
Submitted:
03 July 2024
Posted:
03 July 2024
You are already at the latest version
Abstract
Objectives: To investigate the etiology of amyotrophic lateral sclerosis (ALS) in an adult female patient. Methods: We conducted trio genome sequencing on a 35-year-old woman with progressive weakness in her left upper limb, as well as on her parents. Prior to sequencing, we performed a comprehensive neurological work-up on the patient, including neurological exam, electrophysiology, biomarker assessment, and brain and spinal cord MRI. Results : The propositus experienced weakness and atrophy in her left hand 6 months before the evaluation. This was associated with brisk reflexes and Hoffman sign in the same arm. Electroneuromyography revealed lower motor neuron involvement in three body regions. Neurofilament light chains were elevated in her cerebrospinal fluid. Brain imaging showed asymmetrical T2 hyperintensity of the corticospinal tracts and T2 linear hypointensity of the precentral gyri. Genome trio sequencing identified a likely pathogenic de novo variant in the propositus’s KIF1A gene (NM_001244008.2): c.574A>G, p.(Ile192Val). Discussion: Pathogenic variants in the KIF1A gene have been associated with a wide range of neurological manifestations called KIF1A-associated neurological diseases (KAND). The present report describe a likely pathogenic de novo variant in KIF1A associated with ALS, expanding the phenotypic spectrum of KAND and our understanding of the pathophysiology of ALS.
Keywords:
Amyotrophic Lateral Sclerosis
; Genome sequencing
; KIF1A
; de novo mutation
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the degeneration of upper and lower motor neurons leading to death a median three years after onset. However, a considerable variability in the rate of progression of disease can be observed [1], ranging from slow to acute and dramatic forms. With the growing number of causal genes found to be associated with the disease, which is approaching 40, and the easier access to sequencing technologies, a pathogenic mutation is now identified in 70% of familial ALS cases [2] and up to 10% of sporadic ones [3]. However, due to usual late onset of the disease, interpretation of rare gene variants can be challenging in ALS due to the absence of proven segregation of the suspected variant or by the potential death of the parents of the proband, precluding the discovery of de novo variants.
The kinesin family member 1A gene (KIF1A) encodes a protein that is responsible for the ATP-dependent anterograde axonal transport of synaptic vesicle precursors along the axonal and dendritic microtubules [4]. Initially, biallelic pathogenic mutations in KIF1A were linked to two phenotypes: a recessive form of HSP called spastic paraplegia-30 (SPG30), and the hereditary sensory and autonomic neuropathy type II. Further reports have shown that monoallelic variants in KIF1A are associated with a wide range of neurological manifestations, which are collectively referred to as KIF1A-associated neurological diseases (KAND). These neurological manifestations include pure or complex HSP, Rett-like syndrome, ataxia, epilepsy, PEHO syndrome, or optic atrophy [5].
In this report, we conducted a genome sequencing of a trio consisting of young-onset ALS patient and her parents. This analysis revealed a likely disease-causing new genetic variant in KIF1A that arose spontaneously in the patient. This finding broadens the range of phenotypes associated with KAND and supports the suggested link [6,7] between KIF1A and the pathophysiology of ALS.
