
Submitted:
03 July 2024
Posted:
05 July 2024
You are already at the latest version
Abstract
Most cervical cancer cases are a result of persistent high-risk Human papillomavirus (HPV) infections. Cervical cancer is prevalent in LMICs especially in SSA where it is second most prevalent cancer. In South Africa, it is the second most common cancer affecting women aged between 15 and 44 years. The host immune response has been shown as an important factor in controlling the progression or regression of high-risk HPV infection of the cervix to cervical cancer. The risk of cervical cancer is known to be influenced by the host's genetic diversity, particularly by immune response-regulating genes such as the Human Leukocyte Antigen (HLA) class I and II genes. HLA class I genes present viral peptides to CD8+ T cells which are restricted and pre-programmed for cytotoxic functions. There is very little known about the HLA genes of South African women and how these influence outcome of HPV infections. This review aims to understand the role and influence of HLA class I genes in HPV clearance, persistence, and CD8+ T cell mediated immunity in African women by examining existing literature in this area. Understanding this role can influence and inform therapeutic vaccine design that is population specific.
Keywords:
HPV
; MHC
; HLA Class 1 genes
; cervical cancer
; review.
1. Introduction
Cervical cancer is a major cause of global morbidity and mortality in women, especially in low- and middle-income countries (LMICs) [1,2]. More than 95% of cases of cervical cancer are caused by the high-risk human papillomavirus (HPV), however in majority of cases, HPV infection does not necessarily result in cervical cancer [3]. Cervical cancer from a persistent high-risk HPV infection is a multistep process with different stages of cervical lesions that ultimately progress to cancer if left untreated [4,5,6]. There were more than 650,000 reported diagnoses and 350 000 cervical cancer fatalities in 2022 globally (WHO). The type-specific prevalence of HPV and cervical cancer incidence differ per geographical region especially high-risk HPVs, for example approximately 85% of cervical cancer cases are reported in LMICs such as Sub-Saharan Africa (SSA) where it is the second most prevalent malignancy and leading cause of cancer-related deaths in females [7,8].
As an established causal agent for cervical cancer, HPV is the most common sexually transmitted infection (STI), and most sexually active individuals will acquire this virus at some point in their lives [9,10]. HPVs are DNA viruses with a circular genome of approximately 7.9-8Kbp that contains an early region, encoding the early viral proteins E1, E2, E4, E5, E6, E7, E8, and a late region, encoding L1 and L2 proteins, which are components of the viral capsid [11,12,13]. HPVs have a strict tropism for either cutaneous or mucosal surfaces, as they generally infect the epithelial cells, specifically the basal keratinocytes moreover, they have a strict tropism for either cutaneous or mucosal surfaces [14,15,16,17]. These can be found lining the epithelium of mucosal and genital surfaces [15]. HPVs are categorized as genotypes rather than serotypes because DNA sequence differences between HPV genomes determine whether an HPV has the potential to cause cancer [18,19]. They are classified as high- or low-risk types based on their carcinogenicity [20]. LR-HPVs particularly HPVs 6 and 11 are responsible for nearly all recurrent respiratory papillomas (RRPs) [21], a percentage of low-grade cervical intraepithelial neoplasia (CIN1), vaginal intraepithelial neoplasia (VIN1), and anal intraepithelial neoplasia (AIN1) as well as about 90% of genital warts [22]. HR-HPVs are strongly carcinogenic and linked to malignancies of the anus, vulva, vagina, penis, and cervix; with type 18 and 16 having the most carcinogenic potential [23,24]. This is signified by their ability to integrate well into the host’s cells [24,25,26].
Even though HPV infection can be prevented by vaccination, many HPV-infected women do not benefit from the current prophylactic vaccines especially those in LMICs [27]. In LMICs the vaccine is given only to girls prior sexual debut in the national school-based vaccine rollout, while in high-income countries have implemented gender-neutral vaccination across a wider age group at a cost [28,29,30]. Furthermore, these vaccines are the priciest ever made, making it challenging to implement them in developing nations, where more than 80% of serious illnesses linked to HPV are found [15,30]. Due to the late diagnosis of cervical cancer in underprivileged populations, prophylactic vaccines are unable to stop the development of the disease [31]. This highlights the urgent need for a therapeutic vaccine that safeguards both previously infected women and those who have been exposed to the virus. These vaccines can reduce the incidence of cervical cancer and stop the progression of HPV-related CIN to cervical cancer [32,33].
The great majority of HPV infections are cleared by the immune system over a period of months to years, hence cancer development is not always a result of HPV infection [3,34]. However, HPV infections persist in some women, possibly leading to the development of CIN [10,18]. An adequate and effective immune response encourages spontaneous clearance and elimination of the virus, while a compromised immune response often initiates the progression of the infection to higher grade resulting in cervical lesions [4,35]. Immune response facilitated by HPV- specific CD8+ T cells is known to help facilitate eradicating of HPV infected cells [36].
The host immune response particularly the Human Leukocyte Antigen (HLA) linked cell mediated immune response has an important role in the progression or regression of high- risk HPV infection of the cervix [37,38]. HLA genes are found and encoded for in the major histocompatibility complex (MHC) on the short arm of chromosome 6 (6p21,3) and contain 3,500–4,000 kb of DNA and has over 200 genes [11,39,40]. HLA genes belong to three subgroups: namely, class I (classical: A, B, C, and non-classical: E, F, and G), class II (DR, DQ, and DP), and class III (TNF-α, complement proteins) [41,42]. The HLA genes influence the host immune response by mediating antigen presentation and oversee the controlling of immunological reactions that discriminate between self- and non-self-antigens [39,40,42,43].
The changes in the peptide-binding cleft of the HLA molecule caused by these polymorphisms control how the antigens are presented to and bound to T-cell receptors [42,44,45]. Through mediating antigen presentation and cell-mediated immune responses, the HLA genes have an impact on both the innate and adaptive immune systems by making it easier to identify and eliminate virus-infected cells [46,47]. The HLA genes' high degree of polymorphism affects how T cell responses are activated [42]. Variations in antigen binding and altered immune responses to infections may result from polymorphisms in the HLA molecules in different people [46].
By choosing the peptides produced by the processing of foreign antigens and presenting them on the cell surface for T cell recognition, HLA encourages the activation of an efficient immune response against HPV infected cells [41]. The presentation of a tumour-associated antigen by HLA class I or II genes is necessary for T cell-specific anticancer responses [48]. In order to activate CD8+ and CD4+ T cells, respectively, antigen-presenting cells (APCs) expressing both HLA class I and II must first acquire viral peptides [49].
