
Submitted:
11 July 2024
Posted:
15 July 2024
Read the latest preprint version here
Abstract
Precision metagenomic approaches using Oxford Nanopore Technology (ONT) sequencing has been shown to allow recovery of complete genomes of Escherichia coli O157:H7 from overnight enrichments of agricultural waters. This study tests the applicability of a similar approach for Cronobacter genome recovery from powdered infant formula (PIF) overnight enrichments, where Cronobacter typically dominates the overall microbiome (>90%). In this study, we aimed to test whether using ONT sequencing of overnight PIF enrichments could recover a completely closed Cronobacter genome for further genomic characterization. Ten PIF samples, each inoculated with different Cronobacter strains, covering Cronobacter sakazakii, C. muytjensii, C. dublinensis, C. turicensis, and C. universalis, were processed according to the Bacteriological Analytical Manual (BAM) protocol. qPCR was used for initial screening (detection and quantification) of the overnight enrichments and confirmed that the spiked PIF samples after the overnight enrichment have high levels of Cronobacter (107 to 109 CFU/mL). DNA from overnight PIF enrichments was extracted from the enrichment broth and sequenced using ONT. Results showed that ONT sequencing could accurately identify, characterize, and close the genomes of Cronobacter strains from overnight PIF enrichments in 3 days, much faster than the nearly two weeks required by the current BAM method. Complete genome recovery and species differentiation were achieved. This suggests that combining qPCR with ONT sequencing provides a rapid, cost-effective alternative for detecting and characterizing Cronobacter in PIF, enabling timely corrective actions during outbreaks.

Keywords:
nanopore sequencing
; foodborne pathogen
; complete genomes
; long read sequencing
; Cronobacter
; powdered infant formula
1. Introduction
Precision metagenomics is an approach that customizes the analysis of a metagenomic sample to detect and classify a specific pathogen. Developing culture-independent methods for detecting foodborne pathogens can expedite source tracking and reduce the time to implement corrective measures during suspected case scenarios [1,2,3,4,5,6]. For qualitative analysis of foodborne pathogens, qPCR or metagenomic detection is sufficient to call presumptive positive results, which are then followed by microbiological isolate confirmation and potentially subsequent regulatory actions [7] (https://www.fda.gov/media/102633/download, https://www.fda.gov/media/83177/download). A similar scenario applies to Cronobacter [5,6,8].
Cronobacter in powdered infant formula (PIF) has been associated with infant illnesses and human clinical cases [8,9,10]. Cronobacter was previously classified as one single species, Enterobacter sakazakii. Since 2008, it has been reclassified as a genus with seven identified species [11,12]. Among these species, Cronobacter sakazakii belonging to ST4 is responsible for over 90% of infant illnesses [13]. Potential sources of PIF contamination with Cronobacter spp. during commercial manufacturing include ingredients added after pasteurization, and equipment contamination post-pasteurization, such as spray dryers and fillers [14]. Current Cronobacter spp. detection protocols as outlined by the FDA Bacteriological Analytical Manual (BAM) Chapter 29 [15] employs enrichment in buffered peptone water (BPW), qPCR screening, chromogenic agars for isolation and cultural confirmation by qPCR or biochemical assays. The FDA BAM method does not identify individual species of Cronobacter. Single colony isolation is used for whole genome sequencing (WGS), which confirms the genus identity and determines the species identity and relationship to other Cronobacter strains in the WGS database. This entire process can take approximately two weeks of analysis time.
To expedite the analysis time, we aim to investigate Culture-Independent Diagnostics Tests (CIDT) using sequencing methods for the detection and classification of Cronobacter strains directly from PIF enrichments. This can be accomplished by using short or long-read sequencing. Bertrand et al. (2019) [16] outlined the challenges associated with short-read sequencing, particularly in accurately assembling complex, highly repetitive genomic regions, especially in multi-species environments. They noted that clustering-based species binning lacks precision for strain-level metagenomic assemblies crucial for outbreak investigations. In contrast, Oxford Nanopore Technologies (ONT) (Oxford, United Kingdom) long-read sequencing provides closed genomes while also providing an affordable and portable platform [17].
The limits of assembly of the long-read nanopore sequencing for use as a precision metagenomics technique from enrichments was established previously as requiring a minimum of 107 CFU/ml of overnight enrichment [18]. The main difference between the E. coli precision metagenomic approach from enriched agricultural water and the Cronobacter from PIF is that E. coli levels in enriched agricultural samples can vary greatly, requiring a certain threshold for successful ONT sequencing to recover a complete genome for further characterization. In the case of Cronobacter in PIF we have previously demonstrated that the levels are usually between 107 and 109 CFU/ml in overnight enrichments with more than 90% of the enrichment sample being Cronobacter (our internal study). During the enrichment process, Cronobacter spp. concentration increases to high levels and can be easily quantified by qPCR, according to the BAM Chapter 29 protocol [15]. Therefore, the levels are high enough in the enrichment for the sample to be considered almost as a pure cultured strain.
Despite the benefits of nanopore sequencing, including affordability, portability, long reads, and real-time basecalling, the inherent error rate of nanopore sequencing (when using the fast-calling model with the R9.4.1 flow cell and related library preparation kits) precludes the closed genomes from being used for phylogenic analysis. However, in late 2022 ONT released the R10.4.1 flow cell and the rapid barcoding Kit 24 V14 (RBK-114.24), which, together with a newer basecalling model, showed an increase in accuracy (~Q40) [19,20] (https://rrwick.github.io/2023/12/18/ont-only-accuracy-update.html). Additionally, the ONT protocols have improved to allow successful sequence processing of up to 24 samples (genomes of median ~4.5 Mb) in a single flow cell and still obtain a complete closed bacterial genome with high accuracy (>99.9%) and minimal SNP differences from the reference genome, depending on the species (internal study data not published) [20,21,22]. Consequently, in this pilot study, we aim to test whether we can characterize and close the genomes of up to ten Cronobacter spp. strains that were spiked into PIF and enriched overnight in a single ONT sequencing run.
