
Preprint
Article
β-tocotrienol and δ-tocotrienol as Additional Inhibitors of the Main Protease of Feline Infectious Peritonitis Virus: An In-Silico Analysis
Altmetrics
Downloads
159
Views
72
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Submitted:
05 July 2024
Posted:
08 July 2024
You are already at the latest version
Alerts
Abstract
Feline infectious peritonitis (FIP) is a severe and invariably fatal disease affecting both domestic and wild felines with limited effective therapeutic options available. By considering the significant immunomodulatory effects of vitamin E observed in both animal and human models under physiological and pathological conditions, we have provided a full in-silico investigation of vitamin E and related compounds and their effect on the crystal structure of feline infectious peritonitis virus 3C-like protease (FIPV-3CLpro). This work revealed the β-tocotrienol and δ-tocotrienol analogues as inhibitor candidates for this protein, suggesting their potential as possible drug compounds against FIP or their supplementary use with current medicines against this disease.
Keywords:
Subject: Medicine and Pharmacology - Veterinary Medicine
1. Introduction
Feline coronavirus (FCoV) is a member of the Coronaviridae family, a group of enveloped, single-stranded RNA viruses known for their zoonotic potential and capacity to infect a wide range of hosts, including domestic and wild feline species. FCoV is commonly detected in cats with 50-90% of cats testing positive for FCoV specific antibodies [1,2]. It is frequently encountered in multi-cat environments like shelters and catteries, where close contact and shared resources facilitate transmission. The virus can cause gastrointestinal infections, mainly in young felines, leading to mild clinical symptoms such as diarrhoea, vomiting, mild enteritis, and mild villus atrophy in the small intestine and is referred to as feline enteric coronavirus (FECV) [3,4].
In approximately 5-10% of FCoV infected cats [5,6], mutations in the virus or recombination events within the host can lead to the development of feline infectious peritonitis (FIP), a complex and devastating disease [7,8]. These genetic changes transform the relatively innocuous virus into a virulent pathogen (referred to as feline infectious peritonitis virus, FIPV) capable of evading the host's immune system and causing systemic inflammation [9,10]. A broad spectrum of pathological lesions characterizes FIP, determined by interactions between the FIPV and the host's immune status. Depending on the predominant reaction of the host, the pathological manifestations of FIP are traditionally classified as either the effusive (wet) or non-effusive (dry) form [10,11,12]. In reality, however, most affected individuals will manifest both pathological reactions, even if one predominates over the other (Figure 1). The wet form of FIP is seen in individuals reacting mainly with humoral immunity. This form of the disease is more fulminant, characterized by effusions mainly in the abdominal cavity (peritonitis) and more rarely to the thorax and pericardium [13,14]. Clinically wet forms of FIP are manifested with abdominal swelling, fever, anorexia, and icterus. As humoral immunity appears less appropriate to contain the virus, affected individuals deteriorate rapidly, typically culminating in a fatal outcome.
In contrast, the non-effusive or dry form of the disease is seen in animals reacting primarily with cellular immunity, and it is characterized histologically by granulomas on serous membranes, leptomeninges, and the eyes. Affected animals may show neurological signs, weakness, weight loss, and anorexia. Temporary improvement is occasionally reported in untreated cases of cats with dry forms of FIP; relapse, however, is expected with the disease occurring in waves [15].
Given the clinical significance of FIP and the potential for FCoV to mutate into FIPV, the study of feline coronavirus is paramount to both veterinary medicine and public health research. So far there have been no reports of feline coronavirus transmission to humans but this does not negate the risk of interspecies transmission of the virus. An example of the importance of interspecies transmission cycles can be shown with the recent outbreak of FIP on the island of Cyprus. The virus was reported as a highly pathogenic recombinant between feline coronavirus and canine coronavirus and caused severe illness to spread widely among the feline population. The outbreak was characterized by a 40-fold increase in documented cases compared to the previous years. The diagnosis was confirmed in cats presenting with clinical signs by using RT-qPCR for FCoV on clinical samples (peritoneal, pleural, or cerebrospinal fluid, fine needle aspiration biopsies, or tissue biopsies from granulomatous lesions) [16,17,18]. Strategies to manage and control FCoV infections and ongoing efforts to develop effective vaccines and drugs are therefore crucial for safeguarding feline health and minimizing the risk of transmission to humans in close contact with infected cats.
Computer-aided drug discovery (CADD) holds immense promise in the ongoing battle against feline coronavirus, offering a powerful and innovative approach to identifying potential treatments and antiviral agents. Feline coronavirus, including its severe form, FIP, poses significant challenges to feline health, with limited effective therapeutic options currently available. CADD leverages computational methods, molecular modelling, and simulations to accelerate the drug discovery process [19,20,21]. It allows researchers to screen vast libraries of compounds, predict their binding affinity to viral proteins, and identify potential drug candidates with high specificity, all with reduced time and cost compared to traditional experimental approaches [22,23]. In the context of feline coronavirus, CADD can play a pivotal role in designing novel antiviral drugs by targeting key viral proteins or essential host-virus interactions. By exploring the virus's molecular structure and interactions at the atomic level, CADD can aid in the rational design of compounds that inhibit viral replication or mitigate the progression of FIP [24,25].
Moreover, CADD can facilitate the repurposing of existing drugs, potentially identifying compounds already approved for other diseases that could be effective against FCoV. This approach expedites drug development and leverages existing safety and pharmacological data, reducing the time required for clinical trials [26,27]. As the field of CADD advances, its potential to contribute significantly to developing effective treatments for FCoV becomes increasingly apparent. By harnessing the computational power of CADD, researchers can accelerate the discovery of antiviral agents, potentially improving the prognosis for cats affected by feline coronavirus infections and offering hope for more successful outcomes in the fight against the challenging disease of FIP.
Herein, we provided a full in-silico investigation based on vitamin E and related compounds on their effect on the crystal structure of FIPV-3CLpro against FCoV disease [28]. This work revealed that the β-tocotrienol and δ-tocotrienol molecules could act as inhibitors for this protein, suggesting their potential as possible drug compounds against FIP or their supplementary use with current medicines against this disease [29,30].
2. Computational Tools
In this study, we used all the possible computerized methods that can be performed in computer-aided drug discovery procedures. Virtual screening studies, molecular modelling, molecular docking, molecular dynamics, and density functional theory studies were used in the process. The UCSF CHIMERA molecular modelling system was used for protein visualization and preparation, ChemDraw for ligand design, and AutoDock Vina for molecular docking. Additionally, iGEMDOCK, pharmacological interactions, and virtual screening software were used to evaluate the docking results compared to the inhibitor of the crystal structure of the protease. YASARA software was used for the molecular docking studies, and ORCA (quantum chemistry program) was used for energy minimization of the ligand structures and quantum calculation of the stability and reactivity of the ligand interactions with the amino acid residues. Finally, SwissADME and Swiss Similarity online tools were used to compute physicochemical descriptors and predict ADME parameters, pharmacokinetic properties, druglike nature, and medicinal chemistry friendliness. All the calculations were performed on workstations with CPU power of Intel Core i9, 32GB RAM, and 2x NVIDIA GeForce RTX 3060 GPU support.
3. Computational Methods
Our hypothesis to investigate vitamin E as a potential drug candidate against FCoV infections was based on its antibacterial, antioxidant, anticancer and anti-inflammatory functions. All eight molecules of vitamin E, α, β, γ, δ-tocopherol and α, β, γ, δ-tocotrienol, were selected for in silico evaluation.
Protein structure preparation: The crystal structure of FIPV-3CLpro (PDB: 4ZRO) was retrieved from the protein data bank (www.rcsb.org). Chimera software [31] was utilized to prepare the protein structure. All water molecules were deleted, and the required chain was kept. Heteroatoms excluding the co-crystallized ligand were removed and polar hydrogens were added to the crystal structure. The receptor and the co-crystal ligand were selected to define the binding site and the XYZ coordinates were noted down for further use. The co-crystal ligand was then removed and the protein structure was stored in PDB format for further use.
