
Submitted:
08 July 2024
Posted:
09 July 2024
You are already at the latest version
Abstract
Cancer cells are believed to be recognized and controlled by the immune system of the host, especially the adaptive immune system. Cancers may be initiated by a single gene defect in stem cells or regenerating cells during the replacement of old or injured tissue cells, and additional gene defect(s) then occur. To compensate for missing proteins intracellular homeostasis systems, such as proteostasis, may work with activation of new genes. The risk or predisposing factors in cancers may be related to the number of tissue cell renewals (the turnover time) and the probability of gene defect(s). Oncological symptoms may be associated with substances that are derived from antigens expressed on cancer cells and those derived from damaged cancer cells with corresponding immune reactions. In patients with cancers or other conditions, inadequate immune components to control these substances may result in chronic inflammatory or autoimmune diseases. Cancer cells communicate with host cells, including the immune cells such as T cells, for survival and proliferation and may need assistance from immune cells against intracellular infections or physicochemical insults. T cell-based immune therapeutics have been developed and clinically used in some cancers, especially in advanced cancers. We further discuss the oncogenesis, pathophysiology of cancers, and unresolved issues in tumor immunology and reinterpret the effectiveness of T cell-based therapies under the protein-homeostasis-system hypothesis.
Keywords:
cancer theories
; oncogenesis
; carcinogenesis
; cancer immunology
; T cell-based immunotherapy
; protein-homeostasis-system hypothesis
1. Introduction
Cancers or malignant tumors, including carcinoma, sarcoma, leukemia, and lymphoma, are representative of life-threatening diseases that remain difficult to treat. Cancer can originate from the tissues of any organ, and the clinical nature of individual patients, such as the speed of tumor growth and response to treatment, can differ in cancer originating from the same tissue of the organ. This finding suggests that there may be control system(s) affecting cancer growth, and the immune system of the patient may be associated with cancer growth at least in part [1].
The beginning of modern immunology is based on studies of infectious diseases following the discovery of germs at the end of the 19th century. Since then, the entity of immunology has broadened substantially through the remarkable development of technological methods and branches of biology [2,3]. The immune system of the host distinguishes self and non-self and controls invading pathogens, including bacteria, viruses, parasites, and xenografts that are non-self. Thus, it is suggested that cancer cells may be non-self, but cancer cells can escape the surveillance or regulation of the immune system of the host on occasion [4]. This hypothesis has become a basis of tumor immunology, in which immune cells, especially cytotoxic T cells, can control or kill tumor cells as observed in animal studies and in vitro [5]. Nonetheless, a high number of patients with advanced cancer do not survive despite the activity of the immune system, including cytotoxic T cells and natural killer (NK) cells in the innate immune system, which may act on cancers at the early stages. Moreover, solid cancer cells and metastatic cells require the help of the host’s normal cells such as the cells of blood vessels, interstitial tissue such as fibroblasts, and immune cells, including tissue macrophages, for growth and maintenance of cancers in localized lesions called the tumor microenvironment (TME) [6]. Protein and/or peptide antigens from cancer cells can elicit adaptive immune responses like those from invading pathogens. However, the biological characteristics of cancer cells are far more complex and are different from pathogens since cancer cells can communicate with other host cells. The paradigms in immunology, including distinguishing between self and non-self, have changed. Immune systems control not only large materials such as invading pathogens and foreign cells from other humans or species but also small materials, including pathogen-origin substances such as toxins and pathogen-associated molecular patterns (PAMPs), and substances derived from cells, such as damage (danger)-associated molecular patterns (DAMPs), pathogenic proteins, and peptides [7,8].
Despite the identification of causal pathogens, current immunological concepts have limitations in explaining the pathophysiology of infectious diseases, including influenza and coronavirus disease 2019 (COVID-19), and infection-related immune diseases, including multisystem inflammatory syndrome in children (MIS-C), and Kawasaki disease (KD) [9,10,11]. The same immune components, including T cells, are observed not only in the pathologic lesions of infectious and infection-related immune diseases but also in non-infectious conditions such as trauma and wound healing, organ transplantation, intoxication, allergic diseases, and cancers, suggesting that each component of the immune system may function identically in all conditions including cancers [12,13,14]. It is proposed that organisms can be regarded as a biosystem with an integrated system, known as the protein-homeostasis-system (PHS), for maintaining viability and a healthy state. Similarly, a cancer cell clone could be regarded as an independent biosystem that has biological characteristics different from normal host cells although it is originated from a self-host cell. In the PHS hypothesis, every disease involves an etiological or inflammation-inducing substance associated with disease-onset. The immune system of the host controls the substances to protect cells according to the size and biochemical properties of the substances. The PHS compensates in part for protein deficiencies in the host or within cells in cases of genetic diseases and cancers, respectively [13,14].
In this review, we discuss the unresolved issues in cancers, including etiology, pathophysiology, and immune therapy, through the PHS hypothesis. Given that the PHS hypothesis broadly integrates the pathophysiology of cancers, known pathophysiological or biological characteristics of specific cancers based on current immunological understandings are omitted or reinterpreted.
2. The Origin of Life, Evolution, and Biosystems
The origin of life remains one of the more challenging scientific questions. This tenacious question has been challenged by scientists across many branches of science, including physics, chemistry, and biology [15]. It has been established as the central dogma of molecular biology that life is dependent on the genetic code (DNA), RNA, and protein for its replication and metabolism and persistently evolves to adapt to a changing environment through natural selection [16]. However, every detail of molecular biology can be altered through biological evolution or human engineering (or synthetic biology). For example, the nucleobase 1-aminoadenine was discovered recently. Some phages use this nucleobase instead of adenine (A), one of the 4 basic nucleotides, which also include cytosine (C), guanine (G), and thymine (T) [17]. Also, novel living matter, such as synthetic viruses and bacteria, which do not exist in nature could be synthesized by controlling nonliving matter independently [18]. In the theory of abiogenesis, it is accepted that RNA, the genetic code, proteins, or other precursors as a form of animate matter might be responsible for the beginning of life. For example, non-covalent assemblies of molecules may propagate their compositional information through autocatalytic networks of molecules, achieving the establishment of replication as a characteristic of life. From a systems chemistry perspective, it was proposed that all stable (persistent) replicating systems would tend to evolve toward systems of greater stability. This stability is often enhanced by increasing complexity because any features added to the replicating system that improve replication efficiency will be reproduced [19]. It is plausible that a system that controls replication may in essence be life itself because life is associated with persistent replication and evolution through time. The real nature of the system of replication may be unexplainable like those of certain chemical or physical phenomena observed in nature such as the existence of light and gravity, although the phenomena are reproducible and expressed in mathematical principles. The more complex systems of life, i.e., the biosystems called organisms, have evolved together with the changing environment on the Earth. The embryogenesis of organisms is also a scientific enigma in the biosciences. In the case of humans, one fertilized cell (ovum) completes a newborn composed of trillions of cells in 10 months. Although researchers have revealed in part the genes or mechanisms that are involved in organogenesis in each period of embryogenesis [20,21], it is not yet possible to answer every question such as how or by what the sequential processes of embryogenesis are controlled, and whether the processes are performed by the blueprint encoded in the genome of an individual. How social systems are established, such as the existence of strict classes among species in socially acting animals, including ants, honeybees, and grouping animals, also remains poorly understood. During organogenesis, organ malformations can be induced by defective genes or teratogenic drugs, such as thalidomide, acting on protein metabolisms [22], suggesting that proteins may be major effectors in embryogenesis. However, if a living organism is assumed to be an existing biosystem, it is plausible that a biosystem is created when an ovum and a sperm are combined, and the biosystem uses its own genome to become a neonate and continues its life thereafter. An organism, including a human, is made of matter, but control systems in an organism (a biosystem) such as the systems that perform embryogenesis or maintain homeostasis may not be composed of matter. For instance, the products of literature, music, and fine arts, such as books, musical scores, and paintings are made of matter, but the artistic thinking, creativity, and insight for the works are incorporeal hereditaments of humans that are difficult to define as matter. Although these insights may remain philosophical issues, we have suggested the PHS hypothesis for further understanding of the pathophysiology of the diseases. It is believed that multicellular organisms have evolved from unicellular organisms. All organisms on the Earth, from unicellular organisms to human species, have an integrated system (named the PHS) for maintaining homeostasis and a healthy state. Thus, it could be proposed that each cancer cell is a new biosystem with control systems for viability and homeostasis as in a unicellular organism even though its life is dependent on the host like an invading pathogen
3. Stem Cells, Regenerating Cells, and Cancer Cells
The cells constituting the tissues of each organ are continuously replaced throughout life, and cancer can occur in nearly all human organs. Although the place and mechanisms of tissue replacement in a steady state remains unclear, it can be inferred that the origins of cancer cells are stem cells or regenerating cells, which have some stem cell properties and are involved in tissue replacement or repair. The complex dynamics underlying cancer stem cells have been unraveled through research. There are differences between cancer stem cells and normal stem cells in the mode of division, cell-cycle properties, replicative potential, processing of DNA damage, and the activation/inactivation of cancer-specific molecular pathways [23]. Peripheral stem cells, except a fertilized ovum, are heterogeneous and reside in the bone marrow, adipose/soft tissue, intestines, muscles, and other places and participate in the repair of damaged organ tissues and the possible replacement of older tissues. A reduction in the activity or number of stem cells may be an important feature of the cell senescence associated with the aging of organisms [24]. Most childhood leukemias are abnormal progenitor cells derived from a stem cell in bone marrow, and some solid cancers can express stem cell-origin markers [25], suggesting that cancers may be derived from stem cells. Artificially “induced stem cells” capable of self-renewal can occur through the manipulation of a few transcription factors of genes that are activated during early embryogenesis [26], but it is difficult to achieve in normal cells that are in a steady state. The stem cells that appear during the organogenesis stage of embryogenesis or the repair stage of damaged tissues are proliferated extensively and complete the organ through a precise systemic control system (referred to as the PHS in this review). There may be communication networks across cells that are composed of an organ for the completion of organogenesis or organ tissue repair such as liver cell regeneration in donors for liver transplantation. Advanced solid cancers can form a huge mass (cancer organ) like the structure of a single organ, and its proliferation is assisted by normal support cells including vascular, stromal, and immune cells that are found in the TME. Although cancer and stem cell clones may have a similar mode of gene activation for the initial extensive proliferation in organogenesis or cancer organs, communication networks between cells for stopping proliferation may not occur in cancers because the cells may have different tools for communication across cells. In other words, this process is not controlled by the systemic PHS of the host.