Genetic variation in African populations is the highest among all humans [50]. The genetic variation observed in Africa is broadly correlated with geography [51]. High genetic diversity is another characteristic of African people, which has received little research in the context of HLA genes and their role in HPV and cancer development [52,53]. In South Africa, most studies have been conducted on the relationship between HLA genes and HIV protection and susceptibility where HLA- B*08/08:01, B*18/18:01, B*45/45:01, B*51:01, and B*58:02 have been found to increase susceptibility to HIV infections and even progression to AIDS [54]. However, there is paucity of data on HLA genes and their associations with HPV persistence and HPV disease in African women [55]. Understanding the potential clinical utility of these genes requires expanding research on the oncological implications of HLAs to ethnically diverse groups, including African people.
Therefore, this review aimed to examine existing studies detailing the roles of HLA class I genes and CD8+ T cells and how these roles influence HPV persistence and disease progression in African women.
2. Materials and Methods
2.1. Search Strategy
We searched PubMed, Science direct, and Web of Science, and Google scholar between July 2023 and November 2023 to identify published articles that focused on HPV infections, HPV progression and HLA genes in African women. The literature search was limited to articles published in English. We also conducted a hand search on papers that were specific for each subheading that met the search criteria but did not come up in the search results. The search strategy contained Boolean terms (AND, OR) and other terms of interest such as “Human Papillomavirus” or “HPV” and “HLA” or “MHC” class I genes, and African women. Synonyms for the search terms were used in the search to not exclude important papers. We did not specify by HLA gene class at the start of the search, we however only chose studies that evaluated HLA class I genes. The chosen articles' bibliographies and reference lists were examined for possible links to further relevant research that should be reviewed. From the included studies, abstracts and full-text articles were screened by the reviewer and saved onto a review library on Endnote reference manager version 21 (Clarivate Analytics, Philadelphia, PA, USA). OM independently extracted key study information from the relevant search engines.
2.2. Selection Criteria
Studies were eligible for inclusion if they were conducted in sexually active African women above the age of 18 residing in African countries and infected with HPV. We did not include studies with African women residing outside of Africa and those that are below 18 years of age and not sexually active. Studies that were excluded include those that were 1) not peer-reviewed; 2) not in English; 3) studying HLA class II genes; 4) without African participants in an African setting/ country; 5) those not conducted on women.
There was no date restriction on the publication of the articles chosen. Eligibility was determined independently by one author (OM) through a review of the title and abstract followed by a full-text review. Discrepancies between reviewers were resolved through consensus and involving the review collaborator.
The protocol was registered with PROSPERO before conducting the search under registration number: CRD42023440172.
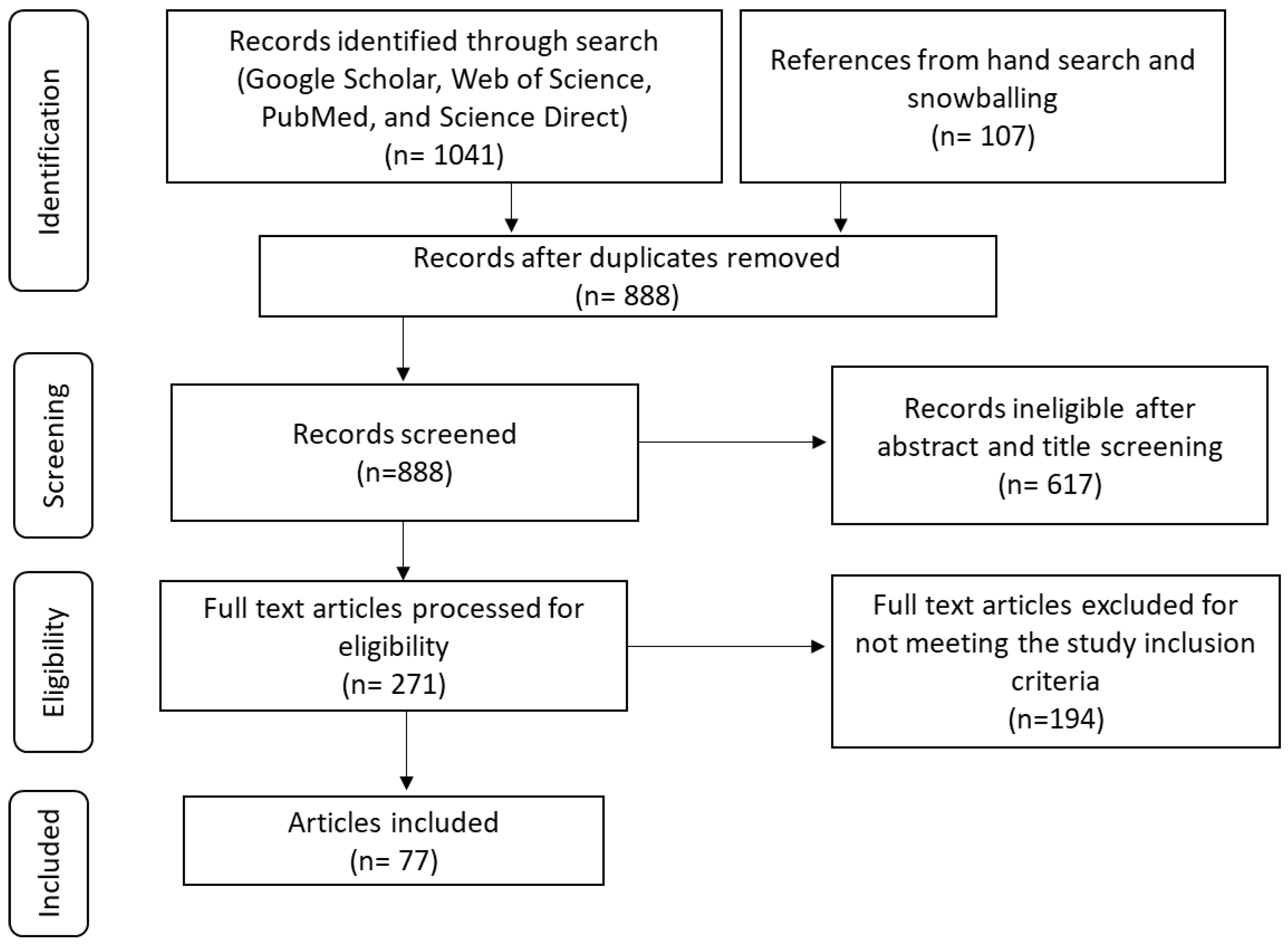
Figure 1.
PRISMA flow diagram during search and selection process.
2.3. Data Extraction
Core study information including title, authors, journal, year published and DOI was extracted onto an Excel spreadsheet. Methodological information was also extracted, including the country where the study was conducted, the aims or objectives, tests used during the study, and demographical participant data such as the age of participants and health status. Result information was also extracted from the selected articles.
