Submitted:
02 July 2024
Posted:
04 July 2024
You are already at the latest version
Abstract
(1) Selecting high-quality varieties with disease resistance by crossbreeding artificially is the most fundamental way to address the damage caused by Calonectria spp in eucalypt plantations. However, understanding the mechanism of the formation of disease-resistant heterosis in eucalypt is crucial for the success of crossbreeding. (2) Two eucalypt hybrids, susceptible EC333 (H1522 × unknown) and resistant EC338 (W1667 × P9060) were screened in our previous study on the infection of the isolates of Calonectria., which causes eucalypt leaf blight. RNA-Seq sequencing was performed on susceptible hybrid EC333, disease-resistant hybrid EC338 (W1667 × P9060), and their parents. A comparison was conducted between the the transcriptomic data sequenced and the reference genome of Eucalyptus grandis, and the The differential expression of genes and functional annotations between hybrids and parents was identified. (3) Gene differential expression analysis shows that there are 3912 differentially expressed genes between EC333 and EC338, with 1631 up-regulated and 2281 down-regulated. The expression trends of differential gene sets in P9060 and EC338 were similar. However, the expression trend of W1767 was opposite that of EC338. The similarity of expression levels and the advantage of stress resistance of E. pellita suggested that genes with significant differences in expression are likely to be related to disease resistance. The GSEA enrichment based on GO annotation revealed that the carbohydrate binding pathway (GO:0030246) is differentially expressed between EC338 and EC333. The gene pathway that differentially expressed between EC338 and EC333, was revealed by GSEA enrichment based on KEGG annotation, is Sesquiterpenoid and Triterpenoid biosynthesis (EGR00909). The Alternative Splicing analysis demonstrated that the AS events gene between EC338 and EC333 is LOC104426602. According to SNP analysis, EC338 has 626 more high-impact mutation loci than the male parent P9060, and 396 more than the female parent W1767, the maternal parent W1767 has 259 more mutation loci in the downstream region than EC338, while the male parent P9060 has 3107 fewer mutation loci in the downstream region than EC338. Additionally, EC338 has 9631 more mutation loci in the exon region than EC333. These findings suggested that the genetic pathways, specific gene regions, and their quantities might play crucial roles in the formation of heterosis linked to the resistance against eucalypt leaf blight. The modules were found in WGCNA, which were strongly and oppositely correlated with EC338 and EC333, such as module MEsaddlebrown is likely to be associated with leaf blight resistance. (4) The present study provides a detailed explanation of the genetic basis for the heterosis of resistance to eucalypt leaf blight, providing a basis for exploring genes related to resistance to eucalypt leaf blight.
Keywords:
eucalypt leaf blight
; heterosis of resistance
; allele specific expression
; genetic basis
1. Introduction
1.1. Background and Significance of Research on Eucalypt Leaf Blight
Eucalypt leaf blight, a significant global epidemic, is triggered by fungi from the Calonectria genus (Ca.) of the Nectriaceae family [1]. Eucalypt (Eucalyptus, Corymbia and Angophora) industrial plantations in Guangdong, Guangxi, Fujian, and Hainan are generally affected by eucalypt leaf blight [2]. Based on the model estimated by Zhu Jianhua et al. (2011), the average annual loss caused by eucalypt leaf blight in Fujian amounts to 51.8 million RM [3]. Therefore, eucalypt leaf blight has become one of the serious factors affecting the investment return and sustainable development of eucalypt plantations in China. The species diversity of the pathogenic bacteria in the genus Calonectria is rich, and 171 species have been reported [4,5]. Six pathogens pose a serious threat to eucalypt plantations in Guangdong and Fujian [6]. The pathogens responsible for eucalypt leaf blight are diverse and variable. Given the diverse and expansive artificial forest environment, as well as the tall and open crown of the trees, implementing effective biological and abiotic control measures for that is a huge challenging. The only solution is to select new resistant genotypes for specific environments and fundamentally solve eucalypt leaf blight.
1.2. About Eucalyptus pellita
The high-temperature and humidity summer season in South China coincides with the outbreak of eucalypt diseases. The superior disease resistance of the hybrid of E. pellita ensures that its growth remains largely unaffected, and accentuating its rapid growth [7]. Our multi-site testing results on hybrid of E. pellita indicated that its rapid growth rate is not inferior to that of hybrid from E. grandis × E. urophylla, and its disease resistance is significantly higher than that of hybrid of E. grandis × E. urophylla in areas severely affected by diseases such as eucalypt leaf blight. For example, the superior hybrid (EC150) of E. urophylla × E. pellita, at 45 months, had an average DBH of 13.7 cm, average height of 18.4 m, and an average volume of 138.2 m³/ha, it is 52.9% -81.5% higher than the control DH3229 in the western Guangdong.
E. pellita has excellent disease resistance and rapid growth, and prefers environments with strong sunlight, humid heat, and abundant rainfall [8,9,10]. E. pellita and its hybrids are one of the ideal tree species for enriching eucalypt plantations species in southern China [11]. Moreover, E. pellita has good hybrid cross-compatibility and trait-complementary with many other eucalypt species [12,13]. It is an important interspecific hybrid parent of eucalypt in South China. In 2007, we evaluated the early introduction (from Australian and Indonesia) of E. pellita provances and families trials in China systematically, and based on the evaluation, more than 200 excellent families were selected and a advanced generation of breeding population for E. pellita were established [14,15].
1.3. Varieties Improvement is the Way to Solve Disease Problems
Selecting parents with different genetic foundations for artificial hybridization and utilizing heterosis is an important way to improve plant growth, biomass, stress resistance, and adaptability [16]. However, the formation mechanism of heterosis in target traits of E. pellita, especially the disease resistance heterosis, has not been studied yet. Therefore, exploring the molecular mechanisms of genetic recombination and gene expression patterns related to the heterosis of E. pellita resistant to eucalypt leaf blight, and exploring gene information related to eucalypt disease resistance, would improve the development efficiency of new disease resistant genotypes, and that’s the most effective way to solve the problems of poor stress resistance (such as eucalypt leaf blight and typhoon resistance) and limited varieties used in eucalypt plantations.
Conducting a transcriptional analysis of differentially expressed genes across various genotypes will facilitate the exploration of the genetic mechanisms underlying heterosis in eucalypt resistance to eucalypt leaf blight. The analysis of transcription levels plays a crucial role in comprehending gene expression patterns, gene architecture, and function. Transcriptome sequencing (RNA sequencing, RNA-Seq)enables the accurate acquisition of comprehensive transcriptional information and high reproducibility [17]. Furthermore, it allows for the detection of subtle transcriptional level differences, thus facilitating a comprehensive and in-depth analysis of differentially expressed genes and their expression patterns between the hybrids and parents. This approach can contribute to a better understanding of the genetic basis underlying the formation of heterosis [18]. In the current study, two eucalypt hybrids, susceptible and resistant, were screened by leaf blight pathogen testing, which had conducted in our precious study. Transcriptome sequencing for two hybrids and their parents was conducted using RNA-Seq, so as to Explore the genetic basis for the formation of heterosis of resistant to eucalypt leaf blight through gene differential expression analysis, and to explore the relevant genes of resistant to eucalypt leaf blight tentatively, finally, providing a theoretical basis for hybrid breeding of eucalypt resistant to leaf blight.
2. Materials and Methods
2.1. Genetic Material
Eucalypt hybrid leaf blight susceptibility testing was conducted via spray inoculation with spore suspension [5]. After 72 hours of inoculation and infection, the susceptibility index (DI) of leaf blight caused by the fungus of the genus Calonectria on the leaves of hybrid eucalyptus was calculated. The percentage of susceptible area of the tested leaves was scanned using the software Leaf Doctor, and the degree of susceptibility was assigned a value from 0 to 5 based on the percentage. The susceptibility index of the tested eucalypt hybrids was calculated after the statistics were completed [19]. This part has been completed in our previous research.
Based on our previous eucalypt leaf blight susceptibility test results [20], eucalypt leaf blight disease-susceptible hybrid EC333 and the disease-resistant hybrid EC338 were screened, and their corresponding parents were found, for a total of 5 eucalypt genotypes used for transcriptome analysis (Table 1). There were three biological replicates for each genotype. During the peak growing season of eucalypt, healthy young leaves with consistent physiological development at the top of the trunk were collected and quickly placed in liquid nitrogen.
2.2. Transcriptome Sequencing and Analysis
RNA Extraction: RNA was isolated utilizing the TRIzol kit (Tiangen, Beijing) and mRNA purification was carried out through oligo-T-linked magnetic beads [21]. The assessment of RNA integrity was conducted with precision employing the Agilent 2100 bioanalyzer, utilizing RNA integrity and total quantity as key reference parameters [22].
Library building and sequencing: First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase(RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, Adaptor with hairpin loop structure were ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 370~420 bp in length, the library fragments were purified with AMPure XP system [23]. Then PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. At last, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired-end reads were generated [24].
Gene Differential Expression Analysis: Raw data (raw reads) of fastq format were firstly processed through fastp software. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. At the same time, Q20, Q30 and GC content the clean data were calculated. All the downstream analyses were based on the clean data with high quality. Reference genome and gene model annotation files were downloaded from genome website directly. Index of the reference genome was built using Hisat2 v2.0.5 and paired-end clean reads were aligned to the reference genome using Hisat2 v2.0.5 [25]. The mapped reads of each sample were assembled by StringTie (v1.3.3b) [26] in a reference-based approach. FeatureCounts v1.5.0-p3 was used to count the reads numbers mapped to each gene. And then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene [27]. (For DESeq2 with biological replicates) Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq2 R package (1.20.0). DESeq2 provide statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P-value <=0.05 found by DESeq2 were assigned as differentially expressed. (For edgeR without biological replicates) Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted by edgeR program package through one scaling normalized factor. Differential expression analysis of two conditions was performed using the edgeR R package (3.22.5). The P values were adjusted using the Benjamini & Hochberg method. Corrected P-value of 0.05 and absolute foldchange of 2 were set as the threshold for significantly differential expression [28].
The correlation of gene expression levels among different eucalypt genotypes is a significant indicator in assessing experimental reliability and sample selection. A higher correlation coefficient indicates greater similarity and closeness in expression patterns. The Encode Project suggests a Pearson correlation coefficient R2 > 0.92 (under optimal sampling and experimental conditions) (ENCODE Project Consortium, 2004). Besides genotype correlation, the correlation between biological replicates during testing (R2 > 0.8) is also vital for the reliability of differential gene analysis. By calculating correlation coefficients based on FPKM values and visualizing them in a heat map, differences between sample groups and duplications within groups can be visually displayed.
Gene Ontology (GO) enrichment analysis of differentially expressed genes was implemented by the clusterProfiler R package, in which gene length bias wascorrected. GO terms with corrected Pvalue less than 0.05 were considered significantly enriched by differential expressed genes. We used clusterProfiler R package to test the statistical enrichment of differential expression genes in KEGG (http://www.genome.jp/kegg/) pathways (Table S2, Figure S1) [29]. The genes were categorized based on their GO annotations according to the subnodes of the three major GO classifications (TERM) (Figure S2).
We use the local version of the GSEA analysis tool http://www.broadinstitute.org/gsea/index.jsp, GO, KEGG data set above were used for GSEA independently. In this investigation, the gene list of E. grandis was retrieved from the database to establish a network, while rMATS (4.1.0) was utilized to scrutinize alternative splicing occurrences.
Alternative Splicing is an important mechanism for regulate the expression of genes and the variable of protein. rMATS(4.1.0) software was used to analysis the AS event. GATK (v4.1.1.0) software was used to perform SNP calling. Raw vcf files were filtered with GATK standard filter method and other parameters (cluster:3; WindowSize:35; QD < 2.0 ; FS > 30.0; DP < 10.
WGCNA (Weighted correlation network analysis) is a systematic biological method used to describe the gene association modes among different samples. It can be used to identify gene sets that are highly synergistic changed, and identify candidate biomarkers or therapeutic targets based on the coherence of gene sets and the correlation between gene sets and phenotypes. The R package WGCNA is a set of functions used to calculate various weighted association analysis, which can be used for network construction, gene screening, gene cluster identification, topological feature calculation, data simulation and visualization. One input file is sample information, that is, a matrix describing the traits of the sample: the traits used for association analysis must be numeric; If it is a regional or categorical variable, it needs to be converted to a 0-1 matrix. The other is gene expression data. For transcriptome sequencing, FPKM can be used as gene expression data [30].
The disease resistant eucalypthybrid is EC338, and the susceptible hybrid is EC333. The paternal parent of EC338 is P9060 (E. pellita), and the maternal parent is W1767 (E. wetarensis). The maternal parent of EC333 is H1522(E. urophylla × E. pellita), the paternal parent of EC333 is unknown, so it was not sequenced together. The target disease resistant genes are most likely to be differentially expressed genes between disease resistant genotype and susceptible genotype, and the disease resistant genes can be traced back to their parents. Therefore, in transcriptome analysis, this study mainly compared the differential gene expression information between disease resistant and susceptible eucalypt hybrids, as well as between hybrids and their parents.
3. Results
3.1. Quality Control of Sequence Data (QC)
3.1.1. Sequencing Data Quality
After performing initial data filtering (Figure S3), examining sequencing error rates, and analyzing the distribution of GC content, high-quality clean reads were obtained for further analysis. From the correspondence between base identification and Phred scores, the evaluation of base calling accuracy in eucalypt samples revealed a Q20 range of 97-98% and a Q30 range of 93-94% (Table S1), indicating reliable data quality with an over 90% correct base identification rate. The sequencing error rate was found to be low, ranging from 0.02 to 0.03, and the GC content distribution was stable around 50% for all samples (Table S1). The results affirm the dependability of the sequencing data and the appropriateness of the refined reads for subsequent analysis.
3.1.2. Mapping Sequencing Information to E. grandis Reference Genome and Its Regional Distribution
Sequencing fragments were randomly interrupted by mRNA. To determine which genes transcribed these fragments, it is necessary to compare the clean reads after quality control to the reference genome of E. grandis [31], a species similar to the material studied in this paper. The transcript sequencing FPKM is paired-end sequencing. A gene is represented by a pair of reads. Perform subsequent quantitative data analysis using the number and percentage of reads aligned to the unique map of the reference genome of E. grandis. The unique map reads of EC338 were 37 971 737. The unique map reads of EC333 were 34 174 054. The clean reads for P9060 and W1767 were 34 581 445 and 37 443 452, respectively. The clean reads for H1522 were 36 358 616 (Table S2).
