
Submitted:
15 July 2024
Posted:
15 July 2024
You are already at the latest version
Abstract
Inflammatory bowel disease (IBD) is an incurable, chronic disorder of the gastrointestinal tract, whose incidence increases every year. Scientific research is constantly delivering new information about the disease and its multivariate, complex etiology. Nevertheless, full discovery and understanding of the complete mechanism of IBD pathogenesis still pose a significant challenge to today's science. Recent studies unanimously confirmed the association of gut microbial dysbiosis with IBD, as well as its contribution to the regulation of the inflammatory process. It transpires that not only the altered composition of pathogenic and commensal bacteria is characteristic of disturbed intestinal homeostasis in IBD, but also of viruses, parasites, and fungi, which are active in the intestine. In fact, the crucial function of microbial metabolome in the human body is altered, which causes a wide range of effects in the host, thus providing a basis for the disease. On the other hand, human genomic and functional research revealed more loci that play an essential role in gut homeostasis regulation, immune response, and intestinal epithelial function. This review aims to organize and summarize currently available knowledge concerning the role and interaction of crucial factors associated with IBD pathogenesis, particularly host genetic composition, intestinal microbiota, and metabolome, with immune regulation.
Keywords:
ulcerative colitis
; Crohn's disease
; gut microbiota
; short-chain fatty acids
; host genetics
; metabolome
1. Introduction
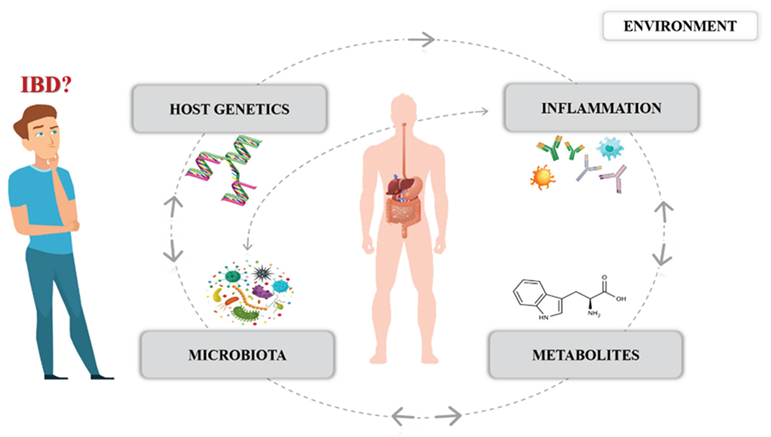
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), affects around 7 million people worldwide. As an inflammatory, chronic, and still incurable disease that permanently reduces the quality of patient’s lives, it currently poses a serious challenge to the medical sector and human society [1,2]. A variety of factors have been implicated in IBD pathogenesis – genetic, immunological, and environmental. Nevertheless, despite the technological advancements in molecular biology, the etiology of IBD is still not fully understood [3]. Recent studies clearly demonstrated that besides the host genetic predispositions, compositional changes in the gut bacterial microbiota are associated with IBD [4]. The fundamental roles of the gut microbial community in terms of host health include maintaining intestinal homeostasis, driving the immune response, and maintaining the function of the epithelial barrier. However, there is a growing body of evidence suggesting that defining the host-microbial relationships, not only including bacteria but also viruses, fungi, parasites, and their specific metabolites, is crucial to understanding IBD pathogenesis. Therefore, we want to systematize, summarize, and discuss the data on IBD pathogenesis, focusing on new insights that pertain to the relationship of microbiota and metabolome to host genetics.
2. Gut Microbiota and IBD
Human gut microbiota constitutes an ecosystem composed of over one hundred trillion various microorganisms, mainly bacteria, but also viruses, fungi, archaea, and parasites. Physiologically, the composition of microbes in the intestine is highly diverse among healthy individuals. However, they are in a symbiotic relationship with the host, forming gut-associated lymphoid tissue (GALT), producing vitamins and metabolites necessary for homeostasis and human well-being. The human intestine constitutes a nutrient-rich environment for the microbial community. This interaction is regulated by environmental factors and host genetics [5]. In IBD patients, a change in the balance between commensal and pathogenic microorganisms in the gut (dysbiosis) is observed, also in the metabolic profile, and finally, in the interaction of microbiota with the host [4]. This microbial imbalance is important because metabolites produced by specific taxa can influence the expression of anti- and pro-inflammatory cytokines, T helper (Th) 1, 2, 17, and regulatory T cells (T-reg) [6]. Studies have revealed that in germ-free mice, a reduced amount of Th 17 cells, lymphocytes, and immunoglobulin (Ig) A in the intestinal mucosa was observed compared to the controls [7,8]. Animal studies confirmed the role of microbiota in the development of inflammatory diseases. They revealed that dysbiotic bacteria transferred to healthy mice induce inflammation. Furthermore, it was proved that gut microbiota obtained from IBD patients alter the immune response and exacerbate UC in mice [10]. At the same time, naive CD4+ lymphocytes transferred from healthy mice to those which lack T and B lymphocytes may induce UC, but the susceptibility depends on the gut microbial composition [11].
2.1. Bacterial Dysbiosis
Alterations in the bacterial composition in the intestine of IBD patients have already been well described. In general, dysbiosis in IBD is observed as a state of a reduced abundance of commensal taxa with a concomitant increase in pathogenic bacteria. Moreover, the intestinal bacterial community shows decreased diversity and is less stable in those patients compared to healthy controls [12]. In a healthy population, the most represented bacterial phyla are Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Verrucomicrobia [13].
The results of global studies indicate that the main shifts at the phylum level include a reduction in Firmicutes and Bacteroidetes in both CD and UC cases [4,14] Moreover, the ratio of Firmicutes/Bacteroidetes is lower, which is a principal indicator of bacterial balance in the gut. Many beneficial taxa are reduced from the Firmicutes phylum, such as Faecalibacterium prausnitzii, Clostridium XIVa, IV, Lactobacillus, Roseburia faecis, producing butyrate and short-chain fatty acids (SCFA), which are of great importance in the host [15]. F. prausnitzii, known as a biomarker of a healthy gut, plays a key role because of its anti-inflammatory metabolite, which inhibits the NF-kB pathway in epithelial cell lines [16]. Verrucomicrobia is also mostly reported to be lowered in IBD patients. The commensal Akkermansia muciniphila belongs to this bacterial phylum and has a pivotal function in the intestine due to ameliorating mucosal inflammation either via microbe-host interactions, which protect the gut barrier function and reduce the levels of inflammatory cytokines, or by improving the microbial community. This probiotic potential of A. muciniphila could make this bacterium an important agent in UC treatment. Deficiency of A. muciniphila in UC patients could be a result of the decreased level of mucins in the gut mucosa, which are the source of energy for these bacteria. Furthermore, A. muciniphila supports the intestinal barrier via its protein Amuc_1100 [17].
On the other hand, in IBD subjects, a marked increase in aerobes is observed. This mainly concerns bacteria from the Proteobacteria and Actinobacteria phyla, including Escherichia coli and Desulfovibrio, which have a pro-inflammatory influence on the intestinal mucosa [18]. This tendency is associated with inflammation, an oxidative process which may induce an overgrowth of bacteria adherent to the colonic mucosa that have a higher tolerance to oxygen [18]. Among IBD patients, the enrichment of mucolytic bacteria is noted with e.g., Ruminococcus gnavus. This process weakens the intestine’s defensive abilities against pathogenic bacteria.
