Submitted:
01 July 2024
Posted:
02 July 2024
You are already at the latest version
Abstract
Saturated fats are widely seen as undesirable components of a healthy diet, as a result of their illusive association with elevated serum cholesterol. The regulation of serum cholesterol is now better understood and polyunsaturated fatty acids rather than saturated fatty acids are responsible. Palmitic acid received further specious allegations of inciting inflammation when in fact it was found to play an auxiliary role as a precursor to ceramide biosynthesis. Studies of arthritic inflammation in lab animals showed dietary saturated fats are anti-inflammatory, whereas polyunsaturated oils are pro-inflammatory. Inflammation plays a role in numerous metabolic diseases, including insulin resistance, fatty liver disease and metabolic syndrome among others. Fat, as triglycerides in adipose tissue, is an efficient way for living organisms to store energy and reduce toxicity of other macronutrients. However, adipose tissue lipolysis releases fatty acids into the blood to provide many organs with an energy supply and spare glucose for brain tissue. Some metabolic disorders result in elevated nonesterified fatty acids in blood, leading to hepatic lipid accumulation, inflammation and insulin resistance. This paper will attempt to clarify the role of saturated fatty acids, and palmitic acid in particular, with regard to certain adverse health conditions.
Keywords:
saturated fatty acid
; nonesterified fatty acids
; palmitic acid
; polyunsaturated fatty acids
; ceramides
; inflammation
; insulin resistance
Introduction
There is a prodigious amount of blame placed on “dietary” saturated fats for a wide range of adverse health conditions; much of it unwarranted or based on misinterpretation of data. This started with the cholesterol-heart disease doctrine that implicated dietary saturated fats as a cause for elevated levels of serum cholesterol in modern societies. The Framingham Heart Study reported that high serum cholesterol (>244 mg/dL) increased risk of coronary heart disease (CHD) compared with low serum cholesterol (<210 mg/dL), although the incidence of CHD for intermediate levels of serum cholesterol (between 210 and 244 mg/dL) was not significantly different from the incidence for those with low serum cholesterol [1]. These results were interpreted to suggest that any increase in serum cholesterol will put a person at greater risk of heart disease. This point of view ignores the fact that the death rate from all causes is greater for people with low serum cholesterol (<180 mg/dL) compared to the normal range of 180–240 mg/dL[2,3,4,5] The Seven Countries Study indicated that there was no increase in death from heart disease until total serum cholesterol exceeded about 250 mg/dL, and death from all causes was greater for lower serum cholesterol in several cohorts of that study, including the Finnish cohorts that had the highest median serum cholesterol of all cohorts [6].
Several studies showed that diets containing predominantly saturated fats in the form of butterfat and coconut oil, with little or no polyunsaturated fatty acids (PUFA) resulted in higher serum cholesterol compared to when the confined subjects were fed diets containing predominantly polyunsaturated vegetable oils [7,8,9,10]. The authors of those studies, and the cholesterol–heart disease community in general, inferred that dietary saturated fats would increase the risk of heart disease, rather than the more correct conclusion that dietary PUFA can lower serum cholesterol compared to diets with relatively little PUFA. These issues have been discussed in several papers [11,12].
The role of PUFA and saturated fatty acids (SFA) in regulation of cholesterol synthesis and lipoprotein metabolism is better understood now [13,14,15]. PUFA bind to a variety of intracellular proteins that regulate expression of genes involved in lipid metabolism, such as peroxisome proliferator-activated receptors (PPARα and PPARγ). All PPARs bind fatty acids, with a strong preference for long chain PUFAs. Activation of PPARα in rats resulted in an increase in liver β-oxidation of PUFA in peroxisomes and subsequent de novo lipogenesis (DNL) to recycle the carbons to palmitic acid (PA) and longer chain SFA as well as oleic acid (OA) and cholesterol [16]. This resulted in increased liver mass relative to body weight due to accumulation of SFA and monounsaturated fatty acids (MUFA) in hepatic triglycerides (TG), as well as a decrease in serum total cholesterol, HDL-cholesterol and TG [16]. On the other hand, selective activation of PPARγ had little effect on liver, but altered adipose tissue lipid metabolism, resulting in no change in serum cholesterol but lower serum TG [16]. See Montaigne et al [17]. for a review of the various actions of PPARs.