The proportion of reads in exonic regions, intronic regions, and intergenic regions of the genome were counted separately. Generally model species with better gene annotation (e.g., human and mouse) have a high proportion of mapping to exonic regions. The distribution of sequencing reads across genomic regions for all eucalypt genotypes in this paper showed that not many reads were mapped to intergenic regions (Figure S4), which might be due to contamination by ncRNAs or a few DNA fragments, or the gene annotations might not be well developed. The reads mapping to the reference genome might have originated from precursor mRNAs or introns stranded by variable splicing events.
3.2. Quantitative Analysis of the Genes Sequenced
The number of reads covered by each newly predicted gene from the start to the end region were counted based on the positional information of the gene mapping on the reference genome of E. grandis. The reads with mapping quality value less than 10, reads on non-composite mapping, and reads with mapping to multiple regions of the genome were filtered out. The quantification of gene expression level was performed for each sample separately and then combined to obtain the expression matrix of all samples [32] to obtain the information of detected genes in eucalypt parents and their hybrids (Table S3).
3.2.1. Detection of Expression Distribution of Genes Sequenced in Different Eucalypt Genotypes
Gene expression quantification using FPKM (expected number of fragments per kilobase of transcript sequence per million base pairs sequenced) is the number of fragments per million fragments per kilobase length from a gene. This metric takes into account the effects of sequencing depth and gene length on the count of fragments and converts the read count number into FPKM. It plots the overall expression distribution of the samples to view the expression level of the samples and compare the overall expression distribution of the different samples. Box plots can be utilized to illustrate the dispersion of a set of data (Figure 1A). Violin plots demonstrate the expression distribution and its probability density, with the inflated portion of the plot indicating the most concentrated region of expression in the entire sample (Figure 1C).
The results of three repeated biological tests indicated that the distribution of gene expression levels among different genotypes were similar. With the exception of a slight difference in the parental W1767, the median and the degree of dispersion of the distribution of gene expression levels of the four genotypes of hybrids EC338, EC333, P9060 and H1522 were relatively similar (Figure 1A, Figure 1C). Therefore, the potential for differential gene screening due to the amount of gene expression can be ruled out. The peak of the FPKM density distribution curve is commonly used to analyze the gene expression level in RNA sequencing data. The peaks of the curves represent the most common FPKM values in the dataset and can provide information about gene expression patterns. FPKM values were calculated by dividing the number of fragments mapped to a gene by the length of the gene and the total number of mapped reads, and then normalizing to the library size.
The density distribution curve is a graphical representation of FPKM values. The peaks of the curves represent the highest frequency FPKM values, indicating the most common gene expression levels in the samples. The distribution of the curves for the five genotypes were generally consistent, with most genes having a log10 (FPKM) between 0 and 5. The first and highest peak of the curve is in close proximity to the origin; however, the density plot in this study had two peaks, indicating that two regions with the highest concentration of gene expression occurred in five genotypes (Figure 1B). This also reflected the two expanded portions of Figure 1C, and in fact Figure 1C was a reexamination and summary of the basis of Figure 1A and Figure 1B.
3.2.2. The Correlation of Gene Expression of Eucalypt Genotypes Sequenced
The gene expression correlation heat map analysis conducted in this study (Figure 2) revealed that the eucalypt hybrid EC333 and EC338 exhibit the highest correlation coefficient (R2 > 0.9), indicating a strong similarity in their expression patterns and close relationship. The high correlations were observed among the three replicate groups of each genotype enhance the reliability of subsequent differential gene analysis.
3.2.3. Principal Component Analysis of Gene Expression
In this study, PCA was carried out on the gene expression values (FPKM) of all sequenced eucalypt genotypes (Figure 3). The expected outcome of PCA is to have samples from different groups dispersed and samples within groups clustered together in the plot. The PCA plot in Figure 3 demonstrated that the five eucalypt genotypes formed distinct clusters, reflecting their genetic relatedness. The eucalypt hybrids were clustered closely together, with a smaller distance between them compared to their distance from the parental genotypes.
3.3. Gene Co-Expression Venn Diagram and Differential Gene Venn Diagram
There were 18 107 differential genes between EC338 and EC333 (Figure 4A), 15 969 differential genes between EC338 and W1767 (Figure 4B). A total of 17 351 differential genes were identified between EC338 and P9060 (Figure 4C). Therefore, target resistance genes might exist in genes shared by these three gene sets. In addition, genes can interact additively or reciprocally, causing the function of recessive disease susceptibility genes in a parent to become visible in their hybrids. The female parent (H1522) of EC333 is not susceptible to leaf blight, while the male parent was unknown, so its susceptibility might be attributed to gene recombination in cross. Then the 17 411 differential genes between EC333 and H1522 (Figure 4D) were hardly to say associated with susceptibility or not. The four pairs of comparison groups described above were distinguished by a common differential gene of 2143 (Figure 5), indicating that these genes were likely associated with resistance to eucalypt leaf blight.
3.4. Gene Expression Analysis (Differential Gene Screening and Clustering)
The results indicated that 1631 genes were up-regulated, while 2281 genes were down-regulated (Figure S5). In the volcano plot, the horizontal axis represents the difference in gene expression, with up-regulated genes depicted to the left of the dashed line as red dots and down-regulated genes as green dots. For samples with biological replicates, the adjusted p-value (padj) must be less than 0.05, which means that both red and green dots in the volcano plot must be above 1.301. Conversely, the smaller the corrected p-value is, the larger the corresponding log10 (p-value) value is, indicating that the more significant the difference between the genes in the comparison combination, and thus the more deviated from the horizontal axis upward, the more worthy of our attention. Among these genes that deviate up the horizontal axis, we need to find the common differential genes in the four pairs of comparative combinations mentioned above (Figure S6).
In this study, the FPKM values of genes are clustered and analyzed using mainstream hierarchical clustering, and genes or samples with similar expression patterns in the heat map are clustered together. The color in each square reflects not the gene expression value, but the value obtained after homogenization (Z-score) of the rows of expression data (range in -3 to 3). Vertical comparison refers to the expression of different genes in the same sample. Horizontal comparison refers to the expression of the same gene in different samples.
The expression results among different eucalypt genotypes revealed that numerous genes exhibited highly significant differential expression in W1767 (E. wetarensis). When the expression level of the differential genes were low in W1767, it is highly expressed in EC338 (Figure 6). All of these genes with large differences in expression between parents and hybrids were likely to be associated with disease resistance and could be the focus for subsequent screening of resistant genes.
3.5. Enrichment Analysis of Differential Genes in Comparison Groups
Given the significant differences in susceptibility to eucalypt leaf blight, the primary focus should be on the differentially expressed genes of the resistance genotype EC338 and the susceptibility genotype EC333, which should be the focus of the analysis of eucalypt leaf blight resistant genes. Furthermore, in order to investigate the inheritance pattern of the parents, the genes shared in the differential expression pathway between EC338 and P9060, EC338 and W1767, and EC333 and H1522 were also examined. This approach was employed in order to analyze the reasons for the formation of disease resistance heterosis, and attempt to identify the key genes related to eucalypt leaf blight resistance.
The GO and KEGG enrichment analysis was conducted to identify the pathways of differentially expressed genes in the comparison group. It is important to note that only pathways with ‘details’ can be extracted to specific genes in Gene Set Enrichment Analysis (GSEA). Therefore, we have primarily selected the pathway genes with ‘details’ after enrichment as the basis for determining the genes related to eucalypt leaf blight resistance.
The GO enrichment analysis indicates that the differentially expressed gene pathways of EC338 (resistant) and EC333 (susceptible) predominantly involve carbohydrate binding, pattern binding, and polysaccharide binding (Table 2 and Figure 7A). These pathways might be key to endowing hybrids with disease resistance advantages, with carbohydrate binding demonstrating the highest probability, because this pathway existed in four pairs of comparison groups, and only carbohydrate binding had details. Terpene synthase activity and Carbon-oxygen lyase activity, acting on phosphates were present in three pairs of genotypically differentially expressed pathways (Table 2, Figure 7B, Figure 7C and Figure 7D). Carbohydrate binding (Table 2, Figure 7A and Figure 7D) and carbon-oxygen lyase activity (Table 2, Figure 7B and Figure 7D) were present in two of the genotype pairs compared.
Figure 7.
Bar (top) and bubble (bottom) plots of the GO analysis for four pairs of differential genes set.
Figure 7.
Bar (top) and bubble (bottom) plots of the GO analysis for four pairs of differential genes set.
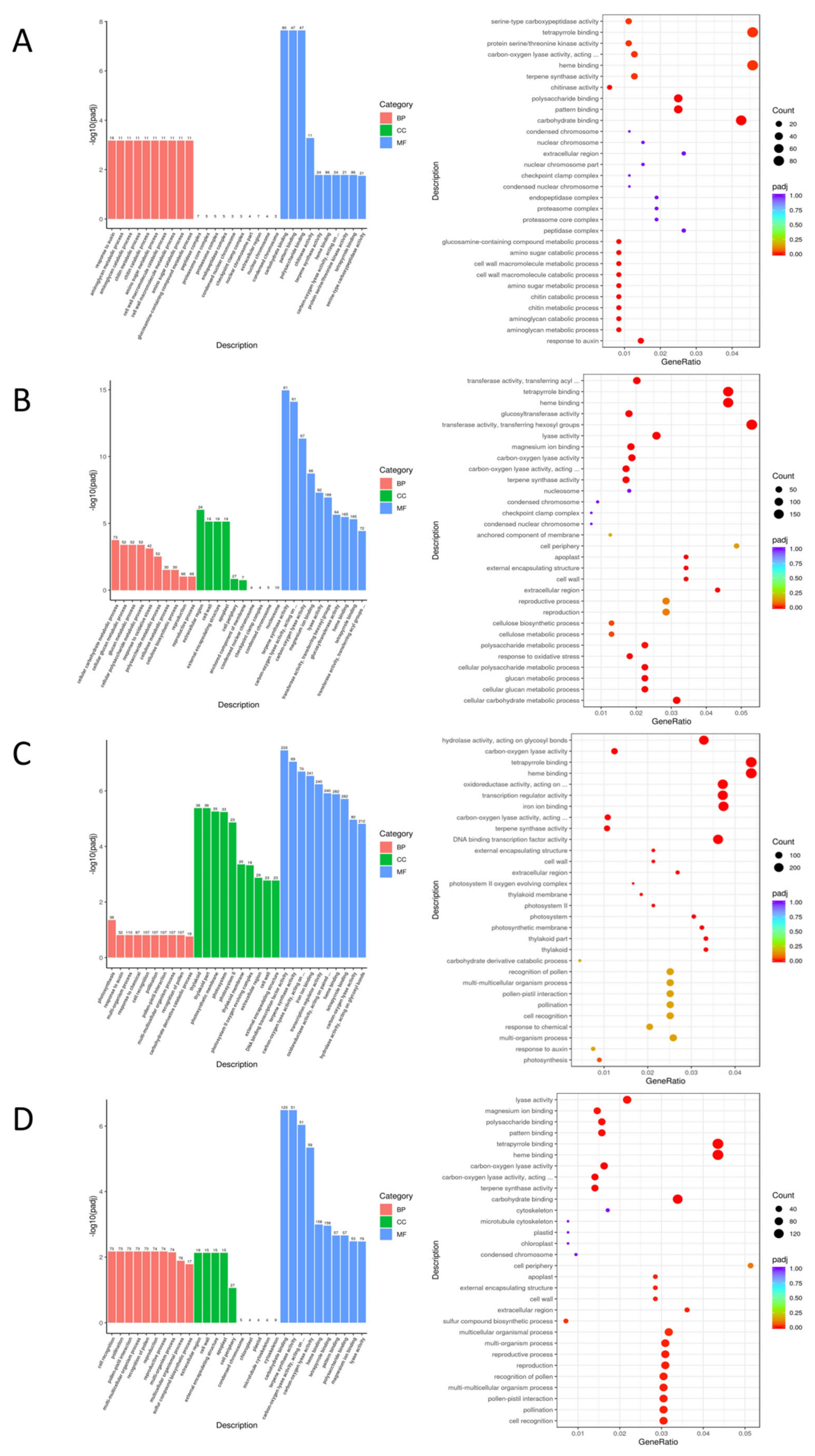
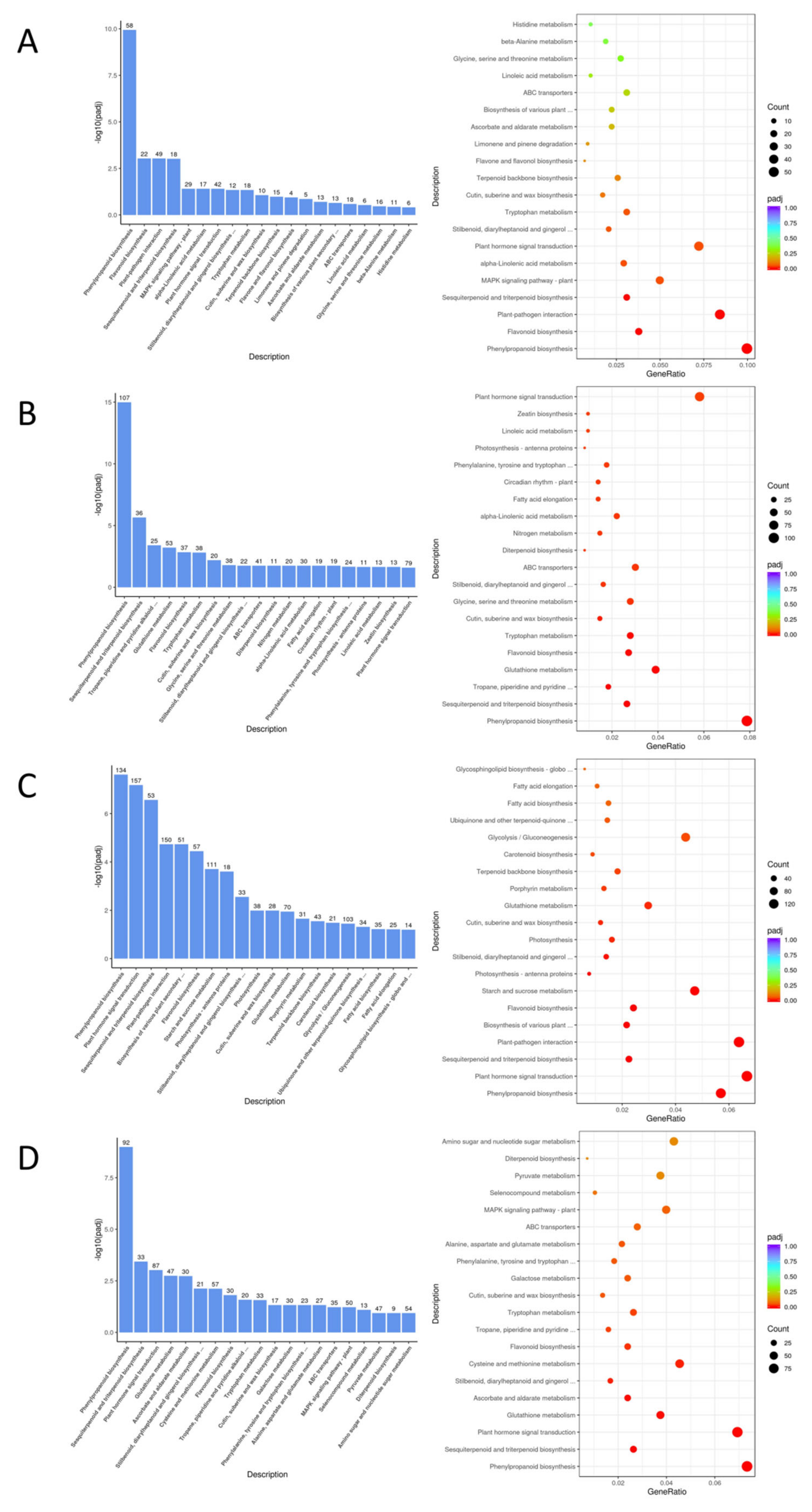
Figure 8.
