
Submitted:
01 July 2024
Posted:
02 July 2024
You are already at the latest version
Abstract
Large-scale genetic studies have identified numerous genetic risk factors that suggest a central role for innate immune cells in susceptibility to Alzheimer’s disease (AD). CD33, an immunomodulatory transmembrane sialic-acid binding protein expressed on myeloid cells, was identified as one such genetic risk factor associated with Alzheimer’s disease. Several studies explored the molecular outcomes of genetic variation at the CD33 locus. It has been determined that the risk variant associated with AD increases the expression of the large isoform of CD33 (CD33M) in innate immune cells and alters its biological functions. CD33 is thought to signal via the interaction of its ITIM domain and the protein tyrosine phosphatase, SHP-1. Here, we utilize different molecular and computational approaches to investigate how AD-associated genetic variation in CD33 affects its interaction with SHP-1 in human microglia and microglia-like cells. Our findings demonstrate a genotype-dependent interaction between CD33 and SHP-1, which may functionally contribute to the AD risk associated with this CD33 variant. We also found that gene-gene interactions impact AD-related traits.
Keywords:
Alzheimer’s disease
; microglia
; CD33
; SHP-1
1. Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disease and the most common form of dementia, characterized initially by short-term memory loss and disorientation, followed by progressive memory loss and cognitive decline [1]. Classical neuropathological features of AD include presymptomatic accumulation of extracellular amyloid beta (Aβ) aggregates which form senile plaques, intracellular aggregation of hyperphosphorylated tau protein causing neurofibrillary tangles (NFTs), and extensive neuronal loss [2,3]. AD has become an urgent global health problem, with an enormous socioeconomic burden. The prevention and treatment of cognitive decline due to AD is a public health priority. However, AD has vast heterogeneity clinically, cellularly, and molecularly, illustrated by resilience to neuropathology, the presence of comorbidities, and genetic background, complicating therapeutic efforts [4]. Thus, there is a pressing need for therapeutics that modify the disease course and reverse the underlying pathology with a more personalized medicine approach. Genetics is one tool we can use to develop personalized targeted therapies.
With the discovery and validation of AD susceptibility loci, we now have more than 70 AD risk factors that give insights into the earliest steps in the pathophysiological processes leading to AD [5,6]. These genome-wide association and sequencing studies have identified and validated several genes, including CD33, that are associated with AD susceptibility and have implicated the innate immune system in disease onset [7,8,9,10,11,12,13]. In the case of CD33, the CD33 rs3865444C risk allele results in increased surface density of full-length CD33 protein on myeloid cells (monocytes, and microglia), as well as a reduced ability to phagocytose amyloid β, leading to accumulation of neuritic amyloid plaques [14]. Further, we have demonstrated that this risk allele leads to greater accumulation of amyloid both in asymptomatic individuals using in vivo amyloid imaging and in post-mortem brain tissue using a detailed quantitation of amyloid pathology [15].
CD33 is also known as Siglec-3 and is a 67-KDa transmembrane glycoprotein expressed on the surface of myeloid cells. It functions as a sialic acid binding receptor and is an inhibitory signaling molecule: its intracellular component contains putative immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that are implicated in the inhibition of cellular activity. The CD33 risk variant leads to decreased splicing of exon 2, which contains the sialic acid binding domain. This splicing suggests that the function of CD33 to bind sialic acid and bring signaling receptors into close proximity to dampen the function of these receptors is critical for AD risk [16]. Understanding how CD33 exerts its downstream effect on signaling proteins in microglia is crucial for our long-term goal of determining whether CD33 or its binding partners are tractable therapeutic targets for AD.
SHP-1 (encoded by PTPN6) is a protein tyrosine phosphatase that has been functionally linked to the inhibitory effects of CD33 signaling; it was found to bind to the ITIM domain of CD33 in both the THP1 and U937 myeloid cell lines after pervanadate treatment, which inhibits tyrosine phosphatases [17]. CD33 knockdown and/or SHP-1 reduction resulted in a shared phenotype of increased SYK phosphorylation and ERK1/2 signaling, inflammatory gene transcription, and phagocytosis of amyloid beta in the human macrophage THP1 cell line and human iPSC microglia [18]. These findings have identified SHP-1 as a potential target to modulate immune activation in AD. However, the influence of the CD33/SHP-1 interaction on the association between CD33 and AD has yet to be explored.
We hypothesize that CD33 and SHP-1 are binding partners and that their interaction in microglia participates in AD risk. In this work, we demonstrate that CD33 and SHP-1 are binding partners in human microglia-like cells in vitro and in human microglia in situ in post-mortem brain tissue. This interaction is increased in a phosphorylation-state and genotype-specific manner in microglia-like cells of the CD33 AD risk variant rs3865444CC. We further demonstrate that there are interactions between CD33 gene expression and PTPN6 gene expression on amyloid, tangles, pathologic AD, and global AD pathology burden.