2. Materials and Methods
Bacterial strains. The bacterial strains used in this study belonged to five different Cronobacter species (Table 1). Bacterial strains were cultured in BD Difco™ brain heart infusion (BHI) broth (Thermo Fisher Scientific, Waltham, MA) and stored in BHI broth with 20% glycerol stocks at -80⁰C.
Artificial contamination and sample processing. The powdered infant formula (PIF) samples were obtained locally and previously tested negative for Cronobacter. For artificial contamination, 25 g of PIF samples were inoculated with ~5 CFU of one strain of Cronobacter and mixed with 225 mL of buffered peptone water (BPW) (Thermo Fisher Scientific), followed by incubation at 36°C for 24 hours. An aliquot of each enrichment was processed as described in Chapter 29 of the BAM as outlined in Figure 1. An additional aliquot of 1 ml was taken from the enrichment and processed as described below for Nanopore sequencing.
DNA extraction and Cronobacter qPCR detection following the BAM procedure. Briefly, after BPW enrichment, 40 mL of enrichment cultures were centrifuged at 3000 × g for 10 min. The resultant pellet was washed and resuspended in PrepMan Ultra and boiled for 5 min. The supernatant was used for qPCR in BAM Chapter 29, which targets the Cronobacter partial macromolecular synthesis operon: the ribosomal protein S21 (rpsU) gene 3'end and the DNA primase (dnaG) gene 5' end [23]. Briefly, two µl of the DNA extract was added to 23 µl master mix containing 0.4 µM Cronobacter forward and reverse primers, 0.15 µM of Internal amplification control (IAC) forward and reverse primers, 0.3 µM of Cronobacter probe, 0.15 µM of IAC probe, 1X iQ qPCR Supermix (BIORAD, Hercules, CA), additional 2.5 U of Taq polymerase and 3 mM of MgCl2 and 50 nM of ROX passive dye. All primers and probes (Supplementary Table S1) employed in this study were purchased from IDT (Coralville, IA, USA).
DNA extraction. Genomic DNA from each spiked sample was extracted using the Maxwell RSC Cultured Cells DNA kit with a Maxwell RSC Instrument (Promega Corporation, Madison, WI) according to manufacturer’s instructions for Gram-negative bacteria with additional RNase treatment. DNA concentration was determined by Qubit 4 Fluorometer (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions.
Whole genome sequencing and assembly. DNA recovered from PIF spiked enrichment samples were sequenced using a GridION nanopore sequencer (Oxford Nanopore Technologies, Oxford, UK). The sequencing libraries were prepared using the Rapid Barcoding Kit 24 V14 (SQK-RBK114.24) and run in FLO-MIN114 (R10.4.1) flow cells, according to the manufacturer’s instructions for 48 hours. There were only two deviations from the original SQK-RBK114.24 library protocol: 1) the starting DNA per samples was 200 ng, and 2) the barcoding was done using 18 µl of sample DNA plus 2 µl of each barcode. The run was live base called using Guppy v 7.1.4 included in the MinKNOW v 23.07.12 software using the super-accurate basecalling model. The initial classification of the reads for each run was done using the “What's in my pot” (WIMP) workflow contained in the EPI2ME cloud service. That workflow allows for taxonomic classification of the reads generated by the GridION sequencing in real time.
The genomes for each spiked sample were obtained by de novo assembly using all nanopore data output per sample using the Flye program v2.9 [24], using the following parameters: --nano-hq, --read-error 0.03, and -i 4. The assembled contigs were classified by taxonomy by Kraken 2 [25] using GalaxyTrakr [26].
in silico MLST and serotyping. The initial analysis and identification of the strains were performed using an in silico Cronobacter spp. MLST approach based on the information available at the MLST website (https://pubmlst.org/organisms/cronobacter-spp). The in silico Cronobacter serotype present in each sample was determined using by batch screening in Ridom SeqSphere+ v9.0.8 (Ridom, Münster, Germany) using the genes described in Wang et al, 2021 [27] and available as part of the CroTrait pipeline (https://github.com/happywlu/CroTrait).
Phylogenetic relationship of the strains by whole genome multilocus typing (wgMLST) analysis. The phylogenetic relationship of the strains was assessed by a whole genome multilocus sequence typing (wgMLST) analysis using Ridom SeqSphere+ v9.0.8. The genome of C. zakazakii ATCC BAA-894 (NC_009778.1) containing 4103 CDSs was used as a reference. The complete closed genomes of these C. zakazakii strains (NC_017933.1 - ES15,
NC_020260.1 - SP291, NZ_CP011047.1 - ATCC 29544, and NZ_CP012253.1 - NCTC 8155) were used for comparison with the reference genome to establish a list of core and accessory genes. Genes that are repeated in more than one copy in any of the two genomes were removed from the analysis as failed genes. A task template then was created that contains both core and accessory genes for this reference Cronobacter sakazakii strain for any future testing (wgMLST scheme). Each individual locus from strain BAA-894 was assigned allele number 1. The assemblies for each individual Cronobacter closed genome in this study were queried against the task template and if the locus was found and was different from the reference genome or any other queried genome already in the database, a new number was assigned to that locus and so on. After eliminating any loci that were missing from the genome of any strain used in our analyses, we performed the wgMLST analysis. These remaining loci were considered the core genome shared by the analyzed strains. We used Nei’s DNA distance method [38] for calculating the matrix of genetic distance, taking into consideration only the number of same/different alleles in the core genes. The resultant genetic distances were used to generate a Neighbor-Joining (NJ) tree. wgMLST uses the alleles number of each locus for determining the genetic distance and builds the phylogenetic tree. The use of allele numbers reduces the influence of recombination in the dataset studied and allows for fast clustering determination of genomes.
Nucleotide sequence accession numbers. The raw ONT data for each individual sample were deposited in GenBank under BioProject accession number PRJNA1117853.
3. Results
- 3.1.1. Cronobacter-Spiked Sample Enrichment Preparation for Nanopore Sequencing
Cronobacter grew in every enrichment culture for all PIF spiked samples. The uninoculated PIF sample continued to test negative following enrichment. Cronobacter concentrations in each enrichment varied from 107 to 109 CFU/mL (Table 2). The BAM qPCR method detected the presence of Cronobacter in each overnight spiked PIF enrichment. A schematic representation of the entire workflow as described in materials and methods is shown in Figure 1. All spiked PIF samples resulted in positive qPCR signal at Cq values of 12.8 to 18.7 (Table 2). The enumeration results and qPCR Cq had an excellent correlation with R2 of 0.93.