In IBD (both UC and CD), general changes in gut microbial composition are common. However, in the case of CD, intestinal microbiota is more altered and unstable compared to UC. Moreover, specific shifts are reported, depending on disease location and activity [12].
2.2. Virome Dysbiosis
Viruses infecting prokaryotes and eukaryotes constitute a part of the gut microbial community. They participate in host ecosystem dynamics and provide antibiotic-resistance genes. The human intestinal virome is stable, personalized and mainly consists of bacteriophages [19]. Certain stress factors can induce the lytic cycle and viral replication. In IBD patients, virome composition changes result from bacterial alterations in the intestine [20]. In a number of independent studies, the Caudovirales phage sequence was detected in both CD and UC samples of stool, mucosal washes, or biopsies [21]. Among Caudovirales phage families, Siphoviridae, Myoviridae, and Podoviridae were present. An increased abundance of the Caudovirales phage in the intestine correlated with decreased bacterial diversity and mucosal inflammation [22]. Furthermore, Ungaro et al. revealed an increased presence of Herpesviridae in the mucosa of CD patients and Hepadnaviridae in UC patients [23]. Recent investigations conducted on germ-free mice demonstrated that Lactobacillus, Bacteroides, and Escherichia infecting phages, as well as phage DNA, enhance the inflammatory process in the gut. There is also evidence that phages infecting eukaryotic cells play a role in IBD pathogenesis, which is due to integration with the host genome and affecting intestinal cells [24].
2.3. Mycobiome Dysbiosis
The contribution of fungi to IBD pathogenesis has been suspected since their pro-inflammatory influence was initially discovered [25]. In fact, mycobiome dysbiosis has been reported extensively in IBD patients. Fungi constitute approximately 0.1% of human intestine microbiota and is mainly composed of the Ascomycota and Basidiomycota phyla, where the most common genera are Saccharomyces, Debaryomyces, Penicillium, Kluyveromyces, and Candida [26].
Mycobiome dysbiosis is characterized by an increased ratio of Basidiomycota to Ascomycota. Higher gut mycobiota diversity was shown in individuals with ileal CD, while decreased diversity was present in cases of UC and colonic CD [26]. Studies proved that an abundance of protective Saccharomyces cerevisiae was detected in IBD subjects at lower levels compared to the controls, in contrast to Candida albicans, Candida tropicalis, Clavispora lusitaniae, Cyberlindnera jadinii, and Kluyveromyces marxianus which were elevated [27]. A different influence of fungi on the inflammation process was discovered. Particularly, Candida albicans can positively modulate susceptibility to inflammation while Saccharomyces boulardii or Saccharomyces cerevisiae has an opposite effect. Knowledge about the interactions between intestinal mycobiota and the human immune system comes mainly from research on Candida and Saccharomyces, however, it has not been fully discovered how the immune system recognizes fungal invasion or colonization in the gut lumen. In particular, the Candida infection significantly raised the plasma IL1β and TNFα levels in the experimental model of colitis [28]. Furthermore, Candida albicans has been observed to expand in active IBD patients compared to IBD in remission [26]. Whereas, a higher abundance of Aspergillus, Wickerhamomyces, Candida, and Sterigmatomyces, and a lower richness of Alternaria, Penicillium, Exophiala, Emericella, Acremonium, Epicoccum, and Trametes was observed by Qiu et al. in UC patients compared to healthy subjects [29].
The evidence of the direct contribution of fungi to IBD includes several mechanisms of action. Particularly, Candida albicans has been proved as one of the most common IBD-associated fungal species because of its virulence factors, such as hyphal formation, toxin secretion, adherence ability, and resistant biofilm formation. This fungal species by secretion of candidalysin, a toxic protein, induce damage to epithelial membrane and intestinal macrophages in the host. Candida albicans has ability to convert from commensal fungi to hyphal structure, but a properly functioning host immune system maintains its nonpathogenic form. Furthermore, it was proved that the interaction of Candida tropicalis with Serratia marcescens and Escherichia coli result in increased production of biofilm [30,31].
The results of different studies indicate a complex relationship between the bacteria and fungi in the gut microbiota, and specific alterations are present in IBD. In a study performed by Sokol et al., a positive correlation was found between the high abundance of protective Saccharomyces and several bacteria such as Bifidobacterium, Blautia, Roseburia, and Ruminococcus in IBD cases [26].
Interestingly, there is growing evidence concerning an association between specific fungal abundance and the genotype of host genes. Such an association was found, for example, between the CLEC7A gene, coding for dectin-1 and an abundance of Malassezia sympodialis, Aspergillus, and Candida in UC, also between the CARD9 gene and an abundance of Saccharomyces cerevisiae, and the TLR1 gene (coding for toll-like receptor 1) and Malassezia sympodialis in IBD [32]. Dectin-1, a transmembrane protein expressed by host macrophages and monocytes, interacts with the β-glucans located in the layer of the fungal cell wall. These proteins are important for host myeloid cell recognition, inflammatory cytokines release, and T-cells development. It was proved, that the interaction between dectin-1 and β-glucans plays an crucial role in response to Candia species, as well as many other fungi [33].
Recent observations supporting the fact that fungal pathogens disturb the host immune system indicate that in CD patients increased antibodies against fungal targets are identified long before diagnosis [34].
2.4. Parasites
Intestinal parasites are well known for their pathogenic potential in the host organism. However, recent studies revealed the commensal role of some of these in the human gut, as in maintaining intestinal homeostasis. Research demonstrated the presence of over 15 different genera of protozoa (amoebozoans, flagellates, ciliates, stramenopiles, and apicomplexans) classified as parasites but belonging to the commensals of the GI tract [36]. The impact of protozoan parasites on the development and progression of IBD has been analyzed with a particular focus on Blastocystis hominis, associated with different gastrointestinal disturbances. Surprisingly, it was observed that the frequency of Blastocystis hominis infection was lower in IBD patients compared to healthy controls, as well as in patients with active UC in contrast to individuals in remission [37]. Additionally, Blastocystis hominis were found to be associated with a higher abundance of Clostridia, Ruminococcaceae, and Prevotellaceae, as well as butyrate-producing Faecalibacterium and Roseburia, but a lower abundance of Enterobacteriaceae. In general, Blastocystis hominis colonization was not linked with colitis-specific dysbiosis but was recognized as a common element of healthy human microbiota acting protectively in gastrointestinal diseases [38]. On the other hand, some studies demonstrated the opposite results, where Blastocystis hominis was in a higher abundance in UC patients compared to the controls [39].
However, numerous protozoan parasites are pathogenic and may induce intestinal inflammation and complicate the course of the disease. Studies prove that Entamoeba histolytica might provoke colitis [40]. Moreover, it is known that Cryptosporidium parvum may exacerbate the course of IBD [41].
Finally, the relationship between parasites and other intestinal microbiota in terms of IBD pathogenesis is a major issue that requires further research and exploration due to insufficient data existing in this area. On the other hand, studies have shown the potential of human helminth in regulating the immune system in IBD. Helminth-derived product therapy is currently being studied in animal colitis models [42].
Currently, microbial dysbiosis in IBD is rather well-established and numerous review studies in this area are available [43]. Nevertheless, understanding these characteristic changes in the composition of the microbiota in patients with IBD, as mentioned in the conclusions of numerous independent world studies, is important in order to fully discover the disease mechanism. It is known that some shifts in taxonomic levels are closely related to the development of intestinal inflammation. However, it is still unclear whether intestinal dysbiosis is the cause or the consequence of inflammation.
