
Submitted:
03 July 2024
Posted:
04 July 2024
You are already at the latest version
Abstract
LCK, a member of the Scr kinase family, is a non-receptor tyrosine kinase involved in immune cell activation, antigen recognition, tumor growth, and cytotoxic response. The enzyme has usually been linked to T lymphocyte activation upon antigen recognition. Lck activation is central to CD4, CD8, and NK activation. However, recently, it has become clearer that activating the enzyme in CD8 cells can be independent of antigen presentation and enhance cytotoxic response. The role of Lck in NK cytotoxic function has been controversial in a similar fashion as the role of the enzyme in CAR T cells. Inhibition of Lck has been mainly to treat hematologic malignancies with a high rate of success. They may be useful in treating other tumor types, and they may be useful to prevent cell exhaustion. New, more selective inhibitors have been documented, and they have shown inter-esting activities not only in tumor growth but in the treatment of autoimmune diseases, asthma, and graft vs host disease. Drug repurposing and bioinformatics can aid in solving several unsolved is-sues on the role of Lck in cancer. In summary, the role of Lck in immune response and tumor growth is not a simple event and requires more research.
Keywords:
Src kinases
; Lck
; hematologic tumors
; solid tumors
; T-cell cytotoxicity
; NK cytotoxicity
; CAR-T cell
; cell proliferation
; cell adhesion.
1. Introduction
Lymphocyte cell-specific protein-tyrosine kinase (Lck) (EC 2.7.10.2), also referred to as Leukocyte C terminal Src kinase (LSK) or p56lck, is a transferase of the Src family kinases (SFKs) [1,2]. In vertebrates, this tyrosine-protein kinase family consists of 8 additional members, subfamily ScrA Src, Yes, Fyn, and Fgr abfamily Scr B Lck, Blk, Hck, and Lyn [2,3]. Their expression varies at the tissue and cellular level, and the variation also aligns with distinct hematopoietic lineages [4]. SFK's role in initiating and regulating intracellular signaling pathways has long been recognized and attributed to tyrosine phosphorylation. This modification, by altering the protein conformation and yielding binding sites for Src homology 2 (SH2) domains and protein tyrosine-binding domains, allows for the regulation of protein activation and protein-protein interaction, respectively. Thus, this effectively enables the SFKs to function in positive and negative signaling cascade regulation [3,4,5].
Lck has been extensively researched in the context of T-cells, their thymic selection and maturation, signaling, and function, and their role in the B-cell lineage's lymphocytic leukemia [3,4,5]. Nevertheless, Lck is also present in tissues [1]. The enzyme is highly expressed in the thymus and then in the appendix, which is higher than in the tonsils, the lymph node, and the spleen. Other tissues include the salivary gland, lung, small intestine, colon, gall bladder, esophagus, urinary bladder, prostate, and cervix [1]. Since the Lck protooncogene is highly active in hematologic and some solid tumors [6], using Lck inhibitors or pan tyrosine kinase inhibitors is a good therapeutic option.
An analysis of Lck structure and function is important to understand the possibility of the enzyme and its modulation in cancer growth, therapy, and immune response. The manuscript provides a general view of the subject.
2. Key Biochemical Elements of Enzyme Activity
Lck is primarily situated at the plasma membrane within the glycolipid-enriched membrane microdomains (GEMs), intimately connected to the TCR-mediated signal transduction [7]. The GEMs are enriched in highly phosphorylated active forms of signaling components of TCR/CD3, T-cell receptor (TCR)–ζ chain-associated 70 kDa tyrosine phosphoprotein (ZAP-70), SH2-domain-containing leukocyte protein of 76 kDa (SLP-76), and phospholipase Cγ1 [8]. The activation of biochemical signaling pathways is triggered by TCR/coreceptor engagement, which governs the outcome of the T cell response in conjunction with signals from costimulatory molecules and cytokine receptors [8]. Lck is the first kinase involved in this process and a fundamental component of T-cell response. Although antigen presentation and TCR engagement are crucial for Lck activation, they are not essential, as was demonstrated in multiple studies where a small pool of Lck was found to be active without antigen presentation [5,6,7,8]; therefore, tight regulation of Lck activity is critical to maintaining physiological T-cell responses [6].
Figure 1 illustrates the critical parts of the enzyme structure [1]. The membrane binding motif is essential for most antigen-dependent lymphocytes. The N-terminal Src homology 4 (SH4) domain is crucial for membrane anchoring. The domain SH4 is always myristylated at glycine position 2 and can be reversibly palmitoylated [8,9,10,11]. Palmitoylation was found to be contingent on myristylation. LCK is palmitoylated on Cys3 and Cys5 by zDHHC2 and zDHHC21 enzymes, critical for its plasma membrane targeting and T-cell activation. Additionally, PLC-γ1 and LAT undergo S-palmitoylation, contributing to T-cell signaling. Amino acids 7-35 are the binding domain to CD4 and CD8α cytoplasmic tails [9,10]. Within the domain, two cysteines are involved in Zn2+ binding to form a complex; “zinc clasp.” This Zn domain seems critical for enzyme activity and T-cell responses [1,10]. The checkpoint inhibitor LAG-3 also competes with the Zn clasp of TCR and Lck [12].
As illustrated in Figure 1, the enzyme's SH4 domain is followed by a Unique domain (UD), domains SH3 and SH2, a short proline-rich linker region, and a catalytic (tyrosine kinase) domain SH1 preceding the short C-terminal tail. SH3 and SH2 domains regulate the catalytic domain's conformational changes through proline-rich and phosphotyrosine-containing motifs. Thus, the phosphorylation of specific residues and the regulatory SH3 and SH2 domains control Lck activity [7,13,14,15]. Lck phosphorylation can be cis-phosphorylated by another molecule or trans-phosphorylation, phosphorylation by another molecule of Lck [12,13]. There is the inhibitory C-terminal residue Y505, an activating residue in the kinase domain Y394; however, there are other phosphorylated residues, residue Y192, and 4 serine residues, S42, S59, S158, and S194 (Figure 1). Phosphorylation at Y192 maintains the structure open, while the serine phosphorylation depends on PKA or PKC kinase activity, S42, and ERK activity, S59 [15,16]. The phosphorylated S59 residue seems involved in mitotic events leading to cell proliferation [16]. It is not clear what the role of the other two serine residues is; however, there seems to be a result of transphosphorylation [15]. In general, the activation mechanisms of LCK can vary depending on the specific signaling pathways and the cellular context.
Wu et al. [17] observed that asparagine directly binds to LCK and functions as a signaling molecule in CD8+ T cells. They reported that the Asn binding site in the Lck structure lies within the catalytic domain (residues 240-320). Asn favors Y394 over Y505 phosphorylation, promoting T-cell activation and memory response [17]. A similar effect on regulatory residues was reported in disulfiram (DSF), which was repurposed and enhanced the anti-tumor immunity of CD8+ T cells. DSF, as a Zinc finger active compound, interacts with Lck through a direct covalent bond via Cysteine 20/23 residue and promotes the Y394 phosphorylation, elevating Lck activation and immobilization of active Lck [18].
The enzyme's activity in physiologic conditions depends on CD45 and Csk. The role of the phosphatase CD45 and the Csk kinase on Lck activity have been studied in detail [19,20,21]. CD45 causes Lck to unfold via dephosphorylation of the Y505, allowing for the trans-autophosphorylation of Y394, leading to the full activation of Lck [14,19]. Its activity is not regulated through the phosphorylation of its kinase domain. Instead, it is allosterically activated when its SH2 domain interacts with SFK-phosphorylated motifs. CD45 can dephosphorylate tyrosine residues in C-terminal Y505 and, in the kinase domain, Y394, and its activity is > 20-fold faster than Lck in an in vitro reconstitution study documented by Hui and Vale [22]. However, Lck phosphorylation on the SH2 domain at tyrosine 192 decreases the regulatory effect of CD45 on Lck [23,24,25], implying that Y192 phosphorylation regulates Lck activity independently of CD45. Courtney and co-workers [24] have shown negative feedback of phosphorylated ZAP70 on Lck due to the phosphorylation of Y192. The phosphorylation renders Lck resistant to CD45 dephosphorylation but unable to be properly activated. The critical issue is that the Lck structure could be open independently of CD45, and the CD45-resistant phosphorylation impairs T-cell response (Figure 3).
Figure 2.
The model represents the different conformational and functional models of Lck. The open structure (A) shows the kinase active domain phosphorylated at Y394. This structure differs from the primed structure (B) in which the enzyme has an active conformation, but the Y394 residue is not phosphorylated. However, this structure can be cis or trans-phosphorylated. The open structure may have the residue Y192 phosphorylated (see Figure 3 for the physiological relevance). Double phosphorylation of Y394 and Y505 is possible, rendering an active enzyme that differs from the closed state on the right (D). It is unclear if the enzyme can be phosphorylated in the three tyrosine residues or if serine phosphorylation affects the kinetics of the active conformation. The figure was done using Bio Render software.
Figure 2.
The model represents the different conformational and functional models of Lck. The open structure (A) shows the kinase active domain phosphorylated at Y394. This structure differs from the primed structure (B) in which the enzyme has an active conformation, but the Y394 residue is not phosphorylated. However, this structure can be cis or trans-phosphorylated. The open structure may have the residue Y192 phosphorylated (see Figure 3 for the physiological relevance). Double phosphorylation of Y394 and Y505 is possible, rendering an active enzyme that differs from the closed state on the right (D). It is unclear if the enzyme can be phosphorylated in the three tyrosine residues or if serine phosphorylation affects the kinetics of the active conformation. The figure was done using Bio Render software.