Database preparation: ZINC, a free database, available in public domain, was used for virtual screening using the filters (biological active molecules, commercially available). The molecules were compressed in SDF format and downloaded from the ZINC database. The downloaded database was filtered and prepared by the Schrodinger suit’s LigPrep [32] module based on their ring size to obtain 400 molecules.
Structure-based virtual screening: PyRx is a virtual Screening platform that can be used in computer aided drug discovery to screen libraries of compounds against potential targets. PuRx 0.8 [33] software (https://pyrx.sourceforge.io) is an open-source tool for virtual screening which has integrated Autodock Vina [34], Python shell, and Open Babel [34] on a single platform. Ligands in SDF format were imported into the dashboard, and the energy was minimized and converted into pdbqt format using Open Babel in PyRx. The prepared protein structure was also loaded to the dashbord and converted into auto dock ligands in pdbqt format for input by PyRx for virtual screening. All the ligands and the macromolecules were defined in the Vina wizard. The auto grid box was set up using grid box dimensions of X=25.00, Y=43.00 and Z=28.27 Å. All the ligands were docked against the 4ZRO protein using the Autodock Vina wizard. For evaluation, the bounded inhibitor of N-(tert-butoxycarbonyl)-L-seryl-L-valyl-N-{(2S)-5-ethoxy-5-oxo-1-[(3S)-2-oxopyrrolidin-3-yl]pentan-2-yl}-L-leucinamide (the co-crystal ligand of 4ZRO protein), was similarly prepared and docked against the protein structure. Based on the binding affinity of vina docking, the top 10 molecules were selected for further studies. Structural analysis and visualization of docking interactions between ligand and active site residues were carried out using Chimera software.
Molecular docking by iGEMDOCK: iGEMDOCK is a graphical environment for recognizing pharmacological interactions and virtual screening. Experimental results show that the success of iGEMDOCK is 78% (root-mean-square derivations below 2.0 angstrom) on 305 protein-compound complexes. The crystal structure of the 4ZRO protein was preprocessed, refined, and energy minimized using the AMBER 96 force field. The top 10 scoring ligands obtained from the visual screening and the co-crystal ligand were prepared using the ORCA software [35]. ORCA is a general-purpose quantum chemistry program package with virtually all modern electronic structure methods. Using the Hartee Fork theory [36], we were able to carry geometry optimizations of the candidate ligands. The scoring function is to reduce the number of false positives:
where Ebind is the empirical binding energy used during the molecular docking; Epharma is the energy of binding-site pharmacophores; Eligpre is a penalty value if the ligand unsatisfied the ligand preferences. Epharma and Eligpre were used to improve the number of true positives by discriminating active compounds. The empirical binding energy (Ebind) is given as:
where Einter and Eintra are the intermolecular and intramolecular energy, respectively, Epenal is a large penalty value if the ligand is out of range of the search box. In this study, Epenal is set to 1000.
Etot = Ebind + Epharma + Eligpre
Ebind = Einter + Eintra + Epenal
Binding free energy estimation: After iGEMDOCK docking, a maestro pose viewer file was generated, containing information about the docked complex of all ligands. This pose information viewer file was taken as input by the Prime [37] module of Schrodinger [38], to estimate the free binding energy of the docked complex. The net binding energy (ΔGbind) was calculated as ΔGbind = EMM + Gsolv - ΤΔS.
Where EMM is the molecular mechanics energy (gas phase); Gsol is the solvation free energy (gas phase) and ΤΔS is the conformational entropy changes during the complex formation.
In silico ADMET studies: ADMET stands for absorption, distribution, metabolism, elimination and toxicity. This profile is essential to rule out late-stage failure in clinical trials. The top ten scoring compounds in virtual screening were accessed for their ADME profile using the Swiss ADME [39] web server (https://www.swissadme.ch/). The SMILES (Simplified Molecular Input Line Entry System) of the chemical compounds were used as an input by the webserver to quickly predict various ADME parameters along with oral absorption, bioavailability score, inhibition of various cytochrome-P enzymes and others. TPSA represents the polarity of the molecule, which affects the absorption of the drug in git and its access to the brain. iLOGP represents the octanol-water partition coefficient, which relies on the free energy of solvation and is calculated with the help of the GB/SA (Generalized-Borne and solvent-accessible surface area) model. ESOL LogS represents aqueous solubility. Cytochrome-P450 isozymes are essential for the metabolism and elimination of drug metabolites. Inhibition of one or more enzymes may cause drug-drug interactions, drug accumulation and adverse effects. PAINS alert helps identify false positives or promiscuous compounds in drug discovery programs. OSIRIS [40] property explorer (https://www.organic-chemistry.org/prog/peo/) was used to study the top 4 hits of tumorigenicity, mutagenicity, irritant and reproductive risks. The SMILES of the compounds were used as an input for the OSIRIS portal, which returned the toxicity results in colour codes (Red for high risk, Orange for medium, and Green for low risk).
3.1. Molecular Dynamics (MD) studies
YASARA software environment [41], is a Molecular Modelling and Molecular Dynamics tool that supports automatic simulation set-up and provides robust calculation of free energies. Using YASARA software we performed molecular dynamics studies, using explicit solvent environment with periodic bounding conditions and an orthorhombic simulation box. The ligand-protein docked complex was fitted inside an orthorhombic box of dimensions 10x10x10 Å. The system was solvated with explicit water molecules and neutralized by adding counter ions like Na+ and Cl- at a concentration of 0.15 M. AMBER96 force field was used for the entire simulation. A standard equilibration protocol minimizes and pre-equilibrates the system before the production run. A production simulation of 100 ns was carried out using normal pressure and temperature (NPT) ensemble at 1.1325 bar pressure and 300 K temperature. At every 10 ps, the new coordinates and energy of the system were saved in the trajectory. A simulation interaction diagram was used to analyze the simulation trajectory and monitor the stability of the complex.
3.2. Density Functional Theory (DFT) studies
Density functional theory studies on B3LYP/6 311++G (d, p) were performed to discriminate the chemical reactivity between vitamin E derivatives. In addition, we were able to calculate the molecular orbitals of the molecules. The value of the energy difference between HOMO and LUMO and the highest occupied molecular orbital (EHOMO) and lowest unoccupied molecular orbital (ELUMO) energies play a significant role in the stability and reactivity of molecules [42,43]. The EHOMO energies of molecules show the molecule’s ability to donate electrons. On the other hand, ELUMO characterizes the ability of the compound to accept electrons. Electronegativity (χ) measures an atom’s power to attract a bonding pair of electrons. Based on the equation χ = −(EHOMO + ELUMO)/2, a larger Δgap always indicates lower chemical reactivity and higher kinetic stability of the investigated species [44,45]. The simultaneous effect of different parameters causes the chemical reactivity of molecules. The distribution and energy of HOMO are important parameters to explain the antioxidant potential of phenolic antioxidants. The electron-donating capacity of the molecule can be predicted by looking at the energy values of HOMO [46].
4. Results
Molecular docking analysis details the interaction energies and specific amino acid residues involved in both van der Waals and hydrogen bond interactions for the complexes formed between β-tocotrienol and δ-tocotrienol with the FIPV-3CLpro protein. The results can be found in Table 1. Figure 2 depicts the FIPV-3CLpro protein and the binding areas of β-tocotrienol and δ-tocotrienol. Figure 3 illustrates the simulation box of the molecular dynamics studies. Additionally, we can see the binding sites of the β and δ-tocotrienol respectively as well as amino acid residue interactions of the two lead candidates of β (green color) and δ-tocotrienol (purple color). The binding pockets of the β (green color) and δ-tocotrienol (purple color) are also depicted in Figure 3. Figure 4 depicts the equations used for the calculations of radius mass, RMSD and RMSF values. Figure 5 includes the whole simulation process for the RMSF values. The remaining work is listed in the supplementary material.
