
Submitted:
28 June 2024
Posted:
08 July 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The main proteinase (Mpro), or 3CLpro, is a critical enzyme in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lifecycle and is responsible for breaking down and releasing vital functional viral proteins crucial for virus development and transmission. As a catalytically active dimer, its dimerization interface has become an attractive target for antiviral drug development. Recent research has extensively investigated the enzymatic activity of Mpro, focusing on its role in regulating the coronavirus replication complex and its significance in virus maturation and infectivity. Computational investigations have identified four druggable pockets, suggesting potential allosteric sites beyond the substrate-binding region. Empirical validation through site-directed alanine mutagenesis targeted residues in both the active and allosteric regions corroborated these predictions. Structural studies of drug target proteins can inform therapeutic approaches, with metadynamics simulations shedding light on the role of H163 in regulating Mpro function and providing insights into its dynamic equilibrium to the wild-type enzyme. Despite the efficacy of vaccines and drugs in mitigating SARS-CoV-2 spread, ongoing viral evolution, selective pressures, and continued transmission pose challenges, potentially leading to resistance mutations. Phylogenetic analyses indicated the existence of several resistance variations predating drug introduction to the human population, emphasizing the likelihood of drug spread. Hydrogen/deuterium-exchange mass spectrometry reveals the structural influence of the mutation, while clinical trials on 3CLPro inhibitors underscore the clinical significance of reduced enzymatic activity and offer avenues for future therapeutic exploration. Understanding the implications of 3CLPro mutations holds promise for shaping forthcoming therapeutic strategies against COVID-19. This review delves into factors influencing mutation rates and identifies areas warranting further investigation, providing a comprehensive overview of Mpro mutations, their categorization, and terminology. Moreover, we examined their associations with clinical outcomes, illness severity, unresolved issues, and future research prospects, including their impact on vaccine efficacy and potential therapeutic targeting.
Keywords:
SARS-CoV-2 Main Protease/3CLPro
; mutations
; vaccine efficacy
; Enzymatic activity
; therapeutic targeting
; Structural alterations
; Antiviral therapy
; Dimerization interface
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) 3C-like protease (3CLpro) is a critical enzyme that breaks down and releases vital functional viral proteins that are necessary for virus development and transmission. Since 3CLpro is a catalytically active dimer, its dimerization interface has become a desirable target for the development of antiviral drugs [1]. The introduction of single amino acid changes at important sites at this interface has been investigated by structural research with the goal of comprehending the complex link between dimerization and catalytic activity [2].
Investigations have been conducted on the effects of single amino acid changes at regions of the 3CLpro dimer interface. Alanine substitutions at residues S1 and E166, in particular, showed interesting effects at site 1, with S1 displaying a twofold increase and E166 exhibiting lower relative activity [3,4]. Mutations in 3CLpro can impact viral replication and protein processing, potentially affecting viral infectivity and pathogenicity. Understanding these mutations is crucial for vaccine development and antiviral drug design, as they may alter the efficacy of therapeutic interventions. Surveillance of 3CLpro mutations is essential for monitoring viral evolution and the emergence of variants with altered phenotypes, informing public health strategies to combat the ongoing COVID-19 pandemic. Furthermore, in recent studies, mutations at site 2, such as S10, E14, and K12, have shown varying results, with some eliminating all activity entirely, while others maintaining approximately 60% of the relative activity [5]. With the exception of S139A, which showed 46% relative activity, mutations at residues R4, E290, and Q299 at site 3 completely eliminated all activity [6]. Remarkably, the relationship between the oligomerization states and catalytic activity of the mutants contradicted conventional thinking on the interaction between dimerization and enzyme performance [7].
In an attempt to investigate the possibility of drug resistance variations, a triple mutant (L50F E166A L167F) with noticeably higher 50% effective concentration (EC50) values for several 3CLpro inhibitors was found [8]. Although these alterations increase resistance, they also result in a significant decrease in enzymatic activity, which raises the possibility of a decrease in viral fitness [9]. Understanding changes in inhibitor/enzyme interactions through structural analysis helped to explain the observed evolution of resistance [10]. These results highlight the intricate relationship between enzyme activity and inhibitor resistance, which advances our knowledge of treatment approaches that target 3CLpro. In another study, the activity of every potential single mutant of SARS-CoV-2 3CLpro was methodically profiled using a thorough mutational scanning technique. The investigation demonstrated the protease's extraordinary flexibility and its capacity to withstand changes in any part of the protein, including the substrate binding site [11,12]. Despite this ability to adapt, some residues essential for protease activity were found, indicating possible new targets for the development of 3CLpro inhibitors in the future [13]. Furthermore, the discovery that the E166V mutation confers resistance against the therapeutic 3CLpro inhibitor nirmatrelvir emphasizes the findings' clinical significance and their implications for drug development concerning the present and potential coronavirus pandemics [14].
To elucidate the mechanism of MPro inhibition, ebselen derivatives were produced in parallel. The results of the investigation showed that adding an electron-withdrawing group (NO2) to the original ebselen inhibitor doubled its inhibitory potency [15]. The specific mechanism of interaction between the novel ebselen derivative and MPro was revealed by structural and biochemical investigations, which demonstrated that MPro was inhibited by the derivative via inhibition of the catalytic Cys145 [16]. In addition, the crystal structure of the catalytically inactive mutant H41N-MPro revealed gatekeeper residues in the substrate binding pocket that prevent substrate binding and shed light on the protein's inhibited state [17]. These results significantly increase our understanding of the mechanism underlying Mpro inhibition and offer important insights into the development of covalent inhibitors [17].Furthermore, the 3CL or nsp5 protease is essential for the maturation of viral proteins during host infection in the setting of SARS-CoV-2 infection [18]. Molecular dynamics simulations were carried out for SARS-CoV-2 3CLpro variants associated with substrates nsp 4|5 and nsp 5|6, including beta and Omicron variants. According to the simulations, conformational changes and substrate binding affinities were not significantly affected by mutations in the 3CLpro versions [19]. Nevertheless, the Beta and Omicron variants showed noticeably high cleavage rates for the nsp4–nsp5 boundary, indicating a possible critical role in viral replication and fitness gain [20]. The importance of Gly143 and Glu166 in substrate recognition was shown by hydrogen bonding investigations, indicating that these residues could be used as possible pharmacophoric centers in the design of inhibitors against beta and Omicron variations [21]. These results imply that mutations may affect the cleavage rate, despite the computational limitations of the study [22]. The goal of this research was to understand how these mutations affect 3CLpro function mechanistically and how they could affect the development of antiviral drugs. These findings could inform future experimental research and drug design approaches.
2. SARS-CoV-2 Main Protease (3CLPro)
3CLPro is a critical enzyme for viral replication, cleaving polyproteins into functional units necessary for the virus's lifecycle (see Figure 1). As a highly conserved and essential component of the virus, 3CLPro is a prime target for antiviral drug development. Inhibitors of 3CLPro have the potential to halt viral replication, making it a focal point in the fight against COVID-19. It is a promising option for antiviral therapy due to its ability to regulate the activities of the coronavirus replication complex [23]. Remarkable substrate-binding site conservation was revealed by the crystal structures of human coronavirus (strain 229E) Mpro, a porcine coronavirus (transmissible gastroenteritis virus [TGEV]) Mpro inhibitor complex, and a homology model for SARS coronavirus (SARS-CoV) Mpro [24]. The capacity of recombinant SARS-CoV Mpro to cleave a TGEV Mpro substrate supports this conservation [24]. A viable path for future drug development is the potential modification of already available rhinovirus 3CLpro inhibitors for the treatment of SARS, according to molecular modeling [25].
Due to its critical function in the proteolytic processing of viral replicase polyproteins, the coronavirus protease nsp5, commonly referred to as MPro or 3CLpro, continues to be the principal target for coronavirus therapy [26]. Considering the variety of known coronaviruses, the critical function of nsp5 in coronavirus biology should be highlighted. This study offers a thorough examination of the structure and function of this protease in relation to different coronaviruses, assesses previous and ongoing attempts to create inhibitors, and suggests innovative strategies for successful therapeutic treatments [27]. This evaluation will direct the development of drugs targeting the coronavirus protease nsp5 given the recent advent of SARS-CoV-2.
As an essential component of the CoV life cycle, 3CLpro breaks down viral polyproteins to produce mature, nonstructural proteins [28,29]. Many 3CLpro inhibitors have been reported for human-related SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 [30]. 3CLpro inhibitors are known to be popular antiviral drugs [31]. Knowing the structural characteristics of 3CLpro and the function of its inhibitors offers important new perspectives for the creation and enhancement of antiviral strategies to treat CoV infections [25].
The 3CLpro enzyme remains essential for regulating coronavirus replication. A study screened a medicinal plant library, identifying possible lead compounds for further optimization in the drug development process to attack COVID-19, by analyzing the sequence and building a 3D homology model of SARS-CoV-2 3CLpro [32,33,34]. This strategy highlights how crucial it is to target 3CLpro to provide effective therapeutic approaches against coronavirus infections.
3. 3CLPro Mutational Landscape
The development of antiviral drugs focused on targeting 3CLpro of SARS-CoV-2. This enzyme is essential for the maturation and infectivity of the virus. Its enzymatic activity has been thoroughly examined in recent research, with an emphasis on the dimerization interface [35]. Previous studies have attempted to elucidate the complex relationship between dimerization and catalytic activity by means of mutational studies at important locations within this interface [36]. Remarkably, the outcomes disproved the widely held notion that catalytic ability requires the dimeric structure as a whole [36]. Significantly, the investigation pinpointed two distinct allosteric locations (R4/E290 and S10/E14) at the interface, providing insight into possible targets for the development of antiviral drugs that target the dimer interface as opposed to the active site [37]. This complex knowledge opens up new possibilities for the development of more potent antiviral drugs by providing insightful information on the adaptability of 3CLpro [38].
Moreover, studies on SARS-3CLpro have added new insights into its enzymatic function. The L141T mutation was used in the investigation to precisely disrupt the helical shape of the inactive monomer [39]. This unexpectedly resulted in the production of an enzymatically active monomer, challenging the generally accepted notion that the dimer structure is a necessary condition for preserving an active conformation[39]. Simple mutations have shown the enzyme to be highly adaptable, underscoring the need for in-depth knowledge of its structural dynamics and creating new avenues for drug creation that may not exclusively rely on its usual dimeric shape [40]. Models of SARS-CoV-2 3CLpro variations, especially those linked to variants of concern (VOCs), have been extremely helpful in understanding the dynamics of enzyme catalysis [10].
Although there were no significant structural alterations caused by these mutations, higher cleavage rates were noted at the borders. Notably, important residues such as Glu166 and Gly143 are essential for substrate recognition [41]. With this finding, the development of antiviral drugs that target beta and Omicron variants can better target these residues as possible pharmacophoric centers [41]. Research on the effects of mutations linked to newly emerging variations of concern (VOCs) provides support for the effectiveness of already available antivirals [42]. AVI-8053 and nirmatrelvir both showed continuous efficacy against MPro mutations (See Figure 2). This minimizes worries about emerging viral variations by demonstrating the continuing viability of MPro as a top-priority antiviral therapeutic candidate [42].
Furthermore, studies further investigated the Pro132His mutation in the Omicron version of the SARS-CoV-2 MPro, delving deeper into the field of allosteric regulation [43]. An allosteric feature was suggested by molecular dynamics simulations, which showed a change in conformational equilibrium and enhanced dynamics in the catalytic site entry loop [44]. The kinetics changed, but the catalytic efficiency remained the same, suggesting possible functional diversification [44]. This thorough knowledge of allosteric mutants becomes important when considering the evolution of viruses and guides the development of potent antiviral drugs that can combat the dynamic nature of the virus and its protease [45].
4. Structural Alterations of 3CLPro
4.1. Insights into How Mutations Affect the Overall Structure of 3CLPro.
3CLpro is an important target for the development of antivirals. Competitive inhibitors targeting the active site were the main focus of early research on the COVID-19 pandemic [46]. On the other hand, four druggable pockets were identified by recent computational investigations, which indicate possible allosteric sites that are not in the substrate-binding region [47]. Site-directed alanine mutagenesis was used to target residues in both the active and allosteric regions to empirically validate these predictions [48]. The outcomes showed that to maintain the thermodynamic stability and catalytic activity of 3CLpro, certain residues in both locations are necessary [49]. This experimental validation established a platform for the development of novel treatment strategies against COVID-19 by bridging the computational and experimental domains.
The structural changes in the SARS-CoV-2 MPro were investigated in relation to its vulnerability to conformational changes associated with redox reactions. A disulfide bond-adopting oxidized conformation point mutant (H163A) was found, indicating a defense mechanism against excessive oxidation [50]. Simulations using metadynamics have shed light on how H163A modifies conformational equilibrium and revealed possible large-scale rearrangements [51]. Comprehending these redox-related alterations improves our knowledge of MPro dynamics and provides novel directions for future SARS-CoV-2 studies [52].
Antiviral drugs, such as nirmatrelvir, can cause target protein alterations, which may enhance viral resistance [53]. Previous research utilized an all-encompassing methodology involving the integrated analysis of naturally occurring mutations with by residue breakdown to identify putative resistance-inducing mutations in 3CLpro [9]. The effects of these mutations were further clarified by molecular dynamics simulations, which showed that some variations had an increased activation free energy and a changed binding affinity [54]. This integrated method helps predict and target potential resistance mechanisms while offering insightful information about the dynamics of drug-protein interactions [45].
A computational investigation to clarify the 3D structure of Omicron 3CLpro was initiated by the appearance of the Omicron variant [55]. Due to changes in chemical interactions caused by recent changes in amino acids, molecular dynamics simulations have revealed possible obstacles to the efficacy of current drugs that target the SARS-CoV-2 primary protease [56]. Targeting conserved areas despite developing variations is crucial, as one study emphasized, and it is important to use computational and in vitro approaches to find novel 3CLpro inhibitors [57]. Comprehensive knowledge of the effects of viral mutations on therapeutic approaches is facilitated by the combination of structural insights and dynamic simulations.
Moreover, to investigate ebselen derivatives as possible inhibitors of MPro/3CLpro, the major protease of SARS-CoV-2, chemical synthesis and knowledge of their modes of interaction are needed [58]. Modifications improved the effectiveness of the inhibition, as demonstrated by LC‒MS data and crystal structures [59]. The binding profile of ebselen-based inhibitors was determined using inhibition experiments and predictions from molecular modeling [60]. Furthermore, the crystal structure of a mutant that is catalytically inactive revealed the inhibited state and highlighted the function of gatekeeper residues [60]. By combining theoretical and experimental methods, this multimodal approach expands our knowledge of possible treatments that target the primary protease of SARS-CoV-2.
4.2. Examination of Changes in Key Structural Motifs and Domains
Delving into the repercussions of mutations within SARS-CoV-2 on the protein structure of MPro and its impact on antiviral therapy efficacy, MPro is one of the key components of SARS-CoV-2 and is crucial for the life cycle of the virus [61,62]. One of the possible reported inhibitors and treatment options for the MPro of SARS-CoV-2 is boceprevir [63]. A recent study examined the effects of mutations in SARS-CoV-2 on the protein structure of MPro and how these alterations affected the affinity of boceprevir, a significant candidate for therapy [64]. Computational tools such as RDP4, MegaX, and mutations were used, while ProMod3 and three-dimensional models assisted in constructing the mutant MPro [65,66]. Structural validation and modeling using qualitative model energy analysis, ProSA, and MolProbity provided insights into the conformational changes induced by these mutations (see Figure 3) [67].
