Submitted:
10 July 2024
Posted:
11 July 2024
You are already at the latest version
Abstract
Indole derivatives are key components of natural products and possesses a wide range of biological and pharmaceutical applications. Here we present the synthesis of a new indole derivative namely 2-(1-ethyl-5-nitro-1H-indole-7-carbonyl) butyl 4-methylbenzenesulfonate (5). The structural elucidation of 5 was accomplished through comprehensive spectroscopic analysis, including Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR), and high-resolution mass spectrometry (HRMS). Our molecular docking study revealed that compound 5 exhibits strong affinity towards Tyrosinase, making it a promising candidate as an antioxidant agent. Keywords: Indole derivative; Tosyl group; Molecular docking; Antioxidant agent.
Keywords:
Indoles
; sulfonamides
; Molecular docking
; Tyrosinase
; Antioxidant
1. Introduction
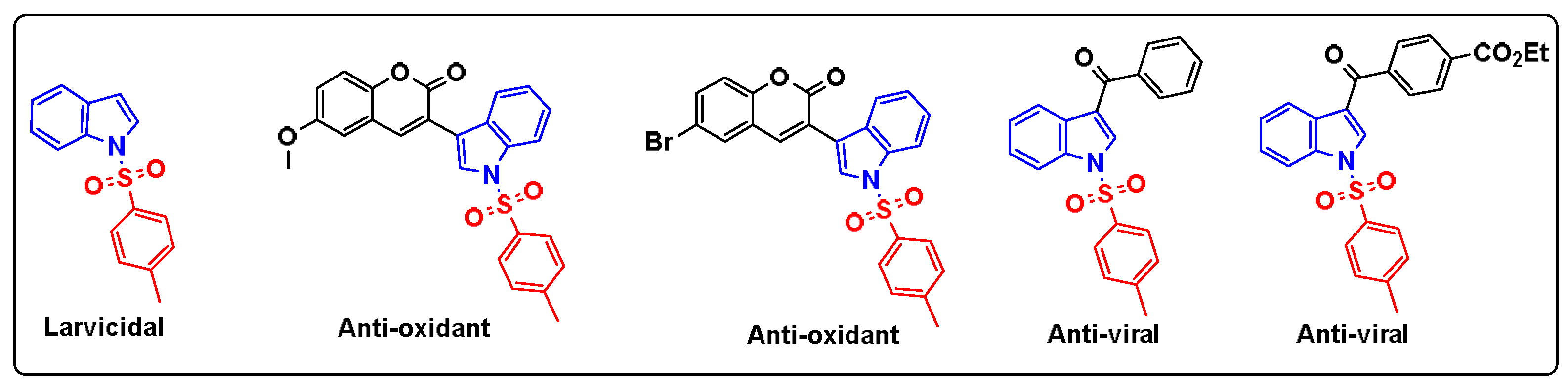
In recent years, heterocyclic compounds have become ubiquitous in pharmaceuticals, appearing in the majority of marketed drugs. Among these, indole derivatives are commonly used in a range array of pharmaceutical applications. This includes their use as anti-cancer agents [1], anti-oxidant agents [2], anti-microbial agents [3] and anti-viral agents [4]. Remarkably, the introduction of the tosyl group on the structure of indole has notably yielded numerous bioactive compounds with different activities such as Larvicidal effect [5], antioxidant activity [6] and anti-viral activity [7] (Figure 1). Based on these findings, we decided to synthesis of a novel indole derivative containing a tosyl moiety and to study its anti-oxidant activity through molecular docking.
2. Results and Discussion
2.1. Synthesis and Characterization
Our targeting tosylated compound was prepared according to the synthesis pathway in Scheme 1. The process of synthesis started with N-alkylation of 5-nitroindazole (1) using the procedure reported by Bortolozzi, R. et al [8]. to give intermediate 2 in a very good yield (98%). This intermediate reacted in the next step with butyryl chloride in the presence of aluminum chloride (AlCl3) to afford the acylated intermediate 3 in a yield of 74%. Thereafter, a condensation reaction using formaldehyde in the presence of K2CO3 was performed to yield 65% of compound 4. Lastly, the hydroxy group of 4 was easily tosylated in soft conditions using p-toluene sulfonyl chloride in the presence of triethyl amine as a base in chloroform leading to compound 5 in very good yield (99%) as illustrated in Scheme 1.
