
Submitted:
13 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
The emergence of drug resistance inducing mutations in Hepatitis C virus (HCV) coupled with genotypic heterogeneity has made targeting NS3/4A serine protease difficult. In this work, we investigated the mutagenic variations in the binding pocket of Genotype 3 (G3) HCV NS3/4A and evaluated ligands for efficacious inhibition. We report mutations at 14 positions within the ligand-binding residues of HCV NS3/4A including H57R and S139P within the catalytic triad. We then modeled each mutational variant for pharmacophore-based virtual screening (PBVS) followed by covalent docking towards identifying a potential covalent inhibitor, i.e. cpd-217. The binding stability of cpd-217 was then supported by molecular dynamic simulation followed by MM/GBSA binding free energy calculation. The free energy decomposition analysis indicated that the resistant mutants alter the HCV NS3/4A-ligand interaction, resulting in an unbalanced energy distribution within the binding site leading to drug resistance. cpd-217 was identified to interact with all NS3/4A G3 variants with significant covalent docking scores. In conclusion, cpd-217 emerges as a potential inhibitor of HCV NS3/4A G3 variants that warrants further in vitro and in vivo studies. The study will pave the way for drug design and development of HCV G3 NS3/4A.
Keywords:
Hepatitis C virus
; MD Simulations
; Covalent inhibitor
; Drug resistance
; Pharmacophore-based virtual screening
1. Introduction
Hepatitis C Virus (HCV) is an enveloped, single-stranded, positive-sense RNA virus of the Flaviviridae family [1]. HCV is the causative agent of liver hepatitis C infection, which has an estimated global prevalence of 2.5% [2]. Chronic HCV infections often lead to liver cirrhosis, hepatocellular carcinomas, and liver failure [2,3,4,5]. HCV isolates exhibit vast genetic heterogeneity [6], based on which the viral variants have been grouped into seven genotypes (G1-G7) and various subtypes [2,7,8,9]. G1 and G3 together account for over 80% of the global infections [10,11]. In particular, G1 has a high prevalence in Europe and North and South America [12] and accounts for 46% of the overall HCV infections [11]. Whereas G3 is more prevalent in Australia and South Asia [12] and is causative of 30% of the global HCV burden [10,11]. Other HCV genotypes (G2 - G7) account for the remaining HCV infections and generally remain highly local concerns [11,13].
The HCV genome is 9.6 kb in length, translating into a precursor polyprotein of ~3000 residues [14]. This polyprotein is then enzymatically processed to yield four structural (core, envelope glycoproteins E1 and E2, and an ion channel p7) and six non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [15]. The structural proteins are processed by host peptidases, while the non-structural proteins are cleaved by viral proteases, NS2 and NS3 [16]. In particular, NS2 is an auto-protease that is responsible for intramolecular cleavage between NS2 and NS3 of HCV polyprotein [17,18]. In the case of NS3, the protein has an N-terminal serine protease domain alongside a C-terminal helicase domain with NTPase activity [19,20,21,22]. NS3, together with its cofactor NS4A, makes a heterodimeric NS3/4A complex that cleaves four scissile peptide bonds (3–4A, 4A–4B, 4B–5A, and 5A–5B) between non-structural proteins [23,24,25]. The multifunctional protein NS3 additionally hydrolyses two signaling proteins, including mitochondrial antiviral signaling protein (MAVS) and C-terminal Toll-interleukin-1 receptor (TIR) domain-containing adaptor inducing IFN-b (TRIF) [26]. Importantly, MAVS and TRIF are responsible for activating the immune response to viral infections [26,27]. NS3/4A-mediated processing of viral and host proteins is central to viral replication and innate immune response, rendering it an ideal therapeutic target [19,20,21,22,28,29].
To date, several NS3/4A targeting direct-acting antivirals (DAAs), including boceprevir [30], telaprevir [31,32], narlaprevir [33], simeprevir, grazoprevir, paritaprevir and glecaprevir have been approved by Food and Drug Administration (FDA) [24,25,34] that act to inhibit viral replication [15,35,36,37,38,39,40]. Three of these marketed drugs are covalent (serine-trap) inhibitors [34]. Nucleophilic attack by the hydroxyl group of Ser139 at NS3 protease catalytic triad on the electrophilic warhead leads to the formation of a stable covalent bond between NS3/4A and inhibitor [33,41,42,43,44]. The nature of the electrophile dictates if covalent inhibition is reversible or irreversible [45]. Covalent NS3/4A inhibitors have demonstrated a good safety profile in addition to being potent in their antiviral activity [46,47]. However, the genetic sequence heterogeneity within NS3/4A impacts the efficiency of NS3/4A inhibitors [24,25]. Consequently, the antiviral response varies significantly between HCV genotypes [48]. NS3/4A of HCV G1 exhibits the highest sustained virological response (SVR) rate (up to 99%) to third-generation NS3/4A inhibitors [19,49,50,51,52]. However, the efficacy of DAAs targeting NS3/4A has been shown to be lower in other genotypes [53,54,55,56,57,58]. Specifically, in G3, a predominant HCV genotype in South Asia, including Pakistan [59] – a low to middle-income country, DAA targeting of NS3/4A has presented a major challenge towards affordable HCV treatment [60,61,62,63]. In addition to decreased SVR rates in response to DAAs [64], NS3/4A G3 is associated with an increased risk of liver cirrhosis and hepatocellular cancer [65].
Besides genotypic variation, polymorphism in NS3/4A is also associated with conferring resistance to approved DAAs [66,67]. These single-site mutations [68] within the ligand-binding pocket of NS3/4A G3 [68,69,70,71] cause resistance in drug binding [66,68,71] leading to the emergence of drug-resistant variants [2,9,72]. Nearly 50% of DAA treatment failures have reported resistance-associated substitutions (RASs) [73,74]. The frequently observed NS3/4A RASs include V36, T54, V55, Q80, R155, A156, and D168 [75]. Specifically, A156F/N/V, V36A+R155K/T, V36M+R155T, V36A/M+A156T, T54A+A156S, T54S+A156S/T, and V36M+T54S+R155K have reported resistance to telaprevir [76]. Therefore, the occurrence of RASs within NS3/4A has challenged the clinical success of approved DAAs [77]. Specifically, D168Q polymorphism has rendered NS3/4A G3 “naturally resistant” to most DAAs [67,69,78]. Moreover, the lack of availability of an NS3/4A G3 crystal structure [79,80] further limits the investigation of genotype-specific mutations [68,81] and their impact on NS3/4A’s interaction with inhibitors [82]. Taken together, NS3/4A G3 remains a difficult-to-treat strain.
In recent decades, in silico drug discovery has revolutionised conventional pharmaceutical development [83,84,85]. Various in silico methods assisted in the discovery of potent antiviral compounds against viruses such as dengue [86], zika [87,88], influenza [89], SARS-coronavirus [90,91] and Ebola [92]. Following this, we have combined in silico techniques [91,93,94,95] to identify small molecules targeting HCV NS3/4A G3. In this work, we have modeled NS3/4A variants by considering G3-specific mutations at fourteen positions within ligand-binding pocket, including residues of the catalytic triad (F43L, H57R, Q80K, R123T, I132L, Y134C/R, S139P, R155G, A156T, V158A, C159V, D168Q, C525W/Y, and Q526H/R) followed by pharmacophore-based virtual screening (PBVS) and covalent docking protocol to identify potential covalent inhibitors. Binding interactions of potential antivirals identified in this study were validated using molecular dynamics (MD) simulations. This study reports a potential anti-HCV compound targeting NS3/4A G3-specific mutations exhibiting strong binding affinity scores (-6.5 to -4.1 kcal/mol) that has the potential to be directly tested for in vitro and in vivo studies for HCV drug development.
2. Materials and Methods
2.1. Sequence Retrieval and Modeling HCV NS3/4A G3
In silico modeling was initiated by retrieving sequences of HCV NS3/4A genotype 3 (G3) from the National Center for Biotechnology Information (NCBI) protein database [96] (see Supplementary Data S1). Inhibitor-bound HCV NS3/4A complexes were retrieved from Protein Data Bank (PDB) [97] for binding site analysis (see Supplementary Table S1). The binding pocket of HCV NS3/4A was investigated using LigPlot+ v.2.1 [98] and Maestro v.11 (Schrödinger, LLC, New York) [99], and the ligand-binding residues were identified (see Supplementary Table S2). Multiple sequence alignment (MSA) was performed using Clustal Omega [100], and the identified interacting residues identified were analysed for mutations within G3-specific sequences retrieved from NCBI protein database [96] (see Supplementary Data S2). A representative sequence for each mutation was selected as a G3-specific variant for modeling (see Supplementary Table S3). ESPript 3.0 was used for MSA visualization [101]. Templates for homology modeling were identified from PDB using Protein Basic Local Alignment Search Tool (BLASTP) algorithm [102] (see Supplementary Table S4). Co-crystallized inhibitor-bound HCV NS3/4A complex (PDB ID: 4A92) was selected as a template. Homology modeling of HCV NS3/4A G3-specific variants was performed using SWISS-MODEL Server [103], an automated homology modeling resource.
2.2. 3D Structure Refinement and Validation
The predicted homology models were refined using GalaxyRefine [104]. Quality assessment of the models was done by using FATCAT [105], MATRAS [106], ERRAT [107], ProSA [108], SAVES Server [109], MolProbity [110], Verify 3D [111], PROCHECK [112] and Maestro [99]. Models with the least number of residues in the disallowed region were selected. The root mean square deviation (RMSD) values were calculated using CHIMERA [113] (see Supplementary Table S5).
2.3. Ligand-Based Pharmacophore Modeling and Virtual Screening
Ligand based pharmacophore modeling was carried out with the Phase module of Maestro [114,115] against HCV NS3/4A. A set of over 100 HCV NS3/4A covalent and non-covalent inhibitors, including marketed drugs reported in literature were retrieved from Protein Data Bank (PDB) (see Supplementary Table S6). The features from these ligands were extracted to generate the ligand-based pharmacophore hypotheses. The compounds were screened from small molecule databases including ZINC [116], MolPort [117], PubChem [118], Mcule [119], and ChEMBL [120]. The screening was performed using the Pharmit web server [121] which uses a pharmacophore and molecular shape search. The resulting hits were refined by applying a series of filters, including Lipinski’s rule (RO5), with at least 2 violations [122]. The oral bioavailability assessment was done using Osiris DataWarrior software [123] and pharmacokinetic and pharmacodynamic properties of the screened ligands were evaluated. These strict criteria removed a huge dataset, and the remaining compounds were utilised for more sophisticated covalent docking. The detailed framework applied in the study for targeting HCV NS3/4A G3 variants using in silico approaches is represented in Figure 1.
2.4. Preparation of HCV NS3/4A and Covalent Docking Protocol
The Protein Preparation Wizard in Maestro [124] was used to prepare modeled HCV NS3/4A G3 variants (G3.v1 - G3.v14). The protein structure was pre-processed by assigning bond orders and adding missing hydrogen atoms using Epik [125] at pH 7.0 ± 2.0. Different tautomeric states were generated for each ligand. Moreover, two alternative stereoisomers were generated per ligand while retaining the specified chiralities. Next, the H-bond network was optimised with PROPKA [126] at pH 7.0. The histidine residues within the active site of serine protease were defined including the histidine of the catalytic triad (His57). Moreover, all water molecules were removed, and a restrained minimization was carried out using the newly optimized OPLS3e force field [99],and the convergence criterion of 0.3 Å RMSD was set for all heavy atoms. The compounds resulting from ligand-based pharmacophore search were processed with the LigPrep module of Maestro [127].
Since the active site of HCV NS3/4A contains a catalytic serine (Ser139), it is possible to target it with compounds that bind covalently. For example, within the crystal structure of HCV NS3/4A protease (PDB ID: 3LON), a ketoamide inhibitor narlaprevir is covalently attached to Ser139 of catalytic triad. Therefore, we carried out a covalent docking protocol to screen the resultant compounds using Schrödinger’s Covalent Docking (CovDock) tool [128] implemented in the Maestro molecular modelling package [99]. In addition to Michael addition reaction, we also explored other reaction types, including nucleophilic addition to double bond or triple bond.
The center of the covalent docking site was defined as the centroid of the catalytic Ser139 residue, and a cube grid centered on this point was set with an edge length of ≤ 20 Å. The compounds were initially docked using the fast-virtual screening mode of CovDock in Maestro and Michael addition reaction to investigate the formation of covalent interaction with Ser139. The compounds that successfully interacted covalently with Ser139 were again docked using a thorough pose prediction mode in Maestro [99] and a maximum number of 100 poses was selected. The cut-off at 2.5 kcal/mol was set to retain poses for further refinement. The poses were ranked using Prime [129] MMGBSA score that employs variable dielectric generalized Born solvation model (VSGB 2.1) [130,131] in the OPLS3e force field [132]. These steps were repeated for other reaction types including nucleophilic addition to a double and a triple bond, nucleophilic substitution, aryl and nitrile activated conjugate addition to the alkyne. The top-ranking compounds were selected for validation using Molecular Dynamics (MD) Simulations.
For G3.v3 with S139P mutation, molecular (noncovalent) docking-based virtual screening was performed to identify potential protease inhibitors. The variant was prepared using a protein preparation wizard in Maestro [99]. Hydrogens were added, and proper bond order and energy minimization were performed. The molecular interaction grid was centered at the active site containing the catalytic triad and was prepared using the receptor grid generation module in Maestro [99]. Molecular docking was performed on selected compounds from pharmacophore-based virtual screening using Glide program of Schrodinger [133]. The resulting lead compounds were analysed for the binding interaction with the catalytic triad.
2.5. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations of each complex were performed to investigate the binding potential of the hit compounds interacting with the residues inside the active site of HCV NS3/4A G3. MD simulations were performed for 100 ns using AMBER 20 simulation package [134]. Simulation trajectories elucidated complex stability, and interaction profiles were investigated. MD simulation protocol as described previously [135,136,137] was implemented however, the length of the production run was increased from 20 to 100 ns. Parameterisation was performed, and a new molecular topology file for each ligand was created. This was initiated from each ligand and ended in covalently bound Ser139. The topology and coordinate files of the complexes were generated using tleap program of AMBER. The Antechamber package of AmberTools was utilised, and parameters were extracted from the GAFF force field (GAFF) [138]. Counter ions were added around the ligand-protease complex to neutralise the charges of each simulation system. The complex was centred in a dodecahedral TIP3P [139] water box with a distance of 10 Å between the solute and the box edge. To maintain constant bond length, covalent bonds were constrained using the SHAKE algorithm [140]. The system was heated and equilibrated after a stepwise minimisation. A production run was performed at 300 K and 1 bar pressure for a period of 100 ns. The time step of 2 fs was set and the trajectory snapshots were saved every 2 ps for onward analysis using the CPPTRAJ program [141] of AMBER.