- 3.1.2. Nanopore Long-Read Sequencing Results
The DNA extracted from 1 ml of each of the 10 spiked PIF overnight enrichment samples were sequenced using ONT for 48 hours. The sequencing output for the run was 17.16 Gb in 3.25 million reads, of which 94% (3 M) passed the quality filter (minimum quality score of 10). Around 600 k reads were unclassified and were discarded. The read length N50 was 8 kb and there was enough data generated for each of the samples at different coverage levels to perform a successful genome assembly and sample characterization (Table 3). Reads below 4000 bp and quality scores below 10 were discarded for downstream analysis resulting in 915,281 remaining reads. The estimated coverage per Cronobacter sample (genome size approximately 4.5 Mb) ranged from 55 – 420 X.
The preliminary Cronobacter species ID and the number of total reads matching each species per spiked PIF sample were determined using Oxford Nanopore EPI2ME “What’s in my pot” (WIMP) workflow analysis (Table 2). Only one species was misclassified by WIMP as C. universalis (EB18) instead of the expected C. turicensis. According to the WIMP analysis 90% to 99.6% of the classified reads per spiked PIF sample belonged to the spiked strain (Table 2). These numbers confirmed what was observed by qPCR for the same samples. According to these values, the concentration of each individual spiked strain ranged from 0.8 x 107 to 0.8 x 109 CFU/ml.
- 3.1.3. Nanopore Long-Read GENOME assembly of Cronobacter PIF Enriched Samples
Since the observed Cronobacter spp. concentrations (Table 2) in spiked PIF overnight enriched sample were similar to the concentrations observed for pure overnight cultures, we proceeded with the assembly using the reads obtained for each sample as we usually do for a pure culture [28,29]. The complete closed genome for each strain was de novo assembled using Flye in our high-performance computing environment (Table 4). The assembly of the reads for each sample resulted in completely closed circular chromosomes and/or plasmids. The G+C mol% of these assemblies were 56.8% to 58.1%, which is within the range of the reported GC content for Cronobacter spp. strains [11]. The genome of some samples (E515 and E603) was composed only of a chromosome without the presence of any extra chromosomal elements. The remainder of the samples carried between 2 to 3 plasmids. Analysis of every contig for each sample with Kraken2 showed that all of them belonged to Cronobacter spp. The chromosome coverage for each sample varied from 65 to 516 X.
- 3.1.4. Multilocus Sequence Typing (MLST) and Serotyping Analysis
in silico MLST analysis (https://pubmlst.org/organisms/cronobacter-spp) showed that most of these strains belonged to eight known sequence types (STs) (ST1, 4, 8, 15, 19, 54, 80, and 81), with E791 belonging to a novel ST profile (151, 20, 111, 90, 195, 212, 239) (Table 5). Interestingly, the closest STs to E791 reported in the Cronobacter MLST database matched in 4 loci and they all belonged to the species C. dublinensis (ST477, 479,576, and 662).
- 3.1.5. wgMLST Analysis of Cronobacter PIF Overnight Enriched samples And Taxa Classification Using a Phylogenetic Tree
The phylogenetic relationship among the 37 Cronobacter spp. genomes (27 completely closed genome representative for each of the seven Cronobacter species publicly available at GenBank - Supplementary Table S1, and the 10 ONT assemblies from the spiked overnight enriched PIF samples) was determined by a custom wgMLST analysis (Figure 2). A total of 3,965 genes were used as templates for the analysis of the Cronobacter spp. strains. Every species was easily distinguished and the wgMLST confirmed their current species definition. wgMLST analysis grouped the 10 PIF spiked samples according to their species and distinguished differences between strains from the same ST (e.g., ST8, differing in at least 34 alleles). This phylogenetic tree also confirmed that sample EB18 was indeed a C. turicensis.
4. Discussion
Considering the importance of powdered infant formula (PIF) safety, accurate detection and classification of Cronobacter spp. in PIF are crucial, particularly during outbreaks. Current methods include qPCR and extensive selective plating before whole genome sequencing (WGS) analysis. This time-consuming process requires nearly two weeks for isolate confirmation. The use of qPCR as a screening tool of the pre-enrichment broth and long-read sequencing analysis of qPCR positive samples, allows detection and full characterization of a Cronobacter strain in 3 to 4 days. Although confirmation by microbiological methods remains the standard, this new method can provide faster information to inform risk management decisions, potentially reducing the risk of illnesses by up to a week.
Since levels of Cronobacter, especially C. sakazakii, in overnight enrichments can reach 107 to 109 CFU/ml and Cronobacter makes up 90.0 – 99.6% of the culture (Table 3), samples from overnight enrichments can be treated as pure isolates and sequenced directly without the need of further selective enrichment or plating steps to isolate the Cronobacter strain from the other microbiota present in the overnight enrichment. Simple ONT sequencing and assembly using all reads has demonstrated in this limited study as adequate to completely close Cronobacter genomes. The data generated from each sample is manageable (compressed fastq files from 0.3 Gb to 1.8 Gb) and can produce a usable genome in about 0.5 to 2 hours post-run in a high-performance environment. This reduces the overall analysis time compared to traditional culture methods and WGS, allowing detection of related strains in a few days (3 days from enrichment to usable data), compared to approximately 2 weeks (Figure 1).
Given the high relative populations of Cronobacter in each enrichment (90.0-99.6%) (Table 3), short-read sequencing could satisfactorily identify and characterize these strains, but a complete genome would not be recovered. Short reads would capture most of the genome, but some fragments and repetitive regions might be missed. In contrast, ONT allows for closing the complete genome (chromosome and/or plasmids) (Table 4). ONT also enables positive identification of most Cronobacter species in the PIF sample within just 1 hour, except for C. turicensis. This highlights a problem with accurate taxa identification using curated databases (In this case RefSeq Database created on 8 June 2021). If the reference genome defining a species is missing in the database, proper identification may not be achieved. Regular updating of databases improves their accuracy and comprehensiveness. Without these regular updates, these databases might lack essential information, leading to incomplete or incorrect identification of microorganisms.
