BCCs was not significantly different from zero.
Figure 5C shows the z-score distribution for the different tumor types when the canonical pathway “LPS/IL1-mediated inhibition of RXR function” was plotted. Again, the SES vs NES datasets were non-informative. For human AK, human SCC and mouse SCCs, the mean z-scores were near or exceeded the activation cutoff of 2.0. In contrast to Figures 5A&B, human BCCs did show a significantly positive mean z-score, although this mean score was well below the activation cutoff.
As PPAR isoforms have significant overlap in target genes, the decrease in canonical PPAR Signaling that is seen in Figure 5A could reflect changes in the activity of one or more of the PPARα, PPARδ, or PPARγ isoforms. Moreover, the decrease in canonical PPARα/RXRα activation could indicate a reduction in PPARα activation. Alternatively, increased activation of the canonical pathway “LPS/IL1-mediated inhibition of RXR function” indicate that RXRα activity is likely reduced, a result that would impact the activity of all three PPAR isoforms. We therefore examined PPAR isotype transcript expression in human sun-exposed skin (SES), AKs, human SCCs and mouse cutaneous SCCs (Figure 5D).
In Figure 5D, mean PPARG expression was increased in SES relative to NES skin by approximately 1.5-fold. In the premalignant AK and malignant BCC and human SCC datasets, this was reversed: mean PPARG expression was reduced by 39.6% in AKs, 62.3% in SCCs and 84.0% in BCCs. This change from non-malignant SES skin was statistically significant for both the SCC and BCC datasets. In mouse SCCs, Pparg expression was also decreased by 84.7% relative to normal skin. The data for PPARA/Ppara in Figure 5D was similar in pattern to that observed for PPARG/Pparg, although to a lesser degree. However, the reduction in PPARA expression for the tumor datasets was not significantly different from that obtained from the SES datasets. In contrast to PPARA and PPARG, PPARD expression was not altered in SES skin or BCCs but PPARD/Ppard was significantly increased in AKs and both human and mouse SCCs.
A limitation of the analysis in Figure 5D is the relatively small number of studies that reported PPAR expression in SES vs NES skin. No PPAR expression data was reported for studies by Kita et al [34] and Zou et al [35] (Table s4). However, both studies limited their reported DEGs to those that met both a statistical threshold and a fold change threshold. The absence of all three PPARs in their listed DEGs indicates that PPARs were only modestly or non-significantly altered in SES vs NES skin. This idea was further supported by obtaining the GTEX dataset for SES (lower leg) and NES (suprapubic) skin and performing a differential expression analysis for the PPARs (Table s4). As expected, the changes in PPAR expression were modest and none were significant. Thus, a significant alteration of PPAR expression is not likely a feature of chronically sun-exposed skin. Thus, a shift to reduced PPARA and PPARG expression is a feature of premalignant AKs as well as the two most common NMSCs (BCC and SCC). For PPARD, this shift is reversed for AKs and SCCs, with a significant increase in expression relative to normal skin.
Interestingly, when we examined Ppara and Ppard expression in our Pparg-/-epi whole transcriptomic dataset, we found that loss of epidermal Pparg resulted in a 60% decrease in Ppara expression (FC = -2.482 (FDR = 1.89E-12)) and a 1.5-fold increase in Ppard expression (FDR=2.66E-04) (not shown). It is important to note that in Pparg-/-epi mice, the observed changes to Ppara expression in the skin are triggered by the loss of Pparg only within keratinocytes.
In the tumor datasets, it is unclear whether the observed changes in PPAR expression are occurring in the tumor epithelium or stromal cells. To address this question, PPAR isotype expression was obtained from a published dataset by Mitsui et al [36]. This study obtained transcriptomic data from AKs and SCC but utilized laser capture microdissection to limit the analysis to the epithelial tissue (Table s4). In this case, the pattern of PPAR expression was similar to the observed changes in Figure 5D. PPARA expression was reduced by 66.6% and 65.7% in SCCs and AKs, respectively (Table S4). PPARG expression was reduced by 58.3% in SCCs and 50.5% in AKs (Table S4). Finally, PPARD expression was increased 2.83-fold in SCCs and 2.48-fold in AKs (Table S4). Thus, this data suggests that alterations in PPAR expression are likely observed in tumor cells themselves. Whether a similar alteration in expression occurs within stromal cells is unclear and requires additional studies.
Since PPAR expression at the transcript level does not necessarily imply PPAR activity, we next performed an upstream regulator analysis in IPA to determine whether alterations in PPAR isoform activity occur in NMSC. In Figure 5E, we plot the predicted mean activation z-scores for PPARα, PPARδ and PPARγ for the tumor datasets. In SES relative to NES skin, the mean z-scores for all three PPARs were not significantly different from zero, indicating that none of the PPARs are predicted to be activated or inhibited in normal chronically sun-damaged skin. In contrast, a trend towards decreased PPARα and PPARγ z-scores is seen in AK lesions while significantly negative z-scores are observed in human SCC, BCCs, and mouse SCCs. In the cases of all three malignancies, the mean z-scores meet or exceed the z-score cutoff of -2.0 that would predict that both PPARα and PPARγ signaling are inhibited and correlate well with the expression data observed in Figure 5D.
While the predicted activity for both PPARα and PPARγ match up well with the expression data, the activity score for PPARδ in Figure 5E is the opposite of what would be predicted simply by assessing PPARD expression in Figure 5D. This is particularly notable for the mouse SCCs, in which PPARD expression is significantly elevated while PPARδ activity is predicted to be inhibited. The discordant results between PPARD expression and PPARδ activity could potentially be explained by reduced RXRα activity. Thus, inhibition of RXR function, such as through LPS or IL1 signaling, could indirectly suppress all RXRα heterodimeric partners, including PPARδ. The idea that all heterotrimeric partners of RXRα might be inhibited is supported by a shift from increased “LXR/RXR Activity” canonical pathway z-scores in sun-damaged skin to a reduction in “LXR/RXR Activity” z-scores for AKs, BCC, and human and mouse SCCs (Figure S5).
Finally, conflicting data is seen in BCCs. Figures 5A and B indicated that PPAR signaling overall and PPARα/RXRα activation are not significantly altered in BCC. Yet, like AKs and SCCs, PPARA & PPARG expression are decreased in Figure 5D and the activity 0f all three PPARs are predicted to be inhibited in Figure 5E. While it is difficult to explain these discrepant results, it is possible that differential epigenetic regulation of PPAR-specific target genes in neoplastic basal cell populations results in a skewed analysis of PPAR activity.
3.7. Increased PPARD Expression Represents a Marker of Myofibroblast Populations Found in Pparg-/-epi Skin
Given that loss of PPAR signaling and PPAR expression is a feature of AKs and SCCs, we examined the expression of the three PPAR isoforms within the individual cell clusters of our scRNAseq dataset. In Figures 6A & B, Ppara and Pparg expression are highly expressed only within the small sebocyte cluster (cluster 19). This is not surprising as PPARα and PPARγ are key to the lipogenesis that is needed to produce the sebum present in mature sebocytes [37]. The importance of PPARγ on sebocyte differentiation is seen in the absence of lipid-filled sebocytes in the dermis of Pparg-/-epi mice [6]. It is therefore not surprising that all of the sebocytes that were identified in the scRNAseq unsupervised cell clustering were found in WT mice (Table S3).
In contrast to Ppara and Pparg, Ppard was highly expressed only in fibroblast clusters with myofibroblast features as well as the smooth muscle cell cluster (Figure 6C). This is somewhat surprising and suggests that high Ppard expression in mouse fibroblasts may represent a marker of myofibroblast differentiation. It might be noted that myofibroblasts are increased in the tumor microenvironment [38]. Thus, future studies are needed to determine whether cancer-associated fibroblasts with myofibroblast features also overexpress Ppard. As noted above, laser capture microdissection data of human AKs and SCCs indicate that increased PPARD occurs within the tumor cell themselves (Table s4 and [36]). Thus, it remains to be seen whether tumor-associated myofibroblasts may be contributing to the increase in PPARD mRNA that was observed in the NMSCs. If so, studies to determine the role of PPARδ in cancer-associated fibroblast formation and function would be of interest.
Unfortunately, scRNAseq lacks the sensitivity to assess the level of PPAR isoform transcripts in the remaining clusters seen in Figures 6A-C. Thus, the absence of measurable Pparg expression in cluster 10 is surprising. ENRICHR analysis indicated that the gene expression signature of cluster 10 cells aligned with both fibroblasts and adipocytes. PPARγ expression is well known to be markedly upregulated during adipocyte differentiation and is necessary for adipogenesis [39]. The absence of increased Pparg expression in this cluster indicates that differentiated adipocytes were not present. This suggests that the adipocyte features in this cluster are reflective of the presence of adipocyte progenitor cells [40] or adipocyte-derived fibroblasts [41]. It might also be noted that the absence of mature adipocytes was also by design, as our cell isolation methodology included low-speed centrifugation to remove less dense lipid-laden adipocytes from our collection. This allowed us to enhance overall cell viability as well as to enrich our cell population for stromal fibroblasts and immune cells.