The structure of compound 5 was then structurally elucidated through spectroscopic techniques. (HRMS) determined its molecular formula to be C22H24N2O6S+, with a molecular ion peak at m/z 445.0154 [M]+. FTIR spectroscopy effectively monitored the conversion of the hydroxy group in compound 4 to the tosyl group in compound 5. In the IR spectrum of compound 4 (Figure 2A), the hydroxy group is indicated by a band at 3451.88 cm⁻¹. This band is absent in the IR spectrum of compound 5 (Figure 2B), confirming the transformation. In the 1H NMR spectrum, the presence of 8 aromatic protons appearing between 7.27 ppm and 9.29 ppm confirms the structure of 5. Furthermore, the presence of a singlet pic in the aliphatic region with an integration of 3 protons corresponding to Ar-CH3 further demonstrates the presence of the tosyl group. As depicted in Table 1 the 13CNMR experiment indicates the presence of 22 carbons, which is in accordance with the structure of 5.
2.2. Antioxidant Activity of Compound 5:
In order to investigate the antioxidant activity of compound 5, we used the crystal structure of tyrosinase from Bacillus megaterium (PDB ID: 3NM8), retrieved from the RSCB protein data bank (https://www.rcsb.org/). This enzyme is a copper-containing glycoprotein recongized for the oxidation of phenol to o-quinone[9] and tyrosine to dopaquinone [10] which is the rate-limiting step in melanin production. As illustrated in Table 2, the compound presented a docking score in the order of -10.86 Kca/mol which suggested that it could bind to the tyrosinase spontaneously indacting a good protein ligand affinity. Enzaymatic inhibition can theroritacly occurs throught the ligand occupency of the enzyme active site by non covalent interactions[11]. Accordingly, compound 5 possesses potent inhibition capacity as it effectively fits into the active site of tyrosinase and forms a plenty of favorable interactions with its amino acid residues. This includes hydrogen bond with Lys47, Π- alkyl and alkyl-alkyl with Ala 44, Ala 40, Lys 47, Thr 267, Ile 139 and Phe 48 as well as an amide- Π stacking interaction with the Ile 39 as shown in Figure 4 B and Figure 4C.
3. Materials and Methods
3.1. General Procedures
All chemicals utilized in this research were procured from commercial suppliers (Alfa Aesar, Sigma Aldrich) and were employed without further purification. The progression of reactions was tracked through thin-layer chromatography (TLC). The determination of melting points was conducted using the Stuart-SMP30 apparatus. FTIR spectra were recorded in the range of 400–4000 cm-1, using a Perkin-Elmer VERTEX 70 spectrophotometer. The 1H and 13C NMR spectra were recorded in appropriate deuterated solvent solution on an Jeol 600 MHz spectrometer at room temperature with chemical shifts reported in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal reference. Mass spectra were acquired using an Exactive Plus Orbitrap mass spectrometer coupled with an LC-MS-MS system.
3.2. Synthesis
Intermediate 2 was prepared as described in [8].
1-(1-Ethyl-5-nitro-1H-indol-7-yl) butan-1-one (3)
AlCl3 (2 mmol) was added dropwise to a solution of 2 (1.5 mmol) in DCM at 0°C and the mixture was stirred for 30 min. After allowing the reaction mixture to rise to ambient temperature, butyryl chloride (1.5 mmol) was added, and the resulting mixture was stirred for 12 h. After total consumption of 1 as indicated by TLC, the resultant mixture was quenched with water (3 mL) and extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with brine and dried over MgSO4, and the solvent was removed under vacuum. The crude was then separated by column chromatography eluting with hexane / DCM (3/2 (v/v)) to afford a yellow solid, Yield: 79%. m.p:113-114 °C. 1H NMR (CDCl3, 600 MHz) δ 9.31 (d, J = 2.3 Hz, 1H), 8.19 (dd, J = 9.1, 2.3 Hz, 1H), 7.90 (s, 1H), 7.40 (d, J = 9.0 Hz, 1H), 4.28 (q, J = 7.3 Hz, 2H), 2.85 (t, J = 7.4 Hz, 2H), 1.82 (h, J = 7.4 Hz, 2H), 1.58 (t, J = 7.4 Hz, 3H), 1.03 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 151MHz) δ 195.32, 143.88, 139.30, 135.69, 125.99, 119.95, 118.86, 118.53, 109.83, 42.32, 41.98, 18.32, 15.29, 14.10. IR (neat cm-1): ν = 1392-1529 (NO2), 1650 (C=O). HRMS (+ESI) m/z: [M+H] + calculated for C14H17N2O3: 261,1161, found, 261,0845.
