
Submitted:
07 July 2024
Posted:
08 July 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID 19 pandemic saw the emergence of various Variants of Concern (VOCs) that took the world by storm, often replacing the ones that preceded them. The characteristic mutant constellations of these VOCs increased viral transmissibility and infectivity. Their origin and evolution remain puzzling. With the help of data mining efforts and the GISAID database, a chronology of 22 haplotypes described viral evolution up until July 23, 2023. Since the 3-dimensional atomic structures of proteins corresponding to the identified haplotypes are not available, ab initio methods were here utilized. Regions of intrinsic disorder proved to be important for viral evolution, as evidenced by the targeted change to the nucleocapsid (N) protein at the sequence, structure, and biochemical levels. The linker region of the N-protein, which binds to the RNA genome and self-oligomerizes for efficient genome packaging, was greatly impacted by mutations throughout the pandemic, followed by changes in structure and intrinsic disorder. Remarkably, VOC constellations acted cooperatively to balance the more extreme effects of individual haplotypes. Our strategy of mapping the dynamic evolutionary landscape of genetically linked mutations to the N-protein structure demonstrates the utility of ab initio modeling and deep learning tools for therapeutic intervention.
Keywords:
COVID-19
; haplotypes
; nucleocapsid protein
; mutation
; pandemic
; protein structure
; recruitment
; variant of concern
; virus evolution
1. Introduction
The Coronavirus Disease 19 (COVID-19) pandemic has led to a total of 775,645,882 reported cases and 7,051,876 deaths worldwide [1]. To put these numbers into perspective the death toll alone is more than the populations of several US states such as Montana, Rhode Island, Delaware, South Dakota, North Dakota, Alaska, the District of Columbia, Vermont, and Wyoming combined [2]. The total number of cases exceeds the population of the entirety of Europe by ~28 million people [3]. This makes COVID-19 one of the deadliest pandemics to hit the human population in the 21st century surpassing the impact of the Middle East Respiratory Syndrome (MERS) (September 2012, Jeddah, Saudi Arabia) and Severe Acute Respiratory Syndrome (SARS) (March 2003, Guangdong, China) coronaviruses [4,5], which had a total death toll of 774 and 949 deaths worldwide respectively [6,7]. This is remarkable because COVID-19 has a much lower mortality rate of 5.19% compared to 13% for SARS and up to 35% for MERS [8]. The disproportionate death tolls amongst these pandemics caused by coronaviruses can be attributed to the asymptomatic phase of COVID-19, which SARS and MERS lack [9]. Asymptomatic patients can carry on with their daily routines without even knowing that they are carriers of similar viral loads as their symptomatic counterparts enabling high transmission rates within a population [10]. This challenges disease control and mitigation as asymptomatic individuals spread disease to the uninfected. The most extreme case of asymptomatic COVID-19 occurrence was the “Diamond Princess” cruise ship population in Japan, which experienced 641 COVID-19 infections out of 3,711 passengers, with over half (328) of those infected being asymptomatic [11]. This makes the personal responsibility of an individual the ‘number one’ factor in preventing the spread of COVID-19 through masking and other precautionary measures [12] and poses an even greater challenge for policymakers to ensure the proper measures are in place to prevent the spread of COVID-19.
The COVID-19 disease is caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), which has a 79.6% sequence similarity to SARS-CoV-1, the causative agent of the first SARS outbreak [13]. Remarkably, 96% of its full-length ~30 kb genome sequence is identical to the sequence of a bat coronavirus [13]. The SARS-CoV-2 genome encodes 29 proteins, including 4 structural proteins crucial to viral function, the Spike (S), Nucleocapsid (N), Membrane (M), and Envelope (E) proteins. The evolution of the S-protein and M-protein and its effects on their structure have been previously reported [14,15]. In contrast, no significant changes were detected in the structure of the E-protein. The N-protein packages the positive-sense RNA genome of the virus forming the ribonucleoprotein structures of the viral capsid. The protein is necessary for viral assembly and RNA synthesis and participates in several cellular processes affecting immunological and cell cycle responses of the host [16]. The first mutations targeting the N-protein were found to be genetically linked via a haplotype (haplotype H2) [17]. The worldwide appearance of H2 very early in the pandemic followed the spread of the first reported haplotype (H5), which harbored the S-protein ‘D614G’ mutation. The N-protein is comprised of 5 distinct regions, two of which are structured and are known as the N-Terminal Domain (NTD) (residues 44-174) and the C-Terminal Domain (CTD) (residues 255-364). These domains are connected together by a disordered linker region (LKR) (residues 175-254) and are flanked by Intrinsically Disordered Regions (IDRs), the N-arm (residues 1-43) and the C-tail (residues 365-419) [18]. The CTD allows the N-protein to self-associate to form dimers while the C-tail mediates higher-order assembly into tetramers [19]. The disordered nature of the IDRs play a crucial role in binding to viral RNA with high cooperativity by enhancing binding affinity and allostery [20]. This increased binding affinity to nucleic acids is the result of the flexibility of disordered regions, which allow for multiple nucleic acid binding sites to associate with the same nucleic acid in an optimized allosteric conformation [21]. Both the NTD and CTD bind to viral RNA to form the nucleoprotein core of the virus [18,22]. The N-protein also interacts with the M-protein to fix ribonucleoproteins to the viral membrane [23]. Aside from its role in viral genome packaging, the N-protein also regulates antiviral immunity by the induction of interferon responses [24]. Following the formation of the ribonucleoprotein core at the ER-Golgi intermediate compartment (ERGIC) where the rest of the structural proteins are assembled, the budding process begins with proteins and viral RNA entering the lumen of secretory vesicles followed by transport of assembled virions out of the host cell via exocytosis [25,26,27]. There is considerable cooperativity between the four structural proteins of SARS-CoV-2. This enables efficient viral development and release, which is also demonstrated by the mutational evolutionary landscape. Many of the mutations observed have been dominated by the S, M, and N proteins throughout the pandemic [14]. These proteins are therefore important targets for vaccine and drug development.
Variants of Concern (VOCs) have been replacing each other since the beginning of the COVID-19 pandemic [28]. Their mutant constellations hold ‘mutations of concern’ that are of immediate priority for surveillance and response. The effect of these mutations on the protein sequence must be linked to effects at 3-dimensional (3D) atomic structure level to dissect the functional significance of individual VOCs and associated haplotypes. Three main strategies model protein structure: homology modeling, fold recognition, and ab initio methodologies [29]. Homology modeling and fold recognition rely on existing sequence and folded structure data and are rather comparative in nature. These methods can be limited in their ability to accurately predict the true 3D atomic structure of novel proteins, especially in molecular systems subjected to fast mutation rates. Ab initio methods, however, do not use pre-existing knowledge. Instead, they build models directly from amino acid sequences and the stoichiometric constraints of those sequences. Such an approach is especially useful for modeling proteins with low homology. AlphaFold2 [30] is the star of the last two biannual ab initio structure prediction experiments (CASP 14 and 15)[31,32]. Its deep learning algorithm makes fast atomic structural predictions with levels of accuracy that are within the margin of error of experimental structure determination methods. Crucially, this reduces reliance on traditional crystallographic and cryo-EM methods that are time-consuming. In the absence of experimental protein structures from VOCs, numerous studies utilized AlphaFold2 to explore the differences between the wild-type (WT) Wuhan strain, which is used as reference, and the emerging variants [15,33,34,35]. However, variant definitions do not reflect the complete viral landscape of SARS-CoV-2. Other groups of mutations can occur in greater frequencies than VOC constellations and VOCs often embody latitude-delimited haplotypes that have their own unique accumulation profiles across climatic zones [14,36]. These haplotypes, which were identified following a study of over 12 million viral proteomes, uncover seasonal patterns of emergence and help link structural conformations to environmental factors revealing an interplay between viral evolution, our environment, and our immune systems [14]. In fact, COVID-19 epidemiological variables exhibit significant negative correlation with temperature and positive correlation with latitude, often peaking around winter months, further indicating the seasonal nature of the virus [36,37,38].