4. Discussion
In this report, we show that loss of epidermal Pparg is sufficient to induce transcriptomic changes that mimic those observed in actinic disease and SCCs. These changes include multiple changes in inflammatory signaling, chemokine expression and immune cell recruitment. Gene set enrichment analysis revealed largely similar activation and inhibition profiles for canonical signaling pathways and diseases and biofunctions. A surprising and informative finding is that there is a switch from increased PPAR signaling in normal sun-exposed skin (SES) to a loss of PPAR signaling in AKs and cutaneous SCCs. This PPAR signaling switch correlates with a similar switch between increased PPARG expression in SES skin to reduced PPARG expression in malignant AK and cutaneous SCC lesions. A similar but less intense change in the pattern of PPARA expression was also observed. In contrast, PPARD expression was increased in both AKs and cutaneous SCCs. This indicates that loss of overall PPAR signaling and an associated loss of PPARG and PPARA expression are important features that distinguish NMSC from sun-damaged skin. To our knowledge, this report is the first to demonstrate that PPAR Signaling the top commonly inhibited canonical signaling pathway in SCC development. We and others have shown that loss of epidermal Pparg promotes both chemical and photocarcinogenesis in mice [9,11,12]. Thus, our data further supports a key role for PPARγ as a tumor suppressor in both human and murine cutaneous SCC formation.
There are multiple potential mechanisms through which PPARγ activity could be lost in cutaneous neoplasia. This includes genetic deletions or inactivating mutations. Since germline loss of one Pparg allele in mice results in an increased susceptibility to chemical carcinogenesis [12], complete loss of PPARγ activity is likely not necessary for increased tumorigenesis. The human PPARG gene is located at the 3p25 chromosomal locus [42]. It is therefore of interest that loss of heterozygosity (LOH) of large portions of 3p were observed in 25% of AKs and 53% of cutaneous SCCs [43]. Another study showed that chromosomal loss of 3p is seen in 53% of SCC and 60% of SCC in situ [44]. LOH for 3p has also been described in 2 of 5 human SCC cell lines (SCC-12 and MET-1, but not SCC-13, SCL-I, or SCL-II) [45]. In addition, LOH of chromosomal locus 3p25 is common in related head & neck SCCs (54%)[46] and laryngeal SCC (60%) [47]. Interestingly, LOH of 3p25 was not seen in any of 10 cases of non-malignant laryngeal squamous metaplasia [47]. As we found that PPARG expression was not reduced in sun-exposed skin, this supports the idea that loss of PPARγ expression and activity is a tumor-specific event. In contrast to PPARG, LOH at chromosomal loci for the PPARA gene (22q) or the PPARD gene (6p) are infrequent (<5%) in cutaneous SCC [43,45,48,49].
Loss of function somatic missense mutations of PPARG have also been described in cancer but are not particularly common [50]. However, as UV is a potent mutagen and UV-induced tumors have the highest reported mutation burden of human cancers [51], it is possible that somatic LOF mutations are more frequent in cutaneous SCC.
An additional potential mechanism for loss of PPARγ signaling in AK and SCCs includes alternative splice variants with dominant negative (dnPPARγ) activity. A number of dnPPARγ splice variants have been shown to be increased in cancer (γORF4 [52], hPPARγ1tr [53], and PPARγΔ5 [54]). In mice, both TNF and LPS suppress Pparg expression and induce PpargΔ5 splice variant expression, providing a mechanism for loss of PPARγ activity through alternative splicing [33]. In the case of hPPARγ1tr, the expression of this splice variant was identified in lung SCC but not adjacent normal tissue [53]. Thus, upregulation of dnPPARγ variants specifically in tumors could also account for reduced PPARγ activity.
Reduced PPARG and PPARA expression have also been documented in some cancers through micro RNAs (miRNAs), long non-coding RNAs (lncRNAs) and promoter hypermethylation.
Using Targetscan 8.0 [55,56], 274 or 502 distinct miRNAs are predicted to target human PPARG or mouse Pparg respectively. While a role for miRNAs in suppressing PPARG expression has not been adequately studied in NMSC, one study demonstrated that miR-27b, miR-130b and miR-138 are all upregulated in colon cancer and correlated negatively with PPARG mRNA and protein expression [57]. In addition, elevation of miR-374a/-128/-130b were seen in 7 cases of human cutaneous SCC relative to non-lesional skin [58]. All three of these miRNAs are predicted to bind to the human PPARG 3’-UTR, although a role in regulating PPARG expression was not addressed.
Like miRNAs, a number of lncRNAs have validated ability to alter PPAR expression [59]. MALAT1 is a known lncRNA regulator of tumor development and is overexpressed in cutaneous SCC [60,61]. MALAT1 is also thought to target PPARG [59]. In competing endogenous RNA networks (ceRNA networks), sequence homology between lncRNAs and miRNAs competes for miRNA target binding to suppress the regulatory function of miRNAs. A study of human cutaneous SCCs examined correlations between 3221 differentially expressed transcripts and 24 differentially expressed lncRNAs (DElncRNA). By incorporating known miRNA targets of the lncRNAs, they were able to predict ceRNA networks that are operational in cutaneous SCC [62]. Of the 24 DElncRNAs that were identified, five were predicted to target PPARG (HCG18, LINC00342, HLA-F-AS1, SNX29P2, POLR214) [62]. In addition, all 5 were down-regulated. Thus, loss of these lncRNAs could in turn promote miRNA-induced suppression of PPARG expression. Consistent with this idea, PPARG expression was decreased in their dataset (Log2FC of -0.91071; FDR 1.73E-07).
Another epigenetic mechanism for potential loss of PPARG expression in skin cancer is promoter hypermethylation. Promoter hypermethylation of the PPARG gene is strongly correlated with a lack of PPARG expression in 30% of primary colorectal cancers as well as poor prognosis [63]. However, it has yet to be determined whether promoter methylation and silencing of PPARG gene expression occurs in NMSC.
Finally, recent studies indicate that PPARγ and NF-κB signaling have a complex and mutually antagonistic relationship. Studies in adipocytes, mesenchymal stem cells and macrophages show that stimulation with LPS or TNFα act to suppress overall PPARG/Pparg transcript expression and target gene expression [33]. Constitutive activation of NF-κB has been described as a common feature of malignancy due to activating mutations of NF-κB transcription factors themselves or upstream regulators [64]. It is possible that loss of PPARG exprersion or activity through any of these epigenetic or genetic mechanisms could result in a self-sustaining negative feedback cycle whereby the initial loss of PPARγ anti-inflammatory a ctivity results in increased inflammatory cytokine production that results in a further degradation of PPARγ signaling within the tumor cells themselves or surrounding stromal cells.
The decrease in PPARα expression suggests that this transcription factor may also play a tumor suppressor role, particularly in mouse SCC. Further studies are needed to clarify the degree to which PPARγ and PPARα activity are suppressed in NMSC and the mechanisms through which this occurs. It would therefore be of interest to determine the degree to which mice lacking epidermal Ppara exhibit transcriptomic changes that mirror those observed in AKs, SCCs, and Pparg-/-epi mice.
Given that PPARγ has an important anti-inflammatory role, it is not surprising that another key finding from our analysis is that inflammatory signaling and immune cell activation are a common feature of Pparg-/-epi mice, AKs and SCCs. This suggests that loss of PPARγ activity acts to promote neoplasia through its ability to induce stromal changes associated with tumorigenesis. This included a prominent accumulation of neutrophils and macrophage cells. It might be noted that mice expressing dominant negative Pparg (dnPparg) in type II pulmonary alveolar cells stimulate the mobilization and recruitment of myeloid cells with MDSC activity [65]. Given that Pparg-/-epi mice have a marked defect in CHS responses, it is tempting to speculate that the myeloid cell infiltrate that we observe exhibits MDSC activity. However, additional functional studies are needed to verify whether these myeloid cells represent MDSCs and contribute to the immune suppression seen in Pparg-/-epi mice.
An interesting feature of our analysis is the increase in PPARD/Ppard during malignant progression. It is possible that whatever mechanism is involved in down-regulating PPARγ and PPARα expression fails to elicit downregulation of PPARδ. Alternatively, since PPAR isoforms have overlapping functions, particularly in cellular energy production [66], the increase in PPARD/Ppard may also serve a compensatory function to mitigate the changes that are caused by loss of the other isoforms in human and mouse tumors. The increase in PPARδ may also promote tumor angiogenesis and progression through its effects on endothelial cells [67].
Another potential explanation for the increase in PPARδ transcripts in AKs and SCCs might be implied by our scRNAseq data from Pparg-/-epi mice. We found that Ppard expression was increased in Pparg-/-epi mice, particularly in fibroblasts expressing myofibroblast markers. It has been reported that PPARδ mediates fibroblast differentiation to profibrotic myofibroblasts by inducing the expression of TGFβ, which in turn induces the expression of alpha smooth muscle actin [68]. While the significance of this finding requires further studies, the presence of myofibroblasts is associated with chronic inflammation, fibrosis, wound healing [69]. Myofibroblast markers are also associated with cancer associated fibroblasts (CAFs) [70]. Thus, the observed increase in PPARD/Ppard expression in the tumor datasets may simply reflect an increase in myofibroblasts that are characteristic of the tumor stroma. Moreover, as PPARγ activation suppresses TGFβ expression and myofibroblast differentiation [71], this may also indicate that PPARγ & PPARδ have opposing actions in myofibroblast differentiation and fibrosis.