2.6. Binding Free Energy Calculations
The binding free energies (ΔGbind) of HCV NS3/4A G3 variants complexed with the most promising hit compounds were calculated using the MM-GBSA method, implemented in AMBER 20. For each system, 10000 snapshots were generated from the last 50 ns stable trajectories with an interval of 5 ps. The total binding free energy was calculated as a sum of solvation free energy (ΔGsol) and the molecular mechanics binding energy (ΔEMM) as given below:
ΔEMM = ΔEint + ΔEele + ΔEvdw
ΔGsol = ΔGpl + ΔGnp
ΔGtotal = ΔEMM + ΔGsol
ΔGbind = ΔEMM + ΔGsol – TΔS
Here, ΔEMM includes electrostatic energy (ΔEele), internal energy (ΔEint), van der Waals energy (ΔEvdw), and the polar (ΔGp) and non-polar (ΔGnp) energy components contributing towards total solvation free energy (ΔGsol). ΔGtotal is the free energy of binding evaluated for both MM-GBSA and MM-PBSA methods after entropic calculations (-TΔS). Per-residue energy decomposition analysis was performed using the MM-GBSA method to estimate the contribution of interacting residues towards ligand-binding. The binding energy was calculated as ΔGresidue using the equation below:
ΔGresidue = ΔEMM + ΔGsol
Here, the ΔGresidue denotes the total energy obtained from sidechain and backbone energy decomposition. Only those amino acids were considered within 8 Å of the active site. The sum of energy contributions from each residue is equal to the system’s overall binding energy [142]. This strategy has been well established for protein–ligand binding studies and for studying protein–protein interactions [95,143].
3. Results and Discussion
3.1. Analysis of Sequence Variation and Molecular Modeling
The results from sequence alignment revealed the catalytic triad of HCV NS3/4A G3 as a site of viral polymorphism due to mutations including H57R and S139P (Figure 2). The analysis further showed mutations within HCV NS3/4A ligand-interacting residues at positions 43, 80, 123, 132, 134, 139, 155, 156, 158, 159, 168, 525 and 526. Of these residues, mutations at positions 80 [144,145,146], 155 [68,147], 156 [148], 123 [66,67] and 168 [149,150,151,152] have been frequently reported as drug resistance-associated substitutions (RASs) in HCV NS3/4A. A representative sequence for each mutation was selected to model the HCV NS3/4A G3 variant (see Supplementary Table S3).
Crystal structure of HCV NS3/4A protease-helicase G1b (PDB ID: 4A92) with the sequence identity in the range 74% - 82% (E-value 0.0) for 15 HCV NS3/4A G3 variants was selected as a template for homology modeling (see Supplementary Table S4). The structure was minimized and used for the HCV3/4A modelling (see Supplementary Figure S1).
The predicted structure quality score was computed using ERRAT [107] and Verify 3D [111] and ranged between 91.33 to 94.85. The MolProbity [110] quality values ranged between 1.50 - 1.61. The superimposition of variants with the template indicated root mean square deviation (RMSD) values in the range 0.28 Å - 0.30 Å (see Supplementary Table S5). The Ramachandran plots generated for each variant model showed model residues >95% in the favored regions (see Supplementary Figure S2). Furthermore, we evaluated the reliability of HCV NS3/4A G3 wildtype (WT) model with its respective templates (PDB IDs; 4B75, 1CU1, 3O8B, 5WDX and 4B6E) and root-mean-square-fluctuations (RMSF) were compared after 100 ns MD simulations. The results indicated no significant fluctuations and the movements of secondary structural elements were analogous to the temaplates, while the catalytic triad remained converged below 1Å (see Supplementary Figure S1).
3.2. Ligand-Based Pharmacophore Search
Next, we incorporated both covalent and non-covalent features of reported HCV inhibitors to build a pharmacophore hypothesis against HCV NS3/4A G3. To identify compounds with the most likely interacting substructures and the potential warheads that can establish covalent bonds with catalytic Ser139, a set of well-defined HCV NS3/4A inhibitors was retrieved (see Supplementary Table S6). Using the PHASE 4.0 module of Schrodinger molecular modeling [99], pharmacophore hypotheses were generated. Five hypotheses were generated, and the hypothesis with the best survival score (5.080) and fitness score was selected. This model consisted of four features (AAHH), including two hydrogen bond acceptors (A4 and A6) and two hydrophobic groups (H14 and H15). The pharmacophore hypothesis was validated with receiver operating characteristic (ROC) analysis [153] to assess its ability to correctly classify compounds as active or inactive [154]. The performance of the pharmacophore hypothesis was further evaluated by the area under the curve (AUC) of the corresponding ROC curve (see Supplementary Figure S3). The validated pharmacophore model AAHH curve (see Supplementary Figure S4) was used to screen databases in the Pharmit web server [121] containing 341,098,760 compounds with 1,606,918,818 conformations. Based upon Lipinski’s rule of five (Ro5), 1,321,302 drug-like compounds were obtained which were further filtered based on ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties in Osiris DataWarrior software [123]. Unique compounds from the filtered 4215 compounds were prepared for docking studies (see Supplementary Table S7).
3.3. Covalent Docking-Based Virtual Screening
Ser139 is a key residue in the active site of HCV NS3/4A, which makes it an attractive target for the design of covalent inhibitors [33,155,156]. Covalent docking-based virtual screening was performed to target the reactive nucleophilic Ser139 to identify compounds with reactive electrophilic moieties. The virtual screening was conducted in two steps, involving i) the selection of candidate ligands based on their relevant conformation such that reactive groups are in close proximity to Ser139; ii) and a virtual chemical reaction between the reactive groups, leading to the formation of a stable covalent bond (S-C) [157,158]. Ligand poses within 5 Å distance cut-off from Ser139 were kept, forming a covalent bond (S-C) according to the reaction type. To identify potential covalent inhibitors from a pharmacophore-based screened library containing chemical warheads, the covalent binding reactions used included Michael addition, nucleophilic addition to a double and a triple bond, nucleophilic substitution, and aryl and nitrile activated conjugate addition to alkyne. The free energies of binding were calculated with the Prime/MM-GBSA method for all the docked poses (see Methods section for details). The compounds with the lowest Prime/MM-GBSA and/or CovDock scores were considered for further molecular inspection.
Overall, a total of 23 compounds from the results of the Michael addition screening, while no compounds could be identified from other reaction types: nucleophilic addition to double bond and nitrile activated conjugate addition to alkyne. Inspection of the structures and binding poses led to the selection of 7 compounds (see Supplementary Table S8 and Supplementary Figure S5). Amongst these, only one inhibitor candidate, CHEMBL569970 (cpd-217: PubChem45485999) (Figure 3) reported high CovDock docking score with all variants incorporating G3-specific mutations and was selected for a detailed interaction analysis. A detailed interaction analysis of cpd-217, and its stability inside the active site of HCV NS3/4A G3 variants, was assessed through 50 ns MD simulations and binding free energies were calculated.
Molecular docking of G3.v1 resulted in 60 compounds (see Supplementary Table S9 and Supplementary Figure S6). Ligand interaction analysis was performed to filter leads. The compound cpd-217 indicated stable interactions, which were validated by molecular dynamics simulation analysis.
3.4. Molecular Insights of Identified Potential Covalent Inhibitor (cpd-217)
The docked complexes of a potential hit with HCV NS3/4A variants were investigated for molecular interactions governed by covalent bond formation. The structures of the lead compound and their molecular interactions with the binding pocket of HCV NS3/4A G3 variants, v1 to v14 (Figure 4A-N), along with the corresponding reaction sites between the chemical warhead of the lead compound and the catalytic Ser139 are displayed in Figure 4.
Overall, cpd-217/ G3 complexes, the catalytic serine (Ser139) was covalently bound to the Michael acceptor warhead (≤ 1.9 Å distance), except HCV NS3/4A G3.v3, which had proline instead of serine at position 139. The residues within the binding pocket [159] of all variants interacting with cpd-217 were compared with the residues interacting with the alpha-ketoamide inhibitor, boceprevir (PDB: 2O8) (see Supplementary Figure S7). Like Boceprevir, cpd-217 formed interactions with Gln41, His57, Ile132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, and Val158. Additionally, cpd-217 interacted with Thr42, Phe43, Val55, Gly58, Asp81, Val158, Cys159, Met485, Phe486, Asp487, Ser488, Val524, Cys525, Gln526, and His528 (Table 1). All variants exhibited R123T (except R123S in G3.v10), I132L, Y134C (except G3.v2) and D168Q mutations of which R123T and D168Q are frequently reported as RASs. Additionally, in G3.v1 with H57R mutation, cpd-217 made five H-bonds with Gln41, Thr42, mutated catalytic His at position 57 to Arg, Leu135 and Arg155. In G3.v2 with Q80K mutation, cpd-217 made four H-bonds with Gln41, His57, Leu135 and Gly137. G3.v3 exhibited catalytic Ser139 mutation to Pro, besides R155G mutation. Cpd-217 made three H-bonds with Asp81, Asp487 and Gln526 and established a salt-bridge interaction with Asp81. In G3.v4 (C525W) and G3.v5 (C525Y), cpd-217 made four H-bonds with Gly137, Ser139 and Ala157. Additionally, a π-π interaction was observed with His57 in G3.v4. In G3.v6 with the F43L mutation, cpd-217 made three H-bonds with residues Thr42, Gly137 and Ala157. Cpd-217 was observed making two H-bonds with Leu137 and Gly137 in G3.v7 (Q526H) and an additional H-bond with His57 in G3.v8 (Y134R). In G3.v9 (V158A), cpd-217 made π-cation interaction with Lys136 besides interacting with Gln41, His57 and Arg155 making three H-bonds. Cpd-217 made four H-bonds with His57, Leu135 and Ser139 besides covalently inhibiting Ser139 in G3.v10 with R123S mutation. In G3.v11 with A156T mutation, no H-bond formation was observed. However, cpd-217 made six H-bonds with Gln41, His57, Leu135 and Ser139 in G3.v12 exhibiting Y134S mutation. A salt-bridge interaction between cpd-217 and Asp81 was observed in G3.v13 with Q526R mutation. Additionally, two H-bonds between cpd-217 and Asp81 and Gly137 were observed. Cpd-217 made five H-bonds with Gln41, Gly137, Ser139 and Ala157 in G3.v14 with Y134T and C159V mutations.
These H-bonds were evinced from the reported crystal complexes, where eight hydrogen bonds are conserved between protease residues (Gly137, Ser138, Ser139, Arg155, Ala157, Ser159) and the viral substrate residues [160] indicating that they played a vital role in binding within wild type (WT) systems [34].
We observed that mutations within the catalytic triad residues: His57Arg and Ser139Pro hindered interaction of cpd-217 with Gly137, a residue of the oxyanion hole, in G3.v1 and v3. Mechanisms of drug resistance due to mutations at positions 80 [144,145,146], 155 [68,147], 156 [148], 123 [66,67] and 168 [149,150,151,152] have been well-investigated [161,162] and the importance of these residues in determining the specificity of inhibitors is substantial. RASs R123T and D168Q in addition to mutation at catalytic triad residue H57R (G3.v1), RASs R123T and Q80K (G3.v2) and RASs R123T and R155G along with mutation at catalytic S139P in G3.v3 inhibited interaction of cpd-217 with Gly137. Moreover, R123T and D168Q along with another RAS A156T weakened the association of cpd-217 with G3.v11 resulting in no H-bond formation or hydrophobic interactions (Figure 5A-N).
However, the success of virtual screening is delineated by finding novel chemical structures, new scaffolds are clearly preferred compared to already known scaffolds. Moreover, due to the ability of covalent inhibitor to bind irreversibly to off target proteins, which could lead to toxicological effects, such as immune responses. We utilized SEA (Similarity Ensemble Approach; http://www.sea.bkslab.org) server to identify the presence of similar chemical scaffolds of cpd-217, based on the set-wise chemical similarities against ChEMBL database. The results indicated no structural similarity with any known anti-viral inhibitors (Supplementary Table S10).. We further predicted the unfavorable side-effects due to off-target effects of known molecules and drugs, SwissTargetPrediction web server (http://www.swisstargetprediction.ch) was utilized which combines different measures of chemical similarity based on both chemical structure (2D) and molecular shape (3D). The cpd-217 was found to have less than 0.1 off-target probability based on cross-validation analysis in ChEMBL databse for human protein ligands (Supplementary Table S11).
3.5. The Stability and Flexibility of cpd-217 through MD Simulation
Molecular dynamics studies have emerged as a reliable method for investigating the stability of protein-ligand complexes [163,164,165,166,167]. Recently, these methods have been combined with more sophisticated binding free energy calculations (e.g. MM/GBSA) to explore the drug resistance mechanisms of HCV resulting from several key mutations of NS3/4A [57,58,168,169]. Here, docking energies expounded only the initial conformation of the cpd-217 at the active site of NS3/4A G3 variants. These complexes were further exploited through MD simulations to predict the most likely binding mode between cpd-217 and the associated residues in all G3 variants (v1 to v14). The present study is primarily involved in the conformation of cpd-217 in G3 variants. Therefore, we were interested in analysing the stability and conformational flexibility of the cpd-217 bound to each G3 variant and corresponding energy contributions of potential residues towards ligand. To elucidate the dynamic stability and to ascertain the rationality of the ligand sampling, root-mean-square deviation (RMSD) and root-mean-sqaure-fluctuation (RMSF) values of protein backbone atoms of each G3 variant and heavy atoms of cpd-217 relative to the respective initial structures were calculated, and RMSD/RMSF trajectories were analysed throughout 100 ns. The representative RMSD and RMSF trajectory plots of each G3 variants with bound cpd-217 are displayed in Figure 6A-N and Figure 7A-D.