2. Materials and Methods
2.1. Isolation of Human Monocytes and Monocyte-Derived Microglia-Like Cells (MDMi) Cell Differentiation
Peripheral blood mononuclear cells (PBMC) were extracted from blood samples from the New York Blood Center (NYBC) cohort using a standard Ficoll protocol (Ficoll-Paque TM PLUS) with Lymphoprep gradient centrifugation (StemCell Technologies). PBMCs were frozen at a concentration of 1 × 107 to 3 × 107 cells/ml in 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich) and 90% (v/v) fetal bovine serum (FBS; Corning). Frozen PBMCs were thawed and washed in 10 ml of phosphate-buffered saline (PBS). CD14+ monocytes were purified from whole PBMCs using anti-CD14+ microbeads (Miltenyi Biotec) and plated at 1.5 × 105 cells in a 96-well plate. To induce the differentiation of MDMi, monocytes were cultured in RPMI-1640 Glutamax (Life Technologies) supplemented with 1% P/S (Lonza), Fungizone (2.5 μg/ml; Life Technologies), and a mixture of the following human recombinant cytokines: macrophage colony-stimulating factor (M-CSF; 10 ng/ml; BioLegend, 574806), granulocyte-macrophage colony-stimulating factor (GM-CSF; 10 ng/ml; R&D Systems, 215-GM-010/CF), nerve growth factor–β (NGF-β; 10 ng/ml; R&D Systems, 256-GF-100), chemokine ligand 2 (CCL2; 100 ng/ml; BioLegend, 571404), and Interleukin-34 (IL-34; 100 ng/ml; R&D Systems, 5265-IL-010/CF) under standard humidified culture conditions (37°C, 5% CO2) for 10 days [19].
2.2. Tyrosine Phosphatase Inhibitor Treatment
MDMi were treated with the protein tyrosine phosphatase inhibitor sodium orthovanadate (pervanadate) immediately before performing proximity ligation assays as described below. A stock solution of 1mM sodium orthovanadate was freshly made with 3% hydrogen peroxide (H2O2) in water. After catalase was added, 100 μM sodium orthovanadate in media was added to the cells and incubated for 15 minutes at 37°C. The untreated control cells were incubated with media without pervanadate.
2.3. Preparation and Immunohistochemistry of Human Brain Tissue
Human post-mortem brain specimens used for immunohistochemistry (IHC) and proximity ligation assay (PLA) were obtained from the New York Brain Bank (NYBB) from the Columbia University Irving Medical Center ADRC and were accrued and processed per its published protocol [20]. Immunohistochemistry was performed on frozen sections of human prefrontal cortex. Sections were fixed in 100% ethanol for 15 minutes at -20°C after thawing them for 10 minutes. Slides were washed three times with 1X PBS. The sections were then blocked with 3% BSA in 1X PBS containing 0.1% Triton-X for 1 hour at room temperature. Primary antibody in 1% BSA in PBS was applied overnight at 4°C. Primary antibodies used were mouse anti-SHP-1, (Thermo Fisher, Cat# MA5-11669); rabbit anti-CD33 (Sigma, Cat# HPA035832). After washing the slides three times with PBS, secondary antibody in PBS was added for 1 hour at room temperature. The secondary antibodies used were goat anti-mouse IgG (H+L) conjugated to Alexa Fluor Plus 488 (Thermo Fisher; Cat # A32723; 1:300) and goat anti-rabbit IgG (H+L) conjugated to Alexa Fluor Plus 555 (Thermo Fisher; Cat# A32732; 1:300). Slides were washed three times and subsequently incubated with 0.3% Sudan Black in 70% ethanol for 10 minutes. After thoroughly washing with PBS, the slides were mounted with ProLong Gold with DAPI (Thermo Fisher, Cat # P36931) (Supplemental Figure 1).
2.4. Proximity Ligation Assay of MDMi and Human Brain Tissue
MDMi and tissue sections were prepared as described below and incubated with the following primary antibodies: Primary antibodies used were mouse anti-SHP-1, (Thermo Fisher, Cat# MA5-11669) and rabbit anti-CD33 ( Sigma, Cat# HPA035832).
MDMi (cells): MDMi were stained in flat-bottom 96-well plates (corning costar #3603). After 10 days of incubation in RPMI-1640 Glutamax supplemented with 1% P/S, Fungizone (2.5 μg/ml), and a mixture of human cytokines (described above), MDMi were centrifuged at 1200 rpm for 5 minutes. Cells were blocked with Fc receptor blocking solution (Human TruStain FcX, Biolegend, 1:20) for 10 minutes on ice. After centrifugation at 1200 rpm for 5 minutes, MDMi were fixed using BD Cytofix/CytoPerm (Thermo Fisher) for 20 minutes on ice. Cells were then washed with BD Perm/Wash (Thermo Fisher) and blocked with PBS containing 5% normal goat serum (Thermo Fisher, 31872), 2% bovine serum albumin (BSA) in PBS. Cells were then incubated with primary antibodies overnight.
Human tissues: Frozen sections of human prefrontal cortex and hippocampus were fixed in 100% ethanol for 15 minutes at -20°C after thawing them for 10 minutes. Slides were washed three times with 1X PBS. The sections were then blocked with 3% BSA in 1X PBS containing 0.1% Triton-X for 1 hour at room temperature. Tissues were then incubated with primary antibodies overnight.