A weakness of our studies is that our mouse model results in embryonic loss of Pparg. Thus, our studies done in adult mice would suffer from long-standing dermal inflammatory changes that would create a new normal homeostatic state. Thus, the proximal events that are first initiated by loss of epidermal PPARγ cannot be assessed. If loss of PPARG is a defining feature of NMSC tumor-stromal interactions, then it would be important to determine the nature of these early signals to identify potential interventional targets. While this would be difficult to do in our current Pparg-/-epi mice, these studies could be performed in floxed Pparg mice crossed with a tamoxifen-inducible Krt14-Cre transgene.
In conclusion, loss of PPARγ expression and activity is a top feature of both Pparg-/-epi mouse skin and AK and SCC transcriptomic datasets. A more modest reduction in PPARα expression and activity is also observed. Single cell analysis also reveals that Pparg-/-epi mouse skin exhibits immune cell infiltrates and myofibroblast differentiation that is indicative of a chronic inflammatory state. This non-biased genomic approach supports previous studies indicating that PPARγ is an important tumor suppressor in cutaneous carcinogenesis leading to actinic disease and squamous cell carcinoma. Specifically, loss of tumor cell-specific PPARγ activity may be necessary for the establishment of the stromal inflammatory microenvironment that is a hallmark of neoplastic disease in the skin.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.Figure S1: Single Cell RNA Sequencing Data Summary for Wildtype (WT) mouse skin. Figure S2: Single Cell RNA Sequencing Data Summary for Pparg-/-epi (KO) mouse skin. Figure S3: Top Diseases and Biofunctions that are common to Pparg-/-epi mice and human BCC. Figure S4: Top Canonical Pathways that are common to Pparg-/-epi mice and human BCC. Figure S5: Canonical Pathway analysis shows tumor-specific reduction in LXR/RXR activation. Table S1: Full list of Diseases & Biofunctions that annotate to the Pparg-/-epi mouse skin DEG dataset. Table S2: Full list of DEGs from the single cell RNA sequencing. Table S3: Cluster identification. Table s4: Published transcriptomic datasets used for analysis.
Author Contributions
Conceptualization, R.L.K.; methodology, R.L.K, X.X., E.D., F.F.; software, R.L.K, H.G.; formal analysis, R.L.K.; writing—original draft preparation, R.L.K.; writing—review and editing, R.L.K.; supervision, R.L.K., Y.L.; project administration, R.L.K.; funding acquisition, R.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Merit Review Award # 1 I01 BX005816 and Merit Review Award # I01 BX003762 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (Institutional Animal Care and Use Committee) of the Indiana University School of Medicine and the Richard L. Roudebush VA Medical Center (protocol 11256, approved on 19 April 2017 and protocol 20006, approved on 3 November 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the corresponding author on request. Some of the data in this report were derived from resources available in the public domain: [See Table s4 for public dataset sources].
Acknowledgments
The authors would like to acknowledge Drs. Jeffrey B. Travers and Matthew J. Turner for their kind and helpful editorial suggestions.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of Nomenclature of Nuclear Receptors. Pharmacol Rev 2006, 58, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Pirat, C.; Farce, A.; Lebègue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.d.; Speca, S.; Berthelot, P.; Desreumaux, P. , et al. Targeting Peroxisome Proliferator-Activated Receptors (PPARs): Development of Modulators. J Med Chem 2012, 55, 4027–4061. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol Ther 2010, 125, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Schmuth, M.; Moosbrugger-Martinz, V.; Blunder, S.; Dubrac, S. Role of PPAR, LXR, and PXR in epidermal homeostasis and inflammation. Biochim Biophys Acta - Mol Cell Biol Lipids 2014, 1841, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-[gamma]. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Konger, R.L.; Derr-Yellin, E.; Zimmers, T.A.; Katona, T.; Xuei, X.; Liu, Y.; Zhou, H.-M.; Simpson, E.R.; Turner, M.J. Epidermal PPARγ Is a Key Homeostatic Regulator of Cutaneous Inflammation and Barrier Function in Mouse Skin. Int J Mol Sci 2021, 22, 8634. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, D.; El Tawdy, A.M.; Metwally, D.; Manar, A.; Rashed, L. Estimation of peroxisomal proliferators-activated receptor g gene expression in inflammatory skin diseases: Atopic dermititis and psoriasis. Our Dermatol Online 2014, 5, 107–112. [Google Scholar] [CrossRef]
- Karnik, P.; Tekeste, Z.; McCormick, T.S.; Gilliam, A.C.; Price, V.H.; Cooper, K.D.; Mirmirani, P. Hair Follicle Stem Cell-Specific PPAR[gamma] Deletion Causes Scarring Alopecia. J Invest Dermatol 2008, 129, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P.; DaSilva, S.C.; Rashid, B.; Martel, K.C.; Jernigan, D.; Mehta, S.R.; Mohamed, D.R.; Rezania, S.; Bradish, J.R.; Armstrong, A.B. , et al. Mice lacking epidermal PPARγ exhibit a marked augmentation in photocarcinogenesis associated with increased UVB-induced apoptosis, inflammation and barrier dysfunction. Int J Cancer 2012, 131, E1055–E1066. [Google Scholar] [CrossRef]
- Konger, R.L.; Derr-Yellin, E.; Travers, J.B.; Ocana, J.A.; Sahu, R.P. Epidermal PPARg influences subcutaneous tumor growth and acts through TNF-a to regulate contact hypersensitivity and the acute photoresponse. Oncotarget 2017, 8, 98184–98199. [Google Scholar] [CrossRef]
- Indra, A.K.; Castaneda, E.; Antal, M.C.; Jiang, M.; Messaddeq, N.; Meng, X.; Loehr, C.V.; Gariglio, P.; Kato, S.; Wahli, W. , et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J Invest Dermatol 2007, 127, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Nicol, C.J.; Yoon, M.; Ward, J.M.; Yamashita, M.; Fukamachi, K.; Peters, J.M.; Gonzalez, F.J. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis 2004, 25, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Konger, R.L. Evidence that peroxisome proliferator-activated receptor γ suppresses squamous carcinogenesis through anti-inflammatory signaling and regulation of the immune response. Mol Carcinog 2019, 58, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Konger, R.L.; Derr-Yellin, E.; Ermatov, N.; Ren, L.; Sahu, R.P. The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism. Molecules (Basel) 2019, 24, 2192. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Wong, K.; Nirschl, C.J.; Souders, N.; Neuberg, D.; Anandasabapathy, N.; Dranoff, G. PPARγ Contributes to Immunity Induced by Cancer Cell Vaccines That Secrete GM-CSF. Cancer Immunol Res 2018, 6, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Ritter, B.; Greten, F.R. Modulating inflammation for cancer therapy. J Exp Med 2019, 216, 1234–1243. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A. , et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 2020, 9. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nature Biotechnology 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., III; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M. , et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.; Wills, Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma'ayan, A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A. , et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016, 44, W90–97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M. , et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Andrade, J. DEApp: an interactive web interface for differential expression analysis of next generation sequence data. Source Code Biol Med 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Qie, C.; Jiang, J.; Liu, W.; Hu, X.; Chen, W.; Xie, X.; Liu, J. Single-cell RNA-Seq reveals the transcriptional landscape and heterogeneity of skin macrophages in Vsir(-/-) murine psoriasis. Theranostics 2020, 10, 10483–10497. [Google Scholar] [CrossRef]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Miyake, T.; Ehsanipour, E.A.; Kelly, P.J.; Becker, L.M.; McGrail, D.J.; Sugimoto, H.; LeBleu, V.S.; Ge, Y.; Kalluri, R. Dermal aSMA+ myofibroblasts orchestrate skin wound repair via b1 integrin and independent of type I collagen production. EMBO J 2022, 41, e109470. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, S.; Aprile, M.; Melillo, D.; Mucel, I.; Giorgetti-Peraldi, S.; Cormont, M.; Italiani, P.; Blüher, M.; Tanti, J.F.; Ciccodicola, A. , et al. TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells. Cells 2021, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Kita, R.; Fraser, H.B. Local Adaptation of Sun-Exposure-Dependent Gene Expression Regulation in Human Skin. PLOS Genet 2016, 12, e1006382. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.-D.; Xu, D.; Deng, Y.-Y.; Wu, W.-J.; Zhang, J.; Huang, L.; He, L. Identification of key genes in cutaneous squamous cell carcinoma: a transcriptome sequencing and bioinformatics profiling study. Ann Transl Med 2021, 9, 1497. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, H.; Suárez-Fariñas, M.; Gulati, N.; Shah, K.R.; Cannizzaro, M.V.; Coats, I.; Felsen, D.; Krueger, J.G.; Carucci, J.A. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol 2014, 134, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Downie, M.M.; Sanders, D.A.; Maier, L.M.; Stock, D.M.; Kealey, T. Peroxisome proliferator-activated receptor and farnesoid X receptor ligands differentially regulate sebaceous differentiation in human sebaceous gland organ cultures in vitro. Br J Dermatol 2004, 151, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Nimma, R.; Kundu, G.; Bulbule, A.; Kumar, T.V.S.; Gunasekaran, V.P.; Tomar, D.; Kumar, D.; Mane, A.; Gill, S.S. , et al. Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene 2021, 40, 2002–2017. [Google Scholar] [CrossRef]