Authorities decided that the entire public needed to lower their intake of saturated fats in the belief that it would decrease incidence of heart disease and reduce deaths in the general population. Numerous studies that attempted to demonstrate that lower dietary saturated fats would decrease heart disease and increase longevity met with great disappointment and were dismissed as flawed in their designs, even by some of the investigators that designed them; most notably the MRFIT study [18,19]. One meta-analysis found no relationship of dietary saturated fat to cardiovascular disease [20]. Other meta-analyses found replacement of dietary SFA with PUFA decreased cardiovascular disease (CVD) events in the selected studies[21,22], whereas replacement with carbohydrate increased CVD events [22]. A meta-analysis of studies that replaced saturated fats and trans fats with vegetable oils found a nonsignificant increase in cardiac events when vegetable oils containing predominantly ω-6 PUFA (e.g., corn, sunflower and safflower oils) were used, whereas vegetable oils containing a mixture of ω-3 and ω-6 PUFA (e.g., soy oil) gave a nonsignificant decrease in cardiac events; comparison of the increase with predominantly ω-6 PUFA to the decrease with a mixture of ω-3 and ω-6 PUFA was significant [23]. The main conclusion from these meta-analyses would be that, in general, replacing saturated fats with either carbohydrate or polyunsaturated vegetable oils is a complex issue with regard to incidence of or death from heart disease. None of these meta-analyses reported overall deaths.
PA is the most abundant SFA in eukaryotic organisms and the most abundant fatty acid overall in the human brain and cell membranes [24]. PA is second only to OA in white adipose tissue (WAT) TG, where PA is generally in the sn-1 position and OA is generally in the sn-2 position of WAT TGs [25]. Consequently, adipose tissue lipolysis results in preferential hydrolysis of PA in the initial step of fatty acid hydrolysis from WAT and subsequent release into the circulation. PA is synthesized in most organisms from acetyl-CoA, via the actions of acetyl-CoA carboxylase and the fatty acid synthase complex, to augment any PA that may be consumed in the diet. The acetyl-CoA precursor is derived from the metabolic breakdown of excess carbohydrates and certain amino acids, as well as from β-oxidation of excess PUFA or long chain FA in hepatic peroxisomes. It is of particular note that the two major essential fatty acids (EFA), linoleic acid (LA) and α-linolenic acid, undergo extensive carbon recycling to other lipids, including PA, stearic acid, OA and cholesterol, as well as to form ketone bodies [26]. PA has numerous important functions in the human body from the time of inception in the womb and throughout life. PA is a major component of lipid membranes and is a precursor for sphingolipids, ceramides, lung surfactant, protein palmitoylation, palmitoylethanolamide (a neuroprotective and anti-inflammatory agent), and longer chain saturated and unsaturated fatty acids [24].
This paper will describe the role of PA (and other FA) in inflammation, insulin resistance (IR) and related physiological processes. The body converts many other macronutrients, such as excess carbohydrate and excess PUFA into PA by de novo lipogenesis and alterations in the intake of SFA, or more specifically PA, will have little influence on the relative levels of PA in the body, and consequently on the numerous physiological processes in which PA takes part.
Saturated Fats and Inflammation
SFA, particularly PA, can augment an inflammatory response and the mechanisms involved have become well understood in the past decade or more. Shi et al [27]. showed that high concentrations (200 μM) of PA in the cell culture medium of mouse macrophages increase release of inflammatory cytokines; MUFA and PUFA did not show this activity. The response to added PA was dependent on toll-like receptor 4 (TLR4). Fatty acid-free bovine serum albumin (BSA) was used to suspend this high concentration of PA in the culture medium, since the solubility of PA in water is only 0.2 μM. The FA-free BSA was reported to have negligible levels of lipopolysaccharide (LPS) that is known to activate TLR4. Suganami et al [28]. reported no significant change in inflammatory cytokines when 100μM PA was added to macrophage cultures containing 2% FA-free BSA, but 200 μM PA gave a significant change. These levels of unesterified PA and BSA are much higher than that found in human blood.