1-(1-Ethyl-5-nitro-1I-indol-7-yl)-2-(hydroxymethyl) butan-1-one (4)
In a vigorously stirred solution of 3 (0.39 mmol) and formaldehyde (1.17 mmol) in EtOH (5mL), a solution of K2CO3 (0.47 mmol) in 1 mL of water and 1 mL of EtOH was added. The resulting mixture was stirred for 24 h at 60 °C. After the total consumption of the starting material, confirmed by thin-layer chromatography (TLC), the solvent was removed, and the crude was extracted three times with ethyl acetate. The resulting organic phases were combined, dried over MgSO4, and then evaporated to dryness. The crude product was subsequently separated by column chromatography, using an eluent composed of DCM/EtOAc (5/2), to obtain a yellow solid, Yield: 57%. m.p: 120-121 °C. 1H NMR (CDCl3, 600 MHz) δ 9.25 (d, J = 2.3 Hz, 1H), 8.15 (dd, J = 9.1, 1.9 Hz, 1H), 7.96 (s, 1H), 7.39 (d, J = 9.0 Hz, 1H), 4.29 (q, J = 7.4 Hz, 2H), 3.93 (ddt, J = 86.1, 10.5, 4.6 Hz, 2H), 3.26 (dd, J = 7.2, 3.9 Hz, 1H), 2.61 (s, 1H OH), 1.76 (ddt, J = 73.1, 14.1, 7.0 Hz, 2H), 1.59 (t, J = 7.4 Hz, 3H), 0.98 (t, J = 7.5 Hz, 3H). 13C NMR (CDCl3, 151 MHz) δ 199.00, 144.07, 139.46, 136.52, 126.04, 119.99, 119.12, 118.84, 109.94, 63.50, 52.03, 42.46, 22.89, 15.21, 12.21. IR (neat cm-1): ν = 1395-1528 (NO2), 1693 (C=O), 3451 (OH). HRMS (+ESI) m/z: [M+H] + calculated for C15H19N2O4: 291.1267, found, 291,0738.
2-(1-Ethyl-5-nitro-1H-indole-7-carbonyl) butyl 4-methylbenzenesulfonate (5)
To a stirred solution of 4 (0.36 mmol) and Et3N (0.72 mmol) in CHCl3 at 0°C, p-toluene sulfonyl chloride (0.54 mmol) was gradually added. The reaction was allowed to progress to room temperature and stirred for 12 h. Upon completion of the reaction, the solvent was evaporated. The crude product underwent extraction three times with DCM (6 mL) and the organic layers were combined, washed with brine, and dried over MgSO4. Subsequently, the solvent was evaporated. Column chromatography eluting with (hexane / DCM (1/2)) yielded the desired compound 9 as a white solid. Yield: 70% White solid. m.p: 151-152 °C.1H NMR (CDCl3, 600 MHz) δ 9.25 (d, J = 2.3 Hz, 1H), 8.22 (dd, J = 9.0, 2.3 Hz, 1H), 7.97 (s, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 9.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 4.35 – 4.12 (m, 4H), 3.55 – 3.40 (m, 1H), 2.43 (s, 3H), 1.82 – 1.54 (m, 5H), 0.90 (t, J = 7.5 Hz, 3H).13C NMR (CDCl3, 151 MHz) δ 194.63, 145.17, 144.10, 139.49, 136.85, 132.48, 129.96, 127.95, 126.00, 120.00, 119.25, 118.82, 110.05, 71.07, 49.35, 42.55, 22.74, 21.73, 15.24, 11.72. IR (neat cm-1): ν = 1397-1529 (NO2), 1645 (C=O). HRMS (+ESI) m/z: [M+H] + calculated for C22H25N2O3: 445.1355, found, 445.0154.
3.3. Molecular Docking
3D structure of tyrosinase (pdb id; 3NM8) was retrieved in PDB format from the RSCB protein data web site ( https://www.rcsb.org/). its preparation was accomplished by BIOVIA Discovery Studio Visualizer by removing water molecules, and unnecessary chains. Thereafter, AutoDockTools (1.5.7) allowed us to add necessary charges and to obtain the PDBQT format. As for the ligand, we used Chemdraw (16.0) to sketch the 2D structure followed by energy optimization using the MM2 algorithm within Chem 3D (16.0) software.
Docking study
Molecular docking was performed using autodock4.0 on the MGL tools 1.5.7 graphical platform. The grid parameters were set in accordance with the methodology outlined by Tamanna. N et al[9]. The best poses were then used for further visualization.
Visualization of docking results
After saving the output PDBQT file of each ligand, BIOVIA Discovery Studio Visualizer was used to study the multiple interactions established between ligands and proteins.