Following overnight incubation with primary antibodies, a humid chamber with water was heated to 37°C. All procedures were performed at 37°C. The cells or tissue sections were washed three times with PBS and subsequently incubated with a plus and minus probe (Sigma; Probe Duolink PLA rabbit PLUS, DUO92002; Probe Duolink PLA mouse MINUS, DUO92004; Probe Duolink PLA mouse PLUS, DUO92001; Probe Duolink PLA goat MINUS, DUO92006; 150 µL/tissue section, 80 µL/well for cells) dissolved in 1X Antibody Diluent buffer (Sigma, provided with kit) for 1.5 hours (tissue sections) or on a shaker (60 rpm) for 1 hour (cells). After one (tissue) or two (cells) washes for 5 minutes with Duolink In Situ Wash Buffer A (Sigma, DUO82049), Duolink Ligation mixture (Sigma, DUO92008) was added for 1.5 hours (tissue, 150µl/section) or 30-40minutes on rotor (cells, 80 µL/well). Samples were washed once (tissue) or twice (cells) for 2 minutes with Wash Buffer A, and then incubated with Duolink Amplification mixture (Sigma, DUO92008) for 1.5 hours in dark (tissue, 150 µL/section; cells, 80 µL/well, on 60 rpm rotator). Then, samples were washed with Duolink In Situ Wash Buffer B (Sigma, DUO82049) for 2 minutes once (tissue) or twice (cells). Subsequently, DUOlink mounting medium with DAPI (Sigma, DUO82040) was added to cells (80 µL/well). After washing with Wash Buffer B, tissue sections were covered with 0.3% Sudan Black for 10 minutes at room temperature, then washed with PBS, and finally mounted with two drops of DUOlink mounting medium with DAPI.
For the PLA and immunohistochemistry, fluorescence was acquired with a confocal laser scanning microscope (LSM 700, Zeiss, Germany) and a Zeiss Axio Observer Z1 fluorescence microscope, running Zen 2012 software (Zeiss). Images were exported to and quantified by using Image J software (NIH, Maryland, USA). PLA interactions were quantified as PLA dots per live DAPI+ nucleated cell.
2.5. Genotyping
For genotyping of the blood or brain samples, DNA was isolated with the PureLink Mini Kit (ThermoFisher) and quantified by nanodrop. Once the DNA was isolated, it was diluted to 25ng/µL. Genotyping for the CD33 SNP rs3865444 was done with the C_1487395_40 assay (ThermoFisher).
2.6. Statistical Analysis
Data are presented as mean ± s.e.m. A comparison of two groups was performed using an unpaired t-test. For multiple-group comparisons, analysis of variance (ANOVA) with post hoc Tukey correction was applied. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA). A p-value of < 0.05 was considered significant.
2.7. Gene-Gene Interaction
Gene-gene interactions were investigated using the ROSMAP RNA-sequencing and GWAS data that have been previously described to examine the interplay between CD33 and PTPN6 using three models [21,22]. All ROSMAP participants were enrolled without known dementia and agreed to detailed clinical evaluation and brain donation at death [23]. Clinical and pathologic methods have been previously described [24,25,26,27,28,29]. Linear regression models for quantitative traits and logistic regression models for binary traits were used to test the association of CD33 gene expression with PTPN6 levels in the dorsolateral prefrontal cortex (DLPFC) with a) neuropathological LOAD status, b) beta-amyloid levels, c) neurofibrillary tangle (NFT) burden, d) global measure of pathology based on the scaled scores of 5 brain regions e) estimated slope of global cognition using longitudinal measurements, f) hippocampal sclerosis, g) TDP43 pathology and h) clinical dementia diagnosis antemortem. The gene expression levels were adjusted for age, sex, RIN score, postmortem interval, and other technical covariates for RNA-sequencing and the residual expression values were used for interaction testing. A quantitative trait loci approach was used to test whether genetic variation in PTPN6 regulates CD33 expression levels and vice versa. Common SNPs (MAF>0.01) were tested for association with residual gene expression levels in a linear regression model adjusting for age, sex, and population substructure variables. Lastly, the R mediation package was used to test whether the association of CD33 expression with clinical AD, AD neuropathology and cognition is mediated by PTPN6 levels. This package was also used to test mediation in the other direction (CD33 mediates PTPN6 association with traits) to establish causality.
3. Results
3.1. CD33 and SHP-1 Interact in Human Microglia-Like Cells in a CD33 Genotype-Sensitive Manner
To investigate the interaction between CD33 and SHP-1, proximity ligation assay (PLA) was performed on primary monocyte-derived microglia-like (MDMi) cell cultures from individuals genotyped for the CD33 AD-associated variant, rs3865444. As PLA detects protein-protein interactions in situ at a distance <40nm, we used this assay to measure the number of interactions per cell [30]. The CD33 AD-associated risk variant, rs3865444CC, increases CD33 surface expression on human monocytes, microglia, and MDMi [14,19]. Here, we demonstrate that CD33 and SHP-1 interact in the PLA assay (Figure 1). However, the number of PLA dots, which represent singular interactions, does not differ between genotypes at the baseline level. As SHP-1 is a phosphatase, we next investigated whether this interaction is sensitive to the phosphorylation state. We treated MDMi with pervanadate, which inhibits phosphatase activity, resulting in globally increased phosphorylation [31]. PLA demonstrated increased CD33-SHP-1 interaction in the context of pervanadate but only in MDMi from individuals homozygous for the risk allele, suggesting that this interaction is both sensitive to the phosphorylation state and to the genetic background of the individual. In the presence of pervanadate, there was a significant increase in the number of CD33-SHP-1 interactions in MDMi from individuals homozygous for the risk variant, rs3865444CC, compared to MDMi from individuals homozygous for the protective variant, rs3865444AA (Figure 1).