Bar (top) and bubble (bottom) plots of KEGG analysis for four differential gene pairs.
The Gene Set Enrichment Analysis (GSEA) enrichment analysis utilizing GO annotation primarily focuses on carbohydrate binding (GO: 0030246) (Figure 9A) of EC333 in EC338 VS EC333, as well as terpene synthase activity (GO: 0010333) (Figure 9B), carbon-oxygen lyase activity, acting on phosphates (GO: 0016838) (Figure 9C) and carbon-oxygen lyase activity (GO: 0016835) (Figure 9D) of EC338 in EC338 VS P9060. The specific differentially expressed genes that were the focus of this pathway were those listed in Table S4, Table S5, Table S6, and Table S7, respectively.
Figure 9.
GSEA enrichment analysis using GO annotations (The top row is Snapshot of enrichment results. The Enrichment Map displays the green ES line graph at the top, revealing the highest peak at the ES value denoting the gene set ES value. In the hits barcode map located in the middle, the leading edge subset represents the genes that make the most significant contribution to the enrichment score, ranging from 0 to the ES value. The top grayscale enrichment of the bottom sorting level value indicates the level of genotype expression. The Distribution plot in the bottom row illustrates that if the final ES value (marked by the black dotted line) is relatively small, the pathway gene set is randomly distributed, whereas a relatively large ES value suggests non-random distribution. Subsequently, the hypothesis regarding substitution is then examined.).
Figure 9.
GSEA enrichment analysis using GO annotations (The top row is Snapshot of enrichment results. The Enrichment Map displays the green ES line graph at the top, revealing the highest peak at the ES value denoting the gene set ES value. In the hits barcode map located in the middle, the leading edge subset represents the genes that make the most significant contribution to the enrichment score, ranging from 0 to the ES value. The top grayscale enrichment of the bottom sorting level value indicates the level of genotype expression. The Distribution plot in the bottom row illustrates that if the final ES value (marked by the black dotted line) is relatively small, the pathway gene set is randomly distributed, whereas a relatively large ES value suggests non-random distribution. Subsequently, the hypothesis regarding substitution is then examined.).
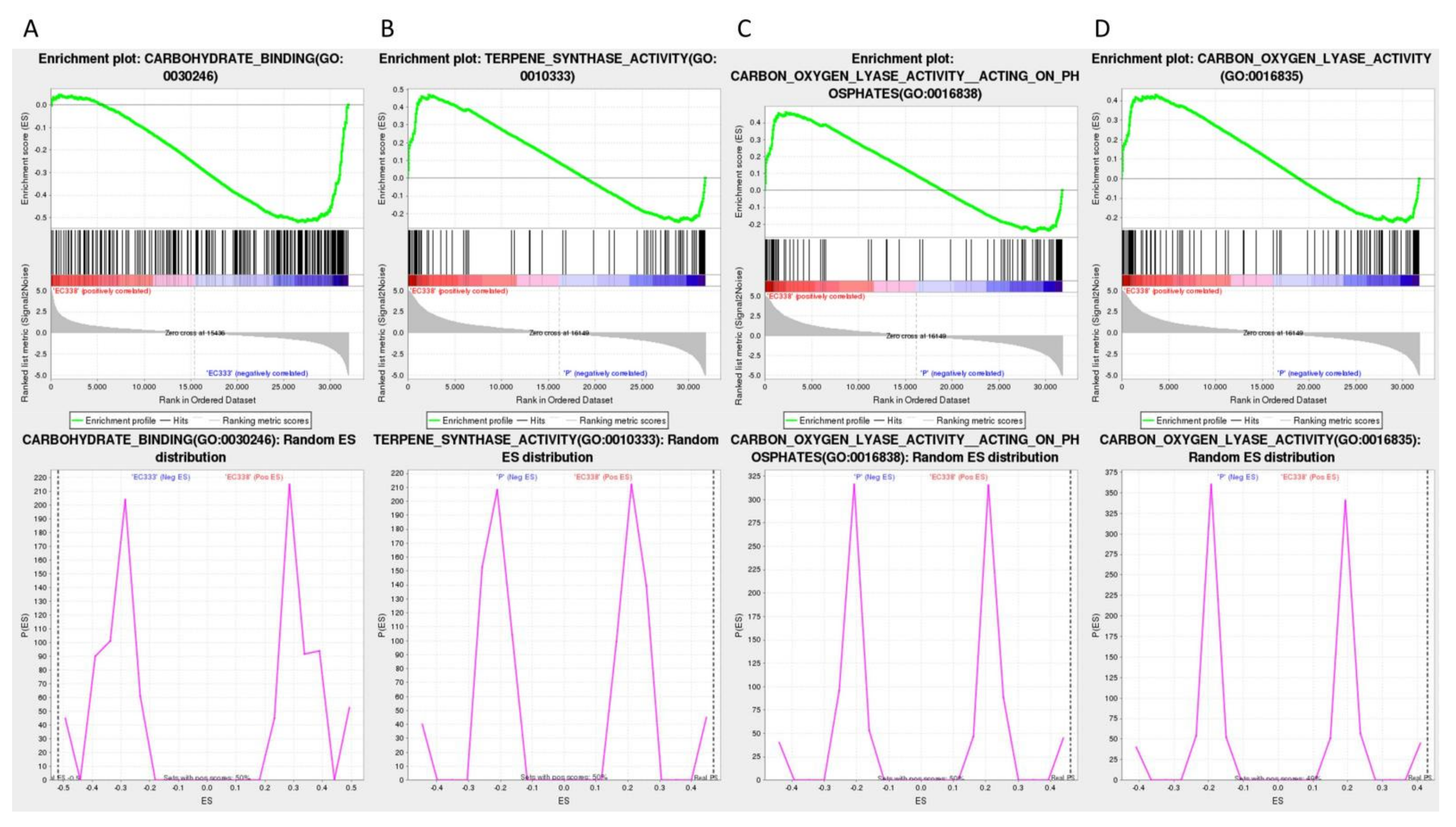
The provided table illustrates that the four differentially expressed gene pathways satisfy the criteria of |NES|>1, NOM p-val<0.05, and FDR<0.25, suggesting promising enrichment outcomes deserving further investigation (Table 3).
The KEGG enrichment analysis revealed that the differentially expressed gene pathways of EC338 (resistant) and EC333 (susceptible) contained phenolylpropanoid biosynthesis, flavonoid biosynthesis, Plant-pathogen interaction, and Sesquiterpenoid and triterpenoid biosynthesis (Table 2 and Figure 8A). These pathways were likely linked to susceptibility to eucalypt leaf blight. Phenylpropanoid biosynthesis (EGR00940) was present in all four pairs of genotype differentially expressed gene pathways, while Sesquiterpenoid and triterpenoid biosynthesis (EGR00909) was found in three pairs of genotype comparisons (Table 2, Figure 8A, Figure 8B and Figure 8C). Flavonoid biosynthesis (EGR00941) and Plant-pathogen interaction (EGR04626) pathways (Table 2 and Figure 8) were present in two pairs of genotype comparisons(Table 2, Figure 8A and Figure 8C).
The GSEA enrichment with KEGG annotation in Table 4 suggested that the main focus is on sesquiterpenoid and triterpenoid biosynthesis (EGR00909) (Figure 10A) of EC338 in EC338 VS EC333, plant-pathogen interaction (EGR04626) (Figure 10C) of W1767 in EC338 VS W1767, as well as phenylpropanoid biosynthesis (EGR00940) (Figure 10B) and Flavonoid biosynthesis (EGR00941) (Figure 10D) of EC338 in EC338 VS W1767 these four pathways. The specific differentially expressed genes that were the focus of this pathway were those listed in Table S8, Table S9, Table S10, and Table S11, respectively.
3.6. Alternative Splicing Event Analysis
The current study identified exon skipping events (SE) with the most significant IncLevelDifference values in the two samples from a pool of the top 20 with the lowest False Discovery Rate (FDR) in four comparison groups: EC338 VS EC333, EC338 VS P9060, EC338 VS W1767, and EC333 VS H1522. It was concluded that the gene LOC104426602 exhibited differential alternative splicing between EC338 and EC333 (Figure 11A), as well as LOC104435785 between EC338 and P9060 (Figure 11B), and LOC104448051 between EC338 and W1767 (Figure 11C). Among various genes with an IncLevelDifference absolute value of 1 in EC333 VS H1522, the exon skipping isoform LOC104418168 was the only one expressed in this differential comparison group (Figure 11D). It is worth noting that LOC104426602, LOC104435785, and LOC104418168 had high count of exon skipping isoforms, whereas LOC104448051 had number of exon inclusion isoforms. The substantial differences in alternative splicing events between the two opposite resistant hybrids could result in variations in transcript and protein structures, as well as functional polymorphisms. Consequently, it was possible that the mentioned four genes are associated with the resistance to eucalypt leaf blight.
3.7. SNP Variation Loci Analysis
P9060 and W1767 exhibited a lower number of silent mutation loci (silent) and missense mutation loci (missense), with counts of 95917 and 98045, respectively. In contrast, other genotypes demonstrated a higher prevalence of functional mutation loci. Missense mutations lead to the substitution of various amino acids, potentially affecting the functionality of the resultant protein. Here we primarily focused on the 2988 distinct missense mutation loci between EC338 (resistant) and EC333 (susceptible). Notably, EC338 showed discrepancies of 12031 and 9903 missense mutation loci compared to its parents (Figure 12A), implying a potential for the emergence of disease resistance-related genes.
INDELs usually cause more significant genetic modifications than SNPs because they involve a larger number of nucleotide sequences. INDELs were used to investigate variant loci between the disease-resistant hybrid EC338 and its parents in present study. It was worth noting that EC338 had 626 more impactful mutation sites than P9060, and 396 more than W1767 (Figure 12B).
The TRANSCRIPT region had the lowest number of nucleotide variations for both INDEL and SNP. Nonetheless, scrutiny of the SNP variation loci demonstrated that the EXON region displayed the highest number of variations across all genotypes. The INDEL variation analysis identified the DOWNSTREAM region had great number of variations. The analysis indicated that EC338 possessed 9631 more mutation loci in the EXON region compared to EC333. Furthermore, the female parent W1767 exhibited 259 additional mutated loci in the DOWNSTREAM region in comparison to EC338, whereas the male parent P9060 displayed 3107 fewer mutated loci in the DOWNSTREAM region than EC338. Gene loci in these regions might be related to eucalypt leaf blight and warrant further exploration.
3.8. Weighted Gene Co-Expression Network Analysis
By setting the correlation coefficient to 0.8 and the soft threshold to 5(Figure 13A), the network was constructed in accordance with the predetermined soft threshold, and the hierarchical clustering of genes within the network was conducted using the dissimilarity between genes to generate a hierarchical clustering tree. Subsequently, the tree was divided into modules using dynamic shearing. The minimum number of genes in the module was 30, merged modules with correlation coefficients greater than 0.75 and anisotropy coefficients less than 0.25 (Figure 13B). Clustered 30 modules based on their eigenvalues. The extraction of the eigenvector genes of each module as the first principal component gene of a particular module also represents the overall level of gene expression within that module. Then performed hierarchical clustering on the characteristic genes of different modules. The positive correlation between MEdarkgrey and MEbrown is the strongest, with a correlation coefficient of 0.55 and a P-value of 0.03 between the eigenvalues of the two modules. The negative correlation between MEdarkgrey and MEgrey is the strongest, with a correlation coefficient of -0.53 and a P-value of 0.04 for the eigenvalues of the two modules. The P-values of the eigenvalues for both pairs of modules were below 0.05, indicating statistical significance.
The two modules with the strongest positive correlation with EC338 were MEpink and METan, with coefficient of 0.485301597 and 0.4389636, respectively. Conversely, the modules MEsaddlebrown and MEred demonstrated the most negative correlations, with coefficient of -0.204643774 and -0.17722817. When considering EC333, the modules MElightgreen and MEsaddlebrown displayed the strongest positive correlations at 0.532894171 and 0.320682081, while modules MEred and MEgreenyellow revealed the most negative correlations at -0.177673509 and -0.170610241 (Figure 14). In theory, the same gene in the module with opposite correlation to the two hybrid species was the gene related to eucalypt leaf blight.
It is crucial that genes shared in modules positively correlated with EC338 and negatively correlated with EC333 Genes shared in modules negatively correlated with EC338 and positively correlated with EC333 were of interest. In this way, of these comparisons, only the MEsaddlebrown module fulfilled the criterion, so only genes 104448928 and others in this module were of interest (Table S12).
4. Discussion
The screening of susceptible and resistant hybrids in this study was conducted through two infection tests on candidate eucalypt hybrids using three Calonectria pathogens. Based on the results of the two infection tests, the resistant genotype EC338 and the susceptible genotype EC333 were screened [20]. Although susceptibility tests had shown differences in the susceptibility index of eucalypt hybrids to different Calonectria pathogens, the relative value of the susceptibility index of the same eucalypt hybrid after being infected twice by three pathogens was basically the same. This basically eliminates the randomness of the susceptibility test results, indicating that the difference in susceptibility of the tested hybrid was due to its inherent genetic differences, that was, the disease resistance was strongly controlled by genetics. Although heterosis is generally controlled by multiple genes and the mechanisms are very complex, the genetic basis for the formation of heterosis varies among different plants, traits, and populations with different genetic backgrounds [33,34]. However, by tracking the parents of disease resistant hybrids and conducting scientific parent combination, it is highly possible to achieve the goal of improvement of disease resistant eucalypt varieties.