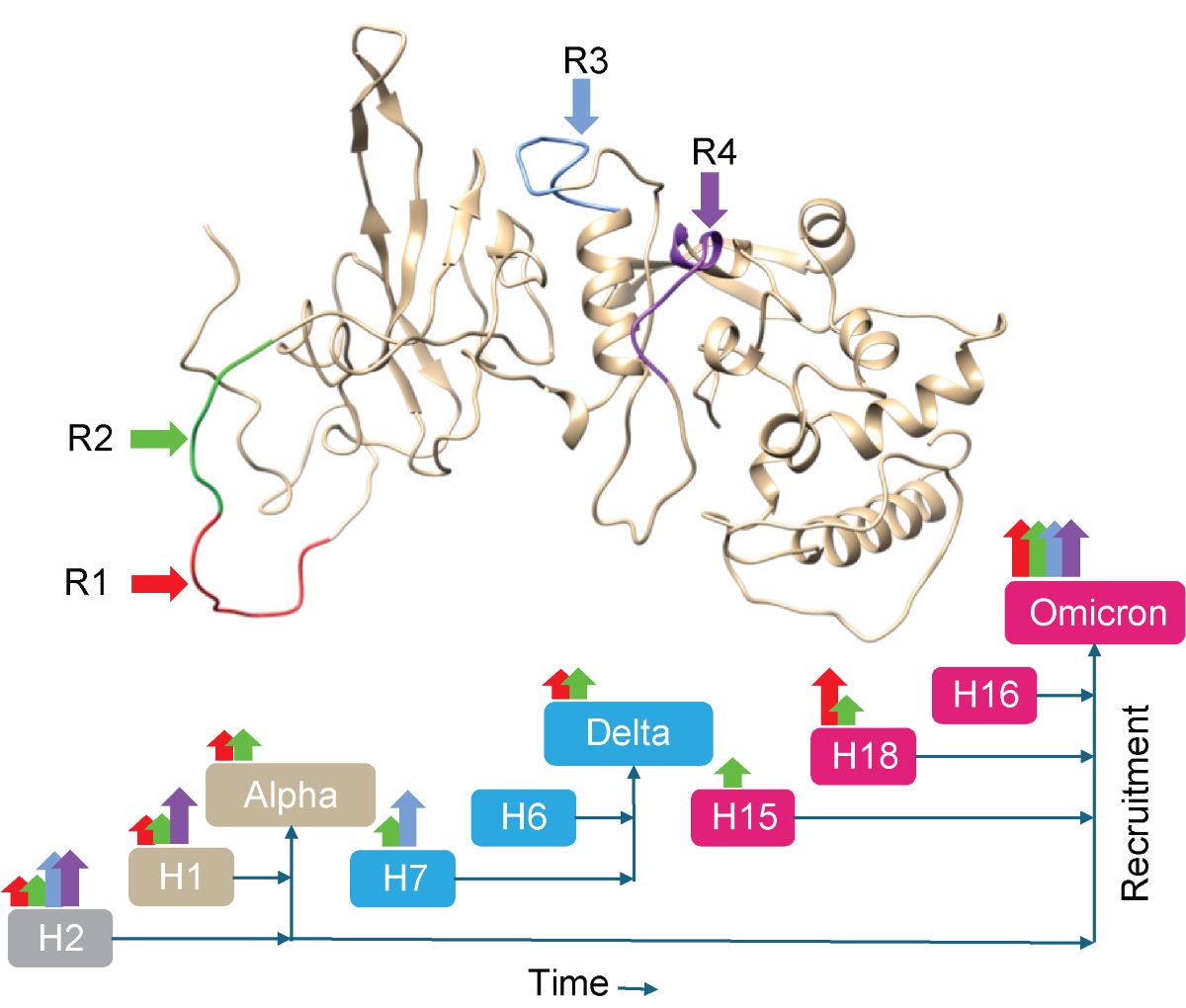
Here we use AlphaFold2 to model the 3D structures of mutant N-protein molecules defining SARS-CoV-2 haplotypes and constellations (Table S1). We also study the effect of mutations on the regions of intrinsic disorder of the molecule. Studying the structural changes observed across the pandemic uncovered patterns of structural recruitment across haplotypes and VOCs indicative of a complex interplay between the virus and its environment that mediates viral evolution. We show the N-protein was impacted by many haplotypes, beginning with H2 and its effects on the LKR region and ending with the rise of the VOC Omicron constellation and cooperative effects on protein structure. Our study highlights the importance of ab initio techniques and the utility of AlphaFold2 for comparative structural studies of proteins without any experimentally determined atomic structures and containing regions of intrinsic disorder known to be difficult to model by traditional means.
2. Materials and Methods
Accelerated ab initio modeling of 3D atomic structures of the N-protein was conducted using the AlphaFold2 pipeline [30] implemented locally in ColabFold without changes or modifications [39]. The output of five ranked structural models was obtained following twelve neural network recycles (processing of predictions through models) that iteratively extracted co-evolutionary information in PDB70 structural templates and multiple sequence alignments (MSAs) for end-to-end training of the deep learning ‘evoformer’ and ‘structure’ multi-layered neural network modules. MSAs were built with fast and sensitive MMseqs2-based homology searches of UniRef100 and a database of environmental sequences. Accuracy was measured with the predicted local distance difference test (pLDDT) and the predicted aligned error (PAE). pLDDT provides a per-residue estimate of prediction confidence based on the LDDT-Cα metric [40]. The expected prediction reliability of a given region or molecule follows pLDDT ‘confidence bands’: >90, models with very high confidence; 90-70, models with confidence, showing good backbone predictions; 70-50, models with low confidence; and <50, models with very low confidence, generally showing ribbon-like structures. pLDDT <60 can be considered a reasonably strong predictor of intrinsic disorder. PAE measures confidence in the relative positions of pairs of residues, which evaluates the cohesiveness of structural modules (e.g., domains).
Structural alignments and visualizations were carried out using Chimera [41]. Reference (corresponding to EPI_ISL_402124) and variant structures were superimposed using the MatchMaker and MatchAlign tools to identify regions with structural divergences. Topological similarities of individual regions or entire molecules were evaluated with average template modeling scores (TM-scores) using USalign [42,43]. N-protein predictions were benchmarked against cryo-EM models of the two structural domains [18]. Besides TM-scores, Global Distance Test – Total Score (GDT-TS) scores were obtained using the LGA (local-global alignment) structure comparative analysis tool with the AS2TS server [44,45,46], which CASP assessors routinely use to evaluate the accuracy of predicted structural models. The data presented in this study for the N-protein are openly available in ModelArchive under accession ma-gca-nprot.
3. Results
3.1. Full-Length Analysis of the N-Protein Using Backbone Root Mean Square Deviations (RMSD)
The N-protein was significantly impacted by four haplotypes (H1, H2, H7, and H18) and VOC Omicron. Mutations affected the structure of four small regions of the molecule. Figure 1 visualizes the amount of structural deviation for each VOC and its corresponding haplotypes with respect to each residue in the N-protein structure. Individual structural alignments from Chimera using the MatchMaker tool were used to extract RMSD headers, which contained the structural deviation per residue in Ångstroms (Å) for the protein backbones. This allowed to ascertain which areas along the length of the N-protein experienced the most change. Using an RMSD threshold of 3 Å (a standard for protein comparison and homologous proteins [47,48], we observed that the N-protein exhibited little change for each haplotype and VOC, aside from affecting the 4 target regions. The most notable change occurred in the LKR region, which is also the region most heavily impacted by amino acid mutations. Haplotypes H2, H1, H7, and VOC Omicron impacted the LKR structure at two specific locations (residues 210-220 and 247-257). Table S1 summarizes the mutations per haplotype/VOC along with their time of origin.
Our results revealed that VOC Alpha and Delta suppressed the individual effects of their constituent haplotypes, whereas VOC Omicron amplified these effects. One notable change occurred in the N-arm and was restricted to H18 and VOC Omicron due to the triple deletion mutation (E31-, R32-, S33-), which significantly changed the structure of the region. In contrast, other haplotypes and VOCs showed more or less the same structure aside from slight shifts in 3D space. VOCs Alpha and Delta behaved antagonistically to the extreme effects of their haplotypes, while VOC Omicron behaved synergistically. However, each VOC tended to inherit non-significant structural changes, though these regions provided little added functional benefit due to their similarity. To further analyze these regions of structural deviation, snapshots of the structural alignment in Chimera were taken and a total of 4 regions of interest (labeled R1-R4) were identified (visualized in Figure 2 (b) and (c)). The first two regions were part of the N-arm and were adjacent to one another. Only H18 and VOC Omicron impacted the N-arm; we observed a shorter loop turn due to the triple deletion in R1 and a slightly tighter conformation of R2 when compared to other haplotypes and VOCs. R3 and R4 were located in the LKR. R3 showed a short alpha helix with the structures of H2, H7 and VOC Omicron being off-axis compared to the rest of the structures. R4 also involved an alpha helix with H1, H2, and VOC Omicron adopting a much longer helix than the rest of the structures and causing them to have a very noticeable shift from the original Wuhan backbone.