All tissue cells in humans have the same genome capable of producing all proteins, as shown in animal reproduction studies using an ovum and somatic cell genome. However, each tissue cell, including a cancer cell, produces and uses limited proteins for maintaining homeostasis by an unknown control system referred to as cell fate [27]. Thus, the expressed and activated genes of cancers are different according to organ tissues. In addition, recently developed single-cell sequencing technologies at the molecular level, including genomic, transcriptomic, epigenomic, proteomic, and metabolomic sequencing have revealed individual variations among cancer cells originating from the same tissue [28]. Furthermore, genetic information in initial cancer cells in leukemias and solid cancers can change continuously during the process of cell proliferation or through insults from anticancer treatment [29,30,31]. Therefore, the state of gene activation such as expression of receptors and production of tumor-specific proteins/peptides could differ in each patient although histopathological findings are similar in cancers from the same organ tissue.
4. Proteostasis, Proteasomes, and Peptides
Proteostasis (or protein homeostasis) is one of the basic concepts in cell biology for survival and the maintenance of homeostasis in the cell. The production, folding, and degradation processes of proteins within the cell are regulated at an appropriate level for the viability of the cell and the entire organism. The processes are controlled by the ubiquitin-proteasome system and the autophagy-lysosome pathways [32,33]. There has been increasing evidence that the disturbance of proteostasis is related to the diseases including neurodegenerative diseases and cancers [34,35]. The proteasomes are in the nucleus and cytoplasm of cells in multicellular animals as well as unicellular organisms, and the type and distribution of proteasomes in cells differ according to the type of tissue [35]. Although the main function of the proteasomes is to control modified or misfolded proteins, various-sized peptides are produced during active proteasomal processes, and some of them have shown biological activity for cells including central nervous system (CNS) cells [36]. The peptidome and proteome between a cell of unicellular organisms such as yeast and cells of human or mice have similar components [37], and thousands of naturally occurring peptides differing in their origin, abundance, and possible functions have been identified in the tissue and biological fluids of vertebrates, plants, insects, fungi, and bacteria [38]. These findings suggest that unknown bioactive peptides are hidden in the peptidomes of different organisms, and peptides may be essential effectors for the viability and homeostasis of the cells [37,38]. Since cells use a limited number of proteins owing to the limited space, peptides have been proposed to be the main effectors acting on biological activities within the cells [12,13,14]. Additionally, not only peptides but also smaller materials, including elements such as oxygen and nitroxide, monoamines such as serotonin and dopamine, neuropeptides, peptide hormones, and biochemicals such as vitamins and fatty acids, play important roles in organisms and cells, but these substances may not be encoded in the genome. Specific cells in organs have their own receptors for these diverse substances, and the substances have variable size and biochemical properties. In general, a specific substance produced and released by specific cells in an organ is used in other cells for systemic homeostasis. Some peptides such as peptide hormones and neuropeptides are released from cells of the endocrine system or CNS for systemic homeostasis [39]. Thus, cells in organisms, including immune cells, may communicate with each other through the substance-receptor-signal transduction pathways [12]. The mechanisms of proteasome inhibitors remain to be further evaluated, but proteasome inhibitors significantly lower the concentration of peptides within cells [40]. Therefore, the effect of proteasome inhibitors could be interpreted as blocking the production of essential peptides for cell survival in certain cells, including multiple myeloma cells. Peptides can be leaked from cells when cells, including cancer cells, are injured by insults from infection, trauma, or other conditions such as cytokine storms. The pathogenic peptides released into the systemic circulation can bind to target cells of organs that have affinitive receptors, and this process initiates T cell-inducing inflammation in the PHS hypothesis, which is discussed later in the review.
5. Immune Responses to Cancers and the PHS Hypothesis
Cancer cells may start from a single-cell clone, proliferate, leave offspring (metastases), and become resistant to anticancer drugs and other treatment, similar to invasive extracellular pathogens. Thus, it is reasonable to propose that there are similar immune mechanisms against cancer cells and external pathogens as non-self. Some concepts of T cell-centered immunology are based on the hypothesis that immunological phenomena observed in vitro could be reproduced in vivo. New concepts in recent tumor immunology, including immunosurveillance, immunoediting, and immune contexture, suggest that immune components in the TME may be associated with tumor growth and dissemination. The initial anti-tumorigenic activity of immune cells tends to change to pro-tumorigenic activity along with tumor growth [41,42,43]. The phenomena could be explained in part through the PHS hypothesis.
As in tumor immunology, T cells are activated through T cell receptor (TCR) bound antigens (peptides) presented through major histocompatibility complexes (MHCs) of antigen-presenting cells (APCs). The activation/suppression of coreceptors, including immune checkpoints, between both cells affect T cell activation. Activated T cells may control the immune disturbances and maintain homeostasis of the immune status against various conditions through the arrangement of T cell subsets, including cytotoxic T cells (CD8+) and helper T cells (CD4+), which include Th1, Th2, and Treg, and cytotoxic CD4+ [44]. Additionally, the pathophysiology of cell injury in infectious diseases can cause pathogen-induced cytopathy and the dysregulation of immune responses [45].
Cancer cells express or release tumor-specific antigens (TSAs). The adaptive immune system of the host can produce specific B cell clones (antibodies) and T cell clones, including cytotoxic T cell clones, against the antigens [5]. Antibody-dependent cellular cytotoxicity (ADCC) by immune cells to cancer cells is observed in vitro, and antibody therapeutics against cancer have been developed, including the anti-CD25 antibody and anti-PD1/PD-1L/CTLA4 antibodies [46]. T cell-deficient mice (nude mice) permit the limited growth of MHC-nonrestricted cancer cells such as human cancer cells, suggesting that T cells can control cancer cells as foreign cells, and this animal model has been used for tumor biology and xenograft research [47]. Cytotoxic T cells and NK cells have shown an ability to kill or suppress various tumor cell lines in vitro and are used clinically in some cancers [48,49]. The mechanisms of cytotoxicity by immune cells involve the release of cytotoxic materials, including perforin and granzymes, and induce apoptosis of target cells via the Fas-Fas ligand pathway [4,48,49]. However, the mechanisms could damage neighboring self-cells, and injured cells could release inflammation-inducing substances if not strictly controlled. Some cytokines and immune mediators, including interleukin (IL)-2 and interferons, can enhance the functions of immune cells, such as T cells and macrophages, in vitro, and these cytokines are used in some cancers to strengthen immunity [50]. While certain soluble immune mediators such as tumor necrosis factor (TNF) can affect tumor cells in vitro, the systemic infusion of TNF can induce severe toxicities including fever, rigors, and pulmonary edema [51]. These known backgrounds could be an obstacle to accepting new concepts, and new immunological observations interpreted by the established dogma could result in contradictions.
The paradigm by which the immune system distinguishes self and non-self has changed. It is now accepted that immune systems react to substances derived from self-cells such as DAMPs as well as those derived from pathogens such as PAMPs [52]. In infectious diseases, the immune systems of mammals control not only pathogens but also substances derived from pathogen-infected cells, including toxins or fragments of pathogens, PAMPs, DAMPs, pathogenic proteins, pathogenic peptides, and other smaller substances not yet identified [12,13,14]. T cells in vivo are composed of heterogeneous T cell subsets, and T cell subsets could be further extended according to the disease being studied [44]. More importantly, activation of T cells (and B cells) can be induced by various mitogens, superantigens, antibodies to T cells, peptides alone, or drugs used in clinical practice [53,54].
There are few intact pathogens in extensively injured organ tissues in virus and mycoplasma infections, including COVID-19 [55,56], suggesting that pathogens such as bacteria and viruses in the blood may be removed effectively by circulating immune cells such as phagocytic cells if they cannot avoid into intracellular space such cases of intracellular pathogen infections. Whereas there has been no evidence of tumor cells engulfed by phagocytes or phagocyte-related immune reactions. Although most cancer cells may express the same MHCs and other receptors for immune reactions and communications with the host cells, some cancer cell lines used in vitro may express different or few self MHCs of the host as non-self. Cytotoxicity can be more readily observed in vitro during direct cell-to-cell interaction between isolated tumor cell lines and T cells or NK cells in vitro, as shown in cell-monolayer techniques. This contrasts with situations in vivo such as in the combined cell mass of solid cancer cells in TME, which are influenced by precise and elaborate systemic control. Heterogenous cytotoxic T cells and NK cells work not only against cancers but also against infectious diseases and other non-infectious conditions under precise systemic control [5,48,49]. Moreover, cancers may act as systemic diseases in which all systems of the host work against insults from the cancer. Not only immune systems but also various intracellular or systemic biological and metabolic systems, including those acting on inflammasomes, apoptosis, autophagy, proteasomes, nutrient and glycolysis pathways, and the epigenetic change, including micro-RNAs, are activated in cancers, similar to the systemic inflammation of infectious diseases or immune-mediated diseases [57,58,59]. It is a reasonable assumption that there are causes (etiological substances) eliciting these diverse biological changes and corresponding control systems against the substances. The biological changes may be useful for detecting diagnostic or prognostic biomarkers and developing targeted therapies for some disorders. On the other hand, they may reflect a specific point in space and time of the disease and need serial examinations during the disease processes, which is an evident limitation of studies. Thus, it is difficult to determine whether biological alterations observed under various conditions, including infections or cancers, could contribute to the development or progression of the disease or they could be secondary or adaptive phenomena, indicating immune reactions to insults caused by the disease [60].
5.1. PHS Hypothesis
We suggest that the PHS hypothesis can address the unresolved pathophysiological issues of the diseases. These diseases include infectious diseases, immune-mediated diseases such as KD and MIS-C, and organ diseases such as kidney and CNS including genetic diseases and cancers [9,10,11,12,13,14]. In brief, there are etiological substances of disease-onset in every disease, and the immune system is one factor in the PHS. For the protection of self-cells, each immune component of the host has the same role in controlling toxic substances according to the size and biochemical characteristics of the substances. The PHS can cope, to some extent, with a protein deficiency in living organisms and within cells. The T cells and B cells of the adaptive immune system control small protein substances. The main function of T cells is to control pathogenic peptides (the size that binds to TCRs) by the production of cytokines or possibly immune peptides. B cells control pathogenic proteins that bind to the B cell receptor (BCR) by antibody production. The innate immune system controls large complex substances such as bacteria, virions, apoptotic and necrotic bodies, transformed cells, and small nonprotein substances. Small nonprotein substances, including elements, monoamines, neuropeptides, polysaccharides, viral RNAs and DNAs, and chemicals (drugs) and biochemicals, are controlled by Toll-like receptor-associated pathways, natural antibodies, the complement system, and immune protein systems that are yet unidentified but could be responsible for prion diseases, Alzheimer’s disease, and amyloidosis [14].
In the PHS hypothesis, the initiation of infectious diseases and infection-associated immune diseases begins with corresponding immune responses against etiological substances derived from infected or damaged self-cells (the focus). The defect of immune components of the host controlling these etiological substances may be associated with chronic inflammatory or autoimmune diseases due to persistent non-specific immune responses. The control of pathogenic peptides may be more crucial than pathogenic proteins for the viability of cells since mammals with T cell deficiency eventually all die through insults from infections or other conditions [12]. The PHS hypothesis insists that the functions of immune cells and immune proteins observed in pathological lesions of various diseases are the same, including cancers {Table 1}.