Studies had shown that the expression or transgenosis of Carbohydrate binding Modules (CBMs) could change cell wall properties and regulate plant growth and development. Such as Keadtidumrongkul et al. found that CBM2a promoted the growth of Arabidopsis thaliana, and the growth promotion of E. camaldulensis was reflected in the increase of plant height, xylem area, xylem fibers, and conduit cells, there was no significant growth-promoting effect on tobacco [35]. But there are also studies indicating that this is not always the case. Such as the overexpression of certain CBMs did not result in enhanced growth of N.tabacum [36]. And in present study, the enrichment analysis based on GO annotation revealed that the differentially expressed gene pathway between the disease-resistant genotype EC338 and the susceptible genotype EC333 was associated with carbohydrate binding (GO: 0030246), indicating that CBMs might also have a correlation with leaf blight resistance in eucalypt.
In this study, GSEA enrichment based on GO annotation revealed a differential pathway of terpene synthetase (TERPENESYNTHASEACTIVITY) in the disease resistant hybrid EC338 compared to its parent E. pellita P9060. Terpenes are critical components of plant defense against herbivores, insects, pathogenic fungi, and bacteria [37,38]. In many forest tree species, terpenes are vital chemical, and physical defense compounds [39,40]. Conifer terpenes are a primary component of a durable, quantitative defense mechanism against stem boring insects and fungal pathogens [41]. Constitutive and induced terpenes are critical for resistance [42,43,44]. This finding might indicate a certain correlation between terpene synthase and resistance to eucalypt leaf blight.
Furthermore, two differential pathways between EC338 and its parent P9060 were found, namely, the carbon-oxygen cleavage activity on phosphates (CARBON-OXYGEN-LYASE-ACTIVITY_ACTING-ON-HOSPHATES) and carbon-oxygen lyase activity (CARBON-OXYGEN-LYASE-ACTIVITY). A study shown that Cold-resistant DEGs-DEPs had abundant hydrolase activities that could act on glycosyl bonds, carbon-oxygen lyase activity, and iron binding [45]. The MCRA proteins from carbon-oxygen lyase family could be classified as fad-containing double bond hydratases and play a role in the virulence of Streptococcus pyogenes M49 [46]. Based on the results of current and other study, it could be found that the above pathways were indeed related to the stress resistance mechanism of plants.
The current study, GSEA enrichment based on KEGG annotation revealed differential pathways in sesquiterpene and triterpenoid biosynthesis (SESQUITERPENOID-AND-TRITERPENOID-BIOSYNTHESIS) between the resistant hybrid EC338 and the susceptible hybrid EC333. Studies had shown that this pathway was related to the stress resistance mechanism of plants. Such as the study on the molecular response mechanism to drought stress in Atractylodes chinensis had revealed 15 single gene encoding enzymes in the biosynthesis pathways of sesquiterpenes or triterpenes [47]. And a finding done by Singh et al. suggested a relationship between sesquiterpenes, phenolics, flavonoids, and the antioxidant capacity of Ferula plants [48]. Moreover, a study had proposed that the synthesis of sesquiterpenes, triterpenes, flavonoids, and phenylpropanoids might play a crucial role in the response to mechanical damage in Aquilaria sinensis [49].
GSEA enrichment based on KEGG annotations also revealed a differential pathway for phenylpropanoid biosynthesis (PHENYLPROPANOID_BIOSYNTHESIS) in the disease-resistant hybrid EC338 compared to its parent. Gan et al. determined that phenylpropanoid biosynthesis might play an important role in salt tolerance in mulberry at proteomic level [50]. Additionally, a research indicated that Pichia galeiformis enhances resistance to diseases and reduces disease incidence and lesion diameters in chalcone synthase by promoting the activation of genes involved in phenylpropanoid biosynthesis [51].
KEGG enrichment revealed that there is a differential plant pathogen interaction pathway (PLANT_PATHOGEN_INTERACTION) between the resistant hybrid EC338 and the susceptible hybrid EC333. Under biotic stress, plants trigger double immunity against pathogens through plant-pathogen interaction mechanisms, i.e. pathogen-associated molecular pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) [52]. Recent research indicates that PTI and ETI work together to enhance plant immunity against pathogens, thereby enhancing the overall defense system of plants [53,54]. Nonetheless, pathogens have developed sophisticated mechanisms to circumvent plant defenses, leading to successful infection by exploiting host susceptibility [55,56]. Whether it involved plant autoimmunity or pathogen evasion, immunity was intricately linked to the plant-pathogen interaction pathway.
In addition, exploring the causes of plant disease resistance heterosis can also evaluate with phenotypic characteristics such as leaf surface wax, cuticle layer, morphological structure, and chlorophyll content. Recent study conducted by Bonora et al. reveals that resistance mechanism of Corymbia citriodora to different pathogens might be related to leaf phenotypic traits, such as the deposition of polyphenols and tannins on the upper/lower epidermis, and the proportion of monoterpenes, steroids, monounsaturated hydrocarbons, and long-chain hydrocarbons [57]. And, increasing the chlorophyll content in cotton can promote photosynthesis and enhance the plant's ability to resist diseases [58]. So, in the subsequent research of this study, in addition to verifying the identified functional genes, attempts could also be made to study the differences in leaf phenotype characteristics between disease resistant and susceptible genotypes, and analyze the reasons for their disease resistance differences from multiple perspectives.
The susceptibility test in the early stage of this study was conducted under controlled conditions on seedlings aged 6 months, using the pathogen of Calonectria to artificially infect and select susceptible hybrids. However, in the 1.5-year field experiment, there was no significant difference in susceptibility between the two hybrids. This might be related to seedling age and environment. As seedling age increases, the temporal expression of genes causes protein structure modification or isomerism, or different proteins interact to resist adverse environments [59].Yang et al. (2022) illustrated, through comparative transcriptomic and proteomic analysis that P. notoginseng mediated resistance to pathogens by regulating the lignin biosynthesis pathway [60]. Therefore, the protein products most likely associated with the gene pathway associated with eucalypt leaf blight discovered in this study can be isolated, and substrate specificity and kinetic parameter analysis could be performed to further explore the formation mechanism of leaf blight resistance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Data quality of transcriptome sequencing of eucalypt hybrids and their parents; Table S2: Presents a comparison of eucalypt hybrids and their parents with the E. grandis reference genome; Table S3: Genes detected in eucalypt parents and their offspring; Table S4: Genes in carbohydrate binding (GO-GSEA); Table S5: Genes in terpene synthase activity (GO-GSEA); Table S6: Genes in carbon-oxygen lyase activity, acting on phosphates (GO-GSEA); Table S7: Genes in carbon-oxygen lyase activity (GO-GSEA); Table S8: Genes in sesquiterpenoid and triterpenoid biosynthesis (KEGG-GSEA); Table S9: Genes in phenylpropanoid biosynthesis (KEGG-GSEA); Table S10: Genes in plant-pathogen interaction (KEGG-GSEA); Table S11: Genes in flavonoid biosynthesis (KEGG-GSEA); Table S12: WGCNA disease resistance related module genes. Figure S1: Schematic of differential gene enrichment analysis; Figure S2: Statistical map of gene classification according to GO annotations for all genes annotated at level 2, png format (bitmap); Figure S3. Sample sequencing data filtering; Figure S4. Distribution of sequenced reads in genomic regions of eucalypt hybrids; Figure S5. Histogram of the number of differential genes for each comparison combination; Figure S6. Volcano of differentially expressed genes for four pairs of genotypes.
Author Contributions
Wanhong Lu and Jianzhong Luo conceived and designed the study; Wanhong Lu, Yan Lin, Anying Huang, Guo Liu oragnised and conducted the trial sampling and managed the samples; Wanhong Lu carried out the laboratory analyses; Wahong Lu and Zhiyi Su collated, managed and analysed data; Zhiyi Su and Wanhong Lu wrote the paper.
Funding
This research was funded by Natural Science Foundation of Guangdong Province, grant number 2021A1515010618. The APC was funded by Natural Science Foundation of Guangdong Province, grant number 2021A1515010618.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to Forest health team from Institute of Fast-Growing Trees, Chinese Academy of Forestry Sciences for the assistance in testing the susceptibility of eucalypt leaf blight in this study, and to dear anonymous reviewers provided numerous suggestions for significantly improving the content of this paper, we greatly appreciate their guidance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, G.Q.; Chen, S.F.; Wu, Z.H.; Zhou, X.D.; Xie, Y.J. Preliminary analyses on diversity and pathogenicity of Calonectria spp. on eucalyptus in China. Chinese Journal of Tropical Crops 2014, 35, 1183–1191. (in Chinese). [Google Scholar]
- Chen, S.F.; Lombard, L.; Roux, J.; Xie, Y.J.; Wingfield, M.J.; Zhou, X.D. Novel species of Calonectria associated with eucalyptus leaf blight in southeast China. Persoonia 2011, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Guo, W.S.; Chen, H.M.; Wu, J.Q.; Chen, Q.Z.; Meng, X.M. Loss estimation of eucalyptus growth caused by of eucalyptus dieback. Forest Pest and Disease 2011, 30, 6–10. (in Chinese). [Google Scholar]
- Liu, Q.L.; Chen, S.F. Two novel species of Calonectria isolated from soil in a natural forest in China. Mycokeys 2017, 26, 25–60. [Google Scholar] [CrossRef]
- Wang, Q.C.; Chen, S.F. Calonectria pentaseptata causes severe leaf disease of cultivated eucalyptus on the Leizhou peninsula of southern China. Plant Dis. 2020, 104, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Q.; Liu, F.F.; Li, J.Q.; Liu, Q.L.; Chen, S.F. Botryosphaeriaceae from eucalyptus plantations and adjacent plants in China. Persoonia 2018, 40, 63–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.B.; Lan, J.; Wang, J.Z.; He, W.L.; Lu, W.H.; Lin, Y.; Luo, J.Z. Population structure and genetic diversity in Eucalyptus pellita based on SNP markers. Front. Plant Sci. 2023, 14, 1278427. [Google Scholar] [CrossRef]
- 8. Harwood, C.E.; Nikles, D.G.; Pomroy, P.C.; et al. Genetic improvement of Eucalyptus pellita in north Queensland, Australia. IUFRO Conference on Siliviculture and Improvement of Eucalypt. BAAPA, Salvador 1997, 1, 219–226.
- Poubel, D.S.; Garcia, R.A.; Latorraca, J.V.F.; Carvalho, A.M. Estrutura Anatômica e Propriedades Físicas da Madeira de Eucalyptus pellita F. Muell (Anatomical structure and physical properties of Eucalyptus pellita F. Muell Wood). Floresta e Ambiente 2011, 18, 117–126. [Google Scholar] [CrossRef]
- Leksono, B.; Kurinobu, S.; Ide, Y. Optimum age for selection based on a time trend of genetic parameters related to diameter growth in seedling seed orchards of Eucalyptus pellita, in Indonesia. J. Forest RES-JPN. 2006, 11, 359–364. [Google Scholar] [CrossRef]
- Qin, L.; Zhao, Y.K.; Huang, R.F.; Lu, J.X.; Luo, J.Z. Genetic variation of growth strains and growth of 8-year-old Eucalyptus pellita. Journal of Central South University of Forestry & Technology (in Chinese). 2008, 28, 58–63(in Chinese).
- Nirsatmanto, A.; Leksono, B.; Kurinobu, S.; Shiraishi, S. Realized genetic gain observed in second-generation seedling seed orchards of Acacia mangium, in South Kalimantan, Indonesia. J. Forest RES-JPN. 2004, 9, 265–269. [Google Scholar] [CrossRef]
- Leksono B, Kurinobu S. Trend of within family-plot selection practised in three seedling seed orchards of Eucalyptus pellita in Indonesia. J. TROP. FOR. SCI. 2005, 18, 121–126.
- Brawner, J.T.; Bush, D.J.; Macdonell, P.F.; Warburton, P.M.; Clegg, P.A. Genetic parameters of red mahogany breeding populations grown in the tropics. AUST. FORESTRY 2010, 73, 177–183. [Google Scholar] [CrossRef]
- Liu, X.H.; Luo, J.Z.; Lu, W.H.; Lin, Y.; Wang, C.B.; Qi, J. Genetic characteristics of 2 consecutive Eucalyptus pellita generations in growth and typhoon resistance. Molecular Plant Breeding 2017, 15, 5103–5111. (in Chinese). [Google Scholar]
- Shang, L.G.; Gao, Z.Y. Qian, Q. Progress in understanding the genetic basis of heterosis in crops. Chin. Bull. Bot. 2017, 52, 10–18. (in Chinese). [Google Scholar]
- Alisoltani, A.; Fallahi, H.; Shiran, B.; Alisoltani, A.; Ebrahimie, E. RNA-Seq SSRs and small RNA-Seq SSRs: New approaches in cancer biomarker discovery. Gene 2015, 560, 34–43. [Google Scholar] [CrossRef]
- Feng, M.F.; Zhao, J.H.; Li, S.C.; Wei, N.; Kuang, B.W.; Yang, X.P. Molecular genetic mechanisms of heterosis in sugarcane cultivars using a comparative transcriptome analysis of hybrids and ancestral parents. Agron. 2023, 13, 348. [Google Scholar] [CrossRef]
- Fang, Z.D. Research methods for plant diseases. China Agriculture Press. 1998. (in Chinese)
- Liang, X.Y.; Lin, Y.; Lu, W.H. Resistance of 14 eucalypt genotypes to Calonectria leaf blight. Eucalypt Science & Technology 2023, 40, 1–10. (in Chinese). [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Cao, Y.; Pau, G.; Lawrence, M.; Wu, T.D.; Seshagiri, S.; Gentleman, R. Prediction and Quantification of Splice Events from RNA-Seq Data. Plos. One 2016, 11, e0156132. [Google Scholar] [CrossRef] [PubMed]
- He, Z.J.; Zhao, X.; Lu, Z.Y.; Wang, H.F.; Liu, P.F.; Zeng, F.Q.; Zhang, Y.J. Comparative transcriptome and gene co-expression network analysis reveal genes and signaling pathways adaptively responsive to varied adverse stresses in the insect fungal pathogen, Beauveria bassiana. J. Invertebr. Pathol. 2018, 151, 169–181.
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomicfeatures. CCF TCBI 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. NAR 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A. Hellsten, U.; Hayes, R.D.; Grimwood, J.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362.