The observed STs in this study agreed with the expected results for the spiked taxa per PIF samples (Table 1 and Table 5). The Cronobacter MLST database currently holds 4,150 strains, with varying numbers for different STs (e.g., 499 strains belong to ST1 (reported as C. sakazakii), 615 strains to ST4 (reported as C. sakazakii), 125 strains to ST8 (reported as C. sakazakii), 1 strain to ST15 (reported as C. sakazakii), 7 strains to ST19 (reported as C. turicensis), 8 strains to ST54 (reported as C. universalis), 7 strains to ST80 (reported as C. dublinensis), and 13 strains ST81 (reported as C. muytjensii). In silico serotype analysis also matched expected results, showing high diversity of serotypes among the samples and within the same species (e.g., two different O types for C. sakazakii strains). ONT rapidly differentiated them using in silico MLST, which is crucial since most Cronobacter sakazakii causing more than 90% of infant illnesses belong to ST4 [13], including recent cases linked to PIF and breast pump equipment caused by ST4 strains [6].
It is important to use qPCR for early screening and quantification to determine the approximate concentration of Cronobacter in PIF enriched samples, which helps estimate the number of samples per flow cell for ONT sequencing. A previous study of ONT sequencing for Culture-Independent Diagnostics Tests (CIDT) of STECs in agricultural water overnight enrichments determined that a minimum of 107 CFU/ml of STEC in a complex mixture was needed for successful genome closure using a single flow cell [18]. This study showed that when the target organism reached concentrations above 107 CFU/ml and 90-99.6% of the sample, up to 10 Cronobacter genomes could be successfully closed with high coverage (100 – 300X). This suggests that more samples could be included, reducing the cost per sample. At current prices, the entire run costs around $860 (flow cell – $700, Sequencing kit library – $110, and DNA extraction – $50), with a cost of 86 USD per sample. Bulk purchasing of flow cells and running 24 samples instead of 10 could reduce the price per sample to around $25. The Cronobacter ONT method from PIF enriched samples has an advantage over STEC enrichments, as a positive PIF sample usually results in a single organism, reducing sample complexity [18] .
The pilot study aimed to test nanopore sequencing for preliminary identification and characterization (MLST, serotyping, and wgMLST) within approximately 3 days from sample collection, comparing results to reference genomes (Table 1 and Figure 2). Sequencing with the new R10.4.1 flow cell showed higher accuracy than R9.4.1 as observed by many others [17,18,30]. Strains with previous sequences in NCBI clustered tightly together, with varied numbers of loci differences (Supplementary Table S1 and Figure 2). For instance, sample E477 (ST8) differed in at least 3 loci from ATCC 29544 (ST1). However, issues such as indels and missing genes highlight the need for cautious interpretation and robust validation of ONT data compared to other sequencing methods like Illumina.
Currently, WGS in bacterial-related diseases focuses on sequencing microorganisms isolated from selective culture plates [6,31,32,33]. Clinical and diagnostic labs are transitioning to Culture Independent Diagnostic Tests (CIDTs) for rapid identification, but CIDTs do not yield physical isolates, hindering outbreak investigations by public health agencies. This in turn prolongs outbreak resolution [34]. Similarly, isolating foodborne bacteria for surveillance or case investigations, such as Cronobacter, takes around 1 week per isolate before WGS can be performed. There's a push for rapid, cost-effective, and highly accurate culture-independent methods for both clinical and food investigations [2,18,34,35]. Previous metagenomic approaches yielded closed STEC genomes but weren't suitable for SNP-based source tracking [18]. This study using newer ONT chemistry (R10.4.1) for Cronobacter identification and subtyping directly from overnight PIF enrichments shows higher quality than earlier results obtained for STEC using the (R9.4.1 flow cells). We accurately identified each spiked strain (MLST and serotype) and subtyped nearly to the strain level (wgMLST). Similar results have been observed by other authors for certain strains of Listeria monocytogenes and STEC [21] and Staphylococcus [22], showing the improvement of ONT sequencing for outbreak investigations. However, protocols for nanopore sequencing, DNA extraction, and library preparation still vary, and validation for repeatability, reproducibility, and robustness is needed before implementation.
5. Conclusions
In conclusion, utilizing ONT for Cronobacter identification and preliminary characterization from PIF samples can expedite the process to approximately 3 days, in contrast to the nearly 2 weeks required by the current FDA methods. Despite high concentrations of Cronobacter in the spiked overnight PIF enrichment, exceeding 107 CFU/ml, a combined approach involving qPCR and nanopore sequencing is advocated in time-sensitive situations to enhance source tracking. qPCR can detect Cronobacter and estimate the concentration in the sample, predicting the adequacy of subsequent nanopore sequencing for complete genome recovery. Additionally, qPCR data can inform the number of samples feasible for sequencing in a single flow cell, reducing costs per sample. Furthermore, additional organisms can be included in the sequencing run when fewer samples are available.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1: Twenty-seven Cronobacter spp. genomes belonging to seven Cronobacter species used for the wgMLST analysis and species definition.
Author Contributions
Conceived and designed the experiments: NGE and YC. Conducted experiments in the lab: HJK. Analyzed the data: NGE. Contributed reagents/materials/analysis tools: NGE and YC. Wrote the paper: NGE and YC. Revision and final draft: YC, HJK, and NGE.
Funding
This research was funded by the Food and Drug Administration Foods Program Intramural Funds”
Data Availability Statement
The raw ONT data for each individual sample were deposited in GenBank under BioProject accession number PRJNA1117853.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brown, E.U. Dessai, S. McGarry and P. Gerner-Smidt. "Use of whole-genome sequencing for food safety and public health in the United States." Foodborne Pathog Dis 16 (2019): 441-50. 10.1089/fpd.2019.2662. https://www.ncbi.nlm.nih.gov/pubmed/31194586.