In previous studies, molecular docking utilizing AutoDock 4.2 elucidated the binding affinity of boceprevir to the active site of MPro, revealing decreased affinity in the presence of mutations. DynOmics facilitated the development of models depicting the functional dynamic structures of MPro [68]. Furthermore, a study identified seven mutations within the MPro of SARS-CoV-2 (L89F, K90R, P108S, A191V, T224A, A234V, and S254F) with consequential effects on the binding affinity of boceprevir [67]. The affinity of the putative protease inhibitor boceprevir decreased as a result of these alterations [69]. When boceprevir was docked to the MPro active site, the binding energies for the wild-type and mutant forms were −10.34 and −9.41 kcal.mol-1, respectively [67]. Elastic network model research revealed Debye–Waller factors, underscoring alterations in the dynamic behavior of mutant MPro compared to that of its wild-type counterpart. These findings underscore the potential challenge posed by mutations in critical pharmacological targets, potentially rendering existing treatments ineffective against SARS-CoV-2 [55,67].
This review illuminates the intricate interplay between mutations in SARS-CoV-2 and the efficacy of pharmacological interventions, particularly focusing on MPro and its interaction with the putative protease inhibitor boceprevir and other antivirals. Through comprehensive computational analyses and modeling techniques, the structural consequences of mutations on MPro were elucidated, shedding light on potential mechanisms underlying altered drug binding. The observed decrease in the affinity of treatments such as boceprevir for mutant MPro highlights the dynamic nature of virus‒host interactions and underscores the need for continuous surveillance and adaptation in therapeutic strategies to combat evolving viral variants. These insights have significant implications for the development of future therapeutics and underscore the importance of understanding viral mutational landscapes in the context of global health emergencies such as COVID-19. The structural alterations in 3CLpro/main protease were elucidated, as were their potential consequences (see Table 1).
5. Consequences for Viral Replication of SARS-CoV-2
5.1. Relationship between 3CLPro Mutations and Viral Replication
Similar to past coronaviruses such as SARS-CoV and MERS-CoV, the advent of SARS-CoV-2 has prompted a worldwide health emergency [70]. Initial drugs that targeted these ancient viruses proved ineffective against SARS-CoV-2, despite their structural similarities [71]. By examining the genomic, proteomic, and pathogenic features of SARS-CoV-2 infection, a thorough investigation has examined mutations and how they affect target proteins [72]. Therapeutic approaches are influenced by structural studies of drug target proteins, such as 3CLpro, which sheds light on the pathophysiology [73]. Significant differences exist between the cytokine profile and inflammatory signaling in SARS-CoV-2 infection, indicating the necessity for customized treatments because the small genetic variation makes current drugs ineffective (see Figure 3) [55].
Although vaccines have been created, finding antivirals against SARS-CoV-2, which has posed a serious threat to world health and prompted the designation of the pandemic, is still very important [74,75]. Since 3CLpro/MPro is involved in the maturation of viral proteins, 3CLpro/MPro, which is encoded by the viral genome, is a viable target for treatment [76]. One effective way to prevent viral replication is to inhibit 3CLpro, and structural research can offer important information for well-considered drug development. To create powerful protease inhibitors to fight COVID-19, this review highlights the significance of structure-guided drug design, which makes use of high-resolution structures and a variety of chemical scaffolds [77].
The MPro of SARS-CoV-2 indicates that it processes viral polyproteins, which is essential for the construction of replicase complexes [78]. One mechanism that has evolved to be evolutionarily conserved among coronaviruses is the cleavage of human MAGED2 by MPro [79]. According to a previous study, the MPro of the beta variant cleaves MAGED2 more effectively than does the MPro of the Omicron variant [79]. By interfering with the connection between the viral nucleocapsid protein and DNA, MAGED2 functionally prevents SARS-CoV-2 replication [79]. MPro inhibits MAGED2, revealing details on the intricate relationship between the virus and its host as well as possible targets for therapeutic intervention [79].
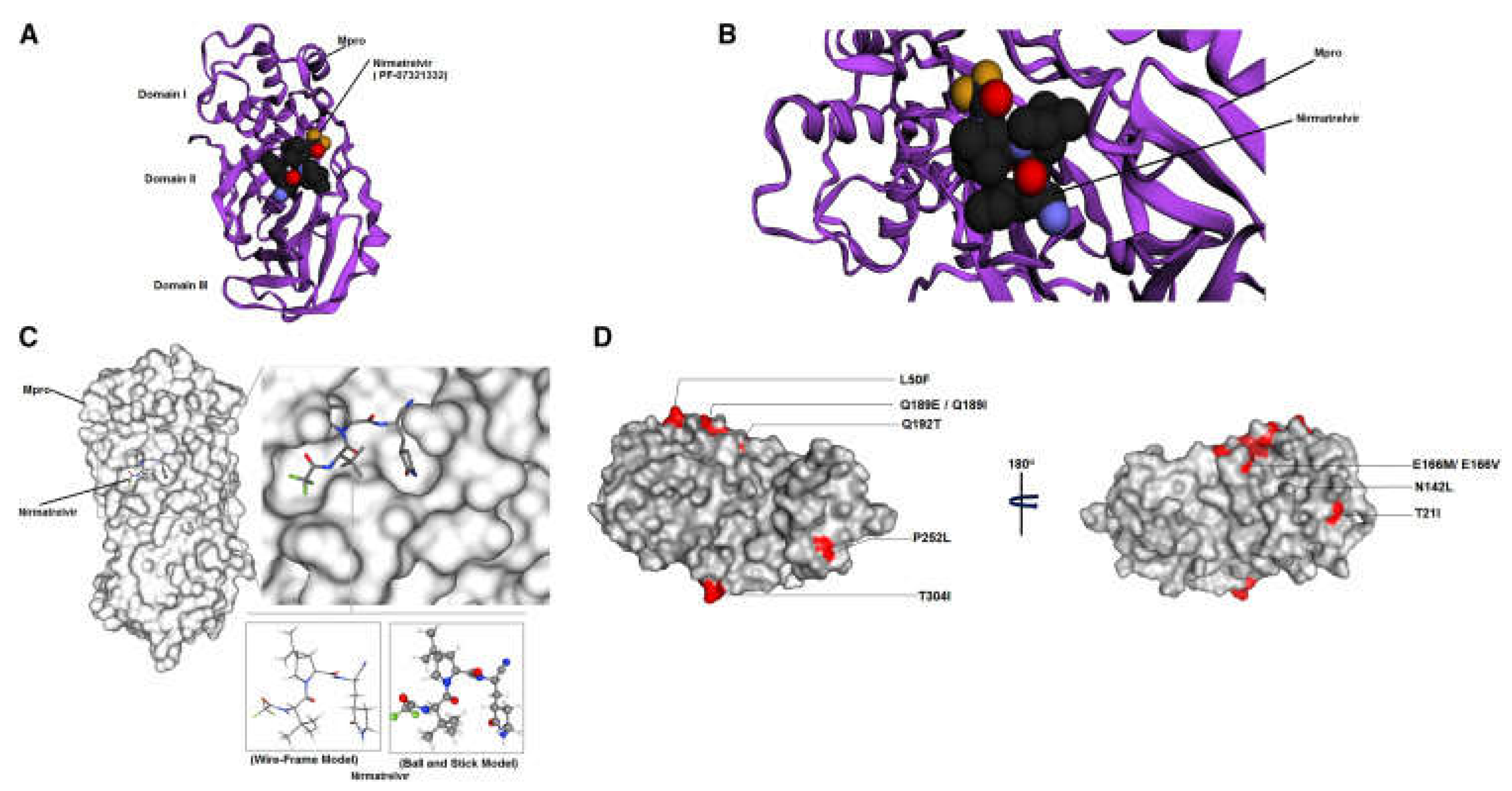
Figure 4.
Nirmatrelvir is shown attached to the MPro complex of SARS-CoV-2 as adapted from its source. These mutations have caused drug resistance, which has been reported from time to time. Several significant mutations have been detected in the SARS-CoV-2 Omicron variant and its subvariants that cause therapeutic escape. The phenomenon of therapeutic escape is of immense concern to scientists. Recently, several mutations have been noted in MPro/3CLpro, which might be responsible for nirmatrelvir resistance. The M49I mutation was discovered in 1,883 genomes in May 2022, with a minor increase in late 2021. Sasi et al. highlighted that the potency of nirmatrelvir is decreased by five mutations, namely, Q189E, Q192T, N142L, Q189I, and E166M. The IC50 of nirmatrelvir against the E166M mutation was reduced by a factor of 24. According to phylogenetic analyses, nirmatrelvir-resistant variants are transmissible and appear to have existed before nirmatrelvir was introduced into the human population. designed specific mutations to hinder nirmatrelvir’s ability to attach to its substrate to study nirmatrelvir resistance. They used 12 wild-type SARS-CoV-2 replicons and one subvariant of the Omicron (BA.1), followed by specific enzymatic assays and cell-based complementation. The results demonstrated that E166V conferred strong nirmatrelvir resistance, approximately 55-fold, with a significant decrease in wild-type replicon fitness (nearly 20-fold) but not in the BA.1 subvariant (2-fold) in both cases. However, L50F improved the fitness of the wild-type replicons (https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531%2823%2900072-0).
Figure 4.
Nirmatrelvir is shown attached to the MPro complex of SARS-CoV-2 as adapted from its source. These mutations have caused drug resistance, which has been reported from time to time. Several significant mutations have been detected in the SARS-CoV-2 Omicron variant and its subvariants that cause therapeutic escape. The phenomenon of therapeutic escape is of immense concern to scientists. Recently, several mutations have been noted in MPro/3CLpro, which might be responsible for nirmatrelvir resistance. The M49I mutation was discovered in 1,883 genomes in May 2022, with a minor increase in late 2021. Sasi et al. highlighted that the potency of nirmatrelvir is decreased by five mutations, namely, Q189E, Q192T, N142L, Q189I, and E166M. The IC50 of nirmatrelvir against the E166M mutation was reduced by a factor of 24. According to phylogenetic analyses, nirmatrelvir-resistant variants are transmissible and appear to have existed before nirmatrelvir was introduced into the human population. designed specific mutations to hinder nirmatrelvir’s ability to attach to its substrate to study nirmatrelvir resistance. They used 12 wild-type SARS-CoV-2 replicons and one subvariant of the Omicron (BA.1), followed by specific enzymatic assays and cell-based complementation. The results demonstrated that E166V conferred strong nirmatrelvir resistance, approximately 55-fold, with a significant decrease in wild-type replicon fitness (nearly 20-fold) but not in the BA.1 subvariant (2-fold) in both cases. However, L50F improved the fitness of the wild-type replicons (https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531%2823%2900072-0).
Inhibitor repurposing or de novo medication discovery has become essential for long-term measures against viruses comparable to the global spread of SARS-CoV-2 and similar viruses in the future [80]. The structural differences between SARS-CoV-2 and the SARS MPro are shown by in-depth simulations of the MPro, suggesting difficulties in the repurposing of drugs [81]. Rapid drug design is hindered by the mutability of the virus and the flexibility of the binding site [82]. The rational design of small-molecule inhibitors is complicated by the lack of recognized stabilizing residues [83]. This extensive analysis offers thorough knowledge of the difficulties in creating viable treatments to combat SARS-CoV-2.
Moreover, the functional implications of SARS-CoV-2 MPro vulnerability to redox-associated conformational changes have been called into doubt by recent research [50]. The crystal structure of an oxidized MPro point mutant (H163A) points to a potential defense mechanism against oxidative stress [50]. The equilibrium shift caused by point mutations was clarified by metadynamics simulations, suggesting directions for novel studies [51]. A comprehension of the ways in which H163 modifies this equilibrium offers insights into the intricate dynamics of SARS-CoV-2 [51]. In addition to identifying possible targets for therapeutic intervention, this research advances our knowledge of virus‒host interactions [84].
5.2. Insights into How Mutations May Influence the Overall Fitness of the Virus
Autoactivation of the main protease (MProWT) from precursor polyproteins is a critical step in the complex process of assembly and maturation of SARS-CoV-2 [85]. A study revealed how mutations affect MPro autoprocessing, which is essential for viral maturation [86]. A mutation in MPro called E290A causes time-dependent autoprocessing at cleavage sites, which affects the generation of dimers. On the other hand, MPro, a precursor including mutations in E290A and R298A, has modified cleavage patterns, resulting in intermediate monomeric products [85]. The process suggested by these insights involves intramolecular cleavage of the N-terminus followed by intermolecular cleavage, which highlights the possibility of developing drugs that specifically target the mostly monomeric MPro precursor (see Figure 5) [85].
Effective antiviral drugs are becoming increasingly necessary as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) evolves and becomes increasingly dangerous [28]. A key pharmacological target is the major protease (MPro), yet resistance issues have been raised [53]. A thorough mutational scan of MPro identified a network of essential amino acids that are mutagenically sensitive, providing prospective sites for inhibitor targeting [48]. Fitness landscapes reveal surface regions that are not active sites that serve as effective targets for inhibitors [45]. Human population-wide clinical variations in MPro show functional competence, indicating high selection pressure and potential for treatment resistance [87]. The MPro mutational guide offers important information for creating inhibitors that have a lower chance of causing viral resistance to evolve [88].
Moreover, concerns over new variations impacting vaccine efficacy were raised during the COVID-19 pandemic, underscoring the need for potent antiviral drugs [89]. Drug development could be centered on the MPro of SARS-CoV-2, a well-known therapeutic target [90]. A promising clinical MPro inhibitor is nirmatrelvir, a peptidomimetic inhibitor [91]. The catalytic competence of MPro variants from different SARS-CoV-2 lineages is comparable to that of the wild type when they are expressed [91]. Notably, nirmatrelvir is still effective against these variations, indicating that its effectiveness is not compromised by changing COVID-19 variants [92].
Among coronavirus enzymes, SARS-CoV-2 3CLpro is an important therapeutic target because it is largely conserved [93]. The mutability and mutation tolerance of 3CLpro are fully understood due to the use of a deep mutational scanning method based on yeast [94]. The flexibility of the protein was demonstrated by the identification of specific residues as unchangeable, which may serve as targets for upcoming inhibitors [95]. Notably, E166V, a mutation that confers resistance to the 3CLpro inhibitor nirmatrelvir, which is used in clinical practice, was detected. [53] This functional map helps with coronavirus drug development methods by providing insights into the biological characteristics of 3CLpro. Specific mutations in 3CLpro can enhance viral replication efficiency, alter protein processing, and improve immune evasion, thereby increasing viral survival and transmissibility. These changes can also lead to the emergence of drug-resistant strains, posing significant challenges for therapeutic interventions and public health management.
6. Host Immune Evasion in SARS-CoV-2
6.1. Exploration of Potential Mechanisms by Which 3CLPro Mutations Contribute to Immune Evasion
3CLPro mutations contribute to immune evasion in SARS-CoV-2, which is crucial for understanding the virus's ability to persist and cause disease. 3CLPro is essential for processing viral polyproteins into functional units necessary for viral replication. Mutations affecting autoprocessing that result in different cleavage patterns are found in MPro, including E290A and E290A/R298A [96]. The population of dimers increases via intramolecular N-terminal cleavage, which is followed by intermolecular C-terminal cleavage [85]. Drug development against SARS-CoV-2 may benefit from the inhibition of mostly monomeric MPro precursors [97]. Mutations in 3CLPro can alter its substrate specificity and processing efficiency, leading to variations in viral protein expression. These variations can obscure viral epitopes from being effectively presented on major histocompatibility complex (MHC) molecules, which reduces the recognition and response of cytotoxic T lymphocytes. This evasion from T-cell surveillance allows the virus to replicate and spread within the host with reduced immune interference. Changes in the 3CLPro structure might influence its interaction with host cell proteins involved in antiviral responses, such as those regulating interferon signaling pathways, thereby further dampening the host's immune defenses.
