
Submitted:
09 July 2024
Posted:
10 July 2024
You are already at the latest version
Abstract
Not occasional phamracokinetic variability and suboptimal exposure have been reported for micafungin in the intensive-care-unit (ICU) patients. This study aims to evaluate the quality of population pharmacokinetic models and provide rational using recommendation of this drug in clinical practice. Monte Carlo simulations were implemented to compare pharmakinetic parameters and probability of target attainment (PTA) towards various Candida species. Body weight, liver function, and SOFA score were the most frequent covariates in the final 16 included studies. micafungin clearance for ICU adults (SOFA≥10 or <10) was 39%–52% and 43%–57% higher, respectively, than non-ICU adults. For Candida infections with MICs <0.016 mg/L, the ICU group had lower proportions achieving PTA > 90% compared to the non-ICU group (2/6 (33%) vs. 4/7 (57%)), and for non-ICU and ICU adults, micafungin dosages of 100 and 150 mg were recommended, respectively. While for C. tropicalis and C. glabrata, 200 and 250 mg were recommended respectively. However, for C. krusei and C. parapsilosis, it failed to reach assumed PTA at MICs of 0.125-0.25 mg/L and 0.125-2 mg/L, respectively. Adjusting micafungin dosage in ICU patients may be necessary. Both ICU and non-ICU adults should modify their dosage regimens based on the Candida species and their respective MICs.
Keywords:
micafungin
; pharmacokinetic/pharmacodynamic
; population pharmacokinetics
; dosing regimen optimization
1. Introduction
Micafungin (MFG) is a semisynthetic, high molecular weight, water-soluble echinocandin [1]. It exerts anti-candida activity by non-competitively inhibiting the synthesis of 1,3-β-D-glucan within the cell wall. This inhibition may also prevent the growth of hyphae in Aspergillus species that are resistant to conventional antifungal agents [2]. Currently, this agent is licensed worldwide for prophylaxis and treatment of invasive Candida infections [3,4].
MFG is minimally absorbed after oral administration and is, thus, only available as an intravenous formulation. It has a high protein-binding rate (approximately 99.8%) and is rapidly distributed to tissues. Furthermore, it exhibits linear pharmacokinetics (PK) and can be used without dosage adjustment under renal impairment and minor-to-moderate hepatic impairment. Moreover, there is no evidence of toxicity even the dosage for adults and neonates exceeds 15 mg/kg and 8mg/kg, respectively [5,6]. Consequently, its outstanding safety and superior efficacy profile render it a first-line treatment option for sensitive Candida infections in pediatric and adult patients.
There is considerable evidence of wide PK variability and suboptimal exposure under standard dosages of MFG, particularly in critically ill patients in the intensive care unit (ICU). First, it is important to note that the physiopathological conditions under which the functions of important organs differ considerably between ICU and non-ICU patients. Such patients frequently require life-support equipment, including hemodialysis (HD), continuous renal replacement therapy (CRRT), or extracorporeal membrane oxygenation (ECMO). Mounting evidence suggests that treatment failure may result from fungal resistance and/or prolonged suboptimal exposure. Consequently, individualized dosage adjustment is of paramount importance in this population.
The clinical outcomes of MFG to treat invasive Candida infections are characterized by pharmacokinetic/pharmacodynamic (PK/PD) indices [7,8,9]. Theoretically, the ratio of the area under the plasma unbound drug concentration-time curve over 24 h to the minimum inhibitory concentration (fAUC24/MIC) at steady state should be a more suitable PK/PD indicator than the ratio of the area under the plasma total drug concentration-time curve over 24 h to the MIC (AUC24/MIC) [9]. However, the latter method is more commonly applied, particularly in the percentage of time above the MIC (T > MIC %). The model proposed by Andes et al. [7] was an inaugural comprehensive study to establish a correlation between clinical and microbiological responses, and the susceptibility cut-off targets of AUC24/MIC were determined to be ≥5000 for C. albicans and C. glabrata, ≥3000 for C. krusei and C. tropicalis, and ≥285 for C. parapsilosis [7,10,11]. It is imperative to enhance the identification and utilization of factors influencing PK/PD to rationalize and optimize MFG dosing schedules. This review aimed to evaluate the quality of different models, summarize the covariates, and provide valuable advice for the rational use of MFG in clinical practice based on the PTA range among various populations, particularly ICU patients, using the simulation results of the included models.
2. Materials and Methods
2.1. Study Identification
Population pharmacokinetic (PPK) studies on MFG were systematically searched in PubMed, Embase, and Web of Science databases from inception to January 30, 2023. Search terms included ‘FK 463′, ‘FK463′, ‘FK-463′, ‘Micafungin Sodium’, ‘Micafungin’, ‘Mycamine’ and ‘population pharmacokinetic*’, ‘pharmacokinetic model*’, ‘nonlinear mixed effect model’, ‘NONMEM’ ‘Pmetrics’, ‘WINNONMIX’, ‘ADAPT’, ‘P-PHARM’, ‘nlmixed’, ‘NLME’, ‘USC PACK’, ‘MONOLIX’. The reference lists of included articles were also reviewed. The literature search was performed by two independent reviewers and inspected by a third author.
All published PPK models of MFG were included if they met the following criteria: (i) study population: patients or healthy subjects; (ii) treatment with MFG as the study drug; and (iii) data analysis: population PK or population PK/PD analysis.
Articles were excluded if they met the following criteria: (I) They were reviews, conference abstracts, or focused on methodology/algorithm/software; (II) They were not published in English; (III) They contained only animal data; (IV) They did not provide sufficient information on the methodology and PPK or PK/PD models.
2.2. Data Extraction
The following information was extracted from the identified models: (1) characteristics and demographics of the study population (e.g., country/race, patients/healthy subjects, age, sex, body weight (BW), Lean Body Weight (LBW), Fat-free Body Weight (FFM), Ideal Body Weight (IBW); (2) study design (e.g., study type, number of included subjects and sampling, dosing regimens, and bio-assay method used); and (3) modeling strategies and final pharmacokinetics of included studies (e.g., software/algorithm, structural model, statistical model, parameter estimates, covariates, between-subject variability (BSV), Inter-occasion variability (IOV) and residual unexplained variability (RUV), model evaluation and model application). (4) PK/PD targets used for simulation; (5) model application and recommended dosage regimens.
2.3. Literature Quality
According to the guidelines established by Kanji et al.[12] and Jamsen et al. [13], a 30-item checklist was introduced to evaluate the quality of the literature, with information to be included when reporting a transparent and accurate clinical PK/PD study. If an item in the checklist was reported in the study, one point was counted in the scoring item; otherwise, no point was counted. The total score for each study was calculated and expressed as a percentage, which was defined as the compliance rate.
2.4. Comparison of Studies
Based on the classifications of patient characteristics in the retrieved studies [14,15,16,17,18,19] ADDIN EN.CITE ts, preschool children, school children, adolescents and adults. The adult virtual scenario was divided into ICU group (SOFA ≥ 10), ICU group (SOFA < 10) and non-ICU group. The additional details are presented in Table S1. MFG was administered as follows: 4 mg/kg in neonates, 2 mg/kg in children younger than 12-year-old, and 100 mg/day in adolescents and adults. The infusion time was set at one hour. All patients received multiple doses and reached a steady state.
The study characteristics and PPK analyses are summarized in a tabular format. Visual predictive distributions (VPDs) of the concentration-time curve at steady state were generated using Monte Carlo simulations based on the reported PPK models from each study [20]. A total of 1000 virtual patients were simulated for each scenario. All simulations were performed using the NONMEM software (version 7.5; ICON Development Solutions, Ellicott City, MD, USA). We employed similarity comparison to ensure the accuracy of the model repository.
Statistical differences between the following PK parameters: clearance from the central compartment (CL), volume of distribution (Vd), and AUC24 were determined among various groups using the t-test. The effects of the included covariates on PK parameters were assessed using forest plots. For continuous covariates, the maximum and minimum values based on the demographic information in the included studies were extracted and scaled to the same range. Regarding binary covariates, each category was similarly disposed. The values of some binary covariates in one model that may be concomitantly used as binary variables in another model refer to the corresponding cut-off values. Each binary and continuous covariate was input to explore its influence on the CL and volume of the central compartment (V1). The upper and lower limits of the parameters were estimated based on the range of the corresponding covariates and were normalized to the median values. Therefore, the effect of each covariate can be shown as the range of the limit to the median value as follows (Equation (1)):
We regarded covariate effects beyond the 80%-125% range as clinically significant, according to the standards employed in bioequivalence studies [21]. All data were analyzed and plotted using R software (version 4.2.1; www.r-project.org; accessed on January 30, 2023).
2.5. Monte Carlo Simulation for the Probability of Target Attainment
The cutoffs of PK/PD indices obtained in animal models have been extrapolated to humans [7]. The susceptibility cut-off targets of AUC24/MIC against various fungal strains provide a critical tool for optimal dosing strategies and definitions of clinically relevant drug resistance. According to EUCAST MIC breakpoints in vitro broth dilution susceptibility testing of Candida spp., each MIC breakpoints of Candida species were chosen (C. albicans ≤ 0.016 mg/L, C. glabrata ≤ 0.032 mg/L, C. krusei ≤ 0.06 mg/L, C. tropicalis ≤ 0.25 mg/L and C. parapsilosis ≤ 2 mg/L), while referring to CLSI standard the MIC breakpoints were ≤ 0.25 mg/L for C. albicans, C. krusei and C. tropicalis, ≤ 0.06 mg/L for C. glabrata and ≤ 2 mg/L for C. parapsilosis [22,23]. We performed Monte Carlo simulations to evaluate the performance of different dosing regimens (100-300 mg/d) and MICs (0.002-4 mg/L) of MFG in ICU and non-ICU adults. PTA ≥ 90% was supposed to generate an optimal dosage regimen of MFG. Sensitivity analysis of the proportions of models sustaining a PTA ≥ 90% was also implemented to recommend optimized dosing regimens.
3. Results
3.1. Study Identification
A PRISMA diagram for this study is shown in Figure 1. A total of 76, 73, and 108 publications were initially selected from PubMed, EMBASE, and Web of Science, respectively. After excluding 79 duplicate records, 178 studies were included. A full-text review was conducted according to the screening criteria and 14 studies were deemed eligible for inclusion. Additional studies were identified from the reference lists of included studies. Finally, 16 articles (17 models) published between 2006 and 2022 were retained.
3.2. Literature Quality
The 30-item checklist and the corresponding risk map about the bias of each study are presented in Table S2 and Figure 2. Given that previous clinical studies identified no potential drug-drug interactions that could alter the PK of MFG, most of the models (13/17) did not identify the use of concomitant medications as covariates. A review of the model development sections of the 16 studies revealed that 13 studies lacked descriptions of the methods for handling missing data. Furthermore, only two studies included a schematic representation of the final model. Over 50% of the studies did not present a plot of concentration over time and/or the effect of concentration. The proportion of participants who adhered to the study protocol ranged from 76.7% to 96.7% with a median adherence rate of 90%. All models, except one, achieved a compliance rate of at least 80%, indicating these studies were of good quality.