Overall, the RMSD values of protein backbone atoms (in all variants) and heavy atoms of cpd-217 remained within 3Å throughout the simulation period, except minor variations (slightly higher than 3Å) were observed in G3.v7 and G3.v9 within the initial 30 ns. The RMSD of backbone atoms of the order of 1 – 3 Å with no significant conformational change confirmed that the system is well equilibrated and cpd-217 remained stable inside the binding pocket of all NS3/4A variants during the simulation period [170,171,172]. Whereas, RMSFs highlighted the flexible regions of G3.WT and G3 variants. There were no pronounced Cα-RMSF differences occurred except few small fluctuations for residues 65 to 72 and 85 to 90. Moreover, no gradual fluctuations were observed in the the catalytic site, which evinced the favourable conformation of cpd-217 inside the binding pocket and depicted convergance of catalytic triad residues (< 1 Å) throughout the simulation period.
3.6. Binding Free Energy Calculated by MM/GBSA Method
The MM/GBSA method has been widely employed in improving the protein-ligand docking results [171,172,173] considering the protein backbone dynamics, electrostatic, van der Waal (vdW), and entropic contributions on the overall binding energy of the complex. The MM/GBSA approach has been widely used in expounding the mechanisms of mutation-induced drug resistance [58,174,175,176,177,178,179,180]. MMGBSA total binding free energy values were extracted from initial frames (avg. of 1000 snapshots from 0.01 to 0.5 ns and referred to as “before MD” or “initial”), and final frames (avg. of 10000 snapshots from last 50 ns, after every 5.0 ps and referred as “after MD”). The total binding free energies (ΔGtotal) and energetic components are tabulated in Table 2. We also calculated the affinity score variation (ΔAS) between wild-type (WT) and G3 variants scores to examine the impact of mutation in overall binding with the cpd-217. To further evaluate the binding stability during simulation, energy shift (ΔΔGshift) was incorporated, which supported the stabilising effect of mutation on ligand binding in protein/ligand complex formation, i.e., more negative value represents more favourable stabilising effect in complex with cpd-217. According to the MMGBSA values during MD simulations (Table 2), the overall binding free energies (ΔGbind) of WT was -14.4 kcal/mol. The major energy contributions to the ligand-binding were due to the favorable van der Waals interactions for WT/cpd-217 complex (ΔEvdw = -41.7 kcal/mol) as compared to electrostatic interactions (ΔEele = -32.8 kcal/mol). In contrast, total solvation energies (ΔGsol) showed unfavorable contributions. These values of WT were compared with the binding energies of all G3 variants. The lower binding energy agrees to a higher affinity towards complex stability and vice versa [173,181]. All G3 variants (v1 to v14) possess few common mutations at Arg123, Ile132 and Asp168, along with other specific mutations, the quantitative information of important residue’s contribution towards cpd-217 binding was exploited through per-residue decomposition analysis (Figure 8).
In Table 2, the ΔGbind of the WT complex was more favorable than the G3 variants, except G3.v2 and v14 (-15.6, and -15.1 kcal/mol). On the contrary, the G3 variants with lower binding affinities indicated that the specific mutations in these variants will trigger drug resistance to a certain extent; this phenomenon has been explicitly elaborated in several molecular modeling assisted drug-resistance studies [53,180,182,183,184,185,186]. Likewise in WT, it was obvious that the ΔEvdw contribution is the main component in the total binding affinity in each variant (ranged from -33.5 to -44.2 kcal/mol), and the following was the ΔEele (ranged from -26.4 to -32.5 kcal/mol). The total solvation energies (ΔGsol) that counteract the electrostatic interactions of all systems were within a difference of < 3 kcal/mol from the WT (36.4 kcal/mol). The contributions of the conformational entropy (-TΔS) in all variants ranged between 15.2 and 25.9 kcal/mol. It was observed that the conformational entropic contributions for the complexes have no impact on the order of the ligand’s free energy of binding (ΔGbind). Therefore, the van der Waals contribution was considered more crucial for cpd-217 interaction with NS3/4A protease and differentiating the binding affinities among these variants.
Amongst the G3 mutants, only G3.v2 and G3.v14 displayed a negative affinity variation (ΔAS) of -1.2 and -0.7 kcal/mol, respectively and depicted slightly higher binding affinity with cpd-217. All other G3 variants displayed a positive affinity variation (ΔAS), as low as +2.8 and as high as +8.1 kcal/mol, and exhibited lower binding affinity than WT. Particularly, the ΔGbind of G3.v4 to G3.v7, and G3.v9 to G3.v11 were approximately the same (ranged from -9.1 to 9.9 kcal/mol, with < -1 kcal/mol variation) and exhibited similar positive correlation (ΔAS ranged from +4.6 to +5.3 kcal/mol). The trend was likely due to the presence of four combined mutations (R123T + I132L + Y134C + D168Q) in these variants (except G3.v10 with R123S mutation), which triggered a similar impact on cpd-217 binding as can be observed from converged protein backbone deviation (<3 Å) (Figure 6). Overall, the variants with R123T + I132L + Y134C + D168Q mutations displayed low binding affinity compared to the WT. More in-depth per-residue decomposition analysis of these variants further supported the underlying impact of these mutations upon ligand binding (Figure 8). Among these G3 variants (G3.v4 to G3.v7, G3.v9 to G3.v11), the interaction energy (ΔGresidue) between cpd-217 and mutated residues R123T (-0.99 to -1.84 kcal/mol, while -0.44 kcal/mol in R123S in G3.v10), I132L (-0.95 to -1.46 kcal/mol), Y134C (-0.23 to -0.63 kcal/mol), D168Q (-0.14 to -0.61 kcal/mol) were less favorable than those in the WT complex (-2.14, -1.56, -0.96, -0.95 kcal/mol). Moreover, R123T + I132L + Y134C + D168Q mutations in these variants (v4 to v7, v9 to v11) further reduced the energy contributions by Gln80 (-0.8 to -1.45 kcal), Asp81 (-0.87 to -1.51 kcal/mol), and Ser139 (-1.72 to -2.13 kcal/mol) as compared to its WT complex (-1.89, -1.56, and -2.36 kcal/mol).
G3.v3 displayed the highest positive affinity variation (ΔAS = +8.1 kcal/mol) and showed the lowest binding affinity of -6.3 kcal/mol among all G3 variants. The underlying impact of G3.v3 on cpd-217 was mainly due to the S139P mutation and combined effects of R155G and D168Q mutations. Note that the essential function of Asp168 is to stabilise the conformation of Arg155 to maintain the favorable interaction between Arg155 and ligand [53,54,58,82,162,168,187,188,189]. On the other hand, Proline residues are known to cause short- and long-range disruptive changes in secondary structural elements by causing steric hindrance [190]. Ser139 is one of the important catalytic residues and substitution of this residue has been reported in altered protease activity [191]. An incorporation of glycine at position 155 completely diminished the important interactions with ligand. At the same time, the substitution of S139P further augmented the impact on cpd-217 binding, as evident from the lowest energy shift (ΔΔSshift) of only -2.3 kcal/mol. The contribution energies by these substituted residues further delineated the binding profile with cpd-217. In G3.v3, the interaction energy was abruptly reduced between cpd-217 and mutated residues Pro130 and Gly155 (-0.46 and +0.86 kcal/mol) compared to Ser139 and Arg155 (-2.36 and -2.45 kcal/mol) in WT complex. Moreover, it also impacted the other catalytic triad residues where the interaction energy (ΔGresidue) of His57 and Asp81 were reduced to -1.56 and -1.14 kcal/mol compared to WT complex (-2.8 and -1.56 kcal/mol). Hence, the binding affinity of G3.v3 was the lowest compared to WT. Likewise, H57R in G3.v1 exhibited a similar impact on the catalytic triad, where the incorporation of the long side chain of Arg57 obstructed the binding site which posed the cpd-217 in different conformation as can be seen from a sudden fluctuation at ~50 ns in Figure 6A, that remained stable afterwards. The Arg57 displayed less interaction energy (-1.42 kcal/mol) as compared to His57 in WT, and the conformation of the side chain of Arg57 further reduced the interaction energy of Asp81 (-0.24 kcal/mol) and Ser139 (-0.84 kcal/mol) as compared to WT complex.
G3.v2 and v14 showed negative affinity variation among all G3 variants and displayed slightly higher binding affinities of -15.6 and 15.1 kcal/mol compared to WT. In G3.v2, the Q80K slightly increased the interaction energy of adjacent Asp81, from -1.56 (WT) to -1.8 kcal/mol, while in G3.v14, the Y134T instead of Y134C (as seen in other G3 variants) along with other mutations triggered a combined effect towards cpd-217 (Table 2). The underlying impact in these two variants (G3.v2 and v14) with bound cpd-217 was evident from the highest energy shift (-8.7 and -9.9 kcal/mol, respectively) during the simulation period, which had a stabilising effect.
To summarize, the structural and pharmacophoric characteristics described here can be utilised to find new leads in compound databases and to design new inhibitors that target G3 variants. This work has supported the idea that further optimization of structural and pharmacophore properties can lead in developing a multifunctional small-molecule inhibitor that targets all of the common G3 specific mutations. Future efforts would be necessary to create novel multi-functional anti-HCV inhibitors to study the structural and pharmacophore properties of cpd-217 that are responsible for its diverse activity against all G3 variants.
4. Conclusions
This work reports the drug-resistant mutations within ligand-binding residues including catalytic triad of HCV NS3/4A. The reported mutations are specific for HCV NS3/4A genotype 3 (G3) prevalent in developing countries, including Pakistan. We report mutations within the catalytic triad residues H57R and S139P and RASs Q80K, R123T, R155G, A156T and D168Q in HCV NS3/4A G3 hinder the interaction of drugs with Gly137 of oxyanion hole. This work highlights the pharmacoinformatics approaches utilised to identify a potential covalent inhibitor of HCV NS3/4A G3 to treat Hepatitis C Virus. Several ligands and FDA-approved marketed drugs were used to generate a pharmacophore with a similar scaffold to screen multiple small molecule libraries. Pharmacophore-based virtual screening (PBVS) followed by covalent docking protocol identified cpd-217 (CHEMBL569970; PubChem45485999) as a potential inhibitor of HCV NS3/4A G3 serine protease. The binding affinity, molecular interactions and stability of binding of the lead compound were investigated by molecular docking protocol and molecular dynamics simulations analysis. The potential warhead identified in this work can serve as a guideline to design covalent inhibitors targeting the catalytic Ser139 considering G3-specific drug-resistant mutations within HCV NS3/4A. The proposed inhibitor may play a key role in expediting the drug discovery process and can be tested in clinical trials to treat HCV. This approach can provide a plethora of energetic information, including the binding free energy between protein and ligand, in addition to enriched structural–dynamical information of protein complex structures in solution. Such information is critical for understanding the nature of protein-ligand interactions and guiding drug design and development, which experimental techniques struggle to readily provide.5. Patents
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1 -S8, Tables S1 – S9
Author Contributions
S.U. and M.F. designed the project and supervised the research; K.I. and M.U.M. developed the pipeline; K.I., M.U.M. and F.S. carried out the in silico analyses; M.U.M carried out the molecular dynamics simulations; K.I., M.U.M, and S.U. wrote the manuscript. S.U., J.F.T., and M.F. obtained funding and provided computational resources. All authors edited and approved the manuscript.
Funding
This work was supported by HEC (21-320SRGP/R&D/HEC/2014, 20-2269/NRPU/R&D/ HEC/12/4792 and 20-3629/NRPU/R&D/HEC/14/585), Ignite (SRG-209), TWAS (RG 14-319 RG/ITC/AS_C), NSERC Alliance (ALLRP 553704-20), and LUMS (STG-BIO-1008, FIF-BIO-2052 and FIF-BIO-0255) grants.
Data and Software Availability
The sequences were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) and structures were downloaded from PDB (https://www.rcsb.org/), which are freely available. For the in silico analyses, several online resources were utilized which are available free for use. Sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) while template for homology modeling was identified using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). For alignment visualization, ESPript 3.0 (https://espript.ibcp.fr/ESPript/ESPript/) was used. Homology modeling was performed using SWISS-MODEL (https://swissmodel.expasy.org/). For structural refinement and assessment webservers including GalaxyRefine (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE), FATCAT (https://fatcat.godziklab.org/), ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/), ProSA (https://prosa.services.came.sbg.ac.at/prosa.php), MolProbity (http://molprobity.biochem.duke.edu/) were used while Verify 3D and PROCHECK were accessed from https://saves.mbi.ucla.edu/. Binding site residues were identified using freely downloadable software including LigPlot (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/download.html) and Chimera (https://www.cgl.ucsf.edu/chimera/download.html) was used for the active site analysis. Small molecule database screening was performed using Pharmit online resource (https://pharmit.csb.pitt.edu/search.html). For pharmacophore design, covalent and molecular docking studies and ligand interaction analysis Maestro, Schrodinger was used that offers a trial learner’s license for use (https://www.schrodinger.com/products/maestro). Molecular dynamics simulations and free energy calculations were performed using AMBERTools Suite (https://ambermd.org/index.php) which is freely available for use.
References
- Lindenbach, B.D.; Rice, C.M. Flaviviridae: the viruses and their replication, vol. 1; Lippincott Williams & Wilkins: Philadelphia, PA, 2001. [Google Scholar]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–40. [Google Scholar] [CrossRef]
- Dan, A.A.; Martin, L.M.; Crone, C.; Ong, J.P.; Farmer, D.W.; Wise, T.; Robbins, S.C.; Younossi, Z.M. Depression, anemia and health-related quality of life in chronic hepatitis C. J. Hepatol. 2005, 44, 491–498. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, S21–S29. [Google Scholar]
- Seeff, L.B. Natural history of chronic hepatitis C. Hepatology 2002, 36, s35–s46. [Google Scholar] [PubMed]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus – 15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2013, 59, 318–327. [Google Scholar] [CrossRef]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a Novel Hepatitis C Virus Genotype From Punjab, India: Expanding Classification of Hepatitis C Virus Into 8 Genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deléage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspé, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Hepatitis C. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 1 May 2021).