3. Metabolomic Changes of Intestinal Microbiota
Once the phenomenon of dysbiosis had been described in IBD patients, studies on changes in the profile of intestinal metabolites began. Global investigations showed disturbances in the metabolic balance in the stools, urine, blood, tissue, and breath of patients [44]. Shifts in the metabolomic profile of IBD patients are a consequence of impairments in gut microbiota composition. In general, functionally important and biologically active metabolites have been observed to be depleted in major IBD cases [45].
3.1. SCFAs
Particularly substantial and beneficial metabolites come in the form of SCFAs, products of microbial fermentation, including acetate, propionate, and butyrate, which have anti-inflammatory properties. Bacteria producing SCFAs (such as Bacteroidetes, Clostridium clusters IV and XIVa, Faecalibacterium. prausnitzii) are reported to be decreased in IBD. SCFAs constitute a source of energy for colonic epithelial cells, modulate intestinal motility, induce the activity of T cells in the colon, and are also responsible for maintaining remission in UC individuals [46]. Acetate and propionate exert extra-intestinal roles, where they act as metabolic substrates for lipogenesis and gluconeogenesis. Studies showed that butyrate leads to the down-regulation of overexpressed inflammatory pathway genes in UC patients [47].
3.2. Bile Acid
Secondary bile acids are products of intestinal microbiota metabolism. They act as signaling factors on nuclear receptors, which control the communication process between intestinal microbiota and the host immune system [48]. Four decades of research was needed to establish IBD’s correlation with bile acid. Research data demonstrated that oral administration of secondary bile acids in mice alleviated the severity of colitis. Bile acid malabsorption was reported to be a common reason for diarrhea in CD and ulcerative colitis patients [49]. Microbial dysbiosis associated with IBD leads to bile acid dysmetabolism and may disturb the anti-inflammatory effect of bile acids [50].
3.3. Hydrogen Sulfide
Hydrogen sulfide is generated by gut microbiota in two biochemical pathways – by sulfate-reducing bacteria (SRB), such as Desulfotomaculum, Desulfosporosinus, Thermodesulfobacterium, Thermodesulfovibrio, Desulfovibrio or by bacteria that ferment sulfur-containing amino acids, such as Fusobacterium nucleatum, Atopobium spp., Gemella sanguinis, Micromonas micros, Streptococcus spp., Actinomyces spp., Eubacterium spp., Veilonella spp., Prevotella spp., Campylobacter spp., and Selenomonas spp. [51]. Studies showed enrichment of hydrogen sulfide producers in CD patients. Furthermore, it is also correlated with the activity of the disease. Individuals with active CD revealed an increase in Enterococcus, Fusobacterium, Haemophilus, Megasphaera, Campylobacter, while Roseburia, Christensenellaceae, Oscillibacter, and Odoribacter were more abundant in subjects with inactive CD [52]. Hydrogen sulfide shows genotoxic and cytotoxic activity at higher concentrations by affecting the genes responsible for cell cycle progression, DNA repair and inflammatory response. Hydrogen sulfide may also inhibit the cytochrome c oxidase activity of mitochondrial respiration [53]. Moreover, functional profiling using shotgun metagenomics confirmed the dysregulation of metabolic pathways involved in different aspects of sulfur metabolism in CD and UC [52]. In IBD, higher hydrogen sulfide generation is confirmed, and disturbed intestinal detoxification of hydrogen sulfide is also proved. There is also evidence that hydrogen sulfide can block the use of butyrate by colonocytes [54].
3.4. Tryptophan
Tryptophan is a precursor for the synthesis of several important bioactive molecules, such as serotonin, melatonin, nicotinamide, and vitamin B3, and it is associated with intestinal inflammation, the epithelial barrier, and the energy homeostasis of the host. This essential amino acid is metabolized in the gastrointestinal tract in three major metabolic pathways, with the participation of kynurenine (KYN), indole, or 5-hydroxytryptamine (5-HT). Tryptophan is involved in the regulation of intestinal inflammation by acting directly or indirectly on the pro/anti-inflammatory cytokines, functions of various immune cells, and intestinal microbial composition [55]. It can be metabolized by the gut microbiota into a range of indole metabolites, some of which can act as ligands for the aryl hydrocarbon receptor (AhR), which has been implicated in IBD pathogenesis [56]. Dysbiosis leads to a loss of microbial activation of tryptophan, causing increased metabolism down the kynurenine pathway. In patients with Crohn’s disease, reduced expression of AhR in inflamed mucosal samples was reported. Recent studies demonstrated that the commensal bacteria Peptostreptococcus russellii could reduce susceptibility to colitis by metabolizing tryptophan to IA (an AhR agonist), improving cell differentiation and reducing inflammatory signals [57].
3.5. Succinate
An increased interest in succinate (tricarboxylic acid cycle intermediate) metabolism in the context of IBD is observed. In the human organism, succinate plays a role as a pro-inflammatory signaling molecule. Furthermore, studies proved it to be a mediator of the macrophage response to lipopolysaccharide [58]. Higher levels of succinate were found in the serum of Crohn’s disease patients, as well as in the fecal samples of both CD and UC subjects [59]. However, a lower level of succinate was shown in the urine of IBD patients, compared to healthy controls. Studies also revealed a decreased abundance of intestinal Phascolarctobacterium in IBD, which is responsible for succinate metabolism [60].
It is already well-documented that these are only a few of the most important metabolites involved in the main metabolic pathways that have an impact on inflammation. Nevertheless, it should be kept in mind that research in this area is still ongoing, and untargeted metabolomics, as well as different small-molecule detection methods combined with functional metagenomics, will provide knowledge about new bioactive molecules relevant to IBD in time [61].
4. Interactions between Host Genes and Microbiota
The impact of genetic factors in IBD conditioning has been known for over three decades. However, the interactions between host genetic variants and the gut microbiota constitute a separate issue. The drivers of IBD in the human genome that have been described include nearly 240 risk loci, many of which are well-known and documented, such as NOD2, ATG16L1, IGRM, CARD9 or IL23R [62]. The basic genetic background of IBD was also described in our previous paper [63]. Currently, the importance of those host genes is highlighted, in that they directly influence microbial colonization in the intestine (The International IBD Genetics Consortium (IIBDGC) et al., 2012) (Figure 1). The latest studies focus on this relationship (including glycosylation level, mucins, and autophagy gene expression level), taking into account this double-sided host-microbe impact as an important part of IBD pathogenesis.
4.1. Glycosylation Genes
The latest research provides evidence that intestinal epithelial glycosylation is an underappreciated process that is linked with all these risk factors of IBD. An association was found between IBD cases and increased expression of truncated O-glycans, as well as altered expression of terminal glycan structures. Epithelial glycans play an important role in regulating the gut microbiota because they provide bacterial ligands, nutrients, and proper colonization of commensal bacteria. Changes in the glycome can be caused by IBD risk genes, glycosyltransferase mislocalization, disturbed glycosyltransferase, and glycosidase expression. What is important to note is that glycome changes are driven by dysbiosis. For example, Salmonella can directly alter human glycan biosynthesis [64]. Impairment of the glycosylation profile leads to disruption of the mucus layer, glycan–lectin interactions, host-microbe interactions, mucosal immunity, and ultimately contributes to IBD pathogenesis [5]. Among the main genes involved in IBD pathogenesis implicated in glycosylation are FUT2, GALC, MANBA, MAN2A1, C1GALT1C1, and TMEM258 [65] (Table 1).