Shi et al [27]. suggested that PA was binding and activating TLR4 to initiate release of inflammatory cytokines. PA alone would not be sufficient to coordinate the molecular interactions of TLR4 and other membrane proteins to initiate release of inflammatory cytokines from immune cells [29]. TLR4 action requires coupling of a TLR4-MD2 membrane protein complex to form dimers and further interactions with other membrane proteins[30], so individual FA chains would not suffice to accomplish this coupling. Nevertheless, authors continue to indicate that SFA activate TLR4 to induce inflammatory changes [31]. Once the membrane complex is established with more suitable coupling agents, such as LPS or fetuin-A found in fetal calf serum used in the culture medium, membrane microdomains known as lipid rafts are involved in signal transduction [32]. PA is a precursor of ceramides found in membrane sphingolipids that make up lipid rafts, and inundating the cells in culture with PA could result in increased production of ceramides to bolster the immune response. Ceramides are an integral part of not only the inflammatory response described above, but are involved in apoptosis, viral and bacterial entry into cells, and inhibition of cell proliferation [33].
It has been disturbing for many years when authors imply that saturated fats can cause or trigger inflammation and cite Shi et al. as their reference [27]. It is important to understand the origins of PA in cells and its roles in cellular metabolism, including synthesis of a wide range of other cellular fatty acids and lipids. PA is the most abundant fatty acid in the mammalian or lean human body and relatively large changes in dietary intake of SFA will have little impact on the relative amount of PA in cell membranes and storage fats. Hence, it is disingenuous to claim that “dietary” SFA can initiate or trigger inflammation. On the other hand, large changes in the levels of consumption of PUFA can have a dramatic effect on their relative levels throughout the body [34]. Changes in the abundance of ω-3 PUFA and ω-6 PUFA in the diet can affect their relative amounts in cell membranes, which may have an impact on their metabolism to bioactive eicosanoids and regulation of the expression of a wide range of proteins in cells. Such changes can have an impact on not only inflammation, but numerous physiological processes [35,36]. In addition, PUFA are highly susceptible to lipid peroxidation and can lead to oxidative stress in various cellular compartments [37].
Animal studies have consistently shown that feeding diets containing primarily saturated fats, with very low levels of PUFA, resulted in less inflammation, whereas adding PUFA to the diet resulted in exacerbation of the inflammation. One study found that adjuvant-treated rats had much less inflammation when fed a diet with beef tallow (very low in PUFA) as the fat source, compared with corn oil as the fat source, whereas a fish oil diet showed an intermediate degree of inflammation [38]. In another study, rats fed an EFA-deficient diet showed relatively little adjuvant-induced inflammation compared to rats fed a control diet, but addition of a corn oil supplement rich in LA to the EFA deficient diet after adjuvant treatment increased the inflammation [39]. Rats fed an EFA-deficient diet starting from the day of adjuvant treatment exhibited 87% less edema in the hind foot pads, compared with control fed animals. Addition of LA to an EFA-deficient diet after adjuvant treatment resulted in increased edema in the foot pads [40]. A study of collagen-induced arthritis found that beef tallow diets resulted in less inflammation than when rats were fed fish oil diets [41]. There was also increased production of IgG and IgE antibodies in fish oil fed rats compared to beef tallow fed rats challenged with egg albumin and alum to induce antibody formation; the increased antibodies resulted in greater active cutaneous anaphylaxis and allergic reactions [42]. Others have found that high dietary intake of LA in rats results in increased levels of standard markers of inflammation [43].
These animal studies showed that dietary saturated fats are not inflammatory, whereas PUFA in the diet exacerbated inflammation, with ω-6 PUFA more inflammatory than ω-3 PUFA. They also indicate that relatively small amounts of LA in the diet are necessary to aggravate the inflammation from various arthritis inducing substances. It would be difficult to do such studies in humans, since there is an abundance of PUFA in the diet of most cultures, resulting in substantial amounts of PUFA in adipose tissue stores. Studies of the effect of diet on arthritic inflammation in humans have primarily focused on comparison of ω-3 PUFA vs ω-6 PUFA;[44] none of the studies compared PUFA with SFA. Those dietary studies generally found that ω-6 PUFA are more inflammatory than ω-3 PUFA, in agreement with some animal studies mentioned above.
Modern Western diets have far more PUFA than are considered necessary to avoid a deficiency. It is estimated from several experimental studies that 0.5% to 1.5% of energy as LA is sufficient to meet the requirements for healthy adults, with perhaps as much as 3% appropriate during pregnancy and lactation or in early childhood development [45]. The average intake of LA in the United States increased from about 6 en% to more than 7 en% between 2000 and 2014, and was more than 21% of total fat intake in 2014 [46].