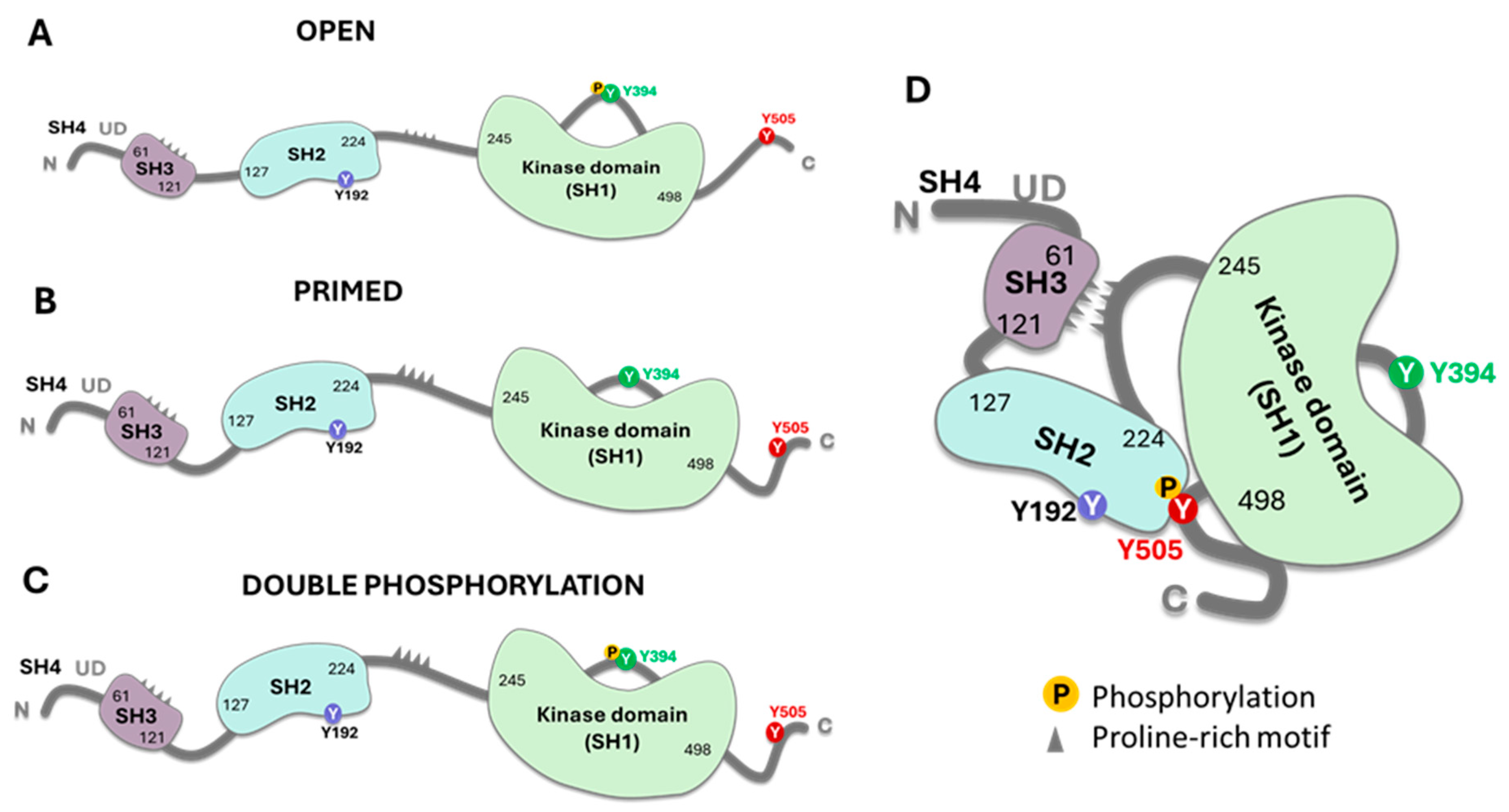
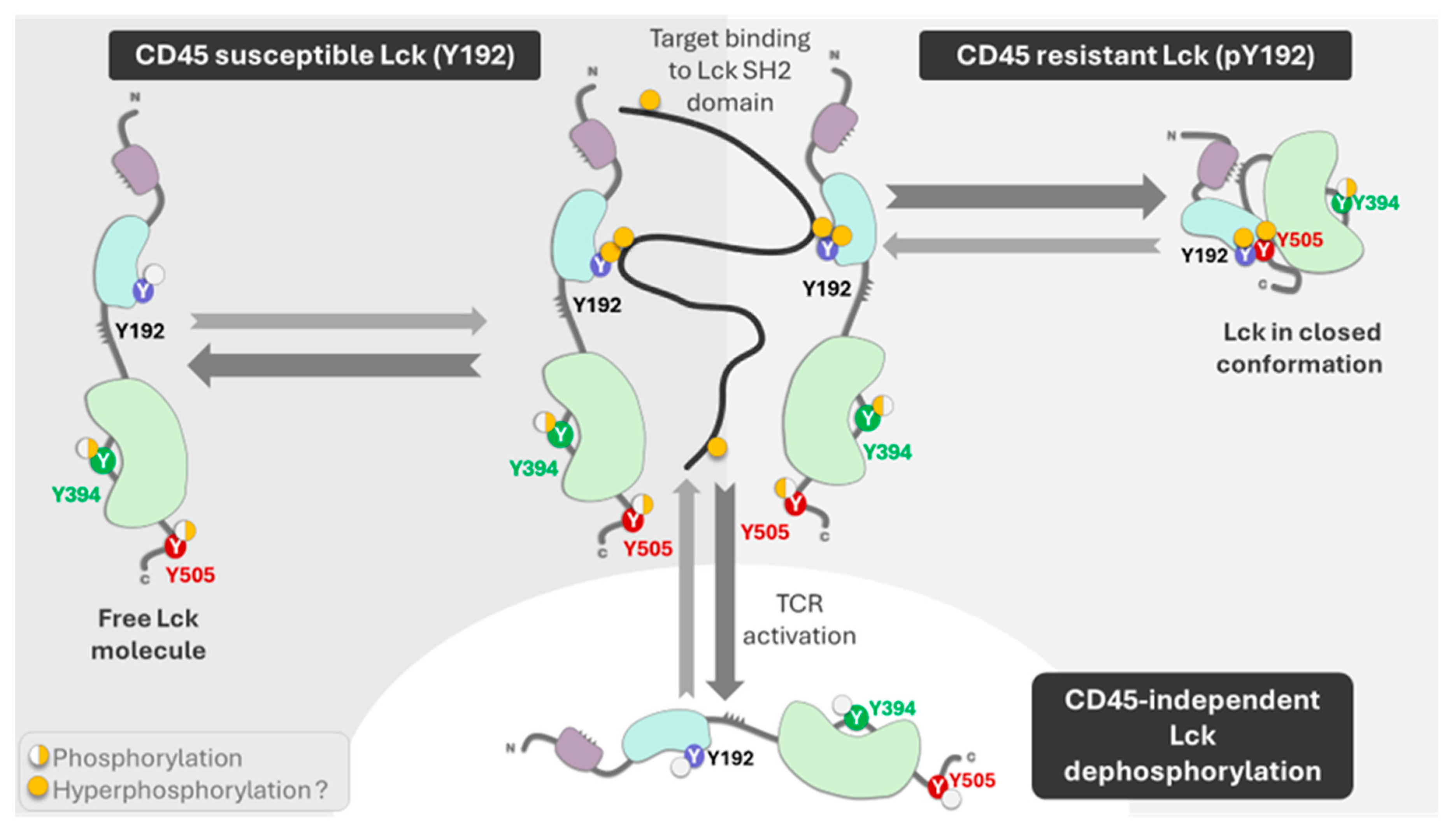
Figure 3.
The figure depicts the role of CD45 and other phosphatases on Lck phosphorylation at residue Y192. The phosphorylation can be dependent (susceptible) to CD45 dephosphorylation, or it can be resistant to phosphorylation and maintain the enzyme in close conformation. The thick lines correspond to the preferential enzyme function, the physiological response. Other phosphatases can dephosphorylate residues Y192, Y384, and Y505 generate a nonfunctional enzyme. The figure was made using Bio Render software.
Figure 3.
The figure depicts the role of CD45 and other phosphatases on Lck phosphorylation at residue Y192. The phosphorylation can be dependent (susceptible) to CD45 dephosphorylation, or it can be resistant to phosphorylation and maintain the enzyme in close conformation. The thick lines correspond to the preferential enzyme function, the physiological response. Other phosphatases can dephosphorylate residues Y192, Y384, and Y505 generate a nonfunctional enzyme. The figure was made using Bio Render software.
The Csk kinase catalyzes the transfer of the phosphate group to the Y505 residue that interacts with the SH2 domain and enables the SH3 domain to engage with the proline-rich linker region; the enzyme goes to a “closed” inactive state [21]. The Csk kinase can bind to phosphatases PTPN12 (PTP-PEST) and immune-cell PTPN22 (LYP/Pep) that promote autoinhibition by dephosphorylating the activation loop [26]. As a cytosolic protein, Csk lacks the membrane-anchoring lipid-binding sites. This raises the need for a recruiter Csk-binding protein to ensure its localization at the plasma membrane for effective negative regulation of Lck activity [21,26]. These proteins include Csk binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains (CBP/PAG1), Fak, Paxillin, and the Dok family [21,26,27]. Once it is open, the likelihood of Y505 trans-autophosphorylation/phosphorylation is low, suggesting that phosphatases are crucial in closing the structure [21,26,27].
Two adaptor proteins, LIME (Lck interacting molecule) and PAG (glycosphingolipid-enriched microdomains), have been involved in Csk inhibition of Lck activity [28,29,30]. LIME is involved in the inflammatory response [28]. The putative mechanism of Lck negative regulation/inhibition stems from PAG1 phosphorylation loss upon TCR stimulus while being abundantly phosphorylated in unstimulated T cells, which allows Csk to dissociate from PAG1/engage with Lck at the membrane, respectively [30]. Both adaptor proteins serve as membrane anchors for the Csk enzyme, facilitating the effect of Csk on the C terminal of Lck and the closure of the enzyme. As depicted in Figure 4, the adaptor proteins recruit Csk in both cases, which is responsible for phosphorylating Lck. The critical issue is the binding of Csk to the phosphorylated residues of both adaptor proteins. Csk interaction with Lck may be modified depending on the interaction with other kinds of kinases depending on the SH2 and SH3 domains.
In severe combined immunodeficiency, ZAP70 cysteine 564 seems to have a regulatory function and is involved in TCR and Lck activation [31]. Cysteine residues outside the catalytic domains of protein tyrosine kinases are critical for enzyme activity. Reversible modifications such as nitrosylation, sulfonation, and glutathionylation have modulated kinase activity [32,33,34,35]. These modifications can be important in enzyme catalytic activity and in enhancing alterations in cell metabolism, leading to malignancy [36].
Signal transduction is induced upon antigen binding to the CD3/TCR receptor in CD4 or CD8 cells in normal physiological conditions [14]. The generation of signal transduction upon Lck activation and the involvement of TCR phosphorylation, ZAP70, and the adaptor protein LAT has been extensively reviewed [14]. New elements indicate that the enzyme may be more active when not attached to the membrane [37]. The enzyme's activity differs in different T-cell subpopulations [38]. The regulatory T cells were shown to have less active Lck than committed T helper or cytotoxic cells [39]. Moreover, with age, Lck activation through co-receptors may be decreased. Co-activating molecules like CD28 may play an important role in this effect [40,41]. Also, cytomegalovirus infection has decreased CD8 activity by affecting CD28 signaling [42], and mice lacking CD28 have higher cytotoxic responses [43]. In the T cell receptor activation dynamic, Lck is more active in CD8 cells mature cells than CD4 mature cells, suggesting that Lck activation is critical in CD8 cytotoxic response [38,44]. Figure 4 illustrates the effect of antigen recognition, Lck activation, and the dynamic of signal transduction. The Figure only represents a partial process in which Lck is involved. The coreceptors also can recruit Lck and other kinases to amplify the antigen-induced signal. On the other hand, CTLA-4 recruits the phosphatase SHP-2, which similarly dephosphorylates Lck as PD1 [36,37] (Figure 5).
The complex DAP10/Syk plays a major role in NK-induced cytotoxic activity. However, the Wiskott–Aldrich Syndrome protein (WASP) phosphorylates Lck at position Y141 and inhibits enzyme activity and NK cell cytotoxicity, suggesting that Lck, besides Syk, is involved in cytotoxic responses independent of TCR and CD/CD8 antigens [45]. Figure 6 illustrates the possible link between Lck and NKG2D receptors for NK cell signal transduction involved with cytotoxicity. Figure 6 illustrates the role of Lck in the induction of CD314.
SHP-1 and SHP-2 recruited by inhibitory receptors are key negative regulators of proximal TCR signaling. They can dephosphorylate multiple crucial cascade elements, including Lck (on the Y394 residue), TCR ITAMs, and ZAP-70 [46,47,48]. The PD-1 (CD279) receptor ligation leads to PAG1 tyrosine-phosphorylation and recruitment of Csk or the phosphorylation of SHP-2, which dephosphorylates Lck, and inhibits it [45-47}. The interactions are critical for cancer chemotherapy [49,50]. Posttranslational modifications may be important in finding new therapeutic targets [51]. In activated T cells, Lck was co-immunoprecipitated with the TRAIL-R/SHP-1 complex [51]. The involvement of TRAIL led to the interruption of proximal TCR signaling, as it hindered the Y394 Lck phosphorylation and dampened the Lck recruitment to the lipid rafts [52]. Thus, TRAIL-R could be a new type of immune checkpoint receptor that can limit T cell activation. The TRAIL-R/SHP-1 axis could be a potential target for treating immune-mediated diseases.
Figure 7 illustrates checkpoint inhibitors' effects on two different inhibition types: the interaction of PD1/PDL-1 or PDL-2 and the inhibitory receptor LAG-3 [53]. LAG-3 is expressed in exhausted cells and binds to the TCR CD4/CD8 cluster, decreasing the pH and binding to the zinc pocket [54]. LAG-3 can bind to different glycan structures, including galectin and α synuclein [53], suggesting it could be an important modulator of tumor-infiltrating lymphocytes. The inhibition of the LAG-3 signal through antibodies has been suggested for therapy, and bi-specific antibodies against PD1 and LAG3 were postulated to have a brother effect in hematological and solid tumors [55].
Lck has been shown to bind to CD2, CD5, CD44, CD48, CD55, CD146 and CD160 [1]. These receptors are involved in several biological responses. CD2/LFA-3 is an interesting complex critical in immunological synapses, and CD2 is preferentially expressed in memory cells [56]. CD2 interacts with Lck and Fyn, forming the lipid raft crucial for T-cell response [57]. CD5 can also be phosphorylated by Lck and is involved in lymphocyte signal transduction, particularly in B lymphocytes [58]. The role of Lck in B cell malignancies is noted but difficult to analyze due to the low proportion of the kinase compared to Btk. It would be interesting to analyze possible scenarios for the role of Lck in B cell physiological responses.
CD44 is a cell surface adhesion receptor upregulated in primed naïve T lymphocytes and highly expressed in T memory cells [59]. Lck is bound to the intracellular site of the intracellular domain side of the receptors, which can be triggered by hyaluronic acid and, with less affinity with other extracellular matrix glycosaminoglycans and proteoglycans [59]. It is also highly expressed in tumors, cancer stem cells, and metastatic sites [60], and different isoforms may have different functions on cell physiology and pathology [61]. CD44 expression in tumor cells could be related to poor survival; however, there are still controversies on the receptor and the possible role of Lck in cell migration and physiological responses.