3. Results
A total of 1148 articles were identified to be eligible for selection of these 260 were duplicates. After title and abstract screening, 617 articles were excluded, and 271 articles were included for full-text review. 194 full-text articles were excluded for not meeting the inclusion criteria, while only 77 articles were eligible for inclusion in the review (Figure 1). All the studies included in the review were restricted to those with female participants, examined HPV infections in African women and other countries for comparison, adaptive immune responses to HPV infections, and the HLA genes associated with HPV infections. Only two studies with approximately 583 female participants specifically evaluated the role of HLA class 1 genes in African women. Most of the studies evaluated HPV infections generally including the HPV life cycle, immune responses to HPV and HLA gene associations with HPV in women from countries other than Africa which was also of interest.
4. Discussion
This review aimed to examine existing studies detailing the known and studied roles of HLA class I genes and CD8+ T cells and how these roles influence HPV persistence and disease progression in African women.
Life cycle of HPV in the cervical epithelium
The lifecycle discussed in this section is with reference to HPV 16 as it is one of the most researched high-risk HPV types, however other high-risk types are also linked to oncogenesis. Infection generally starts when the virus releases viral particles which enter the keratinocytes through microtrauma or abrasion [56]. This is where the viral particles bind to the cellular receptors Heparan sulphate proteoglycans (HSPG) specifically Syndecan-1; and a secondary unknown receptor located on the basement membrane or on the surface of basal layer cells prior to transfer to the basal keratinocyte cell surface [18,57,58,59,60,61]. These receptors can help HPV16 in binding to the extracellular matrix [60]. The virus enters the cells in low viral copy number to not induce viremia and immune response during the non-productive stage and express the viral early genes [34,62]. This process is thought to be mediated by the E1 and E2 HPV oncoproteins [63]. Subsequent to this, the viral DNA enters the productive stage where the DNA is replicated and the viral copy number is amplified to around 50–100 copies per cell; this appears to be independent of the cell cycle [64]. To do this, the machinery responsible for synthesising DNA within the cell must be activated. This is done by the actions of E6 and E7, which permits the amplification of the viral genome in cells that would have otherwise completed the cell cycle [62]. Later, when the basal cells differentiate to form the epithelial suprabasal layer, viral genome replication switches into high copy number mode [65,66]. Then, the virions get released upon epithelial desquamation, causing infection in the neighbouring cells [66].
The expression of viral genes is low during this period of plasmid or episomal maintenance, and in particular, the expression of the oncogenes E6 and E7 is tightly regulated, with hardly any detectable E6/E7 mRNA transcripts [64]. This is important because these oncoproteins are expressed in the later phase of the lifecycle and are thought to be useful in evading immune responses such as apoptosis and programmed cell death activities [66]. The expression of these two genes allows for cell cycle re-entry upon differentiation, providing cellular factors for productive replication [62]. HPV genome can either get integrated with the host genome or stay in an episomal form, with 83% of the HPV-positive cervical cancer cases showing evidence of HPV genome integration into the host cell [66,67]. It is therefore believed that the infected cell exits this early compartment resembling a stem cell and enters the epithelium's proliferating compartment [56]. All viral genes are massively upregulated in expression when the infected keratinocyte enters the differentiating compartment and exits the cell cycle, with early genes E6 and E7 being highly expressed [13]. It is believed that HPV can also infect cells at the squamocolumnar junction, the epithelial reserve cells, and columnar epithelial cells at the cervical transformation zone and endocervix [5,17].
Life cycle deregulation, virus persistence and disease progression
HPV infections usually clear in one to two weeks, but in some people, a lingering infection or a lack of immune clearance can cause the viral life cycle to be disrupted and the viral genome to integrate, which increases the risk of the development of cancer and malignancy [5,18]. It is believed that unregulated long-term or the overexpression of E6 and E7 promotes disease persistence and progression [68,69,70]. The expression of E5 may prevent infected cells from presenting to HLA class I genes, which could allow the infected cells to evade immune surveillance [71]. E5 prevents transport of the HLA class I to the cell surface and retains the complex in the GA [12,72]. Campo et al., 2010 [73], noted that HPV E5 downregulation of HLA class I molecules at the cell surface correlates with poor CD8+T cell responses in E5 expressing cells. Presentation of HPV peptide fragments through the HLA genes is important in mounting an effective cellular mediated immune response against infected cells. These genes as mentioned above control the immune response against viral infections. It has been demonstrated that E7 directly inhibits the HLA class 1 chain [48]. As a result, HLA class I expression is suppressed, which prevents infected cells from being effectively presented to CD8+ T cells [71,74]. By suppressing the expression of the antigen processing machinery components Low molecular weight Protein 2 (LMP2) and Transporter associated with antigen processing 1 (TAP1), E7 is also known to hinder peptide formation and transportation in infected cells [71,75]. This impairs the delivery of HPV peptide fragments to the ER surface-bound HLA class I. TAP1 plays a critical role in presenting HLA class I with the viral antigen and in the presentation of MHC class I antigens, CD8+ and gives HPV a tremendous evasion strategy against the human cellular defence [76].
CIN grades 1 through 3 are usually assumed to be associated with higher levels of E6 and E7 expression, and the neoplastic phenotype is directly caused by these alterations in gene expression [66,77]. In this way, CIN1 lesions can resemble flat warts, which have less cell proliferation in the basal and parabasal layers, and they usually retain the ability to finish the HPV life cycle and create virus particles [17]. When a high-risk HPV infection results in CIN2+ and elevated E6 and E7 expression, the cell is more vulnerable to the accumulation of genetic alterations that progressively aid in the development of cancer [78,79]. It is also believed that the viral deregulation observed in CIN2/3+ promotes the integration of the viral episome into the host cell chromosome, hence further deregulating E6 and E7 expression [67,78]. Cervical cancer contains one or more copies of HPV that are largely integrated randomly into the host chromosome [67]. The viral integration site is often located inside the regulatory E1 or E2 genes [80]. The loss of E6/E7 regulation and integration can promote the development of genetic mistakes that eventually result in cancer as well as the continuous high-level expression of these genes [6]. Cancer can arise because of persistently dysregulated gene expression, as seen in CIN3 and after viral genome integration, which can accumulate secondary genetic alterations in the infected host cell [15,78].
Cell mediated immune responses to HPV infection.