5.2. Oncogenesis in the PHS Hypothesis
Oncogenesis (or carcinogenesis) is not completely understood. Thus, theories or hypotheses are needed to clarify the subject. The somatic cell theories in that a cancer arises from a single defected clone have been accepted, but there are somewhat vague and contradictory phenomena in caners, which cannot be explained by these theories [61]. The tissue organization field theory was proposed in that cancers arise from the default states of proliferation, variation, and organization of cells in the host based on concept of the organicism [62].
In general, it is believed that the transformation of normal cells into cancer cells may be caused by the serial defects of genes over time. However, some cancers, such as leukemia after exposure to excess irradiation, infantile leukemia, or congenital cancers, might be associated with acute multiple gene defects and can occur within a relatively short time [63,64]. Certain cancer cell clones might originate from stem cells or stem cell-like regenerating cells that replace old cells or injured cells. Thus, activated genes and transcription factors in these cells may be similar to those used during the development stage of organogenesis. Given that each cell has an intracellular PHS, i.e., mechanisms for maintaining cell homeostasis such as proteostasis, it is proposed that stem cells or regenerating cells cope with protein deficiency or malfunctional proteins that are essential for viability. If an essential protein is missing, the cells would produce appropriate alternative proteins or necessary peptides as the main biological effectors in cells might be peptides. Cells in some precancerous lesions may be in this stage of oncogenesis, and these cells will also be replaced by new ones. Over time, gene-defective stem cells or replaced cells could be confronted with challenges when alternative proteins, i.e., non-specific proteins or peptides, are toxic for cell homeostasis or additional gene defects occur. Toxic proteins and/or peptides could be controlled within the cell through possibly controlling (immune) proteins or peptides. Additionally, the controlling substances or newly occurring gene defects could be controlled by a proteostasis system with the activation of new genes. During the repeated processes, the gene-defective cells are capable of self-proliferation and can activate new genes against a major gene defect and additional gene defects for homeostasis or proteostasis, and eventually, a cancer cell is complete.
An established cancer cell is composed of complex material with self-identifying markers including MHCs. Thus, the adaptive and innate immune system of the host has limited functions against whole cancer cells. In the PHS hypothesis, the adaptive immune system of the host can only control proteins and peptides expressed on or derived from cancer cells in the same way as they work against virus-infected cells. It is one cue for resolving enigmas in oncology that a major gene or additional gene defects causing oncogenesis even in the same tissue of origin could differ. Thus, major factors of cancer progression are determined by cancer cells themselves that have different intracellular situation of cell homeostasis rather than the host’s immune systems. Subsequent genetic information and clinical course, such as metastasis, drug resistance and the doubling time of the tumor, could be different in each cancer patient regardless of the tissue of origin [Figure 1].
6. MHCs, Cytokine Networks, and Communication of Immune Cells
Each cell of the immune system may communicate with each other through MHCs and cytokine networks to protect cells. MHCs consist of heterogeneous receptor groups and are divided into 2 classes: MHC class I (MHC-I) includes histocompatibility leukocyte antigen (HLA)-A, HLA-B, and HLA-C, and MHC class II includes HLA-DR, HLA-DQ, and HLA-DP [65]. MHCs could be extended into class III by gene clusters of immune proteins such as complement components (Bf, C2, and C4), and into class IV by gene clusters of the TNF family, AIF1, and HSP70 [66]. MHC-I molecules are expressed in almost all cells and are indispensable in embryonic development, tissue repair, and immune reaction in individuals [67]. MHC-I is a major factor in transplantation immunity. Organs with mismatched MHCs-I show a low transplant success and can be attacked by immune cells, mainly CD8+ T cells [68]. On the other hand, immune cells of animals in the same species, including mice and humans, with different MHC-1 do not induce MHC-associated immune responses because of an established the MHC restriction [69]. Recently, it has been found that NK cells recognize and respond to classical and nonclassical MHC-I molecules as well as structural homologs. Innate immune cells in mice have been shown to express MHC-II, which is expressed mainly in immune cells [70]. Additionally, it was reported that decreased or absent MHC- I expression could be associated with an invasive and metastatic tumor phenotype [71]. The findings suggest that MHC-1 may be a self-identifying marker for systemic homeostasis and communication across self-cells. MHC-I may be the receptor of self-peptides; essential peptides for systemic homeostasis that are already expressed in nature with MHC-I in various thymic cells during T cell development. Thus, T cell clones that recognize essential peptides for systemic homeosis are not needed and are removed through “negative selection”. Only T cell clones against possible pathogenic or signaling peptides survive since their main function is a systemic control of peptides in the PHS hypothesis. It is proposed that pneumonia and ARDS in infectious diseases, including influenza, mycoplasma infection, and COVID-19, are caused by pathogenic peptides derived from pathogen-infected cells. The peptides bind to cell receptors, including unbound MHCs or other receptors, of target cells in the lungs. The resulting inflammation occurs as a response of T cells against pathogenic or signaling peptides, possibly through a non-MHC-restricted manner [55,56]. It is believed that a cytokine imbalance such as a cytokine storm is associated with target cell injury in acute infections and other conditions [72]. An excess load of etiological substances (the pathogenic peptides) induces cytokine imbalance such as a cytokine storm because non-specific adaptive immune cells initially act with an excess of cytokines. In transplantation immunology, the PHS hypothesis suggests that non-self MHCs and other foreign receptors expressed on the cells of animals or human tissues could be recognized by B cells as pathological proteins. Pathogenic peptides bound to MHCs or other receptors are recognized by T cells, resulting in immediate graft rejection under the cooperation of other immune components of the host [12]. Conversely, it is plausible that in graft-versus-host disease after bone marrow transplantation, the transplanted immune cells, including T cells and B cells, identify antigens expressed by the target cells of the host as a form of pathogenic peptides or proteins, respectively, and attack them with other immune components [73]. Researchers have reported that HLA loci are associated with the incidence of autoimmune diseases, such as in HLA type B27 in ankylosing spondylitis [74]. MHC-I molecules are receptors for peptides, including essential or pathogenic peptides, and the phenotype of HLA is individualized by polymorphism and decides the shape of binding peptides. Therefore, it is partly conceivable that certain HLA types are associated with the prevalence of certain infectious and immune-mediated diseases [12].
It is a basic immunologic concept that a specific B cell clone (antibody) is produced against a pathogenic protein. Likewise, a pathogenic peptide induces a specific T cell clone against the peptide. This process is dependent on the recombination of BCR and TCR genes. Accordingly, countless members of antigens, but only protein antigens, can be covered by adaptive immune systems. In pathogen infections, T cells and B cells cannot directly control virions or bacteria. T cells or B cells are only associated with protein fragments of pathogens that are processed by APCs and then presented to them. The pathogen-specific B cell (antibodies) and T cell clones appear at least 3-4 days or longer after symptom onset of the disease such as fever and pneumonia in respiratory virus and mycoplasma infections, probably due to a time lag in the recombination of TCR and BCR genes for the production of specific effectors [55,56,60]. The roles of B and T cells in viral infections could be simplified as follows. Virus-specific antibodies induce virus neutralization, prevent viral entry into cells, and help in phagocytosis via Fc receptors. Additionally, the antibodies are involved in a cytopathic effect via ADCC. Cytotoxic T cells (CD8+) recognize virus-origin peptides bound by MHC-I on infected cells and may affect target cells (virus-infected) through the release of intracellular toxic materials and/or apoptosis-inducing mechanisms. Helper T cells (CD4+ cells), including Th1, Th2, and regulatory T cells, recognize antigens bound with MHC-II and are involved in the control of antibody production and immune homeostasis against disease insults [60].
It is noticeable that markedly increased immunoglobulins (IgG, IgM, and IgA) and possibly T cell clones are observed at the early convalescent stage of infectious diseases, including scarlet fever and tuberculosis, and infection-related immune-mediated disease, including acute rheumatic fever and KD; 2-3 folds of serum IgG level are observed in severely affected patients, while the proportion of pathogen-specific antibodies (and T cell clones) is extremely low. These findings suggest that the extensive activation of B cell and T cell clones may be involved in recovery reactions in these diseases through the control of pathogenic proteins and peptides, with other immune components, including platelets [75,76]. Platelets also interact with non-malignant cells in the TME, such as immune cells, fibroblasts, and endothelial cells, and regulate their phenotype [77].
Furthermore, T cells and B cells not only react to protein antigens presented by APCs but also to those that are presented by or bound to other cells. They can also be activated by direct routes such as mitogens, superantigens, drugs, and peptides alone regardless of MHCs as discussed previously. Thus, it is anticipated that the immune responses performed by activated T cell subsets through MHCs and other routes could differ, and that only MHC-restricted immune responses need precise communication between immune cells through secondary signals such as PD-1, CTLA4, and other receptors. Although T cell subsets have been classified by cell markers and cytokine production, the functions of T cells, including cytotoxic T cells, could be elicited or initiated by peptides in MHC-restricted immune reactions. Conversely, it is plausible that T cell subsets might control pathogenic peptides and induce peptide-associated inflammation, manifesting as diverse MHC-restricted immune reactions including cytotoxicity. Numerous peptides and proteins exist in all cells, but their compositions differ according to the tissue cells. When substances within cells are released into the systemic circulation and bind to affinitive receptors of target cells, adaptive immune cells can be activated in a non-MHC-restricted manner. Likewise, substances derived from injured cancer cells, including TSAs, can induce an adaptive immune response with the same mechanism as infectious diseases. On the other hand, cancer cells need to communicate with host cells for survival and growth. Cancer cells, especially those in advanced or larger masses, can encounter situations that require the help of immune cells, including virus invasion, and physical or chemical insults from trauma, heat, irradiation, and anticancer drugs. Thus, cancer cells communicate with the immune cells of the host, including T cells, through MHCs and cytokine networks. Accordingly, cancer cells that are helped by T cells may be affected when this help discontinues. The mechanisms by which cancer cells proliferate, avoid apoptosis, or resist chemotherapy are influenced by immune cells, including tumor-associated macrophages (TAMs), myeloid-derived suppress cells, T cell and B cell subsets, and other immune cells in the TMEs [78].