CD146 (MCAM-melanoma cell adhesion molecule), a cell surface adhesion molecule for Laminin 411, has also been related to Lck activity. In mouse T cells, CD146 deficiency impairs thymocyte development and peripheral activation. CD146 interacts with LCK and can be found in monomeric and dimeric forms in T cells, with the dimerized form increasing after TCR ligation. Dimerized CD146 recruits LCK and promotes LCK autophosphorylation. In tumor models, CD146 deficiency dramatically impairs the antitumor response of T cells [62]. T cells expressing CD146 in humans are mainly responsible for IL-17 production [63]. Inhibition of Lck in the psoriatic mice model has shown improvement [64]. These results generate new elements in which free Lck can bind to different receptors and activate signal transduction signals dependent on the vicinity of other kinases and proteins mainly through the SH2 and SH3 domains, but also with the zinc pocket in the SH4 domain, which may allow the protein to interact independently of the membrane anchors and facilitates lymphocyte activation, T and NK cell.
CD48, a member of the SLAM family of proteins, is a known B and T cell marker involved in cell activation and interaction [65]. The molecule is also expressed in macrophages at inflammatory sites. CD48 can interact with CD2 or CD244 [65]. CD48 cooperates with Lck in the lipid raft, but CD2 may also facilitate the cis interaction of the kinase in the CD2:CD48 binding [66], but also in CD2:CD244 stimulation, leading to a reasonable supposition that Lck can be involved in physiological and pathological responses based on receptor expression. Also, in the immune synapse, it is difficult to conceive which interaction would be prevalent in the priming, activating, or maintaining T cytotoxic responses against tumor cells.
The decay accelerating factor (CD55) is involved in the complement pathway, but it is also a potent activator of naïve CD4 T lymphocytes and is involved in tumor cell growth [67]. The interaction between CD55 and Lck seems to depend upon the lipid rafts formed upon cell binding, which may predispose the interaction of the enzyme with the adaptor protein LIME or the counteraction with the orphan receptor ROR2. Inhibition of Lck sensitizes endometrioid tumors to cisplatin [68]. A similar mechanism can probably be proposed for other epithelial tumors in which cell interaction is important in chemoresistance.
A vital cell marker for CD8 and NK cells related to cytotoxic efficiency is CD160 [68]. There are two isoforms, one transmembrane and another linked to the membrane by GIP. Lck has been linked to the transmembrane form of CD160 [68]. The antigen is also expressed in B-cell hematologic and mammary tumors [70]. It has been indirectly proposed that this interaction in tumors can lead to new therapeutic strategies since higher tumor Lck expression and survival in several tumors [71,72]. However, it is important to note that Lck inhibition has been shown to decrease metastatic potential invasion in oral cancer
In addition to the above receptors and proteins, Lck also binds to other effector molecules such as VAV1, RASA1, and FYB1 and protein kinases, including AXL, RAF1, Fyn, Syk, and PI3K, via its SH3 domain and to the tyrosine-phosphorylated form of KHDRBS1/p70 via its SH2 domain. These bindings highlight the non-unique event of Lck role in cell physiological responses. Figure 8 highlights the effect of Lck activity on multimerization of the Calcium channel transporter TRPM8 in pancreatic and mammary cancer [74]. It is unknown if the up-regulated Lck escapes from normal physiological control; the specific Lck inhibitors may facilitate, as in other tumors, the effect of other drugs to enhance tumor clearance. Modulation of Lck activity entails an intricate interplay of membrane localization, phosphorylation, protein-protein interactions, and the influence of supporting molecules.
3. CAR T Cells and Lck Signaling
In CAR T signaling, as in normal CD8 function, TCR receptors and the recruitment of Lck play a major function in the response of activated CAR-T [75]. Constructing suitable receptors and coreceptors involves Lck, Fyn, and Syk enzymes to potentiate cell activation [75]. In addition, CAR T cells expressing NKG2D antigens are successful in pancreatic cancer. The CAR-T-modified cell decreased the stroma cells surrounding the pancreatic tumor, exposing the neoplastic cells to immune cells and rendering them more susceptible to cytotoxic killing in preclinical models [76,77]. Interestingly, CAR T NKG2D cells have been used in preclinical models to eliminate cell senescence, decreasing the response pack. It is unclear whether the induction of exhaustion markers on CAR-T cells is due to continuous cell activation or absence of stimulating factors.
The partial success of the other CART clinical trials depends on tumor heterogeneity. Acarya S and coworkers [79] recently published the importance of CD28 increasing NK cytotoxicity through the Lck/CD3Z/ZAP70, contrasting with previous reports that Fyn kinase signaling in CAR T cells was more efficient than Lck signaling [80]. Probably, the difference in the construct in which THEMIS, an activator of SHP phosphatase, is recruited is responsible for the effect. [81]. More research may involve properly generating constructs to enhance the effectiveness of the treatment.
4. Lck Genetic Analysis
The first evidence of Lck mutation and lack of protein function came from Goldsmith and Weiss [82], who described the cell Jurkat variant Jcam, which lacks functional Lck due to a deletion in exon 7. However, Oh-Hori and colleagues [83] observed a lack of Lck activity in the T cell line infected with HTLV [83]. Data generated using those cell lines was influential in the T-cell signaling analysis and how the cell activation process was impaired with mutations.
Hauck et al. (2012) reported a case of a patient with a homozygous missense mutation in the LCK gene, resulting in undetectable kinase activity and impaired T-cell receptor (TCR) signaling, leading to severe combined immunodeficiency (SCID) characterized by recurrent infections and poor T-cell function [84]. Similarly, Lanz et al. [85] described a biallelic LCK mutation in a patient with profound T-cell immune deficiency. This mutation affected the protein's stability and function, leading to defective TCR signaling and reduced expression of co-receptors CD4 and CD8, resulting in severe infections and immunodeficiency, highlighting the critical role of Lck in the immune response [85]. Nevertheless, Lui and coworkers [86] analyzed a novel LCK variant (c.1318C>T; P440S) characterized by T cell lymphopenia with a skewed memory phenotype and infant-onset recurrent infections. The partial defect in Lck caused T-cell immunodeficiency and intestinal inflammation. The quantity of transcribed enzyme was enough to allow the maturation of some conventional T cells but not regulatory T cells, which led to intestinal inflammation. Thus, there are probably more Lck mutations involved in tissue dysfunctions and impaired immune responses.
Analyzing the documented structure of the gene, more than 20 SNP have been documented; however, two main rs2124355238, which include a frameshift AC/A variant deletion Ala160fs, and the rs587777335 variant that has 8 missense sub-variants T/C Leu/Pro 2 at position 290. These variants have been analyzed in type 1 diabetes. However, there are other polymorphisms: 2 at position 341, two nonpathogenic, one at position 348, one at 385, and two at position 399. Also, three in the promoter region promoters Lck1-3 and four in the region bind to LIME 1-4 [1].
The functional consequences of LCK variants can be significant, impacting the protein's stability and activity. For example, the C465R mutation disrupts hydrogen bonding within the kinase domain, leading to reduced protein expression and stability. Experiments have confirmed that this mutation affects TCR signaling pathways by causing deficient protein phosphorylation in T-cells expressing the mutant LCK [87]. LCK gene polymorphisms were studied concerning type 1 diabetes (T1D). Hulme et al. [88] identified seven significant single nucleotide polymorphisms (SNPs) but concluded that common LCK polymorphisms are unlikely to play a significant role in T1D susceptibility. In contrast, Zhu et al. [89] found a significant association between the SNP rs10914542 in the LCK gene and T1D. The G allele of rs10914542 was linked to reduced T-cell activation and increased T1D susceptibility.
Since the expression of Lck differs in several tumors, it is unclear what mechanism controls the enzyme's transcription, translation, post-translational, and post-transcription changes. A comprehensive analysis has been published [90]; however, bioinformatics and artificial intelligence analysis of the data suggest that an LCK metagene analysis (a group of genes associated with Lck expression and function) can be beneficial in cancer prognostics based on the published favorable prognosis of the cluster in breast cancer patients [91]. There is a need for standardized clinical trials in which proper data analysis with relevant patient information may provide new avenues of research.
5. Pharmacological Modulation of Lck Activity
Protein kinase inhibitors (PKIs) are crucial in treating various cancers and inflammatory conditions. The list of inhibitors that can affect Lck catalytic activity among other kinases includes dasatinib, imatinib, bosutinib, ponatinib, masitinib, ceritinib, crizotinib, fostamatinib (used on thrombocytopenia), tribanibulin (inhibit tubulin), and sacaritinib (now proposed for non-cancer therapy), as well as the experimental drugs A-770041 and WH-4-23. Even though most inhibitors are not specific, pan tyrosine kinase inhibitors have changed the landscape of targeted cancer therapy [92,93,94,95,96], and the structures and main biological effects have been documented [92,93,94,95,96]. As described earlier, several solid tumors have high expression of Lck, and inhibiting the kinase activity can make the tumor more susceptible to other anti-cancer drugs.
A prediction model for Lck inhibition has been developed, and it is reasonable for assessing enzyme activity in different models based on the structures [97]. The development of new compounds should probably focus on the enzyme's kinase activity, membrane anchor, and zinc pocket elements in the SH4 domain.
Imatinib is used in malignant gastrointestinal stromal tumors (GIST) [96,97,98,99,100]. Imatinib plus binimetinib (MEK kinase inhibitor) were used in the first-line treatment of advanced GIST [101]. Nilotinib is active in KIT-mutant metastatic acral and mucosal melanoma [101]. Bosutinib and pemetrexed (antifolate drug) were administrated in patients with advanced metastatic solid tumors (adenocarcinoma of the lung, adenocarcinoma of the appendix, and urothelial carcinoma) that had progressed on "standard of care" chemotherapy in a phase I study with some success [102]. In patients with advanced pancreatic ductal adenocarcinoma and overexpression of acyl-CoA oxidase-1 (ACOX1), gemcitabine plus masitinib showed a better OS in comparison with gemcitabine plus placebo [103]. Median survival was significantly longer for patients with advanced imatinib-resistant GIST receiving masitinib, followed by post-progression addition of sunitinib when compared against patients treated directly with sunitinib in second-line [103,104]. Also, masitinib is effective as a first-line treatment of advanced GIST with comparable results to imatinib regarding safety and response [105]. Promising results were observed using nilotinib in KIT-driven advanced melanoma, a condition with very few therapeutic options [73,106]. Moreover, Lck was shown to be involved in invasion and metastasis in oral cancer, and the specific inhibitors can be a useful therapy in these patients [107]. In summary, several studies emphasize the effect of tyrosine kinase inhibitors in different solid tumors in which Lck may play an important role. The question also arises of whether the structures have other biological functions that could be used for treatment. As an example of drug repurposing, Barwal A and coauthors showed that ponatinib could bind to PD-1 and inhibit the interaction with PDL-1/2 [108]. There is still room for improvement in cancer therapy using these inhibitors.
Concerning the new structures, A-770041 has been found to inhibit CTV-1 cells (an acute myeloid leukemia cell line) [109], and treatment of human glioma stem cells with A-770041 has crizotinib, resulted in significant inhibition of self-renewal and tumor-sphere formation [110]. WH-4-23, a potent inhibitor for Lck, also inhibits CTV-1 cells [109]. Crizotinib is a TKI that potently inhibits Lck, anaplastic lymphoma kinase (ALK), mesenchymal-epithelial transition (MET), and ROS proto-oncogene receptor 1, tyrosine Kinase (ROS1) {109, 111]. Crizotinib is well tolerated with rapid and durable responses in patients with ALK-positive non-small-cell lung cancer [112] and superior to chemotherapy [113]. Tanaka A and coworkers [114] showed that imatinib selectively depleted effector T reg (eT reg) cells and significantly increased effector/memory CD8+ T cells, enhancing the antitumor response. New structures or drug repurposing can enhance therapeutic efficiency in solid tumors.
It is also essential to address the issue that Lck inhibitors may be beneficial for treating immune-related disorders and not only as an anti-tumor treatment. Several T-cell-dependent responses can be inhibited, such as autoimmunity, inflammatory diseases, and organ transplant rejection [116,117]. This effect could be due to FoxP3-dependent much lower expression of Lck and ZAP-70 in T reg cells compared with other T cells. Imatinib inhibition of Lck further reduced their TCR signal intensity, rendering them selectively susceptible to signal-deprived apoptosis [114]. Other inflammatory diseases, such as lung fibrosis, asthma, rheumatoid arthritis, and diabetes, can be treated with Lck inhibitors [116,117,118]. On the other hand, calcineurin inhibitors target Lck activation in graft vs. host disease [119], generating questions concerning treatment effectiveness and selectivity.
Nilotinib in patients with cGVHD steroid-dependent/refractory cases was associated with a substantial decrease in proinflammatory cytokines, a better outcome of bone marrow transplant [120,121]. Moreover,
Nilotinib has been used to prevent cytomegalovirus (CMV) infection in patients with allogeneic hematopoietic stem cell transplantation by blocking platelet-derived growth factor receptor-alpha, a critical receptor for this virus [122].