Furthermore, 3CLPro mutations may impact enzyme susceptibility to protease inhibitors, which are key components of antiviral therapy. Structural changes caused by these mutations can reduce the binding affinity of inhibitors, rendering treatments less effective and allowing the virus to evade therapeutic interventions. This poses a significant challenge for the development of antiviral drugs, as resistance can develop, necessitating continuous monitoring and adaptation of therapeutic strategies. Additionally, these mutations can enhance viral fitness by improving replication efficiency or by more effectively evading host immune responses (Figure 4).
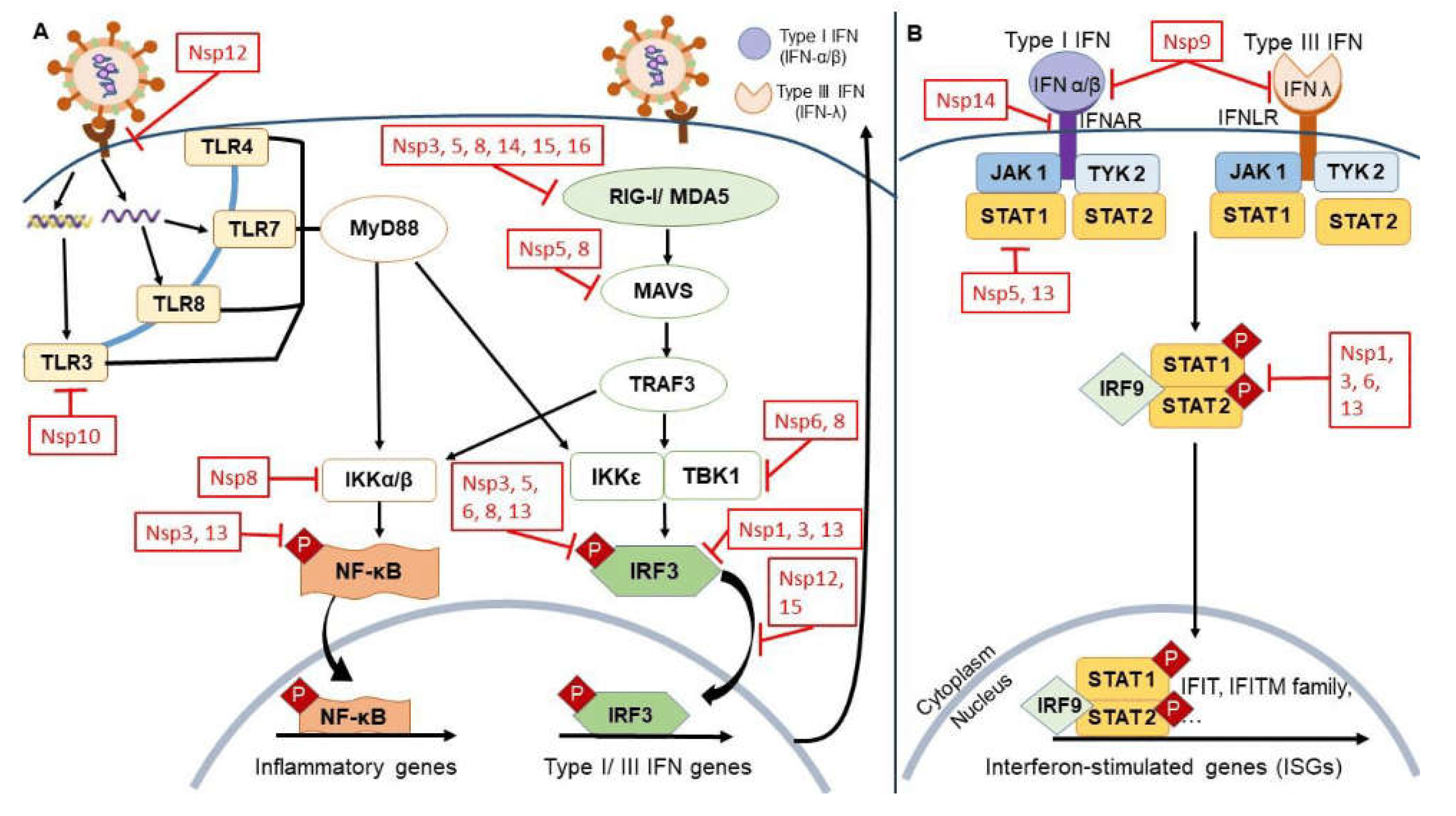
Figure 6.
An overview of the SARS-CoV-2 nonstructural proteins that contribute to host immune escape (in red boxes). (A) Pathogen-associated molecular patterns (PAMPs) of viruses are recognized by various immune cells (macrophages, monocytes, neutrophils, and dendritic and epithelial cells). Upon infection, these immune cells recognize foreign viral antigens and molecules (PAMPs) via pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), thus stimulating cytokines and IFNs and subsequently inducing host immune responses. (B) Type I and type III IFNs are produced and bind to their specific cell surface IFN receptors, thus activating JAK/STAT signaling to promote IFN-stimulated genes (ISGs) to achieve antiviral responses. (Adapted from Park et al. and https://doi.org/10.3390/v14091991).
Figure 6.
An overview of the SARS-CoV-2 nonstructural proteins that contribute to host immune escape (in red boxes). (A) Pathogen-associated molecular patterns (PAMPs) of viruses are recognized by various immune cells (macrophages, monocytes, neutrophils, and dendritic and epithelial cells). Upon infection, these immune cells recognize foreign viral antigens and molecules (PAMPs) via pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), thus stimulating cytokines and IFNs and subsequently inducing host immune responses. (B) Type I and type III IFNs are produced and bind to their specific cell surface IFN receptors, thus activating JAK/STAT signaling to promote IFN-stimulated genes (ISGs) to achieve antiviral responses. (Adapted from Park et al. and https://doi.org/10.3390/v14091991).
Moreover, site-directed mutagenesis verified critical residues in druggable pockets, including allosteric regions, as determined by computational analysis [98]. Changes in the catalytic activity and thermodynamic stability of the 3CLpro active and allosteric regions indicate possible targets for competitive and noncompetitive inhibitors against COVID-19 [6]. By residue analysis is required to identify resistance-conferring mutations and guide inhibitor design techniques since antiviral drugs such as nirmatrelvir, a 3CL protease inhibitor, have the potential to cause mutations [48].
The COVID-19 epidemic emphasizes how important treatment choices are in the face of viral evolution [99]. Since December 2021, MPro has experienced rapid mutational dynamics, especially in the vicinity of the active site, according to genomic data analysis [100]. The development of antiviral resistance is suggested by this increased mutational variability, underscoring the significance of tracking and accounting for these differences in drug development techniques [45].
In an effort to combat COVID-19, the FDA has approved PAXLOVID, a combination therapy that includes nirmatrelvir, which was reported to inhibit 3CLpro activity [101,102]. This approach takes structural and evolutionary aspects into account, emphasizing residues that are susceptible to mutation. The effects go beyond creating broad-spectrum antivirals and fighting drug-resistant virus mutations [53]. Investigating these mechanisms requires a multidisciplinary approach that integrates structural biology to elucidate the effects of specific mutations, virology to understand the impact on viral replication and pathogenesis, and immunology to assess changes in immune recognition. Such comprehensive studies are essential for designing next-generation therapeutics and vaccines that can counteract these evasion strategies and provide robust protection against SARS-CoV-2 and its variants.
6.2. Discussion on the Role of Mutations in Modulating Host‒Virus Interactions
Mutations in the SARS-CoV-2 3CLpro/Mpro gene are pivotal for modulating host‒virus interactions and significantly influence viral replication efficiency and immune evasion. 3CLpro is essential for the processing of viral polyproteins into functional components required for viral replication. Mutations in this protease can modify its substrate specificity and catalytic efficiency, leading to altered viral protein maturation. Such changes can impact the presentation of viral peptides on major histocompatibility complex (MHC) molecules, thereby affecting recognition by cytotoxic T lymphocytes and facilitating immune evasion. Additionally, these mutations might influence protease interactions with host proteins, such as those involved in antiviral signaling pathways, further dampening the host immune response. Structural alterations in 3CLpro due to mutations can also affect the binding affinity and efficacy of protease inhibitors, posing challenges for antiviral drug development. By enhancing the protease’s ability to evade immune detection and resist therapeutic interventions, these mutations contribute to the persistence and pathogenicity of the virus. Understanding the role of 3CLpro mutations in modulating host‒virus interactions is crucial for developing targeted antiviral strategies and improving the effectiveness of therapeutic interventions against SARS-CoV-2.
Due to its conservation and lack of a human analog, dimeric MPro, which is essential for viral proteolytic cleavage, is a top prospect for therapeutic development [64]. Increasing our knowledge of MPro behavior is essential for successful treatment [112]. Like SAR-CoV, all-atom molecular dynamics simulations of fifty mutant MPro dimers from the GISAID database shed light on the behavior of the SARS-CoV-2 MPro [41]. Mutations in residues GLY15, VAL157, and PRO184, the formation of a distinct loop with the D48E variation, and information on substrate binding surface broadening with a noncanonical position for PHE140 are among the new discoveries [41]. Furthermore, the associated compaction dynamics in dual allosteric pockets and the function of certain residues, such as 17 and 128, in dimer stability were clarified [41].
The use of antiviral drugs, such as nirmatrelvir, a covalent inhibitor of the primary protease of SARS-CoV-2 (3CL protease), has raised questions regarding the possibility of drug resistance due to induced mutations [53]. Understanding naturally occurring mutations and noncovalent interactions helps identify possible resistance-inducing mutations in the 3CL protease [12]. The effects of mutations are investigated using QM/MM methods, which emphasize the reduced binding affinity and increased activation free energy of the E166V variation as contributing factors to the observed resistance [122]. These findings may help with the design of inhibitors to address potential resistance mechanisms by revealing the effect of nirmatrelvir on the fitness landscape of the virus.
A crucial target for antiviral therapy is the SARS-CoV-2 3CLpro, a study investigating allosteric locations for reversible noncompetitive inhibitor design, but most related research has focused on competitive inhibitors that target the active site [46]. Four druggable pockets are predicted computationally, and these predictions are validated experimentally by site-directed alanine mutagenesis, which reveals important residues in both the active and allosteric locations. These crucial residues are important because mutations in these residues can reduce or inactivate 3CLpro activity [123]. The basis for developing both competitive and noncompetitive inhibitors as possible COVID-19 treatments is provided by this experimental validation.
Protease inhibitors are powerful antiviral medications, and the U.S. FDA has authorized palovid (nirmatrelvir/ritonavir), the first protease inhibitor against SARS-CoV-2 [124,125]. An engineered chimeric vesicular stomatitis virus (VSV) that relies on the protease was created to find mutations that are resistant to nirmatrelvir [48]. Mutations generated by selective pressure were verified through reintroduction into 3CLpro and assessment using a cellular assay. The predicted alterations show potential emerging resistance variations by alignment with current SARS-CoV-2 sequences [48]. This method helps to characterize the susceptibility of viruses to nirmatrelvir and provides insights into resistance mechanisms. Moreover, these mutations may affect the interaction of PLpro with host cellular proteins, potentially altering the dynamics of viral replication and host cell manipulation. By modulating these interactions, SARS-CoV-2 can achieve a more favorable environment for replication and persistence, highlighting the need for ongoing research to understand the implications of PLpro mutations on viral pathogenicity and immune evasion. Understanding these mutations is critical for the development of antiviral strategies that can effectively target 3CLpro and mitigate its immune evasion capabilities.
7. Antiviral Drug Resistance in SARS-CoV-2
7.1. Evaluation of How Mutations in 3CLPro May Confer Resistance to Existing Antiviral Drugs
Since nirmatrelvir is the first clinical protease inhibitor to be licensed for use against the primary protease of SARS-CoV-2, 3CLpro, it is a powerful antiviral drug [126]. A chimeric vesicular stomatitis virus (VSV) system that relies on 3CLpro autocatalytic processing of a polyprotein for viral replication was created to investigate resistance mechanisms against nirmatrelvir [127]. After applying nirmatrelvir to select for resistance mutations, several independent tests, such as VSV-based systems, cellular assays, biochemical assays, and a recombinant SARS-CoV-2 system, were used to validate the results [128]. Certain mutants exhibited cross-resistance to GC376 and Ensitrelvir. It was previously discovered that these resistance mutations were present in circulating SARS-CoV-2 strains, indicating their existence in the GISAID and NCBI databases [129].
Although vaccines and drugs have helped to reduce the spread of SARS-CoV-2, the ongoing evolution of the virus, growing selective pressures, and continued transmission present difficulties that could result in resistance mutations [130]. Extensive studies on the susceptibility of natural variants of the major protease of SARS-CoV-2 (Mpro/3CLpro) to protease inhibitors have shown that MPro has several single amino acid modifications that confer resistance to nirmatrelvir, the active component of paraclovir [129]. The resistance mutation profile to Ensitrelvir, a clinical-stage inhibitor, is unique[129]. The possibility of these resistant variations spreading is highlighted by phylogenetic analyses, which highlight the fact that several of them existed before these drugs were introduced to the human population[129]. These results highlight the importance of maintaining vigilant resistance and developing a variety of protease inhibitors and antiviral drugs with unique modes of action and resistance profiles to facilitate the development of successful combinatorial treatments.
7.2. Implications for Drug Development Strategies
Given its vital function in virus replication, the primary protease of SARS-CoV-2 (MPro) is an important target for antiviral drug research [131]. Concerns about possible changes in MPro that change its structural and functional features and, as a result, affect the effectiveness of current and future antiviral drug surfaces include the emergence of novel variations of concern (VOCs) [55]. Through the crystal structures of 11 MPro mutants, changes in substrate binding and the rate of cleavage of a viral peptide were revealed by an analysis of 31 mutations spanning five VOCs [132]. Interestingly, mutations affect proteolysis of the immunomodulatory host protein Galectin-8 (Gal-8), which leads to a notable reduction in cytokine release, suggesting changes in host antiviral pathways (see Figure 5) [42]. Mutations linked to the extremely pathogenic Delta VOC and Gamma VOC significantly increased Gal-8 cleavage [42]. Significantly, the IC50s of AVI-8053, an irreversible inhibitor, and nirmatrelvir (Pfizer) demonstrated constant efficacy against all mutations, confirming the ongoing status of Mpro as a top-priority antiviral drug candidate during SARS-CoV-2 evolution [42].