The comparative analysis which outlines the chemical properties, pharmacokinetics, and toxicity profiles of β-tocotrienol and δ-tocotrienol as well as highlighting similarities and differences between the two compounds is shown in Table 2. The endocrine disruption potential of β-tocotrienol (left) and δ-tocotrienol (rights) obtained from the Endocrine Disruptome is found in Figure 6. The orange and yellow coloring indicates medium probability of binding while the green coloring indicates low probability of binding. Both candidate substances show a low probability of binding to the receptors, thus indicating limited toxicity.
5. Discussion
5.1. Virtual Screening and Molecular Docking
The initial virtual screening using PyRx and AutoDock Vina identified several vitamin E derivatives with promising binding affinities to the FIPV-3CLpro (PDB: 4ZRO) protein. Among these, β-tocotrienol and δ-tocotrienol exhibited the highest binding affinities, with docking scores suggesting strong interactions with the active site of the protease. Specifically, the docking scores indicated that these compounds could form stable interactions within the active site, potentially inhibiting the protease activity crucial for FIPV replication. The top ten scoring ligands from the virtual screening were further refined using iGEMDOCK, which offers more detailed insights into the binding interactions. This additional docking study confirmed the high binding affinities of β-tocotrienol and δ-tocotrienol, demonstrating significant interactions with key residues in the FIPV-3CLpro active site. The empirical binding energies (Ebind) calculated by iGEMDOCK further supported these findings, with β-tocotrienol and δ-tocotrienol exhibiting lower Ebind values, indicating stronger binding.
Further binding free energy (ΔGbind) estimations using the Prime module provided additional insights into the stability of the ligand-protein complexes. Both β-tocotrienol and δ-tocotrienol exhibited favorable ΔGbind values, reinforcing their potential as strong inhibitors of FIPV-3CLpro. The consistent results across different docking tools underscore the reliability of these findings, suggesting that β-tocotrienol and δ-tocotrienol could be potent antiviral agents against FIPV.
5.2. ADMET and Molecular Dynamics (MD) Studies
The ADMET profiles of the top ten compounds were assessed to ensure drug-likeness and safety. β-tocotrienol and δ-tocotrienol showed favorable ADME properties, including good oral absorption, high bioavailability, and minimal inhibition of cytochrome-P450 enzymes, reducing the risk of drug-drug interactions. These properties are crucial for the development of any potential therapeutic agent, as they determine the compound's effectiveness and safety in a biological system. The OSIRIS property explorer indicated low risks of tumorigenicity, mutagenicity, irritancy, and reproductive toxicity for these compounds, further validating their potential as safe drug candidates.
MD simulations performed using YASARA demonstrated the stability of the β-tocotrienol and δ-tocotrienol complexes with FIPV-3CLpro over a 100 ns period. The root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) analyses confirmed that both ligand-protein complexes maintained stable interactions throughout the simulation, with minimal deviations indicating a strong and stable binding. The low RMSD and RMSF values observed suggest that these compounds are not only strong binders but also form stable complexes over time, which is essential for effective inhibition.
6. Comprehensive Analysis and Future Directions
The computational studies presented herein provide a comprehensive analysis of vitamin E derivatives as potential inhibitors of FIPV-3CLpro. β-tocotrienol and δ-tocotrienol emerged as the most promising candidates based on multiple criteria, including binding affinity, stability, and ADMET profiles. The strong interactions observed in molecular docking studies were corroborated by binding free energy estimations and MD simulations, highlighting the stability of these complexes. The favorable ADMET profiles suggest that β-tocotrienol and δ-tocotrienol could be developed into safe and effective drugs for FIP treatment. Additionally, their antioxidant properties could provide synergistic benefits when used in combination with existing therapies, enhancing the overall treatment efficacy.
The DFT (Density Functional Theory) analysis further supported the potential of β-tocotrienol and δ-tocotrienol, demonstrating their high reactivity and electron-donating capabilities, which are crucial for effective inhibition of viral proteases. This quantum mechanical analysis provided additional validation of the electronic properties that contribute to the binding efficacy observed in docking studies. The combination of DFT results with empirical binding energies and MD simulation data creates a robust framework for understanding the molecular mechanisms underlying the inhibition of FIPV-3CLpro by these compounds.
This study identified β-tocotrienol and δ-tocotrienol as potent inhibitors of FIPV-3CLpro, providing a strong basis for further experimental validation and development as therapeutic agents against FIP. The integration of various computational methods allowed for a thorough evaluation of these compounds, paving the way for new treatment strategies to combat this fatal feline disease. Future research should focus on in vitro and in vivo validation of these findings, as well as exploring potential formulation and delivery methods to maximize the therapeutic efficacy of β-tocotrienol and δ-tocotrienol in clinical settings.
7. Conclusions
Herein, we have provided a computational evaluation of the vitamin E analogs as inhibitors for the feline infectious peritonitis virus main protease. β-tocotrienol and δ-tocotrienol molecules have a strong binding affinity on the protein based on molecular dynamics evaluation. These results suggest that these compounds are excellent candidates for field trials against FIP disease. Furthermore, our study indicates that vitamin E could be supplemented synergistically with other drugs, such as remdesivir, to improve therapeutic outcomes in feline patients with FIP.
References
- Jiao, Z.; Yan, Y.; Chen, Y.; Wang, G.; Wang, X.; Li, L.; Yang, M.; Hu, X.; Guo, Y.; Shi, Y.; Peng, G. Adaptive Mutation in the Main Protease Cleavage Site of Feline Coronavirus Renders the Virus More Resistant to Main Protease Inhibitors. Journal of Virology 2022, 96. [Google Scholar] [CrossRef] [PubMed]
- Sherding, R.G. Feline Infectious Peritonitis (Feline Coronavirus) 2020, 21, 1–9. 21.