3.2. Genotype-Specific CD33-SHP-1 Interactions in Post-Mortem Human Brain Tissue
We next tested whether SHP-1 colocalizes with the CD33-positive cells in human prefrontal cortex. By utilizing an immunofluorescence technique coupled with confocal microscopy, we found colocalization of SHP-1 and CD33, suggesting these proteins may interact (Supplemental Figure 1). To investigate whether these proteins interact, we performed in situ PLA in frozen post-mortem human brain tissues (prefrontal cortex) from five donors (Supplemental Table 1). We found 18.4 times more PLA signal in the rs3865444CC group compared to the rs3865444AA group (Figure 2), mirroring our genotype-specific findings observed in MDMi. This finding suggests that not only are CD33 and SHP-1 within the same biological complex of 40nm and interacting to signal downstream functions, but their interaction is dictated by genotype in situ. The increased interaction in situ, even without pervanadate treatment, may be due to a modified activation state of SHP-1 in the brain of individuals with the rs3865444CC genotype, suggesting importance of molecular changes throughout the AD brain on microglial signaling.
3.3. CD33 and PTPN6 Gene-Expression Interaction Impacts Risk for Clinical and Pathological Features of AD
To examine the implication of the interaction between CD33 and SHP-1 on disease, we examined gene-gene, SNP-gene, and mediation interactions in a transcriptomics dataset of frontal cortex from the ROSMAP cohort, a longitudinal cohort of aging [23]. To understand the biological significance of this interaction in the context of Alzheimer’s disease, we leveraged existing ROSMAP dorsolateral prefrontal cortex (DLPFC) transcriptomic and genetic datasets and investigated whether the interactions between CD33 and PTPN6 gene expression influence AD clinical and pathological traits. After adjustment for multiple testing correction for testing eight traits, CD33-PTPN6 interaction was significantly associated with amyloid burden, tangles, pathological diagnosis of AD, and global burden of AD pathology. Their interaction was also nominally significant with an antemortem diagnosis of clinical AD (Table 1). This is supportive of the idea that CD33 requires SHP-1 to signal in immune cells. We additionally performed trans-eQTL analysis. We tested the influence of six SNPs in the PTPN6 locus on CD33 expression and 57 SNPs in the CD33 locus and found no trans-eQTLs of gene expression in either gene (Supplemental. 2,3).
We then performed causal mediation analysis to test whether PTPN6 gene expression mediates the association between CD33 gene expression and AD-related traits, or vice versa. PTPN6 gene expression did not mediate any associations between CD33 and any traits (Supplemental Table 4). However, CD33 gene expression was found to mediate the association between PTPN6 gene expression and clinical AD dementia. These findings suggest that PTPN6 gene expression affects CD33 gene expression, and, in turn, the CD33 gene expression affects AD dementia (p=0.008), while PTPN6 gene expression is not directly associated with AD dementia Supplemental Table 5). To examine this further, we used mediation analysis to determine that CD33 gene expression mediated the association of PTPN6 gene expression on AD dementia (Supplemental Tables 2,3).
4. Discussion
In this study, we showed that genotype-dependent interactions between CD33 and SHP-1 in human microglia and microglia-like cells provide novel insights into the molecular mechanisms underlying Alzheimer’s disease (AD) susceptibility. The data demonstrates that the AD-associated CD33 rs3865444CC risk allele modulates the interaction between CD33 and SHP-1 in a phosphorylation-sensitive manner, potentially contributing to altered microglial function in AD. Additionally, we found a significant interaction between CD33 and PTPN6 (the gene encoding SHP-1) gene expression that influence various AD-related traits, suggesting a complex molecular interplay impacting AD pathology.
Recent literature supports the critical role of innate immune cells in AD susceptibility, with CD33 as a prominent genetic risk factor [15,32,33,34,35,36,37,38]. The CD33 risk allele rs3865444C increases full-length CD33 protein expression, leading to impaired microglial internalization of amyloid-β and ultimately amyloid plaque accumulation [16]. Our findings show that the CD33-SHP-1 interaction is phosphorylation-sensitive and genotype-influenced, suggesting that genotype-specific therapeutic strategies may be a relevant approach. Higher CD33-SHP-1 interaction in the brain with the CD33 risk allele may result from chronic activation of CD33, maintaining SHP-1 in an active state, facilitating its downstream signaling, ultimately contributing to AD pathology.
Additionally, utilizing a large transcriptomic dataset, we were able to demonstrate that there are multiple pathological outcomes influenced by the gene-gene interaction of CD33 and PTPN6. Specifically, we find that the interaction of CD33 and PTPN6 modulates the association with amyloid burden, tangles, pathological diagnosis of AD, and global burden of AD pathology. This demonstrates that CD33 and SHP-1/PTPN6 are critical partners of a shared molecular pathway. Despite this strong association with pathology, the gene-gene interaction between CD33 and PTPN6 did not influence cognitive decline, TDP43, Hippocampal sclerosis, and clinical diagnosis of AD, supporting the complexity of AD and microglial genes, as well as the concept that AD is more than buildup of amyloid plaques and tau tangles.
Another interesting contrast between our protein and the gene expression findings in regards to CD33-SHP-1 interaction is the role of genotype. Genetic variation at the CD33 locus does not influence PTPN6 gene expression and genetic variation at the PTPN6 locus does not influence CD33 expression. However, the AD-associated genetic variation at the CD33 locus does influence the interaction of CD33 and SHP-1 at the protein level.
Our study provides important insights, although there are a few limitations to consider. The sample size for the in situ analysis of post-mortem brain tissues was limited to five donors, so expanding the cohort in future studies would help validate our findings and enhance their generalizability. Our study primarily focused on the phosphorylation state of SHP-1 and its interaction with CD33. Other post-translational modifications of SHP-1 or CD33, which might also influence their interaction, were not explored. Comprehensive studies considering various protein modifications are needed. While we demonstrated an interaction between CD33 and SHP-1, the functional consequences of this interaction in the context of microglial activity and AD progression were not fully elucidated. Further studies are needed to link these molecular interactions to specific cellular and pathological outcomes.