7. Associated Factors in Cancers
7.1. Aging
Cancer affects predominantly older individuals and shows a proportional increase in incidence along with increasing age: aging is the most well-known risk factor for cancers. The incidence of each organ in cancer differs, and the cells in each organ have a different replacement cycle. Somatic cells may have limited turnover times in cell division and undergo many replication cycles with telomere shortening. Cancer cells often have paradoxically shorter telomeres compared with those found in normal tissues [79]. It is possible that “old” cells experienced many replacements might have a greater susceptibility to gene mutation through weakened or impaired DNA repair systems and subsequently become cancer cells more easily [80]. Additionally, the ability or function of the immune system, including phagocytic cells such as macrophages and neutrophils, decreases with age in the senescence of the immune system [81]. Older individuals have a long recovery time and show higher mortality when affected with mild infectious diseases, such as influenza and COVID-19, perhaps owing to immune senescence and underlying diseases which are also controlled by immune cells [55,82]. Furthermore, aging is a higher risk factor for neurodegenerative diseases, including Alzheimer’s disease and Parkinson disease, suggesting that these diseases may also be immune-mediated diseases affected by immunosenescence [14]. In general, the growth of cancers in older adults tends to be slower than in younger adults who have more active immune functions against infectious and other insults, suggesting that the host’s control systems against cancer cells may be different from those against pathogen infections.
7.2. Genetic and Epigenetic Factors
Many genetic diseases have a higher risk of specific organ cancers, and there are familial cancers with affected genes, including breast, ovarian, and colorectal cancers [83,84]. The clinical course such as timing of diagnosis, response to treatment, and prognosis can differ somewhat in each patient in the familial cancers. An activation or change in specific genes, including oncogenes and tumor suppressor genes, and somatic mutations, including point mutations, insertions, deletions, amplifications, copy number changes, and translocation of heterogeneous chromosomes, have been observed in cancer cells [85]. Among the genetic changes in cancers, certain genes are commonly activated regardless of the originating tissues, whereas genetic changes in cancers of the same organ tissue can be different. These findings suggest that the establishment of cancer cells may need serial gene defects after a main gene defect and an intracellular homeostasis or proteostasis system against insults from gene defects for cell survival may act as previously proposed. Therefore, the alternation of activated genes and translocation of chromosomes may occur for cell survival and be dependent on the intracellular situations of each cancer cell as an independent biosystem.
The activation of genes (or the production of proteins) is controlled by epigenetic mechanisms without changes in the genetic code. Because the spatial area of a cell is limited, it is proposed that smaller substances, including peptides and epigenetic components such as microRNAs, may have major roles in the homeostasis of the cell. Epigenetic changes have been reported in various cancers [86]. Epigenetic studies have focused on DNA methylation, histone modification, nucleosome remodeling, and RNA-mediated targeting including microRNAs and long non-coding RNAs. It has been accepted that epigenetic changes could be related to the characteristics and prognosis of cancers, and epigenetic components may become a target of cancer treatment [87]. The roles of epigenetic effectors may control the production of proteins, peptides, and other smaller materials needed for systemic and intracellular homeostasis against various insults. It remains unclear whether the change of epigenetic components in cancers acts as causal factors of disease progression or secondary phenomena for disease regression, and epigenetic changes are also observed in other conditions including infection, neurologic diseases, and immune-mediated diseases [88,89].
7.3. Microbiota and Infection
The microbiota refers to an ecologic system of microorganisms living within eukaryotic organisms, including viruses, bacteria, archaea, fungi, and protozoa. These microorganisms reside in the skin, intestines, urogenital tract, upper and lower respiratory tract, and possibly other regions in humans [90]. A homeostatic and interdependent relationship between the microbiota and the host is responsible for the well-being of the host species. There is a strict species-specific relationship between the pathogen strains in the microbiota and the host species. For example, animal species have had their own influenza viruses, coronaviruses, and bacteria strains, including mycoplasma strains, throughout evolution [10,55,91]. Thus, species-specific pathogens, including viruses, could be regarded as the microbiota of a species, but on occasion, they can invade the host. Although viruses in the human microbiota have received less attention, studies on the virome may be important since viruses are major oncogenic pathogens [92].
Dysbiosis is an imbalance in the composition of strains for the interdependent relationship between the host and the microbiota, especially in intestinal bacterial strains. The association between dysbiosis and disease has been reported in various medical fields including oncology [93,94]. It is inferred that the absorption of biological metabolites or toxic substances originating from dysbiotic strains, which could affect genes, is related to the development or progression of disease, including cancer. Additionally, facilitating the invasion of the strains during dysbiosis or affecting the balance between the microflora and the host immune system may be associated with disease [95,96]. A dysbiotic state found in the viruses of the microbiota may also be associated with the immunological, biological, and metabolic homeostasis of the host since most viruses living in bacterial and fungal strains in the microbiota exist in the form of bacteriophage [97].
Chronic pathogen infections are associated with cancers, including hepatitis B virus and hepatitis C virus in hepatocellular carcinoma, human papillomaviruses in cervix cancer, Epstein-Barr virus in head and neck cancer, human immunodeficiency virus on Kaposi’s sarcoma, and H. pylori in gastric cancers [98,99,100,101]. The viruses and possibly intracellular pathogens can live in latently infected cells after initial infection, and repeated reactivation of the pathogens induces target cell injury in the same or other organs. The injury of organ cells may be caused by the cytopathic effect of viruses or more likely by toxic substances derived from injured infected cells with corresponding immune reactions [60]. The host’s repair system should replace injured organ cells. Thus, somatic cells affected by repeated or persistent insults from chronic infections may experience more renewal with shortened turnover time. Stem cells responding to more regenerated processes become vulnerable to gene defects and a higher risk of transforming into cancer cells over time. Patients with immunodeficiencies show a higher risk of cancers due to recurrent or chronic infection, vulnerable cell injury, and shortened turnover time of cell replacement [102,103].
The composition of the microbiota changes continuously after birth, together with immune maturation in individual hosts [104,105]. The strains of the microbiota are also affected by diet and socioeconomic status such as hygiene, antibiotics, and immunotherapy [106]. Thus, the composition of the microbiota can differ between ethnic groups or populations and can be changed by constant deviations in environmental factors. Moreover, the incidence of cancers appears to be different by age, sex, and ethnic groups, suggesting that some cancers might be associated with the microbiota such as dysbiosis or chronic pathogen infections including unidentified pathogens (with bacteriophages) in the microbiota.
7.4. Environmental Factors
The epidemiology of cancers can change along with the changing environment. This also occurs in infectious and infection-related immune-mediated diseases such as KD and inflammatory bowel disease (IBD). The prevalence or incidence rates of some cancers, infectious diseases, and infection-related immune-mediated diseases, including KD and IBD, can differ widely among populations. For example, KD is prevalent in East Asian countries, and IBD and juvenile idiopathic arthritis (JIA) are more prevalent in Western countries [9,107]. Similarly, gastric cancers are more prevalent in East Asian countries whereas colorectal carcinoma is more prevalent in Western countries [108]. In Korea, IBD in children and colorectal cancer in adults were rare diseases a generation ago, but both diseases have increased over time, probably owing to a more westernized style of diet [109]. The epidemiology of cancers may be changing such that colorectal cancer in Western countries is changing as there is an increased occurrence in young adults [110].
Susceptibility to diseases (or different incidence of the diseases), including immune-mediated diseases and cancers, has been explained by genetic factors such as race and ethnicity and environmental factors. However, clinical manifestations of the disease and immune functions against insults from infections or cancers may be similar in individuals across populations. It is proposed that a major environmental factor of susceptibility to infection-related immune-mediated disease, including acute self-limited KD and MIS-C and chronic JIA and IBD, is the different composition of strains in the microbiota between the populations [9,111]. The pathogenic strains in the microbiota colonize first and then invade the host and/or host cells, and etiological or inflammation-inducing substances of the disease are released from the focuses, such as infected cells or cells injured from infectious insults. Thus, the colonization state of etiological agents in the microbiota in populations may be responsible for age, sex, and ethnic predictions in these diseases and possibly cancers. Patients or animals living in more prevalent areas may have a greater chance of being exposed to pathogens originating from the microbiota, whereas genetic differences affecting immune functions in individual hosts decide the disease outcomes [9,111].
Cancers can be affected by other environmental situations where genes can easily be damaged since a cancer cell may be established by serial gene defects after a main gene defect. Acute or chronic exposure to irradiation such as ultraviolet rays and X-rays, chemicals, pollution, and drugs can affect chromosomes and induce gene defects. Cells chronically affected by these conditions may also be associated with rapid turnover of cell replacement with a higher risk of cancer transformation. For example, myeloproliferative neoplasms, including chronic myeloid leukemia, polycythemia vera, essential thrombocythemia, and primary myelofibrosis can induce acute blast transformations during repeated proliferation caused by certain persistent chronic stimulation and/or additional gene defects [112,113].
The phenotype of the clinical severity of infectious diseases, including scarlet fever, acquired immunodeficiency syndrome, pandemic influenza, and immune diseases such as KD, post-streptococcal glomerulonephritis, and Henoch-Schönlein purpura (or IgA vasculitis) has been reported to become milder over time [114,115]. In the view of evolution, there may be a trend to head toward harmonious relationships between species and their microbiota. The incidence of cancer is also influenced by environmental factors such as dysbiosis or infection of the microbiota, and cancer cells could be regarded as a parasitic biosystem living with host like pathogens. Thus, it is possible that some characteristics of cancers in the past and present, including clinical severity and prognosis, would become milder phenotypes since the host and parasitic cells might adapt to each other over time.
7.5. Host and Tumor Factors
Although all patients with advanced cancer do not survive, the clinical course of patients with cancer even in familial cancers differs, suggesting that some host or tumor factors affect the natural course of the disease. Cells of the immune system may play a role in the protection of somatic cells against various insults. Recent studies of tumor immunology have focused on the localized areas of cancers such as the TMEs. However, cancers may be a systemic disease in which all systems of the host work against insults from cancers. Metaphorically, the immune components in the TME are the recruited soldiers of various armies from a whole country at a battlefield for the protection of the nation (the host cells), but they assist the enemy with the markers of troops (cancer cells with the same MHCs and other self-biomarkers) because of the determined or fated role of each army.
The growth of localized or metastatic cancers may be controlled by the systemic immune status at least in part. The immune status of the host is affected by age, composition of microbiota, nutrition, and underlying diseases. Cancer cells themselves do not directly act as toxins to host cells, just as bacteria or viruses themselves are not direct causes of cell damage in infectious diseases [11,12,13,14]. In the PHS hypothesis, the phenotype of an infectious disease or infection-related immune-mediated diseases is decided by substances released from injured or infected cells (the focus), their target cells in organs, and corresponding immune reactions. Accordingly, different pathogens invading cells of the same tissue or cells of different tissue infected by the same pathogen can release different materials as etiological or inflammation-inducing substances. Many patients with advanced cancer show various clinical symptoms, including fever, autoimmune-like manifestations, weight loss, and cachexia, and succumb to infection and/or organ failure. These symptoms may be caused by immune responses to substances derived from cancer cells. The limitation of the immune system against a slowly increasing load of substances could be responsible for death in cancer due to the total exhaustion of immune components as well acute or chronic uncontrollable infectious diseases. Additionally, because the type and load of toxic substances produced by cancer cells could differ in each cancer patient, the clinical course and immune status against the substances could differ in each patient.