- Vojtkovská, V.; Lukešová, G.; Voslářová, E.; Konvalinová, J.; Večerek, V.; Lobová, D. Direct Detection of Feline Coronavirus by Three Rapid Antigen Immunochromatographic Tests and by Real-Time PCR in Cat Shelters. Veterinary Sciences 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Colina, S.E.; Serena, M.S.; Echeverría, M.G.; Metz, G.E. Clinical and molecular aspects of veterinary coronaviruses. Virus Research 2021, 297. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Wittenburg, L.; Yan, V.C.; Theil, J.H.; Castillo, D.; Reagan, K.L.; Williams, S.; Pham, C.D.; Li, C.; Muller, F.L.; Murphy, B.G. An Optimized Bioassay for Screening Combined Anticoronaviral Compounds for Efficacy against Feline Infectious Peritonitis Virus with Pharmacokinetic Analyses of GS-441524, Remdesivir, and Molnupiravir in Cats. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, C.; Liu, X.; Yang, K.; Xu, X.; Yang, H. Crystal Structure of Feline Infectious Peritonitis Virus Main Protease in Complex with Synergetic Dual Inhibitors. Journal of Virology 2016, 90, 1910–1917. [Google Scholar] [CrossRef]
- Dunbar, D.; Babayan, S.A.; Krumrie, S.; Haining, H.; Hosie, M.J.; Weir, W. Assessing the feasibility of applying machine learning to diagnosing non-effusive feline infectious peritonitis. Scientific Reports 2024, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, S.A.; Khan, M.B.; Brassard, R.; Arutyunova, E.; Lamer, T.; Vuong, W.; Fischer, C.; Young, H.S.; Vederas, J.C.; Lemieux, M.J. Crystallization of Feline Coronavirus Mpro With GC376 Reveals Mechanism of Inhibition. Frontiers inChemistry 2022, 10, 1–10. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Liao, G.R.; Lien, A.; Hsu, W.L. Current concepts in the development of therapeutics against human and animal coronavirus diseases by targeting NP. Computational and Structural Biotechnology Journal 2021, 19, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- St. John, S.E.; Therkelsen, M.D.; Nyalapatla, P.R.; Osswald, H.L.; Ghosh, A.K.; Mesecar, A.D. X-ray structure and inhibition of the feline infectious peritonitis virus 3C-like protease: Structural implications for drug design. Bioorganic and Medicinal Chemistry Letters 2015, 25, 5072–5077. [Google Scholar] [CrossRef]
- Zhou, M.; Han, Y.; Li, M.; Ye, G.; Peng, G. Two Inhibitors Against the 3C-Like Proteases of Swine Coronavirus and Feline Coronavirus. Virologica Sinica 2021, 36, 14211430. [Google Scholar] [CrossRef]
- Camero, M.; Lanave, G.; Catella, C.; Lucente, M.S.; Sposato, A.; Mari, V.; Tempesta, M.; Martella, V.; Buonavoglia, A. ERDRP-0519 inhibits feline coronavirus in vitro. BMC Veterinary Research 2022, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Bai, J.; Liu, X.; Wang, M.; Wang, X.; Jiang, P. Tomatidine inhibits porcine epidemic diarrhea virus replication by targeting 3CL protease. Veterinary Research 2020, 51, 1–18. [Google Scholar] [CrossRef]
- Furbish, A.; Allinder, M.; Austin, G.; Tynan, B.; Byrd, E.; Gomez, I.P.; Peterson, Y. First analytical confirmation of drug-induced crystal nephropathy in felines caused by GS-441524, the active metabolite of Remdesivir. Journal of Pharmaceutical and Biomedical Analysis 2024, 247, 116248. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; De Franco, F.; Torquato, P.; Marinelli, R.; Cerra, B.; Ronchetti, R.; Schon, A.; Fallarino, F.; De Luca, A.; Bellezza, G.; Ferri, I.; Sidoni, A.; Walton, W.G.; Pellock, S.J.; Redinbo, M.R.; Mani, S.; Pellicciari, R.; Gioiello, A.; Galli, F. Garcinoic Acid Is a Natural and Selective Agonist of Pregnane X Receptor. Journal of Medicinal Chemistry 2020, 63, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Arutyunova, E.; Khan, M.B.; Fischer, C.; Lu, J.; Lamer, T.; Vuong, W.; van Belkum, M.J.; McKay, R.T.; Tyrrell, D.L.; Vederas, J.C.; Young, H.S.; Lemieux, M.J. N-Terminal Finger Stabilizes the S1 Pocket for the Reversible Feline Drug GC376 in the SARS-CoV-2 Mpro Dimer. Journal of Molecular Biology 2021, 433, 167003. [Google Scholar] [CrossRef]
- Guarnieri, C.; Bertola, L.; Ferrari, L.; Quintavalla, C.; Corradi, A.; Di Lecce, R. Myocarditis in an FIP-Diseased Cat with FCoV M1058L Mutation: Clinical and Pathological Changes. Animals 2024, 14, 1–9. [Google Scholar] [CrossRef]
- Theerawatanasirikul, S.; Kuo, C.J.; Phetcharat, N.; Lekcharoensuk, P. In silico and in vitro analysis of small molecules and natural compounds targeting the 3CL protease of feline infectious peritonitis virus. Antiviral Research 2020, 174, 104697. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Endoh, M.; Fukatsu, H.; Sakurada, H.; Doki, T.; Hohdatsu, T. The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antiviral Research 2017, 145, 96–102. [Google Scholar] [CrossRef]
- Jose, S.; Devi, S.S.; Sajeev, A.; Girisa, S.; Alqahtani, M.S.; Abbas, M.; Alshammari, A.; Sethi, G.; Kunnumakkara, A.B. Repurposing FDA-approved drugs as FXR agonists: a structure based in silico pharmacological study. Bioscience Reports 2023, 43, 1–20. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, X.; Zhong, X.; Hu, W.; Lin, Z.; Zhang, S.; Deng, H.; Lin, W. Prevalence of natural feline coronavirus infection in domestic cats in Fujian, China. Virology Journal 2024, 21, 1–8. [Google Scholar] [CrossRef]
- Chowdhury, R.; Boorla, V.S.; Maranas, C.D. Computational biophysical characterization of the SARS-CoV-2 spike protein binding with the ACE2 receptor and implications for infectivity. Computational and Structural Biotechnology Journal 2020, 18, 2573–2582. [Google Scholar] [CrossRef]
- Fath, M.K.; Naderi, M.; Hamzavi, H.; Ganji, M.; Shabani, S.; ghahroodi, F.N.; Khalesi, B.; Pourzardosht, N.; Hashemi, Z.S.; Khalili, S. Molecular mechanisms and therapeutic effects of different vitamins and minerals in COVID-19 patients. Journal of Trace Elements in Medicine and Biology 2022, 73, 127044. [Google Scholar] [CrossRef]
- August, J.R. Feline Infectious Peritonitis: An Immune-Mediated Coronaviral Vasculitis. Veterinary Clinics of North America: Small Animal Practice 1984, 14, 5. [Google Scholar]