4. Conclusions
This communication presents the synthesis of a novel indole derivative bearing a tosyl group, namely 2-(1-ethyl-5-nitro-1H-indole-7-carbonyl) butyl 4-methylbenzenesulfonate (5). This compound was prepared in 4 steps starting from the commercially available 6-nitroindole 1 using mild conditions. The structural characterization of 5 was achieved using numerous spectroscopic methods including HRMS, FTIR, 1H and 13 NMR. Using molecular docking we showed that compound 5 exhibited good affinity to the tyrosinase (3NM8) suggesting its potent antioxidant activity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H NMR spectrum of 2, Figures S2 and S3: 1H NMR and 13C NMR spectra of 3, Figures S4 and S5: 1H NMR and 13C NMR spectra of 4, Figure S6: 13C NMR spectrum of 5; Figure S7: HRMS spectrum of 5.
Author Contributions
Conceptualization, S.E. and N.E.; methodology, S.E.; software, A.C.; validation, S.E. and N.E.; formal analysis, S.E.; investigation, A.C.; resources, S.E.; data curation, A.C.; writing—original draft preparation A.C.; writing—review and editing, S.E. and N.E.; visualization, S.E. and N.E.; supervision, S.E. and N.E.; project administration, S.E.; funding acquisition, S.E. All authors have read and agreed to the published version of the manuscript
Funding
Research carried out with the assistance of the Hassan 2 Academy of Sciences and Technologies.
Data Availability Statement
All data are included in the manuscript and the Supplementary Materials.
Acknowledgments
The authors are grateful to the Hassan II Academy of Sciences and Techniques and the Euromed University of Fes for funding. The authors are also grateful to the Euromed University of Fes for providing their facilities. AC is grateful for the Euromed University for the scholarship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Prakash, B.; Amuthavalli, A.; Edison, D.; Sivaramkumar, M.S.; Velmurugan, R. Novel Indole Derivatives as Potential Anticancer Agents: Design, Synthesis and Biological Screening. Med. Chem. Res. 2018, 27, 321–331. [Google Scholar] [CrossRef]
- Estevão, M.S.; Carvalho, L.C.; Ribeiro, D.; Couto, D.; Freitas, M.; Gomes, A.; Ferreira, L.M.; Fernandes, E.; Marques, M.M.B. Antioxidant Activity of Unexplored Indole Derivatives: Synthesis and Screening. Eur. J. Med. Chem. 2010, 45, 4869–4878. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Utreja, D.; Ekta; Jain, N.; Sharma, S. Recent Developments in the Synthesis and Antimicrobial Activity of Indole and Its Derivatives. Curr. Org. Synth. 2018, 16, 17–37. [CrossRef]
- Giampieri, M.; Balbi, A.; Mazzei, M.; La Colla, P.; Ibba, C.; Loddo, R. Antiviral Activity of Indole Derivatives. Antiviral Res. 2009, 83, 179–185. [Google Scholar] [CrossRef]
- de Jesus Santos, A.; Macêdo, N.A.; de Holanda Cavalcanti, S.C.; Sarmento, V.H.V.; Moreira Lira, A.A.; dos Santos, C.P.; La Corte Santos, R.; Souza Nunes, R. de Larvicidal Formulation Containing N-Tosylindole: A Viable Alternative to Chemical Control of Aedes Aegypti. Colloids Surfaces B Biointerfaces 2022, 213. [Google Scholar] [CrossRef]
- Wet-osot, S.; Pewklang, T.; Duangkamol, C.; Muangsopa, P.; Ngivprom, U.; Chansaenpak, K.; Ngernsoungnern, A.; Sritangos, P.; Chudapongse, N.; Lai, R.Y.; et al. N-Tosylindole-Coumarin with High Fluorescence Quantum Yield and Their Potential Applications. J. Mol. Struct. 2022, 1260. [Google Scholar] [CrossRef]
- Rana, G.; Kar, A.; Kundal, S.; Musib, D.; Jana, U. DDQ/Fe(NO3)3-Catalyzed Aerobic Synthesis of 3-Acyl Indoles and an In Silico Study for the Binding Affinity of N-Tosyl-3-Acyl Indoles toward RdRp against SARS-CoV-2. J. Org. Chem. 2023, 88, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, R.; Carta, D.; Dal Prà, M.P.; Antoniazzi, G.; Mattiuzzo, E.; Sturlese, M.; Di Paolo, V.; Calderan, L.; Moro, S.; Hamel, E.; et al. Evaluating the Effects of Fluorine on Biological Properties and Metabolic Stability of Some Antitubulin 3-Substituted 7-Phenyl-Pyrroloquinolinones. Eur. J. Med. Chem. 2019, 178, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Narsinghani, T.; Sharma, M.C.; Bhargav, S. Synthesis, Docking Studies and Antioxidant Activity of Some Chalcone and Aurone Derivatives. Med. Chem. Res. 2013, 22, 4059–4068. [Google Scholar] [CrossRef]
- Ando, H.; Kondoh, H.; Ichihashi, M.; Hearing, V.J. Approaches to Identify Inhibitors of Melanin Biosynthesis via the Quality Control of Tyrosinase. J. Invest. Dermatol. 2007, 127, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, S.; Bonneil, É.; Simpson, B.K. Generation of Antioxidative Peptides from Atlantic Sea Cucumber Using Alcalase versus Trypsin: In Vitro Activity, de Novo Sequencing, and in Silico Docking for in Vivo Function Prediction. Food Chem. 2020, 306, 125581. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Biological activities of some tosylindole derivatives.