- Hansson, B.; Rippe, C.; Kotowska, D.; Wasserstrom, S.; Säll, J.; Göransson, O.; Swärd, K.; Stenkula, K. Rosiglitazone drives cavin-2/SDPR expression in adipocytes in a CEBPa-dependent manner. PLoS ONE 2017, 12, e0173412. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: going back to the future. J Lipid Res 2012, 53, 227–246. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S. , et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Costa-Guda, J.; Rosen, E.D.; Jensen, R.T.; Chung, D.C.; Arnold, A. Mutational analysis of PPARG as a candidate tumour suppressor gene in enteropancreatic endocrine tumours. Clin Endocrinol 2005, 62, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Ashton, K.J.; Weinstein, S.R.; Maguire, D.J.; Griffiths, L.R. Chromosomal Aberrations in Squamous Cell Carcinoma and Solar Keratoses Revealed by Comparative Genomic Hybridization. Arch Dermatol 2003, 139, 876–882. [Google Scholar] [CrossRef]
- Ashton, K.J.; Carless, M.A.; Griffiths, L.R. Cytogenetic alterations in nonmelanoma skin cancer: A review. Genes, Chromosomes and Cancer 2005, 43, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer 2002, 99, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Maestro, R.; Gasparotto, D.; Vukosavljevic, T.; Barzan, L.; Sulfaro, S.; Boiocchi, M. Three Discrete Regions of Deletion at 3p in Head and Neck Cancers. Cancer Res 1993, 53, 5775–5779. [Google Scholar] [PubMed]
- Yoo, W.J.; Cho, S.H.; Lee, Y.S.; Park, G.S.; Kim, M.S.; Kim, B.K.; Park, W.S.; Lee, J.Y.; Kang, C.S. Loss of heterozygosity on chromosomes 3p,8p,9p and 17p in the progression of squamous cell carcinoma of the larynx. J Korean Med Sci 2004, 19, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.; Takata, M.; Wu, Y.Y.; Rees, J.L. Genetic change in actinic keratoses. Oncogene 1996, 12, 2483–2490. [Google Scholar]
- Quinn, A.G.; Sikkink, S.; Rees, J.L. Basal Cell Carcinomas and Squamous Cell Carcinomas of Human Skin Show Distinct Patterns of Chromosome Loss1. Cancer Res 1994, 54, 4756–4759. [Google Scholar]
- Ikezoe, T.; Miller, C.W.; Kawano, S.; Heaney, A.; Williamson, E.A.; Hisatake, J.; Green, E.; Hofmann, W.; Taguchi, H.; Koeffler, H.P. Mutational analysis of the peroxisome proliferator-activated receptor gamma gene in human malignancies. Cancer Res 2001, 61, 5307–5310. [Google Scholar]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J. , et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017, 9, 34. [Google Scholar] [CrossRef]
- Aprile, M.; Ambrosio, M.R.; D'Esposito, V.; Beguinot, F.; Formisano, P.; Costa, V.; Ciccodicola, A. PPARG in Human Adipogenesis: Differential Contribution of Canonical Transcripts and Dominant Negative Isoforms. PPAR Res 2014, 2014, 537865. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hwang, J.Y.; Kim, H.J.; Choi, W.S.; Lee, J.H.; Kim, H.J.; Chang, K.C.; Nishinaka, T.; Yabe-Nishimura, C.; Seo, H.G. Expression of a peroxisome proliferator-activated receptor gamma 1 splice variant that was identified in human lung cancers suppresses cell death induced by cisplatin and oxidative stress. Clin Cancer Res 2007, 13, 2577–2583. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front Endocrinol (Lausanne) 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Motawi, T.K.; Shaker, O.G.; Ismail, M.F.; Sayed, N.H. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: the Role of Epigenetics. Sci Rep 2017, 7, 10714. [Google Scholar] [CrossRef]
- Sand, M.; Skrygan, M.; Georgas, D.; Sand, D.; Hahn, S.A.; Gambichler, T.; Altmeyer, P.; Bechara, F.G. Microarray analysis of microRNA expression in cutaneous squamous cell carcinoma. J Dermatol Sci 2012, 68, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Kazeminasab, F.; Baharlooie, M.; Ghaedi, K. Noncoding RNAs Associated with PPARs in Etiology of MAFLD as a Novel Approach for Therapeutics Targets. PPAR Res 2022, 2022, 6161694. [Google Scholar] [CrossRef]
- Li, S.-S.; Zhou, L.; Gao, L.; Wang, Y.-H.; Ding, Z.-H. Role of long noncoding RNA MALAT1 promotes the occurrence and progression of cutaneous squamous cell carcinoma. Nan Fang Yi Ke Da Xue Xue Bao 2018, 38, 421–427. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, J.; Guo, L.; Peng, M. Long non-coding RNA MALAT1 regulates cell proliferation, invasion and apoptosis by modulating the Wnt signaling pathway in squamous cell carcinoma. Am J Transl Res 2021, 13, 9233–9240. [Google Scholar] [PubMed]
- Xu, Y.; Dong, Y.; Deng, Y.; Qi, Q.; Wu, M.; Liang, H.; She, Q.; Guo, Q. Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma. Biology (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Pancione, M.; Sabatino, L.; Fucci, A.; Carafa, V.; Nebbioso, A.; Forte, N.; Febbraro, A.; Parente, D.; Ambrosino, C.; Normanno, N. , et al. Epigenetic Silencing of Peroxisome Proliferator-Activated Receptor γ Is a Biomarker for Colorectal Cancer Progression and Adverse Patients' Outcome. PLoS ONE 2010, 5, e14229. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. NF-κB and Human Cancer: What Have We Learned over the Past 35 Years? Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, G.; Qu, P.; Yan, C.; Du, H. Overexpression of Dominant Negative Peroxisome Proliferator-Activated Receptor-γ (PPARγ) in Alveolar Type II Epithelial Cells Causes Inflammation and T-Cell Suppression in the Lung. Am J Pathol 2011, 178, 2191–2204. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.-F.; Wagner, N. Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.A.; Hwang, J.S.; Yoo, T.; Lee, W.J.; Paek, K.S.; Oh, J.-W.; Park, C.-K.; Kim, J.-H.; Do, J.T.; Kim, J.-H. , et al. Ligand-activated PPARδ upregulates α-smooth muscle actin expression in human dermal fibroblasts: A potential role for PPARδ in wound healing. J Dermatol Sci 2015, 80, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- Procopio, M.G.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.H.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U. , et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 2015, 17, 1193–1204. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Claes, V.; Vallée, A. The Myofibroblast: TGFβ-1, A Conductor which Plays a Key Role in Fibrosis by Regulating the Balance between PPARγ and the Canonical WNT Pathway. Nucl Recept Res 2017, 4, Article. [Google Scholar] [CrossRef] [PubMed]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N. , et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat Commun 2016, 7, 12601. [Google Scholar] [CrossRef] [PubMed]
- McCreery, M.Q.; Halliwill, K.D.; Chin, D.; Delrosario, R.; Hirst, G.; Vuong, P.; Jen, K.Y.; Hewinson, J.; Adams, D.J.; Balmain, A. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat Med 2015, 21, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, R.; Martínez-Cruz, A.B.; Santos, M.; Lorz, C.; Segrelles, C.; Garaulet, G.; Saiz-Ladera, C.; Costa, C.; Buitrago-Pérez, A.; Dueñas, M. , et al. Gene expression profiling of mouse p53-deficient epidermal carcinoma defines molecular determinants of human cancer malignancy. Mol Cancer 2010, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Segura, S.; Gadea, A.; Nonell, L.; Andrades, E.; Sánchez, S.; Pujol, R.; Hernández-Muñoz, I.; Toll, A. Identification of differentially expressed genes in actinic keratosis samples treated with ingenol mebutate gel. PLoS ONE 2020, 15, e0232146. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.L.T.; Tom, L.N.; Quek, X.-C.; Tan, J.-M.; Payne, E.J.; Lin, L.L.; Sinnya, S.; Raphael, A.P.; Lambie, D.; Frazer, I.H. , et al. RNA-seq reveals more consistent reference genes for gene expression studies in human non-melanoma skin cancers. PeerJ 2017, 5, e3631. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Ridgway, R.A.; Cammareri, P.; Treanor-Taylor, M.; Bailey, U.-M.; Schoenherr, C.; Bone, M.; Schreyer, D.; Purdie, K.; Thomson, J. , et al. Driver gene combinations dictate cutaneous squamous cell carcinoma disease continuum progression. Nat Commun 2023, 14, 5211. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Hameetman, L.; Commandeur, S.; Bavinck, J.N.; Wisgerhof, H.C.; de Gruijl, F.R.; Willemze, R.; Mullenders, L.; Tensen, C.P.; Vrieling, H. Molecular profiling of cutaneous squamous cell carcinomas and actinic keratoses from organ transplant recipients. BMC Cancer 2013, 13, 58. [Google Scholar] [CrossRef]
- García-Díez, I.; Hernández-Muñoz, I.; Hernández-Ruiz, E.; Nonell, L.; Puigdecanet, E.; Bódalo-Torruella, M.; Andrades, E.; Pujol, R.M.; Toll, A. Transcriptome and cytogenetic profiling analysis of matched in situ/invasive cutaneous squamous cell carcinomas from immunocompetent patients. Genes Chromosomes Cancer 2019, 58, 164–174. [Google Scholar] [CrossRef]
- Hu, Y.; Li, R.; Chen, H.; Chen, L.; Zhou, X.; Liu, L.; Ju, M.; Chen, K.; Huang, D. Comprehensive analysis of lncRNA-mRNAs co-expression network identifies potential lncRNA biomarkers in cutaneous squamous cell carcinoma. BMC Genom 2022, 23, 274. [Google Scholar] [CrossRef] [PubMed]
- Das Mahapatra, K.; Pasquali, L.; Søndergaard, J.N.; Lapins, J.; Nemeth, I.B.; Baltás, E.; Kemény, L.; Homey, B.; Moldovan, L.-I.; Kjems, J. , et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci Rep 2020, 10, 3637. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Dai, H.; Zhang, X.; Liu, S.; Lin, Y.; Somani, A.K.; Xie, J.; Han, J. Distinct transcriptomic landscapes of cutaneous basal cell carcinomas and squamous cell carcinomas. Genes Dis 2021, 8, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Brooks, Y.S.; Ostano, P.; Jo, S.H.; Dai, J.; Getsios, S.; Dziunycz, P.; Hofbauer, G.F.; Cerveny, K.; Chiorino, G.; Lefort, K. , et al. Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer. J Clin Invest 2014, 124, 2260–2276. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.S.; Peters, S.B.; Kaporis, H.; Cardinale, I.; Fei, J.; Ott, J.; Blumenberg, M.; Bowcock, A.M.; Krueger, J.G.; Carucci, J.A. Genomic Analysis Defines a Cancer-Specific Gene Expression Signature for Human Squamous Cell Carcinoma and Distinguishes Malignant Hyperproliferation from Benign Hyperplasia. J Invest Dermatol 2006, 126, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S. , et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Lo, B.K.; Yu, M.; Zloty, D.; Cowan, B.; Shapiro, J.; McElwee, K.J. CXCR3/ligands are significantly involved in the tumorigenesis of basal cell carcinomas. Am J Pathol 2010, 176, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Jee, B.A.; Lim, H.; Kwon, S.M.; Jo, Y.; Park, M.C.; Lee, I.J.; Woo, H.G. Molecular classification of basal cell carcinoma of skin by gene expression profiling. Mol Carcinog 2015, 54, 1605–1612. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L. , et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef]
Figure 1.