Cancer cells themselves could activate protective mechanisms against external insults, including invading viruses, physical or chemical trauma, and radiation as well as normal cells. Cancer cells can produce mediators such as proinflammatory cytokines and chemo-attractants that promote immune cell infiltration to assist immune cells [116], and they could activate new genes to adapt against these insults. Cancer cells that have undergone activation of many genes show immature histological findings such as anaplasia or chromosome translocation and produce new proteins that are different from normal cells. It is expected that some of these may be toxic to host cells if released or could not be controlled by adaptive immune cells, which could explain the poor prognosis of anaplastic cancers.
The immune cells have receptors to communicate with other immune cells and have a relatively short turnover time compared to somatic cells. In general, immune suppressants, including corticosteroids, intravenous immunoglobulin, and biologics, are effective for the control of adaptive immune cells, especially in rheumatologic and organ transplantation fields. The malignancy of immune cells or hematologic malignancies, including leukemia and lymphoma, have somewhat different clinical, laboratory, and pathological characteristics to solid cancers. Since hematologic cells exist as isolated cells, the receptors and other biomarkers expressed on cancer cells are easier to identify, and some of them are used for the classification of cancers and help to predict prognosis, especially in leukemias [117]. Hematologic malignant cells can also express receptors for immune suppressants such as corticosteroids and can be more vulnerable to anticancer drugs than somatic cells. Moreover, the rapid turnover of malignant cells in the blood and/or in the TME may have less situations to be helped by immune cells and other cells against insults compared to solid cancers. Thus, the rate of complete remission (or cure) would be higher especially in children compared to solid cancers. Although the nature or functional roles of immune cells and somatic cells differ in an organism, hematologic malignancies are a milestone in immune-based therapeutics targeting the receptors expressed on cancer cells.
8. Unresolved Issues in the Immunotherapeutic of Cancers
8.1. Histopathologic Findings and Tumor-Infiltrating Lymphocytes (TILs)
Histopathologic findings in cancers are crucial for confirmative diagnosis, guidance for treatment modalities, and prognosis. Since cancer cells that originate from specific organs express their own TSAs and activated antigens that are expressed rarely in normal cells, detecting antigens can involve various scientific methods, including immunohistochemical stains, next-generation sequencing, nanotechnology, or methods to identify circulating tumor DNA/RNA or exosomes. In addition, imaging techniques such as magnetic resonance imaging and positron emission tomography, have been developed and used for the diagnosis and prognosis of cancers [118,119].
Besides characteristic cancer cells and stromal cells in the TMEs, various immune cells, including neutrophils, eosinophils, subsets of T cell and B cell, NK cells, mast cells, and dendritic cells, and immune proteins, including immunoglobulins and complements, and undetermined immune materials, including amyloid proteins and peptides can also be observed. It is believed that T cells observed around solid cancer lesions and TILs can affect cancer cells. TILs consist of various T cell subsets, including cytotoxic T cells, but the degree of the infiltration of TILs and other immune cells differs in individual patients with cancer in the same tissue of the organ. Furthermore, the administration of enriched TILs to patients has shown limited effectiveness in solid cancers [120]. Other innate immune cells have also been tried for cellular immunotherapies, including dendritic cells, NK cells, lymphokine-activated killer cells, and cytokine-induced killer cells [121,122]. Numerous studies have examined the relationships between immune cells and cancer cells in TMEs, including the pro- or anti-tumorigenic effects of immune cells and immune proteins [1,2,3]. On the other hand, it is an acceptable notion that the histopathological findings of involved organs at any stage of the disease, including TMEs in cancers, can provide information about the state of inflammation. Therefore, these findings can also demonstrate the relationship between etiological or inflammation-inducing substances and the corresponding immune reaction through the PHS hypothesis [14]. The immune components in the TME may work for the protection of cancer cells in the same way as for normal host cells because the main function of the host’s immune system at the cellular level is the protection of self-cells, and cancer cells are a new biosystem expressing self-identification markers including MHCs as discussed previously. Thus, the histopathologic condition in cancer tissues could be described as an inflammation state, and immune cells, including TILs, might be playing a role in protecting cancer cells against insults by recognizing and regulating pathological peptides affecting cancer cells or host cells.
Individual cells in cancers can metastasize to neighboring lymph nodes and other sites through lymphatic or circulatory systems. Awareness of the state of metastasis is critical in deciding the stage of cancer and treatment modality. The blood can be exposed to complex materials from external sources such as whole bacteria, parasites, foreign cells, and transformed self-cells (non-cancer cells), and phagocytes can control them with other immune components as non-self. Additionally, phagocyte-associated histopathological findings, such as hemophagocytosis in macrophage activation syndrome and granuloma formation in tuberculosis or parasite infections, may be an innate immune response when phagocytes cannot completely work against large materials [123,124]. Despite the abundance of phagocytes such as macrophages (monocytes and dendric cells) and neutrophils in the blood or TME, a metastatic cancer cell or a leukemic cell in the blood, lymphatics, and the TME may not trigger the phagocyte-associated immune responses, suggesting that innate immune cells of the host may also not regard the cancer cells as non-self-larger substances.
8.2. Immune Checkpoint Inhibitors (ICIs) and Autoimmune Diseases
Communication between immune cells is crucial for adaptation to any insults such as infection, trauma, and cancers. It is plausible that the major MHC-restricted immune response is precisely controlled through secondary signals such as PD-1, CTLA, and possibly other receptors. ICIs such as monoclonal antibodies and antagonists blocking signals of the receptors are widely used and effective reportedly in anticancer treatment for advanced cancers and some leukemias. Theoretically, it is thought that cytotoxic T cells are more active against cancer cells through the removal of inhibitory signals or preventing T cell exhaustion [125,126]. On the other hand, ICIs may be only effective on cancer cells that express the same MHCs and ligands for PD-1 and CTLA-4. Additionally, ICIs could be ineffective on some cancer cells expressing ligands, and effective on cancer cells without ligands [125]. Furthermore, the effectiveness of ICIs may have confounding factors that are influenced by gender, types of tumors, mutation of genes such as EGFR, Kras, and ALK, and metastases of tumors [127]. The modes of action of binding between the receptors and ligands remains unclear. It is possible that there is a mediating substance between receptors and ligands since most cell-cell interaction might be achieved through substance-receptor-signal transduction pathways.
As previously discussed, it is possible that cells in the “cancer organ” can express ligands for PD-1 or other receptors for communication with immune cells. This is because cancers often use the host’s immune cells, including T cells, in cases of infection or physical and chemical trauma. For further protection from injury, T cells may be activated to control toxic or signaling peptides bound to MHCs or other receptors on cancer cells. Recently, “oncolytic viral therapy” has been introduced for some cancers including brain tumors and bladder cancers, and genetically modified viruses may affect the intracellular homeostasis of cancer cells and induce cell death [128,129]. Given that affected cancer cells may need the assistance of immune cells including T cells, virus therapy is reportedly more effective when treated with immunotherapy [128]. Taken together, the effectiveness of ICIs can be reinterpreted as the inhibitors that block the activation of T cells that are otherwise working for cancer cells.
Patients treated with ICIs could experience the reactivation of autoimmune diseases in a remission state. In general, symptoms of relapsed diseases are mild and respond to corticosteroids though some patients can experience acute severe conditions, and the balance between immune suppression and anti-tumor effects such as the dose and schedule of the drugs remains a major obstacle for individual patients [130]. In the PHS hypothesis, the pathogenic peptides derived from injured cells by initial insults can bind to neighbor cells and/or other organ cells via systemic circulation and can induce cell injury by direct or cytokine storm as previously mentioned. If the production of specific T cell clones or antibodies (B cells) against the pathogenic peptides and proteins is delayed or absent, the continuous activation of non-specific T cells or B cells may be responsible for the occurrence of chronic inflammatory or autoimmune diseases [12,13,14]. Alternatively, continuously activated non-specific T or B cells may protect cells in part against pathogenic peptides or proteins derived from self-cells in autoimmune diseases. Immunosuppressive agents, including corticosteroids and biologics, may partially control the inflammation induced by these activated non-specific T or B cell clones. The modes of action are different in each immune modulator, including corticosteroids and ICIs. Corticosteroids may act on a broad range of T cell clones including non-specific T cell clones, whereas ICIs act on specific T cell clones that are more strictly controlled via immune checkpoints with MHC restriction. Thus, the use of ICIs could elicit drug-induced autoimmune-like diseases and chronic inflammations, and the flare-up of pre-existing autoimmune diseases that have responded to corticosteroids. Corticosteroids induce apoptosis on immature or non-specific T cells, B cells, and eosinophils and are essential drugs for the treatment of leukemias [131]. Additionally, steroid derivatives are effective in some cancers, including breast and prostate cancers [132].
The association between cancers and autoimmune diseases remains to be further evaluated. Patients with cancers, including thymoma, ovarian cancer, small cell lung cancer, and lymphoma, can be affected by autoimmune-like diseases or paraneoplastic syndrome during treatment or even before the diagnosis [133]. Patients with paraneoplastic syndromes can complain of symptoms that include neuropsychiatric, musculoskeletal, and endocrine manifestations [134], suggesting that cancers can affect various organs in a similar way to postinfectious immune-mediated diseases such as extrapulmonary manifestations observed in influenza, mycoplasma infection, and COVID-19. Given that autoimmune diseases and chronic inflammatory diseases are caused by etiological substances originating from injured self-cells in the PHS hypothesis, cancer cells could also be infected by viruses or exposed to physicochemical insults such as trauma, irradiation, and anticancer drugs. Affected cancer cells may adopt defensive strategies, such as the production of interferons and the activation of proteostasis networks. This could allow the release of several substances into the systemic circulation, including TSAs, PAMPs, DAMPs, pathological proteins, and pathological peptides, and the immune system is activated against the substances including those attached to target cells. Tumor lysis syndrome causing renal cell damage is well-known. It is associated with chemotherapy and may be caused by substances from the extensive necrosis of leukemic cells [135].