The major glycosylation gene, FUT2, is expressed in the colon, duodenum, and small intestine. The gene synthesizes an enzyme (fucosyltransferase 2) responsible for attaching fucose sugar residues to proteins and lipids in the host mucosa. Epithelial fucosyltransferase 2 creates a symbiotic environment for the host and commensal bacteria. Reduced FUT2 expression or the lack of a functional variant increases the risk of developing IBD [66]. In the group of mice with Fut2 deficiency, colitis, acute inflammation, large infiltrates, and ulceration of the mucosa were observed. Additionally, the intestinal barrier was disturbed in the animal model, which further exacerbated colon damage. The comparative analysis of the 16S rRNA gene of the microbiota of Fut2+ and Fut2- individuals showed a reduced amount of beneficial bacteria from the Ruminococcaceae and Muribaculaceae family, while the pathogenic microorganisms such as Bilophila, Escherichia, Enterorhabdus were found to be increased in the Fut2- cohort [67].
Other studies in IBD patients showed that the FUT2 variant was associated with a decreased abundance of Escherichia, which usually binds to fucosylated oligosaccharides. The FUT2 gene is also correlated with an increased quantity of Alistipe and Phascolarctobacterium, capable of inducing CD8 cells. The FUT2 gene mutation participates in the pathogenesis of IBD by changing the microbial composition by means of reducing the number of binding sites of adjacent bacteria. Reduced numbers of adhering bacteria may allow the overgrowth of bacteria that induces T-lymphocytes and the intestinal epithelium’s inflammatory process [68].
Another gene implicated in the glycosylation process is MAN2A1, encoding the alpha-mannosidase 2 enzyme, which is correlated with the pathogenesis of UC. Studies show that this gene is responsible for the inflammatory state in connection with the increased recruitment of neutrophils [69].
Furthermore, the C1GALT1C gene coding for galactosyltransferase is also known to have an impact on microbial composition in male mice. Loss of chaperone 1β3GalT from intestinal epithelial cells (IEC) disrupts the mucus layer, leading to dysbiosis and sudden inflammation. Sequencing studies demonstrated an 11-fold reduction in Bacteroides and a 3-fold increase in pathogenic Helicobacter microbes in subjects with IBD [70].
4.2. Autophagy Genes
Recent investigations have identified autophagy-related genes among genetic factors playing a role in IBD etiology [65] (Table 1). Autophagy is crucial for maintaining intestinal homeostasis and the proper immune response. This process was reported to regulate intestinal barrier function by inducing lysosomal degradation of the tight junction protein (claudin 2), thus decreasing epithelial permeability [71]. Dysfunctional autophagy leads to altered epithelial function, dysbiosis, and an aberrant immune response. The GWAS studies of variants identified a group of IBD susceptibility autophagy genes: NOD2, ATG16L1, IGRM, CARD9, and IL23R [72]. Molecular analysis made it possible to formulate a research hypothesis on the significance of the correlation between the genetic aspect of the host and intestinal microbes and to offer an answer to the question: ‘What should be the optimal environment for the appropriate microbial level? The first gene identified was NOD2 (CARD15), which is located in the IBD1 (IBD susceptibility locus 1, OMIM#266600) region on chromosome 16q12.1. The gene is highly expressed in Paneth cells located in the intestinal crypts and partially in the intestinal epithelium [73]. Well-described gene polymorphisms of CD are the following variants: rs2066846 Leu1007fs; rs2066845, Gly908Arg; and rs2066844 Arg702Trp (Table 1). The C allele of rs2066845 carriers significantly influenced the change in the microbiome’s composition through the increased level of fecal bacteria Erysipelotrichaceae [74]. However, the dominant predictor of CD turned out to be the Leu1007fs variant, which causes a frameshift. In effect, it was associated with complications of the disease: 79% of homozygous carriers among patients required hospitalization and ileal surgery [75]. Functional loss from homozygotes leads to a limited regulation of the host’s response to the gut microbes and changes in the microflora. There was a decrease in Clostridia and an increase in Proteobacteria in CD patients. The risk variants were also correlated with altered levels of Faecalibacterium and E.coli [76]. However, similar conclusions in studies with mice showed a high cluster load of Bacteroidetes and Firmicutes in the distal part of the intestine among CD homozygotes. In addition, these studies demonstrated that NOD2 plays an elementary role in the anti-microbial activity of Paneth cells and the proper regulation of the intestinal microflora. In addition, risk alleles predispose to an increase in the pathogenic taxa disrupting the intestinal microbiota - Yersinia, Campylobacter, Citrobacter, Escherichia coli, Helicobacter, Listeria, Mycobacteria, Pseudomonas, or Staphylococcus, Erysipelotrichaceae. It is worth noting that NOD2 interacts with other genes IRGM and ATG16L1, intensifying the process of autophagy. Altered IRGM expression affected the microflora of the healthy group by reducing the abundance of the butyrate-producing genus Roseburia [77]. Studies point out that variant T300A in the ATG16L1 gene leads to impaired autophagy and increased pro-inflammatory cytokine production. CD patients homozygous for variant T300A in the ATG16L1 gene exhibit an increased abundance of pathobionts such as Escherichia coli, Bacteroidesfragilis, and Fusobacteriaceae. The T300A variant of the ATG16L1 gene is also correlated with a reduced amount of the commensal bacterium. These bacteria provide an optimal environment in the intestine and revitalize the host’s mucus layer, thus maintaining its integrity. T300A polymorphism contributes to a reduction in Akermansia muciniphila in IBD patients. In addition, the gene variant led to increased recruitment of Muscipirillum schaedleri, which may propagate colitis in the course of UC [78]. Also, other genes involved in the autophagy process were identified as associated with CD pathogenesis, such as ATG4A, ATG2A, and ATG4D coding for autophagy-related cysteine peptidases [65].
4.3. Mucins
Mucins, large glycoproteins decorated with O-linked glycans, are IBD host-risk factors directly influencing microbiota, which play a significant role in host-microbiome interactions through the ability to gel (moisturize), create chemical barriers, and participate in the cell signaling pathway [79]. HUGO Gene Nomenclature Committee has classified 21 mucin genes so far, of which 13 are expressed in the intestinal epithelium (Table 1) [107].
Some pathogens may down-regulate mucin production as a strategy to disrupt the mucus barrier integrity. In particular, Helicobacter pylori and Clostridioides difficile infections are associated with disrupted mucin synthesis and mucus barrier function. In fact, H. pylori-infected patients indicated a significant decrease in the gene MUC5AC expression level, coding for mucin 5AC. IBD patients exhibit an altered mucus barrier or mucin production. This is supported by some studies, in which animal models with disrupted barrier integrity manifested IBD-like syndromes. Decreased expression of MUC5AC was observed in patients with exacerbated enteritis, and extended molecular studies showed that endoscopic treatment of UC patients increased its expression. However, work is still ongoing to understand the functional role of MUC5AC in inducing inflammation in the pathogenesis of IBD [81]. In 2020, the results of a patient group study showed that the expression of human MUC5AC is correlated with active UC [82]. However, in the control group, 55% of healthy subjects did not express MUC5AC. These results showed that the mucin expression was restricted in healthy, non-inflammatory tissue. The same study was carried out in a mouse model, where a knock-out of Muc5ac was performed. Persistent loss of the functional variant enhanced the interaction of the bacteria with the epithelial host. Sustained contact between Helicobacter pylori and the host intestinal mucosa led to an increase in the level of neutrophilia and then initiated increased production of inflammatory cytokines: interleukin-10 (IL10) and C-X-C motif chemokine ligand 16 (CXCL16) [83]. In conclusion, induction of human MUC5AC expression can directly strengthen the protective barrier through maintaining the integrity and thickness of the mucin layer, which plays a preventive role in inflammation associated with UC [84]. Studies also identified the MUC19 gene as a risk locus for IBD [85].