Single cell sequencing reveals changes in the dermal cell infiltrate in Pparg-/-epi mice. A.) Venn diagram showing the overlap between differentially expressed genes (DEGs) from whole transcriptomic RNA sequencing (RNA-seq) and from single cell RNA sequencing (scRNAseq) of Pparg-/-epi mouse skin. While scRNAseq suffers from reduced sensitivity, thus resulting in a much smaller number of DEGs, 94.94% of the DEGs identified by scRNAseq were also identified as DEGs by whole transcriptomic RNA-seq. B.) Unsupervised cell cluster analysis by uniform manifold approximation and projection (UMAP) depicts 26 cell clusters that are present in both WT and Pparg-/-epi mouse skin. FB: fibroblasts; KC: keratinocytes; Peri: pericytes; Mel: melanocytes; MC: mast cells; PMN: polymorphonuclear cells; Macro: macrocytes; Mono: monocytes; EC: endothelial cells; Adipo: adipocytes; LC: Langerhans cells; TC (g/d): γδ T-lymphocytes; DC: dendritic cells; NK: natural killer cells; NKT: natural killer T-lymphocytes; MyoFB: myofibroblasts; SM: smooth muscle: MDSC; myeloid-derived suppressor cells. C.) Cytokeratin negative stromal cells from either WT or Pparg-/-epi skin were subdivided into CD45 (Ptprc) positive and Ptprc negative (PtprcNeg) cell populations. Ptprc positive immune cells were subdivided into Cd3 positive lymphocyte and Cd3 negative myeloid populations. The different populations are depicted as a percent of total stromal cells. D.) Myeloid populations were further subdivided into granulocytic (PMN), Langerhans cell, and a pooled population of non-granulocytic macrophages, monocytes and dendritic cells (Macro/Mono/DC). Each population is shown as a percent of total myeloid cells.
Figure 1.
Single cell sequencing reveals changes in the dermal cell infiltrate in Pparg-/-epi mice. A.) Venn diagram showing the overlap between differentially expressed genes (DEGs) from whole transcriptomic RNA sequencing (RNA-seq) and from single cell RNA sequencing (scRNAseq) of Pparg-/-epi mouse skin. While scRNAseq suffers from reduced sensitivity, thus resulting in a much smaller number of DEGs, 94.94% of the DEGs identified by scRNAseq were also identified as DEGs by whole transcriptomic RNA-seq. B.) Unsupervised cell cluster analysis by uniform manifold approximation and projection (UMAP) depicts 26 cell clusters that are present in both WT and Pparg-/-epi mouse skin. FB: fibroblasts; KC: keratinocytes; Peri: pericytes; Mel: melanocytes; MC: mast cells; PMN: polymorphonuclear cells; Macro: macrocytes; Mono: monocytes; EC: endothelial cells; Adipo: adipocytes; LC: Langerhans cells; TC (g/d): γδ T-lymphocytes; DC: dendritic cells; NK: natural killer cells; NKT: natural killer T-lymphocytes; MyoFB: myofibroblasts; SM: smooth muscle: MDSC; myeloid-derived suppressor cells. C.) Cytokeratin negative stromal cells from either WT or Pparg-/-epi skin were subdivided into CD45 (Ptprc) positive and Ptprc negative (PtprcNeg) cell populations. Ptprc positive immune cells were subdivided into Cd3 positive lymphocyte and Cd3 negative myeloid populations. The different populations are depicted as a percent of total stromal cells. D.) Myeloid populations were further subdivided into granulocytic (PMN), Langerhans cell, and a pooled population of non-granulocytic macrophages, monocytes and dendritic cells (Macro/Mono/DC). Each population is shown as a percent of total myeloid cells.
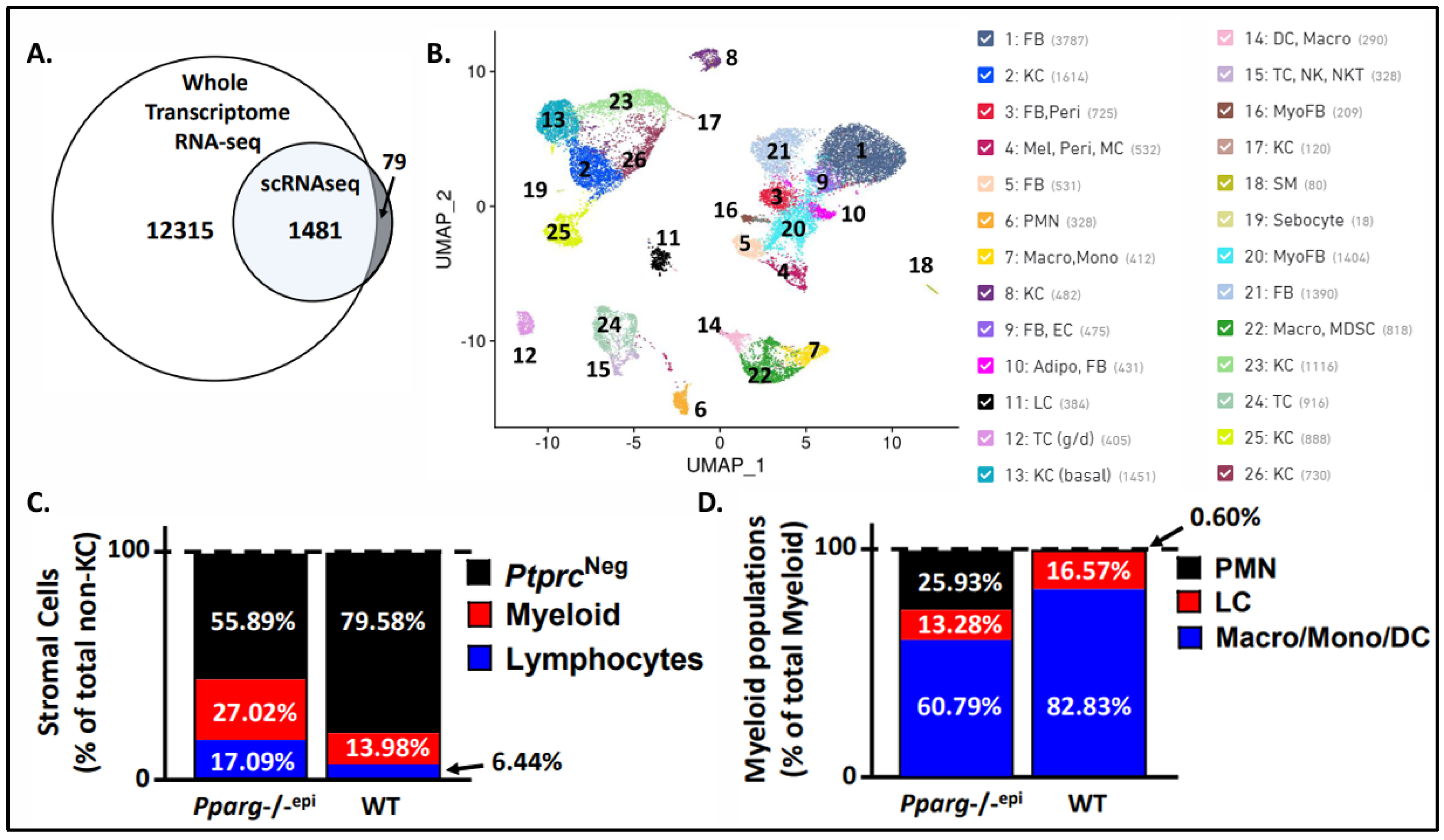
Figure 2.