- Huang, A. D., C. Luo, A. Pena-Gonzalez, M. R. Weigand, C. L. Tarr and K. T. Konstantinidis. "Metagenomics of two severe foodborne outbreaks provides diagnostic signatures and signs of coinfection not attainable by traditional methods." Appl Environ Microbiol 83 (2017): 10.1128/AEM.02577-16. https://www.ncbi.nlm.nih.gov/pubmed/27881416.
- Loman, N. J., C. Constantinidou, M. Christner, H. Rohde, J. Z. Chan, J. Quick, J. C. Weir, C. Quince, G. P. Smith, J. R. Betley, et al. "A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of shiga-toxigenic Escherichia coli O104:H4." JAMA 309 (2013): 1502-10. 10.1001/jama.2013.3231. https://www.ncbi.nlm.nih.gov/pubmed/23571589.
- Maguire, M., P. Ramachandran, S. Tallent, M. K. Mammel, E. W. Brown, M. W. Allard, S. M. Musser and N. Gonzalez-Escalona. "Precision metagenomics sequencing for food safety: Hybrid assembly of shiga toxin-producing Escherichia coli in enriched agricultural water." Front Microbiol 14 (2023): 1221668. 10.3389/fmicb.2023.1221668. https://www.ncbi.nlm.nih.gov/pubmed/37720160.
- Cechin, C. D. F., G. G. Carvalho, C. P. Bastos and D. Y. Kabuki. "Cronobacter spp. In foods of plant origin: Occurrence, contamination routes, and pathogenic potential." Crit Rev Food Sci Nutr 63 (2023): 12398-412. 10.1080/10408398.2022.2101426. https://www.ncbi.nlm.nih.gov/pubmed/35866516.
- Haston, J. C., S. Miko, J. R. Cope, H. McKeel, C. Walters, L. A. Joseph, T. Griswold, L. S. Katz, A. A. Andujar, L. Tourdot, et al. "Cronobacter sakazakii infections in two infants linked to powdered infant formula and breast pump equipment - United States, 2021 and 2022." MMWR Morb Mortal Wkly Rep 72 (2023): 223-26. 10.15585/mmwr.mm7209a2. https://www.ncbi.nlm.nih.gov/pubmed/36862586.
- Archer, D. L. "The evolution of fda's policy on Listeria monocytogenes in ready-to-eat foods in the United States." Current Opinion in Food Science 20 (2018): 64-68. http://www.sciencedirect.com/science/article/pii/S2214799317301728. [CrossRef]
- Li, Y., G. Lin, L. Zhang, Y. Hu, C. Hong, A. Xie and L. Fang. "Genomic insights into Cronobacter spp. recovered from food and human clinical cases in zhejiang province, china (2008-2021)." J Appl Microbiol 134 (2023): 10.1093/jambio/lxad033. https://www.ncbi.nlm.nih.gov/pubmed/36807689.
- Drudy, D., N. R. Mullane, T. Quinn, P. G. Wall and S. Fanning. "Enterobacter sakazakii: An emerging pathogen in powdered infant formula." Clin Infect Dis 42 (2006): 996-1002. 10.1086/501019. https://www.ncbi.nlm.nih.gov/pubmed/16511766.
- Strysko, J., J. R. Cope, H. Martin, C. Tarr, K. Hise, S. Collier and A. Bowen. "Food safety and invasive Cronobacter infections during early infancy, 1961-2018." Emerg Infect Dis 26 (2020): 857-65. 10.3201/eid2605.190858. https://www.ncbi.nlm.nih.gov/pubmed/32310746.
- Joseph, S.H. Sonbol, S. Hariri, P. Desai, M. McClelland and S. J. Forsythe. "Diversity of the Cronobacter genus as revealed by multilocus sequence typing." Journal of clinical microbiology 50 (2012): 3031-39. 10.1128/Jcm.00905-12. https://pubmed.ncbi.nlm.nih.gov/22785185/. [CrossRef]
- Iversen, C., N. Mullane, B. McCardell, B. D. Tall, A. Lehner, S. Fanning, R. Stephan and H. Joosten. "Cronobacter gen. Nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. :Lausannensis subsp. nov. and Cronobacter dublinensis subsp. Lactaridi subsp. nov." Int J Syst Evol Microbiol 58 (2008): 1442-7. 10.1099/ijs.0.65577-0. https://www.ncbi.nlm.nih.gov/pubmed/18523192.
- Hariri, S., S. Joseph and S. J. Forsythe. "Cronobacter sakazakii st4 strains and neonatal meningitis, United States." Emerg Infect Dis 19 (2013): 175-7. 10.3201/eid1901.120649. https://www.ncbi.nlm.nih.gov/pubmed/23260316.
- Ling, N., X. T. Jiang, S. Forsythe, D. F. Zhang, Y. Z. Shen, Y. Ding, J. Wang, J. M. Zhang, Q. P. Wu and Y. W. Ye. "Food safety risks and contributing factors of Cronobacter spp." Engineering 12 (2022): 128-38. 10.1016/j.eng.2021.03.021. https://www.sciencedirect.com/science/article/pii/S209580992100254X.
- Chen, Y. M., N. E.; Liu, K. C.; Mullins, J. S.; Lampel, K.; Hammack, T. "BAM chapter 29: Cronobacter." 2023. https://www.fda.gov/food/laboratory-methods-food/bam-chapter-29-Cronobacter.
- Bertrand, D., J. Shaw, M. Kalathiyappan, A. H. Q. Ng, M. S. Kumar, C. Li, M. Dvornicic, J. P. Soldo, J. Y. Koh, C. Tong, et al. "Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes." Nat Biotechnol 37 (2019): 937-44. 10.1038/s41587-019-0191-2. https://pubmed.ncbi.nlm.nih.gov/31359005/.
- Maguire, M., A. S. Khan, A. A. Adesiyun, K. Georges and N. Gonzalez-Escalona. "Closed genome sequence of a Salmonella enterica serotype senftenberg strain carrying the mcr-9 gene isolated from broken chicken eggshells in trinidad and tobago." Microbiol Resour Announc 10 (2021): e0146520. 10.1128/MRA.01465-20. https://www.ncbi.nlm.nih.gov/pubmed/34042489.