On the other hand, the link between UC and the mucin layer is well established. UC patients display a thinner mucus layer as a result of impaired MUC2 production and secretion. The MUC2 gene induces the production of the MUC2 protein in the gut, which is the major component of the mucin gel layer [86]. Animal model studies also confirmed that the deletion of Muc2 conditioned the reduced thickness of the protective mucin layer and led to the development of colonic inflammation in patients with UC [87]. Current evidence suggests that rare MUC3A alleles can encode mucin proteins with specific conformational changes. Knowing that the processes of O-glycosylation and N-glycosylation depend on the protein’s structure, altering the protein’s conformation can result in reduced glycosylation. Less glycosylation correlates with increased sensitivity to intestinal bacterial proteases and mucin degradation, thereby breaking the continuity of the barrier [88]. MUC9 and MUC20 gene expression was found to be significantly decreased in patients with active UC compared to both the remission group and controls [89].
4.4. Cytokines
It is also known that impaired protective function of the intestinal barrier promotes the synthesis of cytokines. Thus, the complex pathogenesis of IBD includes genes encoding proteins involved in the immune response, such as IL1B, IL6, IL10, IL23, IL23R, and TNFA [90] (Table 1). The latest research from 2022 shows that the decreased level of IL6 contributed to the dysbiosis of the intestinal microflora in mice, increasing the abundance of Gramm-negative bacteria with impaired metabolic pathways. Decreased IL10 expression was found to be associated with a greater abundance of Bacteroides, Prevotella, and Rikenella [91]. Accordingly, an increased serum concentration of interleukin-23 receptor (IL23R) was noted in IBD patients. The disturbed balance of microorganisms leads to the growth and colonization of pathogens, which promotes an increased immune response and the development of IBD. Klebsiella pneumoniae invade intestinal epithelial cells and interact with macrophages to cause the secretion of IL1B and TNFα [92]. Additionally, the disturbing expression of genes responsible for autophagy, which leads to abnormal colonization of AIEC pathobionts, has been described as causing enhanced inflammation and the synthesis of pro-inflammatory cytokine TNFα [93]. We also observed that the species Dorea is negatively correlated with the interferon-gamma (IFNG) response. Moreover, the species mentioned (Streptococcus parasanguinis and Streptococcus australis) were associated with IFNγ production. Other species, such as Streptococcus mitis, Streptococcus oralis, and Streptococcus pneumoniae, were also associated with IL1B overexpression. On the other hand, Bifidobacterium adolescentis was inversely related to the production of TNFα, which highlights the potential cytokine and species specificity in the group of IBD patients [94].

Table 1.
Host genetic variants and their impact on gut microbiota in IBD.
| Disease | Gene, full name |
Genetic variant rs number, minor allel, consequence |
Location |
MAF (%) |
Function/ Impact on the host microbiome |
References |
|---|---|---|---|---|---|---|
| Crohn’s Disease (CD) |
ATG16L1 autophagy related 16 like 1 |
rs12994997 (A) intron variant |
2q14.1 | 39 |
↑The risk allele (A) increases abundance of pathogenic symbionts in the intestinal mucosa: Enterobacteriaceae, Bacteroidaceae, and Fusobacteriaceae. ↑ The protective allele (G) increases the number of commensal bacteria Lachnospiraceae |
[95] |
| rs10210302 (T) 2kb upstream variant |
2q14.2 | 39 | Variant C/C is significantly associated with the protection of IBD patients in the Indian population OR = 0.89 (0.71–1.13) | [96] | ||
| rs2241880 (G) missense variant p. Thr216Ala |
2q14.1 | 40 | A significant difference in the incidence of Listeria monocytogenes and Yersinia enterocolitica pathobionts in patients with CD compared to the control group (p<0.05); ↑ The variant T300A leads to impaired autophagy and increased pro-inflammatory cytokine production. ↑ Patients homozygous for variant T300A in the ATG16L1 gene exhibit an increased abundance of pathobionts such as E. coli, Bacteroidesfragilis, Fusobacteriaceae. Muscipirillum schaedleri, ↓Reduction of Bacteroidetes and Firmicutes |
[97,98] |
||
| rs6754677 (A) intron variant |
2q14.2 | 37 | The homozygous genotype A/A showed a risk of developing CD, is associated with terminal ileitis as well, and is related to autophagy. | [99] | ||
|
NOD2 nucleotide-binding oligomerization domain containing 2 |
rs2066844 missense variant p.Arg702Trp |
16q11.2 | 1 | ↑ Increased level of Enterobacteriaceae family and Helicobacter pylori - a risk factor for colon cancer in CD patients. |
[74] |
|
| rs2066845 missense variant p.Gly908Arg p.Gly908Cys |
16q11.2 | 1 | ↓Reduced of Bacteroidetes and Firmicutes, ↑Additionally, the C allele was significantly associated with an increase in relative abundance in the fecal bacterial family Erysipelotrichaceae. |
[74,76] |
||
| rs2066847 (CC/CCCC) frameshift variant p.Leu1007Pro(fs) |
16q12.1 | 1 | Impaired synthesis of pro-inflammatory cytokines (IL1B) and dendritic cells, leading to deregulation of the host’s antimicrobial defense; ↑ Increased abundance of pathobionts and permeability gut. |
[75] |
||
| - | - | - | ↑Increased pathogenic taxa: Yersinia, Campylobacter, Citrobacter, Escherichia coli, Helicobacter, Listeria, Mycobacteria, Pseudomonas, or Staphylococcus | [100] | ||
|
IRGM immunity related GTPase M |
rs11741861 (G) intron variant |
5q14.3 | 16 | ↓The risk variant reduces the abundance of anaerobic bacteria, butyrate-producing - Roseburia in patients with IBD. The protective barrier mucosa of the colon is compromised and, as a result, inflammation is triggered. |
[77] |
|
| rs13361189 (C) intergenic variant |
5q14.3 | 30 | Alteration in the intensity of inflammation of the intestinal mucosa as a result of the implication of an accelerated immune response | [101] | ||