Given the genotype-dependent nature of the CD33-SHP-1 protein interaction, developing targeted therapies that modulate this interaction may be beneficial. Small molecule inhibitors or biologics that disrupt CD33-SHP-1 signaling in a controlled manner might be a novel therapeutic avenue. Expanding the scope of genetic interactions to include other innate immune receptors and their signaling partners could provide a more comprehensive understanding of the immune dysregulation in AD. Integrating multi-omic data from large cohorts could further elucidate these complex networks. Longitudinal studies tracking the progression of AD in relation to CD33 and SHP-1 interaction dynamics could provide valuable insights into how these interactions evolve with disease progression and influence clinical outcomes.
In conclusion, our study underscores the critical role of understanding the interaction partners and signaling pathways in microglia of genetically associated proteins, contributing significantly to the genetic risk of Alzheimer’s disease. These findings provide valuable insights into the molecular mechanisms underlying AD and suggest potential genotype-specific therapeutic strategies targeting the CD33-SHP-1 pathway. Future research should focus on further elucidating these interactions and exploring therapeutic interventions that can effectively modulate this pathway to mitigate AD progression. Continued investigation in this area holds promise for developing more effective treatments for this devastating disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, LB, MR, ZC, and EMB; Methodology, LB and EMB; Software, LB, MR, AL, and BV; Validation, LB, MR, and ZC; Formal Analysis, LB, MR, and AL; Investigation, LB and EMB; Resources, DB and EMB; Data Curation, LB, MR, and AL; Writing – Original Draft Preparation, LB, MR, ZC, and EMB; Writing – Review & Editing, All authors; Visualization, LB, MR, and AL; Supervision, BV, EMB; Project Administration, EMB and DB; Funding Acquisition, EMB, MR, ZC, and DB. LB, MR, and AL contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the US National Institutes of Health grants RF1AG058852, R01AG043617, F30AG074618, and T32AI148099. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. ROSMAP is supported by P30AG10161, P30AG72975, R01AG15819, R01AG17917, U01AG46152, and U01AG61356.
Institutional Review Board Statement
Both studies, ROS and MAP, were approved by an Institutional Review Board of Rush University Medical Center (ROS IRB# L91020181, MAP IRB# L86121802). Both studies were conducted according to the principles expressed in the Declaration of Helsinki. Each participant signed an informed consent, Anatomic Gift Act, and an RADC Repository consent (IRB# L99032481) allowing her data and biospecimens to be repurposed.
Data Availability Statement
ROSMAP resources can be requested at https://www.radc.rush.edu and www.synpase.org.
Acknowledgments
The authors are grateful to the participants of the NYBC and NYBB for the time and specimens that they contributed. We thank the Columbia University Alzheimer’s Disease Research Center, funded by NIH grant P30AG066462 to S.A. Small (P.I.), and A. Teich for providing biological samples and associated information.
References
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. May 2011;7(3):280-92. [CrossRef]
- Huang HC, Jiang ZF. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer’s disease. J Alzheimers Dis. 2009;16(1):15-27. [CrossRef]
- Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. Apr 2014;71(4):505-8. [CrossRef]
- Duara R, Barker W. Heterogeneity in Alzheimer’s Disease Diagnosis and Progression Rates: Implications for Therapeutic Trials. Neurotherapeutics. Jan 2022;19(1):8-25. [CrossRef]
- Hollingworth P, Harold D, Jones L, Owen MJ, Williams J. Alzheimer’s disease genetics: current knowledge and future challenges. Int J Geriatr Psychiatry. Aug 2011;26(8):793-802. [CrossRef]
- Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. Jan 1 2015;77(1):43-51. [CrossRef]
- Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. Oct 2009;41(10):1088-93. [CrossRef]
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. Oct 2009;41(10):1094-9. [CrossRef]
- Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. May 12 2010;303(18):1832-40. [CrossRef]
- Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. May 2011;43(5):436-41. [CrossRef]
- Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. Jan 10 2013;368(2):117-27. [CrossRef]
- Jin SC, Benitez BA, Karch CM, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. Nov 1 2014;23(21):5838-46. [CrossRef]
- Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. May 2011;43(5):429-35. [CrossRef]
- Bradshaw EM, Chibnik LB, Keenan BT, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. Jul 2013;16(7):848-50. [CrossRef]