- Xiong, Z.Q.; Fan, Y.Z.; Song, X.; Xia, Y.J.; Zhang, H.; Ai, L.Z. Z. Short communication: Genome-wide identification of new reference genes for reverse-transcription quantitative PCR in Streptococcus thermophilus based on RNA-sequencing analysis. J. Dairy Sci. 2020, 103, 10001–10005. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.T.; Yang, N.; Tong, H.; Pan, Q.C.; Yang, X.H.; Tang, J.H.; Wang, J.K.; Li, J.S.; Yan, J.B. Genetic basis of grain yield heterosis in an “immortalized F2” maize population. Theor. Appl. Genet. 2014, 127, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Chen, Y.; Yao, W.; Zhang, C.J.; Xie, W.B.; Hua, J.P.; Xing, Y.Z.; Xiao, J.H.; Zhang, Q.F. Genetic composition of yield heterosis in an elite rice hybrid. PNAS 2012, 109, 15847–15852. [Google Scholar] [CrossRef] [PubMed]
- Keadtidumrongkul, P.; Suttangkakul, A.; Pinmanee, P.; Pattana, K.; Kittiwongwattana, C.; Apisitwanich, S.; Vuttipongchaikij, S. Growth modulation effects of CBM2a under the control of AtEXP4 and CaMV35S promoters in Arabidopsis thaliana, Nicotiana tabacum and Eucalyptus camaldulensis. Transgenic Res. 2017, 26, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Obembe, O.O.; Jacobsen, E.; Timmers, J.; Gilbert, H.; Blake, A.W.; Knox, J.P.; Visser, R.G.F.; Vincken, J.P. Promiscuous, non-catalytic, tandem carbohydrate-binding modules modulate the cell-wall structure and development of transgenic tobacco (Nicotiana tabacum) plants. J. Plant Res. 2007, 120, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Peter, G.F. Breeding and Engineering Trees to Accumulate High Levels of Terpene Metabolites for Plant Defense and Renewable Chemicals. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, V.R.; Krokene, P.; Christiansen, E.; Krekling, T. Anatomical and chemical defenses of conifer bark against bark beetles and other pests. New Phytol. 2005, 167, 353–375. [Google Scholar] [CrossRef]
- King, D.J.; Gleadow, R.M.; Woodrow, I.E. Regulation of oil accumulation in single glands of Eucalyptus polybractea. New Phytol. 2006, 172, 440–451. [Google Scholar] [CrossRef]
- Naidoo, S.; Kulheim, C.; Zwart, L.; Mangwanda, R.; Oates, C.N.; Visser, E.A. Uncovering the defence responses of eucalyptus to pests and pathogens in the genomics age. Tree Physiol. 2014, 34, 931–943. [Google Scholar] [CrossRef]
- Trapp, S.; Croteau, R. Defensive resin biosynthesis in conifers. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 689–724. [Google Scholar] [CrossRef]
- Hodges, J.D.; Nebeker, T.E.; Deangelis, J.D.; Karr, B.L.; Blanche, C.A. Host resistance and mortality: a hypothesis based on southern pine beetle-microorganism-host interactions. Bull. Entomol. Soc. Am. 1985, 31, 31–35. [Google Scholar] [CrossRef]
- Strom, B.L.; Goyer, R.A.; Ingram, L.L.; Boyd, G.D.L.; Lott, L.H. Oleoresin characteristics of progeny of loblolly pines that escaped attack by the southern pine beetle. For. Ecol. Manag. 2002, 158, 169–178. [Google Scholar] [CrossRef]
- Klepzig, K.D.; Robison, D.J.; Fowler, G.; Minchin, P.R.; Hain, F.P.; Allen, H.L. Effects of mass inoculation on induced oleoresin response in intensively managed loblolly pine. Tree Physiol. 2005, 25, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Li, Y.; Sun, L.; Chu, S.Y.; Xu, H.W.; Zhou, X.F. Integration of transcriptomic and proteomic analyses of Rhododendron chrysanthum Pall. in response to cold stress in the Changbai Mountains. Mol. Biol. Rep. 2023, 50, 3607–3616. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.; Liavonchanka, A.; Kamneva, O.; Fiedler, T.; Goebel, C.; Kreikemeyer, B.; Feussner, I. Myosin Cross-reactive Antigen of Streptococcus pyogenes M49 Encodes a Fatty Acid Double Bond Hydratase that plays a role in oleic acid detoxification and bacterial virulence. J. Biol. Chem. 2010, 285, 10353–10361. [Google Scholar] [CrossRef]
- Ma, S.S.; Sun, C.Z.; Su, W.N.; Zhao, W.J.; Zhang, S.; Su, S.Y.; Xie, B.Y.; Kong, L.J.; Zheng, J.S. Transcriptomic and physiological analysis of atractylodes chinensis in response to drought stress reveals the putative genes related to sesquiterpenoid biosynthesis. BMC Plant Biol. 2024, 24, 91. [Google Scholar] [CrossRef]
- Singh, P.P.; Joshi, R.; Kumar, R.; Kumar, A.; Sharma, U. Comparative phytochemical analysis of Ferula assa-foetida with Ferula jaeschkeana and commercial oleo-gum resins using GC-MS and UHPLC-PDA-QTOF-IMS. Food Res. Int. 2023, 164, 112434. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.R.; Du, R.Y.; Wang, Y.; Chen, J.H. RNA-Sequencing reveals the involvement of sesquiterpene biosynthesis genes and transcription factors during an early response to mechanical wounding of aquilaria sinensis. Genes. 2023, 14, 464. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.T.; Lin, Z.W.; Bao, L.J.; Hui, T.; Cui, X.P.; Huang, Y.Z.; Wang, H.X.; Su, C.; Jiao, F.; Zhang, M.J.; et al. Comparative proteomic analysis of tolerant and sensitive varieties reveals that phenylpropanoid biosynthesis contributes to salt tolerance in mulberry. Int. J. Mol. Sci. 2021, 22, 9402. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.L.; Zhang, R.Z.; Li, D.L.; Wang, F. Transcriptomic and coexpression network analyses revealed pine chalcone synthase genes associated with pine wood nematode infection. Int. J. Mol. Sci. 2021, 22, 11195. [Google Scholar] [CrossRef]
- Li, J.B.; Ai, M.T.; Hou, J.; Zhu, P.Q. Cui, X.M.; Yang, Q. Plant-pathogen interaction with root rot of panax notoginseng as a model: Insight into pathogen pathogenesis, plant defence response and biological control. Mol. Plant Pathol. 2024, 25, e13427. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Ahn, H.K.; Ding, P.T.; Jones, J.D.G. Mutual potentiation of plant immunity by cell-surface and intracellular receptors. Nature 2021, 592, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.H.; Jiang, Z.Y.; Bi, G.Z.; Nomura, K.Y.; Liu, M.H.; Wang, Y.P.; Cai, B.Y.; Zhou, J.M.; He, S.Y.; Xin, X.F. Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 2021, 592, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, V.; Tsers, I. Plant susceptible responses: the underestimated side of plant-pathogen interactions. Biol. Rev. Camb. Philos. Soc. 2022, 97, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Seybold, H.; Demetrowitsch, T.J.; Hassani, M.A.; Szymczak, S.; Reim, E.; Haueisen, J.; Lübbers, L.; Rühlemann, M.; Franke, A.; Schwarz, K.; et al. A fungal pathogen induces systemic susceptibility and systemic shifts in wheat metabolome and microbiome composition. Nat. Commun. 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Bonora, F.S.; Nahrung, H.F.; Hayes, R.A.; Scharaschkin, T.; Pegg, G.; Lee, D.J. Changes in leaf chemistry and anatomy of Corymbia citriodora subsp. variegata (Myrtaceae) in response to native and exotic pathogens. Australas. Plant Path. 2020, 49, 641–653. [Google Scholar] [CrossRef]
- Wang, R.; Li, G.; Li, Y. Effects of inducers on relevant resistant substance in colored cotton. Cotton Science. 2005, 17, 107–111 (in Chinese). (in Chinese). [Google Scholar]
- Yin, Y.Q.; Qi, F.; Gao, L.; Rao, S.; Yang, Z.; Fang, W. iTRAQ-based quantitativeproteomic analysis of dark-germinated soybeans in responseto salt stress. RSC Advances. 2018, 8, 17905–17913. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, J.B.; Sun, J.L.; Cui, X.M. Comparative transcriptomic and proteomic analyses to determine the lignin synthesis pathway involved in the fungal stress response in panax notoginseng. Physiol. Mol. Plant P. 2022, 119, 101814. [Google Scholar] [CrossRef]
Figure 1.
Testing the distribution of gene expression levels in eucalypt genotypes (A: Boxplot: A boxplot of gene expression levels, where the horizontal coordinate is the name of the sample (group), and the vertical coordinate is log2(FPKM+1). B: Density: A density plot of gene expression levels, where the horizontal coordinate is log2(FPKM+1), and the vertical coordinate is the density of The gene. C: Violin plot of gene expression levels, where the horizontal coordinate is the name of the sample (group), and the vertical coordinate is log2(FPKM+1). The width of each violin represents the number of genes at that expression level.).
Figure 1.
Testing the distribution of gene expression levels in eucalypt genotypes (A: Boxplot: A boxplot of gene expression levels, where the horizontal coordinate is the name of the sample (group), and the vertical coordinate is log2(FPKM+1). B: Density: A density plot of gene expression levels, where the horizontal coordinate is log2(FPKM+1), and the vertical coordinate is the density of The gene. C: Violin plot of gene expression levels, where the horizontal coordinate is the name of the sample (group), and the vertical coordinate is log2(FPKM+1). The width of each violin represents the number of genes at that expression level.).
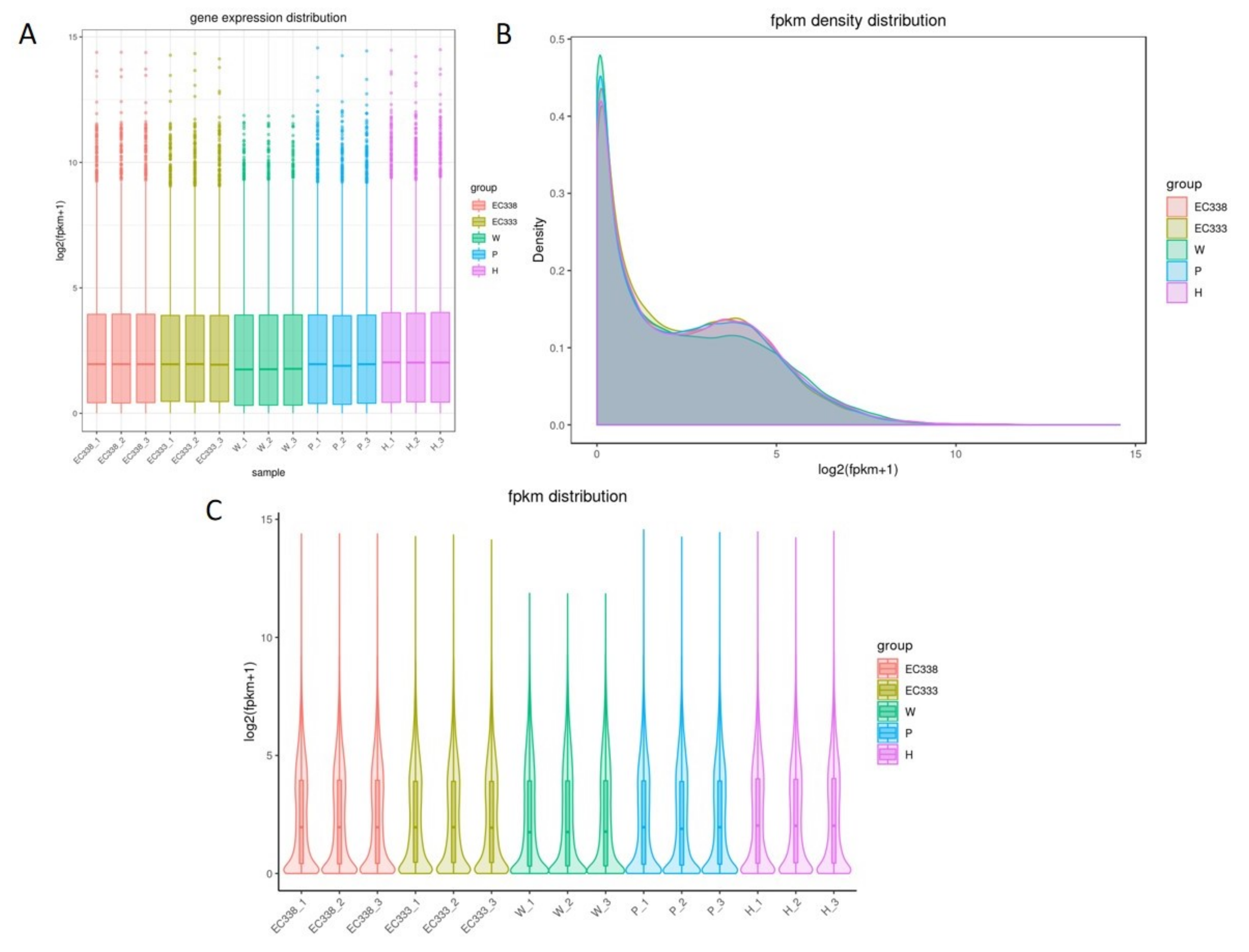
Figure 2.
Correlation heat map of gene expression among various eucalypt genotypes (the horizontal and vertical coordinates are the square of the correlation coefficient between each genotype).
Figure 2.
Correlation heat map of gene expression among various eucalypt genotypes (the horizontal and vertical coordinates are the square of the correlation coefficient between each genotype).