8.3. Chimeric Antigen Receptor T Cell (CAR-T) Therapies
CAR-T therapy is based on the concept that T cells control cancer cells. The T cells are designed artificially by gene manipulation techniques to express receptors for targeting antigens expressed on cancer cells, ideally TSAs. In general, CAR-T clone constructs contain four components: (1) an extracellular target antigen binding domain, (2) a hinge region, (3) a transmembrane domain, and (4) one or more intracellular signaling domains. T cells of cancer patients are isolated, engineered to express receptors for a target antigen, proliferated in vitro, and then infused back into patients [136]. Theoretically, it is a treatment tailored to the cancer of individuals since each cancer cell expresses its own antigens. At present, the treatment is reportedly effective in B cell origin malignancies including B cell lymphoma, B cell leukemia, and multiple myeloma [137]. B cells express receptors for communicating with T cells for MHC-restricted immune responses. Likewise, some clones of B cell malignancies may express the same receptors and MHCs besides TSAs. As discussed previously, there are diverse mechanisms of T cell activation in vitro and in vivo. Although it is believed that T cells recognize protein antigens, classical activation of T cells may be dependent on TCR-recognized peptides (as small proteins) bound to MHCs. The target antigen binding domain of the CAR-T cell is generated using immunoglobulin genes and recognizes proteins, not peptides processed by APCs. Thus, the activation of T cell clones through the CAR may be through an MHC-nonrestricted route, and activated CAR-T cell clones may be non-specific T cells such as those activated by superantigens or monoclonal antibodies targeting receptors of non-specific T cells. Substances, including cytokines, released from activated T cells differ according to T cell subsets with possibly different activation pathways. CARs are typically designed on the nature of target antigens, and most of the target antigens are receptors that are composed of proteins, not peptides. Additionally, the CARs should modulate a signal transduction pathway that can have a serious effect on target cells such as inducing apoptosis or suppressing proliferation. Cells in mammals express various receptors or channels for target substances, including elements, monoamines, neuropeptides, biochemicals, peptides, and proteins such as cytokines. Since peptides as well as smaller materials such as biochemicals and epigenetic components are not encoded in the genome, they are only expressed in a receptor-binding state or isolated forms. In addition, because cells may communicate with other cells through the substance-receptor-signal transduction systems, blocking an essential receptor or signal of target cells may affect the homeostasis of the cells. It is possible that cytokine imbalance caused by extensively activated non-specific CAR-T clones can induce target cell injury and other self-cells. Cytokine-associated cell injury, also known as a cytokine storm, occurs in various conditions including infectious diseases. Thus, this mechanism may be one of the causes of CAR-T toxicities [138]. Certain protein components of receptors expressed on self-cells can induce immune reactions such as the production of autoantibodies if released into systemic circulation as shown in various autoimmune diseases. Thus, artificially created CARs and their fragments could act as pathogenic proteins and peptides and induce inflammation associated with corresponding other adaptive immune cells. Therefore, the mechanism of CAR-T therapy could be interpreted as one of the players that has the role of blocking receptors that are working for the survival or proliferation of target cancer cells.
A variety of immune therapeutics targeting the receptors on cancers, especially hematologic malignancies, have been developed and applied for clinical use, including monoclonal antibodies, antibody-drug conjugates, bispecific T cell engager/ bispecific antibody (bsAb), and cancer vaccines [139,140,141,142,143]. The effectiveness of the therapy depends on the targeting receptors expressed (or biomarkers), including TSAs, and functional characteristics of target receptors for survival and intracellular homeostasis of cancer cells.
8.4. Therapeutic Perspectives
A variety of anticancer drugs have been developed to target cell death or the inhibition of cell proliferation. All cells respond to various toxic stimuli, including cytotoxins, lack of nutrients, physical trauma, and irradiation, but the threshold of cell death or resistance to stimuli differs in each cancer cell as well as in each organ cell. Based on this finding, treatment protocols or guidelines for each organ cancer have been developed and are in the process of being revised. Conventional anticancer therapeutics cannot avoid systemic adverse reactions, but these are inevitable events in improving patient outcomes.
All cells, including cancer cells, are protected by cell membranes and express their own receptors for communicating with other cells. All physiological and pathological phenomena at the molecular level are performed through the binding of substances to the receptors on or in cells. A toxic substance on certain organ cells does not affect others that do not have affinitive receptors. Therefore, it is reasonable and inevitable that the development of treatment modalities through various scientific fields has focused on the receptors of cancer cells [144,145]. The drug-targeted receptors expressed on cancer cells can block intercellular communications through the direct binding to receptors or the blocking of proteins involved in signal transduction pathways of the receptors. T cell-based therapy is an immune therapeutic that has shown some potential in treating diseases, including cancers and possibly immune-mediated diseases. We proposed that ICIs and CAR-T therapies could also be considered as one of the treatments in this category based on the PHS hypothesis. The therapies focused on cancer cells have helped substantially in reducing systemic side effects, but unfortunately, they are used as adjuvant therapy and cannot save all patients at present. It has been revealed that certain targeted therapies for the cancer of a specific organ can be effective in cancers of other organs. This suggests that the characteristics of cancer are determined by each cancer cell, which has different stages of intracellular cell homeostasis, not by the origin of tissues.
In the view of evolution, the development of cancers could be regarded as a natural phenomenon appearing in long-lived multicellular organisms including mammals, because they may have evolved to leave offspring and not to live long as individuals. Humans at present and in the future will live far longer compared to those in the past owing to environmental changes. There might be incomplete control systems against cancer cells in the host of human species because cancers may rarely occur in the far earlier stage of the species and are a host-dependent biosystem occurring mainly in long-lived hosts. Some patients with cancer can be in remission state for a long time with or without treatment including the “spontaneous regression” [61,62] and cancer cells are influenced by internal environment of the host including TMEs, suggesting that there is a control system, including an immune system acting in part, on communication between the host and cancer cells. Thus, control systems against cancer cells could be more adapted over time as cancer cells could be affected by environmental changes as discussed previously.
Researchers believe that cancer can be cured or controlled like autoimmune diseases in the future. Revealing the communication systems between the host and cancer cells may be difficult and time-consuming work since each cancer cell is an independent biosystem with its own homeostasis system. There is a marked complexity of acting substances such as diverse receptors with associated signal transduction systems. From a practical perspective, more advanced drugs that can induce apoptosis or suppress cell proliferation through blocking signals of cancer cells could be developed after the identification of the genetic information and the intracellular homeostasis system of each cancer cell.
9. Conclusions
Although the immune system of the host, especially the adaptive immune system, is believed to recognize and control cancer cells, the concepts are based on hypotheses reasoned from in vitro and ex vivo studies. The T cell-centered immunological concepts have explained only in part the pathophysiology of the diseases including infectious diseases, immune-mediated diseases, and cancers. The PHS hypothesis has provided a common immunopathogenesis of diseases, including infectious diseases, immune-mediated diseases, organ-specific diseases such as kidney and CNS diseases including genetic diseases and cancer [9,10,11,12,13,14]. In this article, we further discuss the oncogenesis and pathophysiology of cancers, and unresolved issues in tumor immunology through the PHS hypothesis. The function of each immune component of the host, including adaptive immune cells, may be similar in all disordered conditions including infectious diseases and cancers. Protein antigens, whether expressed on cancer cells or derived from injured cancer cells, can stimulate the production of specific antibodies (B cell clones) against pathogenic proteins, and specific T cell clones against pathogenic peptides. An inadequate immune response from the host to control these substances may be responsible for chronic inflammatory or autoimmune diseases in patients with cancers as well as other conditions. Additionally, the risk of cancer or predisposing factors is related to the number of tissue cell renewals (the turnover time) and the probability of gene defect(s).
Cancer cells may communicate with host cells, including immune cells, such as TAMs and T cells, for survival and proliferation. Through MHCs and cytokine networks, cancer cells may obtain the help of immune cells, including T cells, to manage external insults such as intracellular infection or physiochemical injuries. Targeted therapeutics blocking the communication between cancer cells and corresponding host cells have been developed and may become a mainstream of future anticancer treatments. T cell-based immune therapeutics, including TILs, ICIs, and CAR-T, are used clinically. We have introduced a new interpretation of the mechanisms used by these therapeutics under the PHS hypothesis. We hope that this review will provide both basic and clinical researchers with new perspectives for resolving the enigmas in tumor immunology and treatment.
Author Contributions