Obesity and Its Associated Disorders
The human body appears to convert excess energy nutrients to fat as a protective mechanism, since sugars are highly reactive and can produce glycated products that result in loss of function for many proteins [47]. Obesity is associated with several life threatening diseases, including heart disease, stroke, IR, type 2 diabetes (T2D), nonalcoholic fatty liver disease (NAFLD), metabolic syndrome and cancer [48]. These diseases are often attributed to the low-grade systemic inflammation that is common in obesity, especially when there is an abundance of abdominal fat [49]. Abdominal or truncal subcutaneous adipose tissue has less adipogenic potential than peripheral adipose tissue and undergoes hypertrophy as fat mass increases. There is insufficient angiogenesis and a hypoxic condition leads to oxidative stress, infiltration of macrophages and subsequent inflammation [50]. The large increase in macrophages and their output of inflammatory cytokines seems to drive the low-grade systemic inflammation and metabolic disturbances that result in IR, T2D and disruption of various metabolic controls [50].
Serum nonesterified FA (NEFA) are associated with IR, but does IR cause the increase in serum NEFA or does the increase in serum NEFA lead to IR? FA are released from adipose tissue in the fasting state, but normally the presence of insulin in the blood after a meal results in inhibition of lipolysis of adipose triglycerides and a decrease in serum NEFA [51]. There may be some spillover of FA from lipoprotein lipase-mediated hydrolysis of triglycerides in chylomicra after a meal, but the postprandial concentrations of NEFA in blood are generally less than during the fasting state [52]. In addition, higher fasting NEFA were associated with lower insulin secretion and greater risk of developing IR [53]. The association of several plasma and red blood cell fatty acids, as well as genetic variations of specific enzymes involved in fatty acid metabolism, with various diseases has been reviewed recently [54]. Many of the fatty acids discussed in that review were not derived from the diet, but were synthesized from other nutrients and/or fatty acids in the diet.
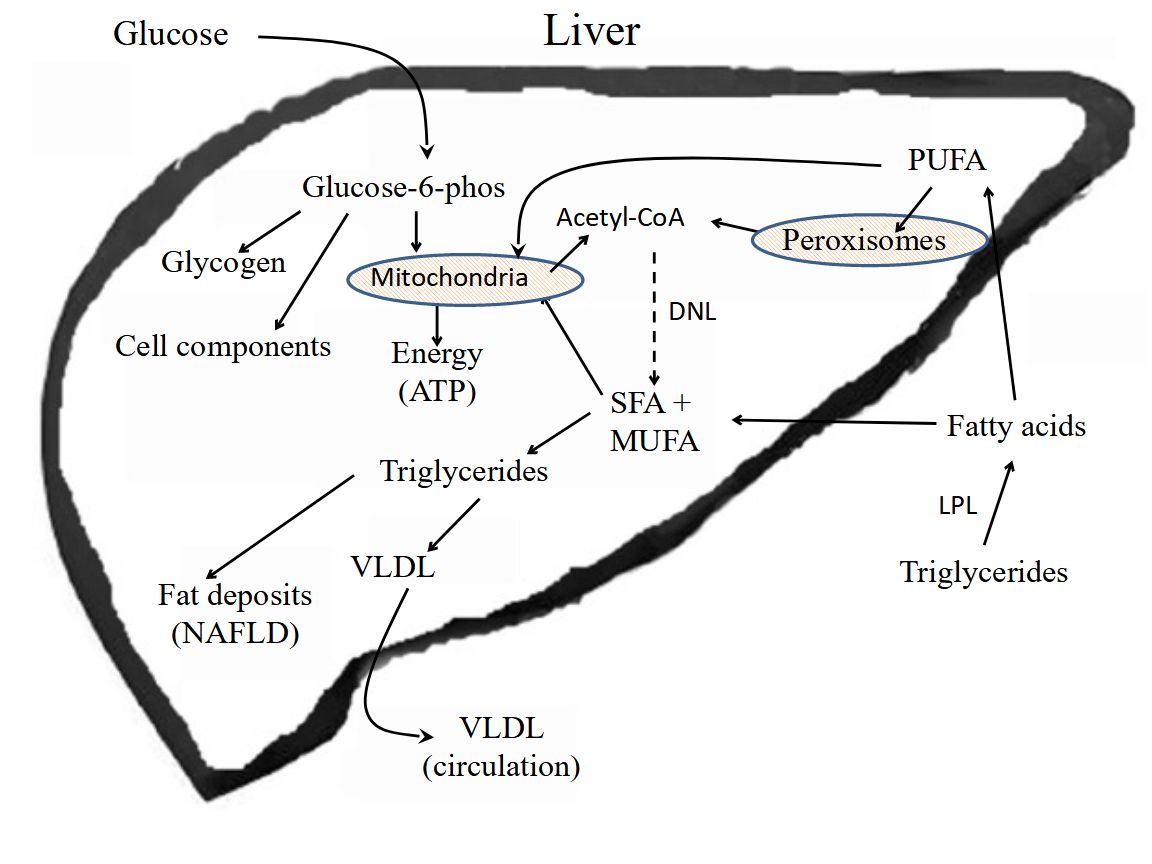
The liver acts as a clearing house for excess nutrients in the blood, converting excess carbohydrates to fatty acids by DNL, removing NEFA from circulation and converting them to TG, and uptake of TG-rich lipoprotein remnants through endocytosis. Under normal conditions the TG are packaged into VLDL and released into the blood [55]. Adipose tissue takes up the bulk of FA from VLDL, which tends to be enriched in SFA and MUFA after processing of nutrients in liver, and stores these fatty acids as TG. However, chronic accumulation of excess TG in hepatocytes can lead to NAFLD, resulting in inflammation in hepatocytes and hepatic IR, as well as muscle IR [56].