- Maguire, M., J. A. Kase, D. Roberson, T. Muruvanda, E. W. Brown, M. Allard, S. M. Musser and N. Gonzalez-Escalona. "Precision long-read metagenomics sequencing for food safety by detection and assembly of shiga toxin-producing Escherichia coli in irrigation water." PLoS One 16 (2021): e0245172. 10.1371/journal.pone.0245172. https://www.ncbi.nlm.nih.gov/pubmed/33444384.
- Sanderson, N. D.K. M. V. Hopkins, M. Colpus, M. Parker, S. Lipworth, D. Crook and N. Stoesser. "Evaluation of the accuracy of bacterial genome reconstruction with oxford nanopore R10.4.1 long-read-only sequencing." Microb Genom 10 (2024): 10.1099/mgen.0.001246. https://www.ncbi.nlm.nih.gov/pubmed/38713194. [CrossRef]
- Lerminiaux, N., K. Fakharuddin, M. R. Mulvey and L. Mataseje. "Do we still need illumina sequencing data? Evaluating oxford nanopore technologies R10.4.1 flow cells and the rapid v14 library prep kit for gram negative bacteria whole genome assemblies." Canadian Journal of Microbiology (2024): 10.1139/cjm-2023-0175. https://pubmed.ncbi.nlm.nih.gov/38354391/.
- Bogaerts, B., A. Van den Bossche, B. Verhaegen, L. Delbrassinne, W. Mattheus, S. Nouws, M. Godfroid, S. Hoffman, N. H. C. Roosens, S. C. J. De Keersmaecker, et al. "Closing the gap: Oxford nanopore technologies R10 sequencing allows comparable results to illumina sequencing for snp-based outbreak investigation of bacterial pathogens." J Clin Microbiol (2024): e0157623. 10.1128/jcm.01576-23. https://www.ncbi.nlm.nih.gov/pubmed/38441926.
- Buytaers, F. E., B. Verhaegen, T. Van Nieuwenhuysen, N. H. C. Roosens, K. Vanneste, K. Marchal and S. C. J. De Keersmaecker. "Strain-level characterization of foodborne pathogens without culture enrichment for outbreak investigation using shotgun metagenomics facilitated with nanopore adaptive sampling." Front Microbiol 15 (2024): 1330814. 10.3389/fmicb.2024.1330814. https://www.ncbi.nlm.nih.gov/pubmed/38495515.
- Seo, K. H. and R. E. Brackett. "Rapid, specific detection of enterobacter sakazakii in infant formula using a real-time pcr assay." J Food Prot 68 (2005): 59-63. 10.4315/0362-028x-68.1.59. https://www.ncbi.nlm.nih.gov/pubmed/15690804.
- Kolmogorov, M., J. Yuan, Y. Lin and P. A. Pevzner. "Assembly of long, error-prone reads using repeat graphs." Nat Biotechnol 37 (2019): 540-46. 10.1038/s41587-019-0072-8.
- Wood, D. E., J. Lu and B. Langmead. "Improved metagenomic analysis with Kraken 2." Genome Biology 20 (2019): 257. 10.1186/s13059-019-1891-0. [CrossRef]
- Gangiredla, J., H. Rand, D. Benisatto, J. Payne, C. Strittmatter, J. Sanders, W. J. Wolfgang, K. Libuit, J. B. Herrick, M. Prarat, et al. "Galaxytrakr: A distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians." BMC Genomics 22 (2021): https://pubmed.ncbi.nlm.nih.gov/33568057/.
- Wang, L., W. Zhu, G. Lu, P. Wu, Y. Wei, Y. Su, T. Jia, L. Li, X. Guo, M. Huang, et al. "In silico species identification and serotyping for Cronobacter isolates by use of whole-genome sequencing data." Int J Food Microbiol 358 (2021): 109405. 10.1016/j.ijfoodmicro.2021.109405. https://www.ncbi.nlm.nih.gov/pubmed/34563883.
- Maguire, M., A. S. Khan, A. A. Adesiyun, K. Georges and N. Gonzalez-Escalona. "Genomic comparison of eight closed genomes of multidrug-resistant Salmonella enterica strains isolated from broiler farms and processing plants in trinidad and tobago." Front Microbiol 13 (2022): 863104. 10.3389/fmicb.2022.863104. https://www.ncbi.nlm.nih.gov/pubmed/35620095.
- Duarte, F., E. Cordero, M. Calderon, A. Godinez, B. Ross, M. Allard and N. Gonzalez-Escalona. "Closed genomes of four multidrug resistance Salmonella enterica serotype infantis isolated in Costa Rrica." Microbiol Resour Announc 13 (2024): e0025723. 10.1128/MRA.00257-23. https://www.ncbi.nlm.nih.gov/pubmed/38019019.
- Chen, Z., D. Kuang, X. Xu, N. Gonzalez-Escalona, D. L. Erickson, E. Brown and J. Meng. "Genomic analyses of multidrug-resistant Salmonella Indiana, Typhimurium, and Enteritidis isolates using minion and miseq sequencing technologies." PLoS One 15 (2020): e0235641. 10.1371/journal.pone.0235641. https://www.ncbi.nlm.nih.gov/pubmed/32614888.
- Hoffmann, M.Y. Luo, S. R. Monday, N. Gonzalez-Escalona, A. R. Ottesen, T. Muruvanda, C. Wang, G. Kastanis, C. Keys, D. Janies, et al. "Tracing origins of the Salmonella Bareilly strain causing a food-borne outbreak in the United States." J. Infect Dis 213 (2016): 502-08. jiv297 [pii];10.1093/infdis/jiv297 [doi]. http://www.ncbi.nlm.nih.gov/pubmed/25995194. Not in File. [CrossRef]
- Bottichio, L., A. Keaton, D. Thomas, T. Fulton, A. Tiffany, A. Frick, M. Mattioli, A. Kahler, J. Murphy, M. Otto, et al. "Shiga toxin-producing Escherichia coli infections associated with romaine lettuce-United States, 2018." Clin Infect Dis 71 (2020): e323-e30. 10.1093/cid/ciz1182. https://www.ncbi.nlm.nih.gov/pubmed/31814028.