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2014, 61, 77–87. [Google Scholar] [CrossRef]
- Cuypers, L.; Ceccherini-Silberstein, F.; Van Laethem, K.; Li, G.; Vandamme, A.; Rockstroh, J.K. Impact of HCV genotype on treatment regimens and drug resistance: a snapshot in time. Rev. Med Virol. 2016, 26, 408–434. [Google Scholar] [CrossRef]
- Murphy, D.G.; Sablon, E.; Chamberland, J.; Fournier, E.; Dandavino, R.; Tremblay, C.L. Hepatitis C Virus Genotype 7, a New Genotype Originating from Central Africa. J. Clin. Microbiol. 2015, 53, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat Rev Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Benureau, Y.; Warter, L.; Malcolm, B.A.; Martin, A. A comparative analysis of the substrate permissiveness of HCV and GBV-B NS3/4A proteases reveals genetic evidence for an interaction with NS4B protein during genome replication. Virology 2010, 406, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Lackner, T.; Müller, A.; Pankraz, A.; Becher, P.; Thiel, H.-J.; Gorbalenya, A.E.; Tautz, N. Temporal Modulation of an Autoprotease Is Crucial for Replication and Pathogenicity of an RNA Virus. J. Virol. 2004, 78, 10765–10775. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C Virus p7 and NS2 Proteins Are Essential for Production of Infectious Virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef]
- Raney, K.D.; Sharma, S.D.; Moustafa, I.M.; Cameron, C.E. Hepatitis C Virus Non-structural Protein 3 (HCV NS3): A Multifunctional Antiviral Target. J. Biol. Chem. 2010, 285, 22725–22731. [Google Scholar] [CrossRef]
- Ezat, A.A.; El-Bialy, N.S.; Mostafa, H.I.A.; Ibrahim, M.A. Molecular Docking Investigation of the Binding Interactions of Macrocyclic Inhibitors with HCV NS3 Protease and its Mutants (R155K, D168A and A156V). Protein J. 2013, 33, 32–47. [Google Scholar] [CrossRef]
- Mostafa, H.I.; El-Bialy, N.S.; Ezat, A.A.; Saleh, N.A.; Ibrahim, M.A. QSAR Analysis and Molecular Docking Simulation of Suggested Peptidomimetic NS3 Protease Inhibitors. Curr. Comput. Aided-Drug Des. 2014, 10, 28–40. [Google Scholar] [CrossRef]
- Failla, C.; Tomei, L.; De Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 1994, 68, 3753–60. [Google Scholar] [CrossRef]
- McCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr Opin Pharmacol. 2016, 30, 84–92. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals. Antiviral Res. 2017, 142, 83–122. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.M.; Ikeda, M.; et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci. 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R.; Ikeda, M.; Lemon, S.M.; Gale, M. Regulation of Interferon Regulatory Factor-3 by the Hepatitis C Virus Serine Protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.; Chevaliez, S.; McHutchison, J.G. The Hepatitis C Virus Life Cycle as a Target for New Antiviral Therapies. Gastroenterology 2007, 132, 1979–1998. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Patel, K. Future therapy of hepatitis C. Hepatology 2002, 36, S245–S252. [Google Scholar] [PubMed]
- Malcolm, B.A.; Liu, R.; Lahser, F.; Agrawal, S.; Belanger, B.; Butkiewicz, N.; Chase, R.; Gheyas, F.; Hart, A.; Hesk, D.; et al. SCH 503034, a Mechanism-Based Inhibitor of Hepatitis C Virus NS3 Protease, Suppresses Polyprotein Maturation and Enhances the Antiviral Activity of Alpha Interferon in Replicon Cells. Antimicrob. Agents Chemother. 2006, 50, 1013–1020. [Google Scholar] [CrossRef]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011, 29, 993–1003. [Google Scholar] [CrossRef]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; et al. Preclinical Profile of VX-950, a Potent, Selective, and Orally Bioavailable Inhibitor of Hepatitis C Virus NS3-4A Serine Protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [Google Scholar] [CrossRef]
- Arasappan, A.; Bennett, F.; Bogen, S.L.; Venkatraman, S.; Blackman, M.; Chen, K.X.; Hendrata, S.; Huang, Y.; Huelgas, R.M.; Nair, L.; et al. Discovery of Narlaprevir (SCH 900518): A Potent, Second Generation HCV NS3 Serine Protease Inhibitor. ACS Med. Chem. Lett. 2010, 1, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.U.; Iman, K.; Khalid, M.F.; Salman, H.M.; Shafi, T.; Rafi, M.; Javaid, N.; Hussain, R.; Ahmad, F.; Shahzad-Ul-Hussan, S.; et al. Evolution of efficacious pangenotypic hepatitis C virus therapies. Med. Res. Rev. 2018, 39, 1091–1136. [Google Scholar] [CrossRef] [PubMed]
- Heintges, T.; Encke, J.; zu Putlitz, J.; Wands, J.R. Inhibition of hepatitis C virus NS3 function by antisense oligodeoxynucleotides and protease inhibitor. J. Med Virol. 2001, 65, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; et al. EASL recommendations on treatment of hepatitis C 2018. J Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Falade-Nwulia, O.; Suarez-Cuervo, C.; Nelson, D.R.; Fried, M.W.; Segal, J.B.; Sulkowski, M.S. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann Intern Med. 2017, 166, 637–648. [Google Scholar] [CrossRef]
- (AASLD) AA for the S of LD. HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Patients Who Develop Recurrent HCV Infection Post Liver Transplantation. Available online: https://www.hcvguidelines.org/unique-populations/post-liver-transplant (accessed on 1 May 2021).
- Organization, WH. Guidelines for the care and treatment of persons diagnosed with chronic hepatitis C virus infection. 2018.
- Lin, K.; Perni, R.B.; Kwong, A.D.; Lin, C. VX-950, a Novel Hepatitis C Virus (HCV) NS3-4A Protease Inhibitor, Exhibits Potent Antiviral Activities in HCV Replicon Cells. Antimicrob. Agents Chemother. 2006, 50, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Hu, Z.; Jiang, X.; Decicco, C.P. Alpha-ketoamides, alpha-ketoesters and alpha-diketones as HCV NS3 protease inhibitors. Bioorg Med Chem Lett. 2000, 10, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.; Victor, F.; Lamar, J.; Johnson, R.; Wang, Q.M.; Glass, J.I.; Yumibe, N.; Wakulchik, M.; Munroe, J.; Chen, S.-H. P4 and P1′ optimization of bicycloproline P2 bearing tetrapeptidyl α-ketoamides as HCV protease inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 5007–5011. [Google Scholar] [CrossRef]
- Telaprevir (Incivek) and boceprevir (Victrelis) for chronic hepatitis C. Med Lett Drugs Ther. 2011, 53, 57–59.
- da Costa Leite, L.F.C. , Mourão, R.H.V., de Lima, M.D.C.A., Galdino, S.L., Hernandes, M.Z., Neves, F.D.A.R., Vidal, S., Barbe, J. and da Rocha Pitta, I.; et al. Synthesis, biological evaluation and molecular modeling studies of arylidene-thiazolidinediones with potential hypoglycemic and hypolipidemic activities. Eur J Med Chem. 2007, 42, 1263–1271. [Google Scholar] [PubMed]
- Isakov, V.; Koloda, D.; Tikhonova, N.; Kikalishvili, T.; Krasavina, E.; Lekishvili, K.; Malaya, I.; Ryska, M.; Samsonov, M.; Tolkacheva, V. Pharmacokinetics of the New Hepatitis C Virus NS3 Protease Inhibitor Narlaprevir following Single-Dose Use with or without Ritonavir in Patients with Liver Cirrhosis. Antimicrob. Agents Chemother. 2016, 60, 7098–7104. [Google Scholar] [CrossRef] [PubMed]
- Mayevskaya, M.V.; Ivashkin, V.T.; Znoyko, O.O.; Klimova, E.A.; Abdurakhmanov, D.T.; Bakulin, I.G.; Bogomolov, P.O.; Burnevich, E.Z.; Galushko, M.Y.; Geyvandova, N.I.; Zhdanov, K.V. Klimova2 DTA. Efficacy and safety of the Russian protease inhibitor narlaprevir at treatment-naive and earlier treated noncirrhotic patients with the 1st genotype chronic hepatitis C (PIONEER study). Russ J Gastroenterol Hepatol Coloproctology 2017, 27, 41–51. [Google Scholar]
- Gane, E.J.; Stedman, C.A.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. New Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Ghalib, R.; Reddy, K.R.; Pockros, P.J.; Ben Ari, Z.; Zhao, Y.; Brown, D.D.; Wan, S.; DiNubile, M.J.; Nguyen, B.-Y.; et al. Grazoprevir–Elbasvir Combination Therapy for Treatment-Naive Cirrhotic and Noncirrhotic Patients With Chronic Hepatitis C Virus Genotype 1, 4, or 6 Infection. Ann. Intern. Med. 2015, 163, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rockstroh, J.K. Summary from AASLD 2015 for Hepatitis C Beyond 95% SVR cure rates: still room for improvement? 2015. Available online: https://www.natap.org/2015/AASLD/AASLD_165.htm (accessed on 1 May 2021).
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. New Engl. J. Med. 2011, 364, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for Previously Untreated Chronic Hepatitis C Virus Infection. New Engl. J. Med. 2011, 364, 2405–2416. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Ban, Y.; Liu, H.; Yao, X. Computational study on the drug resistance mechanism against HCV NS3/4A protease inhibitors vaniprevir and MK-5172 by the combination use of molecular dynamics simulation, residue interaction network, and substrate envelope analysis. J Chem Inf Model. 2014, 54, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Xue, W.; Zhang, W.; Liu, H.; Yao, X. Understanding the drug resistance mechanism of hepatitis C virus NS3/4A to ITMN-191 due to R155K, A156V, D168A/E mutations: a computational study. Biochim Biophys Acta (BBA)-General Subj. 2012, 1820, 1526–1534. [Google Scholar] [CrossRef]
- Özen, A.; Sherman, W.; Schiffer, C.A. Improving the resistance profile of hepatitis C NS3/4A inhibitors: Dynamic substrate envelope guided design. J Chem Theory Comput. 2013, 9, 5693–5705. [Google Scholar] [CrossRef]
- Meeprasert, A.; Hannongbua, S.; Rungrotmongkol, T. Key binding and susceptibility of NS3/4A serine protease inhibitors against hepatitis C virus. J Chem Inf Model. 2014, 54, 1208–1217. [Google Scholar] [CrossRef]
- Guan, Y.; Sun, H.; Li, Y.; Pan, P.; Li, D.; Hou, T. The competitive binding between inhibitors and substrates of HCV NS3/4A protease: a general mechanism of drug resistance. Antiviral Res. 2014, 103, 60–70. [Google Scholar] [CrossRef]
- Xue, W.; Pan, D.; Yang, Y.; Liu, H.; Yao, X. Molecular modeling study on the resistance mechanism of HCV NS3/4A serine protease mutants R155K, A156V and D168A to TMC435. Antiviral Res. 2012, 93, 126–137. [Google Scholar] [CrossRef]
- Anwar, M.I.; Rahman, M.; Hassan, M.U.; Iqbal, M. Prevalence of active hepatitis C virus infections among general public of Lahore, Pakistan. Virol. J. 2013, 10, 351–351. [Google Scholar] [CrossRef]
- Goossens, N.; Negro, F. Is genotype 3 of the hepatitis C virus the new villain? Hepatology 2014, 59, 2403–2412. [Google Scholar] [CrossRef]
- Komatsu, T.E.; Boyd, S.; Sherwat, A.; Tracy, L.; Naeger, L.K.; O’rear, J.J.; Harrington, P.R. Regulatory Analysis of Effects of Hepatitis C Virus NS5A Polymorphisms on Efficacy of Elbasvir and Grazoprevir. Gastroenterology 2017, 152, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Mizokami, M.; Pianko, S.; Mangia, A.; Han, K.-H.; Martin, R.; Svarovskaia, E.; Dvory-Sobol, H.; Doehle, B.; Hedskog, C.; et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: Prevalence and effect on treatment outcome. J. Hepatol. 2017, 66, 910–918. [Google Scholar] [CrossRef]