Scheme 1.
Synthesis pathway of compound 5.
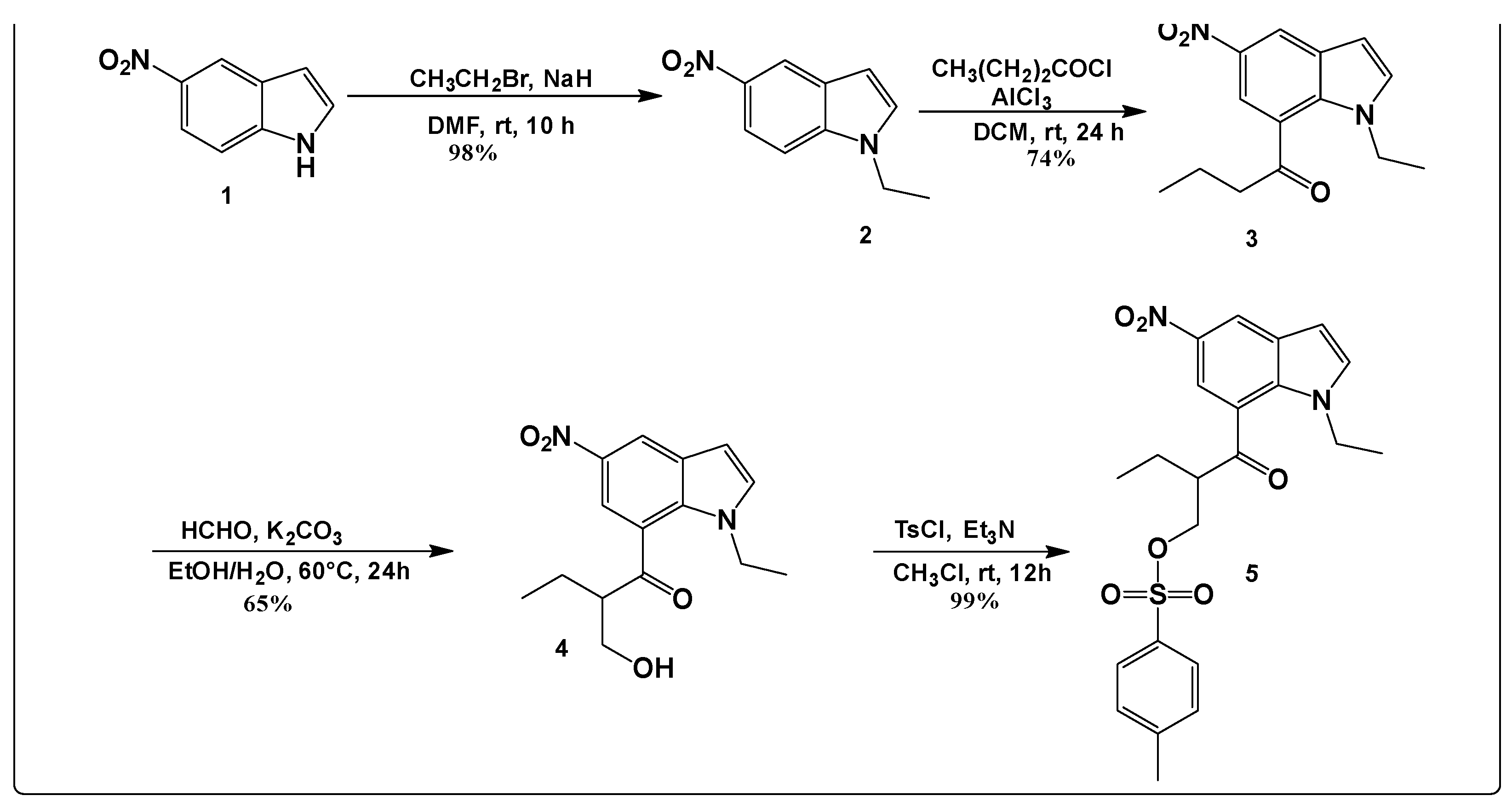
Figure 2.