| rs10065172 (T) missense variant p=Leu105= |
5q14.3 | 30 | ↑Increased susceptibility to CD in individuals of European patients (p = 0.008); Haplotype T/T influenced the binding site of a specific microRNA, causing the deregulation of IRGM-dependent xenophagy bacteria in patients with CD; ↑In addition, the T/T genotype is also associated with an increased level of expression of the cytokine TNFα in the peripheral blood, influencing inflammation. |
[72,102] |
||
| Ulcerative colitis |
MUC13 Mucin 13 |
rs1127233 (G) missense variant Arg503Ser |
3q21.2 | 23 |
Variant correlated with UC p = 0.0003; Disturbed MUC13 gene expression correlated with the NFkB pathway can lead to a loss of membrane integrity and thus permeability. |
[103] |
| Inflammatory Bowel Disease (IBD) |
MUC1 Mucin 1 |
- | - | - | MUC1 codes for the main mucus component which is the physical barrier that protects the intestinal epithelium from intestinal bacteria. MUC1 overexpression and hypoglycosylation have been reported in Muc1-knockout IBD mice showing increased damage to the small intestine following infection with C. jejuni. | [104] |
| rs4072037 (C) synonymous variant p.Thr22= |
1q22 | 37 | ↑Increase in the abundance of Ochrobactrum | [105] | ||
|
MUC2 Mucin 2 |
rs2856111 (T) missense variant Leu58Pro |
11p15.5 | 27 | Reduced gene expression is associated with a thinner mucus layer in UC patients - particularly at the site of inflammation due to the reduction of goblet cells. | [106] | |
| rs11825977(A) missense variant p.Val116Met |
11p15.5 | 12 | Decreased MUC2 mRNA expression increases the risk of inflammation and intestinal dysbiosis. | [103] | ||
|
MUC3A Mucin 3A |
- | 7q22.1 | - | Rare alleles change the conformation of the proteins produced. The conformation affects the glycosylation process, which increases the sensitivity to bacterial proteases, and thus breaks the continuity of the protective gel barrier. |
[88] |
|
|
MUC5AC Mucin 5AC |
rs35783651 (G) missense variant p.Ser221Arg |
11p15.5 | 10 | Protective role by participating in the healing of mucosal epithelial wounds and regulating MGL; H. pylori-infected patients indicated a significant decrease in MUC5AC expression level |
[107] |
|
|
MUC19 Mucin 19 |
rs11564245 (C) missense variant p.Asp803His |
12q12 | 5 | Increased susceptibility to CD in the group of patients. | [3,85] |
|
| rs4768261 (T) missense variant p.Ser1226Phe |
12q12 | 5 | ||||
|
CARD9 Caspase Recruitment Domain Family Member 9 |
rs4077515 (T) missense variant p.Ser12Asn/Ile |
9q21.3 | 37 | Innate immune response to peptidoglycan, a macromolecule in the bacterial cell wall ; Aberrant activation of NF-κB and inflammatory factors in response to Aspergillus fumigates, contributing to intestinal inflammation. |
[108] |
|
| rs10781499 (A) synonymous variant p.Pro42= |
9q34.3 | 37 | ↓Decrease butyrate acetate converting bacteria - Roseburia spp | [77] | ||
| rs10870077 (G) intron variant |
9q34.3 | 37 | ↑Increased risk of UC development by modulating the signaling pathway affecting the inflammatory response. |
[109] |
||
|
FUT2 Fucosyltransferase 2 |
Fut2- | 19q13.33 | - | ↓Reduced beneficial bacteria from the Ruminococcaceae and Muribaculaceae, while the pathogenic microorganisms, such as ↑Bilophila, Escherichia, Enterorhabdus, Alistipe, Phascolarctobacterium were increased in the cohort. |
[67,68] |
|
|
C1GALT1C Core 1 Synthase, Glycoprotein-N-Acetylgalactosamine 3-Beta-Galactosyltransferase 1 |
- | 7p22.1-p21.3 | - | Studies demonstrated an 11-fold reduction of Bacteroides and a 3-fold increase of pathogenic Helicobacter microbes in the IBD cohort. | [70] | |
|
IL1B Interleukin 1- Beta |
- | 2q14.1 | - | Increased ILIB level by attack and colonization of Klebsiella pneumoniae, Streptococcus mitis, Streptococcus oralis, and Streptococcus pneumoniae. | [92] |
|
|
IL6 Interleukin 6 |
- | 1q21.3 | The deficiency of IL6 contributes to the dysbiosis of the gut microbiota and increases the abundance of Gram-negative bacteria. | [110] | ||
|
IL10 Interleukin 10 |
rs1800896 (A) 2kb upstream variant |
1q32.1 | 27 | ↓Loss of IL10 receptor function - induction of inflammation in severe course of UC; Allele A was associated with UC p=0.011 in in Mexican cohort. ↑ Increased susceptibility to fungal infections with Candida albicans ↓Decreased IL10 expression is associated with ↑ increased Bacteroides, Prevotella, and Rikenella |
[91] |
|
|
IL23R Interleukin 23 |
rs1004819 (A) intron variant |
1p.31.3 | 40 | Early age of onset of the disease in the Polish population. | [111] | |
| rs76418789 (A) missense variant p.Gly149Arg |
1p.31.3 | 1 | SNP associated with IBD in the Korean population (p = 0.0096) | [112] | ||
| rs11209026 (A) missense variant p.Arg381Gln |
1p.31.3 | 2 | Protective effect in CD but related to UC; ↑ Increased abundance of Christensenellaceae, Bacteroides caccae, and a ↓ decrease in the commensal bacteria Faecalibacterium prausnitzii |
[113] |
||
| rs2201841 (G) intron variant |
1q11 | 40 | Significant association between polymorphisms and UC, especially in Caucasians. | [113] | ||
|
IFNG Interferon Gamma |
- | 12q15 | - | ↑ Increased level of taxa: Dorea, Streptococcus parasanguinis and Streptococcus australis | [94] |
5. Conclusions
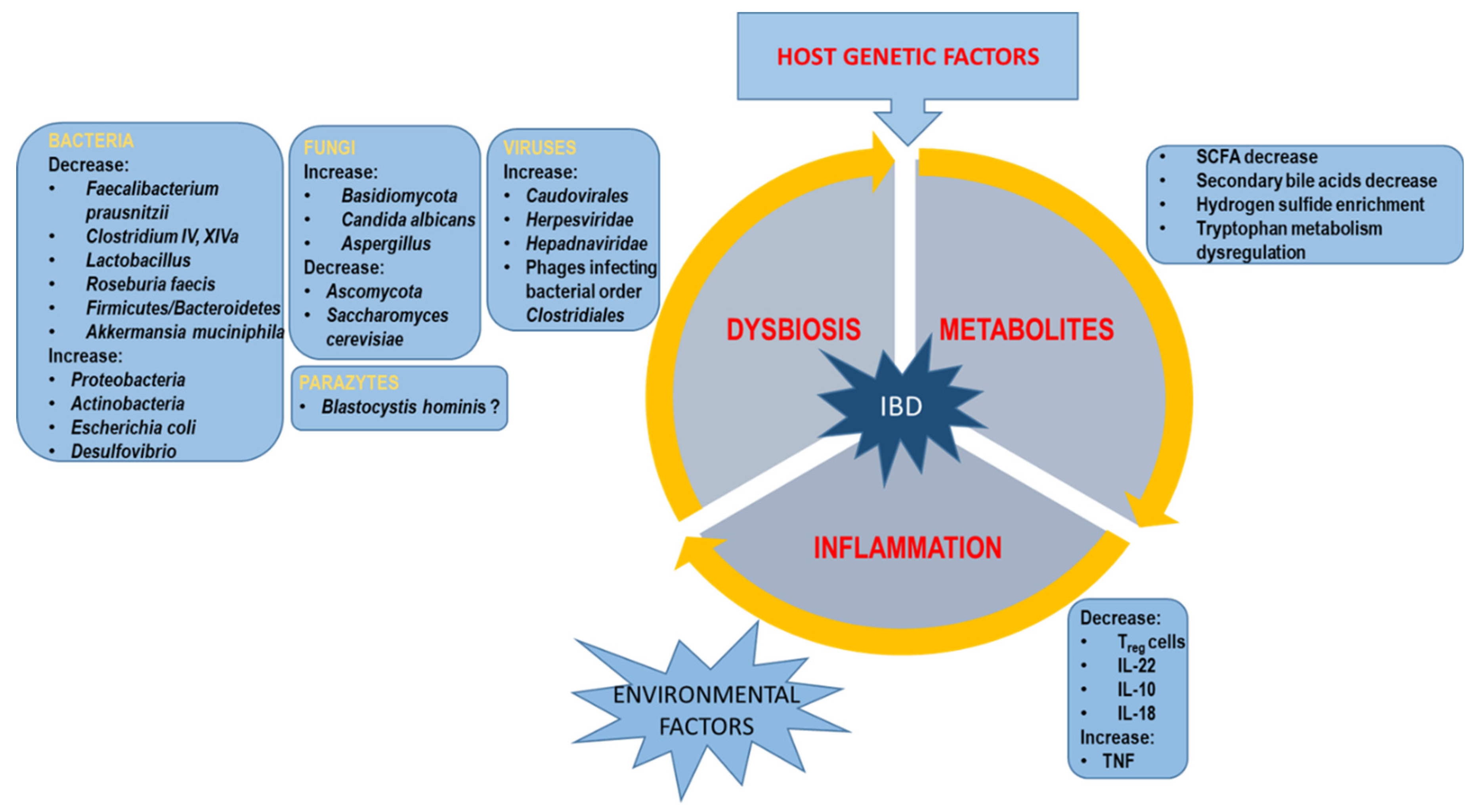
In the last ten years, there has been an intensive development of research on the intestinal microbiota and the metabolome, which has allowed for a broader understanding of the complexity of IBD pathogenesis (Figure 2). However, it is still not entirely clear. NGS technology opens up a new area in microbiota identification, giving scientists a great opportunity to analyze and compare whole microbial communities, including their composition, interactions, and functions. Despite the often inconsistent results of global research, it has been possible to identify the “pathogenic” and “protective” microbiota in the context of IBD development. Similarly, in terms of metabolites, examples of those that intensify inflammation and those that help calm it down, have been identified (Figure 2).