- Harrington, P.R.; Komatsu, T.E.; Deming, D.J.; Donaldson, E.F.; O'Rear, J.J.; Naeger, L.K. Impact of hepatitis C virus polymorphisms on direct-acting antiviral treatment efficacy: Regulatory analyses and perspectives. Hepatology 2017, 67, 2430–2448. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef]
- Soumana, D.I.; Yilmaz, N.K.; Ali, A.; Prachanronarong, K.L.; Schiffer, C.A. Molecular and Dynamic Mechanism Underlying Drug Resistance in Genotype 3 Hepatitis C NS3/4A Protease. J. Am. Chem. Soc. 2016, 138, 11850–11859. [Google Scholar] [CrossRef]
- Romano, K.P.; Ali, A.; Aydin, C.; Soumana, D.; Özen, A.; Deveau, L.M.; Silver, C.; Cao, H.; Newton, A.; Petropoulos, C.J.; et al. The Molecular Basis of Drug Resistance against Hepatitis C Virus NS3/4A Protease Inhibitors. PLOS Pathog. 2012, 8, e1002832. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2015, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Aydin, C.; Gildemeister, R.; Romano, K.P.; Cao, H.; Özen, A.; Soumana, D.; Newton, A.; Petropoulos, C.J.; Huang, W.; et al. Evaluating the Role of Macrocycles in the Susceptibility of Hepatitis C Virus NS3/4A Protease Inhibitors to Drug Resistance. ACS Chem. Biol. 2013, 8, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Gutierrez, J.A. Importance of HCV genotype 1 subtypes for drug resistance and response to therapy. J. Viral Hepat. 2014, 21, 229–240. [Google Scholar] [CrossRef]
- Lahser, F.; Galloway, A.; Hwang, P.; Palcza, J.; Wahl, J.; Robertson, M.; et al. Interim analysis of a 3-year follow-up study of NS5A and NS3 resistance-associated variants (RAVs) after treatment with grazoprevir-containing regimens in patients with chronic hepatitis C virus (HCV) infection. In Hepatology; WILEY-BLACKWELL: Hoboken, NJ, USA, 2016. [Google Scholar]
- Krishnan, P.; Tripathi, R.; Schnell, G.; Reisch, T.; Beyer, J.; Dekhtyar, T.; et al. O057: Long-term follow-up of treatment-emergent resistance-associated variants in NS3, NS5A and NS5B with paritaprevir/r-, ombitasvir-and dasabuvir-based regimens. J Hepatol. 2015, 62, S220. [Google Scholar] [CrossRef]
- Gane, E.J.; Svarovskaia, E.S.; Hyland, R.H.; Stamm, L.M.; Osinusi, A.; Brainard, D.M.; et al. resistance Analysis of Treatment-naïve and Daa-experienced Genotype 1 Patients with and without Cirrhosis Who Received Short-duration Treatment with Sofosbuvir/gs-5816+ Gs-9857: 713. Hepatology 2015, 62, 563A. [Google Scholar]
- Jiang, M.; Mani, N.; Lin, C.; Ardzinski, A.; Nelson, M.; Reagan, D.; Bartels, D.; Zhou, Y.; Nicolas, O.; Rao, B.G.; et al. In Vitro Phenotypic Characterization of Hepatitis C Virus NS3 Protease Variants Observed in Clinical Studies of Telaprevir. Antimicrob. Agents Chemother. 2013, 57, 6236–6245. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Baril, M.; Lamarre, D. Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses 2010, 2, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Poordad, F.; Pol, S.; Asatryan, A.; Buti, M.; Shaw, D.; Hézode, C.; Felizarta, F.; Reindollar, R.W.; Gordon, S.C.; Pianko, S.; et al. Glecaprevir/Pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 2018, 67, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Khattab, M.A.; Ferenci, P.; Hadziyannis, S.J.; Colombo, M.; Manns, M.P.; Almasio, P.L.; Esteban, R.; Abdo, A.A.; Harrison, S.A.; Ibrahim, N.; et al. Management of hepatitis C virus genotype 4: Recommendations of An International Expert Panel. J. Hepatol. 2010, 54, 1250–1262. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Keeffe, E.B. Chronic hepatitis C: genotypes 4 to 9. Clin Liver Dis. 2005, 9, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Cento, V.; Mirabelli, C.; Salpini, R.; Dimonte, S.; Artese, A.; Costa, G.; Mercurio, F.; Svicher, V.; Parrotta, L.; Bertoli, A.; et al. HCV Genotypes Are Differently Prone to the Development of Resistance to Linear and Macrocyclic Protease Inhibitors. PLOS ONE 2012, 7, e39652. [Google Scholar] [CrossRef] [PubMed]
- Courcambeck, J.; Bouzidi, M.; Perbost, R.; Jouirou, B.; Amrani, N.; Cacoub, P.; et al. Resistance of hepatitis C virus to NS3-4A protease inhibitors: mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antivir Ther. 2006, 11, 847. [Google Scholar] [CrossRef]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef]
- Slater, O.; Kontoyianni, M. The compromise of virtual screening and its impact on drug discovery. Expert Opin. Drug Discov. 2019, 14, 619–637. [Google Scholar] [CrossRef]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against Ebola virus. J. Med Virol. 2018, 91, 2029–2048. [Google Scholar] [CrossRef]
- Zhou, Z.; Khaliq, M.; Suk, J.-E.; Patkar, C.; Li, L.; Kuhn, R.J.; Post, C.B. Antiviral Compounds Discovered by Virtual Screening of Small−Molecule Libraries against Dengue Virus E Protein. ACS Chem. Biol. 2008, 3, 765–775. [Google Scholar] [CrossRef]
- Santos, F.R.S.; Nunes, D.A.F.; Lima, W.G.; Davyt, D.; Santos, L.L.; Taranto, A.G.; Ferreira, J.M.S. Identification of Zika Virus NS2B-NS3 Protease Inhibitors by Structure-Based Virtual Screening and Drug Repurposing Approaches. J. Chem. Inf. Model. 2020, 60, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, J.F.-W.; Den-Haan, H.; Chik, K.K.-H.; Zhang, A.J.; Chan, C.C.-S.; Poon, V.K.-M.; Yip, C.C.-Y.; Mak, W.W.-N.; Zhu, Z.; et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antivir. Res. 2017, 145, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.S.; Amaro, R.E.; Xu, D.; Li, W.W.; Arzberger, P.W.; McCammon, J.A. Ensemble-Based Virtual Screening Reveals Potential Novel Antiviral Compounds for Avian Influenza Neuraminidase. J. Med. Chem. 2008, 51, 3878–3894. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Ryu, H.-J.; Lee, S.-H.; Hwang, S.; Breton, V.; Rhee, J.H.; Kim, D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorganic Med. Chem. Lett. 2011, 21, 3088–3091. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, F.; Zhao, Y.; Alvarez, L.; Iliopoulou, M.; Lohans, C.T.; Schofield, C.J.; Padilla-Parra, S.; Siu, S.W.I.; Fry, E.E.; Ren, J.; et al. Structure-Based in Silico Screening Identifies a Potent Ebolavirus Inhibitor from a Traditional Chinese Medicine Library. J. Med. Chem. 2019, 62, 2928–2937. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, Z.; Iman, K.; Iftikhar, N.; Mirza, M.U. Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer: Targets Ther. 2017, 9, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Iman, K.; Mirza, M.U.; Mazhar, N.; Vanmeert, M.; Irshad, I.; Kamal, M.A.; Ikram, N.; Riaz, M. In silico Structure-based Identification of Novel Acetylcholinesterase Inhibitors Against Alzheimer’s Disease. CNS Neurol. Disord. - Drug Targets 2018, 17, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Saadabadi, A.; Vanmeert, M.; Salo-Ahen, O.M.; Abdullah, I.; Claes, S.; De Jonghe, S.; Schols, D.; Ahmad, S.; Froeyen, M. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach. Eur. J. Pharm. Sci. 2020, 155, 105537–105537. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Maestro. Schrödinger Release 2021-3: Maestro; Schrödinger, LLC: New York, NY, 2021. [Google Scholar]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Ye, Y.; Godzik, A. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004, 32, W582–W585. [Google Scholar] [CrossRef]
- Kawabata, T. MATRAS: a program for protein 3D structure comparison. Nucleic Acids Res. 2003, 31, 3367–3369. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- SAVES Server. Available online: https://saves.mbi.ucla.edu/ (accessed on 1 May 2021).
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]
- Eisenberg D, Lüthy R, Bowie JU. [20] VERIFY3D: assessment of protein models with three-dimensional profiles. Methods in enzymology. Elsevier; 1997. pp. 396–404.
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- MolPort. MolPort Database. Available online: https://www.molport.com (accessed on 1 May 2021).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; A Szalai, F. http://Mcule.com: a public web service for drug discovery. J. Chemin- 2012, 4, P17–P17. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- López-López, E.; Naveja, J.J.; Medina-Franco, J.L. DataWarrior: an evaluation of the open-source drug discovery tool. Expert Opin. Drug Discov. 2019, 14, 335–341. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J Chem Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- LigPrep. Schrödinger Release 2021-3: LigPrep; Schrödinger, LLC: New York, NY, 2021. [Google Scholar]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- LLC, S. Prime, Version 3.9; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Zhu, K.; Shirts, M.R.; Friesner, R.A. Improved Methods for Side Chain and Loop Predictions via the Protein Local Optimization Program: Variable Dielectric Model for Implicitly Improving the Treatment of Polarization Effects. J. Chem. Theory Comput. 2007, 3, 2108–2119. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Case DA, Belfon K, Ben-Shalom I, Brozell SR, Cerutti D, Cheatham T, et al. Amber 2020. 2020.
- Mirza, M.U.; Rafique, S.; Ali, A.; Munir, M.; Ikram, N.; Manan, A.; Salo-Ahen, O.M.H.; Idrees, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 37313. [Google Scholar] [CrossRef]
- Jabbar, B.; Rafique, S.; Salo-Ahen, O.M.H.; Ali, A.; Munir, M.; Idrees, M.; Mirza, M.U.; Vanmeert, M.; Shah, S.Z.; Jabbar, I.; et al. Antigenic Peptide Prediction From E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Front. Immunol. 2018, 9, 3000. [Google Scholar] [CrossRef]
- Ikram, N.; Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Salo-Ahen, O.M.H.; Tahir, M.; Qazi, A.; Ahmad, S. Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds. Biomolecules 2019, 9, 124. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U.; Söderhjelm, P. Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model. J. Comput. Chem. 2015, 36, 2114–2124. [Google Scholar] [CrossRef]
- Khan, M.I.; Khan, Z.A.; Baig, M.H.; Ahmad, I.; Farouk, A.-E.; Song, Y.G.; et al. Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: An in silico insight. PLoS One 2020, 15, e0238344. [Google Scholar] [CrossRef]
- Jacobson, I.M.; Dore, G.J.; Foster, G.R.; Fried, M.W.; Radu, M.; Rafalsky, V.V.; Moroz, L.; Craxi, A.; Peeters, M.; Lenz, O.; et al. Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2014, 384, 403–413. [Google Scholar] [CrossRef]
- Lawitz, E.; Matusow, G.; DeJesus, E.; Yoshida, E.M.; Felizarta, F.; Ghalib, R.; Godofsky, E.; Herring, R.W.; Poleynard, G.; Sheikh, A.; et al. Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: A phase 3 study (OPTIMIST-2). Hepatology 2016, 64, 360–369. [Google Scholar] [CrossRef]
- Forns, X.; Lawitz, E.; Zeuzem, S.; Gane, E.; Bronowicki, J.P.; Andreone, P.; Horban, A.; Brown, A.; Peeters, M.; Lenz, O.; et al. Simeprevir With Peginterferon and Ribavirin Leads to High Rates of SVR in Patients With HCV Genotype 1 Who Relapsed After Previous Therapy: A Phase 3 Trial. Gastroenterology 2014, 146, 1669–1679. [Google Scholar] [CrossRef]