A wide range of hepatic insulin sensitivity has been observed in NAFLD patients, which did not show any correlation with hepatic lipid accumulation [57]. Patient hepatic insulin sensitivity correlated with adipose tissue insulin sensitivity in that study, but there was no correlation with muscle insulin sensitivity. The authors suggested that muscle IR was responsible for hepatic lipid accumulation in high hepatic insulin-sensitivity NAFLD patients [57]. Since muscle tissue normally consumes a major portion of blood glucose, insulin resistant muscle tissue would result in an increase in blood glucose that would be taken up by liver and adipose tissue followed by DNL. A study of genetic variants of six genes reported to be associated with NAFLD indicates several metabolites and pathways are involved in NAFLD [58]. Inflammation is a major contributor to hepatic steatosis, which can lead to IR, metabolic syndrome, and eventually to cirrhosis [59].
Many studies have focused on the association of circulating NEFA with IR, T2D and related metabolic disorders, such as NAFLD and metabolic syndrome. The point here is that the abundance of circulating NEFA will be PA and MUFA, because these are the major FA stored in WAT TG. Although PA can augment inflammation through its conversion to ceramides, the major factors that initiate the inflammation are either agents that bind toll-like receptors or pro-inflammatory lipids derived from PUFA. Both types of inflammation initiators can signal release of inflammatory cytokines in various tissues that result in systemic inflammation, which in turn can exacerbate the metabolic dysfunction in liver, adipose tissue and other organs. Cells regulate the levels of PA in cell membranes and other reservoirs, which would mean that ceramide synthesis is regulated by factors other than the availability of PA from diet or other sources [60].
Conclusion
There This brief review had described the role of SFA, and more specifically PA, in several physiological processes. The main point of the review is to alert dietitians and health professionals providing dietary advice to the public about the flawed assertions from the wider health and nutrition authorities that dietary saturated fats should be avoided. There has been much scientific debate regarding the continued push to lower dietary saturated fat intake as described in the Dietary Guidelines for Americans over the past 40 years [61,62]. The basis for that push to lower saturated fat intake was the early studies on diet and serum cholesterol done in the mid-20th century described in the Introduction of this review. We now understand that it is the presence of PUFA in the diet that regulates serum cholesterol levels and SFA do not seem to be involved. The majority of studies that looked at incidence of heart disease or death from heart disease in response to changes in dietary intake of saturated fats showed no effect or the observed effects were inconsistent.
It is disingenuous to claim that “dietary” SFA are triggering or driving systemic inflammation, when it is clear that the SFA involved in sustaining this condition are derived from a range of sources, including conversion of excess dietary carbohydrate to SFA by DNL, or peroxisomal β-oxidation of excess dietary PUFA and subsequent conversion to SFA by DNL. Cells in the body regulate the amount of PA and other FA in cell membranes, and the levels of SFA vary relatively little compared to variations in the levels of PUFA in response to the FA in the diet [34]. A major problem with replacing dietary SFA with PUFA is that PUFA, such as LA, are susceptible to lipid peroxidation that can lead to oxidative stress[37] and promotion of cancer, diabetes and other diseases [63]. There is concern that the high amount of LA in the American diet is responsible for the increase in several chronic diseases [64].