The majority of the N-protein structure was disordered in nature. As shown in Figure 2, the N-arm (light blue), LKR (teal), and C-tail (steel blue) were completely comprised of loops and coils. Conversely, the NTD (lime green) and CTD (medium purple) were structured. Aside from the D63G mutation, no other amino acid mutations occurred in the NTD or CTD regions, helping explain their structural stability. In contrast, the IDRs of the N-protein were the main hotspots for mutational and structural change. Of the 3 IDRs, the LKR saw the most mutations, with 6 positions being impacted compared to 3 in the NTD (triple deletion counted as 1) and 2 in the C-tail. There were few changes in the beta-hairpin of the NTD, which fell under the 3 Å threshold. Figure S1 describes these regions in greater detail, with an atomic-level view of the differences between corresponding atoms of N-proteins from VOCs and haplotypes.
3.2. Regional Analysis with TM Scores Using USalign
TM scores from the Universal Structural Alignment (US-align) program [43] were used to better analyze the structural homology of the four regions of structural deviation (R1–R4) of the N-protein for each haplotype and VOC against the Wuhan structure. This similarity measure is superior to recording single-residue RMSD changes, which are very sensitive to small changes in conformation. The four regions from each haplotype and VOC were sliced out from their original ‘.pdb’ files and compared to the Wuhan regions separately. Figure 3 reports these results as a heatmap with their corresponding TM score in each cell. Darker colors indicate higher structural deviation than lighter colors. Two alignments were used for this analysis, one from Chimera’s superimposition (Figure 3 (a)) and a second further alignment forced by US-align (Figure 3 (b)). This allowed to judge if a structural change was more than just a shift in 3D space impacting instead the morphology of the region altogether. In the heatmap, we revealed numerous values under 0.5, the minimum score required for proteins to be considered in the same fold in the Structural Classification Of Proteins (SCOP) and Class, Architecture, Topology and Homolgous superfamily (CATH) databases [42,50]. There were significant changes observed for the four regions throughout the pandemic. However, when forcing a second alignment in US-align, many values shifted to above 0.7, indicating that those changes were only shifts in 3D space and were most prevalent for R1, which only saw H18 and VOC Omicron retain TM scores below 0.5. In contrast, more than half of the structures had scores lower than or close to 0.5. R2 was similar to R1, but this time, only VOC Omicron retained a value close to 0.5 but had generally lower TM scores than R1. R3 showed very consistent results throughout both alignments, indicating changes in these regions were more morphologically relevant, with H2, H7, and VOC Omicron sharing similar conformations. R4 stood out for its consistency as well, with almost every other structure having a TM score over 0.9 and only H1, H2, and VOC Omicron revealing scores under 0.5 that indicate the targeted nature of this structural inheritance across the length of the pandemic.
3.3. Protein Disorder and pLDDT Scores
Regions R3 and R4 were remarkable because they attained secondary structures in primarily disordered segments. Using the confidence metric of AlphaFold2, the pLDDT score, we showed that these regions had lower values (Figure 4). This is expected. IDRs are known to exhibit lower pLDDT values. In fact, AlphaFold2 now uses pLDDT scores to predict regions of disorder [51]. The lower pLDDT scores of R3 and R4 likely indicate the transient nature of helices, where they exist only briefly and could be part of a Short-Liner-Motif [52,53]. The 3 IDRs in the N-protein had notably lower pLDDT scores than the ordered NTD and CTD regions. The NTD was the most structured region with most of its residues scoring around 90 or 80. Only residues 94 to 98 scored below 70. The CTD was the second most ordered region. It showed lower scores than the NTD but still above 70 for most of its residues. Exceptions included the region spanning residues 312 and 336 and the residues close to the C-tail. Almost the entirety of the residues of the N-arm showed scores below 60. Surprisingly, a small portion of the LKR structure showed scores above the 70 pLDDT threshold in regions spanning residues 223 and 232 and residues 244 and the end of the region. The C-tail had a significant portion above 70, starting from residue 383, which runs towards the end of the structure, with a few smaller regions also scoring 70 towards the start of the C-tail. The N-arm behaved exactly as one would expect from an IDR. However, the LKR and C-tail exhibited some regions of high confidence. Note that IDRs that have high pLDDT scores from AlphaFold2 can undergo conditional folding under certain conditions to interact with other proteins [54]. Thus, the LKR and C-tail regions are potentially involved in molecular interactions.
3.4. Protein Disorder and Binding Capacity across the Pandemic
Protein disorder can play a crucial role in viral function. Therefore, we explored if disorder had changed or evolved during the pandemic. Using the online tool IUPred2A, a combination of Intrinsically Unstructured protein Prediction (IUPred) and ANCHOR [55], we measured the impact of the haplotypes and VOCs on intrinsic disorder and binding capability across the length of the N-protein. In Figure 4, the IUPred scores (a measure of protein disorder) and Anchor2 scores (a measure of binding capability) are plotted for each haplotype and VOC in chronological order from Haplotype H2 (bottom) to VOC Omicron (top). Despite their mutations, H2, H18, and H16 exhibited almost no or negligible change for both the IUPred and Anchor2 scores. Overall, we saw decreases in protein disorder across the pandemic, along with decreases in binding capability for the other haplotypes and the three major VOCs. There were increases in binding capability for H1 and VOC Alpha around residue position 235 due to mutation S235F and for H7 around residue position 203 due to the mutation R203M. Significant changes to protein disorder and binding capability were also limited to the three IDRs, with most of them occurring in the LKR during the H1 to VOC Delta phases of the pandemic. Coversely, the latter half of the pandemic saw most of its changes at the N-arm. VOC Delta and its corresponding haplotypes characteristically impacted the LKR and C-tail, whereas VOC Alpha and its haplotypes targeted the N-arm and LKR. The most notable differences included the N-arm (H1, VOC Alpha, H15, VOC Omicron), LKR (H1, VOC Alpha, H6, VOC Delta), and the C-tail (H7 and VOC Delta). This pattern of changing levels of protein disorder in specific regions of the N-protein indicates the changing needs for viral survival. As with the antagonistic and synergistic recruitment patterns of the various sub-structures along the protein and across the pandemic, we reveal a similar effect with protein disorder and binding capability in these studies. The interplay between the structural conformations and biochemical properties of these regions can help us understand how these different protein structures are adapted to interact with other proteins for effective viral functioning. This can be achieved through for example forthcoming Molecular Docking simulations.
3.5. Benchmarking AlphaFold2 Reference Structures against Experimental Cryo-EM Models
To validate our findings, we compared the AlphaFold2 structures against experimental crystallographic atomic models of the N-protein [18]. These structures were limited to the NTD and the CTD regions as the other regions of the N-protein were very disordered [56] and difficult to model. Since the N-protein has the ability to self-associate and form tetramers mediated by the CTD [19], we benchmarked the AlphaFold2 Wuhan model (with the NTD and CTD regions sliced out for independent comparisons) against the 4 chains of the NTD and the CTD provided by Peng et al. (2020)[18]. These models are available in the PDB database [57,58] with the IDs: 7CDZ for the NTD (doi.org/10.2210/pdb7cdz/pdb) and 7CE0 for the CTD (doi.org/10.2210/pdb7ce0/pdb), each with four chains labeled A to D [18]. Amino-acid Sequence to tertiary Structure (AS2S) [45] and US-align [43] were used to assess the quality of the AlphaFold2 model against the experimentally determined structures, and the results were reported in Table 1. The NTD shows very good structural homology with the experimental structures, obtaining GDT_TS scores around ~90 which can be thought of as the percentage of the protein that was modeled correctly [44], and the TM scores are also very high being above 0.9 for each chain. The AlphaFold2 model of the NTD also remained under an RMSD of 1.5 Å for more than 96% of its structure (126/131 residues) as reported by the AS2S server. These scores indicate very good structural topologies with very confident side chain conformations that can be used for further research and analysis. They also justify the haplotypic analysis of the NTD region. The CTD region, however, had lower GDT_TS and TM scores than the NTD but remained slightly below 70 and ~0.74, respectively, which are still adequate for use in research as both these scores indicate accurate topologies but less confidence with side-chain conformations.
4. Discussion
The N-protein is responsible for binding to the genetic material of the virus (RNA) and packaging it into ribonucleoprotein (RNP) particles [59]. This is achieved through RNA binding at various locations of the N-protein, which spans the N-arm [60], NTD [61], LKR [60], and CTD [20,62,63] regions. The CTD is also responsible for tetramer formation due to its ability to self-associate with the aid of the C-tail region [19]. The LKR plays an important role in RNP packaging [64] and oligomerization of N-protein molecules [65]. Both of these functions are achieved through biochemical and physical changes brought about by the phosphorylation of the region, which introduces significant electrostatic changes that are required to mediate these processes [59]. We observed that the most significant structural changes induced by mutant constellations and haplotypes occurred in the LKR region across numerous phases of the COVID-19 pandemic. In contrast, the NTD only saw significant change (including morphological changes apart from shifts in 3D space) in H18 and VOC Omicron due to the triple deletion. Note that both regions R1 and R2 of the N-arm are part of a single epitope ranging from residues 20-59, and R3 is part of an epitope ranging from residues 211 to 235 [49]. R1 mostly sees significant structural changes in H18 and Omicron as both of those structures harbor the triple deletion which all other structures lack (Figure S1). This suggests that the structural and mutational changes we observed could be due to the immunological pressures acting throughout the pandemic, forcing the virus to evolve to better ensure its survival. R3 is especially important because not only do we observe structural changes, but they are accompanied by the existence of an epitope that also saw two mutations in the region, including G215C and S235F, as well as exhibiting notable decreases in protein disorder (Figure 5). Our findings suggest that structural changes are observed in regions of functional significance that help immune evasion and other viral functions, including oligomerization for effective genome packaging.