- Summa, V.; Ludmerer, S.W.; McCauley, J.A.; Fandozzi, C.; Burlein, C.; Claudio, G.; et al. MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012, 56, 4161–4167. [Google Scholar] [CrossRef]
- Lawitz, E.; Yang, J.C.; Stamm, L.M.; Taylor, J.G.; Cheng, G.; Brainard, D.M.; Miller, M.D.; Mo, H.; Dvory-Sobol, H. Characterization of HCV Resistance from a 3-Day Monotherapy Study of Voxilaprevir, a Novel Pangenotypic NS3/4A Protease Inhibitor. Antivir. Ther. 2017, 23, 325–334. [Google Scholar] [CrossRef]
- Forestier, N.; Susser, S.; Welker, M.W.; Weegink, C.J.; Reesink, H.W.; Zeuzem, S.; et al. Telaprevir resistance mutations in patients with hepatitis C who relapsed after sequential therapy with telaprevir, peg-interferon alfa 2a and ribavirin. In Hepatology; JOHN WILEY & SONS INC: HOBOKEN, NJ, USA, 2007. [Google Scholar]
- Susser, S.; Welsch, C.; Wang, Y.; Zettler, M.; Domingues, F.S.; Karey, U.; Hughes, E.; Ralston, R.; Tong, X.; Herrmann, E.; et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 2009, 50, 1709–1718. [Google Scholar] [CrossRef]
- McPhee, F.; Friborg, J.; Levine, S.; Chen, C.; Falk, P.; Yu, F.; Hernandez, D.; Lee, M.S.; Chaniewski, S.; Sheaffer, A.K.; et al. Resistance Analysis of the Hepatitis C Virus NS3 Protease Inhibitor Asunaprevir. Antimicrob. Agents Chemother. 2012, 56, 3670–3681. [Google Scholar] [CrossRef]
- Lenz, O.; Verbinnen, T.; Lin, T.-I.; Vijgen, L.; Cummings, M.D.; Lindberg, J.; et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother. 2010, 54, 1878–1887. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Zou, J.; Xie, H.-Z.; Yang, S.-Y.; Chen, J.-J.; Ren, J.-X.; Wei, Y.-Q. Towards more accurate pharmacophore modeling: Multicomplex-based comprehensive pharmacophore map and most-frequent-feature pharmacophore model of CDK2. J. Mol. Graph. Model. 2008, 27, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S.; Bogen, S.L.; Arasappan, A.; Bennett, F.; Chen, K.; Jao, E.; Liu, Y.-T.; Lovey, R.; Hendrata, S.; Huang, Y.; et al. Discovery of (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]- 3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]- 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (SCH 503034), a Selective, Potent, Orally Bioavailable Hepatitis C Virus NS3 Protease Inhibitor: A Potential Therapeutic Agent for the Treatment of Hepatitis C Infection. J. Med. Chem. 2006, 49, 6074–6086. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3. 4A serine protease. Infect Disord Targets (Formerly Curr Drug Targets-Infectious Disord. 2006, 6, 3–16. [Google Scholar]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.M.; Venkatraman, S. The discovery and development of boceprevir: A novel, first-generation inhibitor of the hepatitis C virus NS3/4A serine protease. J Clin Transl Hepatol. 2013, 1, 22. [Google Scholar] [PubMed]
- Romano, K.P.; Ali, A.; Royer, W.E.; Schiffer, C.A. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci. 2010, 107, 20986–20991. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Kieffer, T.L.; Bartels, D.; Hanzelka, B.; Müh, U.; Welker, M.; Wincheringer, D.; Zhou, Y.; Chu, H.; Lin, C.; et al. Dynamic Hepatitis C Virus Genotypic and Phenotypic Changes in Patients Treated With the Protease Inhibitor Telaprevir. Gastroenterology 2007, 132, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Muöh, U.; Hanzelka, B.L.; Bartels, D.J.; Wei, Y.; Rao, B.G.; et al. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: sensitivity to telaprevir (VX-950) and interferon α. J Biol Chem. 2007, 282, 22619–22628. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Ahmed, B.; Ashfaq, U.A.; Mirza, M.U. Medicinal plant phytochemicals and their inhibitory activities against pancreatic lipase: molecular docking combined with molecular dynamics simulation approach. Nat. Prod. Res. 2017, 32, 1123–1129. [Google Scholar] [CrossRef]
- Durdagi, S.; Qamar, M.T.U.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Muneer, I.; Tusleem, K.; Abdul Rauf, S.; Hussain, H.M.J.; Siddiqi, A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: a combined ligand and structure-based resources pipeline. Anticancer Drugs 2019, 30, 363–373. [Google Scholar] [CrossRef]
- Guan, Y.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Exploring resistance mechanisms of HCV NS3/4A protease mutations to MK5172: insight from molecular dynamics simulations and free energy calculations. Mol Biosyst. 2015, 11, 2568–2578. [Google Scholar] [CrossRef]
- Wang, H.; Geng, L.; Chen, B.-Z.; Ji, M. Computational study on the molecular mechanisms of drug resistance of Narlaprevir due to V36M, R155K, V36M+R155K, T54A, and A156T mutations of HCV NS3/4A protease. Biochem. Cell Biol. 2014, 92, 357–369. [Google Scholar] [CrossRef]
- Shukla, R.; Tripathi, T. Molecular Dynamics Simulation of Protein and Protein–Ligand Complexes. In Computer-Aided Drug Design; Springer, 2020; pp. 133–161. [Google Scholar]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Pikkemaat, M.G.; Linssen, A.B.; Berendsen, H.J.; Janssen, D.B. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. Des. Sel. 2002, 15, 185–192. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, T.; Wang, W.; Liu, J.S. Detecting and understanding combinatorial mutation patterns responsible for HIV drug resistance. Proc. Natl. Acad. Sci. 2010, 107, 1321–1326. [Google Scholar] [CrossRef]
- Hou, T.; McLaughlin, W.A.; Wang, W. Evaluating the potency of HIV-1 protease drugs to combat resistance. Proteins: Struct. Funct. Bioinform. 2007, 71, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Yu, R. Molecular Dynamics and Free Energy Studies on the Wild-type and Double Mutant HIV-1 Protease Complexed with Amprenavir and Two Amprenavir-Related Inhibitors: Mechanism for Binding and Drug Resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Wang, J.; Hou, T. P-loop Conformation Governed Crizotinib Resistance in G2032R-Mutated ROS1 Tyrosine Kinase: Clues from Free Energy Landscape. PLOS Comput. Biol. 2014, 10, e1003729. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Li, D.; Kang, Y.; Hou, T. Characterizing Drug–Target Residence Time with Metadynamics: How To Achieve Dissociation Rate Efficiently without Losing Accuracy against Time-Consuming Approaches. J. Chem. Inf. Model. 2017, 57, 1895–1906. [Google Scholar] [CrossRef]
- Hou, T.; Zhang, W.; Wang, J.; Wang, W. Predicting drug resistance of the HIV-1 protease using molecular interaction energy components. Proteins: Struct. Funct. Bioinform. 2008, 74, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yao, X.; Wang, C.; Han, J. In Silico Identification of the Potential Drug Resistance Sites over 2009 Influenza A (H1N1) Virus Neuraminidase. Mol. Pharm. 2010, 7, 894–904. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. 2001, 98, 14937–14942. [Google Scholar] [CrossRef]
- Li, N.; Ainsworth, R.I.; Ding, B.; Hou, T.; Wang, W. Using Hierarchical Virtual Screening To Combat Drug Resistance of the HIV-1 Protease. J. Chem. Inf. Model. 2015, 55, 1400–1412. [Google Scholar] [CrossRef]
- Wang, N.X.; Zheng, J.J. Computational studies of H5N1 influenza virus resistance to oseltamivir. Protein Sci. 2009, 18, 707–715. [Google Scholar] [CrossRef]
- He, M.; Li, W.; Zheng, Q.; Zhang, H. A molecular dynamics investigation into the mechanisms of alectinib resistance of three ALK mutants. J. Cell. Biochem. 2018, 119, 5332–5342. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zheng, Q. Multiple Molecular Dynamics Simulations of the Inhibitor GRL-02031 Complex with Wild Type and Mutant HIV-1 Protease Reveal the Binding and Drug-Resistance Mechanism. Langmuir 2020, 36, 13817–13832. [Google Scholar] [CrossRef] [PubMed]
- Soumana, D.I.; Ali, A.; Schiffer, C.A. Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem Biol. 2014, 9, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, N.; Goyal, S.; Wahi, D.; Jain, R.; Jamal, S.; Singh, A.; et al. Molecular principles behind Boceprevir resistance due to mutations in hepatitis C NS3/4A protease. Gene 2015, 570, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, J. Molecular dynamics study on drug resistance mechanism of HCV NS3/4A protease inhibitor: BI201335. Mol Simul. 2015, 41, 674–682. [Google Scholar] [CrossRef]
- MacArthur, M.W.; Thornton, J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991, 218, 397–412. [Google Scholar] [CrossRef]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; et al. Discovery of the HCV NS3/4A protease inhibitor (1 R, 5 S)-N-[3-amino-1-(cyclobutylmethyl)-2, 3-dioxopropyl]-3-[2 (S)-[[[(1, 1-dimethylethyl) amino] carbonyl] amino]-3, 3-dimethyl-1-oxobutyl]-6, 6-dimethyl-3-azabicyclo [3.1. 0] hexan-2 (S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. Journal of medicinal chemistry 2007, 50, 2310–2318. [Google Scholar]
Figure 1.
Framework for in silico analysis of HCV NS3/4A G3. Flowchart summarizing summarising the computational framework for modeling and targeting HCV NS3/4A G3 towards HCV treatment2.2.
Figure 1.
Framework for in silico analysis of HCV NS3/4A G3. Flowchart summarizing summarising the computational framework for modeling and targeting HCV NS3/4A G3 towards HCV treatment2.2.

Figure 2.
Multiple sequence alignment of HCV NS3/4A Genotype 3 variants. Amino acid substitutions at 14 positions are pointed with arrow in blue. The respective mutations at specified positions are mentioned in blue. The conserved residues are highlighted in red. Note: Variant_1 to variant_14 as G3.v1 to G3.v14.
Figure 2.
Multiple sequence alignment of HCV NS3/4A Genotype 3 variants. Amino acid substitutions at 14 positions are pointed with arrow in blue. The respective mutations at specified positions are mentioned in blue. The conserved residues are highlighted in red. Note: Variant_1 to variant_14 as G3.v1 to G3.v14.
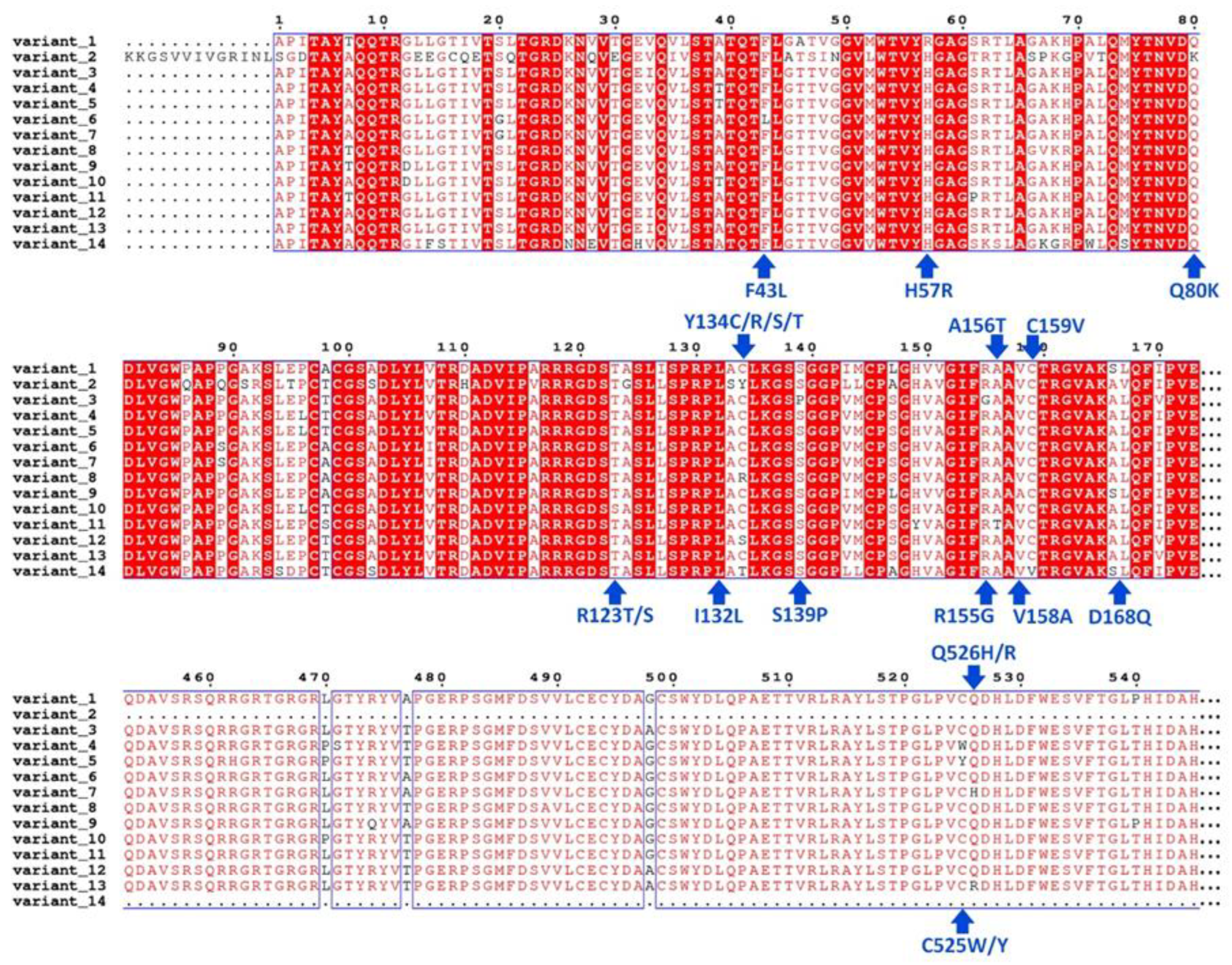
Figure 3.
Chemical structure of CHEMBL569970 (cpd-217; PubChem45485999). The lead indicated binding potential with all HCV NS3/4A variants.
Figure 3.
Chemical structure of CHEMBL569970 (cpd-217; PubChem45485999). The lead indicated binding potential with all HCV NS3/4A variants.
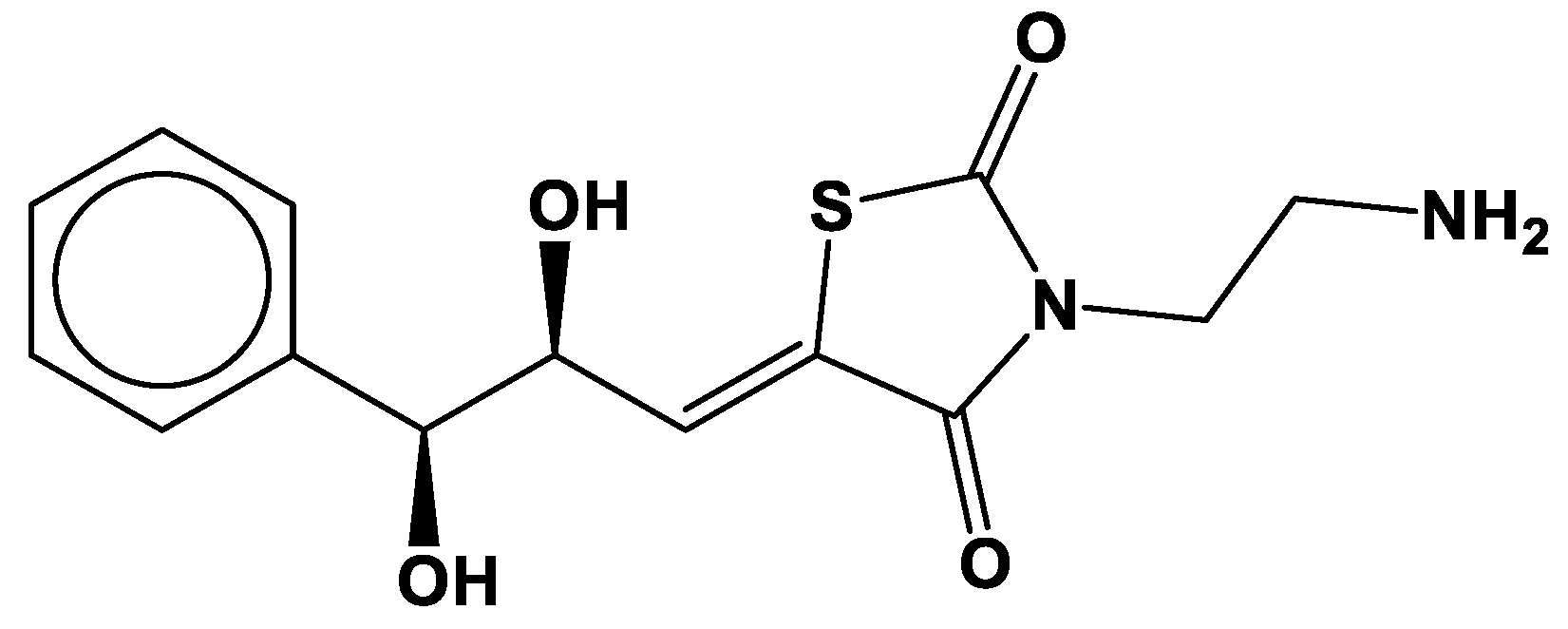
Figure 4.
Surface representation of the binding pocket of HCV NS3/4A G3 variants docked with CHEMBL569970 (cps-217; PubChem45485999). Key interacting residues including Thr42, Leu/Phe43, Val55, Gly58, Asp81, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Met485, Phe486, Gly525, Gln526, Asp527, and Asp528 are labeled. All HCV NS3/4A G3 variant 1 to 14 are highlighted in A-N respectively.
Figure 4.
Surface representation of the binding pocket of HCV NS3/4A G3 variants docked with CHEMBL569970 (cps-217; PubChem45485999). Key interacting residues including Thr42, Leu/Phe43, Val55, Gly58, Asp81, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Met485, Phe486, Gly525, Gln526, Asp527, and Asp528 are labeled. All HCV NS3/4A G3 variant 1 to 14 are highlighted in A-N respectively.