Studies that looked at the influence of SFA vs PUFA on arthritic inflammation in lab animals in vivo consistently found SFA anti-inflammatory compared to PUFA, with ω-6 PUFA more inflammatory than ω-3 PUFA. A sound understanding of the physiology of these systems should lead to well founded dietary recommendations. Dietary PA and other dietary saturated fats are not the culprits in augmenting many of the metabolic abnormalities associated with obesity, inflammation, diabetes and other life-threatening diseases. Dietitians and health care providers need to look more closely at the scientific evidence regarding the beneficial role of SFA in health and nutrition.
Contributions and Conflicts of Interest
The author, Glen D. Lawrence is responsible for the contents of this manuscript and declares no conflicts of interest.
Conflicts of interest
The sole author Glen D. Lawrence reports no conflicts of interest.
References
- Kannel WB, Dawber TR, Kagan A, Revostskie N, Stokes J 3rd. Factors of risk in the development of coronary heart disease--six year follow-up experience. The Framingham Study. Ann Intern Med 1961; 55: 33–50.
- Jacobs D, Blackburn H, Higgins M, Reed D, Iso H, McMillan G et al. Report of the Conference on Low Blood Cholesterol: Mortality Associations. Circulation 1992; 86: 1046–60. [CrossRef]
- Schatz IJ, Kamal M, Yano K, Chen R, Rodriguez BL, Curb JD. Cholesterol and all-cause mortality in elderly people from the Honolulu Heart Program: a cohort study. The Lancet 2001; 358: 351–355. [CrossRef]
- Kirihara Y, Hamazaki K, Hamazaki T, Ogushi Y, Tsuji H, Shirasaki S. The Relationship between Total Blood Cholesterol Levels and All-cause Mortality in Fukui City, and Meta-analysis of This Relationship in Japan. J Lipid Nutr 2008; 17: 67–78. [CrossRef]
- Yi S-W, Yi J-J, Ohrr H. Total cholesterol and all-cause mortality by sex and age: a prospective cohort study among 12.8 million adults. Sci Rep 2019; 9: 1596–1596. [CrossRef]
- Keys A. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Harvard University Press: Cambridge, MA, 1980.
- Hegsted DM, McGandy RB, Myers ML, Stare FJ. Quantitative effects of dietary fat on serum cholesterol in man. Am J Clin Nutr 1965; 17: 281–95. [CrossRef]
- Malmros H, Wigand G. The effect on serum-cholesterol of diets containing different fats. Lancet 1957; 273: 1–7. [CrossRef]
- Bronte-Stewart B, Antonis A, Eales L, Brock JF. Effects of feeding different fats on serum-cholesterol level. Lancet 1956; 270: 521–6.
- Keys A, Anderson JT, Grande F. Prediction of serum-cholesterol responses of man to changes in fats in the diet. Lancet 1957; 273: 959–66.
- Lawrence GD. Dietary fats and health: Dietary recommendations in the context of scientific evidence. Adv Nutr 2013; 4: 294–302. [CrossRef]
- Lawrence GD. Perspective: The Saturated Fat-Unsaturated Oil Dilemma: Relations of Dietary Fatty Acids and Serum Cholesterol, Atherosclerosis, Inflammation, Cancer, and All-Cause Mortality. Adv Nutr 2021; 12: 647–656.
- Le Jossic-Corcos C, Gonthier C, Zaghini I, Logette E, Shechter I, Bournot P. Hepatic farnesyl diphosphate synthase expression is suppressed by polyunsaturated fatty acids. Biochem J 2005; 385: 787–794. [CrossRef]
- Fernandez ML, West KL. Mechanisms by which dietary fatty acids modulate plasma lipids. J Nutr 2005; 135: 2075–2078. [CrossRef]
- Zinöcker MK, Svendsen K, Dankel SN. The homeoviscous adaptation to dietary lipids (HADL) model explains controversies over saturated fat, cholesterol, and cardiovascular disease risk. Am J Clin Nutr 2021; 113: 277–289. [CrossRef]
- Strand E, Lysne V, Grinna ML, Bohov P, Svardal A, Nygard O et al. Short-Term Activation of Peroxisome Proliferator-Activated Receptors alpha and gamma Induces Tissue-Specific Effects on Lipid Metabolism and Fatty Acid Composition in Male Wistar Rats. PPAR Res 2019; 2019: 8047627.
- Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol 2021; 18: 809–823. [CrossRef]
- Multiple Risk Factor Intervention Trial Research Group. Multiple risk factor intervention trial. Risk factor changes and mortality results. Multiple Risk Factor Intervention Trial Research Group. Jama 1982; 248: 1465–77.
- Multiple Risk Factor Intervention Trial Research Group. Mortality rates after 10.5 years for participants in the Multiple Risk Factor Intervention Trial. Findings related to a priori hypotheses of the trial. The Multiple Risk Factor Intervention Trial Research Group. Jama 1990; 263: 1795–801.
- Siri-Tarino PW, Sun Q, Hu FB, Krauss RM. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am J Clin Nutr 2010; 91: 535–46. [CrossRef]
- Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med 2010; 7: e1000252. [CrossRef]
- Jakobsen MU, O’Reilly EJ, Heitmann BL, Pereira MA, Balter K, Fraser GE et al. Major types of dietary fat and risk of coronary heart disease: a pooled analysis of 11 cohort studies. Am J Clin Nutr 2009; 89: 1425–32. [CrossRef]
- Ramsden CE, Hibbeln JR, Majchrzak SF, Davis JM. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. Br J Nutr 2010; 104: 1586–600.
- Carta G, Murru E, Banni S, Manca C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front Physiol 2017; 8: 902. [CrossRef]
- Ahmadian M, Duncan RE, Jaworski K, Sarkadi-Nagy E, Sul HS. Triacylglycerol metabolism in adipose tissue. Future Lipidol 2007; 2: 229–237. [CrossRef]
- Cunnane SC, Ryan MA, Nadeau CR, Bazinet RP, Musa-Veloso K, McCloy U. Why is carbon from some polyunsaturates extensively recycled into lipid synthesis? Lipids 2003; 38: 477–84.
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Inv 2006; 116: 3015–25. [CrossRef]
- Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol 2007; 27: 84–91.
- Murumalla RK, Gunasekaran MK, Padhan JK, Bencharif K, Gence L, Festy F et al. Fatty acids do not pay the toll: Effect of SFA and PUFA on human adipose tissue and mature adipocytes inflammation. Lipids Health Dis 2012; 11: 175. [CrossRef]
- Maeshima N, Fernandez RC. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol 2013; 3: 3. [CrossRef]
- Dimitrov I, Stankova T, Angelova P, Boyadjiev N, Georgieva K, Dimov I et al. Diet-Induced Early Inflammatory Response of Visceral Adipose Tissue in Healthy Male Wistar Rats. Nutrients 2024; 16: 1184. [CrossRef]
- Wong SW, Kwon M-J, Choi AMK, Kim H-P, Nakahira K, Hwang DH. Fatty acids modulate toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem 2009; 284: 27384–27392. [CrossRef]
- Yaribeygi H, Bo S, Ruscica M, Sahebkar A. Ceramides and diabetes mellitus: an update on the potential molecular relationships. Diabet Med 2020; 37: 11–19. [CrossRef]
- Hodson L, Skeaff CM, Fielding BA. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog Lipid Res 2008; 47: 348–380. [CrossRef]
- Calder PC. Fatty acids and inflammation: the cutting edge between food and pharma. Eur J Pharmacol 2011; 668 Suppl 1: S50-58. [CrossRef]
- Levental KR, Malmberg E, Symons JL, Fan Y-Y, Chapkin RS, Ernst R et al. Lipidomic and biophysical homeostasis of mammalian membranes counteracts dietary lipid perturbations to maintain cellular fitness. Nat Commun 2020; 11: 1339. [CrossRef]
- Mortensen MS, Ruiz J, Watts JL. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023; 12: 804. [CrossRef]
- G. D. Lawrence. Effect of dietary lipids on adjuvant-induced arthritis in rats. Nutr Res 1990; 10: 283–290. [CrossRef]
- Denko CW. Modification of adjuvant inflammation in rats deficient in essential fatty acids. Agents Actions 1976; 6: 636–641. [CrossRef]
- Chinn KS, Welsch DJ, Salsgiver WJ, Mehta A, Raz A, Obukowicz MG. Modulation of adjuvant-induced arthritis by dietary arachidonic acid in essential fatty acid-deficient rats. Lipids 1997; 32: 979–988. [CrossRef]
- Prickett JD, Trentham DE, Robinson DR. Dietary fish oil augments the induction of arthritis in rats immunized with type II collagen. J Immunol 1984; 132: 725–9. [CrossRef]
- Prickett JD, Robinson DR, Bloch KJ. Enhanced production of IgE and IgG antibodies associated with a diet enriched in eicosapentaenoic acid. Immunol 1982; 46: 819–826.