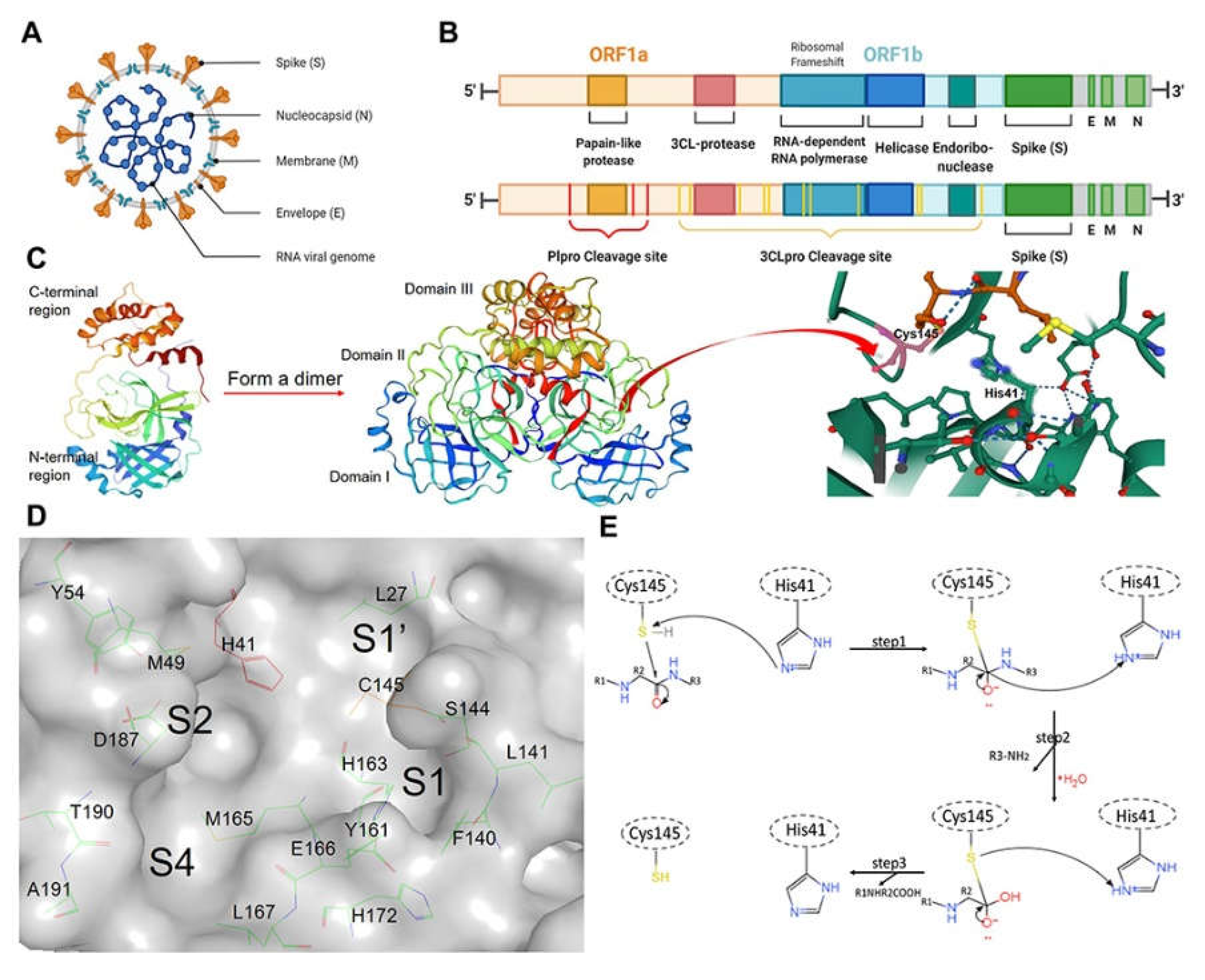
Figure 7.
Redrawn Diagram Progress on SARS-CoV-2 3CLpro inhibitors as adapted from the source: (A) Schematic diagram of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) morphology. (B) Number of cleavage site numbers of papain-like protease (PLpro) and 3C-like proteinase (3CLpro) in the polypeptide chain. (C) SARS-CoV-2 3CLpro 3D structure (monomer and dimer) and the N-terminal catalytic dyad (His41-Cys145) (D). The substrate-binding pocket of SARS-CoV-2 3CLpro included the S1’-S1-S2-S4 core sites. (E) SARS-CoV-2 3CLpro catalytic mechanism (https://doi.org/10.2147/DDDT.S359009).
Figure 7.
Redrawn Diagram Progress on SARS-CoV-2 3CLpro inhibitors as adapted from the source: (A) Schematic diagram of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) morphology. (B) Number of cleavage site numbers of papain-like protease (PLpro) and 3C-like proteinase (3CLpro) in the polypeptide chain. (C) SARS-CoV-2 3CLpro 3D structure (monomer and dimer) and the N-terminal catalytic dyad (His41-Cys145) (D). The substrate-binding pocket of SARS-CoV-2 3CLpro included the S1’-S1-S2-S4 core sites. (E) SARS-CoV-2 3CLpro catalytic mechanism (https://doi.org/10.2147/DDDT.S359009).
In the middle of the worldwide response to the coronavirus pandemic by nontherapeutic means and mass vaccination campaigns, therapeutic alternatives, specifically major protease (MPro) inhibitors for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), continue to be vital [91]. MPro is a relatively conserved target for drugs, even though antiviral agents are susceptible to the impacts of viral mutations [112]. Since December 2021, some countries have authorized the conditional or emergency use of nirmatrelvir, an antiviral that targets the MPro active site; numerous additional inhibitors are currently undergoing clinical study [133]. Accelerated mutational dynamics have been observed in MPro since early December 2021, according to a thorough examination of recent SARS-CoV-2 genomic data [134]. An eight-residue region (R188-G195) showed a considerable increase in mutational diversity close to the active site, which raised concerns regarding the possible emergence of antiviral resistance. This changing diversity calls for constant lead optimization and careful observation in ongoing drug development [134].
Although drugs and vaccinations have helped lessen the severity of the illness and prevent the spread of SARS-CoV-2, problems still exist due to the continuous evolution of the virus and growing selection pressures [135]. An examination of how naturally occurring variations in the MPro respond to protease inhibitors revealed several single amino acid mutations that confer resistance to nirmatrelvir, the active ingredient in Paxlovid. Ensitrelvir (Xocova), another inhibitor in the clinical stage, has shown a unique resistance mutation profile [136]. Phylogenetic analysis revealed that before these drugs were introduced to the human population, resistant variations already existed and were able to propagate[136]. These results highlight the significance of maintaining resistance mutations and creating more protease inhibitors and antiviral drugs with a variety of modes of action and resistance profiles to facilitate successful combinatorial therapy.
8. Experimental Approaches for SARS-CoV-2 3CLPro Mutations
8.2. Overview of the Experimental Techniques Used to Study the Consequences of 3CLPro Mutations
The SARS-CoV-2 MPro has undergone changes, and experimental methods to understand the effects of these mutations have combined structural and computational methods [103]. The vulnerability of MPro to redox-associated conformational changes caused by oxidative stress in cells and the immune system has been the subject of recent studies[50]. The oxidized conformation of an MPro point mutant (H163A), in which the catalytic cysteine forms a disulfide bond, was confirmed by crystallography [50]. Understanding the mechanisms underlying conformational changes in response to oxidative stress is made possible by this structural insight [50]. Furthermore, metadynamics simulations were used to clarify the possible function of H163 in regulating this shift, providing a dynamic view of the equilibrium that is likely present in the wild-type enzyme [50]. Other point mutations have been extensively investigated beyond structural investigations, and their effects on changing the conformational free energy and moving the equilibrium toward the oxidized state were found to be considerable [50]. A thorough understanding of how particular residues, such as H163, affect the conformational dynamics of MPro is made possible by these various experimental techniques, which range from crystallography to computational simulations [104]. These insights may be used to direct focused therapeutic interventions.
Through the integration of crystallography and metadynamics simulations, scientists have investigated the functional implications of mutations in MPro [105]. This combined experimental approach provides new opportunities for studying SARS-CoV-2 and suggests possible targets for drug discovery and therapeutic intervention in addition to improving our understanding of the structural effects of mutations (see Table 3).
9. Future Perspectives on Tackling Similar Pandemics in the Future
Understanding the consequences of 3CLPro mutations offers great potential for influencing upcoming therapeutic approaches in the fight against COVID-19 and future pandemics [106]. SARS-CoV-2 variations have unavoidably emerged as a result of the global increase in infections; significant changes have been found throughout the viral genome, most notably in the spike protein [107]. Mutations such as D614G and N501Y have been linked to changes in virulence and transmissibility [108]. B.1.17, B.1.351, P.1, B.1.617.2, and B.1.1.529 are examples of variants of concern (VOCs) that have sparked worries because of their greater transmissibility, increased severity of the disease, and ability to escape immunity caused by vaccinations and spontaneous infection [109]. In recent studies, a thorough analysis of the landscape of mutations in structural and nonstructural proteins was performed to evaluate how they affect vaccine efficacy, treatments, and diagnostics [110].
Vaccines and antiviral drugs are essential for efficient coronavirus control and treatment methods, particularly for the fatal SARS-CoV-2 virus [111]. The critical function of 3CLpro in viral replication makes it an important target for the development of antiviral drugs [46]. However, comparing and evaluating possible inhibitors is more difficult due to the variety of 3CLpro expression designs and kinetic tests [112]. Examining the various expression designs and assays used to gauge enzymatic activity is the main emphasis of various studies on SARS-CoV-2. To enable direct comparisons across SARS-CoV 3CLpro inhibitors that have been produced globally, this study highlights the significance of standard assay settings and designs and presents a novel Alexa488-QSY7 FRET-based peptide substrate for high-throughput screening [113].
With the 3CLPro inhibitor PF-07321332, recent clinical trials have demonstrated encouraging outcomes, including an 89% decreased risk of hospital admission or death linked to COVID-19 within three days of symptom onset [114]. A study that revealed that patients with a certain SARS-CoV-2 sublineage and a 3CLPro mutation (Pro108Ser) had a milder clinical course than those without the mutation provides support for this important discovery [115]. A 58% decrease in activity was found through enzymatic characterization, which was linked to structural alterations in the substrate-binding region [115]. The influence of the mutation on the structure was revealed by hydrogen/deuterium-exchange mass spectrometry. The mutation was located behind the 108th amino acid residue. The negative effects of affinity tags, nonnative sequences, or low enzyme concentrations on enzymatic activity have been demonstrated by experimental data [115]. In line with the encouraging results of 3CLPro inhibitor clinical trials, studies emphasize the clinical relevance of decreased 3CLPro enzymatic activity and open potential directions for future therapeutic approaches. This review explored the variables affecting mutation rates and suggested areas for further investigation.
Future perspectives in tackling similar pandemics involve a multifaceted approach that integrates advanced scientific research, global collaboration, and robust public health infrastructure. Investment in cutting-edge technologies such as genomic sequencing and artificial intelligence will enhance our ability to rapidly identify and characterize emerging pathogens and their mutations. The establishment of global surveillance networks and data-sharing platforms will facilitate real-time tracking of infectious diseases and enable swift responses to outbreaks. Furthermore, developing broad-spectrum antivirals and vaccines that target conserved viral components can provide preemptive defenses against a range of pathogens. Strengthening public health systems with better preparedness plans, resources, and training is essential for effective crisis management. By fostering international cooperation and leveraging scientific advancements, we can build a resilient framework capable of mitigating the impact of future pandemics, ensuring timely and effective protection of global populations.
10. Conclusion and Importance of Ongoing Research in the Context of SARS-CoV-2 Main Protease Mutations
Recent advancements into SARS-CoV-2 Mpro/3CLpro mutations have shown that it is crucial for understanding how changes in this enzyme may affect viral replication and drug efficacy. By identifying and characterizing these mutations, scientists can better anticipate resistance patterns and improve therapeutic strategies. This research is essential for developing robust antiviral treatments and ensuring long-term control of COVID-19. The Mpro of SARS-CoV-2 is a crucial target for inhibitor design because of its essential function in viral replication and substrate cleavage selectivity [24]. The requirement for the identification and structural characterization of MPro variants is highlighted by ongoing mutations caused by selection pressure and constant neutral drift [116]. Seventy-nine clinically detected MPro mutations were investigated in a thorough analysis that revealed patterns in viral evolution and the distribution of residue substitutions [117]. Differences in cohesion and active site flexibility were found by molecular modeling and protein structure network analysis, which is important for drug discovery projects [118]. Moreover, in another study, 102 naturally occurring MPro mutations were systematically characterized to identify certain residues linked to drug resistance. Drug resistance was validated by X-ray crystal structures and recombinant virus assays, highlighting the significance of maintaining an eye on these hotspots [53]. These findings highlight the importance of continuing research to understand and cope with the changing field of SARS-CoV-2 MPro mutations.
In response to the pressing need for SARS-CoV-2 treatments and to address future pandemics, current research endeavors focus on MPro as a prime candidate for pharmacological targeting [119]. Atomic-level interactions were understood by mechanistic insights obtained from molecular dynamics simulations of ligand-MPro complexes [103]. The identification of critical residues that stabilize ligands in catalytic sites and other pockets emphasizes the significance of a lateral pocket in the enhancement of catalytic sites [120]. Analysis of computational mutations demonstrated that HIS163 contributes to ligand binding [103]. This work provides an important molecular-level understanding for the development of strong inhibitors against SARS-CoV-2 MPro.
Furthermore, the critical significance of SARS-CoV-2 3CLpro and its high degree of conservation among coronaviruses make it an important therapeutic target [24]. The great flexibility and mutation tolerance of 3CLpro were revealed by a methodical profile of every potential single mutant using a yeast-based deep mutational scanning technique [121]. Future inhibitors may target immutable residues as potential targets [95]. The E166V mutation was also found to confer resistance to nirmatrelvir, a 3CLpro inhibitor that is used in clinical practice [53]. With the help of this functional map, researchers may better understand the biological characteristics of 3CLpro and direct the creation of anti-coronavirus drugs.
In conclusion, ongoing research into SARS-CoV-2 3CLpro mutations is crucial for advancing our understanding of the virus's evolution, pathogenicity, and mechanisms of immune evasion. The main protease is essential for the processing of viral polyproteins, which are critical for viral replication and the viral life cycle. Mutations in this protease can significantly alter its enzymatic activity, substrate specificity, and interaction with the host cell machinery. These changes can impact viral replication efficiency, the presentation of viral antigens, and viral susceptibility to protease inhibitors, all of which are pivotal for both the natural immune response and therapeutic interventions. By elucidating how specific mutations affect the structure and function of 3CLpro, researchers can better predict the emergence of more virulent or drug-resistant strains, guiding the development of more effective treatments and vaccines.
The importance of ongoing research in this area cannot be overstated, particularly as SARS-CoV-2 continues to evolve. Continuous monitoring and analysis of 3CLpro mutations will enable the scientific community to overcome potential challenges posed by new variants. This proactive approach is essential for the rapid adaptation of existing therapies and the design of new antiviral drugs that can effectively target mutated proteases. Moreover, understanding the broader implications of these mutations for host‒virus interactions will provide deeper insights into viral pathogenesis and immune evasion strategies. This knowledge is crucial not only for managing the current pandemic but also for preparing for future coronavirus outbreaks, ensuring that global public health responses are informed, effective, and resilient.
Author Contributions
Conceptualization, A.G.-A.M., S.C.U. and H.K.; writing-original draft preparation, A.G.-A.M., N.N.M. and S.C.U.; writing—review and editing, A.G.-A.M., S.C.U., N.A.M., T.W.M., M.G.K., F.T., M.M., R.G.M., N.N., B.S.A., K.J.B. and M.N.; supervision, H.M.K. and R.B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data availability statement: Not applicable.
Acknowledgments
The authors would like to thank the College of Health Sciences, University of KwaZulu-Natal.
Conflicts of interest: The authors declare no conflicts of interest.