2. Case Description
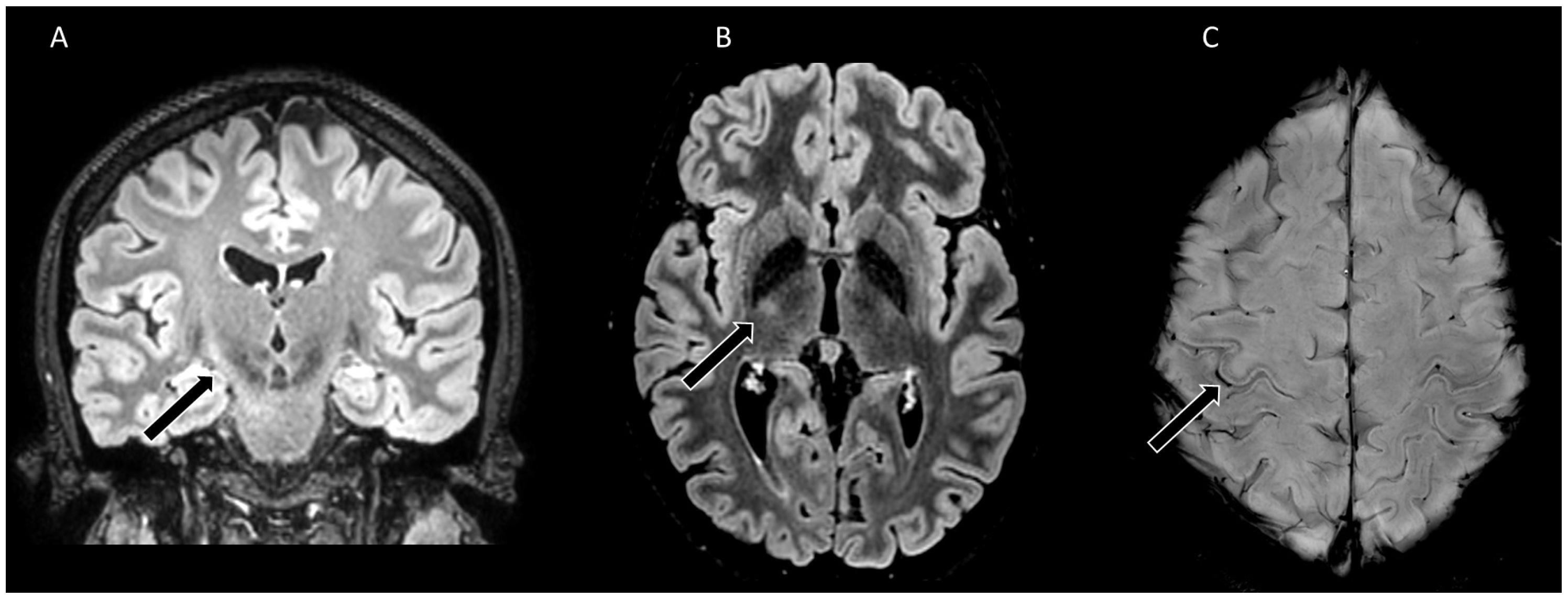
The propositus was a 35-year-old woman; the eldest of two sisters. Her parents did not present any neurological disorder (Figure 1A). She had no significant medical history. She reported a 6-month history of distal left upper limb weakness preceded by cramps, without any sensory disturbance nor pain. Clinical examination found weakness and atrophy of hand muscles (Medical Research Council 2-3/5 in distal and 4/5 in proximal left upper limb muscles). Rare fasciculations were observed on both shoulders and thighs. Deep tendon reflexes were brisk in the four limbs, with left Hoffman sign, indicating upper motor neuron involvement. Needle electromyography found chronic and active denervation in proximal and distal muscles of cervical, thoracic and lumbar regions, with normal sensory conductions. Elevated NfL was found in the CSF (1614 ng/L; normal value <750 ng/L). Spinal cord MRI was unremarkable. Brain MRI was evocative of motor neuron disease, displaying asymmetrical T2 hyperintensity of corticospinal tracts and T2 linear hypointensity of precentral gyri, more pronounced in the right hemisphere (Figure 2). Diagnosis of ALS was made according to El Escorial (definite) and Gold Coast criteria. Riluzole was initiated and the patient enrolled in a clinical trial. After 18 months of follow-up, disease progression was rather slow: left arm weakness aggravated but only minor proximal right upper limb weakness developed, without impairment in walking, respiratory or bulbar symptoms.
Trio genome sequencing revealed a novel heterozygous de novo missense variant in KIF1A in the proband (transcript ID ref: NM_001244008.2: c.574A>G, p.(Ile192Val); Figure 1B). This variant is not in public databases (e.g. gnomAD v4) and is located in the kinesin motor domain, which is known to be prone to mutations. In silico tools, such as Polyphen2, ClinPred and Mistic predicted that the amino acid change (p.Ile192Val) is damaging. Isoleucine 192 is highly conserved across all eukaryotes, including plants (Figure 1C). According to the American College of Medical Genetics (ACMG) criteria [8], the KIF1A variant was classified as probably pathogenic (class 4). None of the other variants identified in this analysis exhibited sufficient evidence to support their involvement in the disease.
3. Discussion
KIF1A has recently been identified as a candidate gene associated with ALS in two separate cohorts from Norway [6] and China [7]. In the Norwegian cohort, rare damaging variants of KIF1A were found in 3/279 ALS patients [6], while in the Chinese cohort, 10/941 ALS patients had these variants [7]. However, these variants were considered to have unknown significance according to the ACMG criteria, and therefore they were deemed unsuitable for definitive genetic counseling for these patients. The present study presents the evidence of a probably pathogenic variant, classified as ACMG class 4, in KIF1A that is associated with ALS. The propositus exhibited classical spinal upper-limb onset ALS associated with typical electrophysiological and MRI features. The only unusual aspects were the young age at onset and the relatively slow progression of the disease. The level of NfL in the CSF, which are known to be correlated with prognosis [9], were found to be 3 times lower the mean levels observed in ALS patients [10].