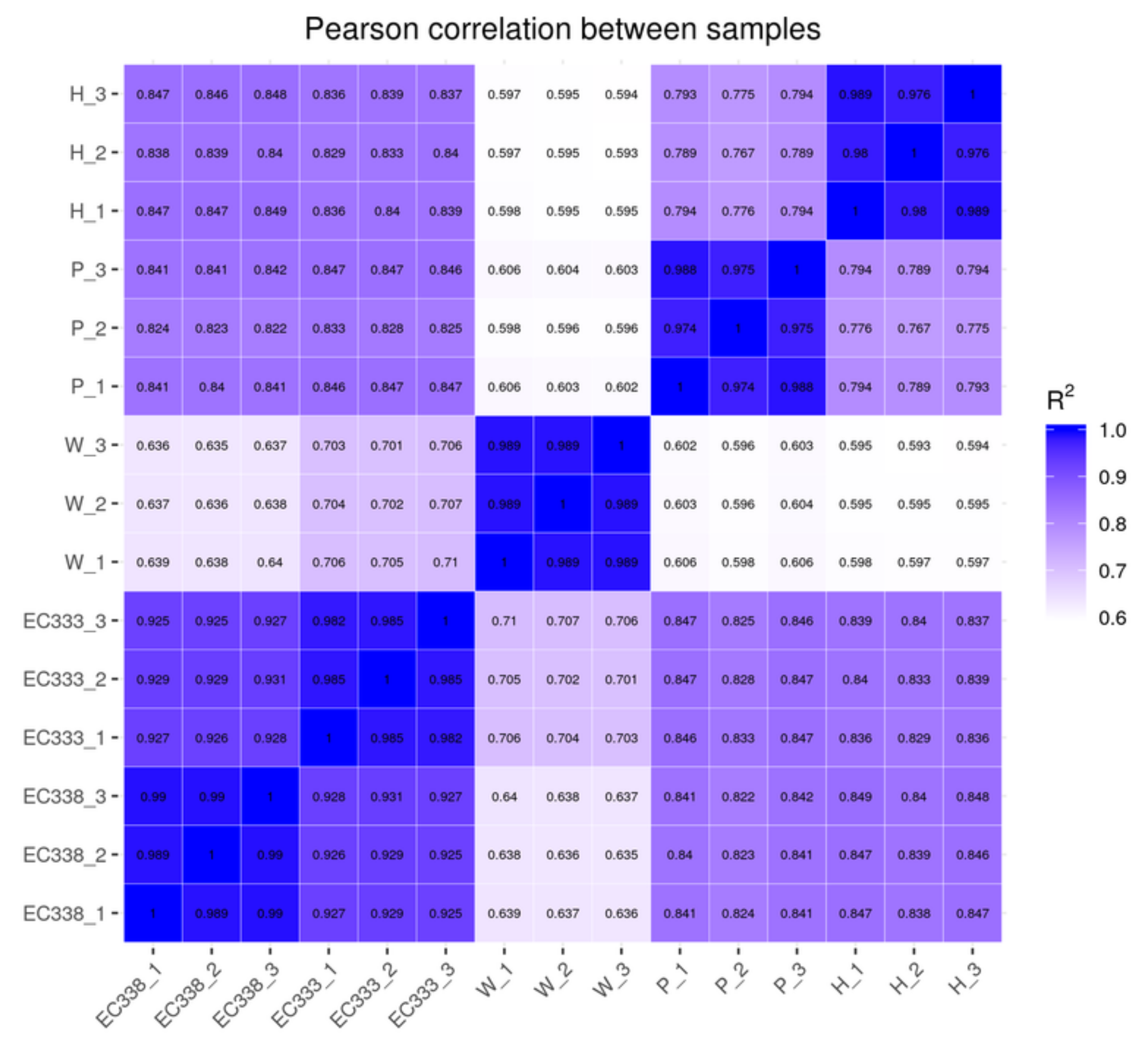
Figure 3.
Principal component analysis plot of gene expression for each eucalypt genotype (A is 2D, B is 3D).
Figure 3.
Principal component analysis plot of gene expression for each eucalypt genotype (A is 2D, B is 3D).
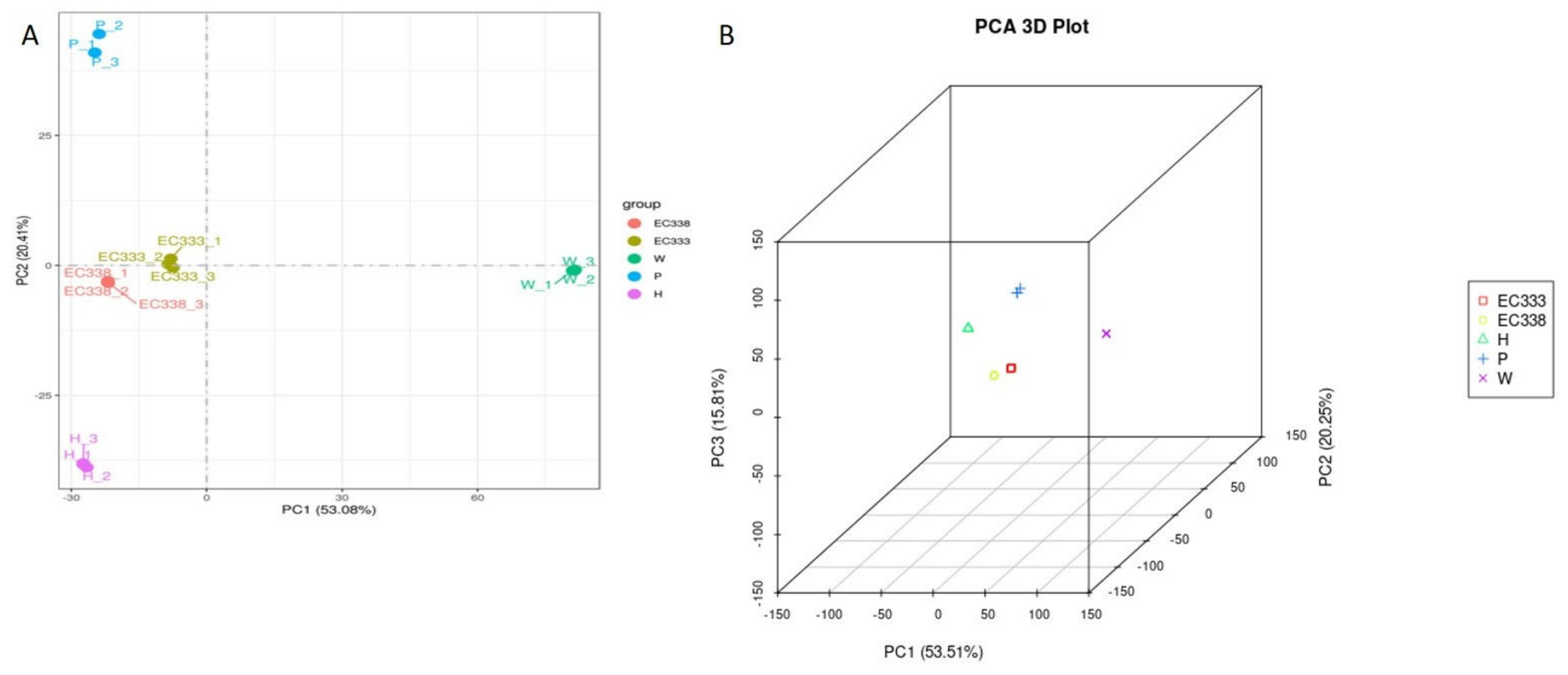
Figure 4.
Gene co-expression Venn diagram in eucalypt genotypes tested.
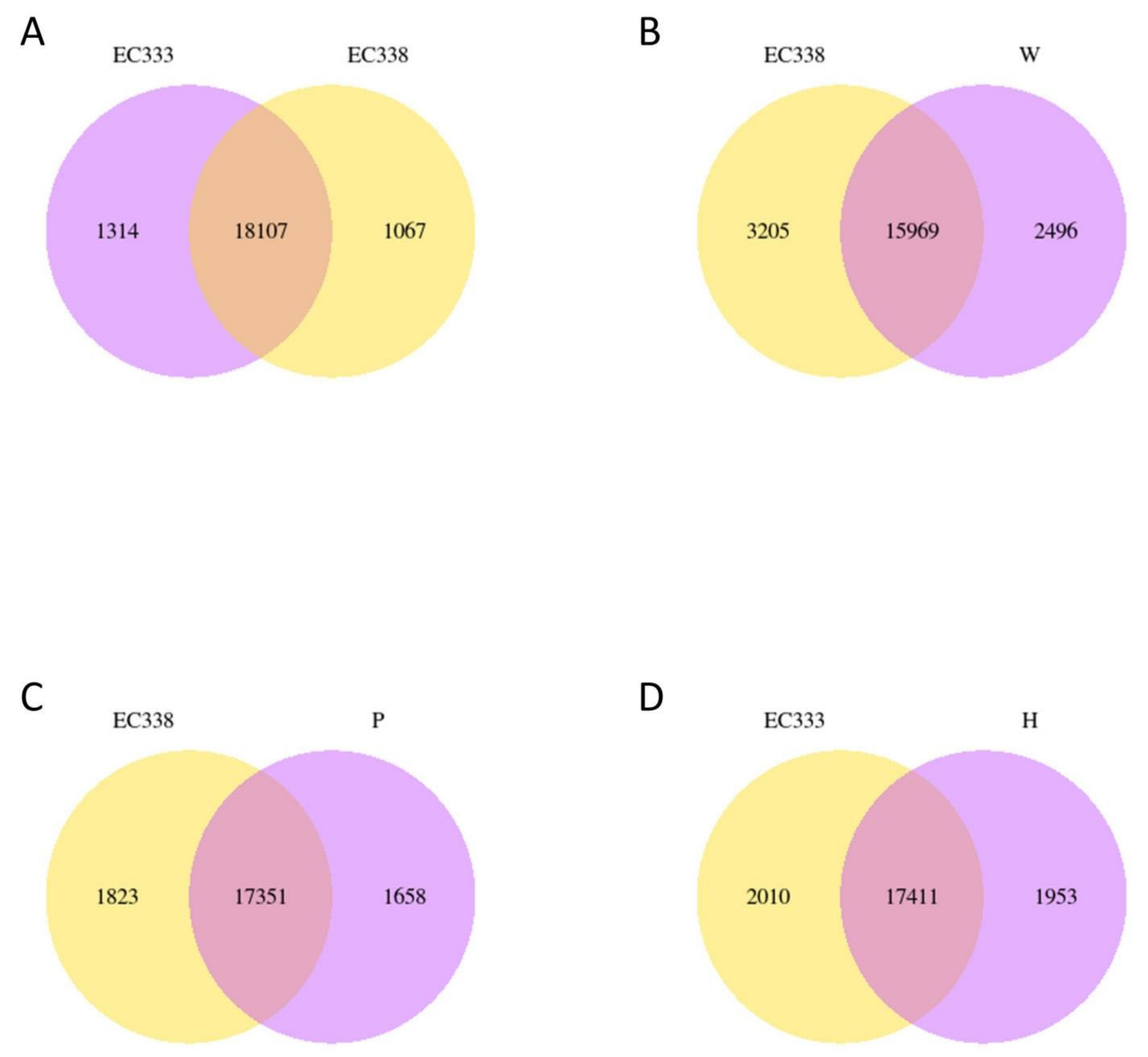
Figure 5.
Differential gene Venn diagram in eucalypt genotypes tested.
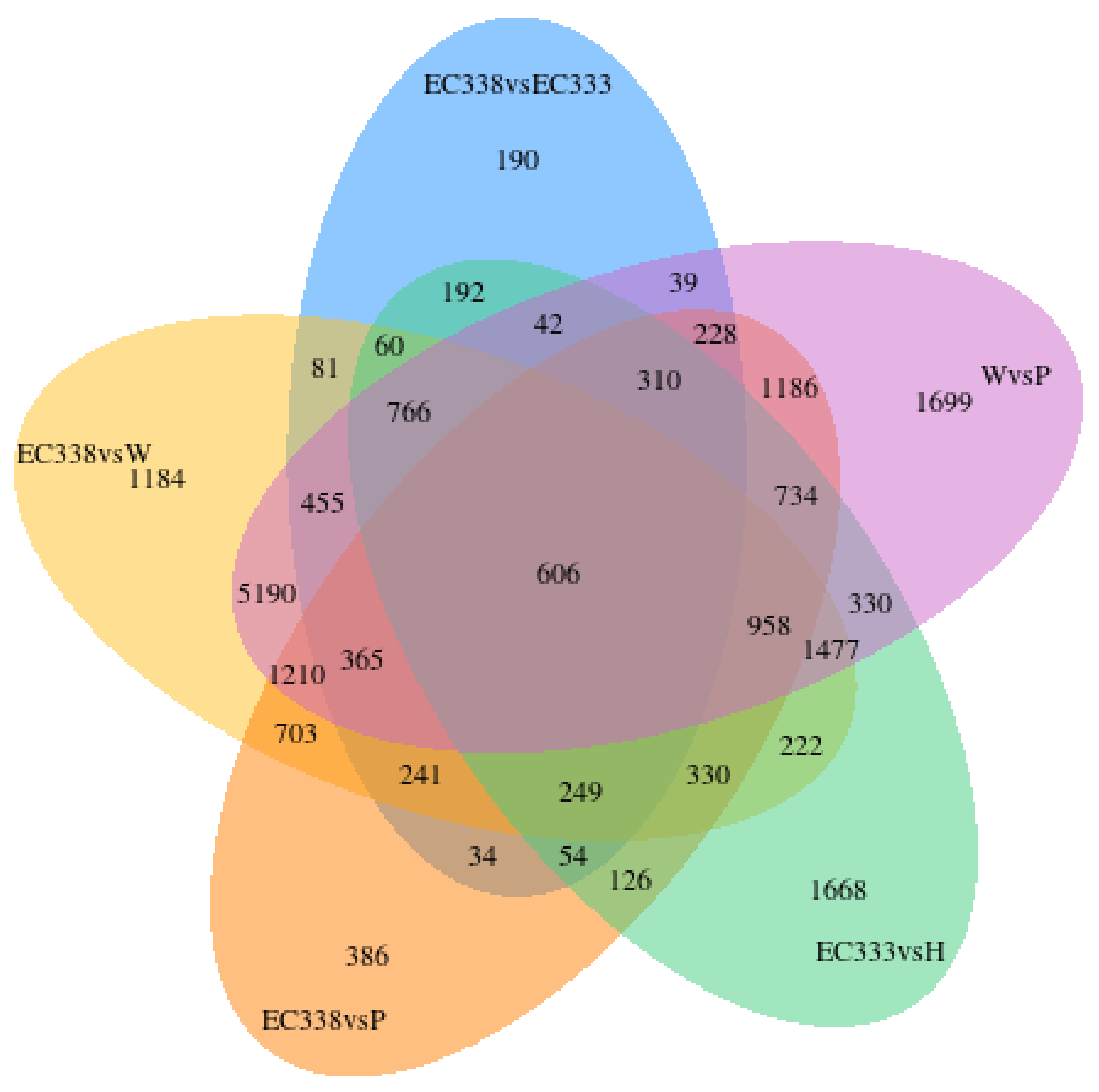
Figure 6.
Heatmap of differentially expressed gene clustering (Horizontal coordinates are sample names, vertical coordinates are values of differentially expressed genes after FPKM normalization. The redder the color, the higher the expression level. The greener, the lower the expression level ).
Figure 6.
Heatmap of differentially expressed gene clustering (Horizontal coordinates are sample names, vertical coordinates are values of differentially expressed genes after FPKM normalization. The redder the color, the higher the expression level. The greener, the lower the expression level ).