References
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. International Journal of Biological Macromolecules 2021, 177, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Reja, A.; Pal, S.; Das, D. Systems chemistry of peptide-assemblies for biochemical transformations. Chemical Society Reviews 2022, 51, 3047–3070. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.S.; Hochberg, G.K.; Thornton, J.W. Simple mechanisms for the evolution of protein complexity. Protein Science 2022, 31, e4449. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K. Evolution of the SARS-CoV-2 proteome in three dimensions (3D) during the first 6 months of the COVID-19 pandemic. Proteins: Structure, Function, and Bioinformatics 2022, 90, 1054–1080. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Hilal, T.; Sunday, N.D.; Khatri, A.; Bürger, J.; Mielke, T.; Belogurov, G.A.; Loll, B.; Sen, R.; Artsimovitch, I. Steps toward translocation-independent RNA polymerase inactivation by terminator ATPase ρ. Science 2021, 371, eabd1673. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Rabeh, W.M. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. Journal of Biological Chemistry 2022, 298. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, F.; Sementa, D.; Jain, A.; Kumar, M.; Tayarani-Najjaran, M.; Kroiss, D.; Ulijn, R.V. Peptide-based supramolecular systems chemistry. Chemical Reviews 2021, 121, 13869–13914. [Google Scholar] [CrossRef] [PubMed]
- Havranek, B.; Demissie, R.; Lee, H.; Lan, S.; Zhang, H.; Sarafianos, S.; Ayitou, A.; Islam, S. Discovery of Nirmatrelvir Resistance Mutations in SARS-CoV-2 3CLpro: A Computational-Experimental Approach. Journal of chemical information and modeling 2023, 63. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Guzmán, C.A.; Andjelkovic, M.; Zinovjev, K.; Ruiz-Pernía, J.J.; Tuñón, I. The impact of SARS-CoV-2 3CL protease mutations on nirmatrelvir inhibitory efficiency. Computational insights into potential resistance mechanisms. Chemical Science 2023, 14, 2686–2697. [Google Scholar] [CrossRef]
- Jochmans, D.; Liu, C.; Donckers, K.; Stoycheva, A.; Boland, S.; Stevens, S.K.; De Vita, C.; Vanmechelen, B.; Maes, P.; Trüeb, B. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. MBio 2023, 14, e02815–e02822. [Google Scholar] [CrossRef]
- Kronenberger, T.; Laufer, S.A.; Pillaiyar, T. COVID-19 therapeutics: small-molecule drug development targeting SARS-CoV-2 main protease. Drug Discovery Today, 2023; 103579. [Google Scholar]
- Mótyán, J.A.; Mahdi, M.; Hoffka, G.; Tőzsér, J. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. International journal of molecular sciences 2022, 23, 3507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.-C. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2023, 613, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, P.; Lenka, D.R.; Batabyal, M.; Pain, P.K.; Kumar, S.; Manna, D.; Kumar, A. Detailed Insights into the Inhibitory Mechanism of New Ebselen Derivatives against Main Protease (Mpro) of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). ACS Pharmacology & Translational Science 2023, 6, 171–180. [Google Scholar] [CrossRef]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nature communications 2021, 12, 3061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nature microbiology 2021, 6, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: open the door for novel therapies. Signal transduction and targeted therapy 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Rangraze, I.; Jhancy, M.; Khan, S. Recent Advances in Understanding COVID-19 Pathophysiology and Therapy: A Review. 2023.
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: Covalent inhibitors structure–activity relationship insights and evolution perspectives. Journal of medicinal chemistry 2022, 65, 12500–12534. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Quantum Chemical Computation of Omicron Mutations Near Cleavage Sites of the Spike Protein. Microorganisms 2022, 10, 1999. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Perera, L.; Tillekeratne, L.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discovery Today, 2021, 26, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. Journal of medicinal chemistry 2020, 65, 2716–2746. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CL(pro), M(pro)) in the age of COVID-19. J Gen Virol 2021, 102. [Google Scholar] [CrossRef] [PubMed]
- Anirudhan, V.; Lee, H.; Cheng, H.; Cooper, L.; Rong, L. Targeting SARS-CoV-2 viral proteases as a therapeutic strategy to treat COVID-19. Journal of medical virology 2021, 93, 2722–2734. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus 2020, 12. [Google Scholar] [CrossRef]
- Wong, N.A.; Saier Jr, M.H. The SARS-coronavirus infection cycle: a survey of viral membrane proteins, their functional interactions and pathogenesis. International journal of molecular sciences 2021, 22, 1308. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.d.A.; Grosche, V.R.; Bergamini, F.R.G.; Sabino-Silva, R.; Jardim, A.C.G. Antivirals against coronaviruses: candidate drugs for SARS-CoV-2 treatment? Frontiers in microbiology 2020, 11, 1818. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. Journal of Virology 2021, 95. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D. SARS-CoV-2 variants and ending the COVID-19 pandemic. The Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Konwar, M.; Sarma, D. Advances in developing small molecule SARS 3CLpro inhibitors as potential remedy for corona virus infection. Tetrahedron 2021, 77, 131761. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front Chem 2021, 9, 819165. [Google Scholar] [CrossRef]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 research using molecular dynamics simulation. The Journal of Physical Chemistry B 2021, 125, 9078–9091. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr Opin Virol 2020, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of pharmaceutical analysis 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Naumovich, V.; Grishina, M.; Potemkin, V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. Journal of biomolecular structure & dynamics 2021, 40, 1–14. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E.; Basu, S. Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Sheik Amamuddy, O.; Verkhivker, G.M.; Tastan Bishop, Ö. Impact of Early Pandemic Stage Mutations on Molecular Dynamics of SARS-CoV-2 M(pro). J Chem Inf Model 2020, 60, 5080–5102. [Google Scholar] [CrossRef]
- Chen, S.A.; Arutyunova, E.; Lu, J.; Khan, M.B.; Rut, W.; Zmudzinski, M.; Shahbaz, S.; Iyyathurai, J.; Moussa, E.W.; Turner, Z.; et al. SARS-CoV-2 M(pro) Protease Variants of Concern Display Altered Viral Substrate and Cell Host Target Galectin-8 Processing but Retain Sensitivity toward Antivirals. ACS Cent Sci 2023, 9, 696–708. [Google Scholar] [CrossRef]
- Sacco, M.D.; Hu, Y.; Gongora, M.V.; Meilleur, F.; Kemp, M.T.; Zhang, X.; Wang, J.; Chen, Y. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res 2022, 32, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.H.; Oliveira, A.S.F.; Schofield, C.J.; Mulholland, A.J.; Duarte, F. Dynamical nonequilibrium molecular dynamics simulations identify allosteric sites and positions associated with drug resistance in the SARS-CoV-2 main protease. JACS Au 2023, 3, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Samant, N.; Schneider-Nachum, G.; Barkan, D.T.; Yilmaz, N.K.; Schiffer, C.A.; Moquin, S.A.; Dovala, D.; Bolon, D.N. Comprehensive fitness landscape of SARS-CoV-2 Mpro reveals insights into viral resistance mechanisms. Elife 2022, 11, e77433. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Communications biology 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Chai, Z.; Wang, Y.; Li, M.; Yu, Z.; Liu, Y.; Lu, S.; Zhang, J. Along the allostery stream: Recent advances in computational methods for allosteric drug discovery. Wiley Interdisciplinary Reviews: Computational Molecular Science 2022, 12, e1585. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Science Translational Medicine 2022, 15, eabq7360. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Frontiers in chemistry 2021, 9, 692168. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Dasari, S.; Barwell, S.A.E.; McLeod, M.J.; Kalyaanamoorthy, S.; Holyoak, T.; Ganesan, A. The H163A mutation unravels an oxidized conformation of the SARS-CoV-2 main protease. Nat Commun 2023, 14, 5625. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Dasari, S.; Barwell, S.; McLeod, M.J.; Kalyaanamoorthy, S.; Holyoak, T.; Ganesan, A. The H163A Mutation Unravels an Oxidized Conformation of the SARS-CoV-2 Main Protease and Opens a New Avenue for Anti-Viral Therapeutic Design. 2022. [Google Scholar] [CrossRef]
- Tucci, A.R.; da Rosa, R.M.; Rosa, A.S.; Augusto Chaves, O.; Ferreira, V.N.S.; Oliveira, T.K.F.; Coutinho Souza, D.D.; Borba, N.R.R.; Dornelles, L.; Rocha, N.S. Antiviral Effect of 5′-Arylchalcogeno-3-aminothymidine Derivatives in SARS-CoV-2 Infection. Molecules 2023, 28, 6696. [Google Scholar] [CrossRef]
- Hu, Y.; Lewandowski, E.M.; Tan, H.; Zhang, X.; Morgan, R.T.; Zhang, X.; Jacobs, L.M.; Butler, S.G.; Gongora, M.V.; Choy, J. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Central Science 2023, 9, 1658–1669. [Google Scholar] [CrossRef]
- Friedman, R. Computational studies of protein–drug binding affinity changes upon mutations in the drug target. Wiley Interdisciplinary Reviews: Computational Molecular Science 2022, 12, e1563. [Google Scholar] [CrossRef]
- Yin, J.; Li, C.; Ye, C.; Ruan, Z.; Liang, Y.; Li, Y.; Wu, J.; Luo, Z. Advances in the development of therapeutic strategies against COVID-19 and perspectives in the drug design for emerging SARS-CoV-2 variants. Computational and structural biotechnology journal 2022, 20, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hegazy, M.-E.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. Journal of biomolecular Structure and Dynamics 2021, 39, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Yadav, S.; Banerjee, S.; Fakayode, S.O.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Mahmud, F.; Whatcott, R.; Thammathong, J. Drug repurposing to identify nilotinib as a potential SARS-CoV-2 main protease inhibitor: insights from a computational and in vitro study. Journal of chemical information and modeling 2021, 61, 5469–5483. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 proteases for COVID-19 antiviral development. Frontiers in Chemistry 2022, 9, 1221. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wei, N.; Jin, L.; Zhang, H.; Luo, J.; Zhang, Y.; Wang, K. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorganic Chemistry 2021, 117, 105455. [Google Scholar] [CrossRef]
- Sahoo, P.; Lenka, D.R.; Batabyal, M.; Pain, P.K.; Kumar, S.; Manna, D.; Kumar, A. Detailed Insights into the Inhibitory Mechanism of New Ebselen Derivatives against Main Protease (Mpro) of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). ACS Pharmacology & Translational Science 2022, 6, 171–180. [Google Scholar]
- Safiabadi Tali, S.H.; LeBlanc, J.J.; Sadiq, Z.; Oyewunmi, O.D.; Camargo, C.; Nikpour, B.; Armanfard, N.; Sagan, S.M.; Jahanshahi-Anbuhi, S. Tools and techniques for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19 detection. Clinical microbiology reviews 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell research 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Frontiers in Chemistry 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, E. Investigation of changes in protein stability and substrate affinity of 3CL-protease of SARS-CoV-2 caused by mutations. Genetics and Molecular Biology 2022, 45, e20210404. [Google Scholar] [CrossRef] [PubMed]
- Mahtarin, R.; Islam, S.; Islam, M.J.; Ullah, M.O.; Ali, M.A.; Halim, M.A. Structure and dynamics of membrane protein in SARS-CoV-2. Journal of Biomolecular Structure and Dynamics 2022, 40, 4725–4738. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, E. Mutations in Main Protease of SARS CoV-2 Decreased Boceprevir Affinity. Brazilian Archives of Biology and Technology 2022, 64. [Google Scholar] [CrossRef]
- Rehman, M.U.; Ali, A.; Ansar, R.; Arafah, A.; Imtiyaz, Z.; Wani, T.A.; Zargar, S.; Ganie, S.A. In Silico molecular docking and dynamic analysis of natural compounds against major nonstructural proteins of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics 2023, 41, 9072–9088. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Parvez, M.S.A.; Azim, K.F.; Imran, M.A.S.; Raihan, T.; Gulshan, A.; Muhit, S.; Akhand, R.N.; Ahmed, S.S.U.; Uddin, M.B. Main protease inhibitors and drug surface hotspots for the treatment of COVID-19: A drug repurposing and molecular docking approach. Biomedicine & Pharmacotherapy 2021, 140, 111742. [Google Scholar]
- Ganesh, B.; Rajakumar, T.; Malathi, M.; Manikandan, N.; Nagaraj, J.; Santhakumar, A.; Elangovan, A.; Malik, Y.S. Epidemiology and pathobiology of SARS-CoV-2 (COVID-19) in comparison with SARS, MERS: An updated overview of current knowledge and future perspectives. Clinical epidemiology and global health 2021, 10, 100694. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nature Reviews Microbiology 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y. Virus‒host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med 2021, 2, 99–112. e117. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E. COVID-19: drug targets and potential treatments. Journal of medicinal chemistry 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; Nahed, A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B. 1.1. 529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environmental research 2022, 209, 112816. [Google Scholar] [CrossRef]
- Kumari, M.; Lu, R.-M.; Li, M.-C.; Huang, J.-L.; Hsu, F.-F.; Ko, S.-H.; Ke, F.-Y.; Su, S.-C.; Liang, K.-H.; Yuan, J.P.-Y. A critical overview of current progress for COVID-19: development of vaccines, antiviral drugs, and therapeutic antibodies. Journal of biomedical science 2022, 29, 68. [Google Scholar] [CrossRef]
- Alzyoud, L.; Ghattas, M.A.; Atatreh, N. Allosteric binding sites of the SARS-CoV-2 main protease: Potential targets for broad-spectrum anti-coronavirus agents. Drug Design, Development and Therapy, 2022; 2463–2478. [Google Scholar]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Fowler, B.; Hong, S.J.; Mohri, H.; Nair, M.S.; et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nature Communications 2022, 13, 1891. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Panwar, U.; Dinesh, D.C.; Boura, E.; Singh, P.; Dubey, V.K.; Singh, S.K. Microsecond MD simulation and multiple-conformation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Frontiers in chemistry 2021, 8, 595273. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Wang, Z.; Wang, P.; Ren, W.; Yu, Y.; Yu, Y.; Yuan, B.; Song, J.; Zhang, X.; Zhang, Y. SARS-CoV-2 main protease cleaves MAGED2 to antagonize host antiviral defense. Mbio 2023, 14, e01373–e01323. [Google Scholar] [CrossRef] [PubMed]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Jha, A.N. A comprehensive review of drug repurposing strategies against known drug targets of COVID-19. Covid 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Aruleba, R.T.; Sibuyi, N.R.S.; Klein, A.; Madiehe, A.M.; Meyer, M. Inhibitory potential of repurposed drugs against the SARS-CoV-2 main protease: a computational-aided approach. Journal of Biomolecular Structure and Dynamics 2022, 40, 3416–3427. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An updated review of computer-aided drug design and its application to COVID-19. BioMed research international 2021, 2021. [Google Scholar] [CrossRef]
- Chatzigoulas, A.; Cournia, Z. Rational design of allosteric modulators: Challenges and successes. Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11, e1529. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus‒host interaction networks as new antiviral drug targets for IAV and SARS-CoV-2. Emerging Microbes & Infections 2022, 11, 1371–1389. [Google Scholar]
- Aniana, A.; Nashed, N.; Ghirlando, R.; Coates, L.; Kneller, D.; Kovalevsky, A.; Louis, J. Insights into the mechanism of SARS-CoV-2 main protease autocatalytic maturation from model precursors. Communications Biology 2023, 6, s42003–s42023. [Google Scholar] [CrossRef]
- Kovalevsky, A.; Coates, L.; Kneller, D.W.; Ghirlando, R.; Aniana, A.; Nashed, N.T.; Louis, J.M. Unmasking the conformational stability and inhibitor binding to SARS-CoV-2 main protease active site mutants and miniprecursor. Journal of Molecular Biology 2022, 434, 167876. [Google Scholar] [CrossRef]
- Duerr, R.; Crosse, K.M.; Valero-Jimenez, A.M.; Dittmann, M. SARS-CoV-2 portrayed against HIV: contrary viral strategies in similar disguise. Microorganisms 2021, 9, 1389. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. International Journal of Molecular Sciences 2020, 21, 3099. [Google Scholar] [CrossRef]
- Nguyen, K.V. Problems associated with antiviral drugs and vaccines development for COVID-19: approach to intervention using expression vectors via GPI anchor. Nucleosides, Nucleotides & Nucleic Acids 2021, 40, 665–706. [Google Scholar]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorganic & Medicinal Chemistry Letters 2022, 62, 128629. [Google Scholar]
- Zhou, Y.; Gammeltoft, K.A.; Ryberg, L.A.; Pham, L.V.; Tjørnelund, H.D.; Binderup, A.; Duarte Hernandez, C.R.; Fernandez-Antunez, C.; Offersgaard, A.; Fahnøe, U. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Science Advances 2022, 8, eadd7197. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Current Opinion in Virology 2021, 49, 36–40. [Google Scholar] [CrossRef]
- Bloom, J.D.; Neher, R.A. Fitness effects of mutations to SARS-CoV-2 proteins. 2023.