3.3. Study Comparison
The characteristics of all included studies are presented in Table 1. All studies were prospectively designed and published between 2006 and 2022. Two models involved healthy adult volunteers, whereas 13 included adult patients. Four models were applied exclusively to pediatric populations: neonates (n = 1), infants (n = 4), preschoolers or schoolchildren (n = 4), and adolescents (n = 4). Of particular concern was that seven models (41%) focused on critically ill populations. One of these models assessed the safety and pharmacokinetics of multiple elevated doses of MFG in preterm neonates with a low BW. Over seven models (41%) have been investigated for hematological malignancies, cancer, and hematopoietic stem cell transplantation. Nine models (53%) were developed using data from phase I, II, III, or IV clinical trials, whereas the remaining studies (47%) enrolled subjects from real-world clinical settings. The median number of participants was 24 (IQR: 13-54.8). A mere nine of the sixteen studies presented data on both BMI and height in conjunction with total body weight (TBW). The remaining seven studies provided only TBW. Roeland et al. [24] calculated LBW. All studies acquired over four plasma samples per patient, except for one study, which did not record an accurate sample size [7].
The final PPK parameters of the included studies are presented in Table 2. All studies employed either a two-compartment model (n=15) or three-compartment model (n=2), accompanied by zero-order infusion and first-order elimination to describe the administration, distribution and disposition procedure of MFG. MFG was administered over a wide range of infusion times (0.5-3 h) [14,16,25].
All studies explored BSV using the exponential model. The BSV values for CL and V1 were 24.1% coefficient of variation (CV) (interquartile range (IQR): 18.1%–32.8%) and 34.3% CV (IQR: 16.7%-51.6%), respectively. RUV was described using additive, proportional, or combined error models. Fourteen studies included proportional errors, with an IQR of 5.6% to 19% CV, and eight included additive errors, with an IQR of 0.0666 to 0.642 mg/L. The IOV) was estimated to be 16.1% CV (IQR: 10%–27.8%) for clearance (CL) and 27%–28.1% CV for central volume of distribution (V1).
In terms of model validation, fifteen models (93.8%) were internally evaluated using more than two methods. However, none of these methods has been validated using an external method. The most commonly employed methods are goodness-of-fit plots (GOF), visual predictive checks (VPC), and prediction-corrected VPC (pcVPC) checks. Although the normalized prediction distribution error (NPDE) method is an effective evaluation tool, it has been employed in only two studies [11,26]. Ten out of the 16 models employed non-parametric bootstrap validations.
Fifteen of the 17 models (88%) conducted Monte Carlo simulations to validate the investigated dosages or to propose new dose recommendations. The PK/PD indicators included AUC24/MIC (n = 9), fAUC24/MIC (n = 1), AUC24 (n = 4), and T>MIC (%) (n = 1). In the context of pediatric patients, five studies [14,16,17,18,19] not only supplemented the most recent labeled dosage regimens but also further validated the feasibility of an intermittent dosing strategy compared to a daily dosing strategy [16,17,19]. Ultimately, the studies concluded that a dose of 15 mg/kg/day in premature neonates nearly equaled 5 mg/kg/day in adults. Chandra et al. [17] proposed that, for pediatric patients with low BW (<30 kg), a dosing strategy of 5 mg/kg twice weekly for the prophylaxis of invasive fungal infections not inferior to the daily dosing schedule. Nevertheless, caution should be exercised when selecting this dosing regimen for pediatric patients with a higher BW (≥30 kg). Didi et al. [16] concluded that 5, 7, 9 mg/kg twice-weekly and flat dosing by weight bands, categorized as follows: in patients with a BW less than 20 kg administering 100 mg, in those with a BW of 20–40 kg, 150 mg and in those with a BW greater than 40 kg, 300 mg, might be alternative strategies for candida prophylaxis.
In 13 models of adult populations, two studies [15,25] demonstrated the rationalization of approved dosages for Candida infections. EW et al. [25] corroborated extending the dosing interval to 300 mg once weekly (3 h infusion) for both prophylaxis and treatment of Candida infections was equivalent to the approved dosage. One study [15] proposed that a dosage of 200-250 mg/d should be initiated to enhance the likelihood of a favorable outcome for Aspergillus infections. In another study, Roeland et al. [24] demonstrated that the current standard dosage is insufficient for obese patients weighing ≥125 kg. Six studies [10,11,26,27,28,29] evaluated the PTA of various intermittent dosing scenarios versus daily dosing regimens in adult ICU patients. Two studies [10,11] indicated that the current dosages were adequate to sustain a PTA ≥ 90% with an MIC not inferior to 0.016 mg/L. However, the daily dose should be increased to 200 mg with an MIC of 0.032 mg/L for adult ICU patients, regardless of Candida species. Zhong et al. [26] demonstrated that the daily dose for ICU patients with a high Sepsis-related organ failure assessment score (SOFA) (>10) should be further increased to 250 and 300 mg to achieve PTA targets, with attenuated MICs of 0.032 and 0.064 mg/L for C. glabrata and C. tropicalis, respectively.
3.4. Visual Predictive Distributions
The concentration-time profiles of MFG in the different virtual populations of pediatric and adult patients are shown in Figure S1 and Figure 3, respectively. First, the model established by Smith et al. [19] demonstrated a greater BSV than any other pediatric subgroup. This may be attributed to the low BW and relatively small sample size, with the added complexity of sourcing samples from critically ill pediatric patients, further amplifying the variability. The inclusion of only one neonatal model precluded intragroup comparison. Second, for children older than four months, the model established by Didi et al. [16] exhibited higher peak concentrations of MFG than the other models within each pediatric group. For adults, all included models displayed similar pharmacokinetic profiles, except for the model by Zhong et al. (2021) [26], which showed higher peak and trough drug concentrations than the other models.

Table 1.
Study characteristics.
| Study (Year) | Study Type | Country /Race |
Study Population | No. of subjects (M/F) |
No. of Samples (Per Person) |
Age (Years) mean±sd median[range] |
Body weight (kg) mean±sd median[range] |
Dosing Regiments | Bioanalytical method [LLOQ, mg/L] |
|---|---|---|---|---|---|---|---|---|---|
| Kenji Tabata et al.(2006)[14] | Phase I, II, III | Japan | Healthy subjects Adult patients Pediatric patients |
82 97 19 |
1353(16.2) 395(4.1) 77(4) |
43.5[0.67-78.0]a 55[19–77]a 6.1±4.8[0.67-15]a |
62.8[45.1-80.6]a 50.3[28-76.4]a 22.0±14.0[7–48] |
2.5-150 mg 12.5-150 mg 1-6 mg/kg |
HPLC-FLD [0.05] |
| Kazuro Ikawa et al.(2009)[15] | prospective | Japan | adult haematology patients | 10(4/6) | 48(4.8) | 63.5+16.2 [30–79] | 55.4±10.3 [46.0–77.4] |
50–300 mg, single dose | HPLC-FLD [0.05] |
| P.B. Smith et al. (2009)[19] | Phase I | America | critically ill preterm neonates > 48 hours | 34(21/13) | NA(>5) | GTA: 26.65[23–39]c PCA: 30.45[26–39]c PTA:26.7[2–82]a |
1.185a [0.54-2.2] | 15 mg qd, 5 days 0.75 mg/kg, 1.5mg/kg, 3.0mg/kg, single dose |
HPLC-MS/MS [0.05] |
| David Andes et al.(2011)[7] | Phase III | North America, Europe, Brazil, India, Thailand, South Africa, Australia | invasive candidiasis or candidemia infectionn | 493(290/203) | NA | 55[13–89]b | 68[28–155]b | 100 -150 mg qd, 14-56 days | NA |
| Emilio Maseda et al. (2014)[27] | prospective | Spain | ICU patients | 10(8/2) | 280(28) | 72±8.2 73.5[54–83] |
69.6±6.3 70.0[61–80] |
100 mg qd | HPLC-UV [0.2] |
| William W. Hope et al. (2015)[18] | Phase I, II | America | treatment or prophylaxis against aspergillus spp. or Candida spp. | 229 | 1919(8.4) | 4 mo to <2 yrs: 1.0±0.4 2–5 yrs: 3.7 ±1.2 6–11 yrs: 9.0 ± 1.5 12–16 yrs: 14.5 ±1.5 |
4 mo to <2 yrs: 7.9 ± 1.7 2–5 yrs: 15.3 ± 4.4 6–11 yrs: 28.9 ±9.0 12–16 yrs: 54.4 ± 17.3 |
0.5, 1, 1.5, 2, 3, 4, 4.5 mg/kg qd | HPLC-FLD [0.05] |
| Lisa C. Martial et al. (2017)[10] | prospective | America | ICU patients | 20(8/12) | 356(17.8) | 68 [20–84] | 76.5 [50–134] | 100 mg qd | HPLC-UV [0.01] |
| Vincent Jullien et al. (2017)[11] | Phase III | France | ICU patients | 100(66/33) | 436(4.4) | 61.4[29.9–92.7] | 84.5[48–141] | 100 mg qd, 14 days | HPLC-FLD [0.2] |
| E. W. Muilwijk et al. (2018)[25] | Phase II | Netherlands | Adult haematology patients | 20(12/8) | ~340(17) | 59.5[38–68] | 86.6[53.5–110.1] | 300 mg q2w or 100 mg qd | HPLC-FLD [0.01] |
| Sharat Chandra et al. (2018)[17] | Phase I | America | HSCT patients and prophylaxis or treatment for fungal disease | 24(6/18) | 267(11.1) | 3.8[0.6-10.4] | 15.4[7.7-30.3] | 5 mg/kg, every 4 days | HPLC-UV [0.05] |
| Roeland E. Wasmann et al. (2019)[24] | Phase IV | Netherlands | Health volunteers (BMI 18.5–25) or obse adults (BMI ≥ 40) | 24(12/12) | ~240 (10) | 31 [22–56]d 51 [35–61]e 46 [24–54]f |
70.8 [61.5–81.5]d 156 [112–184]e 141 [126–180]f |
Morbidly obese subjects: 100 mg or 200mg Normal-weight subjects: 100 mg |
UPLC-FLD [0.01] |
| Silke Gastine et al. (2019)[28] | prospective | Germany | criticall ill adult patients | 36(24/12) | NA(≥9) | 65[22–84] | 94.5[49.9–162] | 100 mg qd | HPLC-FLD [0.1] |
| Zhong Shubai et al. (2019)[26] | prospective | China | Sepsis patients | 32(21/11) | 153(4.8) | 60.1 [23.0–89.0]a | 70.22a [55.0–90.0] | 100, 150, 200mg qd | HPLC-UV [0.2] |
| Iasonas Kapralos et al. (2020)[29] | prospective | Greece | critical ill patients | 14(7/7) | 210 (15) | 61±15 [31–83] | 85±22 [55–130] | 100mg qd | HPLC-FLD [0.059] |
| Saeed Alqahtani et al. (2021a)[30] | prospective | Saudi Arabia | noncancer patients | 9(6/3) | 63(7) | 51.1±19.1 | 69.8±15.7 | 100-150mg qd, two doses | HPLC-UV [0.1] |
| Saeed Alqahtani et al. (2021b)[30] | prospective | Saudi Arabia | cancer patients | 10(6/4) | 70(7) | 47.3±12.3 | 63.4±18.2 | 100mg qd, two doses | HPLC-UV [0.1] |
| Didi Bury et al. (2022)[16] | Phase IV | Netherlands | paediatric patients | 61(34/27) | ~420(>5) | 4.0[1.0–17] | 19.5[8.60–182] | 9 mg/kg (maximum 300 mg), twice-a-week | UPLC-FLD [0.01] |
this data were list as mean or mean±sd [min-max]; b this data were list as median [min-max]; c the unit is week; d classified as group for patients in normal weight administered with 100 mg micafungin; e, f classified as group for patients in obese weight administered with 100 mg, 200 mg micafungin, respectively; * expressed as BMI; PCA/weeks, Postconceptional age; GTA/weeks, Gestational age; PTA/days, Pregnancy termination age.