Superimposition of the IR spectrums of compounds 4 and 5.
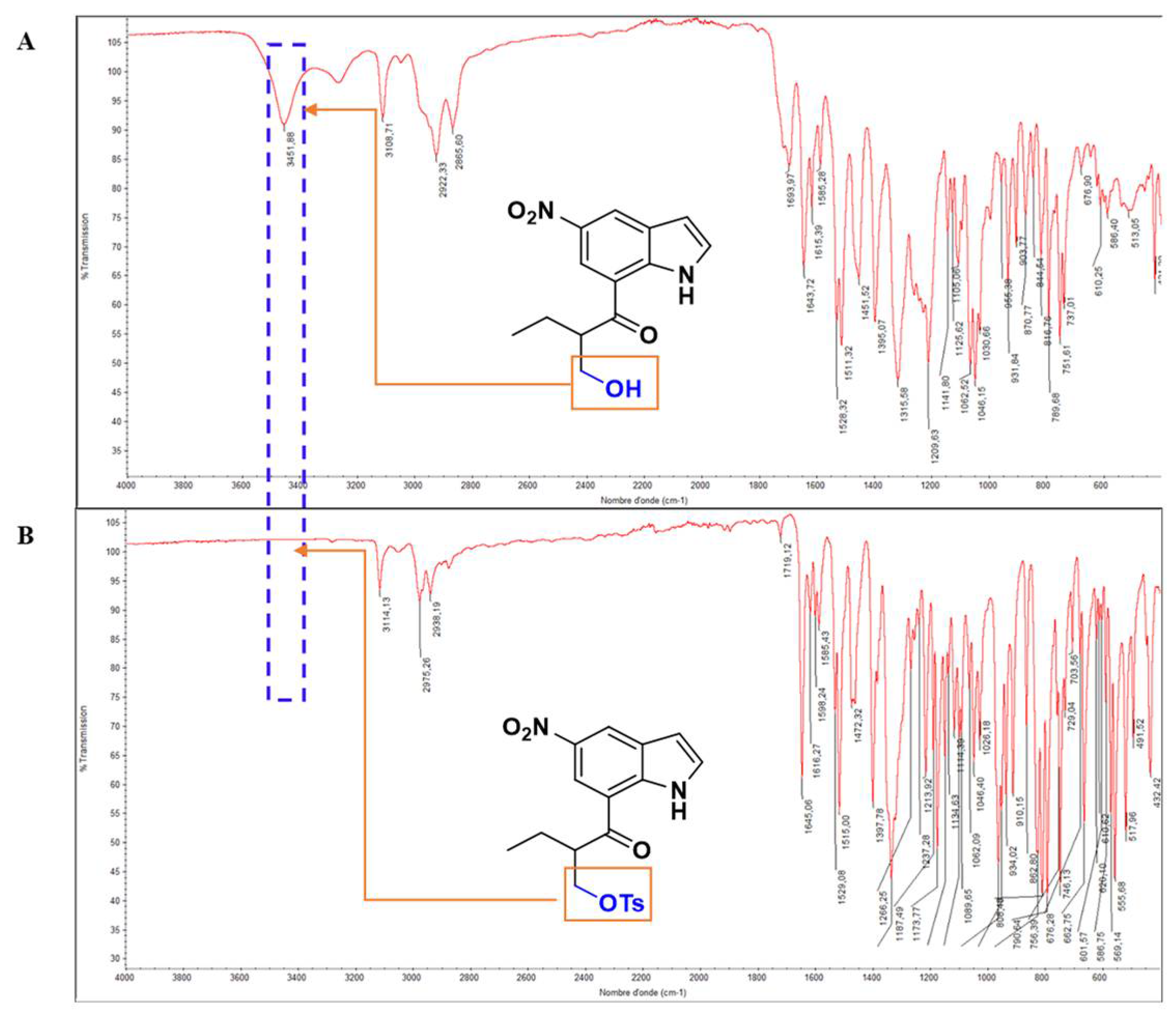
Figure 3.
1H NMR Spectrum of 5 in CDCl3.