- Iketani, S.; Hong, S.J.; Sheng, J.; Bahari, F.; Culbertson, B.; Atanaki, F.F.; Aditham, A.K.; Kratz, A.F.; Luck, M.I.; Tian, R. Functional map of SARS-CoV-2 3CL protease reveals tolerant and immutable sites. Cell Host & Microbe 2022, 30, 1354–1362. e1356. [Google Scholar]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Combinatorial Science 2020, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Feys, J.R. SARS-CoV-2 Mpro inhibitors: achieved diversity, developing resistance and future strategies. Future Pharmacology 2023, 3, 80–107. [Google Scholar] [CrossRef]
- Mensah, J.O.; Ampomah, G.B.; Gasu, E.N.; Adomako, A.K.; Menkah, E.S.; Borquaye, L.S. Allosteric modulation of the main protease (MPro) of SARS-CoV-2 by casticin—insights from molecular dynamics simulations. Chemistry Africa 2022, 5, 1305–1320. [Google Scholar] [CrossRef]
- Irfan, M.; Akhtar, N.; Ahmad, M.; Shahzad, F.; Elavarasan, R.M.; Wu, H.; Yang, C. Assessing public willingness to wear face masks during the COVID-19 pandemic: fresh insights from the theory of planned behavior. International Journal of Environmental Research and Public Health 2021, 18, 4577. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Yang, Q.; Gribenko, A.; Perrin Jr, B.S.; Zhu, Y.; Cardin, R.; Liberator, P.A.; Anderson, A.S.; Hao, L. Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. Mbio 2022, 13, e00869–e00822. [Google Scholar] [CrossRef] [PubMed]
- Reis, S.; Metzendorf, M.-I.; Kuehn, R.; Popp, M.; Gagyor, I.; Kranke, P.; Meybohm, P.; Skoetz, N.; Weibel, S. Nirmatrelvir combined with ritonavir for preventing and treating COVID-19. Cochrane Database of Systematic Reviews 2022. [Google Scholar]
- Chen, S.A.; Arutyunova, E.; Lu, J.; Khan, M.B.; Rut, W.; Zmudzinski, M.; Shahbaz, S.; Iyyathurai, J.; Moussa, E.W.; Turner, Z. SARS-CoV-2 Mpro Protease Variants of Concern Display Altered Viral Substrate and Cell Host Target Galectin-8 Processing but Retain Sensitivity toward Antivirals. ACS Central Science 2023, 9, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.L.; Naik, S.R.; Dingelstad, N.; Lugo, M.R.; Kalyaanamoorthy, S.; Ganesan, A. Molecular dynamics and in silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Scientific Reports 2021, 11, 7429. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Philot, E.; Pantaleão, S.; Torres-Bonfim, N.; Kliousoff, A.; Quiroz, R.; Perahia, D.; Simões, R.; Magro, A.; Scott, A. Unveiling mutation effects on the structural dynamics of the main protease from SARS-CoV-2 with hybrid simulation methods. Journal of Molecular Graphics and Modeling 2023, 121, 108443. [Google Scholar] [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Cheng, L.; Frishcosy, J.; Huzumi, Y.; Qiu, Y.; Schluckbier, T.; Wei, X.; Wei, G.-W. Methodology-centered review of molecular modeling, simulation, and prediction of SARS-CoV-2. Chemical Reviews 2022, 122, 11287–11368. [Google Scholar] [CrossRef]
- Zeng, L.; Li, D.; Tong, W.; Shi, T.; Ning, B. Biochemical features and mutations of key proteins in SARS-CoV-2 and their impacts on RNA therapeutics. Biochemical Pharmacology 2021, 189, 114424. [Google Scholar] [CrossRef] [PubMed]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proceedings of the National Academy of Sciences 2021, 118, e2104241118. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Novel SARS-CoV-2 variants: the pandemics within the pandemic. Clinical Microbiology and Infection 2021, 27, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, J.; Wang, P.; Zhang, Z. The mechanisms of immune response and evasion by the main SARS-CoV-2 variants. Iscience 2022. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Isom, D.G. One year of SARS-CoV-2: How much has the virus changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Mascellino, M.T.; Di Timoteo, F.; De Angelis, M.; Oliva, A. Overview of the main anti-SARS-CoV-2 vaccines: mechanism of action, efficacy and safety. Infection and drug resistance 2021, 3459–3476. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Joyce, R.; Tan, H.; Hu, Y.; Wang, J. SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Accounts of Chemical Research 2022, 56, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacology & Translational Science 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N Engl J Med 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Abe, K.; Kabe, Y.; Uchiyama, S.; Iwasaki, Y.W.; Ishizu, H.; Uwamino, Y.; Takenouchi, T.; Uno, S.; Ishii, M.; Maruno, T. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Scientific reports 2022, 12, 1299. [Google Scholar] [CrossRef]
- Sargsyan, K.; Mazmanian, K.; Lim, C. A strategy for evaluating potential antiviral resistance to small molecule drugs and application to SARS-CoV-2. Scientific Reports 2023, 13, 502. [Google Scholar] [CrossRef] [PubMed]
- Nikhra, V. Living with COVID-19: The Nemesis, the Hubris, and the Elpis. Dynamics 2021, 3, 012. [Google Scholar]
- Chtita, S.; Belhassan, A.; Aouidate, A.; Belaidi, S.; Bouachrine, M.; Lakhlifi, T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs by docking and virtual screening. Combinatorial chemistry & high throughput screening 2021, 24, 441–454. [Google Scholar]
- Bai, B.; Belovodskiy, A.; Hena, M.; Kandadai, A.S.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Khan, M.B.; Arutyunova, E.; Lu, J. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. Journal of Medicinal Chemistry 2021, 65, 2905–2925. [Google Scholar] [CrossRef] [PubMed]
- Yuce, M.; Cicek, E.; Inan, T.; Dag, A.B.; Kurkcuoglu, O.; Sungur, F.A. Repurposing of FDA-approved drugs against active site and potential allosteric drug-binding sites of COVID-19 main protease. Proteins: Structure, Function, and Bioinformatics 2021, 89, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Hong, S.J.; Sheng, J.; Bahari, F.; Culbertson, B.; Atanaki, F.F.; Aditham, A.K.; Kratz, A.F.; Luck, M.I.; Tian, R.; et al. Functional map of SARS-CoV-2 3CL protease reveals tolerant and immutable sites. Cell Host Microbe 2022, 30, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Jochmans, D.; Liu, C.; Donckers, K.; Stoycheva, A.; Boland, S.; Stevens, S.; Vita, C.; Vanmechelen, B.; Maes, P.; Trüeb, B.; et al. The substitutions L50F, E166A and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir; 2022.
- Al Adem, K.; Ferreira, J.C.; Fadl, S.; Mustafa, M.; Rabeh, W.M. Key allosteric and active site residues of SARS-CoV-2 3CLpro are promising drug targets. Biochemical Journal 2023, 480, 791–813. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorganic & medicinal chemistry 2021, 29, 115860. [Google Scholar]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Baghi, H.B.; Nahand, J.S.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to Covid-19 therapy? Biomedicine & Pharmacotherapy 2023, 162, 114367. [Google Scholar]
- Chen, J.-T. Ethnopharmacology and Drug Discovery for COVID-19: Anti-SARS-CoV-2 Agents from Herbal Medicines and Natural Products; Springer Nature: 2023.
- Heilmann, E.; Costacurta, F.; Geley, S.; Mogadashi, S.A.; Volland, A.; Rupp, B.; Harris, R.S.; von Laer, D. A VSV-based assay quantifies coronavirus Mpro/3CLpro/Nsp5 main protease activity and chemical inhibition. Communications Biology 2022, 5, 391. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Volland, A.; von Laer, D. SARS-CoV-2 3CLpro mutations confer resistance to Paxlovid (nirmatrelvir/ritonavir) in a VSV-based, non-gain-of-function system. BioRxiv, 2022; 2022.2007. 2002.495455. [Google Scholar]
- Moghadasi, S.A.; Heilmann, E.; Khalil, A.M.; Nnabuife, C.; Kearns, F.L.; Ye, C.; Moraes, S.N.; Costacurta, F.; Esler, M.A.; Aihara, H.; et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Rella, S.A.; Kulikova, Y.A.; Dermitzakis, E.T.; Kondrashov, F.A. Rates of SARS-CoV-2 transmission and vaccination impact the fate of vaccine-resistant strains. Sci Rep 2021, 11, 15729. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Vanet, A.; Francesconi, V.; Tagliazucchi, L.; Tassone, G.; Venturelli, A.; Spyrakis, F.; Mazzorana, M.; Costi, M.P.; Tonelli, M. Antitarget, Anti-SARS-CoV-2 Leads, Drugs, and the Drug Discovery–Genetics Alliance Perspective. Journal of Medicinal Chemistry 2023, 66, 3664–3702. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir plus ritonavir: first approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Parigger, L.; Krassnigg, A.; Schopper, T.; Singh, A.; Tappler, K.; Köchl, K.; Hetmann, M.; Gruber, K.; Steinkellner, G.; Gruber, C.C. Recent changes in the mutational dynamics of the SARS-CoV-2 main protease substantiate the danger of emerging resistance to antiviral drugs. Frontiers in Medicine 2022, 9, 1061142. [Google Scholar] [CrossRef] [PubMed]
- Cobey, S.; Larremore, D.B.; Grad, Y.H.; Lipsitch, M. Concerns about SARS-CoV-2 evolution should not hold back efforts to expand vaccination. Nature Reviews Immunology 2021, 21, 330–335. [Google Scholar] [CrossRef]
- Moghadasi, S.A.; Heilmann, E.; Khalil, A.M.; Nnabuife, C.; Kearns, F.L.; Ye, C.; Moraes, S.N.; Costacurta, F.; Esler, M.A.; Aihara, H.; et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Sci Adv 2023, 9, eade8778. [Google Scholar] [CrossRef]
Figure 1.
Structure of 3CLpro (SARS-CoV-2) retrieved from RCSB using accession number 6LU7. (A) The domain architecture of three different domains. Domain 1 is magenta, domain 2 is orange, domain 3 is cyan, and connecting loops are gray, while the drug molecule in the active site is shown in yellow as a sphere. (B) The electrostatic potential of 3CLPro and the important active site residues in the sticks, including the two essential residues His41 and Cys145 (catalytic dyad).
Figure 1.
Structure of 3CLpro (SARS-CoV-2) retrieved from RCSB using accession number 6LU7. (A) The domain architecture of three different domains. Domain 1 is magenta, domain 2 is orange, domain 3 is cyan, and connecting loops are gray, while the drug molecule in the active site is shown in yellow as a sphere. (B) The electrostatic potential of 3CLPro and the important active site residues in the sticks, including the two essential residues His41 and Cys145 (catalytic dyad).
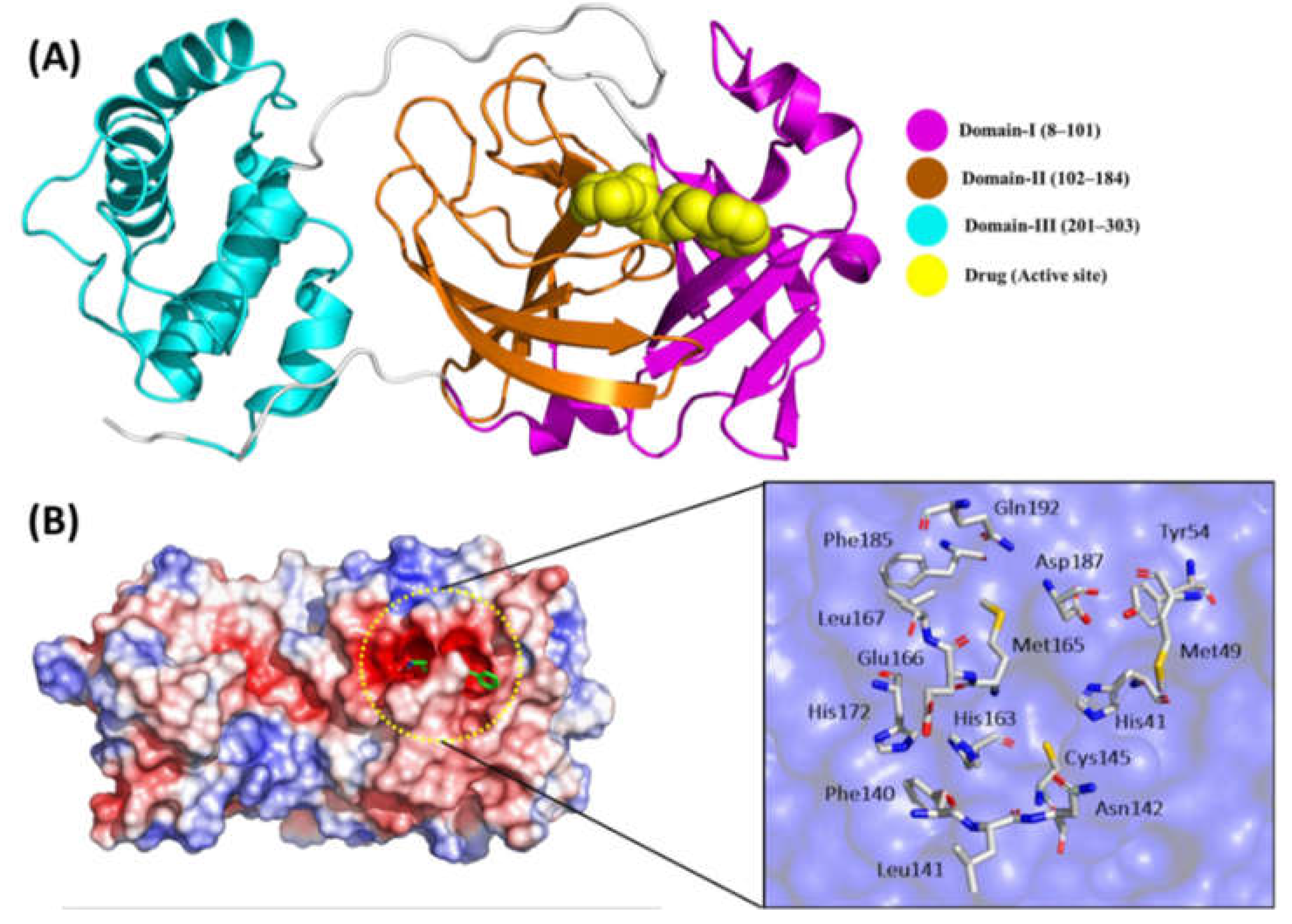
Figure 2.