The weakness of research conducted around the world aimed at characterizing the microbiota associated with IBD is certainly the diverse methodology used to identify the microbiota - analysis of different regions (V1-V9) of the 16S rRNA gene and the diversity of NGS platforms, which affects the final qualitative and quantitative result of identified taxa. Similarly, for methods to identify metabolites associated with IBD, targeted and untargeted compound identification approaches are used. Notably untargeted deep analytical approaches give us the potential to discover new important small molecules.
Among global research on IBD pathogenesis, there is a clearly visible trend to move from microbial profiling into a study of whole genomic sequences of microbes and into host-microbe interactions, which should provide the development of microbiome-based diagnostics and targeted therapy for IBD. Recent studies in this field attempted an integrative analysis of host, microbial, and multi-omics risk factors to find an algorithm for the precise characterization of CD and UC profiles. Understanding the role and pathways of microbial biomarkers in the assessment of disease activity and treatment outcomes is critical to the monitoring and treatment of IBD. The knowledge from this integrative multi–omics analysis is necessary for development of targeting microbiome-based therapeutic approaches, which is a promising strategy to alleviate and cure this inflammatory disease.
One of therapeutic option for IBD patients is fecal microbiota transplantation (FMT) from healthy individuals with rich microbial composition. It allowed for the transfer of a part of the entire ecosystem, not just the microbes. It helps in calming inflammation in patients and is more effective than oral intake of probiotics, but controversial. FMT can be used as therapy in IBD in case the antibiotics are ineffective. A particularly promising probiotic bacteria is Faecalibacterium. prausnitzii, Akkermansia muciniphila or some Roseburia strains.
In recent years, phages–based therapy focused new attention on IBD. It was proved in several clinical trials in adults and children as a safe therapeutic option without adverse events. Moreover, phages constitute a majority of viruses in the human gut. Phages could target potentially disease-causative bacteria, as Ruminococcus gnavus, Escherichia coli, Bacteroides fragilis, or fungi, as C. albicans, enriched in the human IBD intestine.
We are hopeful to observe the progress of scientific research in this area and believe that in the near future IBD will be a curable disease.
Figure 2.
Scheme of factors implemented in IBD pathogenesis.
Author Contributions
OZB gave the concept and wrote the paper; JZS created the table and figures; PE edited the text; KL revised and amended the text; RS and MSZ critically revised the paper. All authors approved the final version of this manuscript.
Funding
This research was funded by the Polish National Science Centre (grant no. 2020/04/X/NZ2/02172) and by the Biocodex Microbiota Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Manuscript based on the public available data in PubMed.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years - United States, 2015, MMWR Morb Mortal Wkly Rep 2016, 65, 1166–1169. [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; Kaplan, G.G. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review, Gastroenterology 2012, 142, 46–54.e42; quiz e30. [CrossRef]
- Kumar, S.; Kumar, A. Microbial pathogenesis in inflammatory bowel diseases, Microbial Pathogenesis 2022, 163, 105383. [CrossRef]
- Li, J.; Butcher, J.; Mack, D.; Stintzi, A. Functional Impacts of the Intestinal Microbiome in the Pathogenesis of Inflammatory Bowel Disease, Inflammatory Bowel Diseases 2015, 21, 139–153. [CrossRef]
- Kudelka, M.R.; Stowell, S.R.; Cummings, R.D.; Neish, A.S. Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD, Nat Rev Gastroenterol Hepatol 2020, 17, 597–617. [CrossRef]
- Buttó, L.F.; Haller, D. Dysbiosis in intestinal inflammation: Cause or consequence, International Journal of Medical Microbiology 2016, 306, 302–309. [CrossRef]
- Littman, D.R.; Rudensky, A.Y. Th17 and Regulatory T Cells in Mediating and Restraining Inflammation, Cell 2010, 140, 845–858. [CrossRef]
- Ayabe, T.; Satchell, D.P.; Pesendorfer, P.; Tanabe, H.; Wilson, C.L.; Hagen, S.J.; Ouellette, A.J. Activation of Paneth Cell α-Defensins in Mouse Small Intestine, Journal of Biological Chemistry 2002, 277, 5219–5228. [CrossRef]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; Bleich, A.; Haller, D. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence, Gut 2016, 65, 225–237. [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; Gevers, D.; McGovern, D.P.B.; Singh, N.; Braun, J.; Jacobs, J.P.; Clemente, J.C.; Grinspan, A.; Sands, B.E.; Colombel, J.-F.; Dubinsky, M.C.; Faith, J.J. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt+ Regulatory T Cells and Exacerbate Colitis in Mice, Immunity 2019, 50, 212–224.e4. [CrossRef]
- Webb, C.R.; den Bakker, H.; Koboziev, I.; Jones-Hall, Y.; Kottapalli, K.R.; Ostanin, D.; Furr, K.L.; Mu, Q.; Luo, X.M.; Grisham, M.B. Differential Susceptibility to T Cell-Induced Colitis in Mice: Role of the Intestinal Microbiota, Inflammatory Bowel Diseases 2018, 24, 361–379. [CrossRef]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. ; Nutrition, IBD and Gut Microbiota: A Review, Nutrients 2020, 12, 944. [CrossRef]
- Jandhyala, S.M. Role of the normal gut microbiota, WJG 2015, 21, 8787. [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; Morgan, X.C.; Kostic, A.D.; Luo, C.; González, A.; McDonald, D.; Haberman, Y.; Walters, T.; Baker, S.; Rosh, J.; Stephens, M.; Heyman, M.; Markowitz, J.; Baldassano, R.; Griffiths, A.; Sylvester, F.; Mack, D.; Kim, S.; Crandall, W.; Hyams, J.; Huttenhower, C.; Knight, R.; Xavier, R.J. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease, Cell Host & Microbe 2014, 15, 382–392. [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; Ferrante, M.; Verhaegen, J.; Rutgeerts, P.; Vermeire, S. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis, Gut 2014, 63, 1275–1283. [CrossRef]