It is important to note that even if most cervical cancer cases are a result of persistent HPV infection, about 80-90% of HPV infections clear spontaneously over time [57]. This clearance can take weeks to months to complete [4,18,81]. As mentioned above, HPV clearance is brought about by a robust immune response whether innate or adaptive [25,56,82]. However complete clearance is possible if an individual is not immunocompromised, which can lead to an undeveloped immune response to HPV [4,9,83]. When an individual first encounters the infection, the host innate immune response becomes the first line of defence against the infection [11,13,34]. HPV causes cell proliferation instead of lysis and does not cause systemic infection, in this way it can escape immune surveillance [25,80]. Clearance is also brought about by a robust CD8+ T cell response against the infection [82]. These cells mainly target infected cells which are presented by the HLA class I genes [84]. CD8+ T cells generally kill these cells using either one of two ways or mechanisms [84]. These are Fas ligand (FasL)-mediated apoptosis induction and granule exocytosis [85]. CTLs also release interferon-γ (IFN-γ) and tumour necrosis factor α (TNF-α) to induce cytotoxicity in the cancer cells [86]. During granule exocytosis, CD8+ T cells release granzymes A and B [87]. Before this, the cells that encounter infected cells produce perforin to make pores in their plasma membrane acting as a path for release of the granzymes to the infected cells [88,89]. The exact mechanisms that CD8+ T cells use to target HPV infected cells and the mediator in this apoptosis is still largely unknown, however research conducted has concluded that most CTL immune responses are usually directed to HPV16 E6 and E7 proteins [90,91,92,93]. From these studies, it can be concluded that persistence is usually brought by a lack of effective CTL response against the E6 protein [48,70,93].
Some studies have found the expression of these cells in HPV infected people; however, it is also unknown if whether these expressed CD8+ T cells can function properly [36,92,94]. Furthermore, factors such as HIV infection which is particularly high in Africa and a strong cofactor for high HPV and cervical cases in SA and SSA can affect the expression CD8+ T cells and disease clearance [95,96,97]. In a similar study by Nakagawa and colleagues [93], they found that HPV persistence is usually brought by a lack of CTL immune response to HPV16 E6 proteins while clearance was seen in patients who had a positive CTL response against the E7 protein. Nevertheless, at least one study using a highly sensitive method detected an HPV-16 E6-/E7-specific memory cytotoxic-T-cell response in all cervical cancer patients [92]. While a T-cell response was only found in half of the patients in a separate study that examined E2-specific T-cell responses in people with low-grade squamous intraepithelial lesions, the presence of an E2-specific T-cell response was associated with the lack of progression that is, regression of the disease [94]. The presence of response was however not correlated with a regression of CIN 2 in most patients that were infected with HPV 16 and this study did not specify whether this response was CD4 T cell or CD8+ T cell specific [94]. Another study where they examined the level of T cell reactivity in people with different stages of HPV infection, found that CD4 T cell responses were more associated with disease clearance when compared to CD8+ T cell responses [98].
HLA class I genes and their role in an HPV infection
HLA class I genes present viral peptides to CD8+ T cells which are restricted and pre-programmed for cytotoxic functions [41,99,100]. Cytotoxic T lymphocytes (CTLs) or CD8+ T cells recognize intracellularly produced viral peptides in the presence of HLA-1 molecules, which result in the destruction of virally infected cells [48,99,100]. Highly effective HPV viral peptide presentation to CD8+ T cells can lead to the mounting of a competent cellular immune response against HPV infections and the cytotoxic clearance of HPV infected cells [101]. Cytotoxic T cells expressing cell-surface CD8 are the most powerful effectors in the anticancer immune response and form the backbone of current successful cancer immunotherapies [82,102]. The CTL response is triggered by endogenous antigens of transformed self-cells presented in the peptide-binding cleft [11]. All nucleated cells have HLA class I genes expressed on their surface and depending on the tissue chosen, these genes exhibit different levels of expression [11,103]. Most research has focused on the HLA class II genes in HPV while little research has been conducted on HLA class I genes and HPV pathogenesis especially in African women in Africa [41,104,105].
The expression or downregulation of these HLA class I genes occurs at various stages of the HPV life cycle [48,72,73]. Both during its trafficking to the cell surface and at the transcriptional level, HLA class I expression is compromised, this expression is mainly due to the processes of the viruses’ oncoproteins [12,81,106]. In a study by Cicchini et al., 2017 [107], they found that HR-HPV E7 expression downregulated the expression of HLA class I genes. HPV proteins also downregulate cell-surface expression of HLA-A and -B, which present viral peptides to cytotoxic T lymphocytes, but does not downregulate the natural killer (NK) inhibitory ligands, which present to all the HLA class 1 genes including HLA-C, -E, and -G [108]. Some studies have reported the correlation of certain HLA class I alleles with persistent infection and cervical cancer. HLA-A*11; A*01; HLA-B*27; -B*35; and -B*44 have all been reported to be associated with HPV-16-positive cervical cancer and persistent HPV-16 infection in European and American women [41,109]. Currently there are two studies that has investigated the distribution of HLA genes and their association with persistent cervical HR-HPV infection in Sub-Saharan Africa [110,111]. In a study conducted on Nigerian women; HLA-A*23:01, HLA-B*53:01, and HLA-C*04:01 were found to be associated with persistent high risk HPV infections; however, it is important to note that this may not be the same in all African women as HLA genes differ significantly due to ancestry and environmental/ geographical conditions [110]. For example; in an earlier study conducted in a group of Kenyan women, it was found that HLA B*35 was associated with increased risk of cervical neoplasia of any grade and/ or persistent HPV infection [111]. It is important to note that the above-mentioned studies are the only of their kind that have been conducted in Africa, this means that in other African countries including South Africa, there is no knowledge of the association and effect of HLA genes (particularly HLA class I) on HPV persistence and cervical cancer. In terms of other HLA class I genes; HLA-E polymorphism has been reported not to have an association with either risk of HPV acquisition or disease persistence [108], while HLA G (and its alleles) expression increases as cervical disease severity increases that is, it is highly expressed in cervical cancer patients than those with CIN 1, 2/ 3 [112].
Notably, epigenetic factors play a key role in gene expression and functionality. As such environmental and geographical conditions affect the expression of genes in certain individuals [113]. While epigenetic alterations serve as a natural mechanism for adaptation to environmental changes, the intricate regulation of gene expression that these modifications foster can have unfavourable effects on an organism, producing an opposite effect from what is anticipated [50]. This can lead to the accumulation of traits that reduce an organism's ability to adapt, which can ultimately result in diseases like cancer [113]. It is plausible that certain generic variants are relevant for some populations while having little or no impact in other populations. For example, HLA-A*02, HLA-A*0201, HLA-B*07, and others are reported to be associated with an increased risk of HPV infection and persistent HPV infections in European women, this was however not a similar case in the studies conducted in African women [110,111]. These factors may also affect the way that diseases manifest in certain communities and the susceptibility of a population to different diseases. In the case of HPV, epigenetic factors are also responsible for the prevalence of certain HPV types over others. For example, in a study by Kuguyo et al., 2021 [96] they found that while genotypes like HPV35 and 52 are said to be common in sub-Saharan African countries including South Africa [114], and Zimbabwe [115,116] while persistent HPV66 is supposedly widespread throughout Northern Africa.