Structure of the SARS-CoV-2 MPro with nirmatrelvir. (a) The domain organization is represented based on the crystal structure of the enzyme complexed with nirmatrelvir (7RFS.pdb). The inhibitor is represented as sticks, the carbon atoms are green, the oxygen atoms are red, and the nitrogen atoms are blue. (b) Enlarged view of the active site showing the most relevant enzyme-inhibitor interactions. The active-site residues forming at least one main-chain- and side-chain-mediated hydrogen bond are colored blue and red, respectively. The residues forming polar interactions are shown in orange. (c) Most relevant inhibitor-binding interactions at the active site. The backbone and side chain-mediated hydrogen bonds are colored blue and red, respectively. The direction of the arrow indicates the donated hydrogen atom. Polar interactions are shown in orange. The colored dots indicate the average number of nonbonded contacts for individual atoms. Orange represents 1 contact, light green represents 2–4 contacts, and dark green represents ≥ 5 contacts. The interactions were mapped based on five crystal structures (7vh8, 7 si9, 7te0, 7rfs, and 7rfw) using PDBSum [62] and LigPlot+ [63].
Figure 2.
Structure of the SARS-CoV-2 MPro with nirmatrelvir. (a) The domain organization is represented based on the crystal structure of the enzyme complexed with nirmatrelvir (7RFS.pdb). The inhibitor is represented as sticks, the carbon atoms are green, the oxygen atoms are red, and the nitrogen atoms are blue. (b) Enlarged view of the active site showing the most relevant enzyme-inhibitor interactions. The active-site residues forming at least one main-chain- and side-chain-mediated hydrogen bond are colored blue and red, respectively. The residues forming polar interactions are shown in orange. (c) Most relevant inhibitor-binding interactions at the active site. The backbone and side chain-mediated hydrogen bonds are colored blue and red, respectively. The direction of the arrow indicates the donated hydrogen atom. Polar interactions are shown in orange. The colored dots indicate the average number of nonbonded contacts for individual atoms. Orange represents 1 contact, light green represents 2–4 contacts, and dark green represents ≥ 5 contacts. The interactions were mapped based on five crystal structures (7vh8, 7 si9, 7te0, 7rfs, and 7rfw) using PDBSum [62] and LigPlot+ [63].
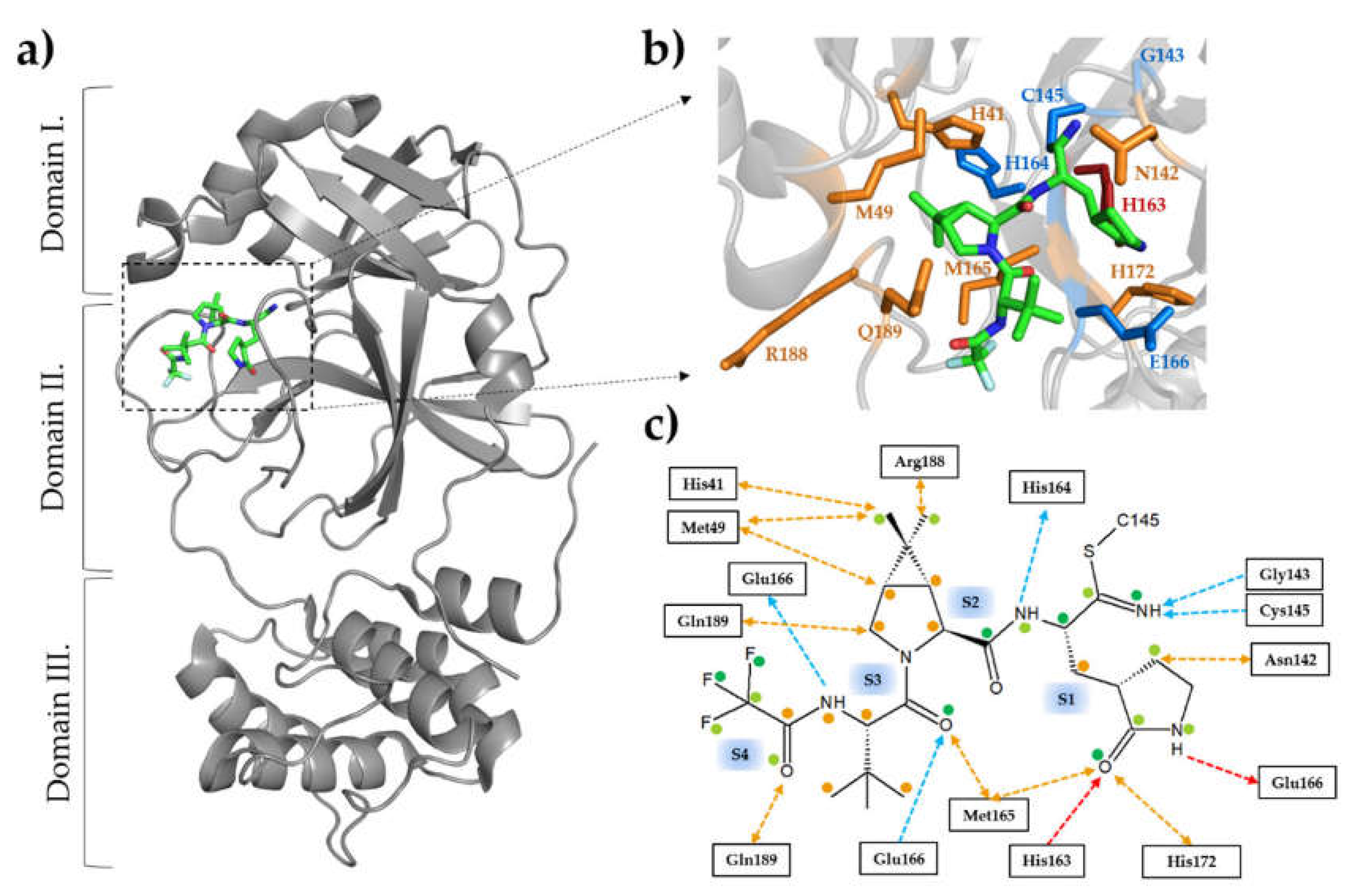
Figure 3.
Sequence and structural analysis of Mpro: (A) A phylogenetic tree was constructed using the maximum likelihood method to infer the closest homologs of SARS-CoV-2 3CLpro. (B) Multiple sequence alignment was performed for the closest homologs of SARS-CoV-2 3CLpro with ≥70% sequence identity. (C) A cartoon representation of the SARS-CoV-2 3CLpro homodimer is shown, with Chain-A (protomer-A) in multicolor and Chain-B (protomer-B) in dark blue. The N-finger, crucial for dimerization and maintaining the active conformation, is highlighted in hot pink. Domain I is colored cyan, domain II is green, and domain III is yellow. The N- and C-termini are labeled, and the residues of the catalytic dyad (Cys-145 and His-41) are highlighted in yellow and labeled. (D) A cartoon representation of the 3CLpro monomer model (Chain/Protomer-A) of SARS-CoV-2 is superimposed with the SARS-CoV 3CLpro structure. The SARS-CoV 3CLpro template is colored cyan, the SARS-CoV-2 3CLpro structure is colored grey, and all identified mutations are highlighted in red. (E) Docking of 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone within the receptor-binding site of SARS-CoV-2 3CLpro is shown, illustrating hydrogen bonds with the catalytic dyad (Cys-145 and His-41). The 3CLpro structure is colored dark blue, the isoflavone is orange, and hydrogen bonds are maroon. (https://www.sciencedirect.com/science/article/pii/S2095177920301271).
Figure 3.
Sequence and structural analysis of Mpro: (A) A phylogenetic tree was constructed using the maximum likelihood method to infer the closest homologs of SARS-CoV-2 3CLpro. (B) Multiple sequence alignment was performed for the closest homologs of SARS-CoV-2 3CLpro with ≥70% sequence identity. (C) A cartoon representation of the SARS-CoV-2 3CLpro homodimer is shown, with Chain-A (protomer-A) in multicolor and Chain-B (protomer-B) in dark blue. The N-finger, crucial for dimerization and maintaining the active conformation, is highlighted in hot pink. Domain I is colored cyan, domain II is green, and domain III is yellow. The N- and C-termini are labeled, and the residues of the catalytic dyad (Cys-145 and His-41) are highlighted in yellow and labeled. (D) A cartoon representation of the 3CLpro monomer model (Chain/Protomer-A) of SARS-CoV-2 is superimposed with the SARS-CoV 3CLpro structure. The SARS-CoV 3CLpro template is colored cyan, the SARS-CoV-2 3CLpro structure is colored grey, and all identified mutations are highlighted in red. (E) Docking of 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone within the receptor-binding site of SARS-CoV-2 3CLpro is shown, illustrating hydrogen bonds with the catalytic dyad (Cys-145 and His-41). The 3CLpro structure is colored dark blue, the isoflavone is orange, and hydrogen bonds are maroon. (https://www.sciencedirect.com/science/article/pii/S2095177920301271).
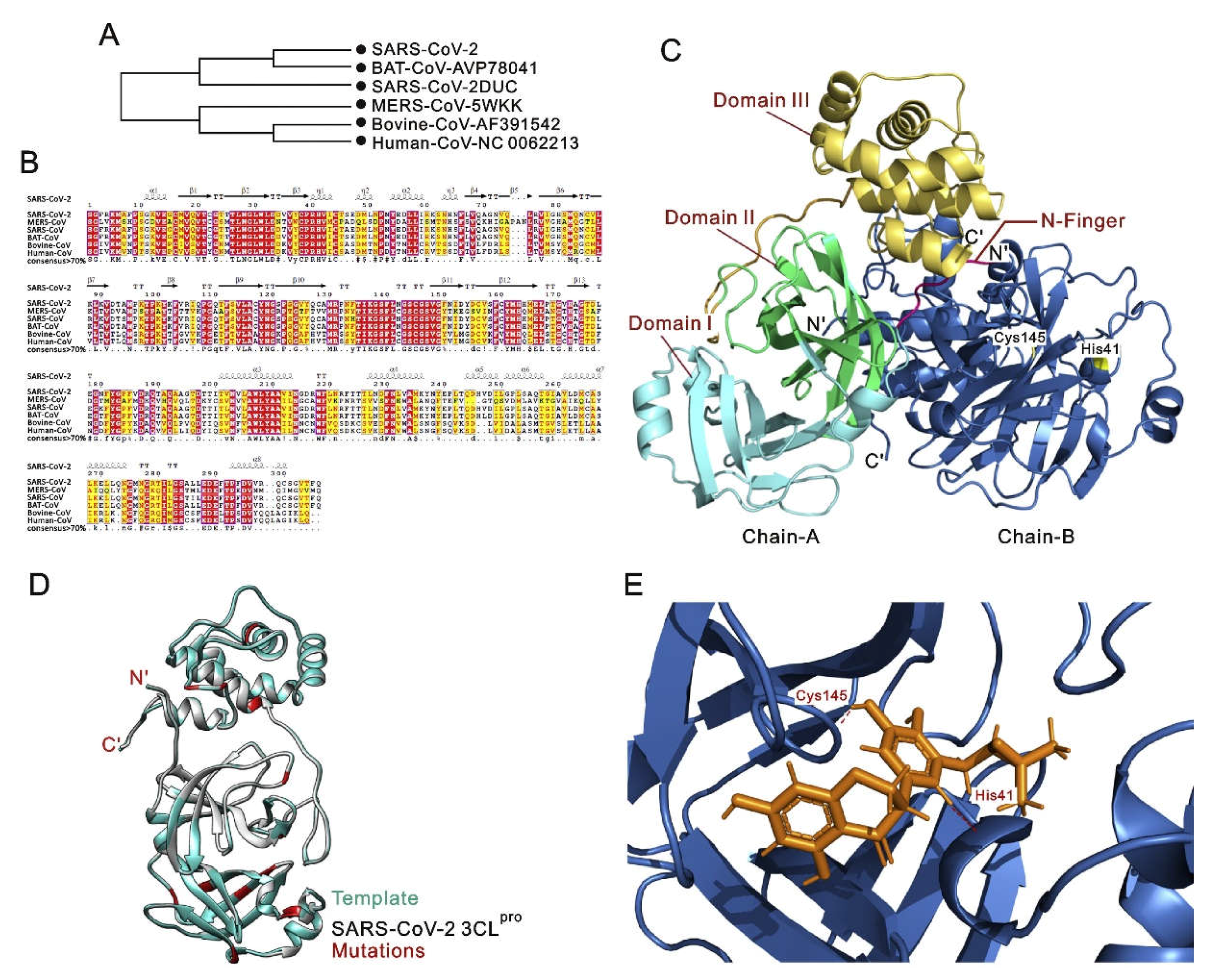
Figure 5.
Nsp5 (3CLpro) structural features of SARS-CoV-2 protease. (a) Monomeric structure of the SARS-CoV-2 nsp5 protease (PDB 6M2N) with the three domains shown. Key structural regions including the N-terminal finger (NF), N-terminal helix (NH) and domain 2-domain 3 interdomain loop (IDL) are labelled. Corresponding locations of MHV nsp5 resistance mutations identified in Deng et al. are labelled with stars [144]. (b) A view of the active site with a consensus cleavage site peptide bound. The catalytic dyad residues are shown. (c) SARS-CoV-2 nsp5 dimer structure with the orientation of the two dimers shown by arrows and the dimerization interface labelled. Green asterisks denote the catalytic site with black sticks shown for the catalytic dyad residues (His41 and Cys145) in both the monomeric (a) and the dimeric structures (c).
Figure 5.
Nsp5 (3CLpro) structural features of SARS-CoV-2 protease. (a) Monomeric structure of the SARS-CoV-2 nsp5 protease (PDB 6M2N) with the three domains shown. Key structural regions including the N-terminal finger (NF), N-terminal helix (NH) and domain 2-domain 3 interdomain loop (IDL) are labelled. Corresponding locations of MHV nsp5 resistance mutations identified in Deng et al. are labelled with stars [144]. (b) A view of the active site with a consensus cleavage site peptide bound. The catalytic dyad residues are shown. (c) SARS-CoV-2 nsp5 dimer structure with the orientation of the two dimers shown by arrows and the dimerization interface labelled. Green asterisks denote the catalytic site with black sticks shown for the catalytic dyad residues (His41 and Cys145) in both the monomeric (a) and the dimeric structures (c).