The N-protein is already a very disordered protein and hence changes are more likely to occur in regions of disorder. However, it is apparent that the NTD and CTD structured regions remain relatively unchanged in sequence and structure except for one mutation (D63G) in the NTD (Figure 1). Interestingly the most common trend observed was that both protein disorder and binding capability actually reduced for most regions of the N-protein throughout the pandemic with the only notable increase being observed in the LKR for H1, H7 and VOC Alpha. The increase of binding capability was inherited from H1 to Alpha. However, the same was not true for VOC Delta and H7. Haplotypes H7 and H8 seemed to cancel out the impact of the R203M mutation on binding affinity and both R203M and G215C removed the increase in binding capability at the LKR (Figure 5). This portrays the complex interactions between different mutations of several haplotypes within their VOC constellations as they can behave synergistically or antagonistically to one another. Despite these changes occurring in the regions of disorder, all of the changes we reported showed a decrease in protein disorder except for one very small portion at the triple deletion site in H18 and VOC Omicron.
R2 and R3, which embody established epitopes [49], were the only two regions that saw structural changes of immunological significance. Both of these regions showed decreases in protein disorder accompanied by decreases in binding capability (Figure 5). This indicates a mechanism of immune evasion where mutations such as the triple deletion in H18 and VOC Omicron, along with the mutations G215C and S235F, acted as a means to bind less effectively with antibodies. These structural changes, along with those in R1 and R4, appear to be recruited throughout the pandemic in various combinations amongst haplotypes and VOC Omicron. This combinatorial strategy highlights the evolutionary landscape of structural exploration that is unfolding in the N-protein, where combinations of smaller structural changes are adopted that benefit viral function. These changes are not limited to phases of the pandemic either. Earlier structures such as those observed in region R4 of haplotypes H1 and H2, were later found in VOC Omicron. In Figure S1 we observe that H1, H2, and Omicron have an alpha helix causing significant differences in atomic distances between corresponding atoms. Similarly, R3 revealed a similar recruitment pattern spanning the entire length of the pandemic, with the same sub-structure being found in H2, H7, and VOC Omicron. H2, H7, and Omicron have an extended alpha helix that is part of the bigger alpha helix downstream of the structure and is slightly shifted from the original axis of the Wuhan structure (Figure S1). H1 and Delta also adopted extended alpha helices in R3 but were not shifted from the original axis. The exploration of various sub-structures at specific regions of a protein can be thought of as a metric of structural entropy where higher numbers of structural conformations indicate higher levels of structural entropy. Most of the changes in the N-protein did not show much variation in the sub-structures but only in the combination of these sub-structures with one another. Since the same structure was recruited at regions R3 and R4 along the length of the pandemic, we consider these to be examples of entropic fixations. Here, structural entropy does not expand as numerous conformations are not being explored, indicating the N-protein adopted a more targeted evolutionary approach when compared to the S-protein [15]. These structural recruitments reveal patterns of synergy and antagonism during the pandemic. VOCs Alpha and Delta behaved antagonistically to the structural effects of their constituent haplotypes undoing their structural impacts. VOC Omicron, in turn, amplified or retained the individual effects of its constituent haplotypes. These effects mimic those found in our study of the S-protein [15].
5. Conclusions
One remarkable finding of our ab initio modeling exercises is that VOC constellations counteracted the more extreme effects of individual haplotypes on protein structure. This reveals a difference in evolutionary pressures for different proteins and showcases how haplotypes can be more or less beneficial for their corresponding VOCs. There also exists a pattern of recruitment where different haplotypes and VOCs adopt similar conformations at specific sites across the pandemic indicating the importance of both structural and mutational entropy [17]. The virus explores various structural changes during the evolutionary process to find the combination of structural conformations that best suit survival and fitness. This strongly suggests that a cooperative activity exists in haplotype-mediated protein communication, which was already made explicit in previously described haplotype-delimited protein interaction networks [14]. The impact of haplotypes H1 and H2 on the N-protein now reveals seasonal effects in mutation accumulation patterns along the length of the pandemic [14]. H1 achieved the highest prevalence in Arctic and Northern temperate regions indicating temperature sensitivity. H2 showed a seasonal pattern of accumulation, which later turned into complete prevalence during the pandemic. This information will be especially valuable for therapeutic interventions and predictive intelligence applications, which could facilitate understanding of protein reformation, de novo protein and construct design, and formation of molecular complexes.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Structural models of regions R1-R4 of high structural change showcasing the atomic level differences for each VOC and haplotype; Table S1: Emergence of VOCs and haplotypes and their mutant constellations.
Author Contributions
Conceptualization, M.A.A. and G.C-A..; methodology, M.A.A. and G.C-A.; validation, M.A.A.; formal analysis, M.A.A. and G.C-A.; investigation, M.A.A.; resources, G.C-A.; data curation, M.A.A.; writing—original draft preparation, M.A.A.; writing—review and editing, M.A.A. and G.C-A.; visualization, M.A.A.; supervision, G.C-A.; project administration, G.C-A.; funding acquisition, G.C-A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the National Institute of Food and Agriculture (ILLU-802-909 and ILLU-483-625), the Office of Research and Office of International Programs, College of Agricultural, Consumer and Environmental Sciences of the University of Illinois at Urbana-Champaign, and the New Frontiers Initiative of the National Center for Supercomputing Applications (NCSA), all to G.C-A
Data Availability Statement
The data presented in this study is openly available in ModelArchive under accession ma-gca-nprot (https://modelarchive.org). Other data and information supporting the findings of this study are either public or available within the article and its supplementary Materials.
Acknowledgments
We gratefully acknowledge all data contributors, i.e., the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based.
Conflicts of Interest
The authors declare that they have no competing interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- WHO COVID-19 Dashboard, COVID-19 Deaths. Available online: https://data.who.int/dashboards/covid19/cases (accessed on 1 July 2024).
- StatsAmerica, States in Profile, Population estimate for 2023, Ranked list. Available online: https://www.statsamerica.org/sip/rank_list.aspx?rank_label=pop1&ct=S18 (accessed on 1 July 2024).
- United Nations, Department of Economic and Social Affairs, Population Division, World Population Prospects. Available online: https://population.un.org/wpp/Download/Standard/Population/ (accessed on 1 July 2024).
- Ramadan, N.; Shaib, H. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): A Review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hodgens, A.; Gupta, V. Severe Acute Respiratory Syndrome. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2022. [Google Scholar]
- Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003 Available online:. Available online: https://www.who.int/publications/m/item/summary-of-probable-sars-cases-with-onset-of-illness-from-1-november-2002-to-31-july-2003 (accessed on 1 July 2024).
- MERS-CoV Worldwide Overview Available online:. Available online: https://www.ecdc.europa.eu/en/middle-east-respiratory-syndrome-coronavirus-mers-cov-situation-update (accessed on 1 July 2024).
- Pormohammad, A.; Ghorbani, S.; Khatami, A.; Farzi, R.; Baradaran, B.; Turner, D.L.; Turner, R.J.; Bahr, N.C.; Idrovo, J. Comparison of Confirmed COVID-19 with SARS and MERS Cases - Clinical Characteristics, Laboratory Findings, Radiographic Signs and Outcomes: A Systematic Review and Meta-analysis. Rev Med Virol 2020, 30, e2112. [Google Scholar] [CrossRef] [PubMed]
- Pustake, M.; Tambolkar, I.; Giri, P.; Gandhi, C. SARS, MERS and CoVID-19: An Overview and Comparison of Clinical, Laboratory and Radiological Features. J Family Med Prim Care 2022, 11, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. New England Journal of Medicine 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, K.; Kagaya, K.; Zarebski, A.; Chowell, G. Estimating the Asymptomatic Proportion of Coronavirus Disease 2019 (COVID-19) Cases on Board the Diamond Princess Cruise Ship, Yokohama, Japan, 2020. Eurosurveillance 2020, 25, 2000180. [Google Scholar] [CrossRef]
- Liu, P.L. COVID-19 Information on Social Media and Preventive Behaviors: Managing the Pandemic through Personal Responsibility. Soc Sci Med 2021, 277, 113928. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Tomaszewski, T.; Ali, M.A.; Caetano-Anollés, K.; Caetano-Anollés, G. Seasonal Effects Decouple SARS-CoV-2 Haplotypes Worldwide 2023.