5. Conclusions
Although cervical cancer is a preventable disease, women in LMICs, especially SSA still bear a disproportional burden of disease and mortalities. Research into the various risk factors linked to the risk of HPV-related disease can contribute to a decrease in the global number of affected women and fatalities. Despite their great effectiveness, the current HPV vaccinations do not offer protection against all high-risk HPV strains, let alone the low-risk strains. Moreover, these vaccinations are only preventative and difficult for women in LMIC to obtain there is low vaccine coverage in areas where the disease is prevalent. Little attention has been paid to understanding the influence of host genes in HPV infections particularly HLA class 1 genes which play a vital role in viral presentation to cytotoxic T cells and virus elimination especially in SSA as evidenced by only two studies conducted in African countries at present time. Research on the relationship between host genetics and HPV disease outcome, especially in Africa will bring light on how the immune system together with host genes is responsible in the clearance of HPV infections in women especially in low-income countries and will contribute to the development of therapeutic vaccines that can help reduce cervical cancer mortality and morbidity in women due to HPV-related cancers.
Author Contributions
Each author made an equal contribution to the work's conception, writing, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health 2020, 8, e191-e203. [CrossRef]
- Hull, R.; Mbele, M.; Makhafola, T.; Hicks, C.; Wang, S.M.; Reis, R.M.; Mehrotra, R.; Mkhize-Kwitshana, Z.; Kibiki, G.; Bates, D.O.; et al. Cervical cancer in low and middle-income countries. Oncol Lett 2020, 20, 2058-2074. [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J Obstet Gynaecol 2020, 40, 602-608. [CrossRef]
- Song, D.; Li, H.; Li, H.; Dai, J. Effect of human papillomavirus infection on the immune system and its role in the course of cervical cancer. Oncol Lett 2015, 10, 600-606. [CrossRef]
- Doorbar, J.; Griffin, H. Refining our understanding of cervical neoplasia and its cellular origins. Papillomavirus Research 2019, 7, 176-179. [CrossRef]
- Gao, G.; Smith, D.I. Human Papillomavirus and the Development of Different Cancers. Cytogenetic and Genome Research 2017, 150, 185-193. [CrossRef]
- Sharma, R.; Aashima; Nanda, M.; Fronterre, C.; Sewagudde, P.; Ssentongo, A.E.; Yenney, K.; Arhin, N.D.; Oh, J.; Amponsah-Manu, F.; et al. Mapping Cancer in Africa: A Comprehensive and Comparable Characterization of 34 Cancer Types Using Estimates From GLOBOCAN 2020. Frontiers in Public Health 2022, 10. [CrossRef]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front Public Health 2020, 8, 552028. [CrossRef]
- Zayats, R.; Murooka, T.T.; McKinnon, L.R. HPV and the Risk of HIV Acquisition in Women. Front Cell Infect Microbiol 2022, 12, 814948. [CrossRef]
- Evans, A.M.; Salnikov, M.; Tessier, T.M.; Mymryk, J.S. Reduced MHC Class I and II Expression in HPV−Negative vs. HPV−Positive Cervical Cancers. Cells 2022, 11, 3911.
- Paaso, A.; Jaakola, A.; Syrjänen, S.; Louvanto, K. From HPV Infection to Lesion Progression: The Role of HLA Alleles and Host Immunity. Acta Cytol 2019, 63, 148-158. [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int J Cancer 2006, 119, 2105-2112. [CrossRef]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of Innate Immunity against Human Papillomavirus (HPV) Infections and Effect of Adjuvants in Promoting Specific Immune Response. Viruses 2013, 5, 2624-2642.
- McBride, A.A. Oncogenic human papillomaviruses. Philos Trans R Soc Lond B Biol Sci 2017, 372. [CrossRef]
- Graham, Sheila V. The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clinical Science 2017, 131, 2201-2221. [CrossRef]
- Ashique, S.; Hussain, A.; Fatima, N.; Altamimi, M.A. HPV pathogenesis, various types of vaccines, safety concern, prophylactic and therapeutic applications to control cervical cancer, and future perspective. Virusdisease 2023, 34, 1-19. [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 Suppl 5, F55-70. [CrossRef]
- Stanley, M.A.; Pett, M.R.; Coleman, N. HPV: from infection to cancer. Biochem Soc Trans 2007, 35, 1456-1460. [CrossRef]
- Burk, R.D.; Chen, Z.; Van Doorslaer, K. Human papillomaviruses: genetic basis of carcinogenicity. Public Health Genomics 2009, 12, 281-290. [CrossRef]
- Stanley, M.A. Immunobiology of papillomavirus infections11Paper presented by invitation at the Second International Conference on Experimental and Clinical Reproductive Immunobiology, Amsterdam, The Netherlands, November 2000. Journal of Reproductive Immunology 2001, 52, 45-59. [CrossRef]
- Fusconi, M.; Grasso, M.; Greco, A.; Gallo, A.; Campo, F.; Remacle, M.; Turchetta, R.; Pagliuca, G.; M, D.E.V. Recurrent respiratory papillomatosis by HPV: review of the literature and update on the use of cidofovir. Acta Otorhinolaryngol Ital 2014, 34, 375-381.
- Ball, S.L.; Winder, D.M.; Vaughan, K.; Hanna, N.; Levy, J.; Sterling, J.C.; Stanley, M.A.; Goon, P.K. Analyses of human papillomavirus genotypes and viral loads in anogenital warts. J Med Virol 2011, 83, 1345-1350. [CrossRef]
- Williamson, A.-L. Recent Developments in Human Papillomavirus (HPV) Vaccinology. Viruses 2023, 15, 1440.