- Gobin, M., J. Hawker, P. Cleary, T. Inns, D. Gardiner, A. Mikhail, J. McCormick, R. Elson, D. Ready, T. Dallman, et al. "National outbreak of shiga toxin-producing Escherichia coli O157:H7 linked to mixed salad leaves, United Kingdom, 2016." Euro Surveill 23 (2018): 10.2807/1560-7917.ES.2018.23.18.17-00197. https://www.ncbi.nlm.nih.gov/pubmed/29741151.
- Carleton, H. A., J. Besser, A. J. Williams-Newkirk, A. Huang, E. Trees and P. Gerner-Smidt. "Metagenomic approaches for public health surveillance of foodborne infections: Opportunities and challenges." Foodborne Pathogens and Disease 16 (2019): 474-79. 10.1089/fpd.2019.2636. https://pubmed.ncbi.nlm.nih.gov/31170005/.
- Pena-Gonzalez, A., M. J. Soto-Giron, S. Smith, J. Sistrunk, L. Montero, M. Paez, E. Ortega, J. K. Hatt, W. Cevallos, G. Trueba, et al. "Metagenomic signatures of gut infections caused by different Escherichia coli pathotypes." Appl Environ Microbiol 85 (2019): 10.1128/AEM.01820-19. https://www.ncbi.nlm.nih.gov/pubmed/31585992.
Figure 1.
Flow diagram for detection, isolation, and full WGS characterization of Cronobacter spp. strains from PIF in Chapter 29 in the BAM.
Figure 1.
Flow diagram for detection, isolation, and full WGS characterization of Cronobacter spp. strains from PIF in Chapter 29 in the BAM.
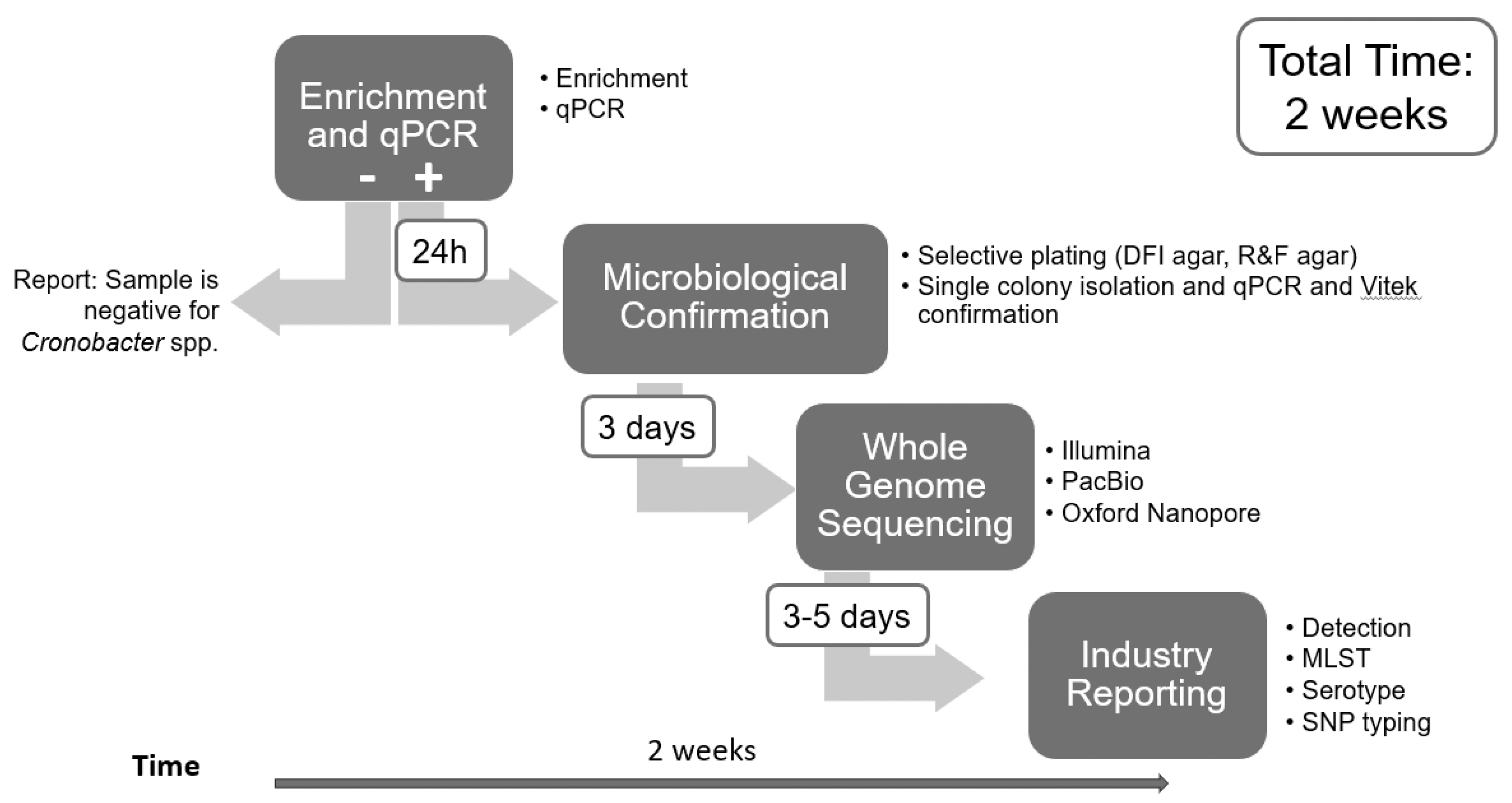
Figure 2.
Neighbor joining (NJ) phylogenetic tree based on gene differences on the 3965 analyzed genes (allele based) of the 10 Cronobacter spp. overnight spiked PIF genomes generated in this study with the other 27 Cronobacter spp. genomes, representing the 7 Cronobacter described species (Supplementary Table S1). Highlighted in green are the 10 samples recovered from the overnight enrichment of spiked PIFs. Next to then are the specie definition, contig numbers per assembly and their ST.
Figure 2.