Table 2.
Final population pharmacokinetic parameters of included studies.
| Study(Year) | Software/ Algorithm |
Compartment | Fixed effect Parameters | Between Subject Variability |
Residual Unexplained Variability |
Model Evaluation |
Model Application | |
|---|---|---|---|---|---|---|---|---|
| Kenji Tabata et al.(2006)[14] | NONMEM FOCE-I |
2 CMT zero-order input first-order elimination |
CL(ml/min) | 13.0+0.228×(BW-2.3)×FIX+0.0345×(PLT-21.6) (IF AGE≥16, FIX=0, IF AGE<16, FIX=1) |
23.80% | 11.00% | GOF; VPC | NA |
| V(L) | 11.2 | |||||||
| Vss(L) | 20.6 | |||||||
| Q(ml/min) | 96.5 | |||||||
| Kazuro Ikawa et al.(2009)[15] | NONMEM FOCE-I |
2 CMT zero-order input first-order elimination |
CL(L/h) | 0.762 | 15.40% | 0.642 mg/L | GOF, boostrap | Assessment of micafungin regimens based on PTA of fAUC24/MIC aganist Aspergillus |
| Vc(L) | 9.25 | 24.60% | ||||||
| Vp(L) | 8.86 | 71.80% | ||||||
| Q(L/h) | 7.02 | 0 FIXED | ||||||
| P Brian Smith et al. (2009)[19] | NONMEM | two compartment zero-order input first-order elimination |
CL(L/h) | 0.0365 | 48.80% | 29.20% | NA | NA |
| FOCE | V(L) | 0.507 | 48.80% | |||||
| Vss(L) | 1.6 | 48.80% | ||||||
| Q(L/h) | 0.0316 | / | ||||||
| David Andes et al.(2011)[7] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.05×(BW/65)0.258 | 36.00% | 19.30% | GOF | Explore the relationship between clinical and micrological response based on PTA for various Candida species. |
| FOCE-I | Vc(L) | 10.2 | 28.30% | |||||
| Vp(L) | 10.3 | 50.50% | ||||||
| Q(L/h) | 6.59 | 84.50% | ||||||
| Emilio Maseda et al. (2014)[27] | NONMEM | two compartment zero-order input first-order elimination |
CL(L/h) | 0.88×(BW/70)0.75 | 20.20% | 1.30% | GOF, bootstrap, VPC |
Evaluate covariate effects; Describe PK in specific population |
| FOCE-I | 22.1% (IOV) | 0.36mg/L | ||||||
| Vc(L) | 12.5 | 8.30% | ||||||
| 28.1% (IOV) | ||||||||
| Vp(L) | 10 | 7.50% | ||||||
| 27.4% (IOV) | ||||||||
| Q(L/h) | 5.03 | / | ||||||
| William W. Hope et al. (2015)[18] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.356×(BW/21.5)0.787×(AST/50)-0.0601×(TBIL/12)-0.0492 | 28.90% | 17.69% | GOF, bootstrap | Evaluate covariate effects; Describe PK in specific population; Identify therapeutic micafungin regimens based on exposure camparible to adult. |
| FOCE-I | Vc(L) | 1.21 | 98.30% | 35.92%a | ||||
| 4.62 | 16.61% | 0.0666 mg/L | ||||||
| Q(L/h) | 5.54 | 123.20% | ||||||
| Lisa C. Martial et al. (2017)[10] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.1 | 40.10% | GOF, bootstrap, pcVPC |
Evaluate covariate effects; Opitimize dosing regimens based on PTA for various Candida species. |
|
| FOCE-I | Vc(L) | 17.6 | 73.20% | |||||
| Vp(L) | 3.63 | 37.0% (IOV) | ||||||
| Q(L/h) | 0.363 | / | ||||||
| Vincent Jullien et al. (2017)[11] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.34×(BW/84)0.59 × 1.14 (if ALB ≤25 g/L) × 0.75 (if SOFA ≥10) | 11.40% | 1.44% | GOF, bootstrap, VPC, NPDE |
Evaluate covariate effects; Analyze the PK/PD in specific population; Evaluate the PTA of current dosing regimen; Opitimize dosing regimens based on PK/PD model. |
| FOCE-I | Vc(L) | 11.8×(BW/84)0.61 × 1.14 (if ALB ≤25 g/L) | 37.81% | |||||
| Vp(L) | 7.68×(BW/84)0.67 × 1.14 (if ALB ≤25 g/L) | 15.00% | ||||||
| Q(L/h) | Q(L/h)=4.67 | 13.90% | ||||||
| EW Muilwijk et al. (2018)[25] | NONMEM | 3 CMT zero-order input first-order elimination |
CL(L/h) | 1.01×(FFM/57.18)0.75 | 21.30% | 7.71% | GOF, bootstrap, VPC |
Evaluated the PK rationale of extending the dosing interval in special population |
| FOCE-I | 9.78% (IOV) | 0.0878 mg/L | ||||||
| V1(L) | 6.26×(FFM/57.18)1 | 48.10% | ||||||
| V2(L) | 6.26×(FFM/57.18)1 | 48.10% | ||||||
| V3(L) | 6.26×(FFM/57.18)1 | 48.10% | ||||||
| 0.809b | ||||||||
| Q1(L/h) | 10.3×(FFM/57.18)0.75 | / | ||||||
| Q2(L/h) | 2.04×(FFM/57.18)0.75 | / | ||||||
| Sharat Chandra et al. (2018)[17] |
NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.78×(BW/70)0.75 | 20.50% | 18% | GOF, pcVPC, boostrap |
Describe PK in specific population; Evaluated the PK rationale of extending the dosing interval of micafungin. |
| FOCE-I | Vc(L) | 13.9×(BW/70) | 31.20% | 0.15mg/L | ||||
| Vp(L) | 5.9×(BW/70) | 0 | ||||||
| Q(L/h) | 1.1×(BW/70)0.75 | 78.30% | ||||||
| Roeland E. Wasmann et al. (2019)[24] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.690×(BW/70)0.74 | 8.10% | 5% | GOF, pcVPC, boostrap |
Evaluate covariate effects; Describe PK in specific population; Opitimize dosing regimens based on PTA in special populations. |
| FOCE-I | Vc(L) | 5.84×(BW/70)1.17 | 12.80% | |||||
| Vp(L) | 6.96×(BW/70)0.71 | / | ||||||
| Q(L/h) | 7.15 | / | ||||||
| Silke Gastine et al. (2019)[28] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.56×0.789 (IF TBIL >4 mg/dL) | 48.90% | 0.26% | GOF, VPC | Evaluate covariate effects; Describe PK in specific population; Evaluate the efficacy of dosing regimen. |
| FOCE-I | Vc(L) | 16.2×0.692 (IF SOFA>10) | 70% | |||||
| Vp(L) | 13.8 | / | ||||||
| Q(L/h) | 14.4 | / | ||||||
| Iasonas Kapralos et al. (2020)[29] |
NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.31 | 19.00% | 14.90% | GOF, boostrap, pcVPC |
Analyze the PK/PD in specific population; Evaluate and optimize dosage regimens. |
| FOCE-I | 45% (IOV) | |||||||
| Vc(L) | 14.2 | 18.00% | ||||||
| 27% (IOV) | ||||||||
| Vp(L) | 12.6 | 51.00% | ||||||
| Q(L/h) | 2.89 | 63.00% | ||||||
| Zhong Shubai et al. (2021)[26] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.76×e((ALT/43)x(-0.268)) | 24.10% | 1.06mg/L | GOF, VPC, Boostrap, NPDE |
Evaluate covariate effects; Evaluated the PK rationale of extending the dosing interval of micafungin. |
| FOCE-I | Vc(L) | 6.7 | 52.80% | |||||
| Vp(L) | 10.2×e(θx(-1.08)) (SOFA score <10, θ=0; SOFA score≥10, θ=1) | 78.87% | ||||||
| Q(L/h) | 4.72 | / | ||||||
| Saeed Alqahtani et al. (2021a)[30] |
Monolix | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.6 | 11.80% | 38.70% | GOF, pcVPC | Describe PK of micafungin; Analyze the PK/PD in specific population; Evaluate the PTA of different dosing regimens within cancaer or within non-cancer populatons. |
| SAEM | Vc(L) | 12 | 7.60% | 0.42mg/L | ||||
| Vp(L) | 2.77 | 20.40% | ||||||
| Q(L/h) | 0.188 | 32.10% | ||||||
| Saeed Alqahtani et al. (2021b)[30] |
Monolix | 2 CMT zero-order input first-order elimination |
CL(L/h) | 1.2 | 34.10% | 45.82% | GOF, pcVPC | Describe PK of micafungin; Analyze the PK/PD in specific population; Evaluate the PTA of different dosing regimens within cancaer or within non-cancer populatons. |
| SAEM | Vc(L) | 10.7 | 7.60% | 0.47mg/L | ||||
| Vp(L) | 3.5 | 36.80% | ||||||
| Q(L/h) | 0.144 | 32.20% | ||||||
| Didi Bury et al. (2022)[16] | NONMEM | 2 CMT zero-order input first-order elimination |
CL(L/h) | 0.678×(FFM/57.19)0.75 | 24.90% | 9% | GOF, pcVPC | Evaluated the PK rationale of extending the dosing interval in special population. |
| FOCE | 10.1% (IOV) | |||||||
| Vc(L) | 7.91×(FFM/57.19) | 34.30% | ||||||
| 87.3%c | ||||||||
| Vp(L) | 9.01×(FFM/57.19) | / | ||||||
| Q(L/h) | 3.50×(FFM/57.19)0.75 | 70.60% |
a Different proportional error applied to patients (n=25) in study 9463-CL-2101 with highly variable trough screen data; b Correlation between inter-individual variability in V1, V2, V3 and inter-individual variability in CL; c Correlation factor between V1 and CL; d ml/min; CMT, compartment; Free-drug AUC/MIC over 24h, fAUC24/MIC; FOCE, First-order conditional estimation; FOCE-I, First-order conditional estimation with interaction; SAE, Stochastic approximation expectation maximization algorithm.
3.5. Pharmacokinetic Parameters
A comprehensive comparison of the simulated CL, V1, and AUC24 of MFG at steady state is shown in Figure 4 and Figure S2-S3 respectively, and the statistical comparison results of pharmacokinetic parameters for various groups versus non-ICU adults are listed in Table 3.
Except for adolescents, significantly lower MFG CL and V1 levels were found in the other pediatric groups than in non-ICU adult patients (p < 0.05). However, the estimated median CL values to per unit BW in neonates, infants, preschool children, school children and adolescents was 1.88, 1.59, 1.37, 1.17, 1.15-folds as high as that of non-ICU adults (Median: 10.97 mL/h/kg, range: 10.36–15.03 mL/h/kg), respectively. No statistical differences were found for CL among schoolchildren, adolescents, and non-ICU adults (p > 0.05). For the ICU population, a visible heterogeneity of PK parameters could be found, the median values of CL at steady state for ICU adults (SOFA≥10) (0.91-1.24 L/h)
3.6. Covariate Effect on Pharmacokinetic Parameters
Five studies, which included body size as a significant covariate, demonstrated an effect on CL with changes exceeding 20% within the normal range. Among all pediatric models, the number of models in which BW explains more than 20% of BSV in CL was as follows: preschool children (n = 2), schoolchildren (n = 3), and adolescents (n = 2). Five of the six adult models indicated that BW affected CL that exceeded 20%. Two models [26,28] suggested that alanine aminotransferase (ALT) and total bilirubin (TBIL) levels within the corresponding ranges affected MFG clearance exceeding 20%. Only one model considered the SOFA score as a covariate for CL. Compared with the reference value, the impact of SOFA score on CL exceeded 20%. Figure 5, Figure S3 and Table S3 illustrated the impact of the significant covariates on the PPK parameters.
3.7. Analysis of Probability of Target Attainment
The PTA of MFG was simulated to assess its applicability (Figure 6 and Figure S4). For C. albicans (MIC = 0.016 mg/L), the number and proportion of models in the ICU group with a typical BW gain of ≥90% were smaller than those in the non-ICU group (2/6 (33%) vs. 4/7 (57%)). In non-ICU adults with low BW, most models (5/7) achieved PTA ≥ 90%; however, once the normal BW was exceeded, 4/7 models maintained the PTA targets. In the ICU group, only half of the models achieved PTA≥90%, and even if BW exceeded the normal range, only 2 models met the PTA criteria. The limited models met the standards for obese populations. For C. glabrata (MIC = 0.032 mg/L), except for the ICU group, neither ICU nor non-ICU adults achieved the assumed PTA target. For C. krusei (MIC = 0.25 mg/L) and C. tropicalis (MIC = 0.064 mg/L), excluding one model in either the ICU or non-ICU group under low BW, none met PTA criteria. Last, for C. parapsilosis (MIC = 2 mg/L), no population achieved PTA ≥90%. Adhering to CLSI standards, none of the models achieved a PTA of 90% at MIC breakpoints across all Candida strains. For patients with suspected or proven infection with C. albicans, sensitivity analysis indicated 100-150 mg/d and 150-200 mg/d of MFG caused relatively adequate exposure for non-ICU and ICU adults when assuming the EUCAST breakpoints. For C. tropicalis and C. glabrata, the counterparts were 200-300 mg/d and ≥250 mg/d, respectively. Nevertheless, a high dose of 300 mg/d did not produce satisfactory PTA, with MIC of 0.125-0.25 mg/L for C. krusei and 1-2 mg/L for C. parapsilosis (Table 4).
4. Discussion
Authors should discuss the results and how they can be interpreted from the perspective of previous studies and of the working hypotheses. The findings and their implications should be discussed in the broadest context possible. Future research directions may also be highlighted.
MFG is a potent echinocandin employed to prevent and treat invasive infections caused by Candida and Aspergillus. Numerous studies have investigated its PK profiles, and several studies have explored the pathophysiological factors affecting exposure variability. To date, this study is the first to systematically summarize the characteristic features of PPK modeling for MFG, and to further explore whether ICU adults should adjust dosing regimens for MFG. Most current studies indicate that age, body size, liver function, and SOFA score are the primary sources of PK variability. Moreover, multiple models have corroborated the rationale for extending dosing intervals based on PK principles. Finally, it was determined that an increase in the maintenance dose by 39%–57% was necessary for ICU adults. Additional dose adjustments were recommended following the assessment of BW, SOFA score, and cultured MICs.
BW accounted for a significant proportion of the variability in CL and volume of distribution (Vd) of MFG. The findings of our study indicate that pediatric patients exhibited lower CL and V1 values than adults. However, CL standardized to allometrically scaled weight is constant in children. Furthermore, the increased BW-adjusted CL and Vd of MFG gradually decreased with age or BW, eventually reaching a plateau comparable to that observed in adults until children exceeded 8 to 9 years of age or reached a BW of 40-50 kg. This finding aligns with the conclusions of Seibel et al. [31] and Hope et al. [18]. This age difference may be attributed to several pathophysiological factors, including reduced blood flow, altered body fat-to-lean mass ratio, and decreased total body water associated with metabolic maturation. Compared to adults, pediatric patients exhibit distinct physiological and pathological characteristics, as well as differences in drug handling processes, which result in substantial PK disparities. This divergence is pronounced in infants whose renal excretion and hepatic metabolism are not yet fully mature. These factors significantly influenced the absorption, distribution, metabolism, and excretion of MFG. These findings theoretically justify the rationale behind the dosage recommendations from approved manufacturing labels. Nevertheless, further studies are necessary to determine optimal dosing strategies for neonates.
MFG clearance was significantly higher in obese adults. Standard dosage might not achieve adequate exposure in this population, indicating the need for dose escalation. The relationship between CL and BW in obese adults is similar to that observed in healthy, normal-weight individuals. Other studies conducted on obese and normal-weight critically ill patients have also shown a comparable relationship between CL and BW [32,33]. A model established by Roeland et al. [24] demonstrated that the typical CL in obese patients weighing 150 kg was 1.76-fold higher than that in normal-weight patients. These findings suggested that a 1.76-fold daily dose may be essential to ensure sufficient PTA against Candida spp. MFG is primarily metabolized by arylsulfatase, catechol-O-methyltransferase, and cytochrome P450 in the liver [34]. The correlation between CL and BW in obese individuals may be attributed to elevated cardiac output, liver blood flow, liver size, and the potential upregulation of arylsulfatase, which primarily participates in the metabolism of sulfate-containing lipids and may be more abundant in obese individuals.
Six studies consistently indicated that ICU adults exhibit higher clearance rates than non-ICU adults, suggesting the necessity of increasing the loading or maintenance dose to achieve the desired PK/PD targets. However, there was substantial heterogeneity among these studies because of large variations. The first reason might be the sample size and capricious physiological and pathological factors among the different cohorts. However, the study with the largest variation in the ICU group was based on a Chinese population, whereas the remaining studies were from other ethnicities, suggesting racial differences in the disposition of MFG. Unfortunately, statistical comparisons among races have been insufficient.
The clearance rate of MFG in ICU patients was unaffected by CRRT or IHD. This can be explained by the high binding affinity of MFG to ALB, which prevents its passage through the filter pores during RRT. This hypothesis is consistent with previous studies that used different RRT systems [35,36]. Consequently, dosage adjustments were not required for patients undergoing CRRT or IHD. The observed variations in CL and Vc among ICU patients between different dates were primarily attributed to hemodynamic instability. One study reported that ICU patients undergoing continuous venovenous hemofiltration exhibited lower inter-individual variability in the clearance and volume of distribution [10]. This discrepancy may be attributed to variations in clinical conditions, as patients undergoing CVVH usually exhibit more stable hemodynamics under fluid retention control and diuresis.
Our study also confirmed that the SOFA score could exert a significant impact on CL, reflecting a broader range of pharmacokinetic variability than any single liver function indicator such as TIBL, ALT, and AST. it is not contradictory that ALT [26], AST [18], and TBIL [18,28] were identified as covariates of CL. This might be because sepsis can induce liver damage through hemodynamic alterations and/or direct or indirect injury to hepatocytes, and liver function represented by the aforementioned sensitive markers might only partially affect sepsis. A TBIL level > 4 mg/dL decreased CL by 21.1%, and a SOFA score > 10 reduced Vc by 30%, highlighting the comprehensive nature of the SOFA score in assessing pharmacokinetic variability. These findings indicate that although routine dosage adjustment for MFG in mild-to-moderate liver dysfunction is not required, dosage adjustment should be considered for ICU patients with severe liver dysfunction (e.g., TBIL > 4 mg/dL). Even if we account for the effect of racial differences, our study indicates at least a 39%–57% higher clearance for ICU adults than for non-ICU adults, providing evidence that an increase in maintenance dose by 39%–57% was required for patients from intensive critical units, and further dose adjustment was recommended after assessment of BW, SOFA score, liver function, and cultured MIC.
These results indicated that albumin levels had a minimal effect on the efficacy of MFG. This conclusion is supported by two lines of evidence. First, half of the included studies evaluated the covariate effects of ALB; however, only one study identified and confirmed its impact on the PK parameters of MFG. This study demonstrated that the impact of ALB on either clearance or Vc was within 14%, a range that might not exert a substantial influence on its exposure. Second, ALB serves as a marker for protein binding. Hypoalbuminemia, which is commonly caused by liver cell injury, systemic inflammation, nephritic syndrome, and malnutrition, has long been a source of perplexity for experts who believe that it might increase the unbound drug concentration, leading to enhanced efficacy and a potential risk of increased adverse reactions [37,38]. However, due to the high protein-binding rate and low hepatic extraction ratio [39], the percentage of unbound drugs is determined by the maximum binding capacity (Bmax) and equilibrium dissociation constant (KD) between the drug molecule and ALB [40]. The unbound MFG concentration remained unaltered in the presence of altered ALB levels. Only one study [26] identified ALB as a covariate of MFG CL; however, most studies employed PK/PD indicators based on total plasma concentration, which are only surrogate markers. Consequently, there was no need to modify the dosage in patients with hypoalbuminemia.
The dosage adjustment recommendations for different Candida species in our study were based on the simulation results using the EUCAST standard. The clinical relevance of the difference in MIC cut-off values between the EUCAST and CLSI breakpoints remains unknown because the EUCAST breakpoints are slightly lower than their CLSI counterparts. However, the current results indicate that for infection with C. albicans at MICs of 0.016 mg/L, 100-150 mg and 150-200 mg/d of MFG are recommended for non-ICU and ICU adults, respectively. In contrast, for C. tropicalis and C. glabrata, at MICs below their EUCAST breakpoints, the recommended doses were 200-300 mg/d and ≥ 250 mg/d, respectively. According to China-SCAN research, non-Candida albicans are more prevalent than C. albicans among ICU patients in China [41]. This finding suggests the necessity of empirically increasing the initial dose to improve clinical outcomes in this special population before reporting microbiological MICs. The simulation dosages could not achieve the assumed PTA targets for C. krusei with an MIC of 0.125-0.25 mg/L and C. parapsilosis with an MIC of 0.125-2 mg/L. Consequently, we postulate that alternative antifungal agents, such as azoles and amphotericin B, may be more efficacious. These findings will be beneficial for optimizing the dosage selection of MFG for infections caused by various Candida species in ICU patients.
This study is subject to several limitations. First, plasma exposure was a surrogate marker of the infection site. It cannot be assumed that treatment failure would occur based on PTA targets calculated by combining the drug concentration levels over time in the central compartment and microbiological MICs. Second, only one model established the covariate effects of the SOFA score on CL, weakening the reliability of the conclusion. Therefore, it is necessary to verify relevant dosing recommendations in clinical cohorts. Finally, further studies are necessary to validate dosing recommendations for neonates.
5. Conclusions
This study identified BW, liver function, and SOFA score as the primary factors influencing PK variation in MFG. One of the most significant findings of our study is the evidence that a 39%–57% increase in MFG dose was required for ICU patients compared with non-ICU patients. Furthermore, both ICU and non-ICU adults should consider adjusting their dosage regimens based on the Candida species and the corresponding MICs. These findings provide valuable insights into the optimization of MFG dosing strategies in different patient populations and clinical situations.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Concentration-time profiles of micafungin at steady state; Figure S2: Distribution of the central volume of micafungin in various typical virtual populations; Figure S3: Distribution of the AUC24 of micafungin in various typical virtual populations; Figure S4: The histogram of the amount of investigated and identified covariates in included studies; Figure S5: The probability target achievement of micafungin for ICU adults or Non-ICU adults against C. Albicans, C. glabrata, C. Krusei, C. tropicalis and C. parapsilosis in included studies; Table S1: Demographic information of simulated patients; Table S2: Checklist for literature quality when reporting a clinical pharmacokinetic study; Table S3: List of tested and significant covariates in the included models; Table S4: The effect of covariates on the range of CL in each study.
Author Contributions
Conceptualization, D.L. and Z.J.; methodology, X.L., X.L., J.M. and Z.J.; software, X.L., X.L. and Z.J.; validation, X.L. and Z.J.; formal analysis, X.L. and X.L.; investigation, X.L., J.M. and Z.J.; resources, X.L. and J.M.; data curation, X.L., X.L., J.M. and Z.J.; writing—original draft preparation, X.L., X.L. and Z.J.; writing—review and editing, X.L., D.L. and Z.J.; visualization, X.L. and Z.J.; Supervision, D.L.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated during and/or analyzed during the current study are available from the published literature.
Acknowledgments
We thank Editage (www.editage.cn) for English language editing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hashimoto, S. Micafungin: A sulfated echinocandin. The Journal of antibiotics 2009, 62, 27–35. [Google Scholar] [CrossRef]
- Denning, D.W. Echinocandin antifungal drugs. Lancet 2003, 362, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Cornely, O.A.; Bassetti, M.; Calandra, T.; Garbino, J.; Kullberg, B.J.; Lortholary, O.; Meersseman, W.; Akova, M.; Arendrup, M.C.; Arikan-Akdagli, S.; et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: Non-neutropenic adult patients. Clinical microbiology and infection : The official publication of the European Society of Clinical Microbiology and Infectious Diseases 2012, 18 (Suppl. S7), 19–37. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clinical infectious diseases : An official publication of the Infectious Diseases Society of America 2016, 62, e1–e50. [Google Scholar] [CrossRef]
- Leroux, S.; Jacqz-Aigrain, E.; Elie, V.; Legrand, F.; Barin-Le Guellec, C.; Aurich, B.; Biran, V.; Dusang, B.; Goudjil, S.; Coopman, S.; et al. Pharmacokinetics and safety of fluconazole and micafungin in neonates with systemic candidiasis: A randomized, open-label clinical trial. British journal of clinical pharmacology 2018, 84, 1989–1999. [Google Scholar] [CrossRef]
- Auriti, C.; Falcone, M.; Ronchetti, M.P.; Goffredo, B.M.; Cairoli, S.; Crisafulli, R.; Piersigilli, F.; Corsetti, T.; Dotta, A.; Pai, M.P. High-Dose Micafungin for Preterm Neonates and Infants with Invasive and Central Nervous System Candidiasis. Antimicrobial agents and chemotherapy 2016, 60, 7333–7339. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.; Ambrose, P.G.; Hammel, J.P.; Van Wart, S.A.; Iyer, V.; Reynolds, D.K.; Buell, D.N.; Kovanda, L.L.; Bhavnani, S.M. Use of Pharmacokinetic-Pharmacodynamic Analyses To Optimize Therapy with the Systemic Antifungal Micafungin for Invasive Candidiasis or Candidemia. Antimicrobial agents and chemotherapy 2011, 55, 2113–2121. [Google Scholar] [CrossRef]
- Andes, D. In vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrobial agents and chemotherapy 2003, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.R.; Diekema, D.J.; Pfaller, M.A.; Marchillo, K.; Bohrmueller, J. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. glabrata in a neutropenic murine candidiasis model. Antimicrobial agents and chemotherapy 2008, 52, 3497–3503. [Google Scholar] [CrossRef]
- Martial, L.C.; Ter Heine, R.; Schouten, J.A.; Hunfeld, N.G.; van Leeuwen, H.J.; Verweij, P.E.; de Lange, D.W.; Pickkers, P.; Brüggemann, R.J. Population Pharmacokinetic Model and Pharmacokinetic Target Attainment of Micafungin in Intensive Care Unit Patients. Clinical pharmacokinetics 2017, 56, 1197–1206. [Google Scholar] [CrossRef]
- Jullien, V.; Azoulay, E.; Schwebel, C.; Le Saux, T.; Charles, P.E.; Cornet, M.; Souweine, B.; Klouche, K.; Jaber, S.; Trouillet, J.-L.; et al. Population pharmacokinetics of micafungin in ICU patients with sepsis and mechanical ventilation. Journal of Antimicrobial Chemotherapy 2017, 72, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Kanji, S.; Hayes, M.; Ling, A.; Shamseer, L.; Chant, C.; Edwards, D.J.; Edwards, S.; Ensom, M.H.; Foster, D.R.; Hardy, B.; et al. Reporting Guidelines for Clinical Pharmacokinetic Studies: The ClinPK Statement. Clinical pharmacokinetics 2015, 54, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Jamsen, K.M.; McLeay, S.C.; Barras, M.A.; Green, B. Reporting a population pharmacokinetic-pharmacodynamic study: A journal’s perspective. Clinical pharmacokinetics 2014, 53, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Katashima, M.; Kawamura, A.; Kaibara, A.; Tanigawara, Y. Population pharmacokinetic analysis of micafungin in Japanese patients with fungal infections. Drug metabolism and pharmacokinetics 2006, 21, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, K.; Nomura, K.; Morikawa, N.; Ikeda, K.; Taniwaki, M. Assessment of micafungin regimens by pharmacokinetic-pharmacodynamic analysis: A dosing strategy for Aspergillus infections. Journal of Antimicrobial Chemotherapy 2009, 64, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Bury, D.; Wolfs, T.F.W.; Ter Heine, R.; Muilwijk, E.W.; Tissing, W.J.E.; Brüggemann, R.J. Pharmacokinetic evaluation of twice-a-week micafungin for prophylaxis of invasive fungal disease in children with acute lymphoblastic leukaemia: A prospective observational cohort study. The Journal of antimicrobial chemotherapy 2022, 77, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Fukuda, T.; Mizuno, K.; Davies, S.M.; Teusink-Cross, A.; Tarin, R.; Marsh, R.A.; Vinks, A.A.; Mehta, P.A. Micafungin antifungal prophylaxis in children undergoing HSCT: Can we give higher doses, less frequently? A pharmacokinetic study. Journal of Antimicrobial Chemotherapy 2018, 73, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.W.; Kaibara, A.; Roy, M.; Arrieta, A.; Azie, N.; Kovanda, L.L.; Benjamin, D.K., Jr. Population Pharmacokinetics of Micafungin and Its Metabolites M1 and M5 in Children and Adolescents. Antimicrobial agents and chemotherapy 2015, 59, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.B.; Walsh, T.J.; Hope, W.; Arrieta, A.; Takada, A.; Kovanda, L.L.; Kearns, G.L.; Kaufman, D.; Sawamoto, T.; Buell, D.N.; et al. Pharmacokinetics of an elevated dosage of micafungin in premature neonates. The Pediatric infectious disease journal 2009, 28, 412–415. [Google Scholar] [CrossRef]
- Duffull, S.B.; Wright, D.F. What do we learn from repeated population analyses? British journal of clinical pharmacology 2015, 79, 40–47. [Google Scholar] [CrossRef]
- Li, Z.R.; Wang, C.Y.; Zhu, X.; Jiao, Z. Population Pharmacokinetics of Levetiracetam: A Systematic Review. Clinical pharmacokinetics 2021, 60, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Castanheira, M.; Messer, S.A.; Rhomberg, P.R.; Jones, R.N. Comparison of EUCAST and CLSI broth microdilution methods for the susceptibility testing of 10 systemically active antifungal agents when tested against Candida spp. Diagnostic microbiology and infectious disease 2014, 79, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Prakash, A.; Meletiadis, J.; Sharma, C.; Chowdhary, A. Comparison of EUCAST and CLSI Reference Microdilution MICs of Eight Antifungal Compounds for Candida auris and Associated Tentative Epidemiological Cutoff Values. Antimicrobial agents and chemotherapy 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Wasmann, R.E.; Smit, C.; ter Heine, R.; Koele, S.E.; van Dongen, E.P.H.; Wiezer, R.M.J.; Burger, D.M.; Knibbe, C.A.J.; Bruggemann, R.J.M. Pharmacokinetics and probability of target attainment for micafungin in normal-weight and morbidly obese adults. Journal of Antimicrobial Chemotherapy 2019, 74, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Muilwijk, E.W.; Maertens, J.A.; van der Velden, W.J.F.M.; ter Heine, R.; Colbers, A.; Burger, D.M.; Andes, D.; Theunissen, K.; Blijlevens, N.M.A.; Bruggemann, R.J.M. Pharmacokinetics of extended dose intervals of micafungin in haematology patients: Optimizing antifungal prophylaxis. Journal of Antimicrobial Chemotherapy 2018, 73, 3095–3101. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhu, X.; Zhao, L.; Song, Y.; Yu, J.; Zheng, Z.; Zang, B. Optimization of Micafungin Dosage for Chinese Patients with Sepsis in the Intensive Care Unit Based on a Population Pharmacokinetic-Pharmacodynamic Analysis. Pharmaceutical research 2021, 38, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Maseda, E.; Grau, S.; Villagran, M.J.; Hernandez-Gancedo, C.; Lopez-Tofino, A.; Roberts, J.A.; Aguilar, L.; Luque, S.; Sevillano, D.; Gimenez, M.J.; et al. Micafungin pharmacokinetic/pharmacodynamic adequacy for the treatment of invasive candidiasis in critically ill patients on continuous venovenous haemofiltration. The Journal of antimicrobial chemotherapy 2014, 69, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Gastine, S.; Lanckohr, C.; Blessou, M.; Horn, D.; Fobker, M.; Bause, D.; Hempel, G.; Ellger, B. Pharmacokinetics of Micafungin in Critically Ill Patients. Scientific reports 2019, 9, 17741. [Google Scholar] [CrossRef]
- Kapralos, I.; Mainas, E.; Neroutsos, E.; Apostolidi, S.; Siopi, M.; Apostolopoulou, O.; Dimopoulos, G.; Sambatakou, H.; Valsami, G.; Meletiadis, J.; et al. Population pharmacokinetics of micafungin over repeated doses in critically ill patients: A need for a loading dose? Journal of Pharmacy and Pharmacology 2020, 72, 1750–1760. [Google Scholar] [CrossRef]
- Alqahtani, S.; Alfarhan, A.; Alsultan, A.; Alsarhani, E.; Alsubaie, A.; Asiri, Y. Assessment of Micafungin Dosage Regimens in Patients with Cancer Using Pharmacokinetic/Pharmacodynamic Modeling and Monte Carlo Simulation. Antibiotics-Basel 2021, 10, 1363. [Google Scholar] [CrossRef]
- Seibel, N.L.; Schwartz, C.; Arrieta, A.; Flynn, P.; Shad, A.; Albano, E.; Keirns, J.; Lau, W.M.; Facklam, D.P.; Buell, D.N.; et al. Safety, tolerability, and pharmacokinetics of Micafungin (FK463) in febrile neutropenic pediatric patients. Antimicrobial agents and chemotherapy 2005, 49, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Maseda, E.; Grau, S.; Luque, S.; Castillo-Mafla, M.P.; Suarez-de-la-Rica, A.; Montero-Feijoo, A.; Salgado, P.; Gimenez, M.J.; Garcia-Bernedo, C.A.; Gilsanz, F.; et al. Population pharmacokinetics/pharmacodynamics of micafungin against Candida species in obese, critically ill, and morbidly obese critically ill patients. Critical care (London, England) 2018, 22, 94. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.G.; Swancutt, M.A.; Gumbo, T. Fractal geometry and the pharmacometrics of micafungin in overweight, obese, and extremely obese people. Antimicrobial agents and chemotherapy 2011, 55, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Wasmann, R.E.; Muilwijk, E.W.; Burger, D.M.; Verweij, P.E.; Knibbe, C.A.; Bruggemann, R.J. Clinical Pharmacokinetics and Pharmacodynamics of Micafungin. Clinical pharmacokinetics 2018, 57, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Kishino, S.; Ohno, K.; Shimamura, T.; Furukawatodo, H. Optimal prophylactic dosage and disposition of micafungin in living donor liver recipients. Clinical transplantation 2004, 18, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Aoyama, T.; Matsumoto, Y.; Ogawa, F.; Yamazaki, H.; Kikuti, A.; Yamamoto, Y. Pharmacokinetics of antifungal agent micafungin in critically ill patients receiving continuous hemodialysis filtration. Yakugaku zasshi : Journal of the Pharmaceutical Society of Japan 2007, 127, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol Aspects Med 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Trainor, G.L. The importance of plasma protein binding in drug discovery. Expert Opin Drug Discov 2007, 2, 51–64. [Google Scholar] [CrossRef]
- Hebert, M.F.; Smith, H.E.; Marbury, T.C.; Swan, S.K.; Smith, W.B.; Townsend, R.W.; Buell, D.; Keirns, J.; Bekersky, I. Pharmacokinetics of micafungin in healthy volunteers, volunteers with moderate liver disease, and volunteers with renal dysfunction. J Clin Pharmacol 2005, 45, 1145–1152. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hoener, B.A. Changes in plasma protein binding have little clinical relevance. Clinical pharmacology and therapeutics 2002, 71, 115–121. [Google Scholar] [CrossRef]
- Guo, F.; Yang, Y.; Kang, Y.; Zang, B.; Cui, W.; Qin, B.; Qin, Y.; Fang, Q.; Qin, T.; Jiang, D.; et al. Invasive candidiasis in intensive care units in China: A multicentre prospective observational study. The Journal of antimicrobial chemotherapy 2013, 68, 1660–1668. [Google Scholar] [CrossRef]
Figure 1.
PRISMA flow diagram for identifying population pharmacokinetics studies of micafungin.
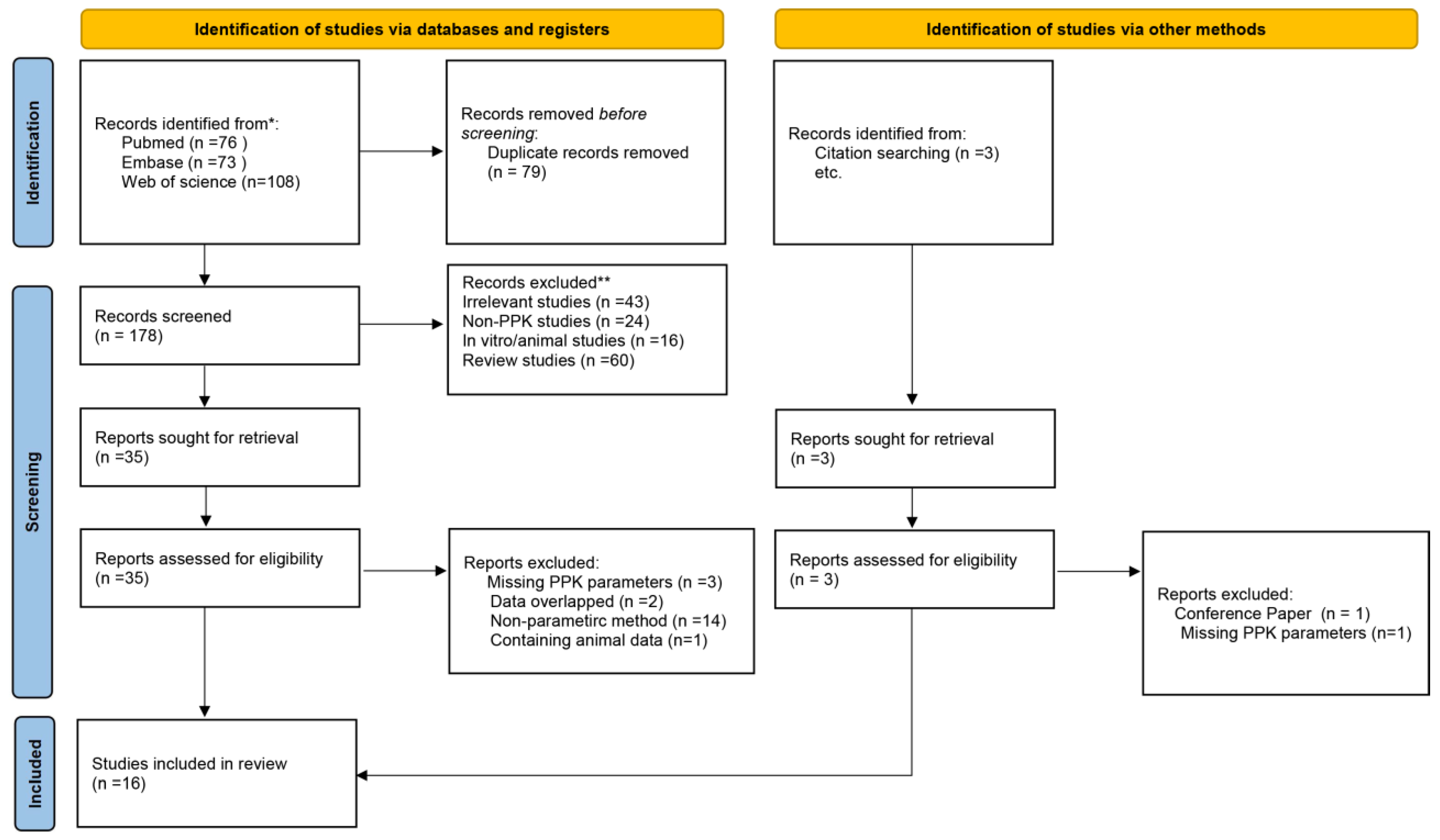
Figure 2.
Risk bias map of the included population pharmacokinetics studies.
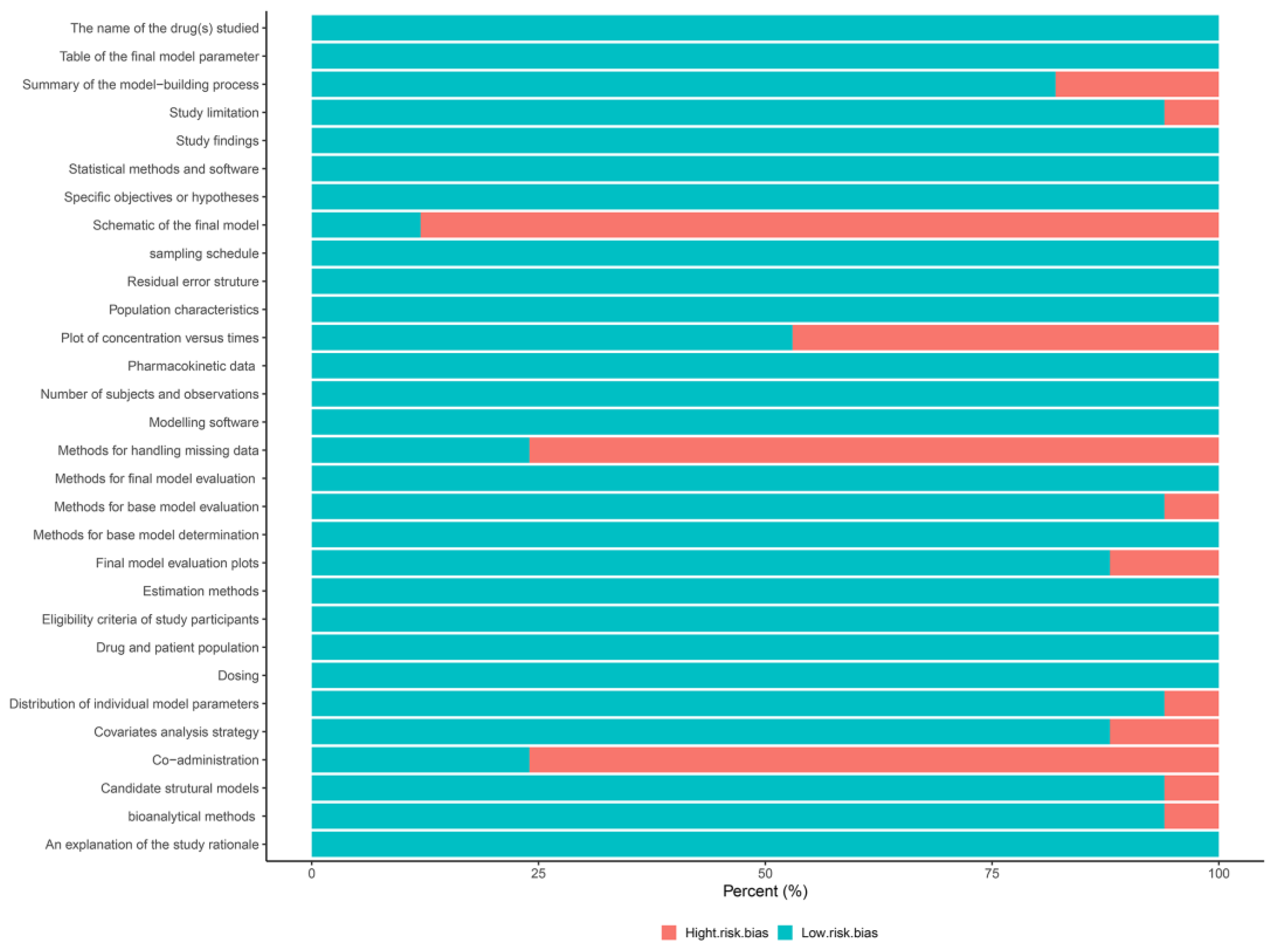
Figure 3.
Concentration-time profiles of micafungin for adults at steady state. The solid orange lines represent the median of the simulated concentration-time profiles and the light orange shadows represent the 5th–95th percentiles of the concentration-time profiles.
Figure 3.
Concentration-time profiles of micafungin for adults at steady state. The solid orange lines represent the median of the simulated concentration-time profiles and the light orange shadows represent the 5th–95th percentiles of the concentration-time profiles.
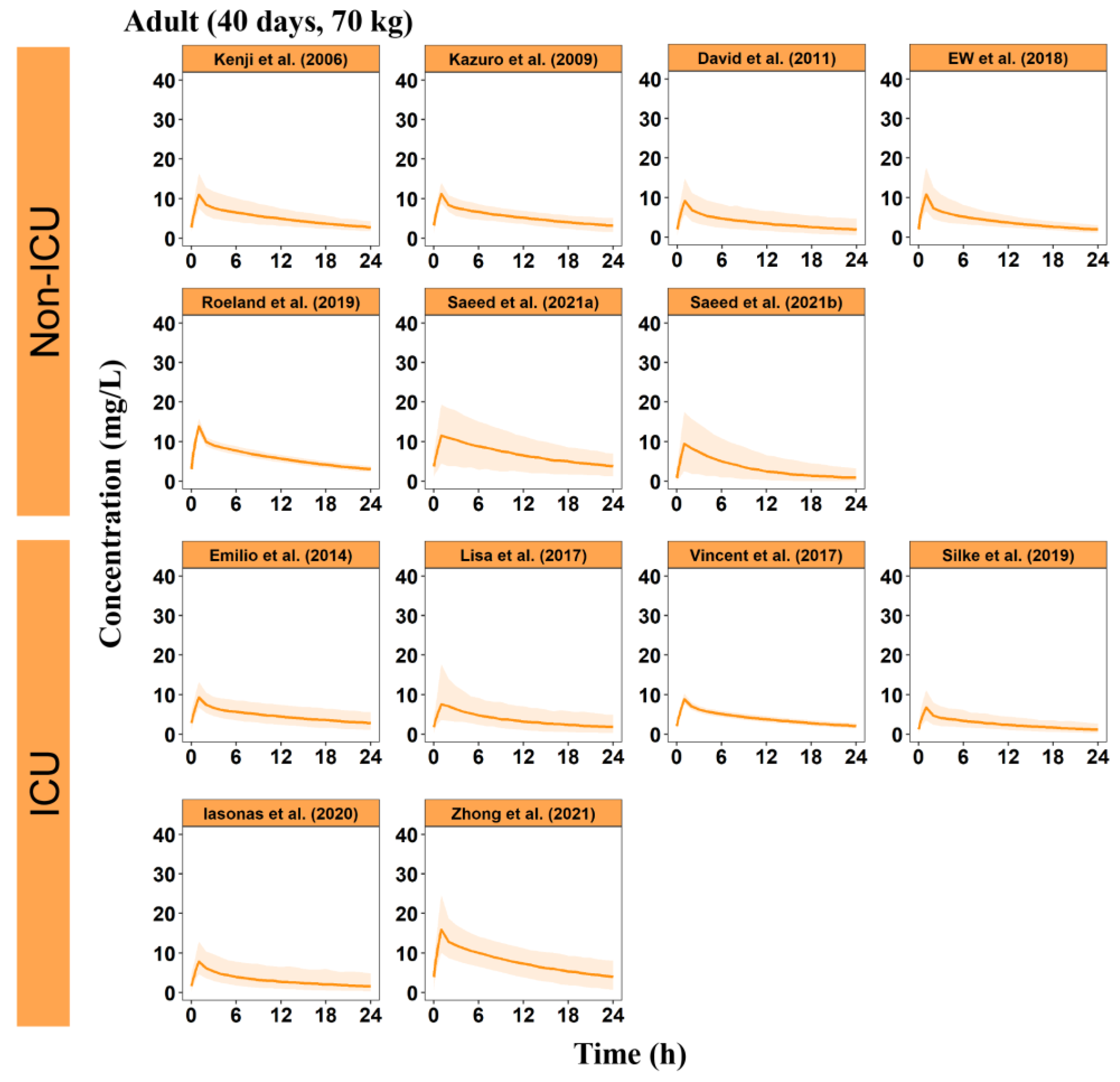
Figure 4.
Distribution of the body weight-adjusted clearance of micafungin in various typical virtual populations.
Figure 4.
Distribution of the body weight-adjusted clearance of micafungin in various typical virtual populations.
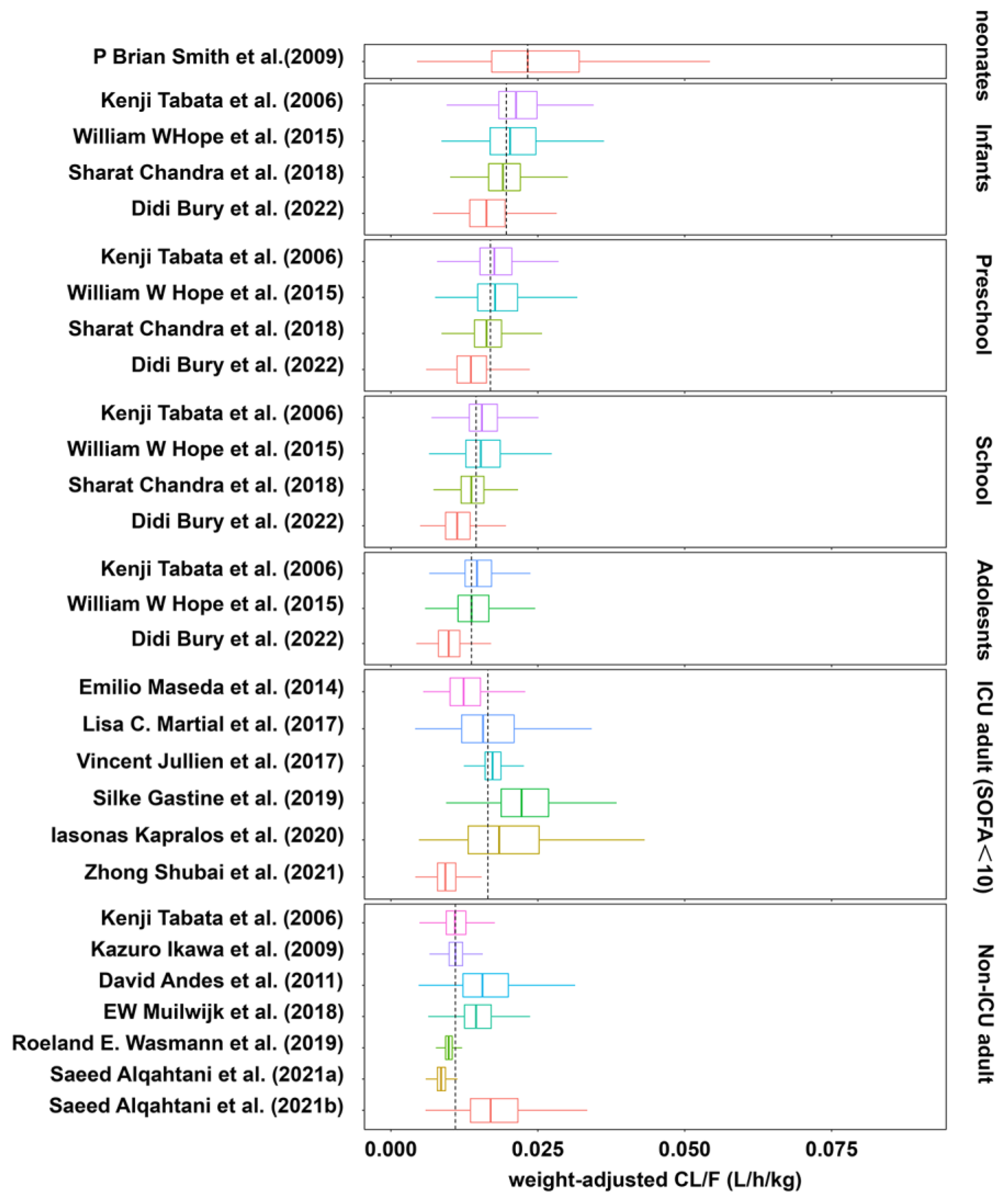
Figure 5.
The effect of covariates on CL of micafungin in included studies. FFM, free-fat mass; PLT, platelet count; ALB, albumin.
Figure 5.
The effect of covariates on CL of micafungin in included studies. FFM, free-fat mass; PLT, platelet count; ALB, albumin.
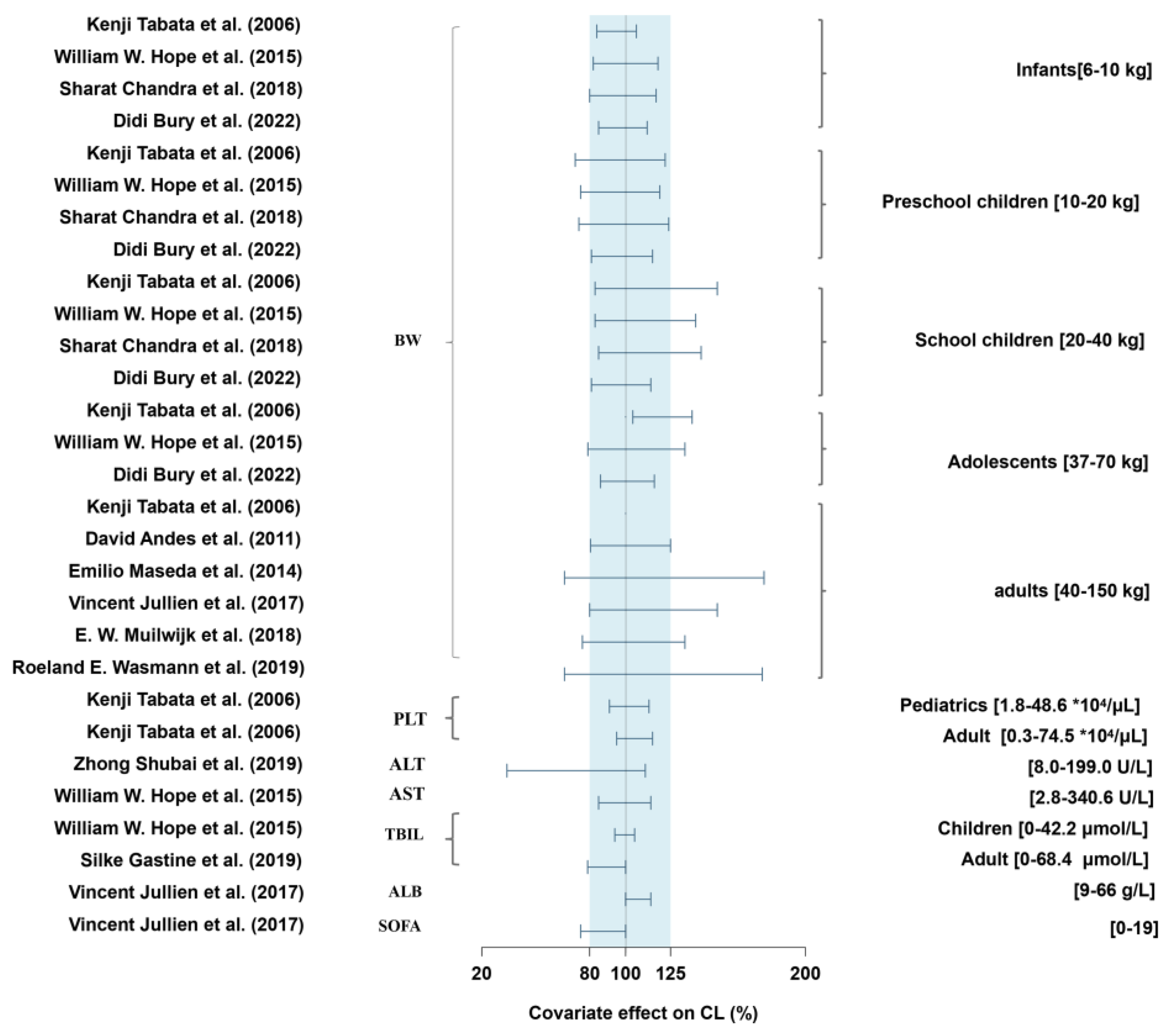
Figure 6.
The PTA of micafungin for ICU adults or Non-ICU adults against Candida spp. The MIC breakpoints for C. Albicans (blue), C. glabrata (pink), C. Krusei (black), C. tropicalis (orange) and C. parapsilosis (green) are marked with dashed and dotted lines in each panel, respectively. A PTA target of 90% is highlighted with red dashed and dotted lines.
Figure 6.
The PTA of micafungin for ICU adults or Non-ICU adults against Candida spp. The MIC breakpoints for C. Albicans (blue), C. glabrata (pink), C. Krusei (black), C. tropicalis (orange) and C. parapsilosis (green) are marked with dashed and dotted lines in each panel, respectively. A PTA target of 90% is highlighted with red dashed and dotted lines.
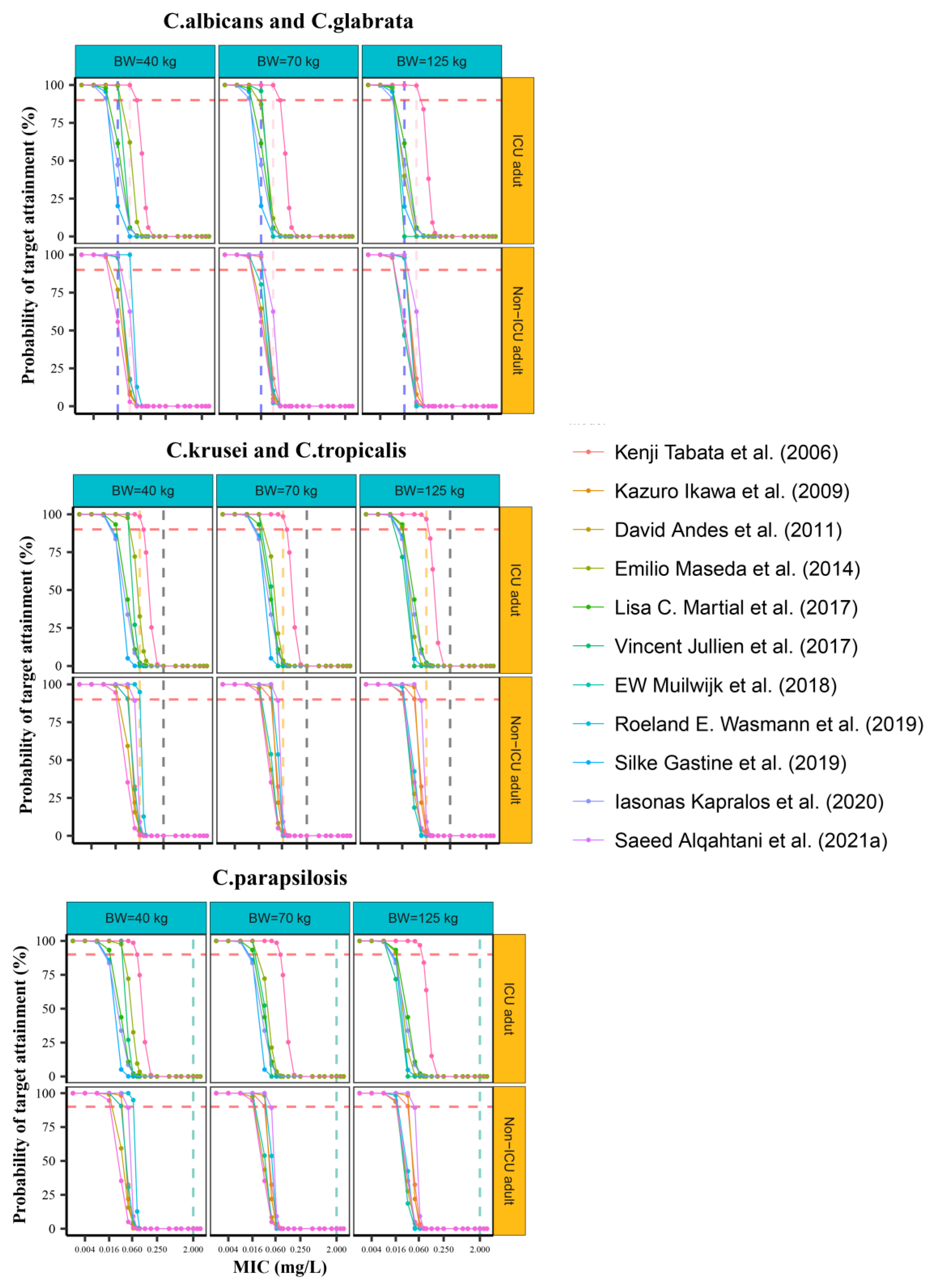
Table 3.
Statistic comparison of pharmacokinetic parameters of micafungin at steady state for various groups towards Non-ICU adults.
Table 3.
Statistic comparison of pharmacokinetic parameters of micafungin at steady state for various groups towards Non-ICU adults.
| Neonates | Infants | Preschool | School | adolescent | ICU adults (SOFA≥10) |
ICU adults (SOFA<10) |
Non-ICU adults | |
|---|---|---|---|---|---|---|---|---|
| BW-adjusted CLa (mL/h/kg) | 23.2 | 19.22 (2.17)** | 16.31 (1.91)* | 13.94 (1.95) | 12.73 (2.56) | 15.85 (4.92) | 16.47 (6.07) | 11.88 (3.03) |
| BW-adjusted CLb (mL/h/kg) | 16.70 (3.78) | 19.92 (2.60)* | ||||||
| CLa (L/h) | 0.03 | 0.16 (0.01)*** | 0.24 (0.03)*** | 0.42 (0.06)** | 0.64 (0.13) | 1.07 (0.34) | 1.10 (0.35) | 0.83 (0.21) |
| CLb (L/h) | 1.17 (0.26) | 1.20 (0.25)* | ||||||
| V1a (L) | 0.49 | 1.43 (0.48)*** | 2.24 (0.95)*** | 3.95 (2.20)** | 4.97 (3.65) | 12.67 (3.77) | 12.29 (4.20) | 8.58 (2.60) |
| V1b (L) | 13.82 (2.81)* | 14.56 (3.16)* | ||||||
| AUC24a (mg*h/L) | 162.53 | 103.96 (60.46) | 123.77 (16.10) | 145.68 (22.41) | 161.85 (36.28) | 128.19 (98.11) | 93.73 (31.87) | 126.01 (31.69) |
| AUC24b (mg*h/L) | 88.55 (19.40) | 85.72 (19.00)* |
Results from all included models, b Results from all included models except Zhong et al. (2021); All values were expressed as mean (standard error) of median values of each models; *p < 0.05, **p < 0.01, ***p < 0.001, comparing with Non-ICU adults. and ICU adults (SOFA <10) (0.92-1.27 L/h) were 39% and 43% higher than non-ICU adults (0.73-1.05 L/h), but significant difference could not be found (p > 0.05), whereas after removing the only model conducted on Chinese race [26], corresponding values for ICU adults (SOFA≥10) (1.03-1.29 L/h) and ICU adults (SOFA <10) (1.10-1.29 L/h) were 52% and 57% higher than non-ICU adults (p < 0.05). This makes it difficult to interpret the unidentified significance between the ICU and non-ICU groups.
Table 4.
Sensitivity analysis of proportions of models achieving PTA > 90% for various dosing regimens.
Table 4.
Sensitivity analysis of proportions of models achieving PTA > 90% for various dosing regimens.
| 50% | 60% | 70% | 80% | |||||
|---|---|---|---|---|---|---|---|---|
| ICU (mg/d) |
Non-ICU (mg/d) |
ICU (mg/d) |
Non-ICU (mg/d) |
ICU (mg/d) |
Non-ICU (mg/d) |
ICU (mg/d) |
Non-ICU (mg/d) | |
| C. albican | 150 | 100 | 150 | 100 | 150 | 150 | 200 | 150 |
| C. glabrata | 250 | 200 | 300 | 250 | 300 | 250 | > 300 | 300 |
| C. krusei | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 |
| C. tropicalis | 250 | 200 | > 300 | 200 | > 300 | 300 | > 300 | > 300 |
| C. parapsilosis | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 | > 300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