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Frontiers in Public Health 2021, 8. [CrossRef]
- Stanley, M.A.; Sterling, J.C. Host responses to infection with human papillomavirus. Curr Probl Dermatol 2014, 45, 58-74. [CrossRef]
- Ashique, S.; Hussain, A.; Fatima, N.; Altamimi, M.A. HPV pathogenesis, various types of vaccines, safety concern, prophylactic and therapeutic applications to control cervical cancer, and future perspective. VirusDisease 2023, 34, 172-190. [CrossRef]
- Ebrahimi, N.; Yousefi, Z.; Khosravi, G.; Malayeri, F.E.; Golabi, M.; Askarzadeh, M.; Shams, M.H.; Ghezelbash, B.; Eskandari, N. Human papillomavirus vaccination in low- and middle-income countries: progression, barriers, and future prospective. Front Immunol 2023, 14, 1150238. [CrossRef]
- Bardají, A.; Mindu, C.; Augusto, O.J.; Casellas, A.; Cambaco, O.; Simbine, E.; Matsinhe, G.; Macete, E.; Menéndez, C.; Sevene, E.; et al. Awareness of cervical cancer and willingness to be vaccinated against human papillomavirus in Mozambican adolescent girls. Papillomavirus Res 2018, 5, 156-162. [CrossRef]
- Bruni, L.; Saura-Lázaro, A.; Montoliu, A.; Brotons, M.; Alemany, L.; Diallo, M.S.; Afsar, O.Z.; LaMontagne, D.S.; Mosina, L.; Contreras, M.; et al. HPV vaccination introduction worldwide and WHO and UNICEF estimates of national HPV immunization coverage 2010–2019. Preventive Medicine 2021, 144, 106399. [CrossRef]
- Asempah, E. HPV vaccine and cervical cancer policy and policymaking research interest in sub-Saharan Africa: A scoping review. Journal of Cancer Policy 2020, 26, 100258. [CrossRef]
- El-Zein, M.; Richardson, L.; Franco, E.L. Cervical cancer screening of HPV vaccinated populations: Cytology, molecular testing, both or none. J Clin Virol 2016, 76 Suppl 1, S62-s68. [CrossRef]
- Conageski, C. Human Papillomavirus Vaccines. Clin Obstet Gynecol 2023, 66, 433-447. [CrossRef]
- Cheng, L.; Wang, Y.; Du, J. Human Papillomavirus Vaccines: An Updated Review. Vaccines (Basel) 2020, 8. [CrossRef]
- Sasagawa, T.; Takagi, H.; Makinoda, S. Immune responses against human papillomavirus (HPV) infection and evasion of host defense in cervical cancer. J Infect Chemother 2012, 18, 807-815. [CrossRef]
- Bedoya, A.M.; Jaramillo, R.; Baena, A.; Castaño, J.; Olaya, N.; Zea, A.H.; Herrero, R.; Sanchez, G.I. Location and density of immune cells in precursor lesions and cervical cancer. Cancer Microenvironment 2013, 6, 69-77.
- Hewavisenti, R.V.; Arena, J.; Ahlenstiel, C.L.; Sasson, S.C. Human papillomavirus in the setting of immunodeficiency: Pathogenesis and the emergence of next-generation therapies to reduce the high associated cancer risk. Frontiers in Immunology 2023, 14. [CrossRef]
- Muntinga, C.L.P.; de Vos van Steenwijk, P.J.; Bekkers, R.L.M.; van Esch, E.M.G. Importance of the Immune Microenvironment in the Spontaneous Regression of Cervical Squamous Intraepithelial Lesions (cSIL) and Implications for Immunotherapy. J Clin Med 2022, 11. [CrossRef]
- Sheu, B.-C.; Chang, W.-C.; Lin, H.-H.; Chow, S.-N.; Huang, S.-C. Immune concept of human papillomaviruses and related antigens in local cancer milieu of human cervical neoplasia. Journal of Obstetrics and Gynaecology Research 2007, 33, 103-113. [CrossRef]
- Mosaad, Y.M. Clinical Role of Human Leukocyte Antigen in Health and Disease. Scandinavian Journal of Immunology 2015, 82, 283-306. [CrossRef]
- Janeway Jr, C.A. Immunobiology the immune system in health and disease. Artes Medicas 1997.
- Bhaskaran, M.; Murali, S.V.; Rajaram, B.; Krishnasamy, S.; Devasena, C.S.; Pathak, A.; Ravi, V.; Swaminathan, K.; Ayyappa, A.; Vedhantham, S.; et al. Association of HLA-A, -B, DRB, and DQB Alleles with Persistent HPV-16 Infection in Women from Tamil Nadu, India. Viral Immunol 2019, 32, 430-441. [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Frontiers in Immunology 2017, 8. [CrossRef]
- Holoshitz, J. The quest for better understanding of HLA-disease association: scenes from a road less travelled by. Discovery medicine 2013, 16, 93.
- Djaoud, Z.; Parham, P. HLAs, TCRs, and KIRs, a Triumvirate of Human Cell-Mediated Immunity. Annual Review of Biochemistry 2020, 89, 717-739. [CrossRef]
- Jiang, N.; Yu, Y.; Wu, D.; Wang, S.; Fang, Y.; Miao, H.; Ma, P.; Huang, H.; Zhang, M.; Zhang, Y.; et al. HLA and tumour immunology: immune escape, immunotherapy and immune-related adverse events. Journal of Cancer Research and Clinical Oncology 2023, 149, 737-747. [CrossRef]
- Espinoza, H.; Ha, K.T.; Pham, T.T.; Espinoza, J.L. Genetic Predisposition to Persistent Human Papillomavirus-Infection and Virus-Induced Cancers. Microorganisms 2021, 9, 2092.
- Xu, H.-H.; Yan, W.-H.; Lin, A. The Role of HLA-G in Human Papillomavirus Infections and Cervical Carcinogenesis. Frontiers in Immunology 2020, 11. [CrossRef]
- Gameiro, S.F.; Zhang, A.; Ghasemi, F.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection. Viruses 2017, 9. [CrossRef]
- Ekanayake Weeramange, C.; Shu, D.; Tang, K.D.; Batra, J.; Ladwa, R.; Kenny, L.; Vasani, S.; Frazer, I.H.; Dolcetti, R.; Ellis, J.J.; et al. Analysis of human leukocyte antigen associations in human papillomavirus–positive and –negative head and neck cancer: Comparison with cervical cancer. Cancer 2022, 128, 1937-1947. [CrossRef]
- Gomez, F.; Hirbo, J.; Tishkoff, S.A. Genetic variation and adaptation in Africa: implications for human evolution and disease. Cold Spring Harb Perspect Biol 2014, 6, a008524. [CrossRef]
- Fan, S.; Kelly, D.E.; Beltrame, M.H.; Hansen, M.E.B.; Mallick, S.; Ranciaro, A.; Hirbo, J.; Thompson, S.; Beggs, W.; Nyambo, T.; et al. African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biology 2019, 20, 82. [CrossRef]
- Pfennig, A.; Petersen, L.N.; Kachambwa, P.; Lachance, J. Evolutionary Genetics and Admixture in African Populations. Genome Biology and Evolution 2023, 15. [CrossRef]
- Adolf, I.C.; Almars, A.; Dharsee, N.; Mselle, T.; Akan, G.; Nguma, I.J.; Nateri, A.S.; Atalar, F. HLA-G and single nucleotide polymorphism (SNP) associations with cancer in African populations: Implications in personal medicine. Genes & Diseases 2022, 9, 1220-1233.