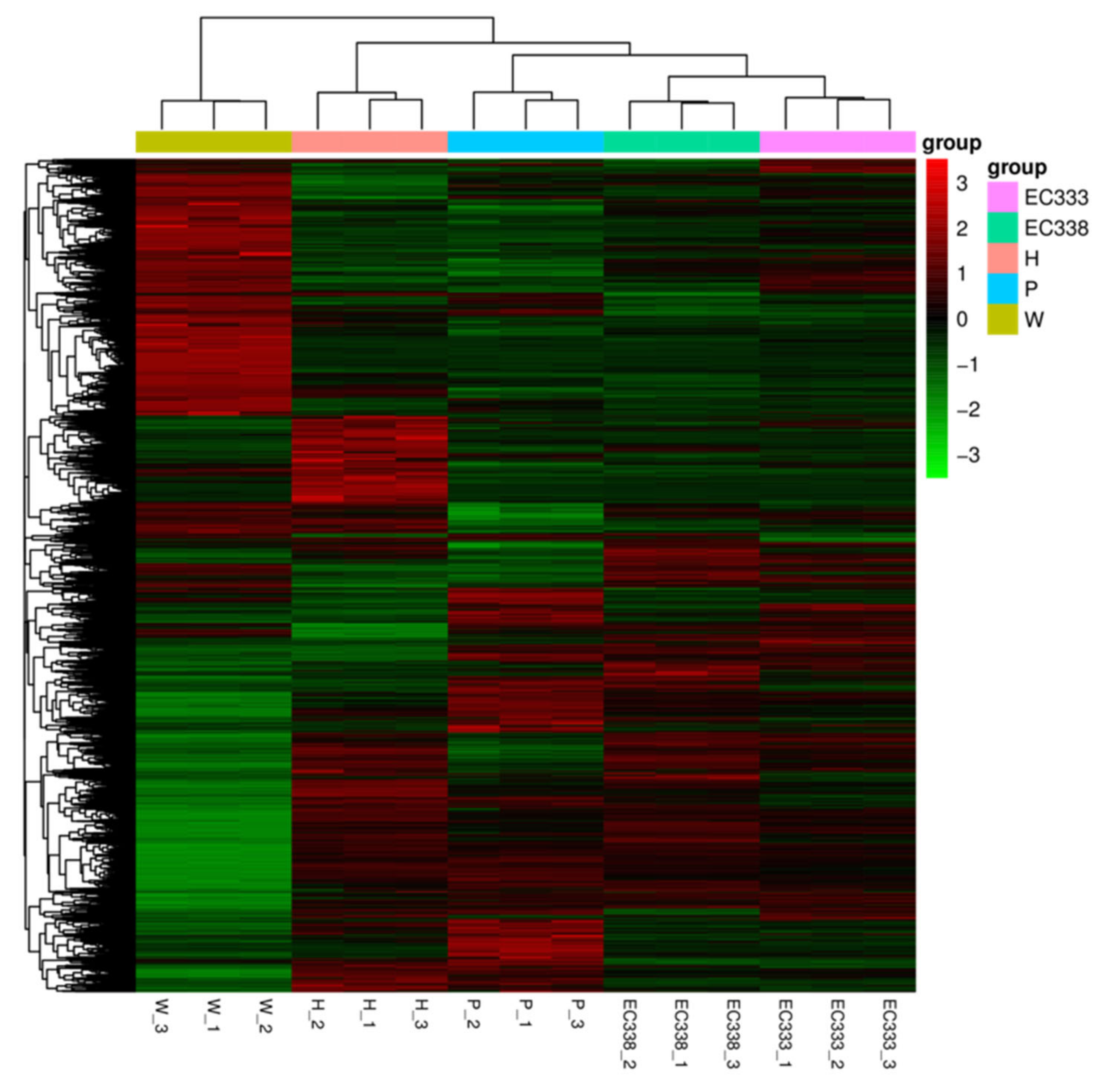
Figure 10.
GSEA enrichment analysis using KEGG annotations (here the legends same as Figure 9.).
Figure 10.
GSEA enrichment analysis using KEGG annotations (here the legends same as Figure 9.).
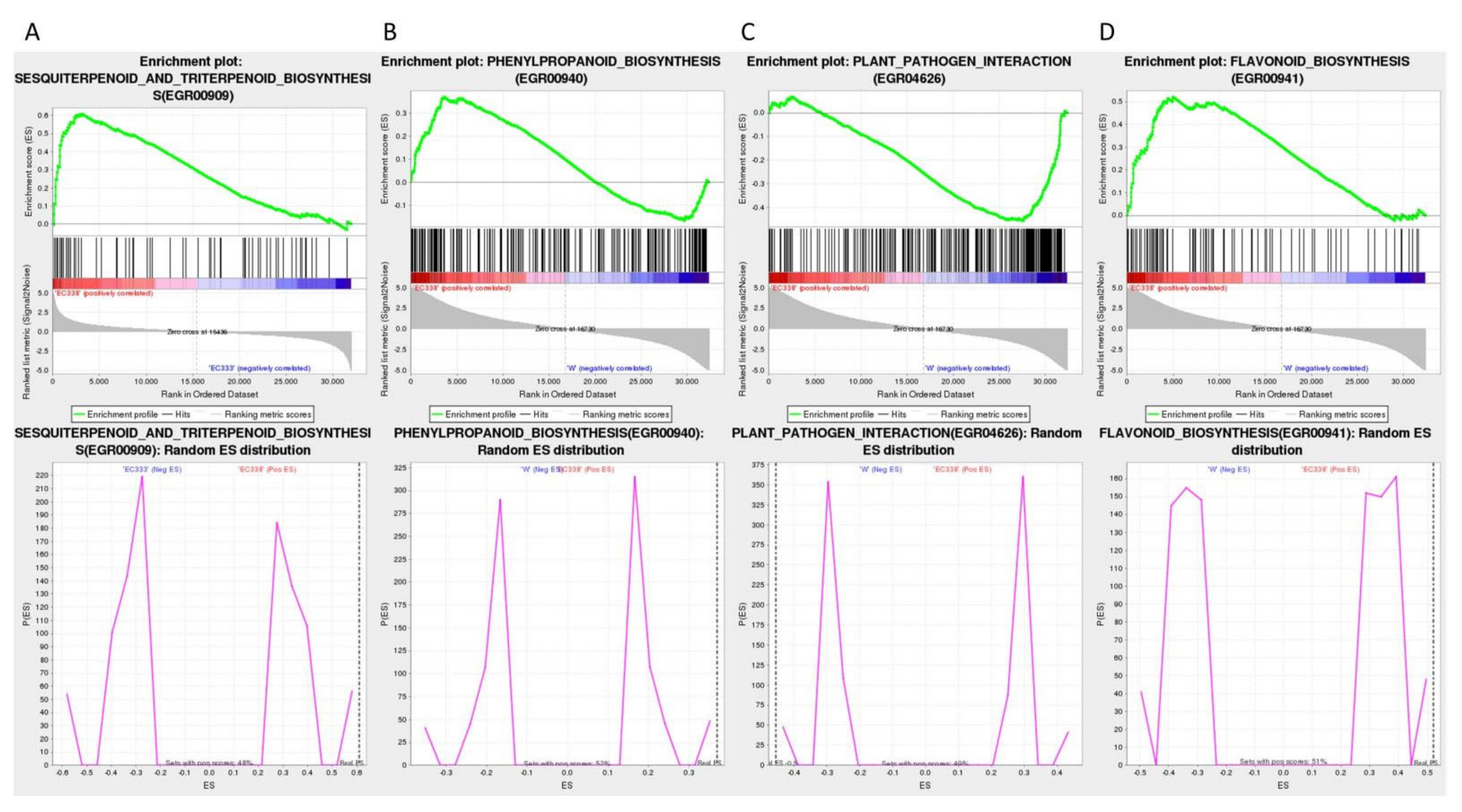
Figure 11.
SE type of Alternative Splicing.
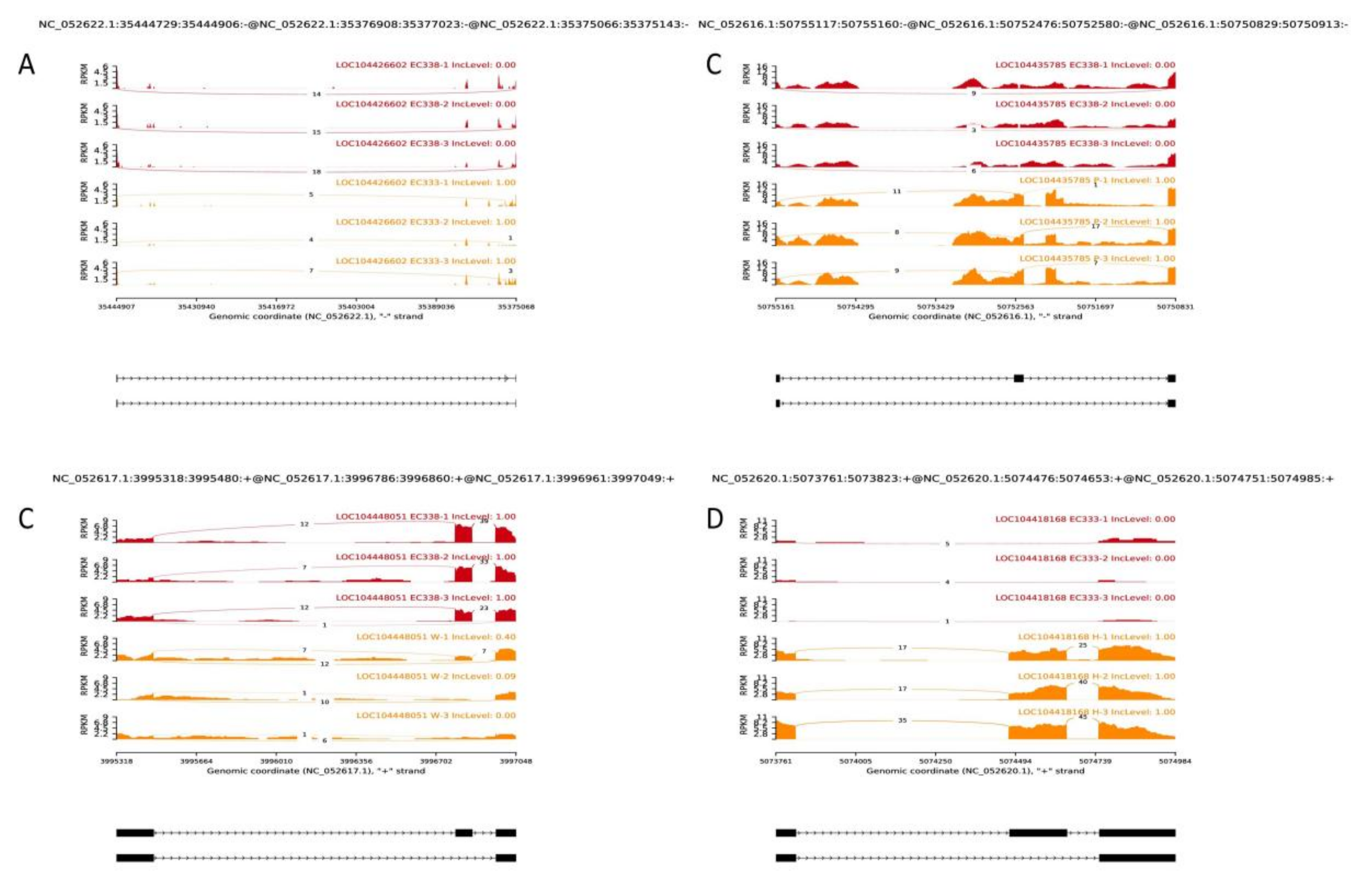
Figure 12.
SNP variant loci region statistics (The horizontal coordinates represent the samples. A: The histogram of each sample variant loci categorized based on function (SNP_function). B: The histogram of that by impact (INDEL_impact). C: The histogram of that by region (INDEL_region). D: The histogram of that by region (SNP_region).).
Figure 12.
SNP variant loci region statistics (The horizontal coordinates represent the samples. A: The histogram of each sample variant loci categorized based on function (SNP_function). B: The histogram of that by impact (INDEL_impact). C: The histogram of that by region (INDEL_region). D: The histogram of that by region (SNP_region).).
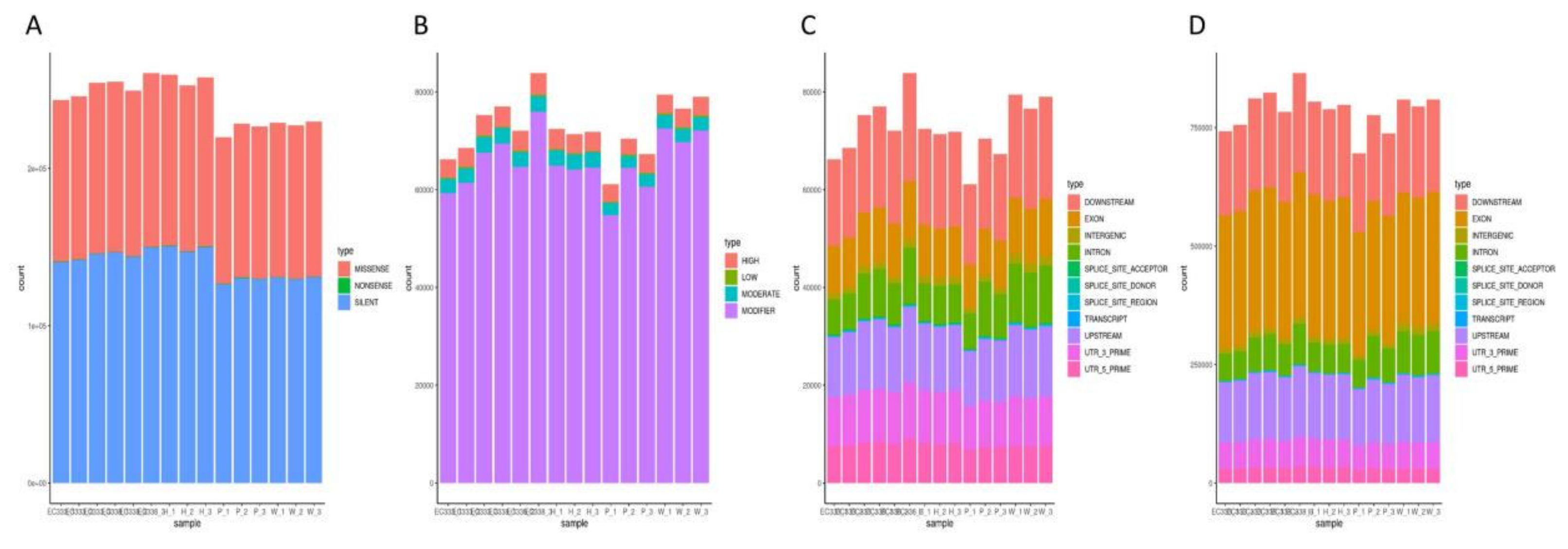
Figure 13.