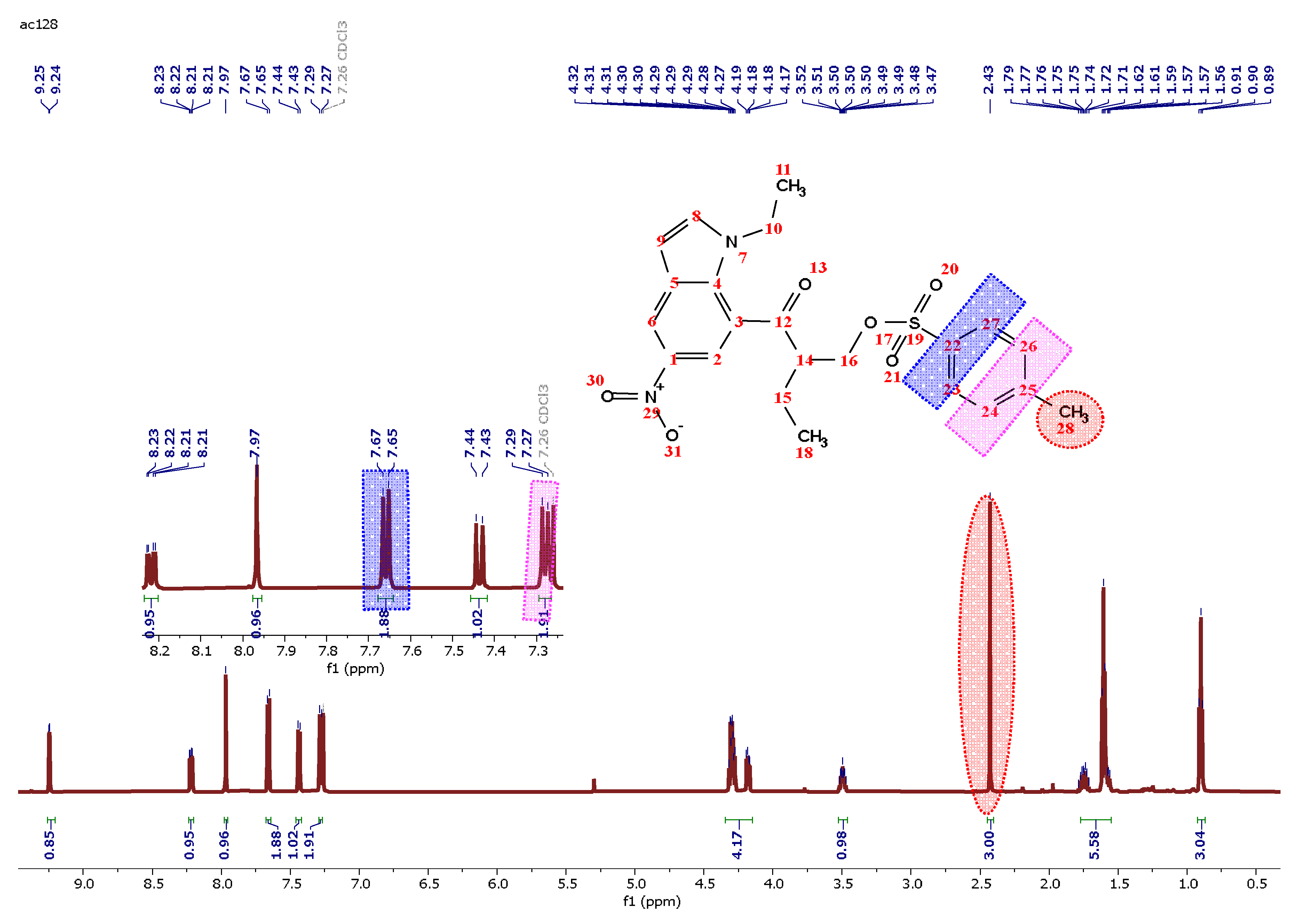
Figure 4.
(A) Compound 5 fits in the active site of tyrosinase (B) and (C) 3D and 2D representations of interaction residues (D) and (E) surface representations of 5 withing tyrosinase active site.
Figure 4.
(A) Compound 5 fits in the active site of tyrosinase (B) and (C) 3D and 2D representations of interaction residues (D) and (E) surface representations of 5 withing tyrosinase active site.