- Marchix J, Choque B, Kouba M, Fautrel A, Catheline D, Legrand P. Excessive dietary linoleic acid induces proinflammatory markers in rats. J Nutr Biochem 2015; 26: 1434–1441. [CrossRef]
- Hurst S, Zainal Z, Caterson B, Hughes CE, Harwood JL. Dietary fatty acids and arthritis. Prostaglandins Leukot Essent Fatty Acids 2010; 82: 315–318.
- Cunnane SC. Problems with essential fatty acids: time for a new paradigm? Prog Lipid Res 2003; 42: 544–568.
- Raatz SK, Conrad Z, Jahns L. Trends in linoleic acid intake in the United States adult population: NHANES 1999-2014. Prostaglandins Leukot Essent Fatty Acids 2018; 133: 23–28. [CrossRef]
- Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022; 11: 1312. [CrossRef]
- Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 2017; 127: 1–4. [CrossRef]
- Patel P, Abate N. Body fat distribution and insulin resistance. Nutrients 2013; 5: 2019–2027. [CrossRef]
- Adeva-Andany MM, Domínguez-Montero A, Adeva-Contreras L, Fernández-Fernández C, Carneiro-Freire N, González-Lucán M. Body Fat Distribution Contributes to Defining the Relationship between Insulin Resistance and Obesity in Human Diseases. Curr Diabetes Rev 2024; 20: e160823219824. [CrossRef]
- Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie 2016; 125: 259–266. [CrossRef]
- Collins SM, Broadney MM, Ghane N, Davis EK, Jaramillo M, Shank LM et al. Free Fatty Acids as an Indicator of the Nonfasted State in Children. Pediatrics 2019; 143: e20183896. [CrossRef]
- Salgin B, Ong KK, Thankamony A, Emmett P, Wareham NJ, Dunger DB. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J Clin Endocrinol Metab 2012; 97: 3302–3309. [CrossRef]
- Chaaba R, Bouaziz A, Ben Amor A, Mnif W, Hammami M, Mehri S. Fatty Acid Profile and Genetic Variants of Proteins Involved in Fatty Acid Metabolism Could Be Considered as Disease Predictor. Diagnostics (Basel) 2023; 13: 979. [CrossRef]
- Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab 2021; 50: 101238. [CrossRef]
- Kato K, Takamura T, Takeshita Y, Ryu Y, Misu H, Ota T et al. Ectopic fat accumulation and distant organ-specific insulin resistance in Japanese people with nonalcoholic fatty liver disease. PLoS One 2014; 9: e92170. [CrossRef]
- Shigiyama F, Kumashiro N, Furukawa Y, Funayama T, Takeno K, Wakui N et al. Characteristics of hepatic insulin-sensitive nonalcoholic fatty liver disease. Hepatol Commun 2017; 1: 634–647. [CrossRef]
- Fernandes Silva L, Vangipurapu J, Oravilahti A, Männistö V, Laakso M. Plasma Metabolite Signatures in Male Carriers of Genetic Variants Associated with Non-Alcoholic Fatty Liver Disease. Metabolites 2023; 13: 267. [CrossRef]
- Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 2017; 127: 55–64.
- Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 2010; 688: 1–23. [CrossRef]
- Krauss RM, Kris-Etherton PM. Public health guidelines should recommend reducing saturated fat consumption as much as possible: NO. Am J Clin Nutr 2020; 112: 19-24. [CrossRef]
- Kris-Etherton PM, Krauss RM. Public health guidelines should recommend reducing saturated fat consumption as much as possible: YES. Am J Clin Nutr 2020; 112: 13–18. [CrossRef]
- Jaganjac M, Zarkovic N. Lipid Peroxidation Linking Diabetes and Cancer: The Importance of 4-Hydroxynonenal. Antioxid Redox Signal 2022; 37: 1222–1233. [CrossRef]
- Mercola J, D’Adamo CR. Linoleic Acid: A Narrative Review of the Effects of Increased Intake in the Standard American Diet and Associations with Chronic Disease. Nutrients 2023; 15: 3129. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