- Kipar, A.; Meli, M.L. Feline Infectious Peritonitis: Still an Enigma? Veterinary Pathology 2014, 51, 505–526. [Google Scholar] [CrossRef]
- Attipa, C.; Gunn-Moore, D.; Mazeri, S.; Epaminondas, D.; Lyraki, M.; Hardas, A.; Loukaidou, S.; Gentil, M. Concerning feline infectious peritonitis outbreak in Cyprus. Veterinary Record 2023, 192, 449–450. [Google Scholar] [CrossRef]
- Attipa, C.; et al. Emergence and spread of feline infection peritonitis due to a highly. Biorxiv, 2023; 1–23. [Google Scholar]
- Mavra, A.; Petrou, C.C.; Vlasiou, M.C. Ligand and Structure-Based Virtual Screening in Combination, to Evaluate Small Organic Molecules as Inhibitors for the XIAP Anti-Apoptotic Protein: The Xanthohumol Hypothesis. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Verbrugghe, A.; Janssens, G.P. J.; Van de Velde, H.; Cox, E.; De Smet, S.; Vlaeminck, B.; Hesta, M. Failure of a dietary model to affect markers of inflammation in domestic cats. BMC Veterinary Research 2014, 10, 1–9. [Google Scholar] [CrossRef]
- Candellone, A.; Badino, P.; Gianella, P.; Girolami, F.; Raviri, G.; Saettone, V.; Meineri, G. Evaluation of antioxidant supplementation on redox unbalance in hyperthyroid cats treated with methimazole: A blinded randomized controlled trial. Antioxidants 2020, 9. [Google Scholar] [CrossRef]
- Timmons, R.M.; Webb, C.B. Vitamin E supplementation fails to impact measures of oxidative stress or the anaemia of feline chronic kidney disease: a randomised, double-blinded placebo control study. Veterinary Medicine and Science 2016, 2, 117–124. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Petrou, C.C.; Sarigiannis, Y.; Pafiti, K.S. Density functional theory studies and molecular docking on xanthohumol, 8-prenylnaringenin and their symmetric substitute diethanolamine derivatives as inhibitors for colon cancer-related proteins. Symmetry 2021, 13. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Pati, K.S. (n.d.). Screening Possible Drug Molecules for Covid-19. The Example of Vanadium (III/IV/V) Complex Molecules with Computational Chemistry and Molecular Docking. [CrossRef]
- Ioannou, K.; Vlasiou, M.C. Metal-based complexes against SARS-CoV-2. BioMetals 2022, 35, 639–652. [Google Scholar] [CrossRef]
- Vlasiou, M.C. Computer-aided Drug Discovery Methods for Zoonoses. Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry 2023, 22, 131–132. [Google Scholar] [CrossRef]
- El-Assaad, A.M.; Hamieh, T. SARS-CoV-2: Prediction of critical ionic amino acid mutations. Computers in Biology and Medicine 2024, 178, 108688. [Google Scholar] [CrossRef]
- Liu, X.; Ren, Y.; Qin, S.; Yang, Z. Exploring the mechanism of 6-Methoxydihydrosanguinarine in the treatment of lung adenocarcinoma based on network pharmacology, molecular docking and experimental investigation. BMC Complementary Medicine and Therapies 2024, 24, 1–18. [Google Scholar] [CrossRef]
- Zhou, C.; Yan, L.; Xu, J.; Hamezah, H.S.; Wang, T.; Du, F.; Tong, X.; Han, R. Phillyrin: an adipose triglyceride lipase inhibitor supported by molecular docking, dynamics simulation, and pharmacological validation. Journal of Molecular Modeling 2024, 30. [Google Scholar] [CrossRef]
- Mattson, N.M.; Chan, A.K. N.; Miyashita, K.; Mukhaleva, E.; Chang, W.H.; Yang, L.; Ma, N.; Wang, Y.; Pokharel, S.P.; Li, M.; Liu, Q.; Xu, X.; Chen, R.; Singh, P.; Zhang, L.; Elsayed, Z.; Chen, B.; Keen, D.; Pirrotte, P.; … Chen, C.W. A novel class of inhibitors that disrupts the stability of integrin heterodimers identified by CRISPR-tiling-instructed genetic screens. Nature Structural and Molecular Biology 2024, 31, 465–475. [Google Scholar] [CrossRef]
- Ludwig, V.; da Costa Ludwig, Z.M.; Modesto, M. de A.; Rocha, A.A. Binding energies and hydrogen bonds effects on DNA-cisplatin interactions: a DFT-xTB study. Journal of Molecular Modeling 2024, 30, 1–9. [Google Scholar] [CrossRef]
- Khachatryan, H.; Matevosyan, M.; Harutyunyan, V.; Gevorgyan, S.; Shavina, A.; Tirosyan, I.; Gabrielyan, Y.; Ayvazyan, M.; Bozdaganyan, M.; Fakhar, Z.; Gharaghani, S.; Zakaryan, H. Computational evaluation and benchmark study of 342 crystallographic holo-structures of SARS-CoV-2 Mpro enzyme. Scientific Reports 2024, 14, 1–14. [Google Scholar] [CrossRef]
- Tendongmo, H.; Kogge, B.F.; Tamafo Fouegue, A.D.; Tasheh, S.N.; Tessa, C.B. N.; Ghogomu, J.N. Theoretical screening of N-[5′-methyl-3′-isoxasolyl]-N-[(E)-1-(-2-thiophene)] methylidene]amine and its isoxazole based derivatives as donor materials for bulk heterojunction organic solar cells: DFT and TD-DFT investigation. Journal of Molecular Modeling 2024, 30, 1–16. [Google Scholar] [CrossRef]
- Vlasiou, M. In Silico Techniques Used for the Evaluation of Anticancer Metal-Based Compounds. Anti-Cancer Agents in Medicinal Chemistry 2022, 23, 490–491. [Google Scholar] [CrossRef]
- Vlasiou, M. In Search of Antiviral Metal-Based Drugs. The Open Medicinal Chemistry Journal 2021, 15, 30–31. [Google Scholar] [CrossRef]
- Vlasiou, M.C. Structural characterization of two novel, biological active chalcone derivatives, using density functional theory studies. Biointerface Research in Applied Chemistry 2021, 11, 15051–15057. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Pafiti, K.S. Spectroscopic evaluation of Zn (II) complexes with drug analogues: Interactions with BSA and the pH effect on the drug-Zn (II) system. Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy 2020, 241, 1–9. [Google Scholar] [CrossRef]
Figure 1.
Abdominal cavity in situ, exhibiting a large amount of straw-colored effusion fluid, polymerized fibrin (black arrows), and granulomas on the intestinal serosa (red arrows). .
Figure 1.
Abdominal cavity in situ, exhibiting a large amount of straw-colored effusion fluid, polymerized fibrin (black arrows), and granulomas on the intestinal serosa (red arrows). .
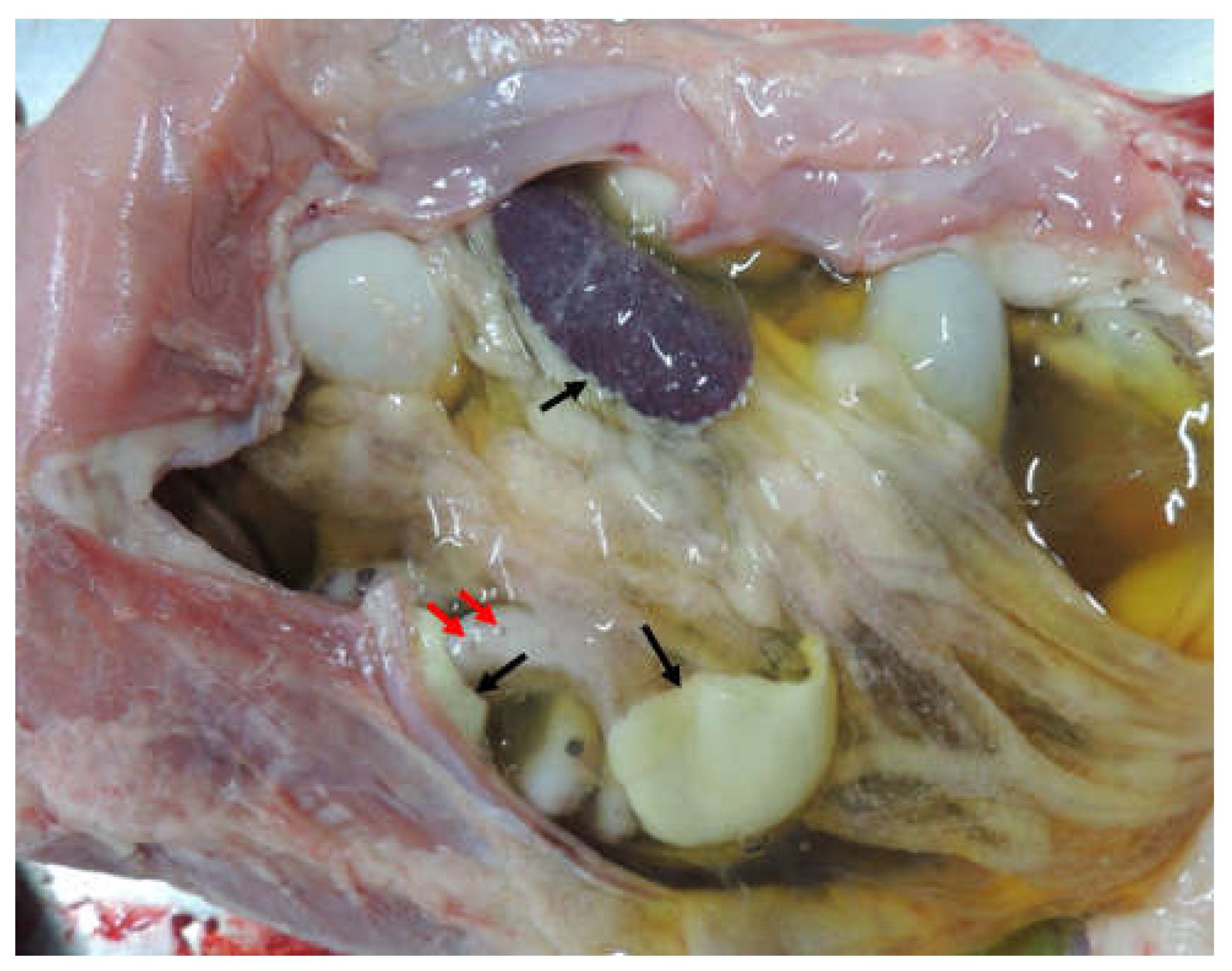
Figure 2.
Binding sites of β and δ-tocotrienol respectively on the protein.