KIF1A can be added to the list of ALS-causing genes that affect cytoskeletal dynamics, joining ALS2, DCTN1 and KIF5A [11]. Drawing a connection between KIF1A and another member of the kinesin family, KIF5A, is an intriguing prospect. Expression of ALS-associated KIF5A mutant leads to the formation of cytoplasmic aggregates, inclusions of mitochondria, and synaptic vesicles that are toxic to motor neurons through a gain-of-function mechanism [12,13,14]. While the connection between KIF1A mutations and the characteristic extranuclear aggregates of TDP-43 in ALS is not yet understood, other kinesins, such as DCTN1, have been reported to disrupt the dynamics of stress granules and promote the formation of TDP-43 cytoplasmic aggregation in cultured cells [15].
The catalytic core of KIF1A (C351) is the crucial domain for highly processive movement16 and confers KIF1A with the ability to move more than five times as processively as dimeric conventional kinesin [17]. Interestingly, isoleucine 192 is located in the alpha-5-helix of this catalytic core. Numerous mutations have been identified in the motor domain of KIF1A as a cause of KAND, with both loss-of-function and gain-of-function effects on axonal transport [18]. The elucidation of the mechanisms underlying neurodegeneration and the association with ALS pathological markers, including TDP-43 aggregates, can be further advanced through the future development of models such as motor neurons derived from patient induced pluripotent stem cells or Drosophila. This approach has already been successfully employed in the case of KIF5A [12,13].
As clinical trials in the nano-rare disease KAND are currently underway with antisense oligonucleotides targeting KIF1A [19], the present report emphasizes the importance of systematic trio sequencing in sporadic ALS. In the event of de novo mutations, this sequencing approach can upgrade the level of pathogenicity of suspected variants from unknown significance to probably pathogenic.
4. Methods
The patient and her parents provided informed written consent for genome sequencing and publication of findings. Peripheral venous blood was collected from the family trio. Whole-genome sequencing was performed following the recommendations of 2025 French genomic medicine initiative. Genomic DNA extracted from whole blood was sequenced according to standard procedures for a PCR-Free genome on a NovaSeq6000 instrument (Illumina). Sequencing data were aligned to the GRCh38p13 full assembly using Burrows-Wheeler Aligner version 0.7+. The sequencing achieved an average target depth of approximately 42x, with over 98% of the genome covered at a depth greater than 20x. Variants were called by several algorithms, including GATK4+, Bcftools1.10+, Manta1.6+, and CNVnator0.4+, and annotated using the variant effect predictor. Detected variants were prioritized using in-house procedures. Commercially available ELISA kits were used for the measurement of neurofilament light chain (NfL) concentration in cerebrospinal fluid (CSF; Uman Diagnostics). MRI was performed on a clinical 3.0-T Philips Ingenia system (Philips Healthcare).
Author Contributions
Conceptualization: E.B. and C.G.; software: C.G.; validation: E.B., M.H., C.R., P.L. and C.G.; formal analysis: C.R. and P.L.; investigation: E.B., F.C., M.H. ; writing—original draft preparation: E.B.; writing—review and editing: E.B, C.G., A.B., C.R. and P.L..; visualization : E.B., C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient and her parents to publish this paper.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Acknowledgments
This research was made possible through access to the data generated by the 2025 French Genomic Medicine Initiative. We thank Philip Robinson (DRS, Hospices Civils de Lyon) for help in manuscript preparation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17(5):423-433. [CrossRef]
- Feldman EL, Goutman SA, Petri S, et al. Amyotrophic lateral sclerosis. Lancet. 2022;400(10360):1363-1380. [CrossRef]
- Ruf WP, Boros M, Freischmidt A, et al. Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun. 2023;5(3):fcad152. Published 2023 May 9. [CrossRef]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81(5):769-780. [CrossRef]
- Vecchia SD, Tessa A, Dosi C, et al. Monoallelic KIF1A-related disorders: a multicenter cross sectional study and systematic literature review [published correction appears in J Neurol. 2021 Oct 15;:] [published correction appears in J Neurol. 2023 Apr;270(4):2345-2346]. J Neurol. 2022;269(1):437-450. [CrossRef]
- Olsen CG, Busk ØL, Holla ØL, et al. Genetic overlap between ALS and other neurodegenerative or neuromuscular disorders. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(1-2):177-187. [CrossRef]
- Liao P, Yuan Y, Liu Z, et al. Association of variants in the KIF1A gene with amyotrophic lateral sclerosis. Transl Neurodegener. 2022;11(1):46. Published 2022 Oct 26. [CrossRef]
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. [CrossRef]
- E, Demattei C, Lehmann S, et al. Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur J Neurol. 2020;27(2):251-257. [CrossRef]
- Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry. 2016;87(1):12-20. [CrossRef]
- Nicolas A, Kenna KP, Renton AE, et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. 2018;97(6):1268-1283.e6. [CrossRef]
- Soustelle L, Aimond F, López-Andrés C, Brugioti V, Raoul C, Layalle S. ALS-Associated KIF5A Mutation Causes Locomotor Deficits Associated with Cytoplasmic Inclusions, Alterations of Neuromuscular Junctions, and Motor Neuron Loss. J Neurosci. 2023;43(47):8058-8072. Published 2023 Nov 22. [CrossRef]
- Pant DC, Parameswaran J, Rao L, et al. ALS-linked KIF5A ΔExon27 mutant causes neuronal toxicity through gain-of-function. EMBO Rep. 2022;23(8):e54234. [CrossRef]
- Nakano J, Chiba K, Niwa S. An ALS-associated KIF5A mutant forms oligomers and aggregates and induces neuronal toxicity. Genes Cells. 2022;27(6):421-435. [CrossRef]
- Ueda T, Takeuchi T, Fujikake N, et al. Dysregulation of stress granule dynamics by DCTN1 deficiency exacerbates TDP-43 pathology in Drosophila models of ALS/FTD. Acta Neuropathol Commun. 2024;12(1):20. Published 2024 Feb 4. [CrossRef]
- Kikkawa M, Okada Y, Hirokawa N. 15 A resolution model of the monomeric kinesin motor, KIF1A. Cell. 2000;100(2):241-252. [CrossRef]
- Okada Y, Hirokawa N. A processive single-headed motor: kinesin superfamily protein KIF1A. Science. 1999;283(5405):1152-1157. [CrossRef]
- Chiba K, Kita T, Anazawa Y, Niwa S. Insight into the regulation of axonal transport from the study of KIF1A-associated neurological disorder. J Cell Sci. 2023;136(5):jcs260742. [CrossRef]
- Crooke ST. A call to arms against ultra-rare diseases. Nat Biotechnol. 2021;39(6):671-677. [CrossRef]
Figure 1.
Identification of KIF1A mutation. (A) Pedigree of the affected family. (B) Visualization of whole-genome sequencing alignments using Integrative Genome Viewer software for the proband and her parents. The sequence of the KIF1A gene is displayed in reverse. The de novo heterozygous c.574A>G mutation in the KIF1A gene is indicated by an arrow. (C) Amino acid sequence comparison of the KIF1A motor domain and orthologous proteins from various species. Identical amino acids to the human KIF1A sequence are shown in bold. The missense mutation is indicated by an arrow on top of the mutated amino acid Isoleucine (I) 192 highlighted in yellow.
Figure 1.
Identification of KIF1A mutation. (A) Pedigree of the affected family. (B) Visualization of whole-genome sequencing alignments using Integrative Genome Viewer software for the proband and her parents. The sequence of the KIF1A gene is displayed in reverse. The de novo heterozygous c.574A>G mutation in the KIF1A gene is indicated by an arrow. (C) Amino acid sequence comparison of the KIF1A motor domain and orthologous proteins from various species. Identical amino acids to the human KIF1A sequence are shown in bold. The missense mutation is indicated by an arrow on top of the mutated amino acid Isoleucine (I) 192 highlighted in yellow.
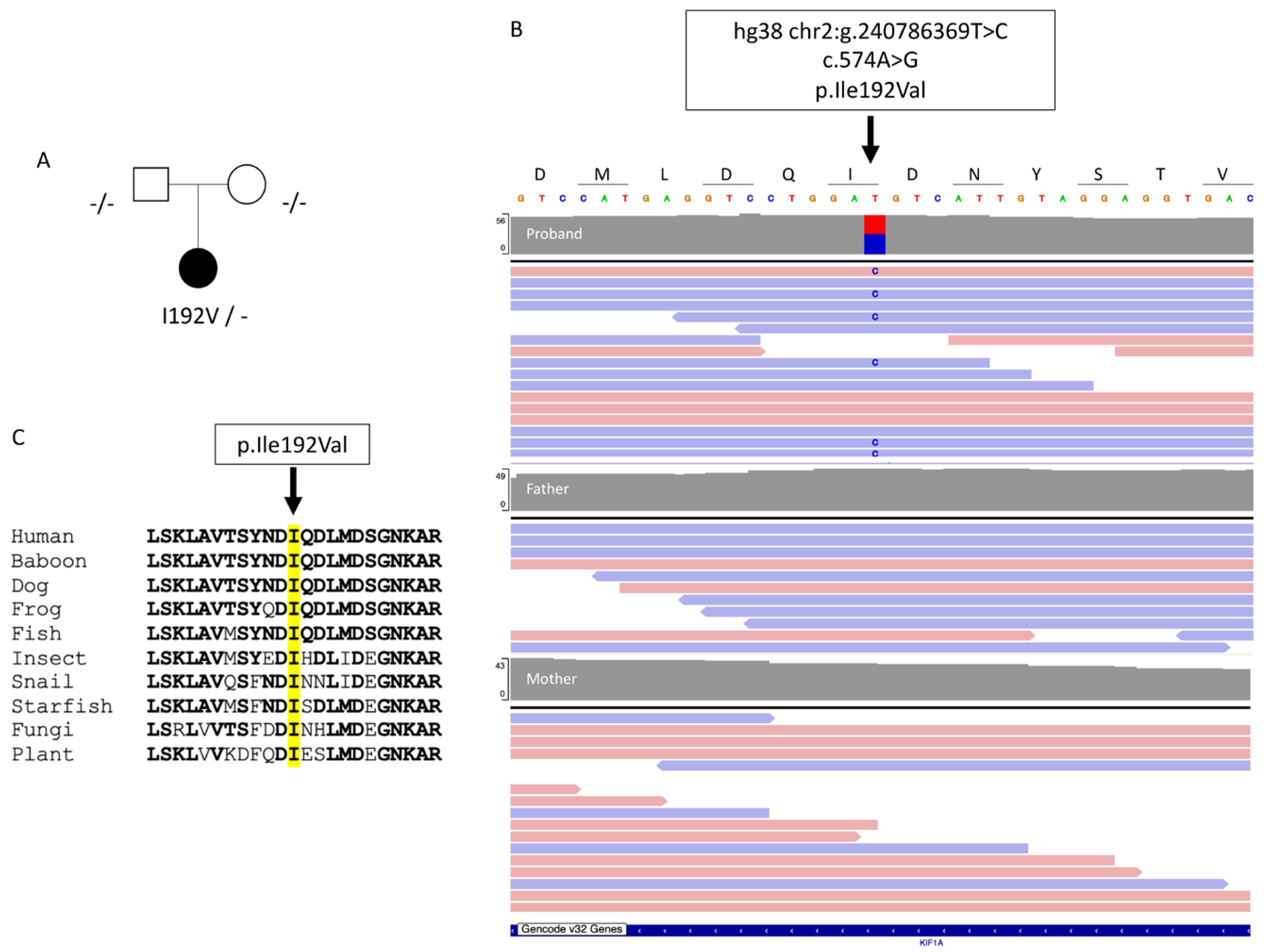
Figure 2.
Brain MRI of a 35-year-old woman with ALS harboring a de novo probably pathogenic c.574A>G KIF1A mutation. Coronal (A) and axial (B) FLAIR sequences revealed asymmetrical hypersignal of the corticospinal tracts (black arrow indicating the right corticospinal tract). Axial SWI sequence (C) displayed linear hyposignal of the right precentral gyrus (black arrow). Abbreviations: FLAIR: Fluid-attenuated inversion recovery, SWI: Susceptibility weighted imaging.
Figure 2.
Brain MRI of a 35-year-old woman with ALS harboring a de novo probably pathogenic c.574A>G KIF1A mutation. Coronal (A) and axial (B) FLAIR sequences revealed asymmetrical hypersignal of the corticospinal tracts (black arrow indicating the right corticospinal tract). Axial SWI sequence (C) displayed linear hyposignal of the right precentral gyrus (black arrow). Abbreviations: FLAIR: Fluid-attenuated inversion recovery, SWI: Susceptibility weighted imaging.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