Neighbor joining (NJ) phylogenetic tree based on gene differences on the 3965 analyzed genes (allele based) of the 10 Cronobacter spp. overnight spiked PIF genomes generated in this study with the other 27 Cronobacter spp. genomes, representing the 7 Cronobacter described species (Supplementary Table S1). Highlighted in green are the 10 samples recovered from the overnight enrichment of spiked PIFs. Next to then are the specie definition, contig numbers per assembly and their ST.


Table 1.
Metadata of the strains used in the spiking experiment in this study.
| Samples | Cronobacter species | Source | Country of Origin | Available at NCBI |
|---|---|---|---|---|
| E477 | sakazakii | Human (throat) | Unknown | ATCC 29544 |
| E515 | dublinensis | Water | Switzerland | NA |
| E603 | muytjensii | Unknown | unknown | ATCC 51329 |
| E604 | sakazakii | Clinical | Canada | SK90 |
| E791 | dublinensis | Human (blood) | USA | CDC 5960-70 |
| E797 | universalis | Water | UK | NCTC 9529 |
| E825 | sakazakii | Human (breast abscess) | USA | NA |
| EB13 | sakazakii | Neonate (meningitis) | Switzerland | NA |
| EB18 | turicensis | Neonate (meningitis) | Switzerland | NA |
| E899 | sakazakii | Clinical | USA | NA |
NA- not available.
Table 2.
Taxonomic classification of the spiked PIF samples and qPCR values obtained from the overnight enrichments.
Table 2.
Taxonomic classification of the spiked PIF samples and qPCR values obtained from the overnight enrichments.
| Samples | Cronobacter Taxa expected | Cronobacter Taxa observed WIMPa | Cronobacter Taxa observed by phylogenetic tree | qPCR CT value | Estimated Cronobacter CFU/mlb | Cronobacter ONT % DNA sample |
|---|---|---|---|---|---|---|
| E477 | sakazakii | sakazakii | sakazakii | 12.8 | 7.9E+08 | 97.0 |
| E515 | dublinensis | dublinensis | dublinensis | 16.0 | 1.6E+07 | 90.0 |
| E603 | muytjensii | muytjensii | muytjensii | 12.9 | 6.3E+08 | 99.0 |
| E604 | sakazakii | sakazakii | sakazakii | 13.6 | 1.6E+08 | 90.0 |
| E791 | dublinensis | dublinensis | dublinensis | 17.0 | 1.6E+07 | 92.0 |
| E797 | universalis | universalis | universalis | 13.4 | 7.9E+08 | 99.6 |
| E825 | sakazakii | sakazakii | sakazakii | 12.9 | 1.0E+08 | 97.1 |
| EB13 | sakazakii | sakazakii | sakazakii | 14.4 | 3.2E+08 | 98.2 |
| EB18 | turicensis | universalis | turicensis | 18.7 | 7.9E+06 | 90.4 |
| E899 | sakazakii | sakazakii | sakazakii | 17.7 | 2.0E+07 | 95.4 |
aWIMP (“What's in my pot” workflow in Epi2me) identified sample EB18 as composed by 3 different taxa: universalis (59%), sakazakii (20.4%), and malonaticus (11%). bin the overnight enrichment.
Table 3.
Nanopore sequencing run statistics.
| Samples | Total reads | Total Mb | Estimated coverage Cronobacter genome all reads (X) | Reads above 4000 bp | Total Mb above 4000 bp | Estimated coverage Cronobacter genome > 4kb reads (X) |
|---|---|---|---|---|---|---|
| E477 | 228,000 | 1,069 | 238 | 82,998 | 732 | 163 |
| E515 | 70,096 | 354 | 79 | 26,066 | 248 | 55 |
| E603 | 523,730 | 2,665 | 592 | 203,038 | 1,889 | 420 |
| E604 | 127,684 | 643 | 143 | 47,322 | 455 | 101 |
| E791 | 257,927 | 1,148 | 255 | 85,301 | 752 | 167 |
| E797 | 278,012 | 1,382 | 307 | 105,162 | 969 | 215 |
| E825 | 210,799 | 1,010 | 224 | 77,642 | 697 | 155 |
| EB13 | 239,315 | 1,228 | 273 | 91,575 | 870 | 193 |
| EB18 | 319,323 | 1,648 | 366 | 125,660 | 1,168 | 260 |
| E899 | 191,946 | 941 | 209 | 70,517 | 647 | 144 |
Table 4.
Assembly statistics for each PIF spiked sample in this study.
| Samples | Contig No. | %GC content | Genome size (bp) | Genome coverage (X) |
|---|---|---|---|---|
| E477 | 3 | 56.6 | 4507829; 93905; 53449 | 136; 156; 287 |
| E515 | 1 | 57.9 | 4487108 | 65 |
| E603 | 1 | 57.7 | 4305928 | 516 |
| E604 | 3 | 56.6 | 4412859; 117865; 52143 | 115; 123;179 |
| E791 | 2 | 58.1 | 4349860; 166041 | 208; 239 |
| E797 | 2 | 57.9 | 4075540; 129777 | 273; 306 |
| E825 | 3 | 56.8 | 4257543; 97419; 53456 | 185; 163; 260 |
| EB13 | 3 | 56.7 | 4347023; 131190; 31203 | 214; 265; 1269 |
| EB18 | 4 | 57.2 | 4384296; 144804; 53716; 44722 | 283; 357; 552; 375 |
| E899 | 2 | 56.7 | 4340415; 53472 | 176; 284 |
Table 5.
MLST and serotyping of the spiked PIF assembled samples.
| Samples | STa | Cronobacter Taxa by ST | Serotypeb |
|---|---|---|---|
| E477 | 8 | sakazakii | SO1 |
| E515 | 80 | dublinensis | DO2 |
| E603 | 81 | muytjensii | MuO2 |
| E604 | 15 | sakazakii | SO2 |
| E791 | novel | dublinensis | DO1a |
| E797 | 54 | universalis | UO1 |
| E825 | 8 | sakazakii | SO1 |
| EB13 | 1 | sakazakii | SO1 |
| EB18 | 19 | turicensis | TO1 |
| E899 | 4 | sakazakii | SO2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