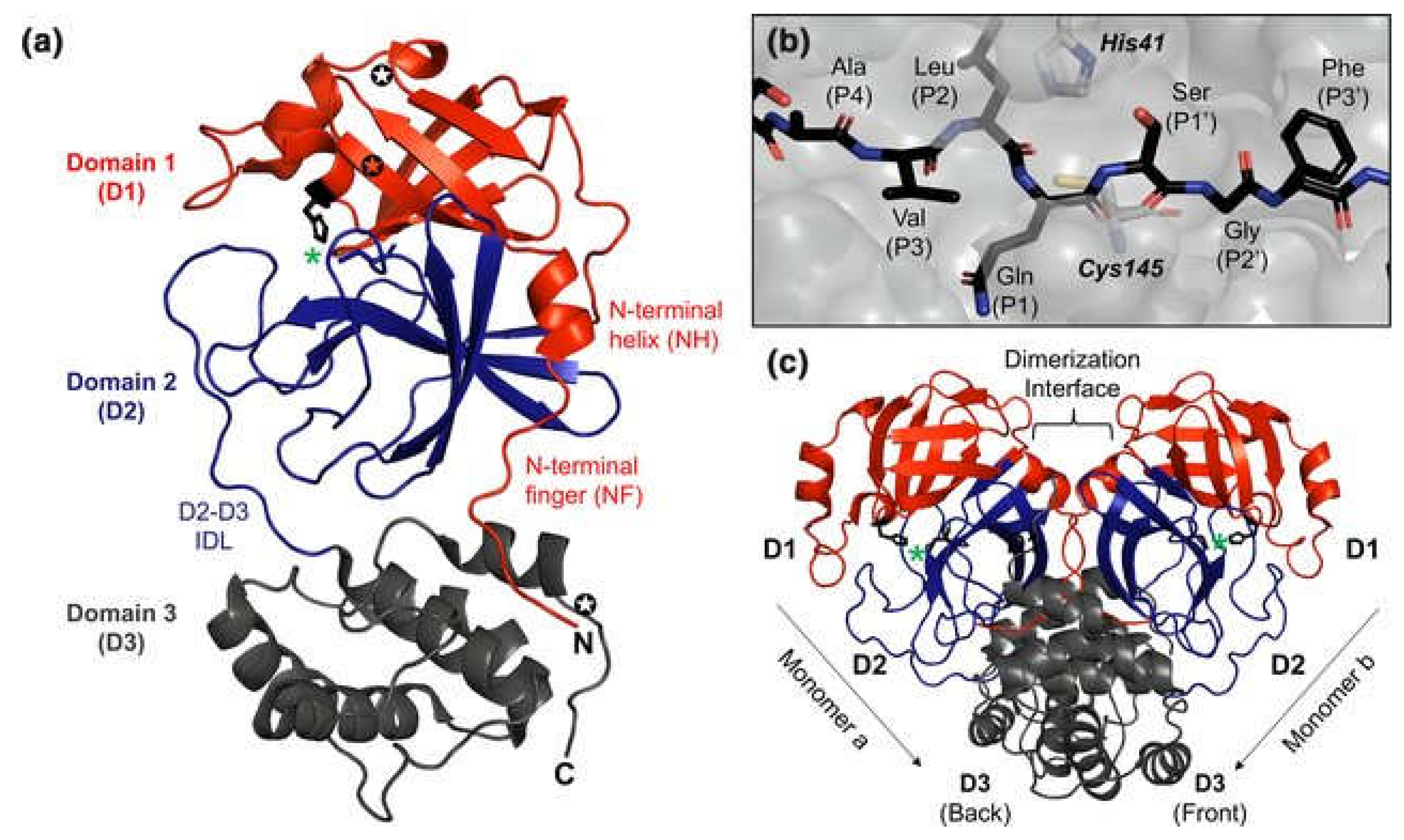

Table 1.
Outline of the structural alterations of the 3CLPro.
| Structural Alterations | Description | Potential Consequences | Research Implications | Ref. |
|---|---|---|---|---|
| Active Site | Analysis of changes in residues crucial for substrate binding and catalysis, including mutations that may affect substrate specificity or enzymatic activity. Structural modifications in the active site can impact inhibitor binding and efficacy. | Mutations in the active site may lead to altered substrate specificity, reduced enzymatic activity, or resistance to inhibitors. | Understanding alterations in the active site provides insights into the design of specific inhibitors targeting mutant enzymes. | Kidera, A., Moritsugu, K., Ekimoto, T. and Ikeguchi, M., 2021. Allosteric regulation of 3CL protease of SARS-CoV-2 and SARS-CoV observed in the crystal structure ensemble. Journal of Molecular Biology, 433(24), p.167324, Al Adem, K., Ferreira, J.C., Fadl, S., Mustafa, M. and Rabeh, W.M., 2023. Key allosteric and active site residues of SARS-CoV-2 3CLpro are promising drug targets. Biochemical Journal, 480(11), pp.791-813. |
| Substrate Binding Pocket | Examination of modifications in the pocket's shape, size, and residues involved in substrate recognition. Alterations in the substrate binding pocket can influence substrate affinity, catalytic efficiency, and enzyme-substrate interactions. Understanding changes in this region is crucial for predicting the impact on enzymatic function and drug binding. | Changes in the substrate binding pocket may affect substrate recognition, leading to altered enzymatic activity or decreased inhibitor binding. | Knowledge of substrate binding pocket alterations aids in the design of novel inhibitors with improved binding affinity and specificity. | Ferreira, J.C., Fadl, S. and Rabeh, W.M., 2022. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. Journal of Biological Chemistry, 298(6), Al Adem, K., Ferreira, J.C., Fadl, S., Mustafa, M. and Rabeh, W.M., 2023. Key allosteric and active site residues of SARS-CoV-2 3CLpro are promising drug targets. Biochemical Journal, 480(11), pp.791-813. |
| Catalytic Residues | Assessment of alterations in residues directly involved in catalysis, such as those participating in nucleophilic attack and formation of the enzyme-substrate complex. Changes in catalytic residues can disrupt enzymatic activity, leading to loss of function or altered kinetics. Identifying mutations in these residues provides insights into the mechanisms underlying enzyme dysfunction. | Mutations in catalytic residues can impair enzymatic activity, resulting in reduced substrate turnover or altered reaction kinetics. | Understanding the effects of catalytic residue mutations helps elucidate the molecular basis of enzyme dysfunction and guides the development of therapeutic strategies targeting these mutations. | Ferreira, J.C., Fadl, S., Ilter, M., Pekel, H., Rezgui, R., Sensoy, O. and Rabeh, W.M., 2021. Dimethyl sulfoxide reduces the stability but enhances catalytic activity of the main SARS-CoV-2 protease 3CLpro. The FASEB Journal, 35(8), Kidera, A., Moritsugu, K., Ekimoto, T. and Ikeguchi, M., 2021. Allosteric regulation of 3CL protease of SARS-CoV-2 and SARS-CoV observed in the crystal structure ensemble. Journal of Molecular Biology, 433(24), p.167324. |
| Dimerization Interface | Investigation of changes in residues forming the dimer interface, affecting enzyme activity and stability. The dimerization interface plays a crucial role in maintaining enzyme structure and function. Mutations in this region can disrupt dimer formation, leading to monomerization or altered dimer stability. Understanding alterations in the dimer interface is essential for elucidating the impact on enzyme oligomerization and function. | Mutations at the dimerization interface may disrupt enzyme dimerization, leading to decreased enzyme stability or altered catalytic activity. | Knowledge of dimerization interface alterations informs strategies for stabilizing enzyme dimers or disrupting aberrant dimerization as therapeutic interventions. | Ferreira, G.M., Kronenberger, T., Tonduru, A.K., Hirata, R.D.C., Hirata, M.H. and Poso, A., 2022. SARS-CoV-2 Mpro conformational changes induced by covalently bound ligands. Journal of Biomolecular Structure and Dynamics, 40(22), pp.12347-12357, Tekpinar, M. and Yildirim, A., 2022. Impact of dimerization and N3 binding on molecular dynamics of SARS-CoV and SARS-CoV-2 main proteases. Journal of Biomolecular Structure and Dynamics, 40(14), pp.6243-6254. |
| Allosteric Sites | Analysis of modifications in sites distal from the active site, influencing enzyme activity through allosteric regulation. Changes in allosteric sites can allosterically modulate enzyme function, altering substrate binding affinity or catalytic activity. Identifying alterations in these sites provides insights into potential allosteric regulatory mechanisms and their impact on enzyme function. | Mutations in allosteric sites may affect enzyme regulation, leading to altered substrate binding or catalytic activity in response to regulatory signals. | Understanding alterations in allosteric sites elucidates mechanisms of enzyme regulation and provides opportunities for developing allosteric modulators to modulate enzyme activity. | Chan, H.H., Oliveira, A.S.F., Schofield, C.J., Mulholland, A.J. and Duarte, F., 2023. Dynamical nonequilibrium molecular dynamics simulations identify allosteric sites and positions associated with drug resistance in the SARS-CoV-2 main protease. JACS Au, 3(6), pp.1767-1774, Al Adem, K., Ferreira, J.C., Fadl, S., Mustafa, M. and Rabeh, W.M., 2023. Key allosteric and active site residues of SARS-CoV-2 3CLpro are promising drug targets. Biochemical Journal, 480(11), pp.791-813. |
| Domain Arrangement | Examination of changes in the arrangement or conformation of structural domains, affecting enzyme function and stability. Alterations in domain arrangement can disrupt domain-domain interactions, affecting enzyme stability or catalytic efficiency. Understanding modifications in domain arrangement provides insights into structural changes that may impact enzyme function and stability. | Changes in domain arrangement may affect enzyme stability, alter domain-domain interactions, or disrupt catalytic activity. | Knowledge of domain arrangement alterations informs strategies for stabilizing enzyme structure or designing domain-specific inhibitors to target mutant enzymes. | Kidera, A., Moritsugu, K., Ekimoto, T. and Ikeguchi, M., 2021. Allosteric regulation of 3CL protease of SARS-CoV-2 and SARS-CoV observed in the crystal structure ensemble. Journal of Molecular Biology, 433(24), p.167324, Gasparini, P., Philot, E.A., Pantaleão, S.Q., Torres-Bonfim, N.E.S.M., Kliousoff, A., Quiroz, R.C.N., Perahia, D., Simões, R.P., Magro, A.J. and Scott, A.L., 2023. Unveiling mutation effects on the structural dynamics of the main protease from SARS-CoV-2 with hybrid simulation methods. Journal of Molecular Graphics and Modeling, 121, p.108443. |
| Overall Structure | Assessment of global structural changes, including alterations in secondary structure elements and overall folding pattern. Changes in overall structure can impact enzyme stability, substrate binding, and catalytic activity. Analyzing alterations in overall structure provides insights into the structural basis of enzyme dysfunction and the potential effects on enzymatic function. | Modifications in overall structure may lead to protein misfolding, decreased stability, or loss of enzymatic function. | Understanding alterations in overall structure aids in identifying structural determinants of enzyme dysfunction and guides the development of strategies to restore enzyme function or stability. | Chen, Y.W., Yiu, C.P.B. and Wong, K.Y., 2020. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research, 9, Diessner, E.M., Takahashi, G.R., Cross, T.J., Martin, R.W. and Butts, C.T., 2023. Mutation effects on structure and dynamics: adaptive evolution of the SARS-CoV-2 main protease. Biochemistry, 62(3), pp.747-758. |
Table 3.
Experimental techniques used to study the consequences of 3CLPro mutations.
| Experimental Technique | Description | Advantages | Limitations | Ref. |
|---|---|---|---|---|
| X-ray Crystallography | Determines the three-dimensional structure of proteins, including mutant forms of 3CLPro, by analyzing the diffraction pattern of X-rays passing through protein crystals. Provides detailed atomic resolution information about protein structure. | Provides high-resolution structural data, allowing precise visualization of mutant 3CLPro conformations. | Requires protein crystallization, which can be challenging for some proteins and mutants. The technique is also time-consuming and requires access to specialized equipment and expertise. | Jaskolski, M., Dauter, Z., Shabalin, I.G., Gilski, M., Brzezinski, D., Kowiel, M., Rupp, B. and Wlodawer, A., 2021. Crystallographic models of SARS-CoV-2 3CLpro: In-depth assessment of structure quality and validation. IUCrJ, 8(2), pp.238-256. |
| Cryo-Electron Microscopy (Cryo-EM) | Utilizes electron microscopy to visualize biological samples, including mutant 3CLPro proteins, at cryogenic temperatures. Provides high-resolution images of protein structure, offering insights into conformational changes caused by mutations. | Enables visualization of protein structures in near-native states, including flexible regions and large protein complexes. | Requires expensive equipment and significant expertise. Image processing and analysis can be complex, and resolution may be lower compared to X-ray crystallography for some samples. | Kaniyala Melanthota, S., Banik, S., Chakraborty, I., Pallen, S., Gopal, D., Chakrabarti, S. and Mazumder, N., 2020. Elucidating the microscopic and computational techniques to study the structure and pathology of SARS-CoVs. Microscopy research and technique, 83(12), pp.1623-1638. |
| Mass Spectrometry | Identifies and quantifies proteins, peptides, and posttranslational modifications in mutant 3CLPro samples. Enables the characterization of protein structure, stability, and interactions, as well as the detection of mutation-induced alterations. | Highly sensitive and versatile technique for analyzing protein samples, including mutant forms of 3CLPro. | Requires specialized equipment and expertise. Data analysis can be complex, particularly for large proteins and complex samples. Sample preparation and handling may affect results. | Abe, K., Kabe, Y., Uchiyama, S., Iwasaki, Y.W., Ishizu, H., Uwamino, Y., Takenouchi, T., Uno, S., Ishii, M., Maruno, T. and Noda, M., 2022. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Scientific reports, 12(1), p.1299. |
| Enzyme Activity Assays | Measures the catalytic activity of mutant 3CLPro enzymes by monitoring substrate turnover or product formation. Assesses the impact of mutations on enzyme function, substrate specificity, and catalytic efficiency. | Provides direct assessment of mutant 3CLPro functionality, allowing quantitative analysis of enzymatic activity. | May require optimization for specific mutants and conditions. The results may be influenced by assay conditions, substrate choice, and enzyme purification methods. | Hu, Y., Lewandowski, E.M., Tan, H., Zhang, X., Morgan, R.T., Zhang, X., Jacobs, L.M., Butler, S.G., Gongora, M.V., Choy, J. and Deng, X., 2023. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS central science, 9(8), pp.1658-1669. |
| Circular Dichroism Spectroscopy (CD) | Analyzes the secondary structure of mutant 3CLPro proteins by measuring the differential absorption of circularly polarized light. Provides information about protein folding, stability, and conformational changes induced by mutations. | Rapid and nondestructive technique for studying protein secondary structure and stability. | Limited to analyzing protein secondary structure and may not provide detailed information about tertiary or quaternary structure. Requires careful interpretation of results. | Abe, K., Kabe, Y., Uchiyama, S., Iwasaki, Y.W., Ishizu, H., Uwamino, Y., Takenouchi, T., Uno, S., Ishii, M., Maruno, T. and Noda, M., 2022. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Scientific reports, 12(1), p.1299. |
| Fluorescence Spectroscopy | Studies the structural and dynamic properties of mutant 3CLPro proteins by monitoring changes in fluorescence emission upon ligand binding or conformational transitions. Offers insights into protein stability, folding, and interaction dynamics. | Sensitive method for detecting changes in protein structure and dynamics. Can be used to study protein‒ligand interactions and conformational changes induced by mutations. | Requires fluorescent labeling of proteins, which may affect protein function. Data interpretation can be complex, and results may be influenced by environmental factors. | Moghadasi, S.A., Esler, M.A., Otsuka, Y., Becker, J.T., Moraes, S.N., Anderson, C.B., Chamakuri, S., Belica, C., Wick, C., Harki, D.A. and Young, D.W., 2022. Gain-of-signal assays for probing inhibition of SARS-CoV-2 Mpro/3CLpro in living cells. MBio, 13(3), pp.e00784-22. |
| Molecular Dynamics Simulations | Uses computational models to simulate the behavior and dynamics of mutant 3CLPro proteins at the atomic level over time. Predicts protein structure, flexibility, and interactions, elucidating the effects of mutations on protein stability and function. | Allows exploration of mutant 3CLPro behavior and interactions at atomic resolution, providing insights into dynamic processes. | Requires computational resources and expertise. The results may be influenced by force field parameters, simulation length, and initial protein conformation. Interpretation can be challenging. | Gasparini, P., Philot, E.A., Pantaleão, S.Q., Torres-Bonfim, N.E.S.M., Kliousoff, A., Quiroz, R.C.N., Perahia, D., Simões, R.P., Magro, A.J. and Scott, A.L., 2023. Unveiling mutation effects on the structural dynamics of the main protease from SARS-CoV-2 with hybrid simulation methods. Journal of Molecular Graphics and Modeling, 121, p.108443. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