Authors contributed to the study conception, material collection, and writing. The final draft of the manuscript was written by KYL, and all authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank Drs. Soo-Ho Lee, Sung-Ho Lee, and Jae-Kyun Hur for the assistant tasks on this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Gordon, S.; Imhof, B.A.; Núñez, G.; Bousso, P. Élie Metchnikoff (1845-1916): celebrating 100 years of cellular immunology and beyond. Nat. Rev. Immunol. 2016, 16, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Golstein. P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer. G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: a renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Rhim, J.-W.; Kang, J.-H. Kawasaki disease: laboratory findings and an immunopathogenesis on the premise of a “protein homeostasis system”. Yonsei Med. J. 2012, 53, 262–275. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Rhim, J.-W.; Kang, J.-H. Hyperactive immune cells (T cells) may be responsible for acute lung injury in influenza virus infections: a need for early immune-modulators for severe cases. Med. Hypotheses 2011, 76, 64–69. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Kang, J.-H.; Lee, K.-Y. Etiological and pathophysiological enigmas of severe coronavirus disease 2019, multisystem inflammatory syndrome in children, and Kawasaki disease. Clin. Exp. Pediatr. 2022, 65, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y. A common immunopathogenesis mechanism for infectious diseases: the protein-homeostasis-system hypothesis. Infect. Chemother. 2015, 47, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y. A unified pathogenesis for kidney diseases, including genetic diseases and cancers, by the protein-homeostasis-system hypothesis. Kidney Res. Clin. Pract. 2017, 36, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y. Common immunopathogenesis of central nervous system diseases: the protein-homeostasis-system hypothesis. Cell Biosci. 2022, 12, 184. [Google Scholar] [CrossRef] [PubMed]
- Malaterre, C.; Jeancolas, C.; Nghe, P. The Origin of Life: What Is the Question? Astrobiology 2022, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, X.; Wei, Y.; Cheng, Y.; Guo, Y.; Khudyakov, I.; et al. A widespread pathway for substitution of adenine by diaminopurine in phage genomes. Science 2021, 372, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.E.; Bashor, C.J.; Collins, J.J. A brief history of synthetic biology. Nat. Rev. Microbiol. 2014, 12, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Pross, A.; Pascal, R. The origin of life: what we know, what we can know and what we will never know. Open Biol. 2013, 3, 120190. [Google Scholar] [CrossRef]
- Valet, M.; Siggia, E.D.; Brivanlou, A.H. Mechanical regulation of early vertebrate embryogenesis. Nat. Rev. Mol. Cell. Biol. 2022, 23, 169–184. [Google Scholar] [CrossRef]
- Wu, Z.; Guan, K.L. Hippo Signaling in Embryogenesis and Development. Trends Biochem. Sci. 2021, 46, 51–63. [Google Scholar] [CrossRef]
- van Gelder, M.M.; van Rooij, I.A.; Miller, R.K.; Zielhuis, G.A.; de Jong-van den Berg, L.T.; Roeleveld, N. Teratogenic mechanisms of medical drugs. Hum. Reprod. Update 2010, 16, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Verga Falzacappa, M.V.; Ronchini, C.; Reavie, L.B.; Pelicci, P.G. Regulation of self-renewal in normal and cancer stem cells. FEBS J. 2012, 279, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Li, D. Senescent Stem Cell Dysfunction and Age-Related Diseases. Stem Cells Dev. 2023, 32, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Cheng, L.; Du, J.; Peng, Y.; Allan, R.W.; Wei, L.; et al. Diagnostic utility of novel stem cell markers SALL4, OCT4, NANOG, SOX2, UTF1, and TCL1 in primary mediastinal germ cell tumors. Am. J. Surg. Pathol. 2010, 34, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, M.; Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370, 20140367. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Luo, M.; Wang, R. Identifying critical regulatory interactions in cell fate decision and transition by systematic perturbation analysis. J. Theor. Biol. 2024, 577, 111673. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Samuel, N.; Hudson, T.J. Translating genomics to the clinic: implications of cancer heterogeneity. Clin. Chem. 2013, 59, 127–137. [Google Scholar] [CrossRef]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef]
- Yoon, K.A.; Woo, S.M.; Kim, Y.H.; Kong, S.Y.; Lee, M.K.; Han, S.S.; et al. Comprehensive Cancer Panel Sequencing Defines Genetic Diversity and Changes in the Mutational Characteristics of Pancreatic Cancer Patients Receiving Neoadjuvant Treatment. Gut Liver 2019, 13, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell. Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Hommen, F.; Bilican, S.; Vilchez, D. Protein clearance strategies for disease intervention. J. Neural Transm. (Vienna) 2022, 129, 141–172. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.V.; Karpov, V.L. Proteasomes and several aspects of their heterogeneity relevant to cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Türker, F.; Cook, E.K.; Margolis, S.S. The proteasome and its role in the nervous system. Cell. Chem. Biol. 2021, 28, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Yang, C.; Castro, L.M.; Tashima, A.K.; Ferro, E.S.; Moir, R.D.; et al. Analysis of the yeast peptidome and comparison with the human peptidome. PLoS ONE 2016, 11, e0163312. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, I.; Ivanov, V.; Fesenko, I. Peptidome: Chaos or Inevitability. Int. J. Mol. Sci. 2021, 22, 13128. [Google Scholar] [CrossRef]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef]
- Hoyer, D.; Bartfai, T. Neuropeptides and neuropeptide receptors: drug targets, and peptide and non-peptide ligands: a tribute to Prof. Dieter Seebach. Chem. Biodivers. 2012, 9, 2367–2387. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1117. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Page`s, C.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Mohamed Khosroshahi, L.; Rokni, M.; Mokhtari, T.; Noorbakhsh, F. Immunology, immunopathogenesis and immunotherapeutics of COVID-19; an overview. Int. Immunopharmacol. 2021, 93, 07364. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Cancer Biol. 2020, 64, 1–12. [Google Scholar] [CrossRef]
- Pelosi, A.C.; Scariot, P.P.M.; Garbuio, A.L.P.; Kraemer, M.B.; Priolli, D.G.; Masselli Dos Reis, I.G.; et al. A systematic review of exercise protocols applied to athymic mice in tumor-related experiments. Appl. Nutr. Metab. 2023, 48, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Kamperschroer, C.; Frank, B.; Il, C.; Lebrecd, H.; Mitchell-Ryane, S.; Molinierf, B.; et al. Current approaches to evaluate the function of cytotoxic T-cells in non-human primates. J. Immunotoxicol. 2023, 20, 2176952. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.; Arlen, P.M.; Tsang, K.Y. Potentiation of natural killer cells to overcome cancer resistance to NK cell-based therapy and to enhance antibody-based immunotherapy. Front. Immunol. 2023, 14, 1275904. [Google Scholar] [CrossRef]
- Rahman, T.; Das, A.; Abir, M.H.; Nafiz, I.H.; Mahmud, A.R.; Sarker, R.M.; et al. (2023) Cytokines and their role as immunotherapeutics and vaccine adjuvants: The emerging concepts. Cytokine 2023, 169, 156268. [Google Scholar] [CrossRef]
- Balkwil, I.F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Patel S (2018) Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [CrossRef]
- Meng, X.; Yerly, D.; Naisbitt, D.J. Mechanisms leading to T-cell activation in drug hypersensitivity. Curr. Opin.Allergy Clin. Immunol. 2018, 18, 317–324. [Google Scholar] [CrossRef]
- Deacy, A.M.; Gan, S.K.; Derrick, J.P. Superantigen Recognition and Interactions: Functions, Mechanisms and Applications. Front. Immunol. 2021, 12, 731845. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y.; Rhim, J.-W.; Kang, J.-H. Immunopathogenesis of COVID-19 and early immunomodulators. Clin. Exp. Pediatr. 2020, 63, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y. Pneumonia, acute respiratory distress syndrome, and early immune-modulator therapy. Int. J. Mol. Sci. E: pii. [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2012, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cel.l Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Kang, H.-M.; Lee, K.-Y. Pathophysiology of Virus Infections: The Protein-Homeostasis-System Hypothesis. Preprints 2023, 2023072131. [Google Scholar] [CrossRef]
- Smithers, D.W. An attack on cytologism. Lancet 1962, 1, 493–499. [Google Scholar] [CrossRef]
- Soto, A.M.; Sonnenschein, C. The cancer puzzle: Welcome to organicism. Prog. Biophys. Mol. Biol. 2021, 165, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; et al. ; Children’s Oncology Group. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Drabent, P.; Fraitag, S. Malignant Superficial Mesenchymal Tumors in Children. Cancers (Basel) 2022, 14, 2160. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Gruen, J.R.; Weissman, S.M. Human MHC class III and IV genes and disease associations. Front. Biosci. 2001, 6, D960–972. [Google Scholar] [CrossRef] [PubMed]
- Gill, T.J. 3rd; Siew, S.; Kunz, H.W. Major histocompatibility complex (MHC)-linked genes affecting development. J. Exp. Zool. 1983, 228, 325–345. [Google Scholar] [CrossRef] [PubMed]
- McManigle, W.; Pavlisko, E.N.; Martinu, T. Acute cellular and antibody-mediated allograft rejection. Semin. Respir. Crit. Care Med. 2013, 34, 320–335. [Google Scholar] [CrossRef]
- Zinkernagel, R.M. The Nobel Lectures in Immunology. The Nobel Prize for Physiology or Medicine, 1996. Cellular immune recognition and the biological role of major transplantation antigens. Scand. J. Immunol. 1997, 46, 421–436. [Google Scholar] [PubMed]
- Robinette, M.L.; Colonna, M. Innate lymphoid cells and the MHC. HLA 2016, 87, 5–11. [Google Scholar] [CrossRef]
- Bubeník, J. Tumour MHC class I downregulation and immunotherapy (Review). Oncol. Rep. 2003, 10, 2005–2028. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Cron, R.Q. The Multifaceted Immunology of Cytokine Storm Syndrome. J. Immunol. 2023, 210, 1015–1024. [Google Scholar] [CrossRef]
- Ball, L.M.; Egeler, R.M. Acute GvHD: pathogenesis and classification. Bone Marrow Transplant. 2008, 41, S58–64. [Google Scholar] [CrossRef]