- Mellet, J.; Tshabalala, M.; Agbedare, O.; Meyer, P.W.A.; Gray, C.; Pepper, M. Human leukocyte antigen (HLA) diversity and clinical applications in South Africa. South African Medical Journal 2019, 109, 29. [CrossRef]
- Adebamowo, S.N.; Adeyemo, A.; Adebayo, A.; Achara, P.; Alabi, B.; Bakare, R.A.; Famooto, A.O.; Obende, K.; Offiong, R.; Olaniyan, O.; et al. Genome, HLA and polygenic risk score analyses for prevalent and persistent cervical human papillomavirus (HPV) infections. European Journal of Human Genetics 2024. [CrossRef]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev 2012, 25, 215-222. [CrossRef]
- Stanley, M. Immunology of HPV Infection. Current Obstetrics and Gynecology Reports 2015, 4, 195-200. [CrossRef]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. Febs j 2009, 276, 7206-7216. [CrossRef]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; Da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: a review of binding and entry. Journal of virology 2013, 87, 6062-6072.
- Horvath, C.A.; Boulet, G.A.; Renoux, V.M.; Delvenne, P.O.; Bogers, J.P. Mechanisms of cell entry by human papillomaviruses: an overview. Virol J 2010, 7, 11. [CrossRef]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 2008, 82, 12565-12568. [CrossRef]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9. [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 2011, 85, 8981-8995. [CrossRef]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol 2010, 117, S5-10. [CrossRef]
- Paaso, A.; Koskimaa, H.M.; Welters, M.J.P.; Grenman, S.; Syrjanen, K.; van der Burg, S.H.; Syrjanen, S. Cell mediated immunity against HPV16 E2, E6 and E7 peptides in women with incident CIN and in constantly HPV-negative women followed-up for 10-years. Journal of Translational Medicine 2015, 13. [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Frontiers in Microbiology 2020, 10. [CrossRef]
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virol 2011, 6, 45-57. [CrossRef]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 2011, 19, 33-39. [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int J Mol Sci 2018, 19. [CrossRef]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892.
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus Immune Evasion Strategies Target the Infected Cell and the Local Immune System. Front Oncol 2019, 9, 682. [CrossRef]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R., Jr.; Venuti, A. hrHPV E5 oncoprotein: immune evasion and related immunotherapies. J Exp Clin Cancer Res 2017, 36, 71. [CrossRef]
- Campo, M.S.; Graham, S.V.; Cortese, M.S.; Ashrafi, G.H.; Araibi, E.H.; Dornan, E.S.; Miners, K.; Nunes, C.; Man, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137-142. [CrossRef]
- Senba, M.; Mori, N. Mechanisms of virus immune evasion lead to development from chronic inflammation to cancer formation associated with human papillomavirus infection. Oncol Rev 2012, 6, e17. [CrossRef]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000, 19, 4930-4935. [CrossRef]
- Einstein, M.H.; Leanza, S.; Chiu, L.G.; Schlecht, N.F.; Goldberg, G.L.; Steinberg, B.M.; Burk, R.D. Genetic variants in TAP are associated with high-grade cervical neoplasia. Clinical Cancer Research 2009, 15, 1019-1023.
- Wilting, S.M.; Steenbergen, R.D.M. Molecular events leading to HPV-induced high grade neoplasia. Papillomavirus Research 2016, 2, 85-88. [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Reviews in Medical Virology 2015, 25, 2-23. [CrossRef]
- Pešut, E.; Đukić, A.; Lulić, L.; Skelin, J.; Šimić, I.; Milutin Gašperov, N.; Tomaić, V.; Sabol, I.; Grce, M. Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge. Viruses 2021, 13. [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog 2017, 13, e1006211. [CrossRef]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Research 2017, 231, 21-33. [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. British Journal of Cancer 2021, 124, 359-367. [CrossRef]
- Reusser, N.M.; Downing, C.; Guidry, J.; Tyring, S.K. HPV Carcinomas in Immunocompromised Patients. J Clin Med 2015, 4, 260-281. [CrossRef]
- Wang, C.; Xiong, C.; Hsu, Y.-C.; Wang, X.; Chen, L. Human leukocyte antigen (HLA) and cancer immunotherapy: HLA-dependent and-independent adoptive immunotherapies. Ann. Blood 2020, 5, 10.21037.
- Cassioli, C.; Baldari, C.T. The Expanding Arsenal of Cytotoxic T Cells. Frontiers in Immunology 2022, 13. [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. Journal of Cellular Physiology 2019, 234, 8509-8521. [CrossRef]
- Martina, J.A.; Wu, X.S.; Catalfamo, M.; Sakamoto, T.; Yi, C.; Hammer, J.A., 3rd. Imaging of lytic granule exocytosis in CD8+ cytotoxic T lymphocytes reveals a modified form of full fusion. Cell Immunol 2011, 271, 267-279. [CrossRef]
- Li, K.; Qiu, J.; Pan, J.; Pan, J.P. Pyroptosis and Its Role in Cervical Cancer. Cancers (Basel) 2022, 14. [CrossRef]
- Wang, J.L.; Hua, S.N.; Bao, H.J.; Yuan, J.; Zhao, Y.; Chen, S. Pyroptosis and inflammasomes in cancer and inflammation. MedComm 2023, 4, e374.
- Nakagawa, M.; Stites, D.P.; Farhat, S.; Sisler, J.R.; Moss, B.; Kong, F.; Moscicki, A.-B.; Palefsky, J.M. Cytotoxic T lymphocyte responses to E6 and E7 proteins of human papillomavirus type 16: relationship to cervical intraepithelial neoplasia. Journal of Infectious Diseases 1997, 175, 927-931.
- Bontkes, H.J.; De Gruijl, T.D.; van den Muysenberg, A.J.; Verheijen, R.H.; Stukart, M.J.; Meijer, C.J.; Scheper, R.J.; Stacey, S.N.; Duggan-Keen, M.F.; Stern, P.L. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes in women with cervical neoplasia. International journal of cancer 2000, 88, 92-98.
- Valdespino, V.; Gorodezky, C.; Ortiz, V.; Kaufmann, A.M.; Roman-Basaure, E.; Vazquez, A.; Berumen, J. HPV16-specific cytotoxic T lymphocyte responses are detected in all HPV16-positive cervical cancer patients. Gynecologic oncology 2005, 96, 92-102.