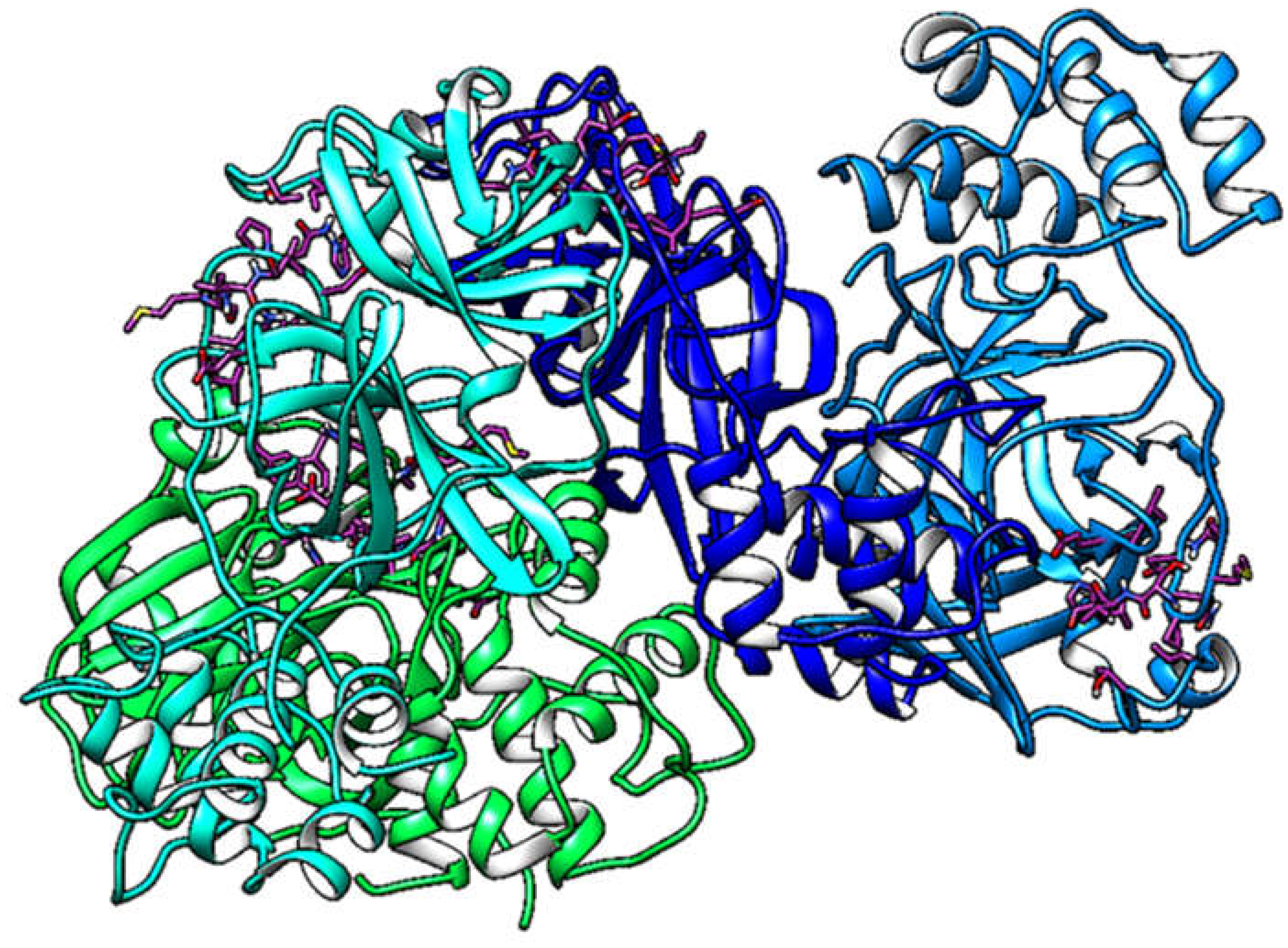
Figure 3.
Simulation box of the molecular dynamics studies (A). Binding sites of the β and δ-tocotrienol respectively (B). Amino acid residue interactions of the two lead candidates. β (green color) and δ-tocotrienol (purple color) (C). Binding pocket of the β (green color) and δ-tocotrienol (purple color) (D).
Figure 3.
Simulation box of the molecular dynamics studies (A). Binding sites of the β and δ-tocotrienol respectively (B). Amino acid residue interactions of the two lead candidates. β (green color) and δ-tocotrienol (purple color) (C). Binding pocket of the β (green color) and δ-tocotrienol (purple color) (D).
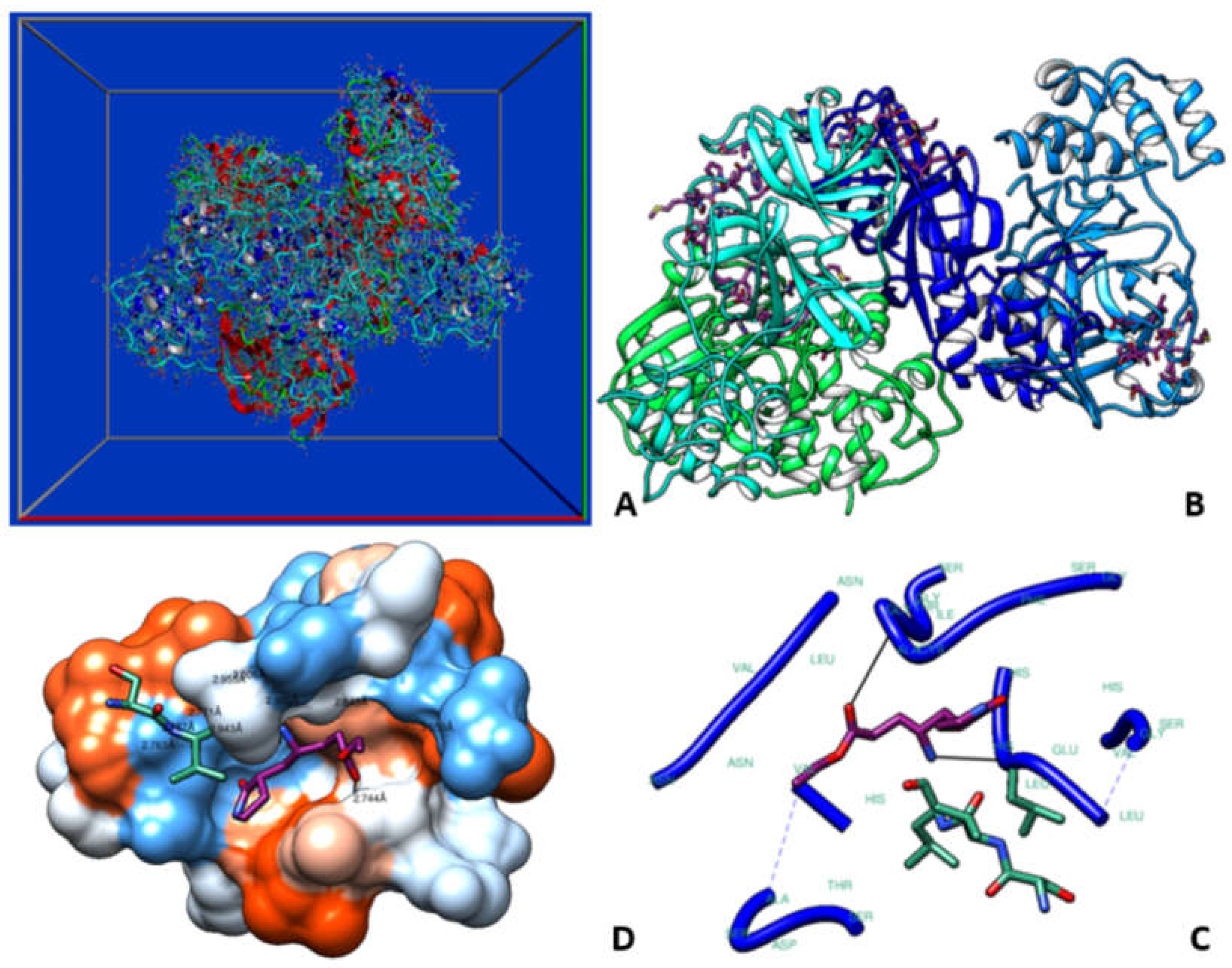
Figure 4.