- Bradshaw EM, Chibnik LB, Keenan BT, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. Jul 2013;16(7):848-50. [CrossRef]
- Raj T, Ryan KJ, Replogle JM, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. May 15 2014;23(10):2729-36. [CrossRef]
- Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, Freeman SD. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J Biol Chem. Apr 23 1999;274(17):11505-12. [CrossRef]
- Wissfeld J, Nozaki I, Mathews M, et al. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia. Jun 2021;69(6):1393-1412. [CrossRef]
- Ryan KJ, White CC, Patel K, et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med. Dec 20 2017;9(421). [CrossRef]
- Vonsattel JP, Del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. May 2008;115(5):509-32. [CrossRef]
- De Jager PL, Ma Y, McCabe C, et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data. Aug 07 2018;5:180142. [CrossRef]
- Ng B, White CC, Klein HU, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. Oct 2017;20(10):1418-1426. [CrossRef]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis. 2018;64(s1):S161-S189. [CrossRef]
- Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. Jun 27 2006;66(12):1837-44. [CrossRef]
- Bennett DA, Wilson RS, Schneider JA, et al. Natural history of mild cognitive impairment in older persons. Neurology. Jul 23 2002;59(2):198-205. [CrossRef]
- Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology. 2006;27(3):169-76. [CrossRef]
- Oveisgharan S, Yang J, Yu L, et al. Estrogen Receptor Genes, Cognitive Decline, and Alzheimer Disease. Neurology. Apr 04 2023;100(14):e1474-e1487. [CrossRef]
- Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol. Jan 2019;85(1):114-124. [CrossRef]
- Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age-related neuropathologies? Brain. Aug 17 2021;144(7):2166-2175. [CrossRef]
- Fredriksson S, Gullberg M, Jarvius J, et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. May 2002;20(5):473-7. [CrossRef]
- Fantus IG, Kadota S, Deragon G, Foster B, Posner BI. Pervanadate [peroxide(s) of vanadate] mimics insulin action in rat adipocytes via activation of the insulin receptor tyrosine kinase. Biochemistry. Oct 31 1989;28(22):8864-71. [CrossRef]
- Chan G, White CC, Winn PA, et al. CD33 modulates TREM2: convergence of Alzheimer loci. Nat Neurosci. Nov 2015;18(11):1556-8. [CrossRef]
- Dos Santos LR, Pimassoni LHS, Sena GGS, et al. Validating GWAS Variants from Microglial Genes Implicated in Alzheimer’s Disease. J Mol Neurosci. Jun 2017;62(2):215-221. [CrossRef]
- G N S HS, Marise VLP, Satish KS, et al. Untangling huge literature to disinter genetic underpinnings of Alzheimer’s Disease: A systematic review and meta-analysis. Ageing Res Rev. Nov 2021;71:101421. [CrossRef]
- Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. May 22 2013;78(4):631-43. [CrossRef]
- Gu X, Dou M, Cao B, Jiang Z, Chen Y. Peripheral level of CD33 and Alzheimer’s disease: a bidirectional two-sample Mendelian randomization study. Transl Psychiatry. Oct 03 2022;12(1):427. [CrossRef]
- Li WW, Wang Z, Fan DY, et al. Association of Polygenic Risk Score with Age at Onset and Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease in a Chinese Cohort. Neurosci Bull. Jul 2020;36(7):696-704. [CrossRef]
- Malik M, Chiles J, Xi HS, et al. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum Mol Genet. Jun 15 2015;24(12):3557-70. [CrossRef]
Figure 1.
CD33 and SHP-1 interact in a genotype dependent manner. (a) A representative confocal image of MDMi cells stained for Proximity Ligation Assays (PLA). The proximity ligation puncta (red dots) indicated by arrows represent the protein-protein interaction between CD33 and SHP-11. DAPI (blue) is used to counterstain the nucleus. (b) Quantification of the PLA dots shows a significant increase in the PLA counts in the CD33 CC groups (n=4) when the cells are treated with pervanadate compared to the CD33 AA group (n=4). Data represented as mean ± SEM. ***p < 0.001, *p<0.01, Two-way ANOVA followed by Tukey’s multiple comparisons test. Scale bar: 50 µm.
Figure 1.
CD33 and SHP-1 interact in a genotype dependent manner. (a) A representative confocal image of MDMi cells stained for Proximity Ligation Assays (PLA). The proximity ligation puncta (red dots) indicated by arrows represent the protein-protein interaction between CD33 and SHP-11. DAPI (blue) is used to counterstain the nucleus. (b) Quantification of the PLA dots shows a significant increase in the PLA counts in the CD33 CC groups (n=4) when the cells are treated with pervanadate compared to the CD33 AA group (n=4). Data represented as mean ± SEM. ***p < 0.001, *p<0.01, Two-way ANOVA followed by Tukey’s multiple comparisons test. Scale bar: 50 µm.