- Nakagawa, M.; Stites, D.P.; Patel, S.; Farhat, S.; Scott, M.; Hills, N.K.; Palefsky, J.M.; Moscicki, A.-B. Persistence of Human Papillomavirus Type 16 Infection Is Associated with Lack of Cytotoxic T Lymphocyte Response to the E6 Antigens. The Journal of Infectious Diseases 2000, 182, 595-598. [CrossRef]
- Woo, Y.L.; van den Hende, M.; Sterling, J.C.; Coleman, N.; Crawford, R.A.F.; Kwappenberg, K.M.C.; Stanley, M.A.; van der Burg, S.H. A prospective study on the natural course of low-grade squamous intraepithelial lesions and the presence of HPV16 E2-, E6- and E7-specific T-cell responses. International Journal of Cancer 2010, 126, 133-141. [CrossRef]
- Adebamowo, S.N.; Famooto, A.; Dareng, E.O.; Olawande, O.; Olaniyan, O.; Offiong, R.; Adebamowo, C.A. Clearance of Type-Specific, Low-Risk, and High-Risk Cervical Human Papillomavirus Infections in HIV-Negative and HIV-Positive Women. J Glob Oncol 2018, 4, 1-12. [CrossRef]
- Kuguyo, O.; Dube Mandishora, R.S.; Thomford, N.E.; Makunike-Mutasa, R.; Nhachi, C.F.B.; Matimba, A.; Dandara, C. High-risk HPV genotypes in Zimbabwean women with cervical cancer: Comparative analyses between HIV-negative and HIV-positive women. PLoS One 2021, 16, e0257324. [CrossRef]
- Mbuya, W.; Held, K.; Mcharo, R.D.; Haule, A.; Mhizde, J.; Mnkai, J.; Mahenge, A.; Mwakatima, M.; Sembo, M.; Mwalongo, W.; et al. Depletion of Human Papilloma Virus E6- and E7-Oncoprotein-Specific T-Cell Responses in Women Living With HIV. Frontiers in Immunology 2021, 12. [CrossRef]
- Steele, J.C.; Mann, C.H.; Rookes, S.; Rollason, T.; Murphy, D.; Freeth, M.G.; Gallimore, P.H.; Roberts, S. T-cell responses to human papillomavirus type 16 among women with different grades of cervical neoplasia. British Journal of Cancer 2005, 93, 248-259. [CrossRef]
- Li, X.C.; Raghavan, M. Structure and function of major histocompatibility complex class I antigens. Curr Opin Organ Transplant 2010, 15, 499-504. [CrossRef]
- Hopkins, J.R.; MacLachlan, B.J.; Harper, S.; Sewell, A.K.; Cole, D.K. Unconventional modes of peptide–HLA-I presentation change the rules of TCR engagement. Discovery Immunology 2022, 1. [CrossRef]
- Bagarazzi, M.L.; Yan, J.; Morrow, M.P.; Shen, X.; Parker, R.L.; Lee, J.C.; Giffear, M.; Pankhong, P.; Khan, A.S.; Broderick, K.E.; et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med 2012, 4, 155ra138. [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front Immunol 2018, 9, 14. [CrossRef]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 2013, 105, 1172-1187. [CrossRef]
- Zhao, M.; Qiu, L.; Tao, N.; Zhang, L.; Wu, X.; She, Q.; Zeng, F.; Wang, Y.; Wei, S. HLA DRB allele polymorphisms and risk of cervical cancer associated with human papillomavirus infection: a population study in China. European journal of gynaecological oncology 2013, 34, 54-59.
- Othmane, Y.B.; Ghazouani, E.; Mezlini, A.; Lagha, A.; Raïs, M.; Kochkar, R.; Zidi, S.; Afrit, M.; Mota-Vieira, L.; Loueslati, B.Y. HLA class II susceptibility to cervical cancer among Tunisian women. Bulletin du cancer 2012, 99, E81-E86.
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. International Journal of Cancer 2018, 142, 224-229. [CrossRef]
- Cicchini, L.; Blumhagen, R.Z.; Westrich, J.A.; Myers, M.E.; Warren, C.J.; Siska, C.; Raben, D.; Kechris, K.J.; Pyeon, D. High-Risk Human Papillomavirus E7 Alters Host DNA Methylome and Represses HLA-E Expression in Human Keratinocytes. Sci Rep 2017, 7, 3633. [CrossRef]
- Ferguson, R.; Ramanakumar, A.V.; Richardson, H.; Tellier, P.P.; Coutlée, F.; Franco, E.L.; Roger, M. Human leukocyte antigen (HLA)-E and HLA-G polymorphisms in human papillomavirus infection susceptibility and persistence. Hum Immunol 2011, 72, 337-341. [CrossRef]
- Mora, M.J.; de los Ángeles Bayas-Rea, R.; Mejía, L.; Cruz, C.; Guerra, S.; Calle, P.; Sandoval, D.M.; Galarza, J.M.; Zapata-Mena, S. Identification of human leukocyte antigen in precancerous and cancerous cervical lesions from Ecuadorian women. Infection, Genetics and Evolution 2022, 105, 105365. [CrossRef]
- Adebamowo, S.N.; Adeyemo, A.A. Classical HLA alleles are associated with prevalent and persistent cervical high-risk HPV infection in African women. Hum Immunol 2019, 80, 723-730. [CrossRef]
- Muchiri, L.W.; Sekadde-Kigondu, C.B.; Estambale, B.; Temmerman, M.T. Risk association between human leucocyte antigens (HLA) and cervical neoplasia in Kenyan women. African Journal of Pharmacology and Therapeutics 2012, 1.
- Dong, D.D.; Yang, H.; Li, K.; Xu, G.; Song, L.H.; Fan, X.L.; Jiang, X.L.; Yie, S.M. Human Leukocyte Antigen-G (HLA-G) Expression in Cervical Lesions: Association With Cancer Progression, HPV 16/18 Infection, and Host Immune Response. Reproductive Sciences 2010, 17, 718-723. [CrossRef]
- Da Silva, M.L.R.; De Albuquerque, B.; Allyrio, T.; De Almeida, V.D.; Cobucci, R.N.O.; Bezerra, F.L.; Andrade, V.S.; Lanza, D.C.F.; De Azevedo, J.C.V.; De Araújo, J.M.G.; et al. The role of HPV-induced epigenetic changes in cervical carcinogenesis (Review). Biomed Rep 2021, 15, 60. [CrossRef]
- Mbulawa, Z.Z.A.; Phohlo, K.; Garcia-Jardon, M.; Williamson, A.L.; Businge, C.B. High human papillomavirus (HPV)-35 prevalence among South African women with cervical intraepithelial neoplasia warrants attention. Plos One 2022, 17. [CrossRef]
- Mudini, W.; Palefsky, J.M.; Hale, M.J.; Chirenje, M.Z.; Makunike-Mutasa, R.; Mutisi, F.; Murahwa, A.; Mario, A. Human Papillomavirus Genotypes in Invasive Cervical Carcinoma in HIV-Seropositive and HIV-Seronegative Women in Zimbabwe. J Acquir Immune Defic Syndr 2018, 79, e1-e6. [CrossRef]
- Fitzpatrick, M.B.; Hahn, Z.; Mandishora, R.S.D.; Dao, J.; Weber, J.; Huang, C.; Sahoo, M.K.; Katzenstein, D.A.; Pinsky, B.A. Whole-Genome Analysis of Cervical Human Papillomavirus Type 35 from rural Zimbabwean Women. Scientific Reports 2020, 10, 7001. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