- Ali, M.A.; Caetano-Anollés, G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants. Biology 2024, 13, 134. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, Y.; Zhou, H.; Sun, C.; Zhang, S. The SARS-CoV-2 Nucleocapsid Protein: Its Role in the Viral Life Cycle, Structure and Functions, and Use as a Potential Target in the Development of Vaccines and Diagnostics. Virol J 2023, 20, 6. [Google Scholar] [CrossRef]
- Tomaszewski, T.; DeVries, R.S.; Dong, M.; Bhatia, G.; Norsworthy, M.D.; Zheng, X.; Caetano-Anollés, G. New Pathways of Mutational Change in SARS-CoV-2 Proteomes Involve Regions of Intrinsic Disorder Important for Virus Replication and Release. Evol Bioinform Online 2020, 16, 1176934320965149. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 Nucleocapsid and Their Perspectives for Drug Design. The EMBO Journal 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and Self-assembly of the SARS-CoV-2 Nucleocapsid Protein. Protein Sci 2020, 29, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Hsu, Y.-L.; Chang, Y.-H.; Chao, F.-A.; Wu, M.-C.; Huang, Y.-S.; Hu, C.-K.; Huang, T.-H. Multiple Nucleic Acid Binding Sites and Intrinsic Disorder of Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein: Implications for Ribonucleocapsid Protein Packaging. J Virol 2009, 83, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Thompson, E.B. Intrinsic Disorder as a Mechanism to Optimize Allosteric Coupling in Proteins. Proceedings of the National Academy of Sciences 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T. The SARS Coronavirus Nucleocapsid Protein – Forms and Functions. Antiviral Research 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat Commun 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- de Haan, C.A.M.; Rottier, P.J.M. Molecular Interactions in the Assembly of Coronaviruses. Adv Virus Res 2005, 64, 165–230. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography 2020, 2020.06.23.167064.
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The Intracellular Sites of Early Replication and Budding of SARS-Coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Chapter 11 - Protein Structure Prediction. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press, 2022; pp. 177–188 ISBN 978-0-323-89775-4.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XIV. Proteins: Structure, Function, and Bioinformatics 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XV. Proteins: Structure, Function, and Bioinformatics 2023, 91, 1539–1549. [Google Scholar] [CrossRef]
- Ford, C.T.; Jacob Machado, D.; Janies, D.A. Predictions of the SARS-CoV-2 Omicron Variant (B.1.1.529) Spike Protein Receptor-Binding Domain Structure and Neutralizing Antibody Interactions. Front. Virol. 2022, 2. [Google Scholar] [CrossRef]
- Kilim, O.; Mentes, A.; Pál, B.; Csabai, I.; Gellért, Á. SARS-CoV-2 Receptor-Binding Domain Deep Mutational AlphaFold2 Structures. Sci Data 2023, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jian, X.; Syed, A.A.S.; Fahira, A.; Zheng, C.; Zhu, Z.; Wang, K.; Zhang, J.; Wen, Y.; Li, Z.; et al. Structural Comparison and Drug Screening of Spike Proteins of Ten SARS-CoV-2 Variants. Research 2022, 2022. [Google Scholar] [CrossRef]
- Tomaszewski, T.; Gurtler, V.; Caetano-Anollés, K.; Caetano-Anollés, G. Chapter 8 - The Emergence of SARS-CoV-2 Variants of Concern in Australia by Haplotype Coalescence Reveals a Continental Link to COVID-19 Seasonality. In Methods in Microbiology; Pavia, C.S., Gurtler, V., Eds.; Covid-19: Biomedical Perspectives; Academic Press, 2022; Vol. 50, pp. 233–268.
- Burra, P.; Soto-Díaz, K.; Chalen, I.; Gonzalez-Ricon, R.J.; Istanto, D.; Caetano-Anollés, G. Temperature and Latitude Correlate with SARS-CoV-2 Epidemiological Variables but Not with Genomic Change Worldwide. Evol Bioinform Online 2021, 17, 1176934321989695. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N.; Caetano-Anollés, G. Worldwide Correlations Support COVID-19 Seasonal Behavior and Impact of Global Change. Evolutionary Bioinformatics 2023, 19, 11769343231169377. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J Comput Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-Align: Universal Structure Alignments of Proteins, Nucleic Acids, and Macromolecular Complexes. Nat Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A. LGA: A Method for Finding 3D Similarities in Protein Structures. Nucleic Acids Res 2003, 31, 3370–3374. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Zhou, C.E.; Slezak, T.; Kuczmarski, T.; Rama, D.; Torres, C.; Sawicka, D.; Barsky, D. AS2TS System for Protein Structure Modeling and Analysis. Nucleic Acids Res 2005, 33, W111–115. [Google Scholar] [CrossRef] [PubMed]
- LGA - Protein Structure Comparison Facility Available online:. Available online: http://proteinmodel.org/AS2TS/LGA/lga.html (accessed on 2 July 2024).
- Chothia, C.; Lesk, A.M. The Relation between the Divergence of Sequence and Structure in Proteins. The EMBO Journal 1986, 5, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.A.; Finkelstein, A.V.; Skolnick, J. What Is the Probability of a Chance Prediction of a Protein Structure with an Rmsd of 6 å? Folding and Design 1998, 3, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Olsen, K.S.; Gentry, K.M.; Sambade, M.; Beck, W.; Garness, J.; Entwistle, S.; Willis, C.; Vensko, S.; Woods, A.; et al. Landscape and Selection of Vaccine Epitopes in SARS-CoV-2. Genome Med 2021, 13, 101. [Google Scholar] [CrossRef]
- TM-Score: Quantitative Assessment of Similarity between Protein Structures Available online:. Available online: https://zhanggroup.org/TM-score/ (accessed on 1 July 2024).
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A Structural Biology Community Assessment of AlphaFold2 Applications. Nat Struct Mol Biol 2022, 29, 1056–1067. [Google Scholar] [CrossRef]
- Bruley, A.; Mornon, J.-P.; Duprat, E.; Callebaut, I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules 2022, 12, 1467. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Choy, W.-Y.; Karttunen, M. AlphaFold2: A Role for Disordered Protein/Region Prediction? Int J Mol Sci 2022, 23, 4591. [Google Scholar] [CrossRef]
- Alderson, T.R.; Pritišanac, I.; Kolarić, Đ.; Moses, A.M.; Forman-Kay, J.D. Systematic Identification of Conditionally Folded Intrinsically Disordered Regions by AlphaFold2. Proc Natl Acad Sci U S A 120, e2304302120. [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Research 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat Commun 2021, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Research 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bank, R.P.D. RCSB PDB: Homepage Available online:. Available online: https://www.rcsb.org/ (accessed on 1 July 2024).
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef]
- Peng, T.-Y.; Lee, K.-R.; Tarn, W.-Y. Phosphorylation of the Arginine/Serine Dipeptide-Rich Motif of the Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein Modulates Its Multimerization, Translation Inhibitory Activity and Cellular Localization. The FEBS Journal 2008, 275, 4152–4163. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural Basis of RNA Recognition by the SARS-CoV-2 Nucleocapsid Phosphoprotein. PLoS Pathog 2020, 16, e1009100. [Google Scholar] [CrossRef]
- Takeda, M.; Chang, C.; Ikeya, T.; Güntert, P.; Chang, Y.; Hsu, Y.; Huang, T.; Kainosho, M. Solution Structure of the C-Terminal Dimerization Domain of SARS Coronavirus Nucleocapsid Protein Solved by the SAIL-NMR Method. Journal of Molecular Biology 2008, 380, 608–622. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chang, C.; Chang, Y.-W.; Sue, S.-C.; Bai, H.-I.; Riang, L.; Hsiao, C.-D.; Huang, T. Structure of the SARS Coronavirus Nucleocapsid Protein RNA-Binding Dimerization Domain Suggests a Mechanism for Helical Packaging of Viral RNA. Journal of Molecular Biology 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chen, C.-M.M.; Chiang, M.; Hsu, Y.; Huang, T. Transient Oligomerization of the SARS-CoV N Protein--Implication for Virus Ribonucleoprotein Packaging. PLoS One 2013, 8, e65045. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.K.; Lal, S.K. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein Is Phosphorylated and Localizes in the Cytoplasm by 14-3-3-Mediated Translocation. Journal of Virology 2005, 79, 11476–11486. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Regions of structural deviation. The vertical axis represents each of the haplotypes and variants, impacting the N-protein, arranged in chronological order from earliest to latest, up until the rise of VOC Omicron. The horizontal axis represents the amino acid positions along the length of each of the N-protein molecules. The horizontal bars representing the haplotypes and VOC constellations are colored in hues of light blue and thistle in an alternating fashion whereas VOCs Alpha, Delta, and Omicron are colored yellow, green, and purple, respectively. When a residue along the length of any of these protein molecules crosses the 1Å, 2 Å, 3 Å, and 5 Å RMSD, then that position is colored in dark blue, light blue, orange, and red, respectively. The area labelled ‘Epitopes’ includes reside ranges for important epitopes found along the length of the N-protein as reported by Smith et al., (2021)[49]. The horizontal bar labeled ‘N-regions’ indicates all positions of important regions/domains along the N-protein: N-arm (1-43), NTD (44-174), LKR (175-254), CTD (255-364), and C-tail (365-419). The horizontal bar labeled SS indicates the position of alpha helices (brown), beta sheets (dark red), and coils (beige). The horizontal bar labeled pLDDT represents the confidence level of AlphaFold2 for each residue along the S-protein. On top of the graph are the individual mutations that comprise each haplotype and variant.