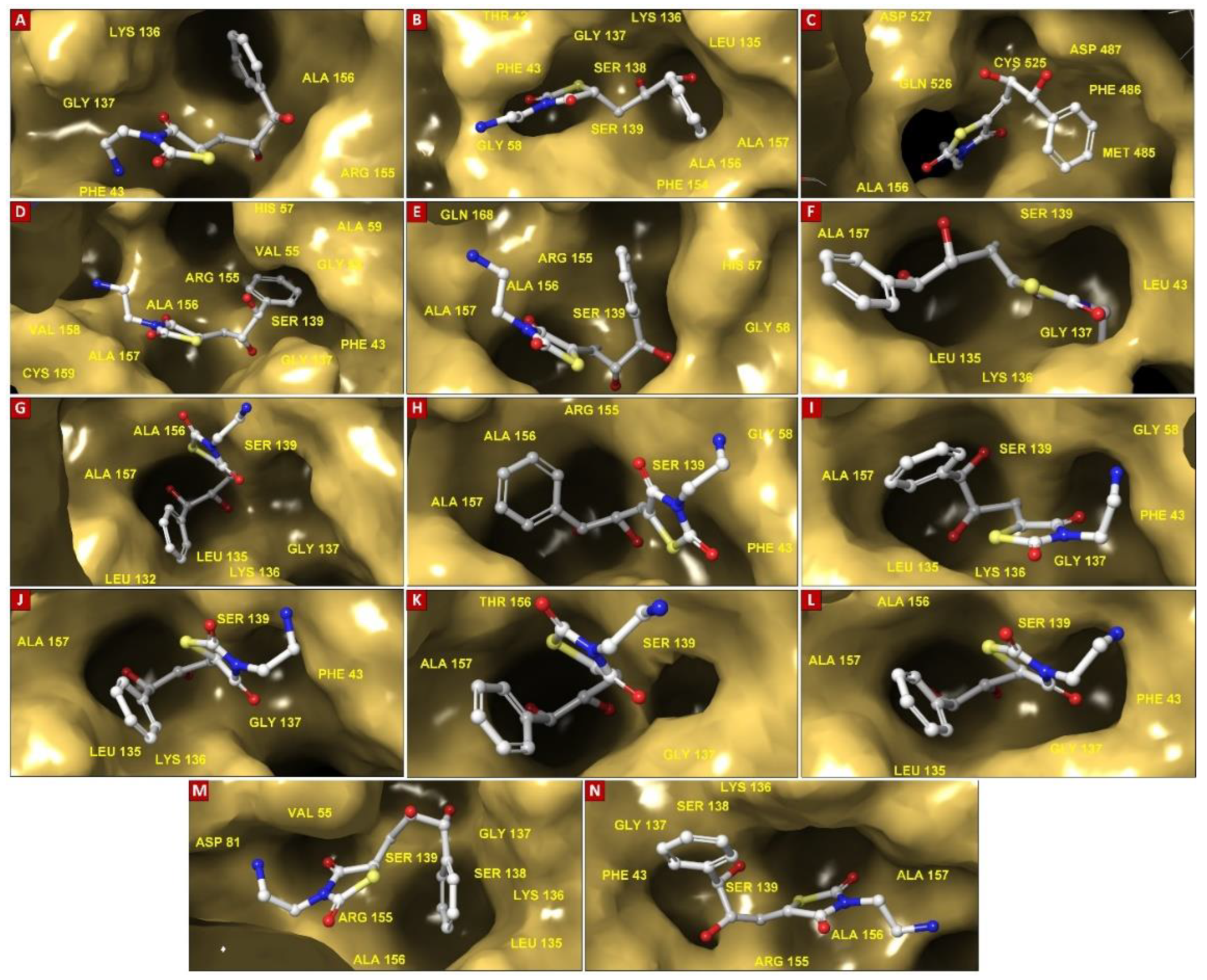
Figure 5.
Ligand interaction diagrams of CHEMBL569970 (PubChem45485999) with residues inside the binding pocket of HCV NS3/4A G3 variants. Residues making hydrogen bond (H-bond) interactions include Gln41, Thr42, His/Arg57, Asp81, Leu135, Gly137, Ser139, Arg155, Ala157, Asp487, and Gln526. Asp81 is involved in making salt bridge in G3.v3 and 13. Lys136 (G3.v10) and H57 (G3.v4) is involved in hydrophobic interactions including Pi-cation and Pi-Pi stacking, respectively. All HCV NS3/4A G3 variants 1 to 14 are highlighted in A-N respectively.
Figure 5.
Ligand interaction diagrams of CHEMBL569970 (PubChem45485999) with residues inside the binding pocket of HCV NS3/4A G3 variants. Residues making hydrogen bond (H-bond) interactions include Gln41, Thr42, His/Arg57, Asp81, Leu135, Gly137, Ser139, Arg155, Ala157, Asp487, and Gln526. Asp81 is involved in making salt bridge in G3.v3 and 13. Lys136 (G3.v10) and H57 (G3.v4) is involved in hydrophobic interactions including Pi-cation and Pi-Pi stacking, respectively. All HCV NS3/4A G3 variants 1 to 14 are highlighted in A-N respectively.
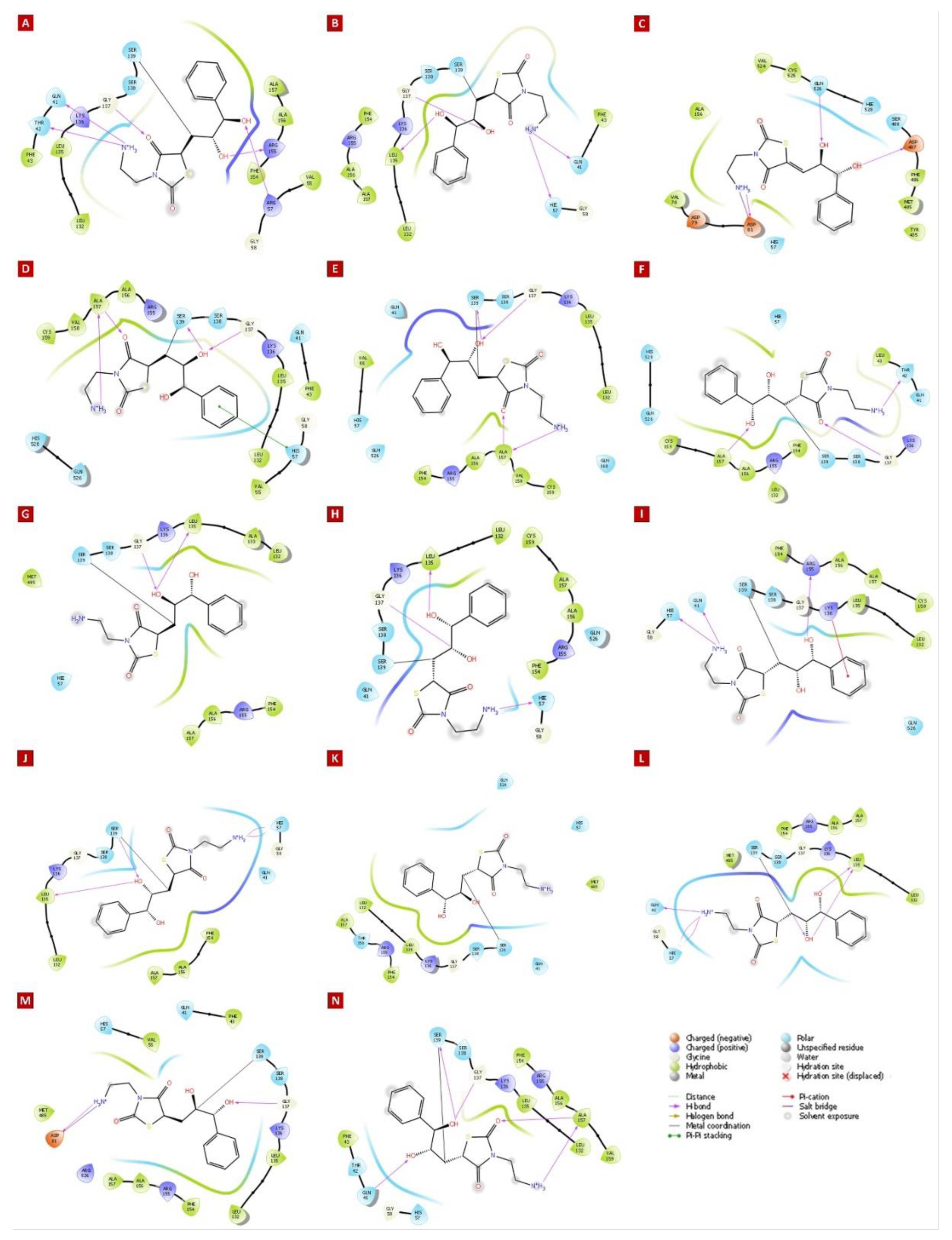
Figure 6.
The root-mean-square deviation (RMSD) of the backbone atoms (CA, N, C) of the HCV NS3/4A G3 variants and heavy atoms of cpd-217 relative to the initial structure.
Figure 6.
The root-mean-square deviation (RMSD) of the backbone atoms (CA, N, C) of the HCV NS3/4A G3 variants and heavy atoms of cpd-217 relative to the initial structure.
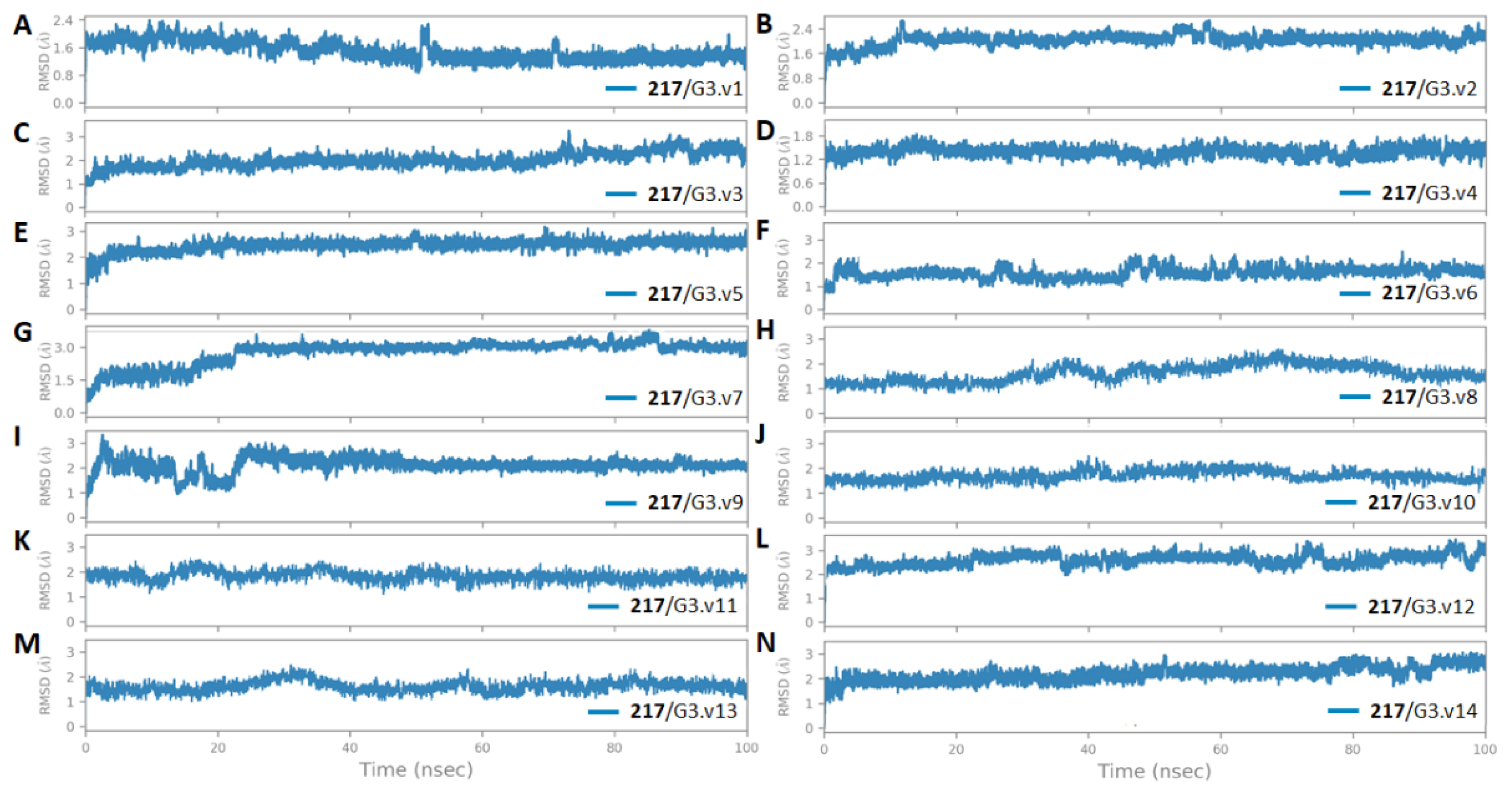
Figure 7.
Root-mean-square deviation of HCV NS3/4A G3 wildtype (WT) with all G3 varaints. The MD-simulated HCV NS3/4A G3.WT (yellow) is superimposed on G3 variants (cornflower blue), G3.v1 – v7 in (A) and G3.v8 – v14 in (B), after 100 ns. While the NS4A is colored orange red and catalytic triad residues are represented in ball and stick representation (yellow). The RMSF plots of HCV NS3/4A G3.WT (black line) with G3.v1 – v7 in (C), and G3.v8 – v14 in (D) are displayed. The residue numbers along x-axis and fluctuations in Å along y-axis, while catalytic triad residues are highlighted red.
Figure 7.
Root-mean-square deviation of HCV NS3/4A G3 wildtype (WT) with all G3 varaints. The MD-simulated HCV NS3/4A G3.WT (yellow) is superimposed on G3 variants (cornflower blue), G3.v1 – v7 in (A) and G3.v8 – v14 in (B), after 100 ns. While the NS4A is colored orange red and catalytic triad residues are represented in ball and stick representation (yellow). The RMSF plots of HCV NS3/4A G3.WT (black line) with G3.v1 – v7 in (C), and G3.v8 – v14 in (D) are displayed. The residue numbers along x-axis and fluctuations in Å along y-axis, while catalytic triad residues are highlighted red.