Equations used in Radius mass calculations and RMSD and RMSF values calculations.
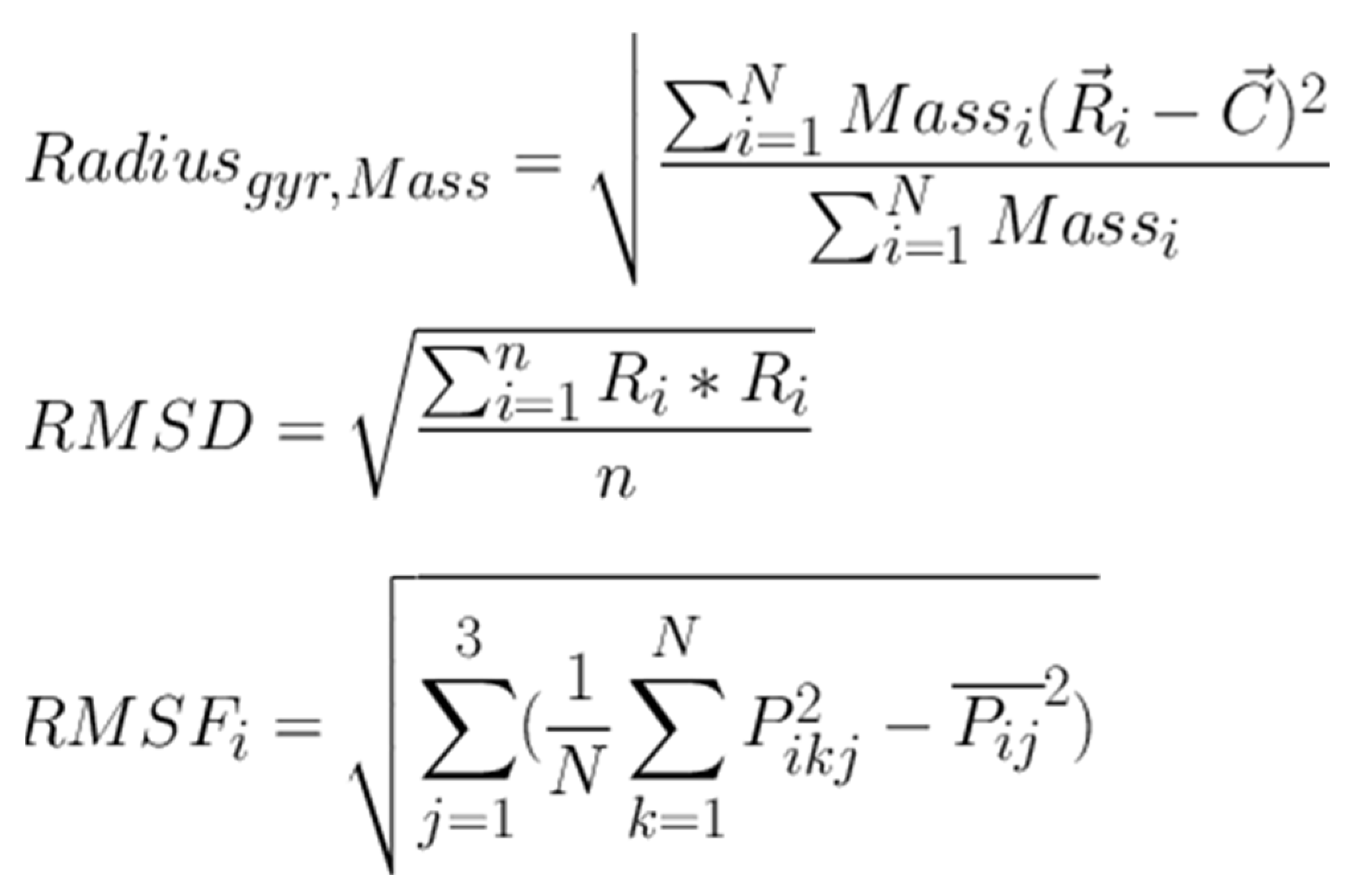
Figure 5.
RMSF values through the amino acid residues interactions (Molecule A: β-tocotrienol, Molecule B: δ-tocotrienol).
Figure 5.
RMSF values through the amino acid residues interactions (Molecule A: β-tocotrienol, Molecule B: δ-tocotrienol).
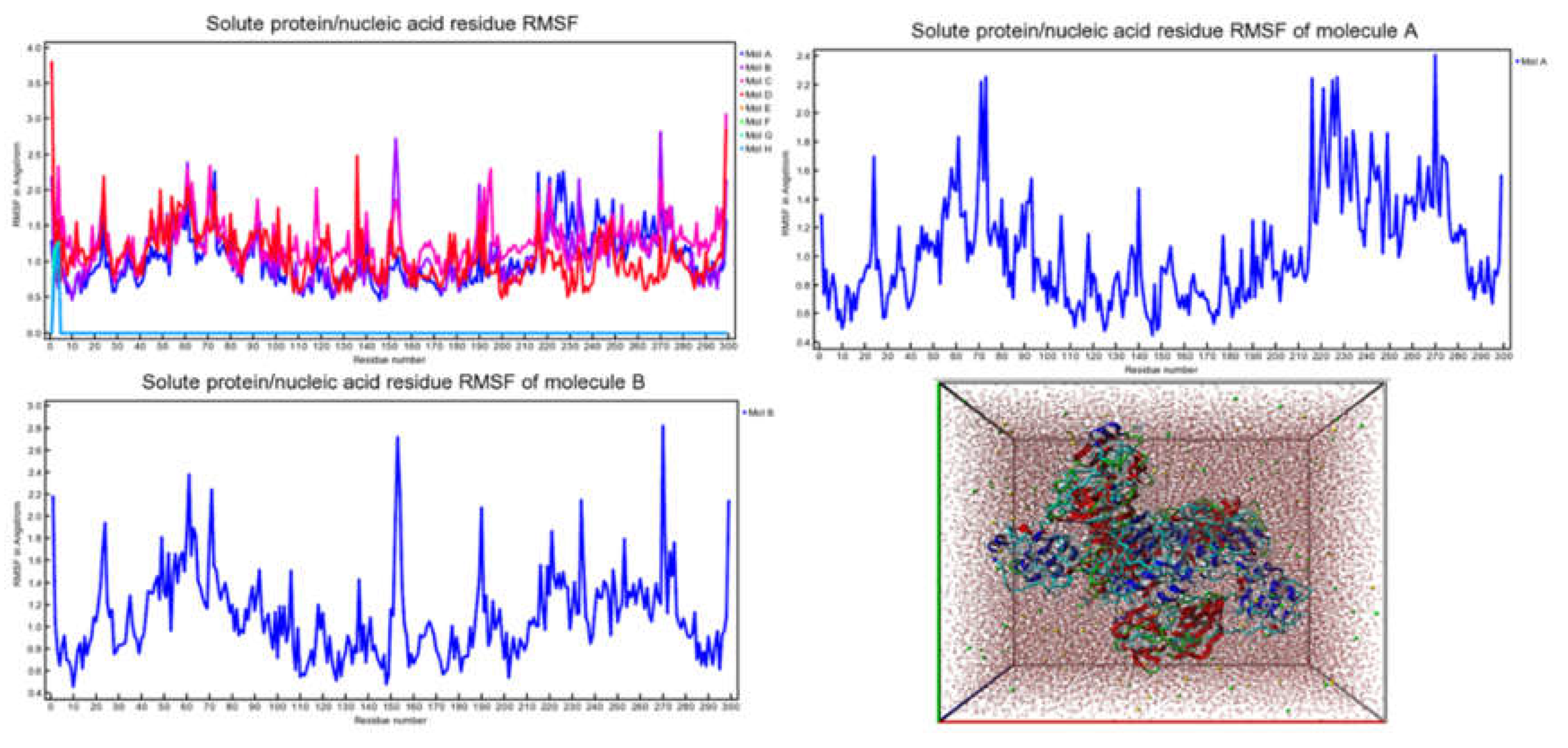
Figure 6.