Figure 1.
Regions of structural deviation. The vertical axis represents each of the haplotypes and variants, impacting the N-protein, arranged in chronological order from earliest to latest, up until the rise of VOC Omicron. The horizontal axis represents the amino acid positions along the length of each of the N-protein molecules. The horizontal bars representing the haplotypes and VOC constellations are colored in hues of light blue and thistle in an alternating fashion whereas VOCs Alpha, Delta, and Omicron are colored yellow, green, and purple, respectively. When a residue along the length of any of these protein molecules crosses the 1Å, 2 Å, 3 Å, and 5 Å RMSD, then that position is colored in dark blue, light blue, orange, and red, respectively. The area labelled ‘Epitopes’ includes reside ranges for important epitopes found along the length of the N-protein as reported by Smith et al., (2021)[49]. The horizontal bar labeled ‘N-regions’ indicates all positions of important regions/domains along the N-protein: N-arm (1-43), NTD (44-174), LKR (175-254), CTD (255-364), and C-tail (365-419). The horizontal bar labeled SS indicates the position of alpha helices (brown), beta sheets (dark red), and coils (beige). The horizontal bar labeled pLDDT represents the confidence level of AlphaFold2 for each residue along the S-protein. On top of the graph are the individual mutations that comprise each haplotype and variant.
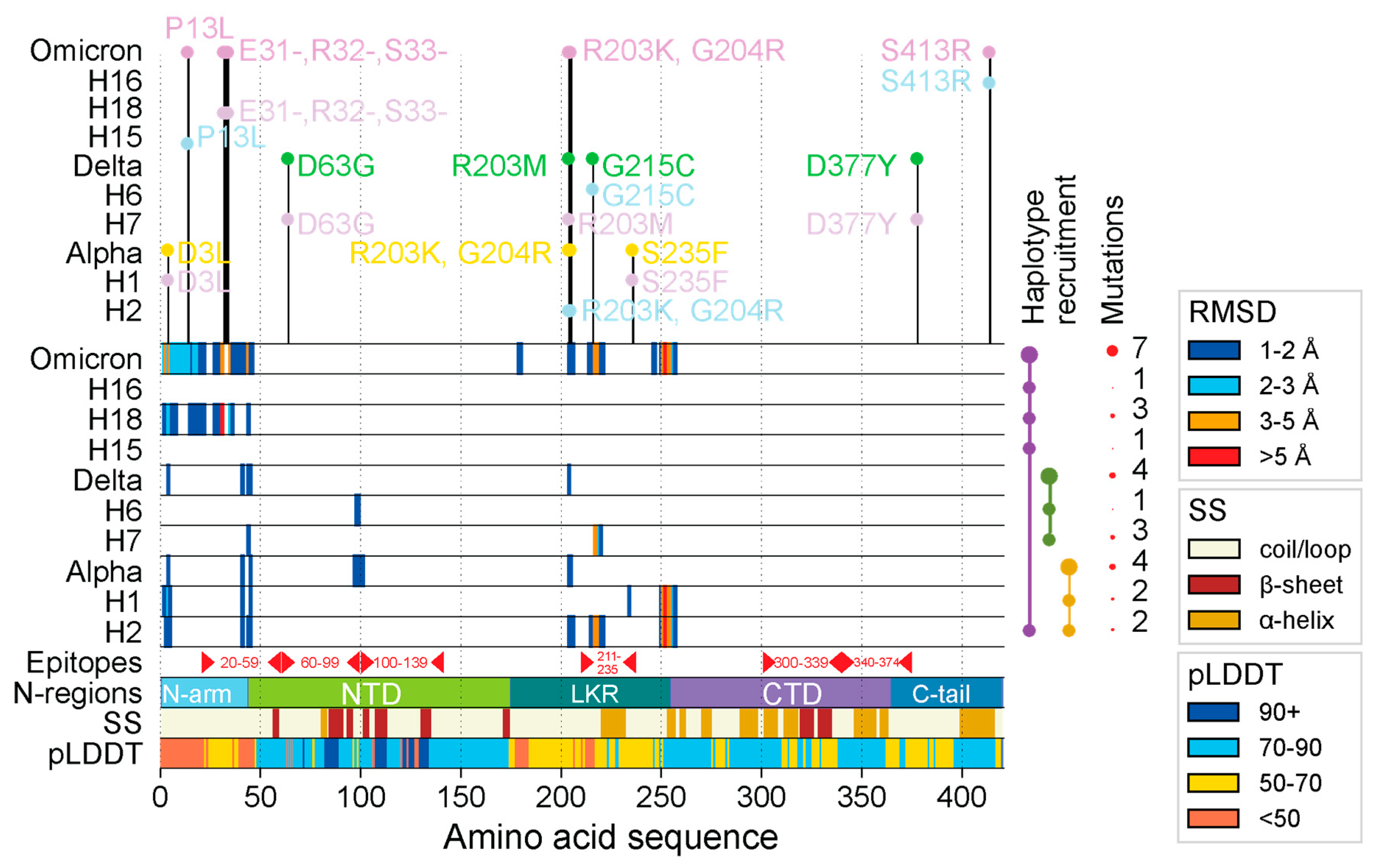
Figure 2.
Three-dimensional (3D) models of molecular change at the atomic level in regions of structural deviation. (a) The N-protein modeled by local ColabFold for the Wuhan strain with its 5 structural domains color-coded as N-arm (light blue), NTD (lime green), LKR (teal), CTD (purple), and C-tail (steel blue), the same color scheme as the N-regions bar in Figure 1. Areas that saw structural deviation crossing the 3 Å threshold for each haplotype and VOC have been highlighted in red. (b) All haplotypes and VOCs were superimposed onto the Wuhan structure using Chimera and are color-coded according to the top right index. (c) Using data from Figure 1, all residues that surpassed the 3 Å threshold and other regions from further inspection in Chimera were translated into 4 regions of structural deviation (R1 to R4). Model snapshots of these regions were taken in Chimera to ensure all of the superimposed structures and their corresponding residues were adequately captured for each region. The location of regions in the amino acid sequence is indicated in red. Each variant and haplotype structure is color-coded according to the index.
Figure 2.
Three-dimensional (3D) models of molecular change at the atomic level in regions of structural deviation. (a) The N-protein modeled by local ColabFold for the Wuhan strain with its 5 structural domains color-coded as N-arm (light blue), NTD (lime green), LKR (teal), CTD (purple), and C-tail (steel blue), the same color scheme as the N-regions bar in Figure 1. Areas that saw structural deviation crossing the 3 Å threshold for each haplotype and VOC have been highlighted in red. (b) All haplotypes and VOCs were superimposed onto the Wuhan structure using Chimera and are color-coded according to the top right index. (c) Using data from Figure 1, all residues that surpassed the 3 Å threshold and other regions from further inspection in Chimera were translated into 4 regions of structural deviation (R1 to R4). Model snapshots of these regions were taken in Chimera to ensure all of the superimposed structures and their corresponding residues were adequately captured for each region. The location of regions in the amino acid sequence is indicated in red. Each variant and haplotype structure is color-coded according to the index.
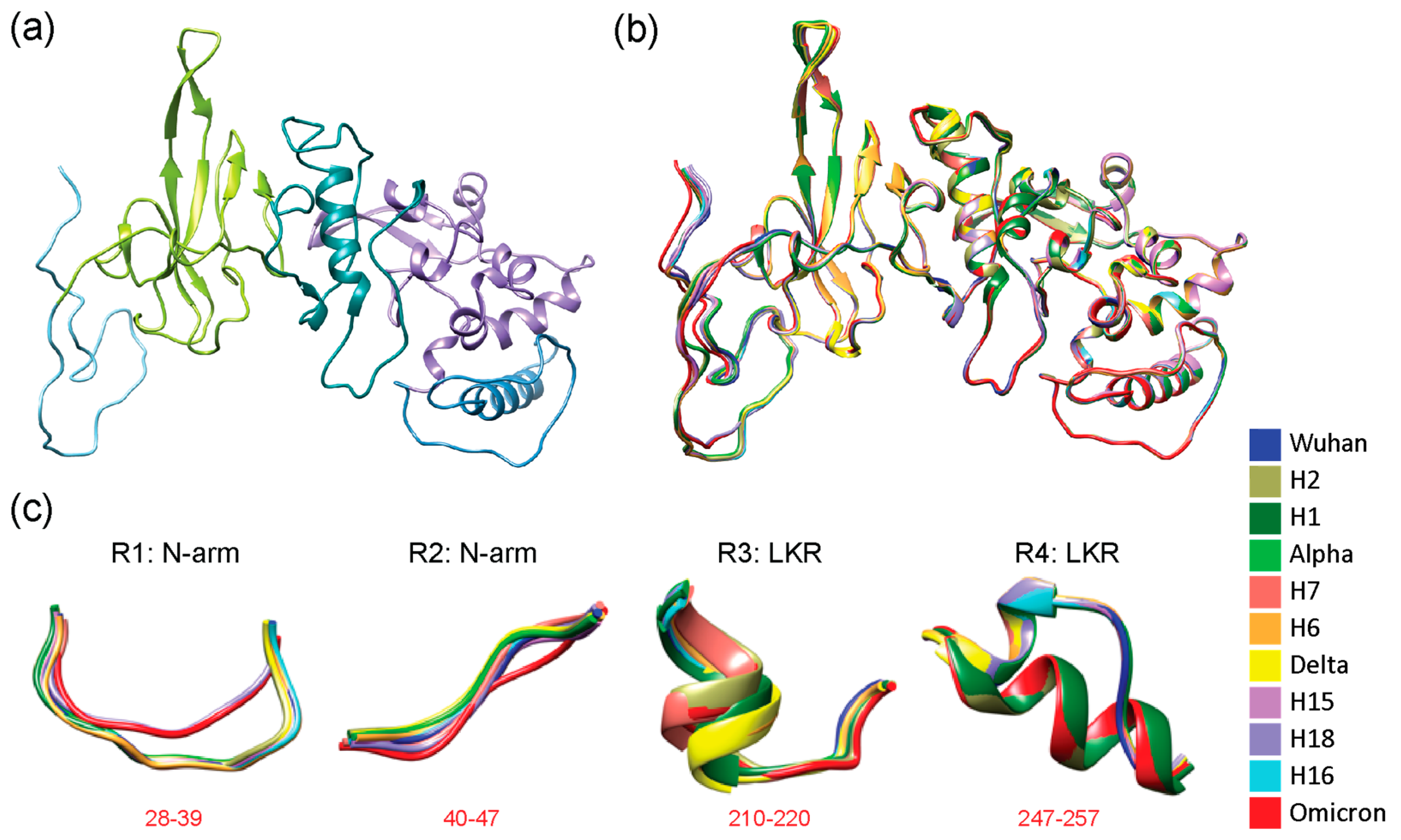
Figure 3.
Heatmap of TM scores (ranging 0–1) of regions of structural domains. The 4 regions of structural deviation (R1-R4) were used to slice the corresponding regions from each file and obtain TM scores for each VOC and haplotype with USalign, using the Wuhan reference molecule as the template. The horizontal axis is arranged in chronological order depicting changes across the timeline of the pandemic, and the vertical axis represents the 5 structural domains described in Figure 3. All TM scores falling under the TM threshold of 0.5 are colored in light cream. (a) TM scores using alignments from Chimera superimposition. (b) TM scores using USalign alignments. (c) Difference between heatmaps in (a) and (b).
Figure 3.
Heatmap of TM scores (ranging 0–1) of regions of structural domains. The 4 regions of structural deviation (R1-R4) were used to slice the corresponding regions from each file and obtain TM scores for each VOC and haplotype with USalign, using the Wuhan reference molecule as the template. The horizontal axis is arranged in chronological order depicting changes across the timeline of the pandemic, and the vertical axis represents the 5 structural domains described in Figure 3. All TM scores falling under the TM threshold of 0.5 are colored in light cream. (a) TM scores using alignments from Chimera superimposition. (b) TM scores using USalign alignments. (c) Difference between heatmaps in (a) and (b).
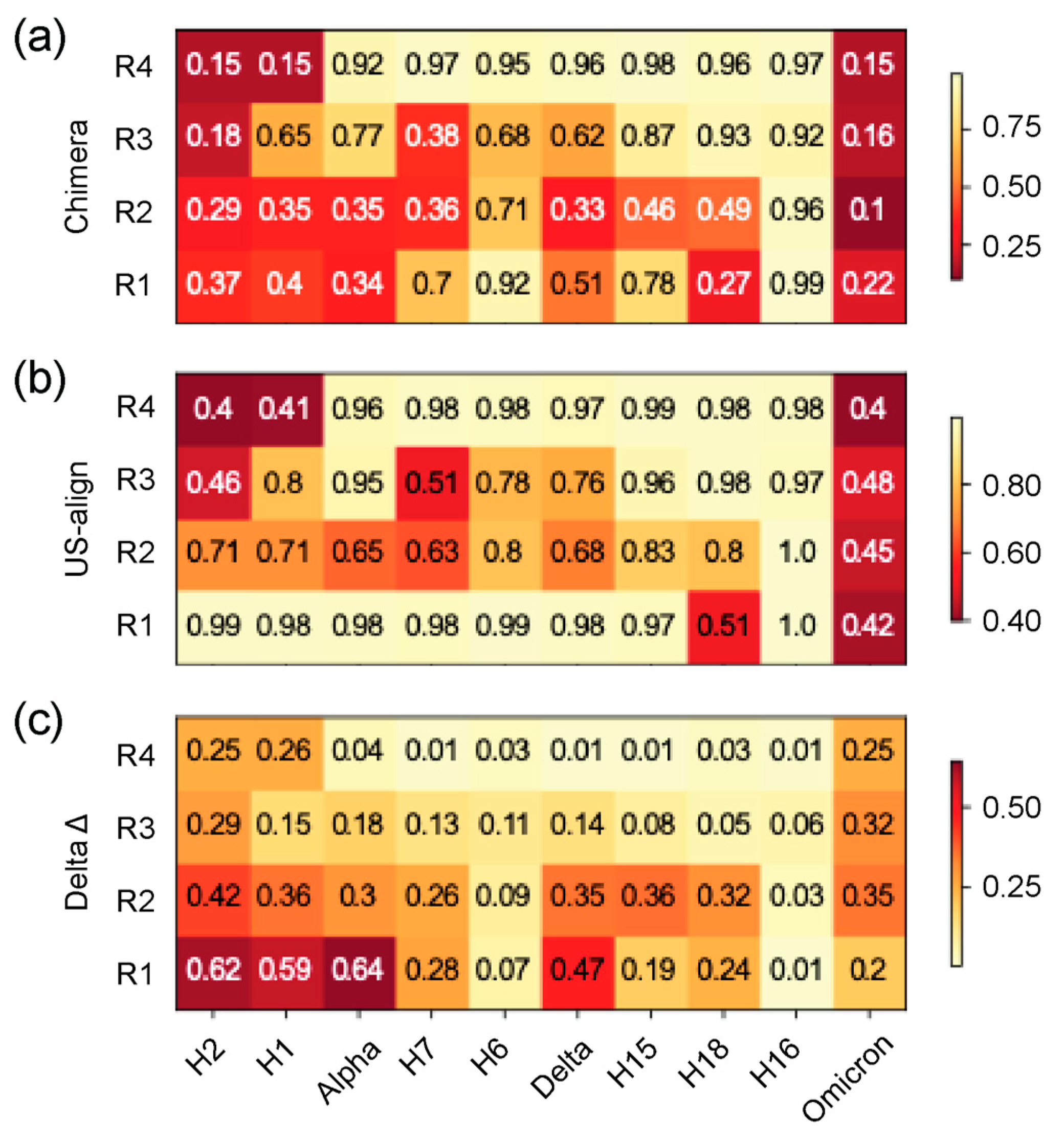
Figure 4.
Per-residue pLDDT scores of each AlphaFold2 top-ranked modeled 3D structure.