6. Conclusions
The role of the enzyme in the adaptative immune response has been studied; however, there are still questions concerning the importance of enzyme activity in specific subpopulations and the importance of the sequence of events involving immune synapsis, the recruitment of membrane-free or bound enzyme and the role of Lck activation and inhibition by protein-protein interaction aside from CD45 and Csk. This information is crucial in understanding the immune response against tumors and how its Lck modulation can lead to a practical cytotoxic function, T cell exhaustion, or the generation of T regulatory cells. Interesting but unsolved questions have been observed in CAR-T and CAR-NK cells, as well as cell cytotoxic function and exhaustion, which need to be resolved. Figure 9 summarizes the regulation of Lck activity in different conditions. Since lipid rafts and modifications modify the enzyme's location and function, only partial information has been found in some items, like boundary lipids.
On the other hand, the role of Lck in cancer is complex. In hematological tumors, the role of Lck and other kinases is directly linked to tumor growth. In solid tumors, progression and metastasis have been related to Lck activity. However, Lck overexpression in solid tumors is related to increased survival, and the treatment with pan TK inhibitors or Lck inhibitors is crucial for the increase in susceptibility to standard treatments, showing that the enzyme activity and not only its presence can be involved in tumor growth.
New interesting tyrosine kinase inhibitor molecules have been developed that show promise for cancer treatment. Lck inhibitors may also modulate T-cell responses in acute and chronic inflammatory responses. Drug repurposing and chemical modifications of known structures can open new avenues of research for understanding disease mechanisms and new therapeutic targets for more efficient and specific effects.
Author Contributions
Conceptualization, J.B.D.S., M.H., J.V.G.; investigation, V.V., H.D., A.P. A. K., J.V.G., J.B.D.S..; resources, J.B.D.S., M.H.; writing—original draft preparation, V.V., H.D., A.P. A. K., J.V.G.; writing—review and editing, J.B.D.S., M.H.; supervision, J.B.D.S.; project administration, J.B.D.S., M.H.; funding acquisition, J.B.D.S., M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by SALVAGE (OP JAK)—Saving Lives through Research in the Field of Early Detection and Prevention of Cancer: Molecular, Genomic, and Social Factors. Registration number: Czech Ministry of Education, Youth, and Sports CZ.02.01.01/00/22_008/0004644. and PerMed Personalised Medicine: From Translational Research into Biomedical Applications from the Technology Agency of the Czech Republic (PerMed, project number TN02000109).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- P06239 · LCK_human https://www.uniprot.org/uniprotkb/P06239/entry. Assessed June 9, 2024.
- Serfas, M. S.; Tyner, A. L. BRK, SRM, FRK, and SRC42A form a distinct family of intracellular SRC-Like tyrosine kinases. Oncology Research 2003, 13(6), 409–419. [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene. 2004 Oct 18;23(48):7906-9. [CrossRef]
- Palacios, E. H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene, 2004;23(48), 7990 8000. [CrossRef]
- Levin, S. E.; Weiss, A. Non-Receptor Tyrosine Kinases in T Cell Antigen Receptor Function. Handbook of Cell Signaling (Second Edition), 2009; Chapter 68. pp 507-516. Editor Bradshaw R.A.; Dennis, E.A. Academic Press. [CrossRef]
- Bommhardt, U.; Schraven, B.; Simeoni, L. Beyond TCR Signaling: Emerging Functions of Lck in Cancer and Immunotherapy. Int J Mol Sci. 2019;20(14):3500. [CrossRef]
- Filipp, D.; Ballek, O.; Manning, J. Lck, Membrane Microdomains, and TCR Triggering Machinery: Defining the New Rules of Engagement. Front Immunol. 2012 Jun 12;3:155. [CrossRef]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. 2021 Dec 13;6(1):412. [CrossRef]
- Porciello, N.; Cipria, D.; Mai, G.; Lanz, A.L.; Milanetti, E.; Grottesi, A.; Howie, D.; Cobbold, S.P.; Schermelleh, L.; He, H.T.; D'Abramo, M.; Destainville, N.; Acuto, O,; Nika, K. Role of the membrane anchor in the regulation of Lck activity. J Biol Chem. 2022 Dec;298(12):102663. [CrossRef]
- Kocyła, A.M.; Czogalla, A.; Wessels, I.; Rink, L.; Krężel, A. A combined biochemical and cellular approach reveals Zn2+-dependent hetero- and homodimeric CD4 and Lck assemblies in T cells. Structure. 2024 Mar 7;32(3):292-303.e7. [CrossRef]
- Chen, Y.; Li, Y.; Wu, L. Protein S-palmitoylation modification: implications in tumor and tumor immune microenvironment. Front Immunol. 2024 Feb 13;15:1337478. [CrossRef]
- Mariuzza, R.A.; Shahid, S.; Karade, S.S. The immune checkpoint receptor LAG3: Structure, function, and target for cancer immunotherapy. J Biol Chem. 2024 May;300(5):107241. [CrossRef]
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; Höfer, T.; Viola, A.; Acuto, O. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity. 2010 Jun 25;32(6):766-77. [CrossRef]
- Fernández-Aguilar, L.M.; Vico-Barranco, I.; Arbulo-Echevarria, M.M.; Aguado, E. A Story of Kinases and Adaptors: The Role of Lck, ZAP-70 and LAT in Switch Panel Governing T-Cell Development and Activation. Biology (Basel). 2023 Aug 24;12(9):1163. [CrossRef]
- Bozso, S.J.; Kang, J.J.H.; Nagendran, J. The role of competing mechanisms on Lck regulation. Immunol Res. 2020 Oct;68(5):289-295. [CrossRef]
- Kesavan, K.P.; Isaacson, C.C.; Ashendel, C.L.; Geahlen, R.L.; Harrison, M.L. Characterization of the in vivo sites of serine phosphorylation on Lck identifying serine 59 as a site of mitotic phosphorylation. J Biol Chem. 2002 Apr 26;277(17):14666-73. [CrossRef]
- Wu, J.; Li, G.; Li, L.; Li, D.; Dong, Z.; Jiang, P. Asparagine enhances LCK signalling to potentiate CD8+ T-cell activation and anti-tumour responses. Nature Cell Biology, 2021; 23(1), 75–86. [CrossRef]
- Wang, Q.; Zhu, T.; Miao, N.; Qu, Y.; Wang, Z.; Chao, Y.; Wang, J.; Wu, W.; Xu, X.; Xu, C.; Li, X.; Wang, F. Disulfiram bolsters T-cell anti-tumor immunity through direct activation of LCK-mediated TCR signaling. EMBO Journal, 2022: 41(16). [CrossRef]
- Rheinländer, A.; Schraven, B.; Bommhardt, U. CD45 in human physiology and clinical medicine. Immunol Lett. 2018 Apr;196:22-32. [CrossRef]
- Inderberg, E.M.; Mensali, N.; Oksvold, M.P.; Fallang. L.E.; Fåne, A.; Skorstad, G.; Stenvik, G.E.; Progida, C.; Bakke, O.; Kvalheim, G.; Myklebust, J.H.; Wälchli, S. Human c-SRC kinase (CSK) overexpression makes T cells dummy. Cancer Immunol Immunother. 2018 Apr;67(4):525-536. [CrossRef]
- Zhu, S.; Wang, H.; Ranjan K, Zhang D. Regulation, targets and functions of CSK. Front Cell Dev Biol. 2023 Jun 16;11:1206539. [CrossRef]
- Hui, E., & Vale, R. D. (2014). In vitro membrane reconstitution of the T cell receptor proximal signaling network. Nature Structural & Molecular Biology, ,1(2), 133. [CrossRef]
- Kästle, M., Merten, C., Hartig, R.; Kaehne, T.; Liaunardy-Jopeace, A.; Woessner, N. M.; Schamel, W. W. A.; James, J. R., Minguet, S., Simeoni, L., Schraven, B. Tyrosine 192 within the SH2 domain of the Src-protein tyrosine kinase p56Lck regulates T-cell activation independently of Lck/CD45 interactions. Cell Comm Signal, 2020: 18(1). [CrossRef]
- Courtney, A. H.; Amacher, J.; Kadlecek, T. A.; Mollenauer, M.; Au-Yeung, B. B.; Kuriyan, J., Weiss, A. A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Molecular Cell, 2017; 67(3), 498-511.e6. [CrossRef]
- Kästle, M.; Merten, C.; Hartig, R.; Plaza-Sirvent, C.; Schmitz, I.; Bommhardt, U.; Schraven, B.; Simeoni, L. Type of PaperY192 within the SH2 Domain of Lck Regulates TCR Signaling Downstream of PLC-γ1 and Thymic Selection. Int J Mol Sci. 2022 Jun 30;23(13):7271. [CrossRef]
- Brian, B. F., 4th; Sjaastad, F. V.; Freedman, T. S. SH3-domain mutations selectively disrupt Csk homodimerization or PTPN22 binding. Scientific reports, 2022; 12(1), 5875. [CrossRef]
- Okada, M. Regulation of the SRC family kinases by Csk. Int J Mol Sci, 2012; 8(10), 1385–1397. [CrossRef]
- Hur, E.M.; Son, M.; Lee, O.H.; Choi, Y.B.; Park, C.; Lee, H.; Yun, Y. LIME, a novel transmembrane adaptor protein, associates with p56lck and mediates T cell activation. J Exp Med. 2003 Nov 17;198(10):1463-73. [CrossRef]
- Ventimiglia, L.N.; Alonso, M.A. The role of membrane rafts in Lck transport, regulation and signalling in T-cells. Biochem J. 2013 Sep 1;454(2):169-79. [CrossRef]
- Strazza, M.; Azoulay-Alfaguter, I.; Peled, M.; Adam, K.; Mor, A. Transmembrane adaptor protein PAG is a mediator of PD-1 inhibitory signaling in human T cells. Communications Biol, 2021: 4. [CrossRef]
- Schultz, A.; Schnurra, M.; El-Bizri, A.; Woessner, N. M.; Hartmann, S. E.; Hartig, R.; Minguet, S.; Schraven, B.; Simeoni, L. A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling. Cells, 2022; 11(17), 2723. [CrossRef]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; Gong, Y.; Deng, H.; Lu, X. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci U S A. 2018 Oct 2;115(40):10094-10099. [CrossRef]
- Mohanasundaram, K.A.; Haworth, N.L.; Grover, M.P.; Crowley, T.M.; Goscinski, A.; Wouters, M.A. Potential role of glutathione in evolution of thiol-based redox signaling sites in proteins. Front Pharmacol. 2015 Mar 10;6:1. [CrossRef]
- Nakamura, K.; Hori, T.; Yodoi, J. Alternative binding of p56lck and phosphatidylinositol 3-kinase in T cells by sulfhydryl oxidation: implication of aberrant signaling due to oxidative stress in T lymphocytes. Mol Immunol. 1996 Jul;33(10):855-65. [CrossRef]
- Lasser, S. A.; Ozbay Kurt, F. G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nature reviews. Clinical oncology, 2024; 21(2), 147–164. [CrossRef]
- Rudd, C.E. How the Discovery of the CD4/CD8-p56lck Complexes Changed Immunology and Immunotherapy. Front Cell Dev Biol. 2021 Mar 15;9:626095. [CrossRef]
- Liang, Y.; Ye, L. Bound to be perfect: Lck and T cell co-receptors. Nat Immunol. 2023 Jan;24(1):5-7. [CrossRef]
- Horkova, V.; Drobek, A.; Mueller, D.; Gubser, C.; Niederlova, V.; Wyss, L.; King, C.G.; Zehn, D.; Stepanek, O. Dynamics of the Coreceptor-LCK Interactions during T Cell Development Shape the Self-Reactivity of Peripheral CD4 and CD8 T Cells. Cell Rep. 2020 Feb 4;30(5):1504-1514.e7. [CrossRef]
- Qin, Z.; Hou, P.; Lin, H.; Chen, M.; Wang, R.; Xu, T. Inhibition of Lck/Fyn kinase activity promotes the differentiation of induced Treg cells through AKT/mTOR pathway. Int Immunopharmacol. 2024 Jun 15;134:112237. [CrossRef]
- Le Page, A,; Dupuis, G.; Larbi, A.; Witkowski, J.M., Fülöp T. Signal transduction changes in CD4+ and CD8+ T cell subpopulations with aging. Exp Gerontol. 2018 May; 105:128-139. [CrossRef]
- Tedeschi, V.; Paldino, G.; Kunkl, M.; Paroli, M.; Sorrentino, R.; Tuosto, L.; Fiorillo, M.T. CD8+ T Cell Senescence: Lights and Shadows in Viral Infections, Autoimmune Disorders and Cancer. Int J Mol Sci. 2022;23(6):3374. [CrossRef]
- Vieira Braga, F.A.; Hertoghs, K.M.; van Lier, R.A.; van Gisbergen, K.P. Molecular characterization of HCMV-specific immune responses: Parallels between CD8(+) T cells, CD4(+) T cells, and NK cells. Eur J Immunol. 2015 Sep;45(9):2433-45. [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016 May 17;44(5):973-88. [CrossRef]
- Paprckova, D.; Niederlova, V.; Moudra, A.; Drobek, A.; Pribikova, M.; Janusova, S.; Schober, K.; Neuwirth, A.; Michalik, J.; Huranova, M.; Horkova, V.; Cesnekova, M.; Simova, M.; Prochazka, J.; Balounova, J.; Busch, D.H.; Sedlacek, R.; Schwarzer, M.; Stepanek, O. Self-reactivity of CD8 T-cell clones determines their differentiation status rather than their responsiveness in infections. Front Immunol. 2022 Oct 6;13:1009198. [CrossRef]
- Huang, L.; Zhu, P.; Xia, P.; Fan, Z. WASH has a critical role in NK cell cytotoxicity through Lck-mediated phosphorylation. Cell Death Dis 2016; 7, e2301. [CrossRef]
- Moore, E.K.; Strazza, M.; Mor, A. Combination Approaches to Target PD-1 Signaling in Cancer. Front Immunol. 2022 Jul 14;13:927265. [CrossRef]
- Chiang, G. G.; Sefton, B. M. Specific Dephosphorylation of the Lck Tyrosine Protein Kinase at Tyr-394 by the SHP-1 Protein-tyrosine Phosphatase. J Biol Chem, 2001; 276(25), 23173-23178. [CrossRef]
- Poirier, A.; Ormonde, J. V. S.; Aubry, I.; Abidin, B. M.; Feng, C.; Martínez-Córdova, Z.; Hincapie, A. M.; Wu, C.; Pérez-Quintero, L. A.; Wang, C.; Gingras, A.; Madrenas, J.; Tremblay, M. L. The induction of SHP-1 degradation by TAOK3 ensures the responsiveness of T cells to TCR stimulation. Science Signaling, 2024; 17(817). [CrossRef]
- Celis-Gutierrez, J.; Blattmann, P.; Zhai, Y.; Jarmuzynski, N.; Ruminski, K.; Grégoire, C.; Ounoughene, Y.; Di Fiore, F.; Aebersold, R.; Roncagalli, R.; Gstaiger, M.; Malissen, B. (). Quantitative interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Reports, 2019; 27(11), 3315-3330.e7. [CrossRef]
- Li, K.; Yuan, Z.; Lyu, J.; Ahn, E.; Davis, S.J.; Ahmed, R.; Zhu, C. PD-1 suppresses TCR-CD8 cooperativity during T-cell antigen recognition. Nat Commun. 2021 May 12;12(1):2746. [CrossRef]
- Wang, R.; He, S.; Long, J.; Wang, Y.; Jiang, X.; Chen, M.; Wang, J. Emerging therapeutic frontiers in cancer: insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp Hematol Oncol. 2024 Apr 23;13(1):46. [CrossRef]
- Chyuan, I.T.; Liao, H.J.; Tan, T.H.; Chuang, H.C.; Chu, Y.C.; Pan, M.H.; Wu, C.S.; Chu, C.L.; Sheu, B.C.; Hsu, P.N. Association of TRAIL receptor with phosphatase SHP-1 enables repressing T cell receptor signaling and T cell activation through inactivating Lck. J Biomed Sci. 2024 Mar 27;31(1):33. [CrossRef]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3's Enigmatic Mechanism of Action. Front Immunol. 2021 Jan 8;11:615317. [CrossRef]
- Guy, C.; Mitrea, D.M.; Chou, P.C.; Temirov, J.; Vignali, K.M.; Liu, X.; Zhang, H.; Kriwacki, R.; Bruchez, M.P.; Watkins, S.C.; Workman, C.J.; Vignali, D.A.A. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat Immunol. 2022 May;23(5):757-767. [CrossRef]
- Luke, J.J.; Patel, M.R.; Blumenschein, G.R. Hamilton, E.; Chmielowski, B.; et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med 2023; 29, 2814–2824 (). [CrossRef]
- Binder, C.; Cvetkovski, F.; Sellberg, F.; Berg, S.; Paternina Visbal, H.; Sachs, D.H.; Berglund, E.; Berglund, D. CD2 Immunobiology. Front Immunol. 2020 Jun 9;11:1090. [CrossRef]
- Nunes, R.J.; Castro, M.A.; Gonçalves, C.M.; Bamberger, M.; Pereira, C.F.; Bismuth, G.; Carmo, A.M. Protein interactions between CD2 and Lck are required for the lipid raft distribution of CD2. J Immunol. 2008 Jan 15;180(2):988-97. [CrossRef]
- Burgueño-Bucio, E.; Mier-Aguilar, C.A.; Soldevila, G. The multiple faces of CD5. J Leukoc Biol. 2019 May;105(5):891-904. [CrossRef]
- Baaten, B.J.; Li, C.R.; Bradley, L.M. Multifaceted regulation of T cells by CD44. Commun Integr Biol. 2010 Nov;3(6):508-12. [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol. 2017 Mar 7;5:18. [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018 May 10;11(1):64. [CrossRef]
- Duan, H.; Jing, L.; Jiang, X.; Ma, Y.; Wang, D.; Xiang, J.; Chen, X.; Wu, Z.; Yan, H.; Jia, J.; Liu, Z.; Feng, J.; Zhu, M.; Yan, X. CD146 bound to LCK promotes T cell receptor signaling and antitumor immune responses in mice. J Clin Invest. 2021 Nov 1;131(21):e148568. [CrossRef]
- Raychaudhuri, S.K.; Abria, C.; Raychaudhuri, S.P. Phenotype and pathological significance of MCAM+ (CD146+) T cell subset in psoriatic arthritis. Mol Biol Rep. 2021 Oct;48(10):6787-6796. [CrossRef]
- Al-Harbi, N.O.; Ahmad, S.F.; Almutairi, M.; Alanazi, A.Z.; Ibrahim, K.E.; Alqarni, S.A.; Alqahtani, F.; Alhazzani, K.; Alharbi, M.; Alasmari, F.; Nadeem, A. Lck signaling inhibition causes improvement in clinical features of psoriatic inflammation through reduction in inflammatory cytokines in CD4+ T cells in imiquimod mouse model. Cell Immunol. 2022 Jun;376:104531. [CrossRef]
- McArdel, S.L.; Terhorst, C.; Sharpe, A.H. Roles of CD48 in regulating immunity and tolerance. Clin Immunol. 2016 Mar;164:10-20. [CrossRef]
- Li, B.; Lu, Y.; Zhong, M.C.; Qian, J., Li, R.; Davidson, D.; Tang, Z.; Zhu, K.; Argenty, J.; de Peredo, A.G.; Malissen, B.; Roncagalli, R.; Veillette, A. Cis interactions between CD2 and its ligands on T cells are required for T cell activation. Sci Immunol. 2022 Aug 5; 7(74):eabn6373. [CrossRef]
- Bharti, R.; Dey, G.; Lin, F.; Lathia, J.; Reizes, O. CD55 in cancer: Complementing functions in a non-canonical manner. Cancer Lett. 2022 ;ec 28;551:215935. [CrossRef]
- Saygin, C.; Wiechert, A.; Rao, V.S.; Alluri, R.; Connor, E.; Thiagarajan, P.S.; Hale, J.S., et al.. CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors. J Exp Med. 2017 Sep 4;214(9):2715-2732. [CrossRef]
- Giustiniani, J.; Bensussan, A.; Marie-Cardine, A. Identification and characterization of a transmembrane isoform of CD160 (CD160-TM), a unique activating receptor selectively expressed upon human NK cell activation. J Immunol. 2009 Jan 1;182(1):63-71. [CrossRef]
- Oumeslakht, L.; Aziz, A.I.; Bensussan, A.; Ben Mkaddem, S. CD160 receptor in CLL: Current state and future avenues. Front Immunol. 2022 Nov 7;13:1028013. [CrossRef]
- Zhan, F.; He, L.; Yu, Y.; Chen, Q., Guo, Y., Wang, L. A multimodal radiomic machine learning approach to predict the LCK expression and clinical prognosis in high-grade serous ovarian cancer. Sci Rep 2023; 13, 16397 (). [CrossRef]
- Wang, F.; Zheng, A.; Zhang, D.; Zou, T.; Xiao, M.; et al. Molecular profiling of core immune-escape genes highlights LCK as an immune-related prognostic biomarker in melanoma. Front Immunol. 2022 Oct 20;13:1024931. [CrossRef]
- Weiße, J.; Rosemann, J.; Müller, L.; Kappler, M.; Eckert, A. W.; Glaß, M., et al. Identification of lymphocyte cell-specific protein-tyrosine kinase (LCK) as a driver for invasion and migration of oral cancer by tumor heterogeneity exploitation. Mol Cancer 2021; 20, 88 . [CrossRef]
- Huang, Y.; Li, S.; Liu, Q; et al. The LCK-14-3-3ζ-TRPM8 axis regulates TRPM8 function/assembly and promotes pancreatic cancer malignancy. Cell Death Dis 2022; 13, 524. [CrossRef]
- Honikel, M.M.; Olejniczak, S.H. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules. 2022 Sep 15;12(9):1303. [CrossRef]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother Adv. 2021 Aug 13;1(1):ltab018. [CrossRef]
- Czaplicka, A.; Lachota, M.; Pączek, L.; Zagożdżon, R.; Kaleta, B. Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence. Cells. 2024 Jan 4;13(1):101. [CrossRef]
- Deng, Y.; Kumar, A.; Xie, K.; Schaaf, K.; Scifo, E.; Morsy, S.; Li, T.; Ehninger, A.; Bano, D.; Ehninger, D. Targeting senescent cells with NKG2D-CAR T cells. Cell Death Discov. 2024 May 4;10(1):217. [CrossRef]
- Acharya, S.; Basar, R.; Daher, M.; Rafei, H.; Li, P.; Uprety, N, et al.. CD28 costimulation augments CAR signaling in NK cells via the LCK/CD3Z/ZAP70 signaling axis. Cancer Discov. 2024 Jun 20. [CrossRef]
- Wu, L.; Brzostek, J.; Sakthi Vale, P.D.; Wei, Q.; Koh, C.K.T.; et al.. CD28-CAR-T cell activation through FYN kinase signaling rather than LCK enhances therapeutic performance. Cell Rep Med. 2023 Feb 21;4(2):100917.. [CrossRef]
- Zhang, J.; Jiang, Z.; Zhang, X.; Yang, Z.; Wang, J.; Chen, J.; et al. THEMIS is a substrate and allosteric activator of SHP1, playing dual roles during T cell development. Nat Struct Mol Biol. 2024 Jan;31(1):54-67. [CrossRef]
- Goldsmith, M.A.; Weiss A. Isolation and characterization of a T-lymphocyte somatic mutant with altered signal transduction by the antigen receptor. Proc Natl Acad Sci U S A. 1987 Oct;84(19):6879-83. [CrossRef]
- Oh-Hori, N.; Koga, Y.; Yoshida, H.; Morita, M.; Kimura, G.; Nomoto, K. Human T-cell leukemia virus type-I-infected T-cell lines scarcely produce p56lck, whether or not they express lck mRNA. Int J Cancer. 1990 Aug 15;46(2):315-9. [CrossRef]