- Pedersen, S.J.; Maksymowych, W.P. The Pathogenesis of Ankylosing Spondylitis: an Update. Curr. Rheumatol. Rep. 2019, 21, 58. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Lee, J.S. Immunoglobulin G has a role for systemic protein modulation in vivo: a new concept of protein homeostasis. Med. Hypotheses 2006, 67, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Oh, J.H.; Rhim, J.W.; Lee, K.-Y. Correlation between elevated platelet count and immunoglobulin levels in the early convalescent stage of Kawasaki disease. Medicine (Baltimore) 2017, 96, e7583. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Jurasz, P. The role of platelets in the tumor microenvironment: From solid tumors to leukemia. Biochim. Biophys. Acta 2016, 1863, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Jackaman, C.; Tomay, F.; Duong, L.; Abdol Razak, N.B.; Pixley, F.J.; Metharom, P.; et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res. Rev. 2017, 36, 105–116. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Doan, J.V.; Ising, M.E.; Evans, N.R.; et al. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H. Mechanisms and impacts of chromosomal translocations in cancers. Front. Med. 2012, 6, 263–274. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2019, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; et al. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Liptáková, A.; Čurová, K.; Záhumenský, J.; Visnyaiová, K.; Varga, I.I. Microbiota of female genital tract - functional overview of microbial flora from vagina to uterine tubes and placenta. Physiol. Res. 2022, 71, S21–S33. [Google Scholar] [CrossRef]
- Borkenhagen, L.K.; Salman, M.D.; Ma, M.J.; Gray, G.C. Animal influenza virus infections in humans: A commentary. Int. J. Infect. Dis. 2019, 88, 113–119. [Google Scholar] [CrossRef]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wergo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Chen, J.; Domingue, J.C.; Sears, C.L. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin. Immunol. 2017, 32, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.P.; Fox, B.W.; Chao, P.H.; Schroeder, F.C.; Sengupta, P.A. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature 2020, 583, 415–420. [Google Scholar] [CrossRef]
- Clinton, N.A. : Hameed, S.A.; Agyei, E.K.; Jacob, J.C.; Oyebanji, V.O.; Jabea, C.E. Crosstalk between the Intestinal Virome and Other Components of the Microbiota, and Its Effect on Intestinal Mucosal Response and Diseases. J. Immunol. Res. 2022, 2022, 7883945. [Google Scholar] [CrossRef]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef] [PubMed]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Alipour, M. Molecular Mechanism of Helicobacter pylori-Induced Gastric Cancer. J. Gastrointest. Cancer 2021, 52, 23–30. [Google Scholar] [CrossRef]
- Kebudi, R.; Kiykim, A.; Sahin, M.K. Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr. Pediatr. Rev. 2019, 15, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Vangipuram, R.; Tyring, S.K. AIDS-Associated Malignancies. Cancer Treat. Res. 2019, 177, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Selma-Royo, M.; Cortés-Macías, E.; Sánchez, G.; Collado, M.C. Gut Microbiota Development in Infants and Children. World Rev. Nutr. Diet 2022, 124, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Maggini, S.; Pierre, A.; Calder, P.C. Immune Function and Micronutrient Requirements Change over the Life Course. Nutrients 2018, 10, 1531. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y.; Han, J.W.; Lee, J.S. Kawasaki disease may be a hyperimmune reaction of genetically susceptible children to variants of normal environmental flora. Med. Hypotheses 2007, 69, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Ang, T.L.; Fock, K.M. Clinical epidemiology of gastric cancer. Singapore Med. J. 2014, 55, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Eun, C.S. Inflammatory bowel disease in Korea: epidemiology and pathophysiology. Korean J. Int. Med. 2022, 37, 885–894. [Google Scholar] [CrossRef]
- Araghi, M.; Soerjomataram, I.; Bardot, A.; Ferlay, J.; Cabasag, C.J.; Morrison, D.S.; et al. Changes in colorectal cancer incidence in seven high-income countries: a population-based study. Lancet Gastroenterol. Hepatol. 2019, 4, 511–518. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Kang, H.M.; Han, J.W.; Lee, K.-Y. A Presumed Etiology of Kawasaki Disease Based on Epidemiological Comparison With Infectious or Immune-Mediated Diseases. Front. Pediatr. 2019, 7, 202. [Google Scholar] [CrossRef]
- Shahin, O.A.; Chifotides, H.T.; Bose, P.; Masarova, L.; Verstovsek, S. Accelerated Phase of Myeloproliferative Neoplasms. Acta Haematol. 2021, 114, 484–499. [Google Scholar] [CrossRef]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer 2015, 5, e366. [Google Scholar] [CrossRef]
- Kil, H.R.; Yu, J.W.; Lee, S.C.; Rhim, J.-W.; Lee, K.-Y. Changes in clinical and laboratory features of Kawasaki disease noted over time in Daejeon, Korea. Pediatr. Rheumatol. Online J. 2017, 15, 60. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Lee, Y.T.; Kang. H.M.; Suh, J.S.; Lee, K.-Y. Changes in clinical features in Henoch-Schönlein purpura during three decades: an observational study at a single hospital in Korea. Clin. Rheumatol. 2019, 38, 2811–1818. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Mullighan, C.G. Molecular markers in ALL: Clinical implications. Best Pract. Res. Clin. Haematol. 2020, 33, 101193. [Google Scholar] [CrossRef] [PubMed]
- Hofman, P.; Badoual, C.; Henderson, F.; Berland, L.; Hamila, M.; Long-Mira, E.; et al. Multiplexed immunohistochemistry for molecular and immune profiling in lung cancer-just about ready for prime-time? Cancers (Basel) 2019, 11, E283–118. [Google Scholar] [CrossRef]
- Sarhadi, V.K.; Armengol, G. Molecular Biomarkers in Cancer. Biomolecules 2022, 12, 1021. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Cheng, M.; Ma, J.; Chen, Y.; Zhang, J.; Zhao, W.; Zhang, J.; et al. Establishment, characterization, and successful adaptive therapy against human tumors of NKG cell, a new human NK cell line. Cell. Transplant. 2011, 20, 1731–1746. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K.; Behrens, E.M. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr. Opin. Rheumatol. 2014, 26, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb). 2011, 91, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.; Oronsky, B.; Reid, T.R. Commentary on oncolytic viruses: past, present, and future. J. Immunother. Cancer 2023, 11, e007905. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Li, L.; Mo, T.; Na, J.; Qian, Z.; Fan, D.; et al. Oncolytic viral vectors in the era of diversified cancer therapy: from preclinical to clinical. Clin. Transl. Oncol. 2022, 24, 1682–1701. [Google Scholar] [CrossRef] [PubMed]
- Klavdianou, K.; Melissaropoulos, K.; Filippopoulou, A.; Daoussis, D. Should We be Afraid of Immune Check Point Inhibitors in Cancer Patients with Pre-Existing Rheumatic Diseases? Immunotherapy in Pre-Existing Rheumatic Diseases. Mediterr. J. Rheumatol. 2021, 32, 218–226. [Google Scholar] [CrossRef]
- McColl, A.; Michlewska, S.; Dransfield, I.; Rossi, A.G. Effects of glucocorticoids on apoptosis and clearance of apoptotic cells. ScientificWorldJournal 2007, 7, 1165–1181. [Google Scholar] [CrossRef]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA Damage Response in Prostate and Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8273. [Google Scholar] [CrossRef]
- Itani, M.; Goldman Gollan, Y.; Ezell, K.; Mohanna, M.; Sabbagh, S.; Mears, C.; et al. Thymoma and Myasthenia Gravis: An Examination of a Paraneoplastic Manifestation. Cureus 2023, 15, e34828. [Google Scholar] [CrossRef]
- Farina, A.; Villagrán-García, M.; Vogrig, A.; Zekeridou, A.; Muñiz-Castrillo, S.; Velasco, R.; et al. Neurological adverse events of immune checkpoint inhibitors and the development of paraneoplastic neurological syndromes. Lancet Neurol. 2024, 23, 81–94. [Google Scholar] [CrossRef]
- Rahmani, B.; Patel, S.; Seyam, O.; Gandhi, J.; Reid, I.; Smith, N.; et al. Current understanding of tumor lysis syndrome. Hematol. Oncol. 2019, 37, 537–347. [Google Scholar] [CrossRef]
- Ramesh, P.; Hui, H.TY.L.; Brownrigg, L.M.; Fuller, K.A.; Erber, W.N. Chimeric antigen receptor T-cells: Properties, production, and quality control. Int. J. Lab. Hematol. 2023, 5, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Wu, X.; Zhang, Y.; Zeng. L.; Dong, Y.; Liu, R.; et al. Adverse events and efficacy of second-round CAR-T cell therapy in relapsed pediatric B-ALL. Eur. J. Haematol. 2024, 112, 75–82. [Google Scholar] [CrossRef]
- Sliwkowski, M.X.; Mellman, I. Antibody therapeutics in cancer. Science 2013, 341, 1192–1198. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-Drug Conjugates-A Tutorial Review. Molecules 2012, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy: are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of oncogenesis and pathophysiology of cancers under the protein-homeostasis-system (PHS) hypothesis.
Figure 1.
Schematic diagram of oncogenesis and pathophysiology of cancers under the protein-homeostasis-system (PHS) hypothesis.
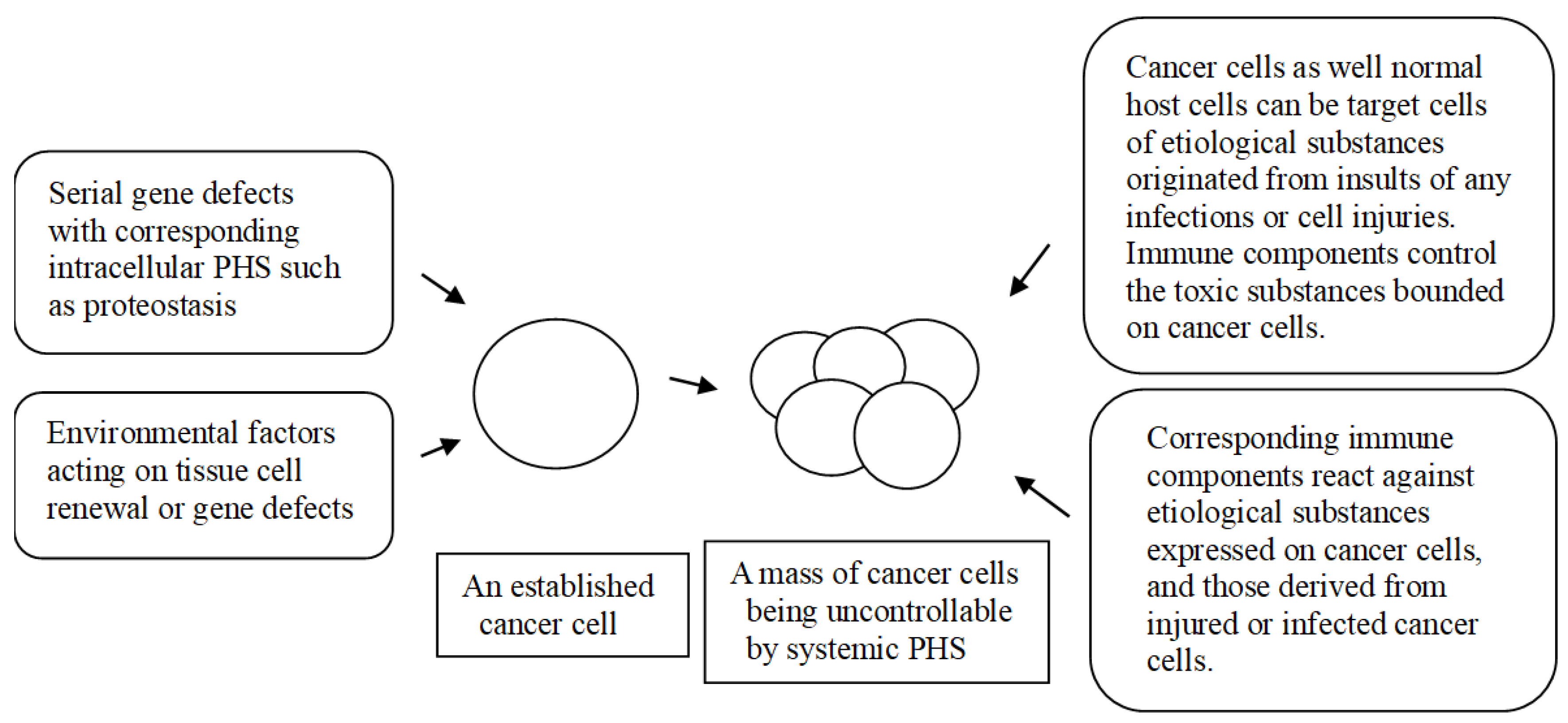

Table 1.
Immune effectors against cancers in the PHS hypothesis.
| Immune effectors (or events) |
Main functions |
| Adaptive immune system | |
| T cells | Control of pathogenic peptides targeted to cancer cells or host cells via MHC-restricted (TCR-associated) and non-MHC-restricted manners |
| B cells | Control of pathogenic proteins targeted to cancer cells or host cells via MHC-restricted (BCR-associated) and non-MHC-restricted manners |
| Innate immune system | |
| Natural killer cells | Control of transformed cells such as virus-infected cells and tumor cells in MHC-non-restricted manner(?) |
| Tissue macrophage-linaeged cells | Antigen presentation to adaptive immune cells in MHC-restricted immune responses. Possible control of communications between cancers cells and host cells such as stromal cells in TMEs |
| Phagocytes (neutrophils and circulating monocyte/macrophages) | Control of large complex substances such as viruses, bacteria, parasite, and apoptotic & necrotic bodies associated with infected or injured cancer cells and normal cells |
| Mast cells, basophils, eosionphils | These cells are activated by substances that are associated with receptors on the cells and control the substances through inflammation involved in these cells |
| Unidentified innate immune components against small non-protein toxic materials | There are non-protein toxic or inflammation-inducing substances, including elements, monoamins, neuropeptides, LPS, RNAs, DNAs, chemicals, and biochemicals. TLR-associated immune responses, natural antibodies, and immun proteins and/or peptides control these diverse substances. The immune proteins, including PrP gene products and other amyloid proteins, control pathogenic monoamine metabolites or neuropeptides especially in CNS [14] |
| Production of alternative proteins in genetic diseases and cancers | The systemic and intracellular PHS control in part insults from a protein deficiency or malfunctional protein in organ tissues or within a cell |
MHC, major histocompatibility complex; TCR, T cell receptor; BCR, B cell receptor; TME, tumor microenvironment; LPS: lipopolysaccharide; TLR, Toll-like receptor; PrP, prion; CNS, central nervous system.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