Fibroblast clusters expressing myofibroblast gene markers are increased in Pparg-/-epi mouse skin. A-C.) Violin plots showing the log 2 fold change (Log2FC) of the following genes that were expessed in both cytokeratin negative & CD45 (Ptprc) negative stromal cell clusters: (A) alpha-1 type I collagen (Col1a1), (B) S100 calcium-binding protein A4 (S100a4), (C) Smooth muscle actin 2 (Acta2). D.) The percent of cluster 16 & 20 fibroblasts that are enriched in the expression of myofibroblast (MyoFB) markers as a percent of total non-immune stromal cells are shown for both WT and Pparg-/-epi mouse skin.
Figure 2.
Fibroblast clusters expressing myofibroblast gene markers are increased in Pparg-/-epi mouse skin. A-C.) Violin plots showing the log 2 fold change (Log2FC) of the following genes that were expessed in both cytokeratin negative & CD45 (Ptprc) negative stromal cell clusters: (A) alpha-1 type I collagen (Col1a1), (B) S100 calcium-binding protein A4 (S100a4), (C) Smooth muscle actin 2 (Acta2). D.) The percent of cluster 16 & 20 fibroblasts that are enriched in the expression of myofibroblast (MyoFB) markers as a percent of total non-immune stromal cells are shown for both WT and Pparg-/-epi mouse skin.
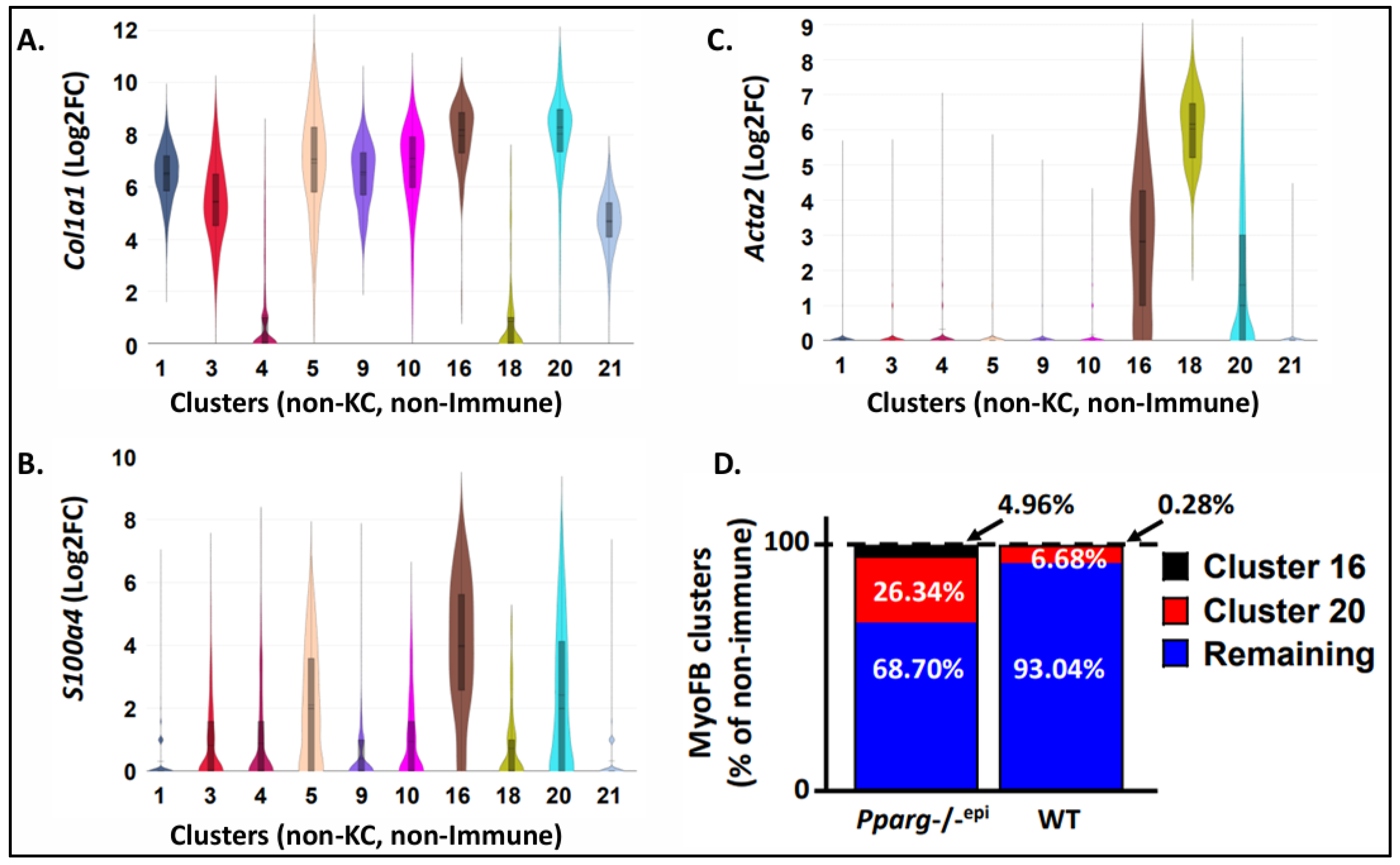
Figure 3.
Heat map showing top common Diseases and Biofunctions that are mapped to the differentially expressed genes found in Pparg-/-epi mice, human actinic keratoses (AK), human squamous cell carcinoma (SCC) and mouse SCCs. Differentially expressed genes from the whole transcriptomic RNA sequencing (RNAseq) and single cell RNA sequencing (scRNAseq) were obtained for Pparg-/-epi relative to WT mice. A.) The DEGs from these two datasets, as well as DEGs obtained from publicly available human AK and SCC tumor databases (Table S4) were uploaded for Qiagen’s Ingenuity Pathway Analysis. A comparison analysis was performed and the top 25 Diseases & BioFunction that were mapped to the various databases are shown and ranked by activation z-score. B.) Comparison heat map showing the top 25 Diseases & BioFunctions that are common to both the DEGs from Pparg-/-epi mouse skin and mouse SCC transcriptomic datasets.
Figure 3.
Heat map showing top common Diseases and Biofunctions that are mapped to the differentially expressed genes found in Pparg-/-epi mice, human actinic keratoses (AK), human squamous cell carcinoma (SCC) and mouse SCCs. Differentially expressed genes from the whole transcriptomic RNA sequencing (RNAseq) and single cell RNA sequencing (scRNAseq) were obtained for Pparg-/-epi relative to WT mice. A.) The DEGs from these two datasets, as well as DEGs obtained from publicly available human AK and SCC tumor databases (Table S4) were uploaded for Qiagen’s Ingenuity Pathway Analysis. A comparison analysis was performed and the top 25 Diseases & BioFunction that were mapped to the various databases are shown and ranked by activation z-score. B.) Comparison heat map showing the top 25 Diseases & BioFunctions that are common to both the DEGs from Pparg-/-epi mouse skin and mouse SCC transcriptomic datasets.
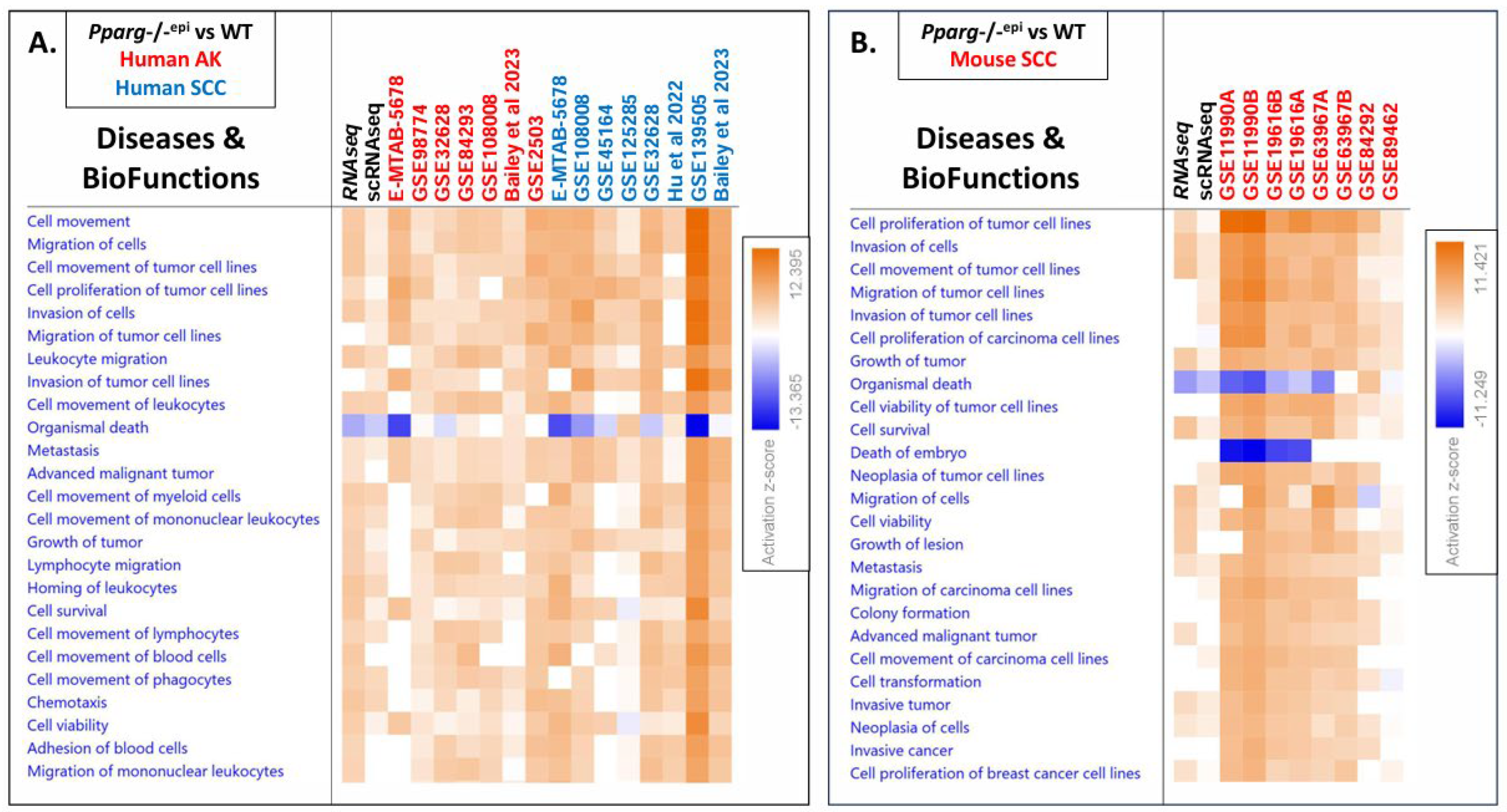
Figure 4.