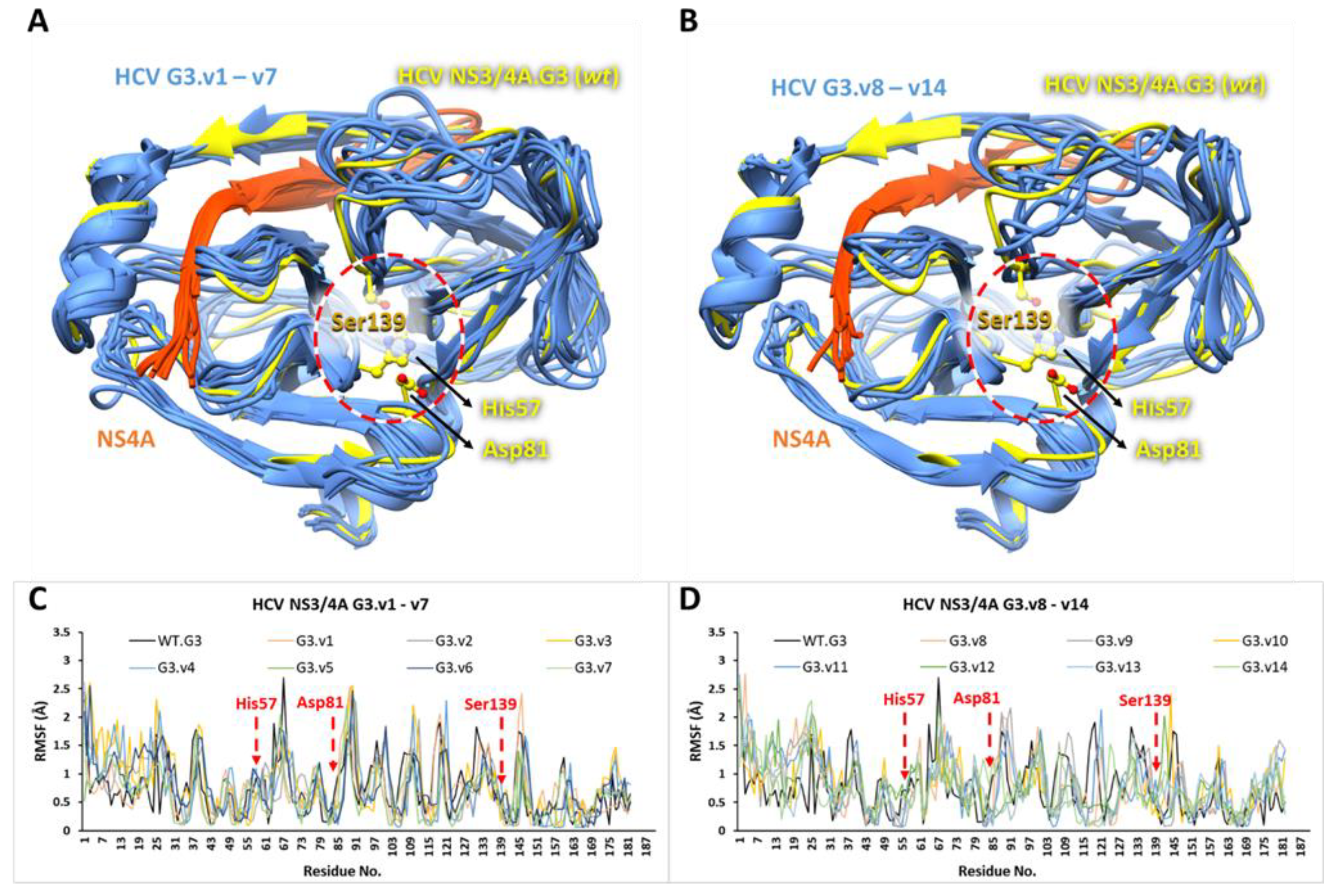
Figure 8.
Per-residue decomposition analysis of the potential binding site residues of HCV NS3/4A G3 variants in the presence of cpd-217. The colour codes are represented for each variant, while wild type (PDB ID: 4A92) is colored black. The mutated residues in corresponding variants are highlighted in red. The values are measured in kcal/mol.
Figure 8.
Per-residue decomposition analysis of the potential binding site residues of HCV NS3/4A G3 variants in the presence of cpd-217. The colour codes are represented for each variant, while wild type (PDB ID: 4A92) is colored black. The mutated residues in corresponding variants are highlighted in red. The values are measured in kcal/mol.
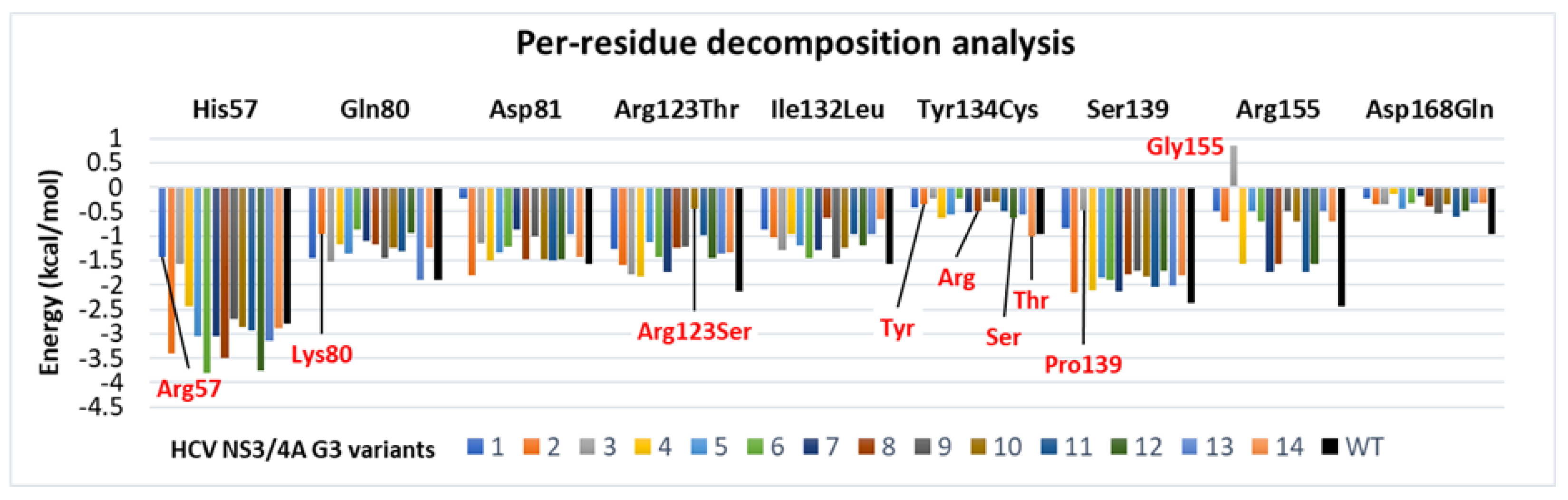

Table 1.
Pharmacokinetic and pharmacodynamics properties of CHEMBL569970 (cpd-217; PubChem45485999) along with post-docking analysis and binding interactions with HCV NS3/4A G3 variants (v1 to v14).
Table 1.
Pharmacokinetic and pharmacodynamics properties of CHEMBL569970 (cpd-217; PubChem45485999) along with post-docking analysis and binding interactions with HCV NS3/4A G3 variants (v1 to v14).
| Molecule Name | RMSD | Druglikeness | Mutagenic | Tumorigenic | cLogP | cLogS | Polar Surface Area | Reproductive Effective | Irritant |
| cpd-217 | 0.121 | -0.402 | none | none | -0.488 | -1.886 | 129.16 | none | none |
| Docking Score and Binding Interactions of cpd-217 | |||||||||
| HCV NS3/4A G3 Variant No. | Docking Score (kcal/mol) | Mutations within NS3/4A G3 sequences | Ligand-binding Residues | Covalent Bond with Ser139 | No. of H-bonds | H-Bond (residues involved) | salt bridge (residues involved) | Pi-cation interaction (residues involved) | Pi-Pi interaction (residues involved) |
| 1 | -6.588 | His57Arg, Arg123Thr, Ile132Leu, Tyr134Cys, Asp168Gln | Gln41, Thr42, Phe43, Val55, Arg57, Gly58, Leu132, Leu135, Lys136, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157 | Yes | 5 | Gln41, Thr42, Arg57, Leu135, Arg155 | - | - | - |
| 2 | -5.308 | Gln80Lys, Arg123Thr, Ile132Leu, Asp168Gln | Gln41, Phe43, His57, Gly58, Leu132, Leu135, Lys136, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157 | Yes | 4 | Gln41, His57, Leu135, Gly137 | - | - | - |
| 3 | -5.447 | Arg123Thr, Ile132Leu, Tyr134Cys, Ser139Pro, Arg155Gly, Asp168Gln | His57, Val78, Asp79, Asp81, Ala156, Met485, Phe486, Asp487, Ser488, Val524, Cys525, Gln526, His528 | No (S139P) | 3 | Asp81, Asp487, Gln526 | Asp81 | - | - |
| 4 | -6.397 | Arg123Thr, Ile132Leu, Tyr134Cys, Asp168Gln, Cys525Trp | Gln41, Phe43, Val55, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Arg155, Ala156, Ala157, Val158, Cys159, Gln526, His528 | Yes | 4 | Gly137, Ser139, Ala157 | - | - | His57 |
| 5 | -5.986 | Arg123Thr, Ile132Leu, Try134Cys, Asp168Gln, Cys525Tyr | Gln41, Val55, His57, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Val158, Cys159, Gln168 | Yes | 4 | Gly137, Ser139, Ala157 | - | - | - |
| 6 | -5.101 | Phe43Leu, Arg123Thr, Ile132Leu, Tyr134Cys, Asp168Gln | Gln41, Thr42, Leu43, His57, Gly58, Leu132, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Cys159, Gln526, His528 | Yes | 3 | Thr42, Gly137, Ala157 | - | - | - |
| 7 | -5.395 | Arg123Thr, Ile132Leu, Tyr134Cys, Asp168Gln, Gln526His | His57, Leu132, Ala133, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Met485 | Yes | 2 | Leu135, Gly137 | - | - | - |
| 8 | -5.630 | Arg123Thr, Ile132Leu, Tyr134Arg, Asp168Gln | Gln41, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Cys159, Gln526 | Yes | 3 | His57, Leu135, Gly137 | - | - | - |
| 9 | -5.184 | Arg123Thr, Ile132Leu, Tyr134Cys, Val158Ala, Asp168Gln | Gln41, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Cys159, Gln526 | Yes | 3 | Gln41, His57, Arg155 | - | Lys136 | - |
| 10 | -4.722 | Arg123Ser, Ile132Leu, Tyr134Cys, Asp168Gln | Gln41, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Ala156, Ala157 | Yes | 4 | His57, Leu135, Ser139 | - | - | - |
| 11 | -4.169 | Arg123Thr, Ile132Leu, Tyr134Cys, Ala156Thr, Asp168Gln | Gln41, His57, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Thr156, Ala157, Met485, Gln526 | Yes | 0 | - | - | - | - |
| 12 | -5.288 | Arg123Thr, Ile132Leu, Tyr134Ser, Asp168Gln | Gln41, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Met485 | Yes | 6 | Gln41, His57, Leu135, Ser139 | - | - | - |
| 13 | -5.392 | Arg123Thr, Ile132Leu, Tyr134Cys, Asp168Gln, Gln526Arg | Gln41, Phe43, Val55, His57, Asp81, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Met485, Arg526 | Yes | 2 | Asp81, Gly137 | Asp81 | - | - |
| 14 | -6.153 | Arg123Thr, Ile132Leu, Tyr134Thr, Cys159Val, Asp168Gln | Gln41, Thr42, Phe43, His57, Gly58, Leu132, Leu135, Lys136, Gly137, Ser138, Ser139, Phe154, Arg155, Ala156, Ala157, Val159 | Yes | 5 | Gln41, Gly137, Ser139, Ala157 | - | - | - |
Table 2.
Predicted MMGBSA calculations throughout 100 ns for HCV NS3/4A G3 variants complexed with cpd-217.
Table 2.
Predicted MMGBSA calculations throughout 100 ns for HCV NS3/4A G3 variants complexed with cpd-217.
| HCV | MMGBSA energy components (kcal/mol) |
Energy Shift ΔΔGshifta |
Variation (ΔAS)b | ||||||||
| Before MD | After MD (Last 50 ns) | ||||||||||
| ΔGtol | ΔEele | ΔEvdw | ΔEMM | ΔGsol | -TΔS | ΔGbind | |||||
| G3.v1 | -5.7 | -32.5 | -44.2 | -76.7 | 39.1 | 25.9 | -11.6 | -5.9 | 2.8 | ||
| G3.v2 | -6.9 | -29.1 | -43.2 | -72.3 | 36.4 | 20.3 | -15.6 | -8.7 | -1.2 | ||
| G3.v3 | -4.0 | -27.3 | -37.1 | -64.4 | 35.4 | 22.7 | -6.3 | -2.3 | 8.1 | ||
| G3.v4 | -4.4 | -32.4 | -38.1 | -70.5 | 38.2 | 22.4 | -9.9 | -5.5 | 4.5 | ||
| G3.v5 | -3.8 | -31.1 | -42.3 | -73.4 | 39.5 | 24.7 | -9.2 | -5.5 | 5.1 | ||
| G3.v6 | -4.1 | -29.1 | -40.2 | -69.3 | 37.2 | 23.0 | -9.1 | -5.0 | 5.3 | ||
| G3.v7 | -3.8 | -26.4 | -36.6 | -63.0 | 38.2 | 15.2 | -9.6 | -5.8 | 4.8 | ||
| G3.v8 | -3.8 | -31.0 | -36.4 | -67.4 | 37.2 | 19.0 | -11.2 | -7.4 | 3.2 | ||
| G3.v9 | -3.1 | -27.4 | -33.5 | -60.9 | 36.4 | 15.3 | -9.2 | -6.2 | 5.2 | ||
| G3.v10 | -3.0 | -28.1 | -37.1 | -65.2 | 36.1 | 20.0 | -9.1 | -6.1 | 5.3 | ||
| G3.v11 | -4.7 | -30.5 | -35.5 | -66.0 | 38.2 | 18.0 | -9.8 | -5.1 | 4.6 | ||
| G3.v12 | -4.4 | -31.4 | -40.1 | -71.5 | 39.1 | 21.0 | -11.4 | -7.0 | 3.0 | ||
| G3.v13 | -5.0 | -27.4 | -39.2 | -66.6 | 37.6 | 18.5 | -10.5 | -5.5 | 3.9 | ||
| G3.v14 | -5.2 | -32.1 | -43.3 | -75.4 | 38.2 | 22.1 | -15.1 | -9.9 | -0.7 | ||
| WT | -5.7 | -32.8 | -41.7 | -74.5 | 36.4 | 23.7 | -14.4 | -8.6 | 0.0 | ||
Note: a ΔΔGbind represents the energy shift, which a complex attained throughout 100 ns. A lower value indicates a higher binding affinity and favourable stabilising impact over the simulation period. b ΔAS represents affinity score variation. For binding energy, lower values represent more affinity with the protein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