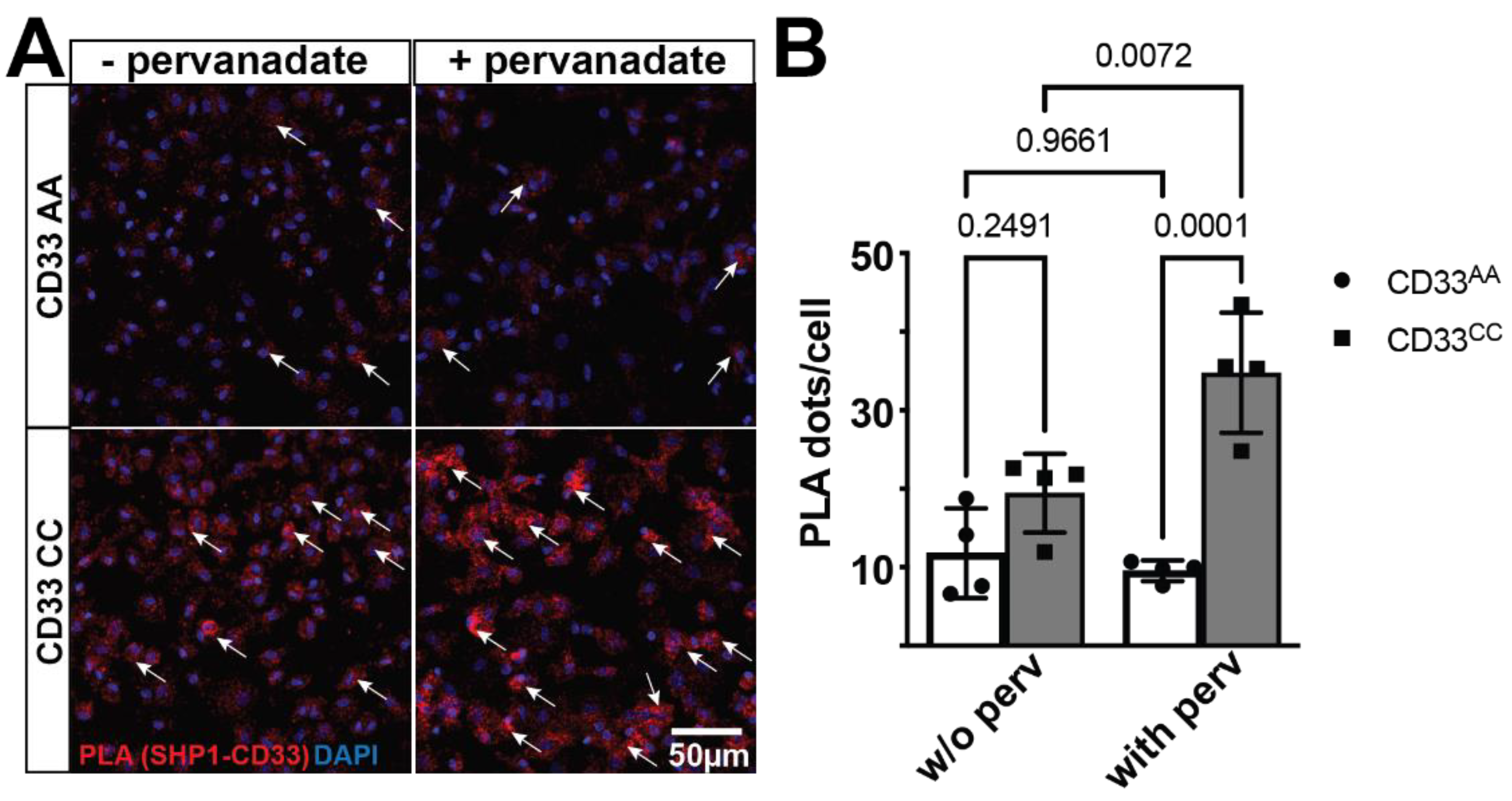
Figure 2.
CD33 and SHP-1 interact in situ in a CD33 genotype-dependent manner. (a) A representative confocal image of postmortem human prefrontal cortex stained for Proximity Ligation Assays (PLA). The proximity ligation puncta (red dots) indicated by arrows represent the protein-protein interaction between CD33 and SHP-1. DAPI (blue) is used to counterstain the nucleus. (b) Quantification of the PLA dots show a significant increase in the PLA counts in the CD33 CC group (n=16 tissue sections, 11.36 ± 1.956) when compared to the CD33 AA groups (n=10 tissue sections, 0.6185 ± 0.1482). Data represented as mean ± SEM. ***p < 0.001, Unpaired t-test. Scale bar: 100 µm.
Figure 2.
CD33 and SHP-1 interact in situ in a CD33 genotype-dependent manner. (a) A representative confocal image of postmortem human prefrontal cortex stained for Proximity Ligation Assays (PLA). The proximity ligation puncta (red dots) indicated by arrows represent the protein-protein interaction between CD33 and SHP-1. DAPI (blue) is used to counterstain the nucleus. (b) Quantification of the PLA dots show a significant increase in the PLA counts in the CD33 CC group (n=16 tissue sections, 11.36 ± 1.956) when compared to the CD33 AA groups (n=10 tissue sections, 0.6185 ± 0.1482). Data represented as mean ± SEM. ***p < 0.001, Unpaired t-test. Scale bar: 100 µm.
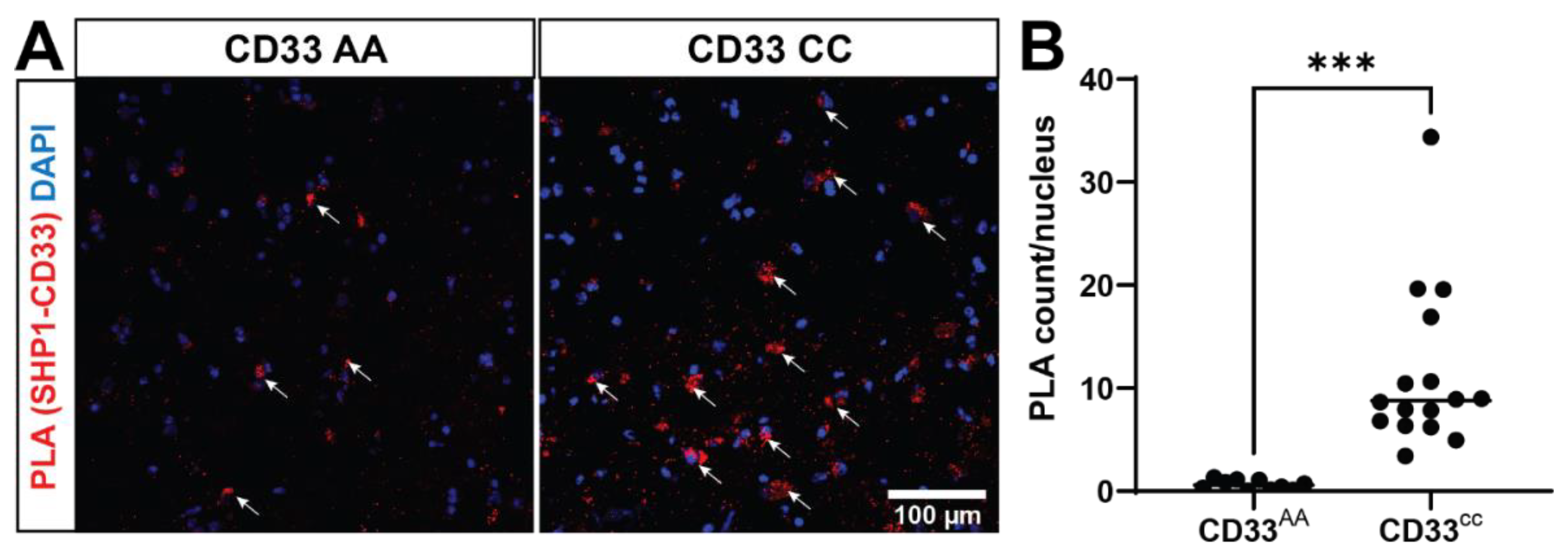

Table 1.