Module division in WGCNA analysis (A: Soft threshold plot; in the left plot, the horizontal coordinate is the soft threshold and the vertical coordinate is the correlation between connectivity k and p(k). The right plot is the soft threshold versus the average connectivity of the network. B: Module hierarchical clustering tree; the branches are gene modules and the leaves are genes.Merged colors are the modules with dissimilarity coefficients less than 0.25 merged with colors to represent the different modules.C: Inter-module correlation heatmap; the top part of the plot is the clustering based on the module's eigenvalues, and the values of the vertical coordinates correspond to the similarity between different modules. The upper part is the clustering based on the module eigenvalues, the value of the vertical coordinate is the similarity between different modules. The lower part is the clustering heatmap between different modules, each row and column in the graph represents one module. The darker the color (redder) in the box, the stronger the correlation; the lighter the color of the box, the weaker the correlation.).
Figure 13.
Module division in WGCNA analysis (A: Soft threshold plot; in the left plot, the horizontal coordinate is the soft threshold and the vertical coordinate is the correlation between connectivity k and p(k). The right plot is the soft threshold versus the average connectivity of the network. B: Module hierarchical clustering tree; the branches are gene modules and the leaves are genes.Merged colors are the modules with dissimilarity coefficients less than 0.25 merged with colors to represent the different modules.C: Inter-module correlation heatmap; the top part of the plot is the clustering based on the module's eigenvalues, and the values of the vertical coordinates correspond to the similarity between different modules. The upper part is the clustering based on the module eigenvalues, the value of the vertical coordinate is the similarity between different modules. The lower part is the clustering heatmap between different modules, each row and column in the graph represents one module. The darker the color (redder) in the box, the stronger the correlation; the lighter the color of the box, the weaker the correlation.).
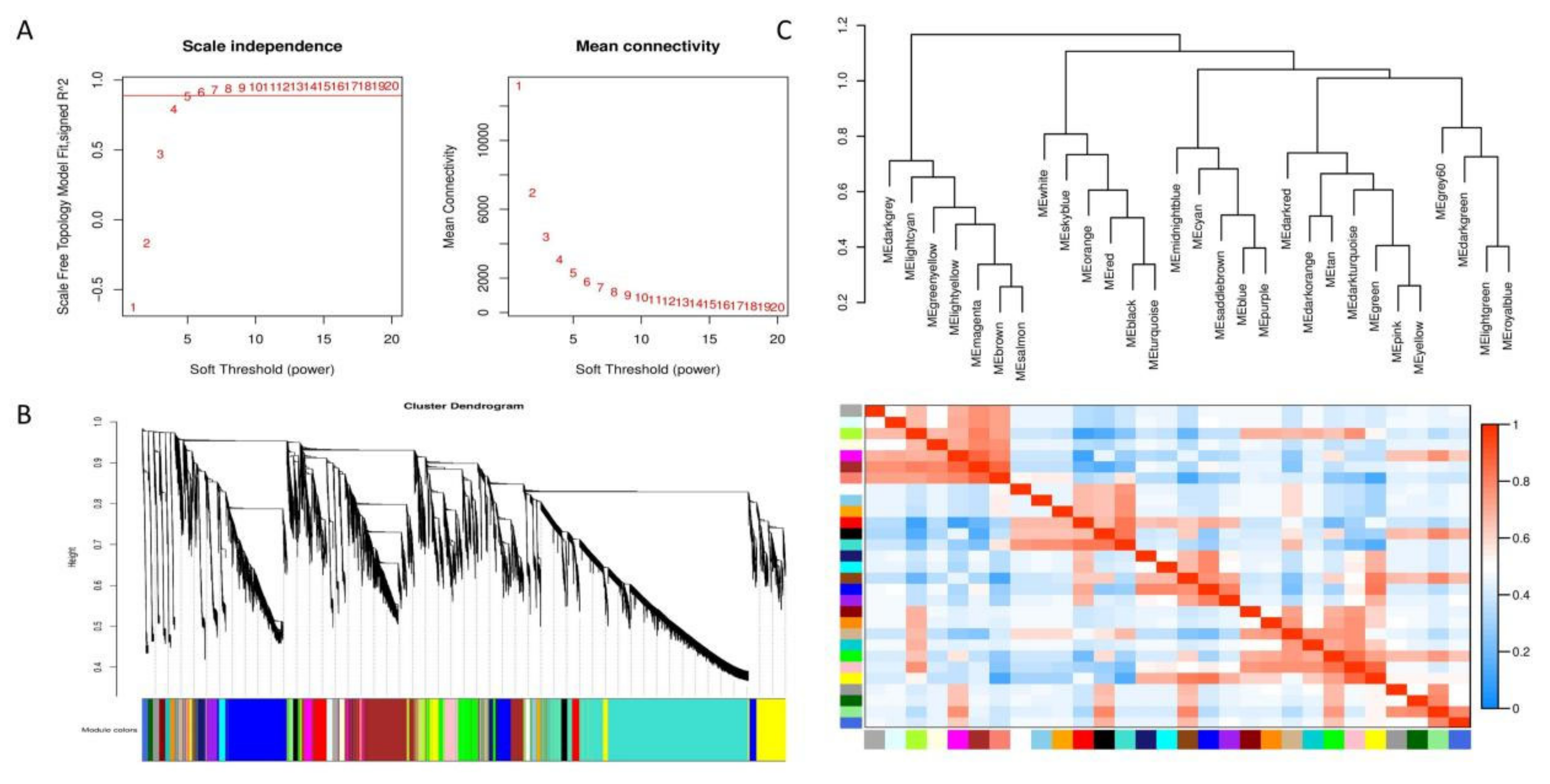
Figure 14.
Heatmap of genotype and inter-module correlations in eucalypt genotypes (The horizontal axis, known as the abscissa, denotes the sample, while the vertical axis, referred to as the ordinate, represents the module. Each cell in the grid contains a numerical value indicating the correlation between the module and the sample. A value closer to 1 signifies a more robust positive correlation between the module and the sample, whereas a value nearing -1 indicates a stronger negative correlation. The degree of negative correlation strength is proportional to the value. Additionally, the significance level, denoted by the value in parentheses, reflects the strength of significance, with smaller values indicating higher significance.).
Figure 14.
Heatmap of genotype and inter-module correlations in eucalypt genotypes (The horizontal axis, known as the abscissa, denotes the sample, while the vertical axis, referred to as the ordinate, represents the module. Each cell in the grid contains a numerical value indicating the correlation between the module and the sample. A value closer to 1 signifies a more robust positive correlation between the module and the sample, whereas a value nearing -1 indicates a stronger negative correlation. The degree of negative correlation strength is proportional to the value. Additionally, the significance level, denoted by the value in parentheses, reflects the strength of significance, with smaller values indicating higher significance.).
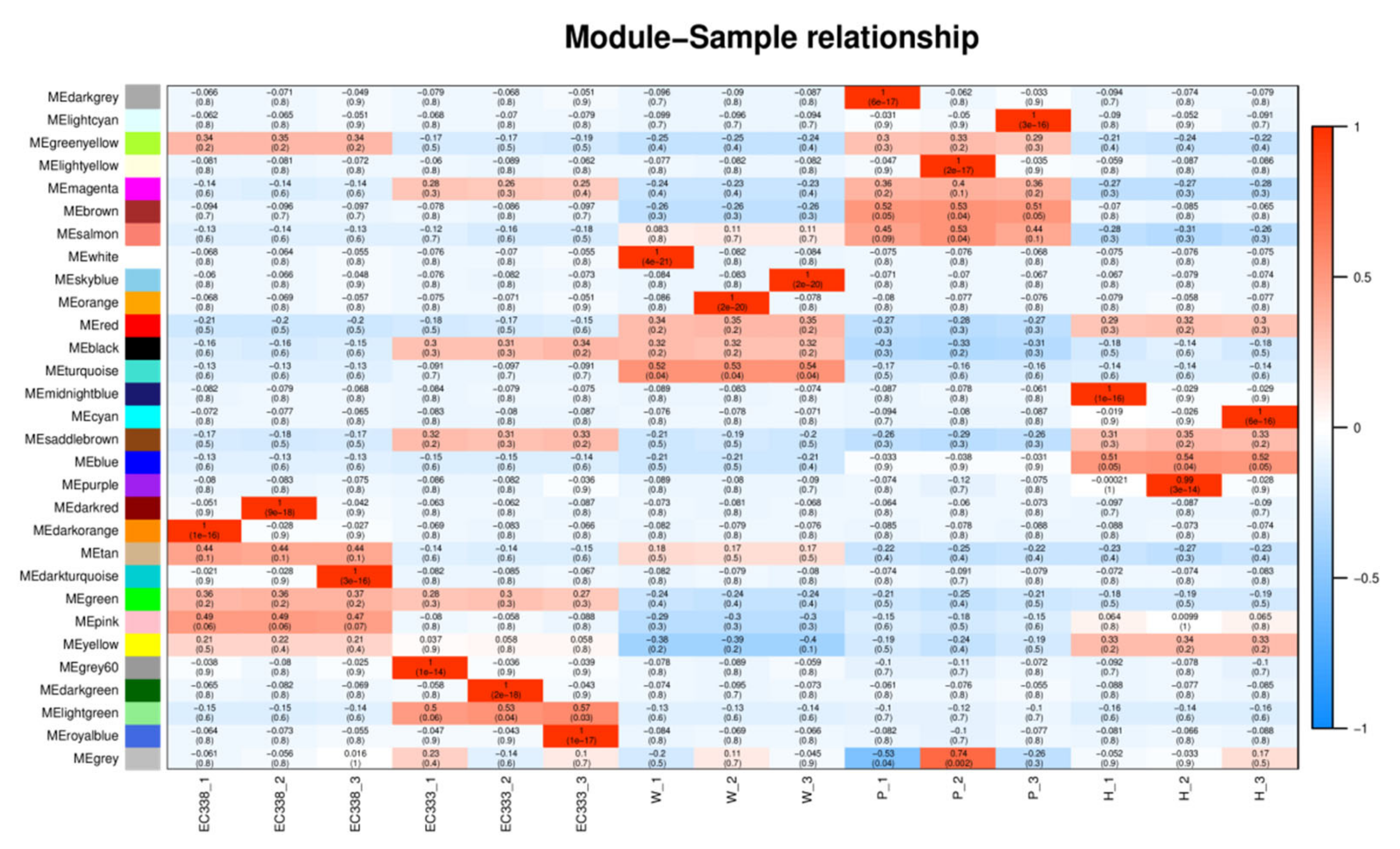

Table 1.
Eucalypt leaf blight susceptible and resistant genotypes and their parents.
| Hybrids | Susceptibility | Female | Species | Male | Species |
|---|---|---|---|---|---|
| EC338 | resistant | W1767 | E. Wetarensis | P9060 | E. pellita |
| EC333 | susceptible | H1522 | E. urophylla × E. pellita | Unknown | E. urophylla |
Table 2.
GO and KEGG enrichment analysis of four pairs of differential genes.
| Comparisons | GO | KEGG |
|---|---|---|
| EC338 VS EC333 | Carbohydrate binding, Pattern binding, Polysaccharide binding (−log10(padj)>6) | Phenylpropanoid biosynthesis, Flavonoid biosynthesis, Plant-pathogen interaction, Sesquiterpenoid and triterpenoid biosynthesis (−log10(padj)>2.5) |
| EC338 VS P9060 | Terpene synthase activity, Carbon-oxygen lyase activity, acting on phosphates, Carbon−oxygen lyase activity (−log10(padj)>10) | Phenylpropanoid biosynthesis, Sesquiterpenoid and triterpenoid biosynthesis (−log10(padj)>5) |
| EC338 VS W1767 | DNA binding transcription factor activity, Terpene synthase activity, Carbon-oxygen lyase activity, acting on phosphates, Iron ion binding, Tanscription regulator activity (−log10(padj)>6) | Phenylpropanoid biosynthesis, Plant hormone signal transduction, Sesquiterpenoid and triterpenoid biosynthesis, Plant-pathogen interaction, Biosynthesis of various plant secondary …, Flavonoid biosynthesis (−log10(padj)>4) |
| EC333 VS H1522 | Carbohydrate binding, Terpene synthase activity, Carbon-oxygen lyase activity, acting on phosphates, Carbon-oxygen lyase activity (−log10(padj)>4) |
Phenylpropanoid biosynthesis (−log10(padj)>7.5) |
Table 3.
Significance test for differential gene analysis in GSEA enrichment using GO-annotated.
| Differential genotypes | Genotypes | Name | Size | ES | NES | NOM p-val |
FDR q-val | FWER p-val | Rank at max | Leading edge |
|---|---|---|---|---|---|---|---|---|---|---|
| EC338 VS EC333 | EC333 | Carbohydrate binding (GO:0030246) | 286 | -0.519 | -1.580 | 0 | 0.171 | 0.75 | 5450 | tags=40%, list=17%, signal=48% |
| EC338 VS P9060 | EC338 | Terpene synthase activity (GO:0010333) | 86 | 0.469 | 1.952 | 0 | 0.201 | 0.188 | 2191 | tags=36%, list=7%, signal=39% |
| EC338 | Carbon-oxygen lyase activity, acting on phosphates (GO:0016838) | 89 | 0.462 | 1.939 | 0 | 0.149 | 0.188 | 2191 | tags=35%, list=7%, signal=37% |
|
| EC338 | Carbon-oxygen lyase activity (GO:0016835) | 112 | 0.429 | 2.024 | 0 | 0.098 | 0.047 | 3561 | tags=35%, list=11%, signal=39% |
Table 4.
Significance test for differential gene analysis in GSEA enrichment using KEGG-annotated.
| Differential genotypes | Genotypes | Name | Size | ES | NES | NOM p-val | FDR q-val | FWER p-val | Rank at max | Leading edge |
|---|---|---|---|---|---|---|---|---|---|---|
| EC338 VS EC333 | EC338 | Sesquiterpenoid and triterpenoid biosynthesis (EGR00909) | 65 | 0.610 | 1.714 | 0 | 0.066 | 0.093 | 3071 | tags=35%, list=10%, signal=39% |
| EC338 VS W1767 | EC338 | Phenylpropanoid biosynthesis (EGR00940) | 224 | 0.369 | 1.900 | 0 | 0.048 | 0.048 | 3594 | tags=27%, list=11%, signal=30% |
| W1767 | Plant-pathogen interaction (EGR04626) | 281 | -0.455 | -1.501 | 0 | 0.178 | 0.599 | 4752 | tags=37%, list=15%, signal=43% | |
| EC338 | Flavonoid biosynthesis (EGR00941) | 89 | 0.521 | 1.455 | 0 | 0.103 | 0.760 | 4985 | tags=42%, list=15%, signal=49% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