- Hauck, F.; Randriamampita, C.; Martin, E.; Gerart, S.; Lambert, N.; Lim, A.; Soulier, J.; Maciorowski, Z.; Touzot, F.; Moshous, D.; et al. Primary T-Cell Immunodeficiency with Immunodysregulation Caused by Autosomal Recessive LCK Deficiency. J Allergy Clin Immunol 2012, 130, 1144-1152.e11. [CrossRef]
- Lanz, A.-L.; Erdem, S.; Ozcan, A.; Ceylaner, G.; Cansever, M.; Ceylaner, S.; Conca, R.; Magg, T.; Acuto, O.; Latour, S.; et al. A Novel Biallelic LCK Variant Resulting in Profound T-Cell Immune Deficiency and Review of the Literature. J Clin Immunol 2023, 44, 1. [CrossRef]
- Lui, V.G.; Hoenig, M.; Cabrera-Martinez, B.; Baxter, R.M.; Garcia-Perez, J.E., et al.. A partial human LCK defect causes a T cell immunodeficiency with intestinal inflammation. J Exp Med. 2024 Jan 1;221(1):e20230927. [CrossRef]
- Lanz, A.L.; Erdem, S.; Ozcan, A.; Ceylaner, G.; et al. Novel Biallelic LCK Variant Resulting in Profound T-Cell Immune Deficiency and Review of the Literature. J Clin Immunol. 2023 Dec 15;44(1):1. [CrossRef]
- Hulme, J.S.; Barratt, B.J.; Twells, R.C.J.; Cooper, J.D.; Lowe, C.E.; Howson, J.M.M.; et al. Association Analysis of the Lymphocyte-Specific Protein Tyrosine Kinase (LCK) Gene in Type 1 Diabetes. Diabetes 2004, 53, 2479–2482. [CrossRef]
- Zhu, Q.; Wang, J.; Zhang, L.; Bian, W.; Lin, M.; Xu, X.; Zhou, X. LCK Rs10914542-G Allele Associates with Type 1 Diabetes in Children via T Cell Hyporesponsiveness. Pediatr Res 2019, 86, 311–315. [CrossRef]
- Han, M.; Li, Y.; Guo, Y.; Zhu, W.; Jiang, J. Integrative and Comprehensive Pan-Cancer Analysis of Lymphocyte-Specific Protein Tyrosine Kinase in Human Tumors. Int J Mol Sci. 2022 Nov 13;23(22):13998. [CrossRef]
- Bai, F.; Jin, Y.; Zhang, P.; Chen, H.; Fu, Y.; Zhang, M.; Weng, Z.; Wu K. Bioinformatic profiling of prognosis-related genes in the breast cancer immune microenvironment. Aging (Albany NY). 2019 Nov 12;11(21):9328-9347. [CrossRef]
- Elkamhawy, A.; Ali, E.M.H.; Lee, K. New horizons in drug discovery of lymphocyte-specific protein tyrosine kinase (Lck) inhibitors: a decade review (2011-2021) focussing on structure-activity relationship (SAR) and docking insights. J Enzyme Inhib Med Chem. 2021 Dec;36(1):1574-1602. [CrossRef]
- Roskoski, R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol Res. 2024 Feb;200:107059. [CrossRef]
- Musumeci, F.; Schenone, S. Unlocking Potential and Limits of Kinase Inhibitors: The Highway to Enhanced Cancer Targeted Therapy. Pharmaceutics. 2024 May 7;16(5):625. [CrossRef]
- Bommhardt, U.; Schraven, B.; Simeoni, L. Beyond TCR Signaling: Emerging Functions of Lck in Cancer and Immunotherapy. Int J Mol Sci. 2019 Jul 16;20(14):3500. [CrossRef]
- Zhang, J.; Wu, Y.J.; Hu, X.X.; Wei, W. New insights into the Lck-NF-κB signaling pathway. Front Cell Dev Biol. 2023 Feb 24;11:1120747. [CrossRef]
- Cheng, Y.; Ji, C.; Xu, J.; Chen, R.; Guo, Y.; Bian, Q.; Shen, Z.; Zhang, B. LCK-SafeScreen-Model: An Advanced Ensemble Machine Learning Approach for Estimating the Binding Affinity between Compounds and LCK Target. Molecules. 2023 Nov 1;28(21):7382. [CrossRef]
- Schindler, C.G.; Armbrust, T.; Gunawan, B.; Langer, C.; Füzesi, L.; Ramadori, G. Gastrointestinal stromal tumor (GIST) -- single center experience of prolonged treatment with imatinib. Z Gastroenterol. 2005 Mar;43(3):267-73. [CrossRef]
- Schlemmer, M.; Bauer, S.; Schütte, R.; Hartmann, J.T.; Bokemeyer, C.; Hosius, C, Reichardt, P. Activity and side effects of imatinib in patients with gastrointestinal stromal tumors: data from a German multicenter trial. Eur J Med Res. 2011 May 12;16(5):206-12. [CrossRef]
- Lam, T.J.R.; Udonwa, S.A.; Masuda, Y.; Yeo, M.H.X.; Farid Bin Harunal Ras, M., Goh, B.K.P. A systematic review and meta-analysis of neoa;djuvant imatinib use in locally advanced and metastatic gastrointestinal stromal tumors. World J Surg. 2024 May 17. [CrossRef]
- Karim, N.A.; Ullah, A.; Wang, H.; Shoukier, M.; Pulliam, S.; Khaled, A.; Patel, N.; Morris, J.C. A Phase I Study of the Non-Receptor Kinase Inhibitor Bosutinib in Combination with Pemetrexed in Patients with Selected Metastatic Solid Tumors. Curr Oncol. 2022 Dec 3;29(12):9461-9473. [CrossRef]
- Deplanque, G.; Demarchi, M.; Hebbar, M.; Flynn, P.; Melichar, B.; Atkins, J.; Nowara, E.; Moyé, L. et al. A randomized, placebo-controlled phase III trial of masitinib plus gemcitabine in the treatment of advanced pancreatic cancer. Ann Oncol. 2015 Jun;26(6):1194-1200. [CrossRef]
- Adenis, A.; Blay, J.Y.; Bui;Nguyen, B.; Bouché, O.; Bertucci, F.; et al.. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: a randomized controlled open-label trial. Ann Oncol. 2014 Sep;25(9):1762-1769. [CrossRef]
- Le Cesne, A.; Blay, J.Y.; Bui, B.N.; Bouché, O.; Adenis, A. et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur J Cancer. 2010 May;46(8):1344-51. [CrossRef]
- Larkin, J.; Marais, R.; Porta, N.; Gonzalez de Castro, D.; Parsons, L.; Messiou, C.; et al. Nilotinib in KIT-driven advanced melanoma: Results from the phase II single-arm NICAM trial. Cell Rep Med. 2024 Mar 19;5(3):101435. [CrossRef]
- Mishra R. Oral tumor heterogeneity, its implications for patient monitoring and designing anti-cancer strategies. Pathology, research and practice, 2024; 253, 154953. [CrossRef]
- Barnwal, A.; Das, S.; Bhattacharyya, J. Repurposing Ponatinib as a PD-L1 Inhibitor Revealed by Drug Repurposing Screening and Validation by In Vitro and In Vivo Experiments. ACS Pharmacol Transl Sci. 2023 Jan 12;6(2):281-289. [CrossRef]
- Li, L.; Cui, Y.; Shen, J.; Dobson, H.; Sun, G. Evidence for activated Lck protein tyrosine kinase as the driver of proliferation in acute myeloid leukemia cell, CTV-1. Leuk Res. 2019 Mar;78:12-20. [CrossRef]
- Zepecki, J.P.; Snyder, K.M.; Moreno, M.M.; Fajardo, E.; Fiser, A.; Ness, J.; Sarkar, A.; Toms, S.A.; Tapinos, N. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene. 2019 Mar;38(10):1734-1750. [CrossRef]
- Harada, D.; Isozaki, H.; Kozuki, T.; Yokoyama, T.; Yoshioka, H.; Bessho, A. et al. Crizotinib for recurring non-small-cell lung cancer with EML4-ALK fusion genes previously treated with alectinib: A phase II trial. Thorac Cancer. 2021 Mar;12(5):643-649. [CrossRef]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M. , et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012 Oct;13(10):1011-9. [CrossRef]
- Cruz, B.D.; Barbosa, M.M.; Torres, L.L.; Azevedo, P.S.; Silva, V.E.A.; Godman, B.; Alvares-Teodoro, J. Crizotinib Versus Conventional Chemotherapy in First-Line Treatment for ALK-Positive Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Oncol Ther. 2021 Dec;9(2):505-524. [CrossRef]
- Tanaka, A.; Nishikawa, H.; Noguchi, S.; Sugiyama, D.; Morikawa, H.; Takeuchi, Y. et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J Exp Med. 2020 Feb 3;217(2):e20191009. [CrossRef]
- Burchat, A.; Borhani, D.W.; Calderwood, D.J.; Hirst, G.C.; Li, B.; Stachlewitz, R.F. Discovery of A-770041, a src-family selective orally active lck inhibitor that prevents organ allograft rejection. Bioorg Med Chem Lett. 2006 Jan 1;16(1):118-22. [CrossRef]
- Kumar Singh, P.; Kashyap, A.; Silakari, O. Exploration of the therapeutic aspects of Lck: A kinase target in inflammatory mediated pathological conditions. Biomed Pharmacother. 2018 Dec;108:1565-1571. [CrossRef]
- Kagawa, K.; Sato, S.; Koyama, K.; Imakura, T.; Murakami, K.; Yamashita, Y.; Naito, N.; Ogawa, H.; Kawano, H.; Nishioka, Y. The lymphocyte-specific protein tyrosine kinase-specific inhibitor A-770041 attenuates lung fibrosis via the suppression of TGF-β production in regulatory T-cells. PLoS One. 2022 Oct 27;17(10):e0275987. [CrossRef]
- Alqarni, S.A.; Bineid, A.; Ahmad, S.F.; Al-Harbi, N.O.; Alqahtani, F.; Ibrahim, K.E.; Ali, N.; Nadeem, A. Blockade of Tyrosine Kinase, LCK Leads to Reduction in Airway Inflammation through Regulation of Pulmonary Th2/Treg Balance and Oxidative Stress in Cockroach Extract-Induced Mouse Model of Allergic Asthma. Metabolites. 2022 Aug 25;12(9):793. [CrossRef]
- Carter, N.M.; Pomerantz, J.L. Calcineurin inhibitors target Lck activation in graft-versus-host disease. J Clin Invest. 2021 Jun 1;131(11):e149934. [CrossRef]
- Olivieri, A.; Mancini, G.; Olivieri, J.; Marinelli Busilacchi, E.; Cimminiello, M. et al. Nilotinib in steroid-refractory cGVHD: prospective parallel evaluation of response, according to NIH criteria and exploratory response criteria (GITMO criteria). Bone Marrow Transplant. 2020 Nov;55(11):2077-2086. [CrossRef]
- Srour, M.; Alsuliman, T.; Labreuche, J.; Bulabois, C.E.; Chevallier, P. et al. Nilotinib efficacy and safety as salvage treatment following imatinib intolerance and/or inefficacy in steroid refractory chronic graft-versus-host-disease (SR-cGVHD): a prospective, multicenter, phase II study on behalf of the Francophone Society of Bone Marrow Transplantation and Cellular Therapy (SFGM-TC). Bone Marrow Transplant. 2023 Apr;58(4):401-406. [CrossRef]
- Lin, C.T.; Hsueh, P.R.; Wu, S.J.; Yao, M.; Ko, B.S.; Li, C.C.; Hsu, C.A.; Tang, J.L.; Tien, H.F. Repurposing Nilotinib for Cytomegalovirus Infection Prophylaxis after Allogeneic Hematopoietic Stem Cell Transplantation: A Single-Arm, Phase II Trial. Biol Blood Marrow Transplant. 2018 Nov;24(11):2310-2315. [CrossRef]
Figure 1.
A general scheme of Lck with the molecule's different domains and critical parts. The key points are the proline SH domains, the proline motif, the phosphorylation sites (S for serine and Y for tyrosine), and the link to the membrane (myristylation or palmitoylation). In the closed state of the enzyme, it refers to phosphorylated at position Y505. Image generated using Bio Render software.
Figure 1.
A general scheme of Lck with the molecule's different domains and critical parts. The key points are the proline SH domains, the proline motif, the phosphorylation sites (S for serine and Y for tyrosine), and the link to the membrane (myristylation or palmitoylation). In the closed state of the enzyme, it refers to phosphorylated at position Y505. Image generated using Bio Render software.
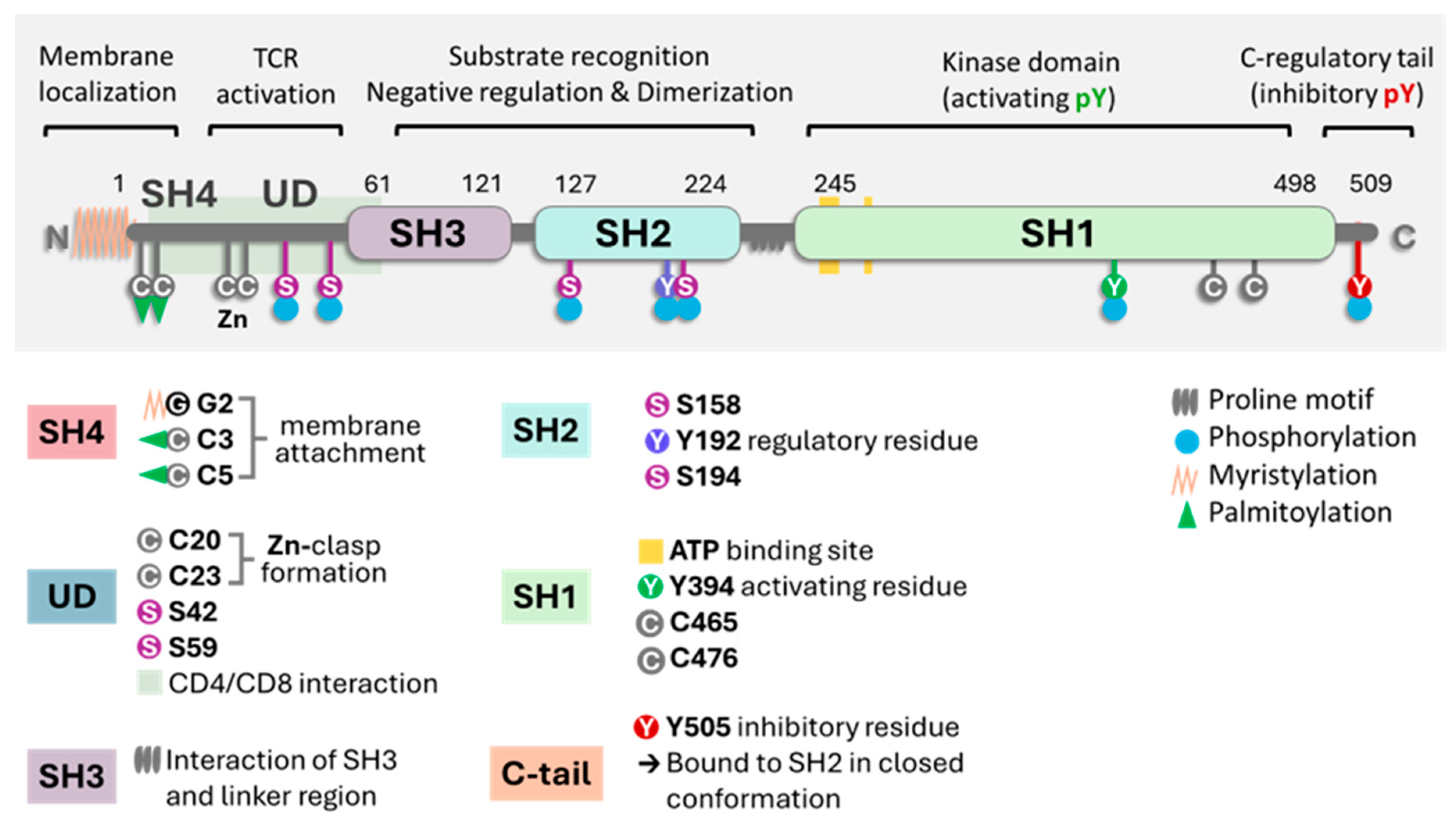
Figure 4.
The figure illustrates the importance of the adaptor proteins on Csk anchoring and Lck interaction with Fyn and other kinase and Csk-induced closure of the Lck open structure. The effect of the interaction of Csk linked by phosphorylated residue on the SH2 domain of PAG or LIME generated two different physiological responses. The figure was made using Bio Render software.
Figure 4.
The figure illustrates the importance of the adaptor proteins on Csk anchoring and Lck interaction with Fyn and other kinase and Csk-induced closure of the Lck open structure. The effect of the interaction of Csk linked by phosphorylated residue on the SH2 domain of PAG or LIME generated two different physiological responses. The figure was made using Bio Render software.
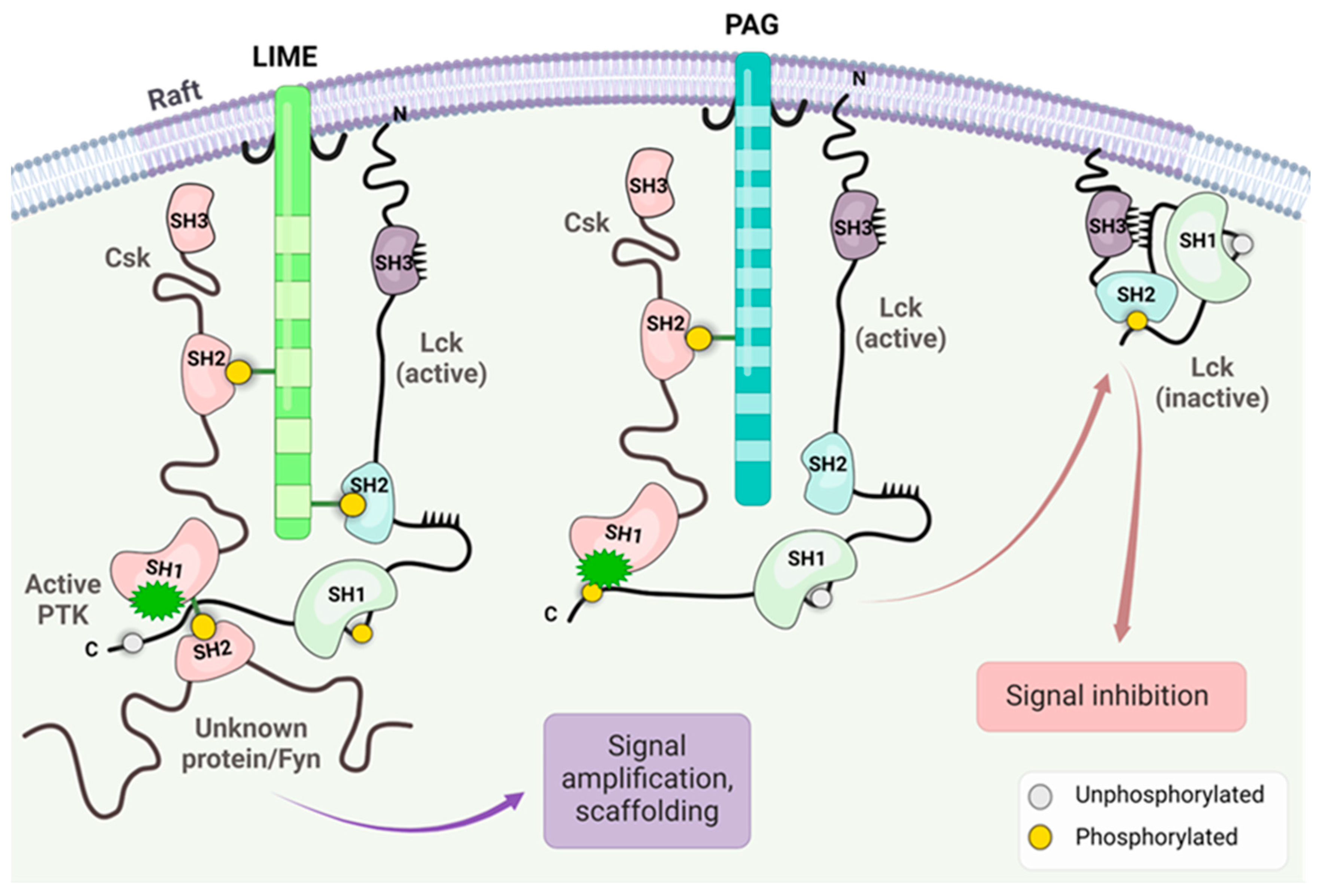
Figure 5.
Signal transduction induced in CD8 upon antigen binding. Part A of the figure illustrates the response induced by a low antigen level compared to a normal level in part B of the Figure. The critical element is Lck involvement in TCR γ chains, the phosphorylation of ZAP70, and the phosphorylation of the adaptor protein LAT, activating signaling pathways downstream.
Figure 5.
Signal transduction induced in CD8 upon antigen binding. Part A of the figure illustrates the response induced by a low antigen level compared to a normal level in part B of the Figure. The critical element is Lck involvement in TCR γ chains, the phosphorylation of ZAP70, and the phosphorylation of the adaptor protein LAT, activating signaling pathways downstream.
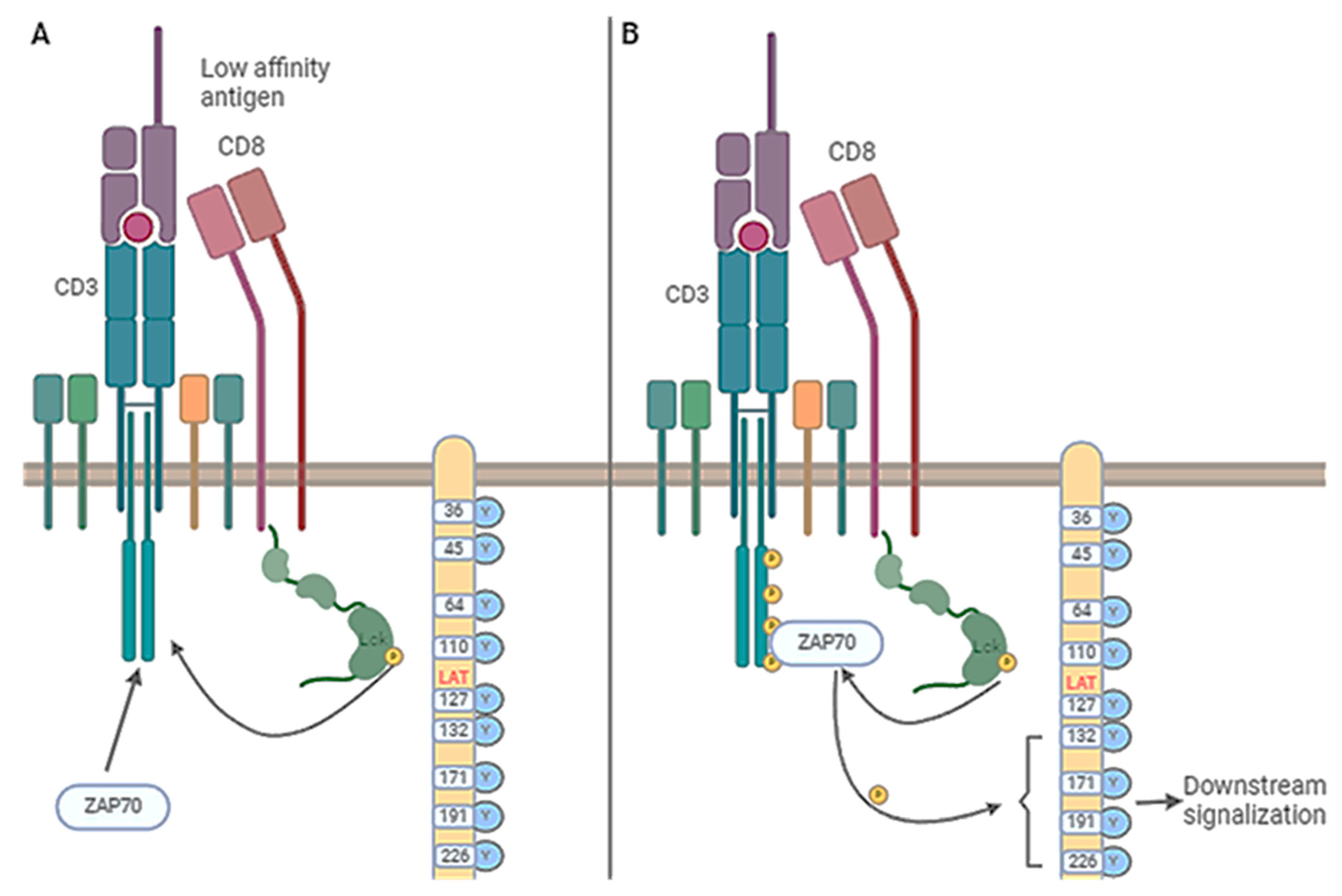
Figure 6.
The figure depicts the interaction between the killing receptor NKG2D (CD314) and Lck and Fyn. The interaction is dependent on DAP10 and DOC8. CD314 is expressed in NK and CD8 cells. Phosphorylation of Lck by the WASP protein prevents the cytotoxic response of NK and CD8 cells [45].
Figure 6.
The figure depicts the interaction between the killing receptor NKG2D (CD314) and Lck and Fyn. The interaction is dependent on DAP10 and DOC8. CD314 is expressed in NK and CD8 cells. Phosphorylation of Lck by the WASP protein prevents the cytotoxic response of NK and CD8 cells [45].