Heat maps showing top common Canonical Pathways that are mapped to the differentially expressed genes found in Pparg-/-epi mice, human actinic keratoses (AK), human squamous cell carcinoma (SCC) and mouse SCCs. A&B.) The data analyzed in Figure 3 was further analyzed for common Canonical Pathways using IPA. Canonical Pathways in Figure A and/or B are marked as follows: “PPAR Signaling” is marked by the red arrow. “LPS/IL-1 Mediated inhibition of RXR Function” is marked by a black arrow. The green arrow marks “PPARα/RXRα Activation” and the blue arrow marks “LXR/RXR Activation”. A.) The top 30 Canonical Pathways that were mapped to the Pparg-/-epi mouse and human AK & SCC databases are shown and ranked by activation z-score. B.) The top 30 Canonical Pathways that are common to Pparg-/-epi mouse skin, human SCC and mouse SCC transcriptomic datasets are shown (ranked by z-score). .
Figure 4.
Heat maps showing top common Canonical Pathways that are mapped to the differentially expressed genes found in Pparg-/-epi mice, human actinic keratoses (AK), human squamous cell carcinoma (SCC) and mouse SCCs. A&B.) The data analyzed in Figure 3 was further analyzed for common Canonical Pathways using IPA. Canonical Pathways in Figure A and/or B are marked as follows: “PPAR Signaling” is marked by the red arrow. “LPS/IL-1 Mediated inhibition of RXR Function” is marked by a black arrow. The green arrow marks “PPARα/RXRα Activation” and the blue arrow marks “LXR/RXR Activation”. A.) The top 30 Canonical Pathways that were mapped to the Pparg-/-epi mouse and human AK & SCC databases are shown and ranked by activation z-score. B.) The top 30 Canonical Pathways that are common to Pparg-/-epi mouse skin, human SCC and mouse SCC transcriptomic datasets are shown (ranked by z-score). .
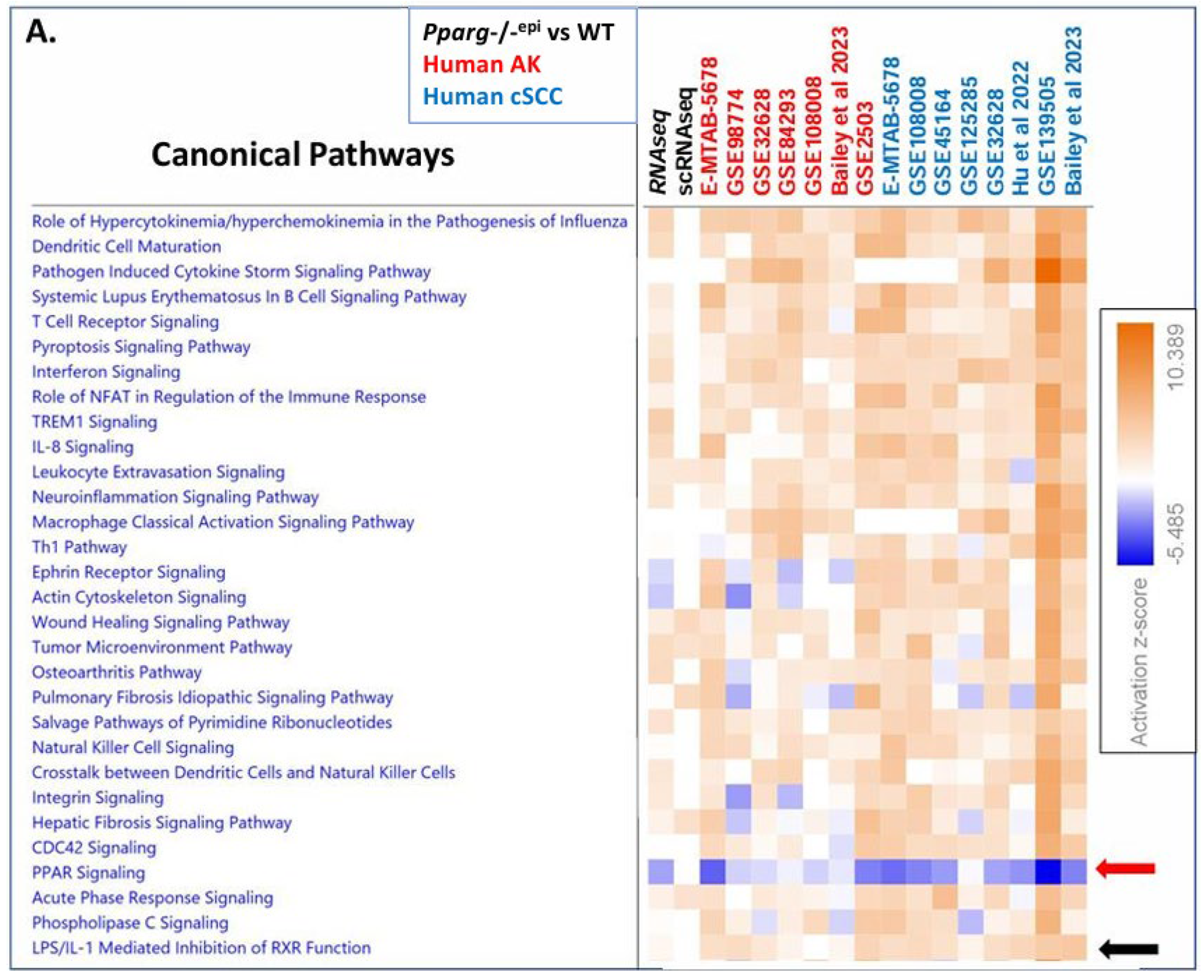
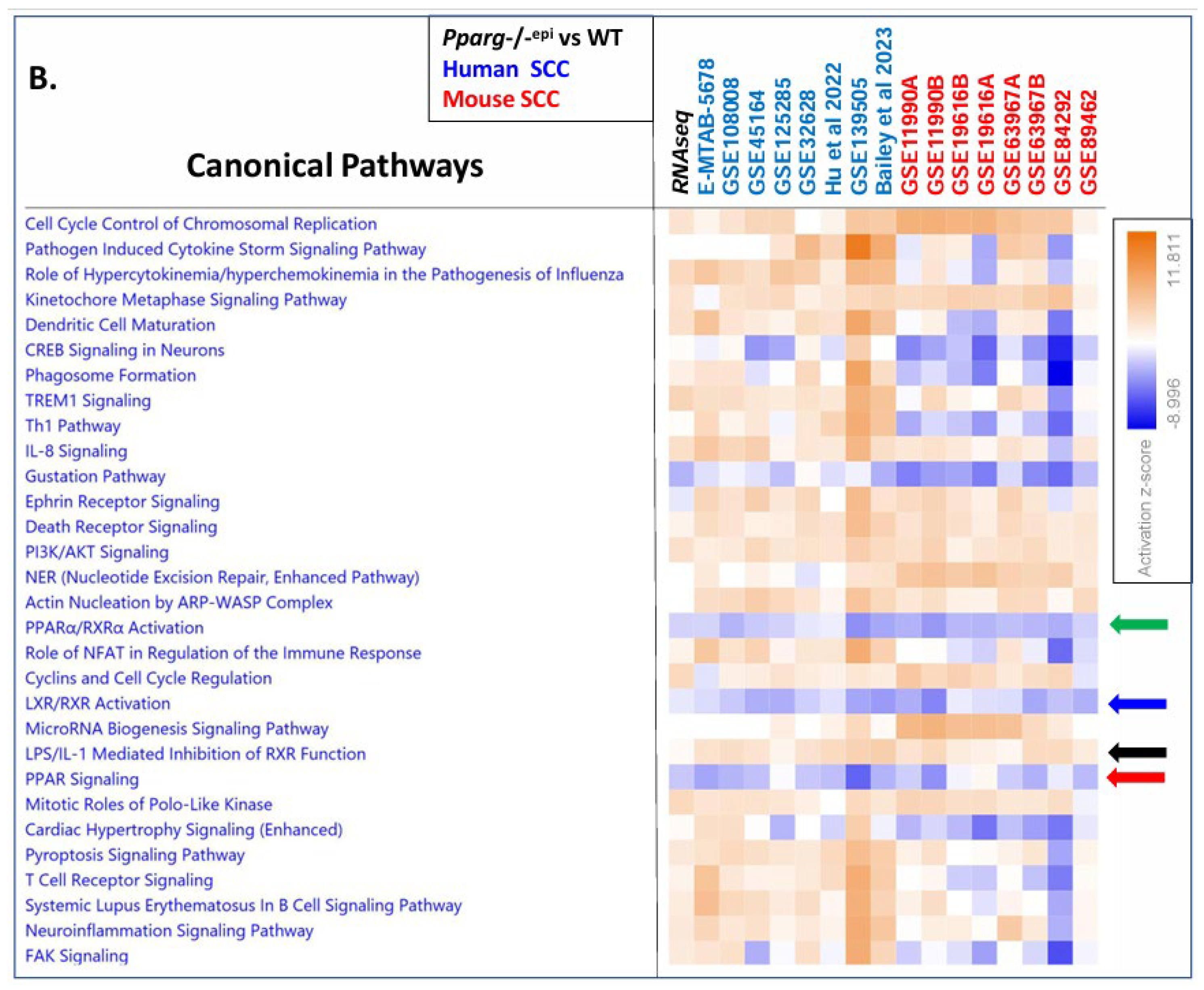
Figure 5.