Test for the interaction between CD33 gene expression (gx) and PTPN6 gene expression on trait.
Table 1.
Test for the interaction between CD33 gene expression (gx) and PTPN6 gene expression on trait.
| Trait | Variable | b | se | t | p |
|---|---|---|---|---|---|
| Amyloid | Intercept | 1.742 | 0.039 | 44.546 | 1.45E-246 |
| CD33 gx | 0.070 | 0.056 | 1.236 | 0.217 | |
| PTPN6 gx | 0.022 | 0.073 | 0.297 | 0.766 | |
| CD33 gx:PTPN6 gx | -0.210 | 0.063 | -3.349 | 8.39E-04 | |
| Tangles | Intercept | 2.347 | 0.047 | 49.816 | 4.20E-282 |
| CD33 gx | 0.077 | 0.068 | 1.137 | 0.256 | |
| PTPN6 gx | -0.034 | 0.088 | -0.383 | 0.701 | |
| CD33 gx:PTPN6 gx | -0.247 | 0.076 | -3.276 | 1.09E-03 | |
| Pathologic AD | Intercept | 0.644 | 0.072 | 8.889 | 6.18E-19 |
| CD33 gx | 0.099 | 0.104 | 0.958 | 0.338 | |
| PTPN6 gx | 0.038 | 0.135 | 0.280 | 0.780 | |
| CD33 gx:PTPN6 gx | -0.359 | 0.114 | -3.136 | 1.72E-03 | |
| AD dementia | Intercept | 0.163 | 0.084 | 1.945 | 0.052 |
| CD33 gx | 0.294 | 0.125 | 2.358 | 0.018 | |
| PTPN6 gx | -0.202 | 0.156 | -1.289 | 0.197 | |
| CD33 gx:PTPN6 gx | -0.299 | 0.146 | -2.054 | 0.040 | |
| Cognitive decline | Intercept | -0.016 | 0.003 | -4.804 | 1.78E-06 |
| CD33 gx | -0.007 | 0.005 | -1.499 | 0.134 | |
| PTPN6 gx | 0.001 | 0.006 | 0.217 | 0.828 | |
| CD33 gx:PTPN6 gx | 0.003 | 0.006 | 0.612 | 0.541 | |
| TDP-43 | Intercept | -0.711 | 0.077 | -9.259 | 2.05E-20 |
| CD33 gx | 0.190 | 0.113 | 1.679 | 0.093 | |
| PTPN6 gx | -0.081 | 0.145 | -0.562 | 0.574 | |
| CD33 gx:PTPN6 gx | -0.296 | 0.140 | -2.113 | 0.035 | |
| Hippocampal sclerosis | Intercept | -2.260 | 0.120 | -18.778 | 1.14E-78 |
| CD33 gx | 0.314 | 0.183 | 1.712 | 0.087 | |
| PTPN6 gx | -0.244 | 0.229 | -1.067 | 0.286 | |
| CD33 gx:PTPN6 gx | -0.279 | 0.227 | -1.230 | 0.219 | |
| Global AD pathology burden | Intercept | 0.760 | 0.021 | 35.835 | 2.32E-186 |
| CD33 gx | 0.019 | 0.031 | 0.637 | 0.525 | |
| PTPN6 gx | 0.033 | 0.040 | 0.831 | 0.406 | |
| CD33 gx:PTPN6 gx | -0.123 | 0.034 | -3.613 | 3.16E-04 |
Table 2.
Causal Mediation Analysis where the exposure is PTPN6 gene expression, the mediator is CD33 gene expression, and the outcome is AD dementia.
Table 2.
Causal Mediation Analysis where the exposure is PTPN6 gene expression, the mediator is CD33 gene expression, and the outcome is AD dementia.
| b | Lower 95% CI | Upper 95% CI | p-value | |
|---|---|---|---|---|
| Average Causal Mediation Effect (ACME) | 0.055 | 0.012 | 0.100 | 0.008 |
| Average Direct Effect (ADE) | -0.042 | -0.119 | 0.040 | 0.278 |
| Total Effect | 0.014 | -0.045 | 0.070 | 0.666 |
| Proportion Mediated (PM) | 4.092 | -22.768 | 27.120 | 0.670 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