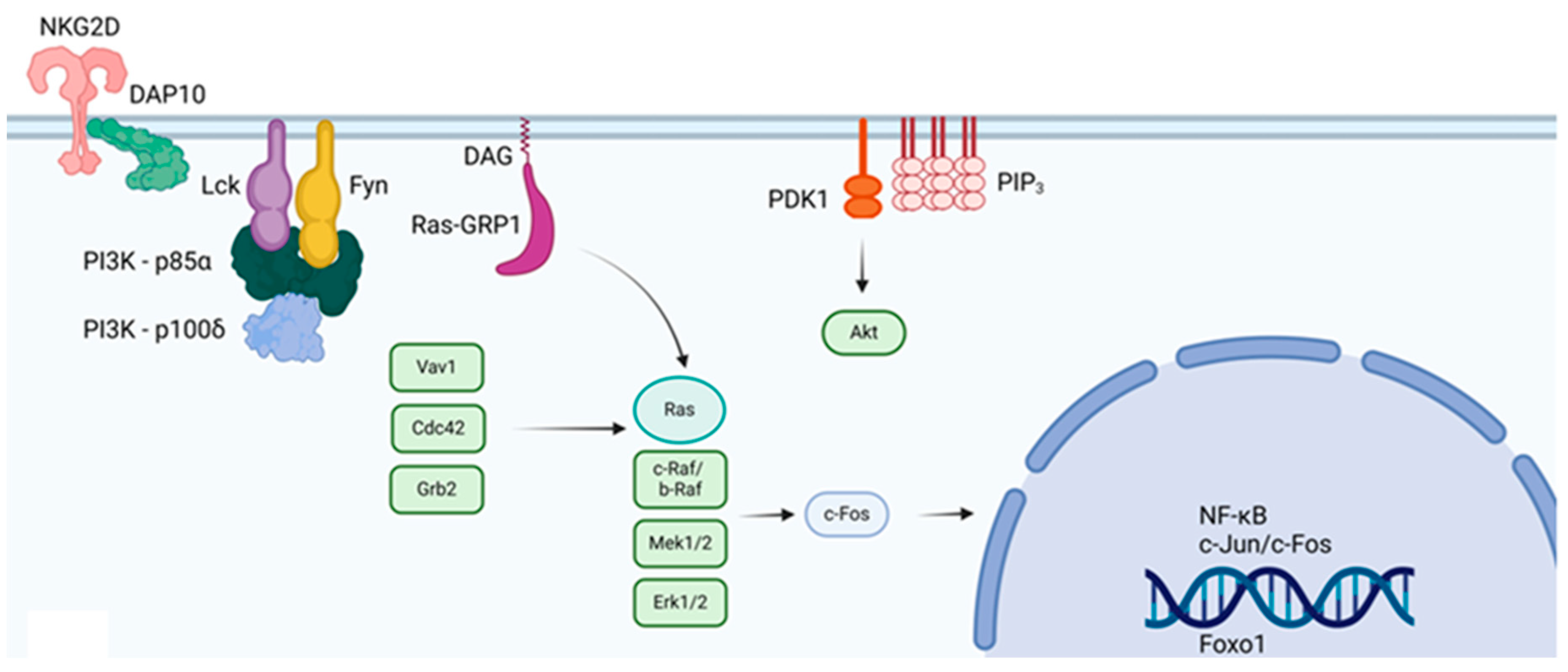
Figure 7.
Role of the Inhibitor Receptors on Lck activation. This figure illustrates two different processes. On the left, SHP-2 activation induced by PD-1/PDL-1/2 ligation prevents the phosphorylation of Lck and, consequently, the signal generated by TCR/ZAP70/LAT phosphorylation. On the right side, the figure illustrates the effect of LAG-3, the intracellular part of the protein that competes for Zn and blocks the interaction of the Zn domain on LcK and CD4/CD8 receptors, not allowing the enzyme to be activated. Inhibition of PD1/PDL1/PDL2 and LAG-3 (checkpoint inhibitors) impedes the inhibition of Lck and restores the signal transduction required for a proper T-cell response.
Figure 7.
Role of the Inhibitor Receptors on Lck activation. This figure illustrates two different processes. On the left, SHP-2 activation induced by PD-1/PDL-1/2 ligation prevents the phosphorylation of Lck and, consequently, the signal generated by TCR/ZAP70/LAT phosphorylation. On the right side, the figure illustrates the effect of LAG-3, the intracellular part of the protein that competes for Zn and blocks the interaction of the Zn domain on LcK and CD4/CD8 receptors, not allowing the enzyme to be activated. Inhibition of PD1/PDL1/PDL2 and LAG-3 (checkpoint inhibitors) impedes the inhibition of Lck and restores the signal transduction required for a proper T-cell response.
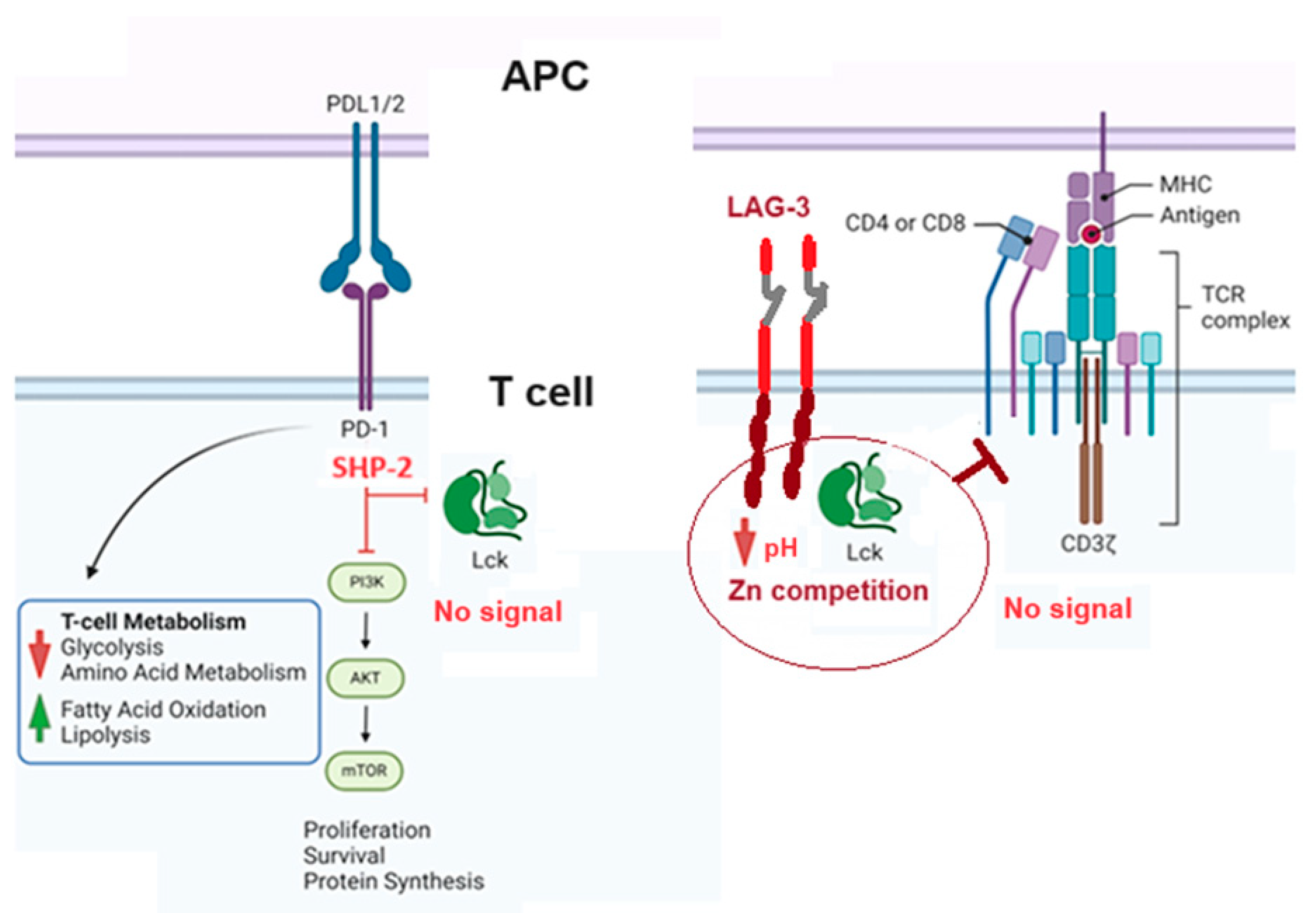
Figure 8.
The figure illustrates the interaction between TRPM8, a calcium channel, and the activity of Lck. Lck phosphorylates the Y1022 residue, which, in the presence of 14-3-3ζ, forms a multimeric complex in pancreatic and mammary tumor cells. Protein tyrosine kinase inhibitors, saracatinib, and dasatinib can block this event. Pre-treatment with PTK inhibitors generates susceptibility of the tumor to cisplatin. Tumors that express Lck can be targeted by the PTK inhibitors, enhancing tumor susceptibility to standard treatment. The figure was modified from Huang Y et al. [74]. The Bio Render software was used to make the figure.
Figure 8.
The figure illustrates the interaction between TRPM8, a calcium channel, and the activity of Lck. Lck phosphorylates the Y1022 residue, which, in the presence of 14-3-3ζ, forms a multimeric complex in pancreatic and mammary tumor cells. Protein tyrosine kinase inhibitors, saracatinib, and dasatinib can block this event. Pre-treatment with PTK inhibitors generates susceptibility of the tumor to cisplatin. Tumors that express Lck can be targeted by the PTK inhibitors, enhancing tumor susceptibility to standard treatment. The figure was modified from Huang Y et al. [74]. The Bio Render software was used to make the figure.
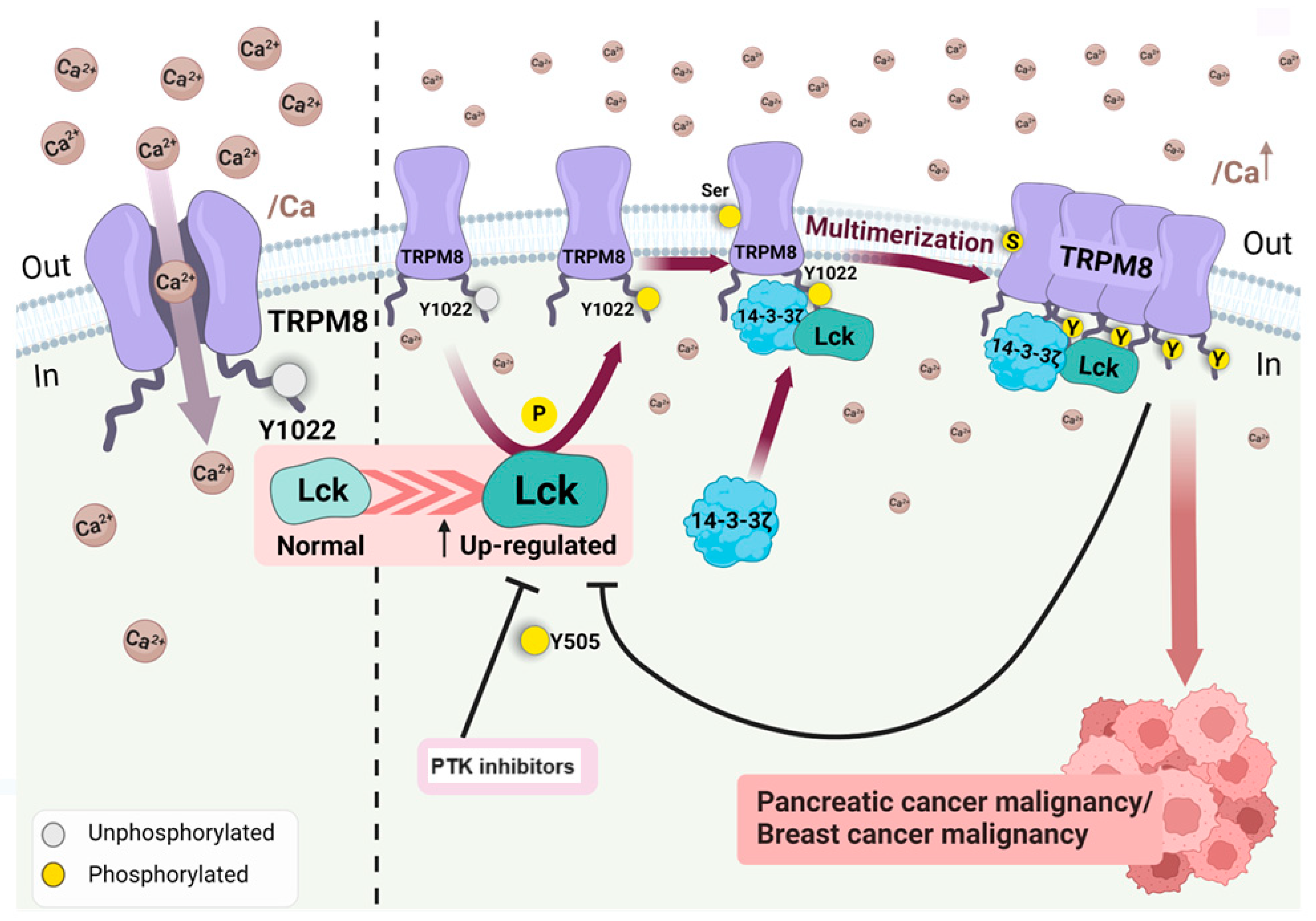
Figure 9.
Represents an overview of compounds and events that affect Lck activity.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