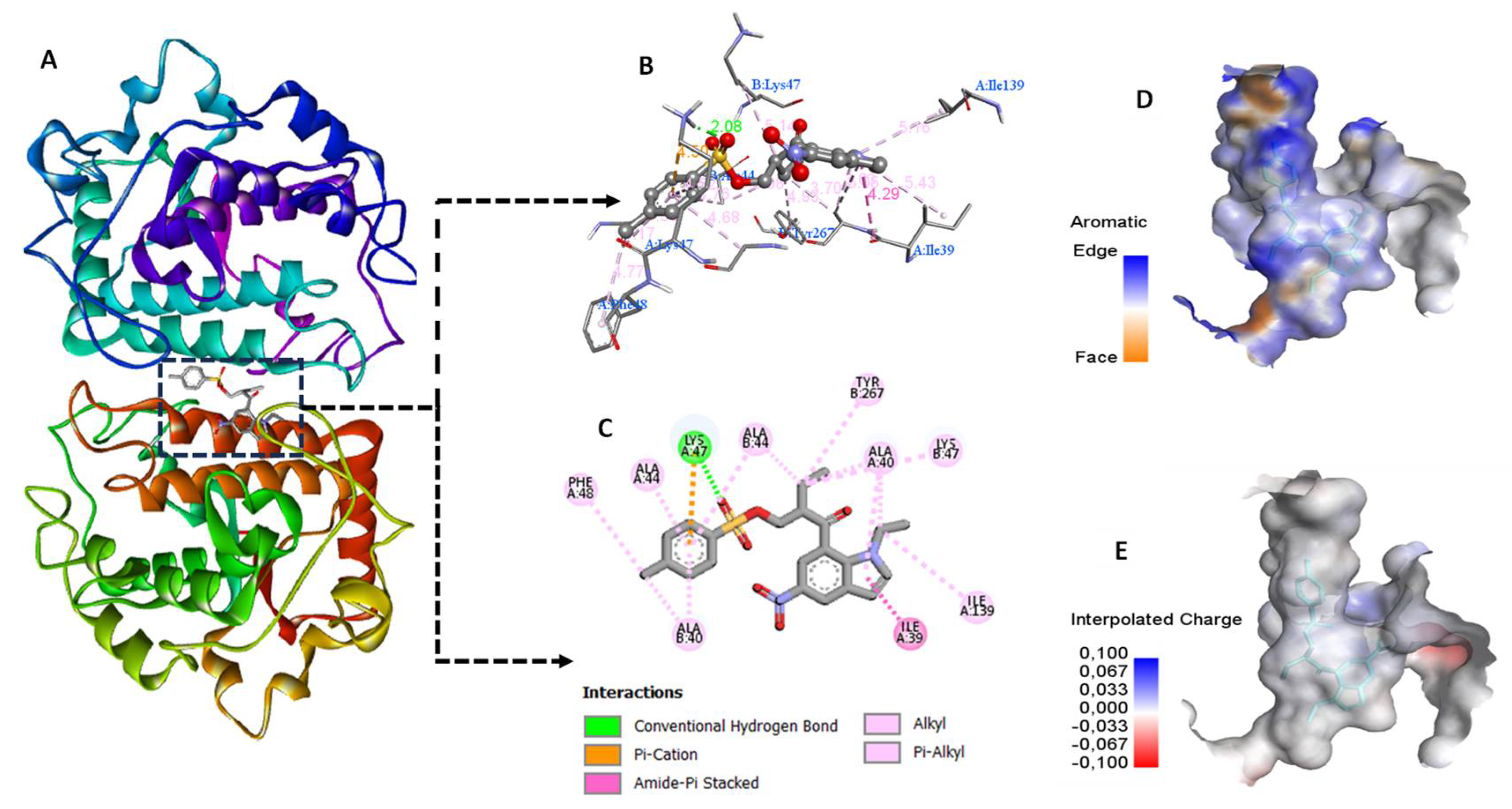

Table 1.
13C NMR data of compound 5.
| Shift (ppm) | Number of C | Class |
| 11.72 | 1 | s |
| 15.24 | 1 | s |
| 21.73 | 1 | s |
| 22.73 | 1 | s |
| 42.55 | 1 | s |
| 49.35 | 1 | s |
| 71.07 | 1 | s |
| 110.05 | 1 | s |
| 118.82 | 1 | s |
| 119.25 | 1 | s |
| 120.00 | 1 | s |
| 126.00 | 1 | s |
| 127.95 | 2 | s |
| 129.96 | 2 | s |
| 132.48 | 1 | s |
| 136.85 | 1 | s |
| 139.49 | 1 | s |
| 144.10 | 1 | s |
| 145.17 | 1 | s |
| 194.63 | 1 | s |
Table 2.
Docking results of compound 5.
| compound | Docking Score (Kcal/mol) | Interactions | Interacting residues |
| P5 | -10.86 | H-bond Π- alkyl Alkyl-alkyl Amide- Π stacking |
Lys 47 Ala 44, Ala40, Lys 47, Thr267 Ile 139, Phe 48 Ile 39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