Transcriptomic analysis indicates that PPAR signaling is inhibited, while PPARγ (and PPARα) expression and activity are suppressed in human AKs, human SCCs and mouse SCCs. A-C.) Following GSEA, activation z-scores were obtained for each of the different datasets for the following canonical pathways: (A.) PPAR Signaling, (B.) PPARα/RXRα Activation, and (C.) LPS/IL1-mediated Inhibition of RXR. The plots depict the mean & SEM of the activation z-scores for the different Canonical Pathways. The hashmark lines represent the cutoff for predicted activation (2.0 activation z-score) or predicted inhibition (-2.0) of the respective canonical pathway. For the hSES dataset: no predictive z-score was provided for these canonical pathways (ND = No data). D.) For each dataset, the mRNA expression for each of the three different PPAR isoforms were obtained as Log2 fold change (Log2FC). The data shown is the mean and SEM of the Log2FC. E.) After uploading the DEG data from each tumor or hSES dataset for IPA analysis, activation z-scores were obtained for PPARα, PPARδ and PPARγ. The data shown is the mean and SEM of the activation z-score for each PPAR isoform. The hashmark line represents the cutoff for predicted inhibition (-2.0) of the respective PPAR isoform. Blue asterisks = Significantly different from zero. One sample T-test. Red asterisks = Significantly different from hSES expression (1-way ANOVA). Black asterisks = Significantly different from each other (1-way ANOVA). *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Figure 5.
Transcriptomic analysis indicates that PPAR signaling is inhibited, while PPARγ (and PPARα) expression and activity are suppressed in human AKs, human SCCs and mouse SCCs. A-C.) Following GSEA, activation z-scores were obtained for each of the different datasets for the following canonical pathways: (A.) PPAR Signaling, (B.) PPARα/RXRα Activation, and (C.) LPS/IL1-mediated Inhibition of RXR. The plots depict the mean & SEM of the activation z-scores for the different Canonical Pathways. The hashmark lines represent the cutoff for predicted activation (2.0 activation z-score) or predicted inhibition (-2.0) of the respective canonical pathway. For the hSES dataset: no predictive z-score was provided for these canonical pathways (ND = No data). D.) For each dataset, the mRNA expression for each of the three different PPAR isoforms were obtained as Log2 fold change (Log2FC). The data shown is the mean and SEM of the Log2FC. E.) After uploading the DEG data from each tumor or hSES dataset for IPA analysis, activation z-scores were obtained for PPARα, PPARδ and PPARγ. The data shown is the mean and SEM of the activation z-score for each PPAR isoform. The hashmark line represents the cutoff for predicted inhibition (-2.0) of the respective PPAR isoform. Blue asterisks = Significantly different from zero. One sample T-test. Red asterisks = Significantly different from hSES expression (1-way ANOVA). Black asterisks = Significantly different from each other (1-way ANOVA). *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.

Figure 6.
Cell clusters with high level expression of the three PPAR isoforms. A-C.) The expression of Ppara (A), Pparg (B) and Ppard (C) are shown for all of the individual cell clusters obtained following scRNAseq of skin cells (combined cells isolated from both WT & Pparg-/-epi mouse skin). Both Ppara and Pparg expression were highly enriched in the sebocyte cluster 19. In contrast, Ppard expression was enriched in myofibroblasts (clusters 16 & 20) and smooth muscle cells (cluster 18).
Figure 6.
Cell clusters with high level expression of the three PPAR isoforms. A-C.) The expression of Ppara (A), Pparg (B) and Ppard (C) are shown for all of the individual cell clusters obtained following scRNAseq of skin cells (combined cells isolated from both WT & Pparg-/-epi mouse skin). Both Ppara and Pparg expression were highly enriched in the sebocyte cluster 19. In contrast, Ppard expression was enriched in myofibroblasts (clusters 16 & 20) and smooth muscle cells (cluster 18).
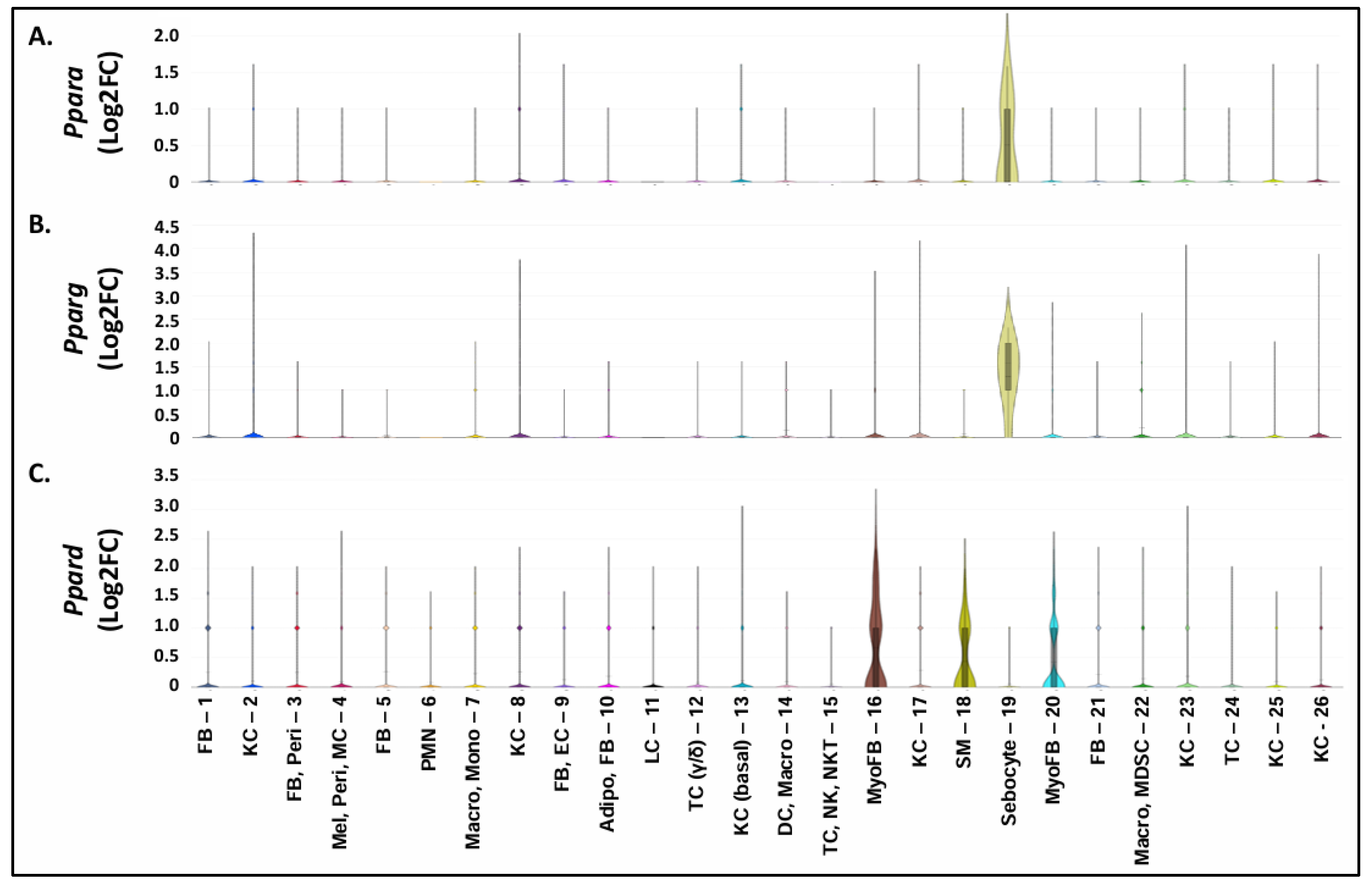
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