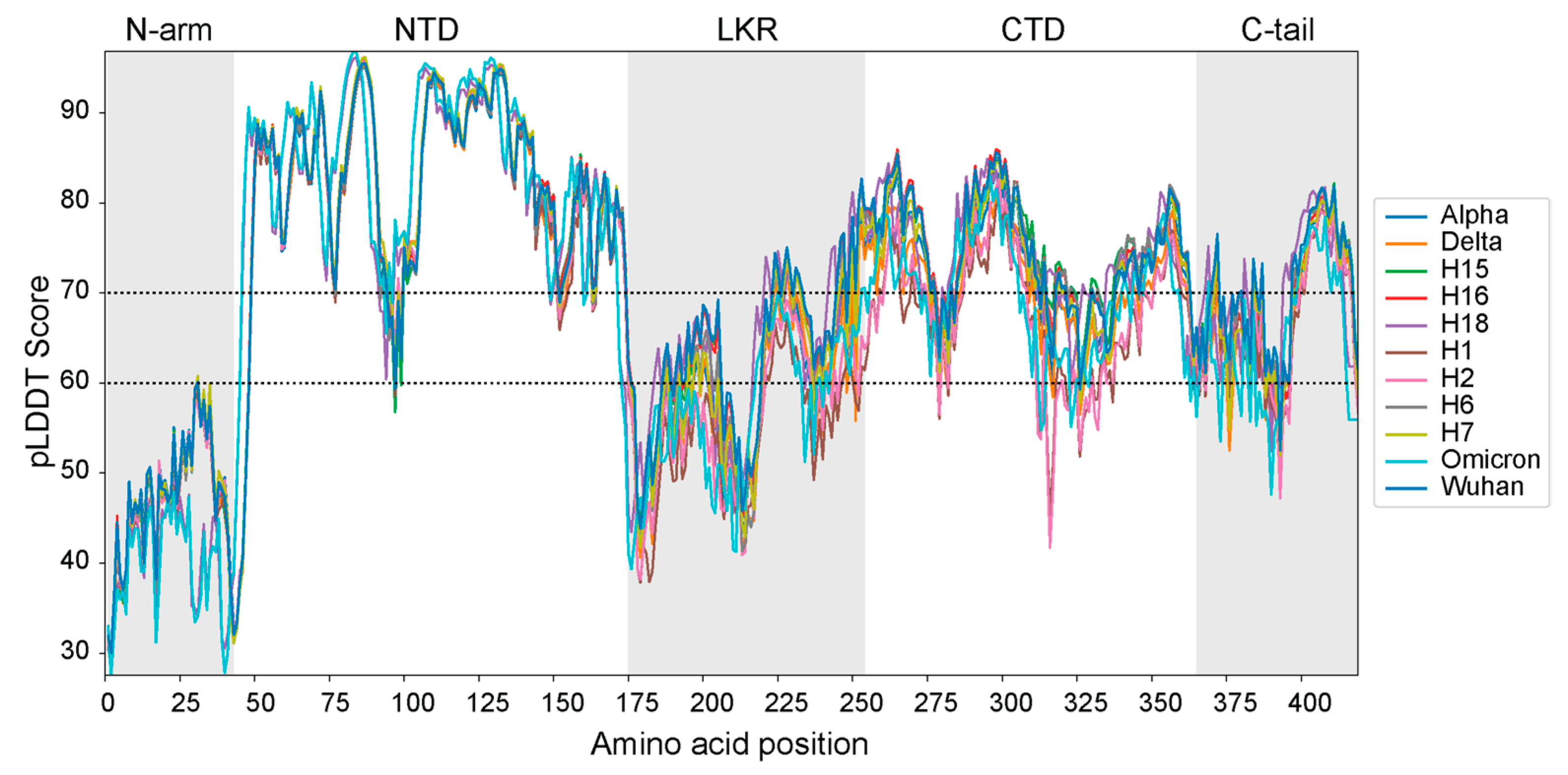
Figure 5.
Intrinsic disorder and binding capability across the pandemic.
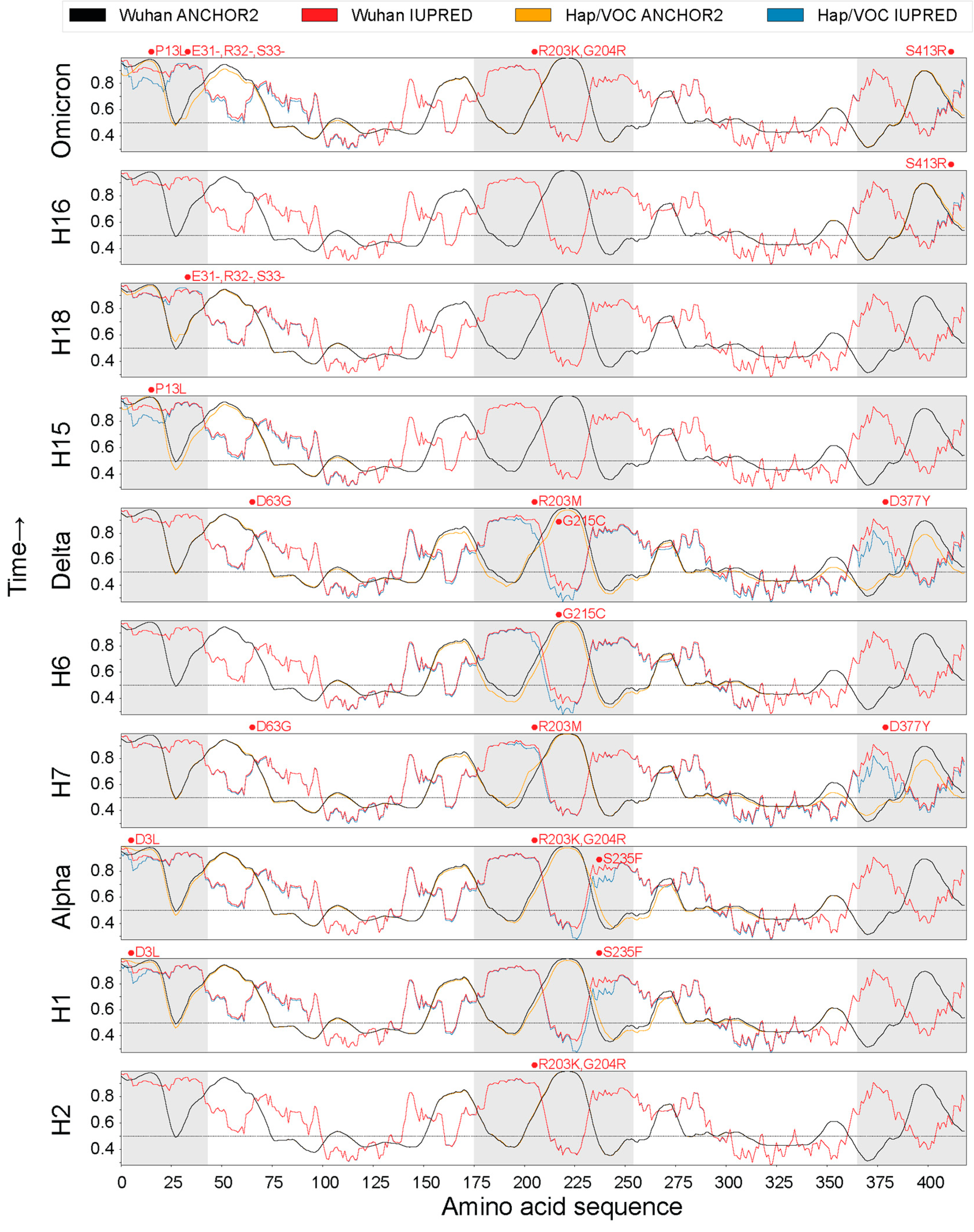

Table 1.
Benchmarking results.
| State-Chain | GDT-TS (AS2S) | TM Score/L (US-Align Server) | Superimposed RMSD < 5 Å (AS2S) |
|---|---|---|---|
| NTD-A (7CDZ) | 91.99*(128/131) = 89.88 |
*L1: 0.93398, L2: 0.91375 |
1.293/128 |
| NTD-B (7CDZ) | 90.35*(127/131) = 87.59 |
0.92756, 0.90091 |
1.366/127 |
| NTD-C (7CDZ) | 95.24*(126/131) = 91.60 |
0.95983, 0.92447 |
0.941/126 |
| NTD-D (7CDZ) | 94.26*(122/131) = 87.78 |
0.93300, 0.90592 |
1.1108/122 |
| CTD-A (7CE0) | 69.73*(109/110) = 69.10 |
0.74156 | 2.508/109 |
| CTD-B (7CE0) | 69.50*(109/110) = 68.87 |
0.73968 | 2.519/109 |
| CTD-C (7CE0) | 69.39*(107/110) = 67.50 |
0.73391 | 2.476/107 |
| CTD-D (7CE0) | 68.93*(107/110) = 67.05 |
0.73302 | 2.490/107 |
*L1 represents the length of the AlphaFold2 reference (Wuhan WT) model superimposed onto all other cryo-EM structures.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