Endocrine disruption potential of β-tocotrienol (left) and δ-tocotrienol (right) obtained from the Endocrine Disruptome. Orange and yellow coloring indicates medium probability of binding while green coloring indicates low probability of binding.
Figure 6.
Endocrine disruption potential of β-tocotrienol (left) and δ-tocotrienol (right) obtained from the Endocrine Disruptome. Orange and yellow coloring indicates medium probability of binding while green coloring indicates low probability of binding.
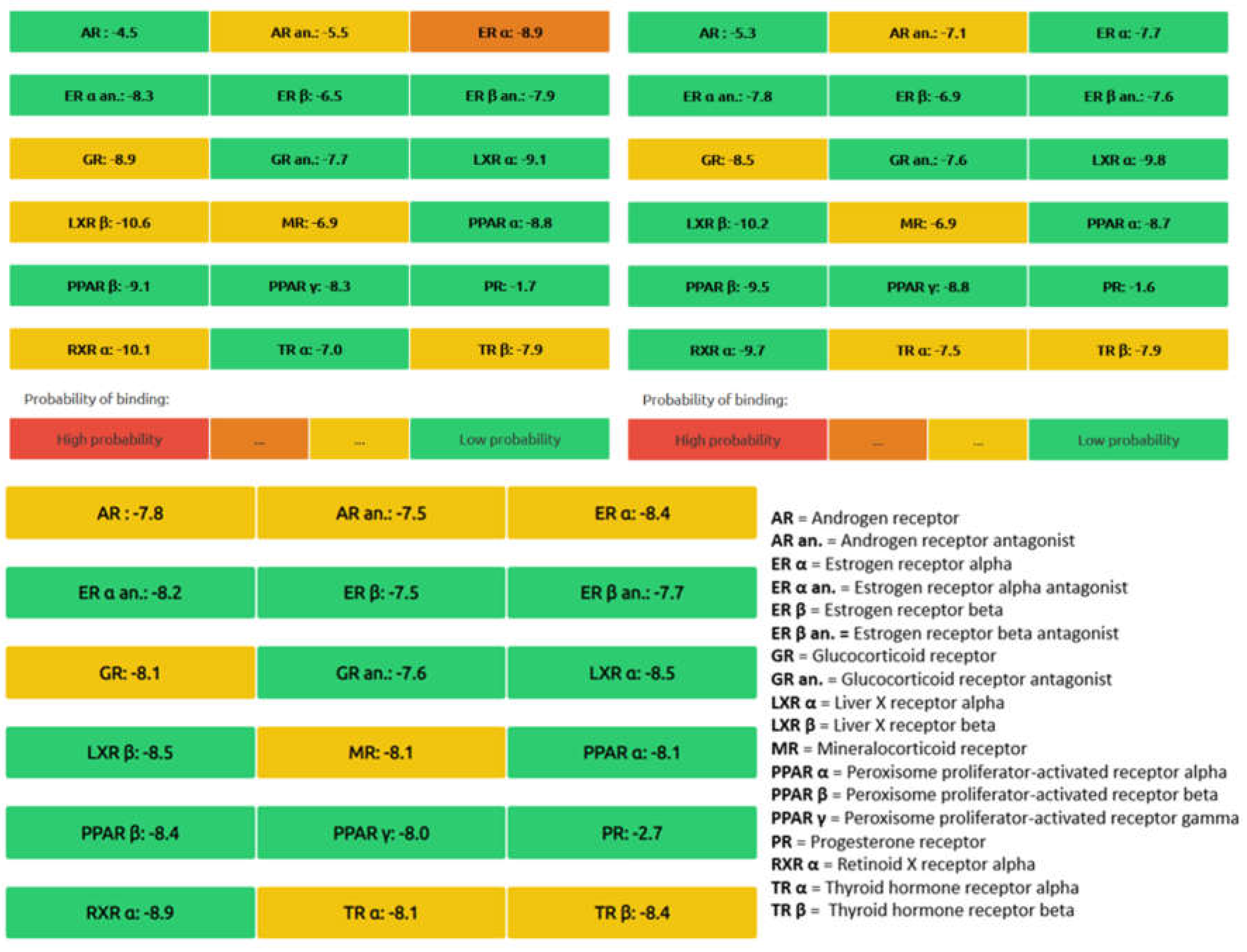

Table 1.
Energies and amino acid residues of the two most promising candidates (β-tocotrienol and δ-tocotrienol) on the FIPV-3CLpro protein.
Table 1.
Energies and amino acid residues of the two most promising candidates (β-tocotrienol and δ-tocotrienol) on the FIPV-3CLpro protein.
| Complex | Total Energy (KJ/mole) | Energy VDW (KJ/mole) | Energy Hbond (KJ/mole) | Amino Acid Residue Hbonds | Amino Acid Residue VDW interactions |
|---|---|---|---|---|---|
| β-tocotrienol- FIPV-3CLpro | -94.92 | -91.42 | -3.5 | PHE 111 | MET 6, GLN 8, GLU 291, ARG 294 |
| δ-tocotrienol- FIPV-3CLpro | -98.23 | -86.32 | -11.9 | PHE 111, VAL 127 | GLY 122, GLN 8, PHE 111, ASN 112, GLU 291, ARG 294, VAL 299 |
Table 2.
ADMET studies for β and δ tocotrienol.
| Formula | C28H42O2 | C27H40O2 | Formula | C28H42O2 | C27H40O2 |
|---|---|---|---|---|---|
| Molecular weight | 410.63 g/mol | 396.61 g/mol | Total Clearance | 0.814 log ml/min/Kg | 0.847 log ml/min/Kg |
| Num. of heavy atoms | 30 | 29 | Renal OCT2 substrate | No | No |
| Num. of aromatic heavy atoms | 6 | 6 | AMES toxicity | No | No |
| Num. of rotatable bonds | 9 | 9 | Max. tolerated dose | 0.45 log mg/Kg/day | 0.729 log mg/Kg/day |
| Num. of H-bond acceptors | 2 | 2 | hERG I Inhibitor |
No | No |
| Num. of H-bond donors | 1 | 1 | hERG Inhibitor |
Yes | Yes |
| Molar refractivity | 132.88 | 127.2 | Oral rat acute toxicity (LD50) | 2.18 mol/Kg | 1.945 mol/Kg |
| Log Po/w | 5.14 | 5.35 | Oral rat chronic toxicity (LOAEL) | 2.967 log mg/Kg_bw/day | 2.956 log mg/Kg_bw/day |
| Log S | -7.57 | -7.26 | Hepatotoxicity | No | No |
| GI absorption | Low | Low | Skin Sensitisation | No | No |
| BBB permeant | No | No | T. Pyriformis toxicity | 1.127 log ug/L | 1.182 log ug/L |
| P-gp substrate | Yes | Yes | Minnow toxicity | -2.5 mM | -4.247 mM |
| CYP1A2 inhibitor | No | No | No | No | |
| CYP2C19 inhibitor | No | No | |||
| CYP2C9 inhibitor | No | No | |||
| CYP2D6 inhibitor | No | No | |||
| CYP3A4 inhibitor | No | Yes | |||
| Log Kp (skin permeation) | -2.46 cm/s | -2.63 cm/s | |||
| Lipinski | Yes, 1 violation: MLOGP>4.15 | Yes, 1 violation: MLOGP>4.15 | |||
| Bioavailability Score | 0.55 | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated