
Submitted:
09 July 2024
Posted:
10 July 2024
You are already at the latest version
Abstract
Eotaxin-3 is a key chemokine with a relevant role in eosinophilic esophagitis, a rare chronic immune/antigen-mediated inflammatory disorder. Eotaxin-3 is a potent activator of eosinophil emergence and migration, which may lead to allergic airway inflammation. We investigated by bioinformatics tools the protein structure and the possible effects of the known variations reported in public databases. Following a procedure already established, we created a 3D model of the whole protein and modelled the structure of 105 protein variants due to known point mutations. The effects of the amino acid substitution at the level of impact on protein structure, stability, and possibly function, were detected by the bioinformatics procedure and described in detail. A web application was implemented to browse the results of the analysis and visualize the 3D models, with the opportunity of downloading the models and analyze by the own software. Among 105 amino acid substitutions investigated, the study evidenced in 44 cases at least one change in any of the investigated structural parameters. Other 6 variations are also relevant, although a structural effect is not detected by our analysis, because they substituted amino acids highly conserved, which suggests a possible function role. All these variations should be the object of particular attention, as they may induce loss of functionality in the protein.
Keywords:
eosinophilic esophagitis
; protein variants
; protein structural analysis
; protein modelling
; web application
1. Introduction
Eosinophilic gastroenteritis (EGE) is a rare chronic immune/antigen-mediated inflammatory disorder. EGE has historically been described as an eosinophilic disease involving more than one gastrointestinal tract, including the esophagus in 77% of cases. Recently, an international consensus has systematized the various nosographic entities and brought them together under the general term eosinophilic gastrointestinal diseases (EGID) due to the disorder's assorted clinical presentation [1]. EGID are conditions characterized by an excessive infiltration of eosinophils, which can affect any part of the digestive tract. Specific forms of EGID include eosinophilic gastritis, eosinophilic gastroenteritis, eosinophilic esophagitis (EoE), and eosinophilic colitis, depending on the anatomical tracts involved.
EoE is a chronic inflammatory condition of the esophagus commonly associated with symptoms of esophageal dysfunction. This disease is primarily caused by abnormal eosinophil infiltration in the esophageal mucosa, along with the contribution of several other types 2 inflammatory mediators [2]. EoE is considered the most common EGID. The estimated incidence in adults and children is 7.0 and 5.1 per 100,000/year, respectively, with an overall prevalence estimate of 34 per 100,000/year [3]. The disease is more common in males across all age groups, with a male-to-female ratio of 3:1. The peak incidence occurs between ages 20 and 40 years, with up to 22% of patients undergoing upper endoscopy for non-obstructive dysphagia and more than 50% of patients referred for food bolus impaction receiving a diagnosis of EoE. Yet, EoE symptoms have a significant impact on quality of life, often causing psychological distress. While EoE was initially thought to be rare, its incidence and prevalence have been rapidly increasing in both children and adults worldwide. This increase prompts the question of whether EoE is truly a new disease or simply a newly recognized condition. Besides, there is a relevant epidemiological question about whether the rise in EoE cases is due to the increased allergenicity of foods or the increased susceptibility of individuals to environmental factors. Notably, there has been a significant global increase in the prevalence of allergies in recent decades. Accordingly, most diagnosed adult patients have comorbid conditions allergic rhinitis, asthma, IgE-mediated food allergies, and atopic dermatitis [4,5], while higher rates of hypersensitivity to food antigens are found in children.
EoE diagnosis is based on three main parameters that are common across all age groups. These include symptoms of esophageal dysfunction, the presence of at least 15 eosinophils per High Power Field (HPF) or 60 eosinophils per mm2 on biopsies taken during endoscopy, and the exclusion of other non-EoE disorders that may cause or contribute to esophageal eosinophilia. These disorders include eosinophilic gastritis, gastroenteritis or colitis with esophageal involvement, gastroesophageal reflux disease (GERD), achalasia and other esophageal motility disorders, hypereosinophilic syndrome, Crohn's disease with esophageal involvement, fungal or viral infections, connective tissue disorders, hypermobility syndromes, autoimmune disorders and vasculitis, dermatologic conditions with esophageal involvement (e.g., pemphigus), drug hypersensitivity reactions, pill-induced esophagitis, "graft-versus-host disease," and Mendelian disorders. It's worth noting that only about 30% of cases show increased eosinophils in peripheral blood in laboratory tests.
EoE pathogenesis involves a complex interaction of genetic and environmental factors, including diet and antigenic load, but a full understanding remains elusive [2] with evidence suggesting that allergies play a significant role in EoE. First, special diets that remove certain foods often help. Second, EoE often occurs with other allergies like eczema and asthma. Third, researchers can cause EoE in animals by exposing them to allergens. Finally, molecules involved in allergies, like IL-5 and IL-13, seem required for the inflammation in EoE.
Twin studies indicate that the aetiology of EoE is also influenced by genetic factors, with a higher disease concordance observed in monozygotic twins compared to dizygotic twins. Overall, EoE is believed to be influenced by a combination of genetic factors, with known risk variations having a modest impact on the overall risk of the disease. The risk variants associated with EoE patients are supposed to primarily influence the regulation of gene expression and hundreds of genes show altered levels in the esophagus of affected individuals suggesting that the disorder has a complex genetic component. Furthermore, the significant variations in IL-13 in esophageal cells strongly suggest that this cytokine plays a crucial role in regulating genes associated with disease development [6].
In genome-wide association studies, genetic variants at three loci 5q22 [TSLP/WDR36], 2p23 [CAPN14], and 11q13 [LRRC32/EMSY] have consistently been identified. In other studies, significant associations were found at 12q13 (STAT6), 19q13 (ANKRD27), and 16p13 (CLEC16A). Other studies have identified PTEN, TGFBR1/TGFBR2/PBN, and IL5/IL13 as crucial risk loci for EoE. Finally, evidence from epidemiologic studies strongly suggests that EoE has a substantial genetic component, likely as a result of gene-environment interactions, particularly those involving early-life exposures [7].
A key EoE chemokine for eosinophil chemotaxis is eotaxin-3 (CCL26; C-C Motif Chemokine Ligand 26), a potent activator of eosinophil emergence and migration, potentially triggering allergic airway inflammation. Its ability to attract eosinophils is stronger as compared to other chemokines [13], representing one of the most highly induced genes in the esophageal epithelium and peripheral blood during active inflammation in EoE patients [14,15]. Similar to EoE, epithelial cells from both healthy individuals and GERD patients express eotaxin-3 upon stimulation by TH2 cytokines [16], thus embodying the role of a critical driver of eosinophil migration to the esophageal tissue. Located on chromosome 7q, the CCL26 gene encodes a protein produced by vascular endothelial and lung epithelial cells upon stimulation by IL-4 or IL-13. Eotaxin-3 belongs to a group of three chemokines that activate the CCR3 receptor, leading to calcium mobilization and the recruitment of eosinophils and basophils from peripheral blood to inflammation sites [17]. Remarkably, eotaxin-3 also exhibits antimicrobial activity. In atopic diseases, it likely contributes to eosinophil accumulation. Additionally, research suggests a role for eotaxin-3 in promoting epithelial-mesenchymal transition and even tumor growth and invasion [18].
According to information available in public databases (see below under Methods), eotaxin-3 is subjected to point variations that may alter the correct function of this protein. In the past years, we have established a bioinformatics procedure to generate an integrated database and a web application to gather and distribute information regarding predicted structural and functional impacts of genetic variations on several proteins [19], and possibly to assess their connection with the clinical outcomes of the related diseases. We freely shared these results with the scientific community through our web platform [19]. This allowed researchers worldwide to access relevant information for their specific research. We decided to develop a new web application that will also gather relevant information about eotaxin-3 and the potential effects of its amino acid known substitutions, to contribute to the understanding of the protein structure and function and possible impairment due to its single nucleotide variation with consequent amino acid substitution. We investigated the eotaxin-3 point variations listed in the UniProt database for the possible effects on protein structure and stability by integrating bioinformatics analysis of the wild-type protein structure, molecular modelling of the single amino acid substitutions and evaluation of the effects in comparison to the wild-type protein. The results are described in this article and available via a web interface for free access to any interested research team. The potential applications include the possible interest in specific variations as potential markers of disease conditions.
2. Materials and Methods
2.1. Protein models Construction
General information for eotaxin-3 sequence and its variations were retrieved by the UniProt database [20], where eotaxin-3 is reported with UniProt code Q9Y258. The PDB database [21] reports only two NMR structures (PDB code: 1G2S and 1G2T) both, as expected, lacking the first 23 residues corresponding to the signal peptide. A complete AlphaFold model [22] is instead available (identifier: AF-Q9Y258-F1), although with a low confidence score for the N- and C- terminal part of the protein. Due to the lower quality of the AlphaFold model compared to the experimental structure and the incompleteness of this latter, we decided to build by Modeller9.22 [23] novel ten 3D models exploiting as templates both the structures, the experimental one and the AlphaFold prediction. PDB structure with code 1G2S was preferred to one of the 20 NMR models present in the PDB file 1G2T because it represents their energy-minimized average structure [24]. Models were assessed by ProSA-web [25] for the calculation of the Z-score, ProCheck [26] for the analysis of the Ramachandran plot provided by SAVES v6.0 platform, Q-MEAN provided by SWISS-MODEL [27], and the RMSD calculated by PyMol [28] and using 1G2S as a reference structure. Among the ten models built, the one that reaches the best quality parameters has been chosen as the reference 3D model for the wild-type protein and has been used as the base for the variant models construction by Modeller9.22, exploiting the python script mutate_model.py (available from the Modeller web site: https://salilab.org/modeller/wiki/Mutate_model). Graphical representations of the protein have been obtained by Discovery Studio (Dassault Systèmes) and Ligplus [https://www.ebi.ac.uk/thornton-srv/software/LigPlus/].
Modelled variants are all the missense mutations available for each protein in the UniProt database in the disease and variants section (https://www.uniprot.org, accessed on 24 June 2024). In particular, 105 mutations were modelled and analyzed for eotaxin-3.
2.2. Prediction of the Variant Impact
Once the mutant models had been obtained, the impact of these variants on the structure and function of exotoxin-3 was predicted using an automated procedure for performing a series of structural analysis tools, as has been done in previous and similar work [19, 29]. More in-depth, wild-type and mutant models were analyzed and the results obtained were compared, by DSSP [30] to investigate variation in the secondary structure, HBPLUS [31] to highlight the difference in terms of H-bonds formation and NACCESS [32] to detect the change in the solvent accessibility of each residue. Furthermore, a script generated in-house allows the detection and comparison of the salt-bridge between positive and negative residues. In addition, the effect of these mutations on the stability of the protein was analysed using a consensus method, which included the results of five stability predictors releasing the one achieved by the majority. The five different web servers are MAESTROweb [33], INPS-3D [34], PoPMuSiC [35], DynaMut2 [36], and DUET [37]. When at least 3 out of 5 predictors gave the same result overcoming the cutoff of significance of each method [38] the mutation was classified as “more stable” or “less stable”, while it was classified as “uncertain” when the consensus of the predictors was not achieved.
2.3. Database and Web Interface
The results of all the analyses made on wild-type and mutant models were stored in a database, freely accessible through a web interface accessible at the following URL: http://www.protein-variants.eu/eotaxin-3-protein-db/. In addition, the database collects information about the nucleotide substitution determining the variation (from the UniProt entry), exon organization (from NCBI Reference Sequence entry: NG_015989.1), and the amino acid conservation score (calculated by the Consurf server [39]). The web application is a generalization of the tool described in our previous works [19, 29].
3. Results and Discussion
3.1. Protein Modelling of Human Eotaxin-3 and Its Variants
The study of human eotaxin-3 experimental structures found on RCSB PDB highlighted several missing residues in the N-terminal region corresponding to the signal peptide. Therefore, in order to obtain a complete 3D structure of the protein, a full chain model was created, based on the structure identified by the PDB code 1G2S, and the predicted AlphaFold model: AF-Q9Y258-F1. The model obtained (Figure 1) has a complete chain that includes also the regions deleted or not visible in the experimental structures and offers, as a benefit over the AlphaFold model, quality improvement in terms of energetic and stereochemical properties (See Table 1).
The resulting model of the full wild-type eotaxin-3 was used to generate in silico the 3D models of the missense variants of the protein. Those models were then analyzed and compared to the wild-type in order to obtain information on the variants' impact, in terms of structure, stability, and possibly protein functionality. Data obtained were collected in the free database available at http://www.protein-variants.eu/eotaxin-3-protein-db/. For each variant, the database provides a detailed analysis of the structural parameters detectable in the mutated amino acid and compared with the wild-type protein, using convenient side-by-side tables. Each variant can be viewed on the web application via a 3D viewer, just as the user has the option of downloading the 3D model in PDB format and analyzing it with their own bioinformatics tools.
In the following paragraphs, the predicted effects of the main variations are described.
3.2. Overview of the Effects of Amino Acid Variations on Eotaxin-3
The 105 eotaxin-3 variants analyzed evidenced in 44 cases at least one change in any of the investigated structural parameters. Among them, 18 cases affect an amino acid with a conservation score equal to 7 or better, suggesting an important role for that residue, at the structural or functional level. Moreover, further 6 variants affect an amino acid with a conservation score equal to 7 or better, although no structural relevant change is observed by our analysis.
The overall variants analyzed in this work indicate that the secondary structure of the protein is affected only in three cases, the solvent accessibility in ten, while the salt bridges are modified with a gain only in one case. The H-bond interactions vary in 30 cases, while the predicted protein stability decreases in 22 cases. The complete summary of the impact for each variant at the level of the analyzed properties (i.e., secondary structure, solvent accessibility, stability, H-bonds, salt bridges) is reported in Supplementary Table S1.
The loss or gain of interactions as H-bonds or salt bridges can lead to a change in protein stability, and in our analysis, there are 14 cases in which the loss of stability can be related to the loss of interactions. However, there are 8 mutations predicted to affect the protein stability without any detected effect on the other parameters investigated. In further paragraphs, some of these cases are analyzed in detail.
3.3. Effects on Disulfide Bonds, Secondary Structure, Salt Bridges and H-bonds
Eotaxin-3 is characterized by the typical Greek key structure present in all the chemokines, stabilized by two disulfide bonds. In particular, the immature protein starts with a signal peptide composed of 23 amino acids, after which there is an N-loop, that will compose the N-terminal portion of the mature form, here the first two Cys are located close together (Cys33 and Cys34). A beta sheet, composed of three strands connected by two turns called 30s and 40s, follows the N-terminal portion; a final helix closes the structure. In the 30s loop, it is located at the third Cys (Cys57), while the fourth Cys (Cys73) is located at the end of the third strand in the third loop connecting the last strand to the helix, named 50s loop. In the case of eotaxin-3, the two disulfide bonds occur between Cys33-Cys57 and Cys34-Cys73. The preservation of this structure has a crucial role in the correct recognition of the chemokines by the specific receptors [40]. In our analysis, there are one variation that substitutes Cys33, four substitute Cys34, and one substitutes Cys73, resulting in all six cases in the loss of a disulfide bond, with a potential destabilizing effect on the protein stability. On the other side, five amino acid substitutions add a new Cys in the protein, with a potential effect on the protein stability in terms of the opportunity of creating unexpected disulfide bonds, with an effect similar to the “ruffled” conformation of the Anfinsen’s experiment. Two of these five variants do not evidence any other effect on the other parameters investigated.
Only three mutations affect the secondary structure and address two residues located one after another, i.e. Leu15 and Leu16. In the details, Leu15His, Leu16Pro, and Leu16Met affect the helix constituting the signal peptide that in physiological conditions is cleaved from the mature portion of the protein during its secretion from the cell. Perturbation of the correct conformation of this signal peptide could lead to a defect in protein activation and secretion resulting in a protein lacking its correct function.
Despite the low number of mutations that seem to affect the secondary structure, noteworthy are also the variants that insert proline in place of other residues in a portion of the structure interested by turns or loops (Ala23Pro, Ser30Pro). Actually, the backbone portion of the proline leads to a reduction of the conformational space for phi-psi angles and may induce a distortion of the interested secondary structure. Due to the particular nature of this protein, its turns and loops orientation is crucial for its function. Ala23Pro moreover is the last amino acid composing the signal peptide its altered orientation may also result in a defect in the cut of the mature form. The mutations Leu12Pro and Gln82Pro instead, although located in two alpha-helices, do not destroy the secondary structure, but it cannot be excluded that the distortion that they induce in the helices could have an impact on the protein function, as well as the loss of the backbone H-bond, due to the nitrogen peculiarity of the proline, may destabilize the helix conformation.
One mutation, Lys78Glu, seems to constitute a very significant change because it replaces a positive charge with a negative one. In particular, Lys78 in the wild-type form is located in the 50s loop and creates an H-bond with Val81. The substitution of Lys by Glu determines the formation of two novels H-bonds and one salt bridge. In particular, the mutation Lys78Glu may make H-bonds with His75, Trp80 and Val81 and create a salt bridge with H39 that in the wild-type makes no interactions. In details, Val81 interactions remain unchanged, His75 (ND1 atom) gains an interaction with Glu78 (OE2 atom) while in the wild-type has no interactions, Trp80 does not change its interactions with Lys83 and Tyr84 but gains an H-bond between its NE1 atom and OE1 atom of Glu78 (see Figure 2).
The acquisition of novel bonds by amino acids previously not directly involved in the intra-chain network is not always an improvement for the protein; actually, these amino acids could be involved in interactions with other proteins or with the CXC chemokine receptors and their engagement in other bonds could makes them less available for functional inter-chains interactions.
The most altered parameter is the H-bond interaction, with a loss or gain in 30 variations, and in 14 cases there is also a loss in terms of protein stability, due to the general contribution of H-bonds to the conformational stability.
3.4. Variations Affecting Protein Stability and Protein Function
There are 22 variants that are less stable than the wild-type protein. Among them, seven (Cys34Phe, Trp44Cys, Tyr50Asp, Ala61Thr, Phe64Cys/Leu, Ile85Thr) affect only protein stability without apparent effects on the other properties studied, ten (Cys34Ser, Pro43Ser, Thr53Ser, Val62Gly, Ile63Arg, Thr66Ala, Thr74Asn, Trp80Ser/Leu, Tyr84Asn) affect protein stability and vary their H-bond interactions, one (Val47Ala) affects only stability and solvent accessibility, while four (Cys34Tyr/Arg, Trp80Gly/Arg) affect solvent accessibility, H-bond interactions, and protein stability (Figure 3). Fourteen of these 22 variants involve buried residues; Tyr50, Trp84, and Ile85 are instead partially exposed residues, while Trp44 and Pro43 are totally exposed. Cys34Thr/Arg and Trp80Gly/Arg substitute a buried and a partially exposed residue, respectively, with totally exposed residues; while Val47Ala induces a change from buried to partially exposed.
In the case of all Cys34 mutations, the impact on protein stability can be due to the breaking of one of the two disulfide bonds. The other mutations instead map at the interface among the beta-sheet and the C-terminal alpha helix, except for Pro43, Thr53, Ala61 and Thr66. Seven variants involve the substitution of apolar residues with polar ones and three from polar not charged residues to charged ones. The possible effect on stability is therefore explained also for those variants that seem not to affect the other properties analyzed. The change in polarity jointly with the substitution of several aromatic residues at the interface by residues smaller and strongly different in shape can lead to a possible loss of buried hydrophobic interaction with a possible effect on stability and consequent subtle structural rearrangements. Structural preservation is in fact fundamental for eotaxin-3 recognition by the CCR3 receptor. Moreover, studies of site-directed mutagenesis have indicated that the N-loop region, i.e. the loop following the second cysteine, of chemokines and the 40s loop are important for receptor binding, therefore mutation on Pro43 and Thr66 could alter CCR3 binding [24].
Among the 105 missense variants analyzed, 11 show the annotation of moderate impact category assigned by NCI-TCGA (Leu12Met, Ser27Asn, Tyr37His, Arg48Gln, Ser49Arg, Thr53Ser, Arg60Trp, Ala61Val, Trp80Leu, Gln93His, Leu94Val), that indicate them as non-disruptive variants although they might affect protein functionality. Among these mutations, the ones for which our analyses reveal structural effects are Arg48Gln, Ser49Arg, Thr53Ser, and Trp80Leu. Arg48Gln seems to induce a change in solvent accessibility raising the exposition to the solvent, Ser49Arg modifies the H-bond network by creating additional H-bonds with Tyr84 and Ile85. Thr53Ser causes a loss of stability and a perturbation of the H-bond interactions. Moreover, it is a highly conserved residue together with Ala61 and Trp80. Trp80, located at the beginning of the C-terminal helix, seems to be a crucial residue, highly conserved. It is the object of four mutations, and its change has a strong impact on several features. In the wild-type, Trp80 makes hydrophobic interactions with Val81, His39, Tyr84, and Lys78 and can make an H-bond with Lys83 or Tyr84 respectively (Figure 4A). Its mutation into Gly raises the relative solvent accessibility and destroys the possible H-bond with Lys83 and all the hydrophobic interactions losing all the interaction among the C-terminal helix and the N-loop region (Figure 4B), probably essential for the structural compactness. Similarly, the mutation into Ser leads to the loss of the entire hydrophobic interactions even if it preserves the ability to make one of the two H-bonds (Figure 4C). Trp80Arg instead modifies the interaction network, losing the possible H-bond with Lys83, preserving the one with Tyr84, and maintaining only the hydrophobic interaction with His39 (Figure 4D). Trp80Leu, judged as of moderate impact by NCI-TCGA, preserves the hydrophobic interaction with His39, loses the ones with Val81, Tyr84, and Lys78 but creates a novel interaction with Pro41; however, it loses the possibility to create an H-bond with Lys83. In this case, the interaction with the N-loop seems in part preserved but there is a loss of interaction with the remaining part of the C-terminal helix giving probably more flexibility to this portion and less compactness as well (Figure 4E).
4. Conclusions
Eotaxin-3 protein is closely associated with diseases characterized by eosinophilic inflammation, playing a crucial role in the recruitment and activation of eosinophils in inflamed areas. The observation of the possible effects of amino acid variations on the protein at the structural or functional level makes it possible to highlight what variations could be of particular attention as potential markers of disease conditions. Our study highlighted a structural or functional impact for at least 50 out of 105 amino acid substitutions. Our interest in EoE led us to study this protein, but other proteins could also be studied using the same approach that aims to gain observation on the possible effects of point mutations on proteins in terms of impact on the structure and stability, and possibly function. Therefore, we plan to extend the web application to other proteins related to EoE. The integration of this information may open up new opportunities for the diagnosis and therapy of diseases related to eosinophil-rich inflammation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, A.F., G.I. and A.M.; Methodology, A.F. and A.d.A.; Formal Analysis, D.G., A.d.A. and A.F.; Investigation, D.G., A.d.A. and A.F.; Data Curation, A.F., A.M., A.d.A.; Writing—Original Draft Preparation, A.F., A.M., D.G., P.I. and G.I.; Writing—Review & Editing, A.F., A.M., D.G., A.d.G., P.I. and G.I.; Supervision, A.F.; Funding Acquisition, G.I., A.M., A.F.. All authors have read and agreed to the published version of the manuscript.
Funding
This paper was partially supported by the PRIN-2022 PNRR project (The biomarker landscape for diagnosis and follow-up of eosinophilic esophagitis: from bench to bedside; project code: P2022BCXTK). This work was also supported by the CNR project NUTRAGE FOE-2021 DBA.AD005.225. A.M. was partially supported by funds from BANCA D’ITALIA.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Acknowledgments
We thank the financial support provided by the National Research Council’s NUTRAGE project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dellon, E.S.; Gonsalves, N.; Abonia, J.P.; Alexander, J.A.; Arva, N.C.; et al. International Consensus Recommendations for Eosinophilic Gastrointestinal Disease Nomenclature. Clin Gastroenterol Hepatol 2022, 20, 2474–2484.e3. [Google Scholar] [CrossRef]
- Furuta, G.T.; Katzka, D.A. Eosinophilic Esophagitis. N Engl J Med 2015, 373, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, W.E.; Barendsen, M.E.; Warners, M.J.; van Rhijn, B.D.; Verheij, J.; et al. Emerging incidence trends of eosinophilic esophagitis over 25 years: Results of a nationwide register-based pathology cohort. Neurogastroenterol Motil 2021, 33, e14072. [Google Scholar] [CrossRef] [PubMed]
- Benninger, M.S.; Strohl, M.; Holy, C.E.; Hanick, A.L.; Bryson, P.C. Prevalence of atopic disease in patients with eosinophilic esophagitis. Int Forum Allergy Rhinol 2017, 7, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.A.; Wu, S.Z.; Ibrahim, O.; Bena, J.; Rizk, M.; et al. Prevalence of atopic comorbidities in eosinophilic esophagitis: A case-control study of 449 patients. J Am Acad Dermatol 2017, 76, 559–560. [Google Scholar] [CrossRef]
- Blanchard, C.; Mingler, M.K.; Vicario, M.; Abonia, J.P.; Wu, Y.Y.; et al. IL-13 involvement in eosinophilic esophagitis: Transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007, 120, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kuhl, J.T.; Martin, L.J.; Langefeld, C.D.; Dellon, E.S.; et al. Early-life environmental exposures interact with genetic susceptibility variants in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol 2018, 141, 632–637.e5. [Google Scholar] [CrossRef] [PubMed]
- Bagnasco, D.; De Ferrari, L.; Bondi, B.; Candeliere, M.G.; Mincarini, M.; et al. Thymic Stromal Lymphopoietin and Tezepelumab in Airway Diseases: From Physiological Role to Target Therapy. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Sims, J.E.; Williams, D.E.; Morrissey, P.J.; Garka, K.; Foxworthe, D.; et al. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J Exp Med 2000, 192, 671–680. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; et al. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front Immunol 2018, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, P.P.; Tan, Z.; Page, B.D.G.; Hedtrich, S. TSLP as druggable target - a silver-lining for atopic diseases? Pharmacol Ther 2021, 217, 107648. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Na, H.G.; Choi, Y.S.; Bae, C.H.; Song, S.Y.; et al. C-C Motif Chemokine Receptor 3-Mediated Extracellular Signal-Regulated Kinase 1/2 and p38 Mitogen-Activated Protein Kinase Signaling: Promising Targets for Human Airway Epithelial Mucin 5AC Induction by Eotaxin-2 and Eotaxin-3. Int Arch Allergy Immunol 2023, 184, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Wang, N.; Stringer, K.F.; Mishra, A.; Fulkerson, P.C.; et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006, 116, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.A.; Hoyt, K.J.; Mounzer, C.; Ivester, H.M.; Barnes, B.H.; et al. Expression profiling identifies key genes and biological functions associated with eosinophilic esophagitis in human patients. Front Allergy 2023, 4, 1239273. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.; Zhang, X.; Huo, X.; Yu, C.; Zhang, Q.; et al. Omeprazole blocks eotaxin-3 expression by oesophageal squamous cells from patients with eosinophilic oesophagitis and GORD. Gut 2013, 62, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Tanita, K.; Ohuchi, K.; Sato, Y.; Lyu, C.; et al. Increased serum CCL26 level is a potential biomarker for the effectiveness of anti-PD1 antibodies in patients with advanced melanoma. Melanoma Res 2020, 30, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Shi, H.; Liu, D.; Zhang, Z.; Luo, W.; et al. Association of CCL11, CCL24 and CCL26 with primary biliary cholangitis. International Immunopharmacology 2019, 67, 372–377. [Google Scholar] [CrossRef] [PubMed]
- d'Acierno, A.; Scafuri, B.; Facchiano, A.; Marabotti, A. The evolution of a Web resource: The Galactosemia Proteins Database 2. 0. Hum Mutat 2018, 39, 52–60. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Research 2018, 46, 2699. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; et al. The Protein Data Bank. Nucleic Acids Research 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Mayer, K.L.; Mayer, M.R.; Stone, M.J. NMR Solution Structure and Backbone Dynamics of the CC Chemokine Eotaxin-3. Biochemistry 2001, 40, 7820–7831. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available at: http://www.pymol.org/pymol.
- Biancaniello, C.; D’Argenio, A.; Giordano, D.; Dotolo, S.; Scafuri, B.; et al. Investigating the Effects of Amino Acid Variations in Human Menin. Molecules 2022, 27, 1747. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J. Satisfying Hydrogen Bonding Potential in Proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef]
- Hubbard, S.; Campbell, S.; Thornton, J. Molecular recognition: Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 1991, 220, 507–530. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P.; et al. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform 2011, 12, 151. [Google Scholar]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Del Prete, E.; Scafuri, B.; Facchiano, A. Performance of Web tools for predicting changes in protein stability caused by mutations. BMC Bioinformatics 2021, 22, 345. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Kufareva, I. Chemokines and their receptors: Insights from molecular modeling and crystallography. Current Opinion in Pharmacology 2016, 30, 27–37. [Google Scholar] [CrossRef]
Figure 1.
3D model of eotaxin-3. The image shows the backbone of the complete sequence, represented as a ribbon. The N-terminal signal peptide (yellow) is followed by the region with an N-loop, a central beta-sheet, whose beta-strands (in cyan) are connected by three turns (30s, 40s, 50s loop) and a C-terminal alpha-helix (in red). Two disulfide bonds occur between C33-C57 and C34-C73 (shown in balls and sticks representation). Image has been generated by Discovery Studio software.
Figure 1.
3D model of eotaxin-3. The image shows the backbone of the complete sequence, represented as a ribbon. The N-terminal signal peptide (yellow) is followed by the region with an N-loop, a central beta-sheet, whose beta-strands (in cyan) are connected by three turns (30s, 40s, 50s loop) and a C-terminal alpha-helix (in red). Two disulfide bonds occur between C33-C57 and C34-C73 (shown in balls and sticks representation). Image has been generated by Discovery Studio software.
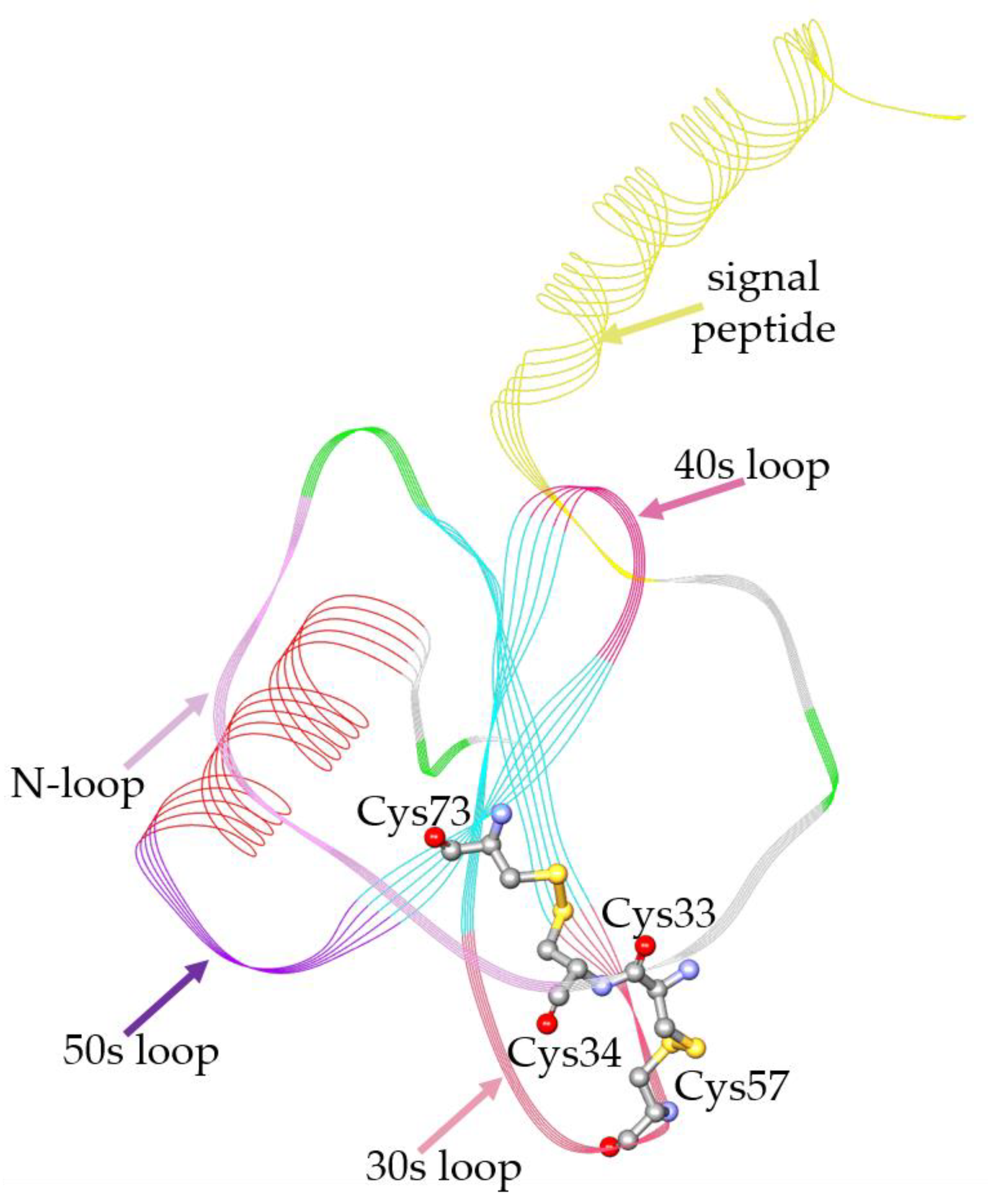
Figure 2.
Detail of the interactions formed by the wild-type residue Lys78 (panel A) and the mutated residue Glu78 (panel B). Green dotted lines correspond to H-bonds interaction, violet lines to the peptide bonds of the protein backbone, the red dotted lines to the salt bridge, while residues showed as red arcs are involved in hydrophobic interactions. Images are generated by Ligplus software.
Figure 2.
Detail of the interactions formed by the wild-type residue Lys78 (panel A) and the mutated residue Glu78 (panel B). Green dotted lines correspond to H-bonds interaction, violet lines to the peptide bonds of the protein backbone, the red dotted lines to the salt bridge, while residues showed as red arcs are involved in hydrophobic interactions. Images are generated by Ligplus software.
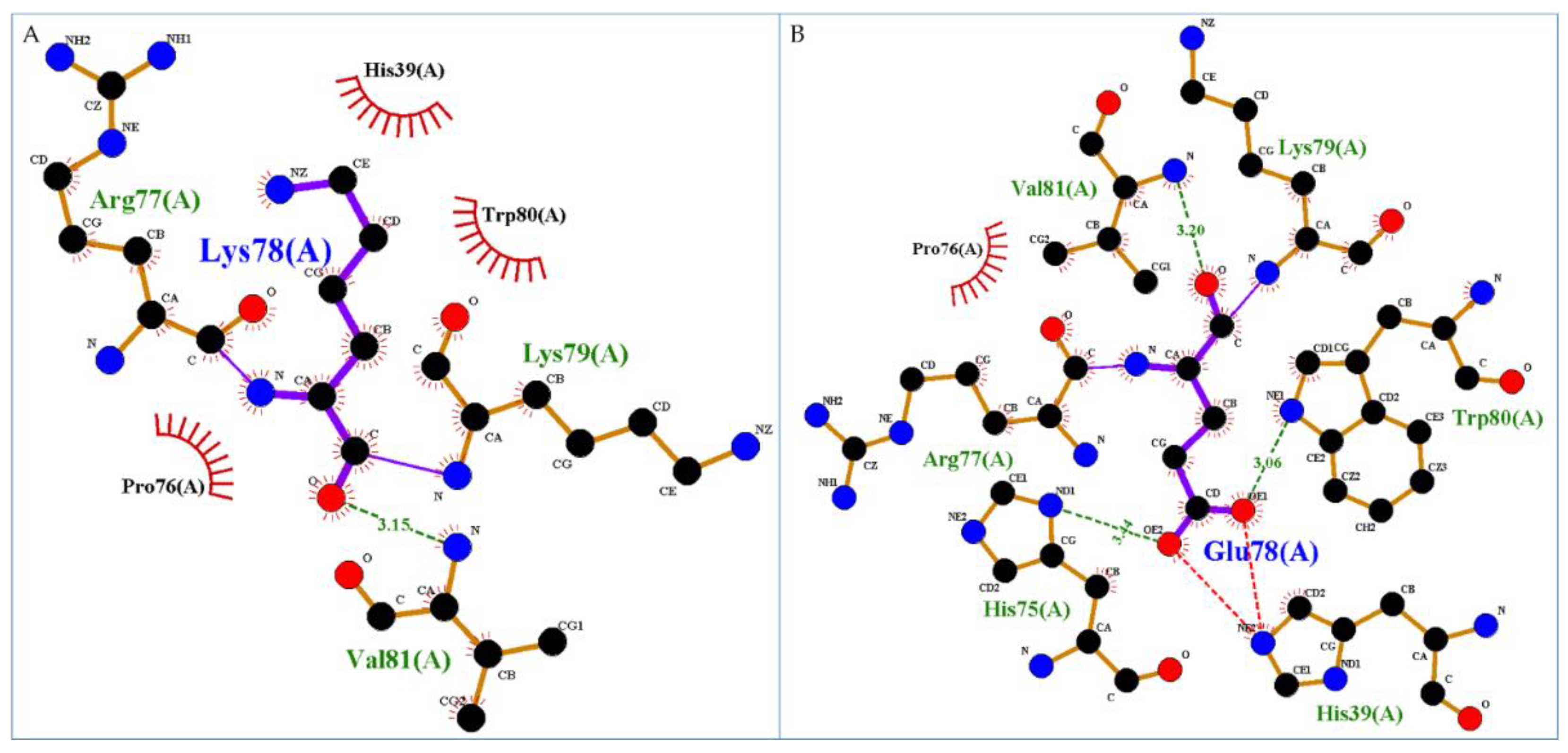
Figure 3.
Structural position in the eotaxin-3 model of wild-type residues affected by mutations predicted as destabilizing and visualization of their mutations. Affected residues are shown as magenta ball and stick models. In overlap the mutated residues are represented in sticks: green for the mutations affecting only stability, yellow for the variants involving stability and modification of the H-bond interactions, orange for mutations affecting stability and solvent accessibility, blue for those mutations with the highest impact (i.e. affecting stability, solvent accessibility and H-bond network). The disulfide bonds are highlighted in cyan sticks. Image has been generated by Discovery Studio software.
Figure 3.
Structural position in the eotaxin-3 model of wild-type residues affected by mutations predicted as destabilizing and visualization of their mutations. Affected residues are shown as magenta ball and stick models. In overlap the mutated residues are represented in sticks: green for the mutations affecting only stability, yellow for the variants involving stability and modification of the H-bond interactions, orange for mutations affecting stability and solvent accessibility, blue for those mutations with the highest impact (i.e. affecting stability, solvent accessibility and H-bond network). The disulfide bonds are highlighted in cyan sticks. Image has been generated by Discovery Studio software.
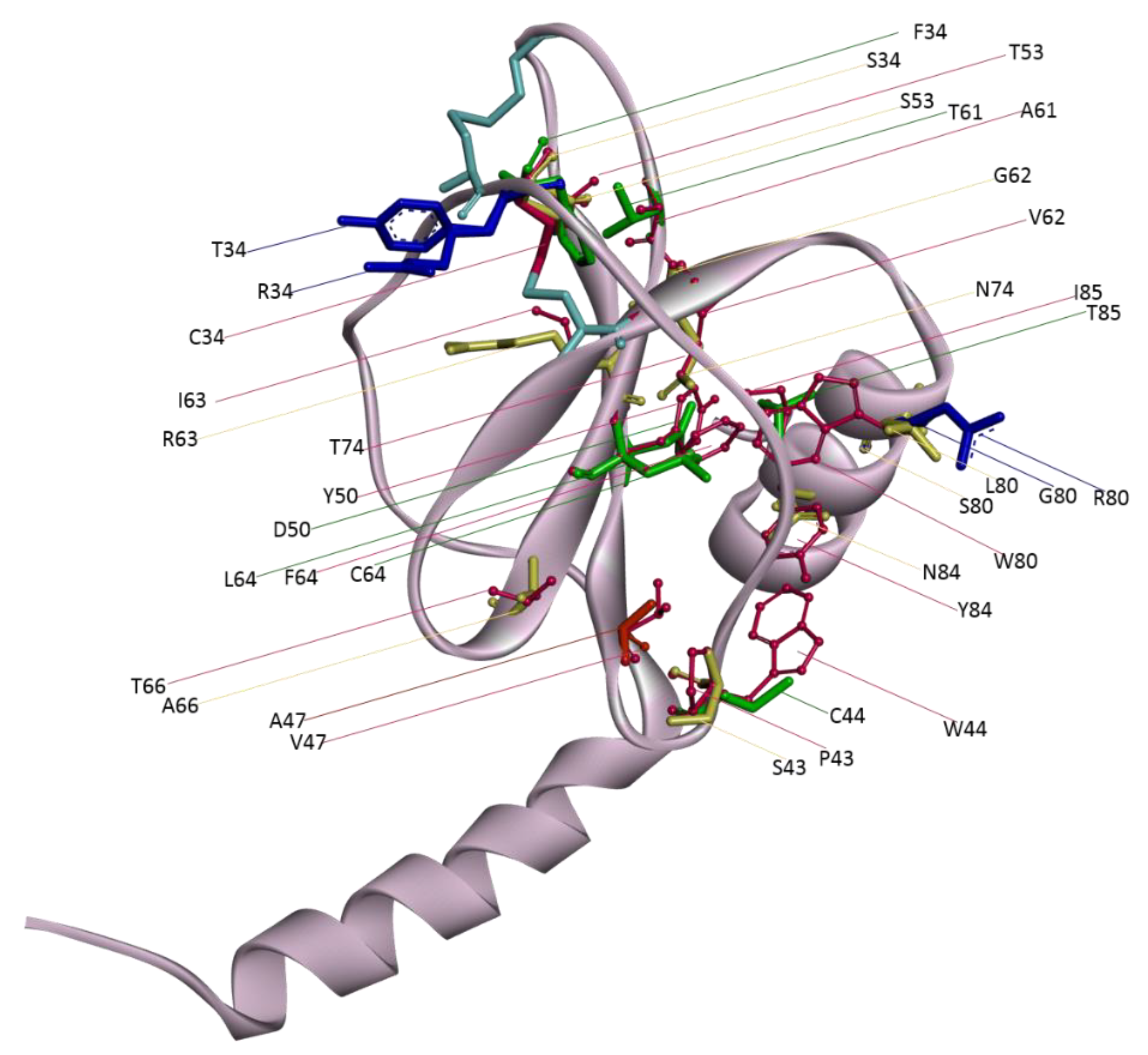
Figure 4.
Details of the intra-chain interactions formed by Trp80 (panel A) or by its substituents: i.e. Gly80 (panel B), Ser80 (panel C), Asp80 (Panel D), Leu80 (Panel E). Residue 80 is always represented in magenta sticks, while the residues interacting with W80 in the wild-type are shown in blue sticks. Residues interacting with residue 80 are labelled. Hydrophobic interactions are shown as purple dotted lines, while H-bond interactions are visible as green dotted lines. Protein is represented in line ribbon and colored by secondary structure. Image has been generated by Discovery Studio software.
Figure 4.
Details of the intra-chain interactions formed by Trp80 (panel A) or by its substituents: i.e. Gly80 (panel B), Ser80 (panel C), Asp80 (Panel D), Leu80 (Panel E). Residue 80 is always represented in magenta sticks, while the residues interacting with W80 in the wild-type are shown in blue sticks. Residues interacting with residue 80 are labelled. Hydrophobic interactions are shown as purple dotted lines, while H-bond interactions are visible as green dotted lines. Protein is represented in line ribbon and colored by secondary structure. Image has been generated by Discovery Studio software.
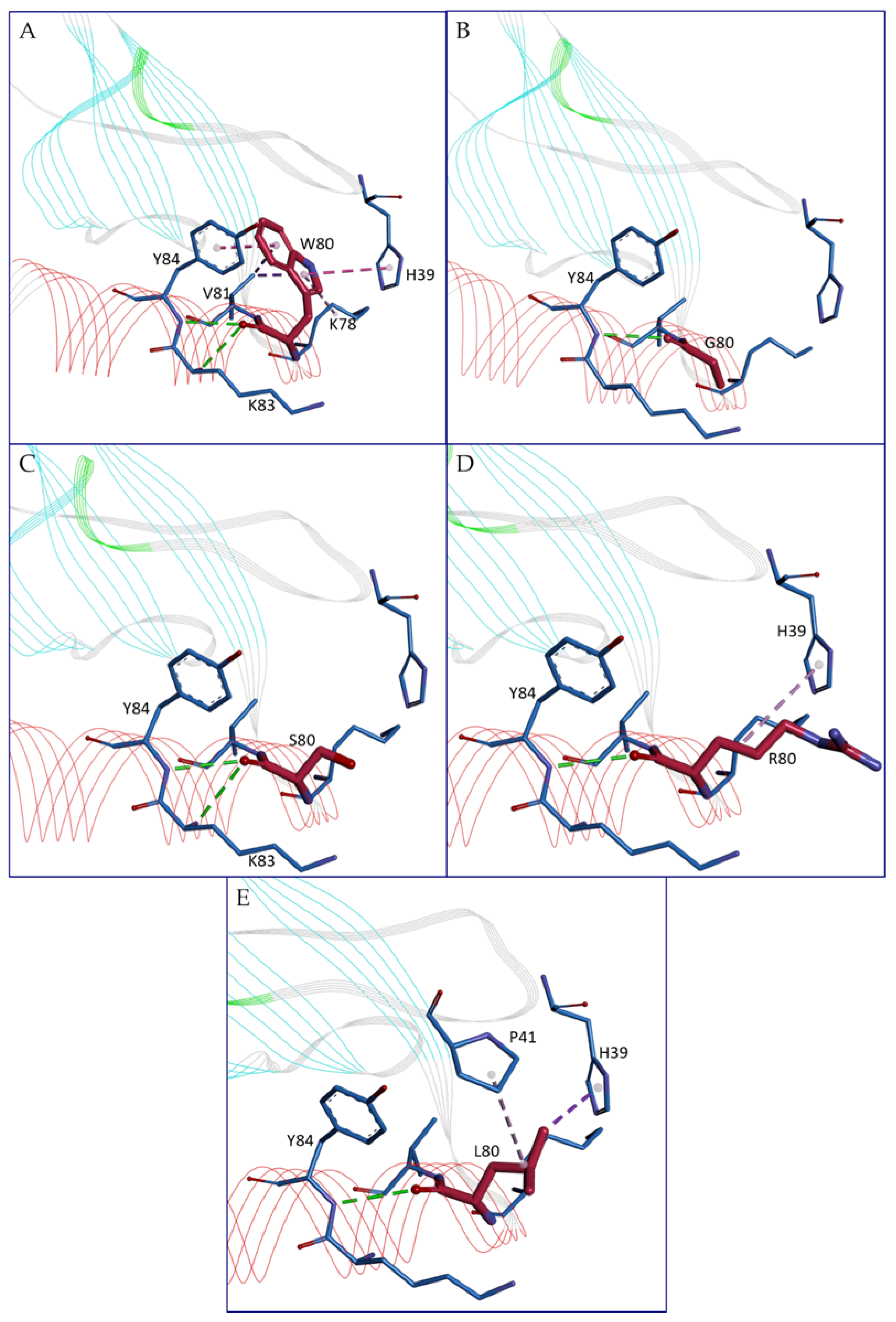

Table 1.
Quality parameters of the 3D structures of eotaxin-3. The caption “full” indicates the calculation of the parameters on the entire chain of the protein including the regions deleted or not visible in the experimental structures, the caption partial instead refers to the parameters obtained on models cut in such a way to present only the residues visible by the crystallographic structures. Tools and details for the calculation of the parameters are described in the Methods section.
Table 1.
Quality parameters of the 3D structures of eotaxin-3. The caption “full” indicates the calculation of the parameters on the entire chain of the protein including the regions deleted or not visible in the experimental structures, the caption partial instead refers to the parameters obtained on models cut in such a way to present only the residues visible by the crystallographic structures. Tools and details for the calculation of the parameters are described in the Methods section.
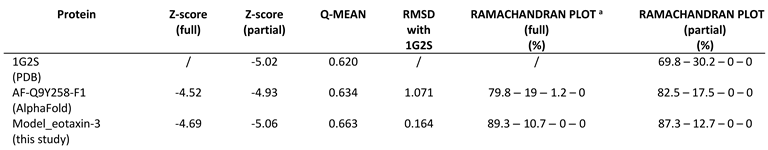 |
a The Ramachandran plot analysis by PROCHECK reports the percentage of residues with phi/psi angles in four regions defined as: most favoured – allowed – generously allowed – disallowed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