- Zhang, M.; Qiu, X.; Zhang, H.; Yang, X.; Hong, N.; Yang, Y.; Chen, H.; Yu, C. Faecalibacterium prausnitzii Inhibits Interleukin-17 to Ameliorate Colorectal Colitis in Rats, PLoS ONE 2014, 9, e109146. [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; Myridakis, A.; Delzenne, N.M.; Klievink, J.; Bhattacharjee, A.; van der Ark, K.C.H.; Aalvink, S.; Martinez, L.O.; Dumas, M.-E.; Maiter, D.; Loumaye, A.; Hermans, M.P.; Thissen, J.-P.; Belzer, C.; de Vos, W.M.; Cani, P.D. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice, Nat Med 2017, 23, 107–113. [CrossRef]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; Slomski, R.; Skrzypczak-Zielinska, M. Dysbiosis of gut microbiota in Polish patients with ulcerative colitis: a pilot study, Sci Rep 2021, 11, 2166. [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; Guerin, E.; Velayudhan, V.; Ross, R.P.; Hill, C. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific, Cell Host Microbe 2019, 26, 527–541.e5. [CrossRef]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; Hill, C. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease, Cell Host & Microbe 2019, 26, 764–778.e5. [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease, Journal of Pediatric Gastroenterology & Nutrition 2019, 68, 30–36. 68. [CrossRef]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; Sung, J.J.Y.; Yu, J.; Chan, F.K.L.; Cao, Q.; Sheng, J.-Q.; Ng, S.C. Gut mucosal virome alterations in ulcerative colitis, Gut 2019, 68, 1169–1179. [CrossRef]
- Ungaro, F.; Massimino, L.; D’Alessio, S.; Danese, S. The gut virome in inflammatory bowel disease pathogenesis: From metagenomics to novel therapeutic approaches, United European Gastroenterol. J. 2019, 7, 999–1007. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; Porter, N.; Martens, E.; O’Connell, R.; Jacob, V.; Scherl, E.; Crawford, C.; Stephens, W.Z.; Casjens, S.R.; Longman, R.S.; Round, J.L. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis, Cell Host & Microbe 2019, 25, 285–299.e8. [CrossRef]
- Iliev, I.D.; Leonardi, I. Fungal dysbiosis: immunity and interactions at mucosal barriers, Nat Rev Immunol 2017, 17, 635–646. [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; Cosnes, J.; Seksik, P.; Langella, P.; Skurnik, D.; Richard, M.L.; Beaugerie, L. Fungal microbiota dysbiosis in IBD, Gut 2017, 66, 1039–1048. [CrossRef]
- Knox, N.C.; Forbes, J.D.; Peterson, C.-L.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiome in Inflammatory Bowel Disease: Lessons Learned From Other Immune-Mediated Inflammatory Diseases, Am J Gastroenterol 2019, 114, 1051–1070. [CrossRef]
- Zwolinska-Wcislo, M.; Brzozowski, T.; Budak, A.; Kwiecien, S.; Sliwowski, Z.; Drozdowicz, D.; Trojanowska, D.; Rudnicka-Sosin, L.; Mach, T.; Konturek, S.J.; Pawlik, W.W. Effect of Candida colonization on human ulcerative colitis and the healing of inflammatory changes of the colon in the experimental model of colitis ulcerosa, J Physiol Pharmacol 2009, 60, 107–118.
- Qiu, X.; Ma, J.; Jiao, C.; Mao, X.; Zhao, X.; Lu, M.; Wang, K.; Zhang, H. Alterations in the mucosa-associated fungal microbiota in patients with ulcerative colitis, Oncotarget 2017, 8, 107577–107588. [CrossRef]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Höfs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; Murciano, C.; Blagojevic, M.; Thavaraj, S.; Förster, T.M.; Hebecker, B.; Kasper, L.; Vizcay, G.; Iancu, S.I.; Kichik, N.; Häder, A.; Kurzai, O.; Luo, T.; Krüger, T.; Kniemeyer, O.; Cota, E.; Bader, O.; Wheeler, R.T.; Gutsmann, T.; Hube, B.; Naglik, J.R. Candidalysin is a fungal peptide toxin critical for mucosal infection, Nature 2016, 532, 64–68. [CrossRef]
- Kasper, L.; König, A.; Koenig, P.-A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; Hube, B. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes, Nat Commun 2018, 9, 4260. [CrossRef]
- The International IBD Genetics Consortium (IIBDGC). Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease, Nature 2012, 491, 119–124. [CrossRef]
- Taylor, P.R.; Brown, G.D.; Reid, D.M.; Willment, J.A.; Martinez-Pomares, L.; Gordon, S.; Wong, S.Y.C.; Receptor, G. ; Dectin-1, Is Predominantly Expressed on the Surface of Cells of the Monocyte/Macrophage and Neutrophil Lineages, The Journal of Immunology 2002, 169, 3876–3882. [CrossRef]
- Miyoshi, J.; Sofia, M.A.; Pierre, J.F. The evidence for fungus in Crohn’s disease pathogenesis, Clin J Gastroenterol 2018, 11, 449–456. [CrossRef]
- Eichenberger, R.M.; Ryan, S.; Jones, L.; Buitrago, G.; Polster, R.; de Oca, M.M.; Zuvelek, J.; Giacomin, P.R.; Dent, L.A.; Engwerda, C.R.; Field, M.A.; Sotillo, J.; Loukas, A. Hookworm Secreted Extracellular Vesicles Interact With Host Cells and Prevent Inducible Colitis in Mice, Front. Immunol. 2018, 9, 850. [Google Scholar] [CrossRef]
- Hamad, I.; Raoult, D.; Bittar, F. Repertory of eukaryotes (eukaryome) in the human gastrointestinal tract: taxonomy and detection methods, Parasite Immunol 2016, 38, 12–36. [CrossRef]
- Coskun, A.; Malatyali, E.; Ertabaklar, H.; Yasar, M.B.; Karaoglu, A.O.; Ertug, S. Blastocystis in ulcerative colitis patients: Genetic diversity and analysis of laboratory findings, Asian Pacific Journal of Tropical Medicine 2016, 9, 916–919. [CrossRef]
- The Blastocystis Investigation Group, C. Audebert, G. Even, A. Cian, A. Loywick, S. Merlin, E. Viscogliosi, M. Chabé, Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota, Sci Rep 2016, 6, 25255. [Google Scholar] [CrossRef]
- Tai, W.-P.; Hu, P.-J.; Wu, J.; Lin, X.-C. Six ulcerative colitis patients with refractory symptoms co-infective with Blastocystis hominis in China, Parasitol Res 2011, 108, 1207–1210. [CrossRef]
- Verstockt, B.; Vermeire, S.; Van Assche, G.; Ferrante, M. When IBD is not IBD, Scandinavian Journal of Gastroenterology 2018, 53, 1085–1088. [CrossRef]
- Vadlamudi, N.; Maclin, J.; Dimmitt, R.A.; Thame, K.A. Cryptosporidial infection in children with inflammatory bowel disease, Journal of Crohn’s and Colitis 2013, 7, e337–e343. [CrossRef]
- Arai, T.; Lopes, F. Potential of human helminth therapy for resolution of inflammatory bowel disease: The future ahead, Experimental Parasitology 2022, 232, 108189. [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: causation or correlation? , Nat Rev Gastroenterol Hepatol 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Tefas, C.; Ciobanu, L.; Tanțău, M.; Moraru, C.; Socaciu, C. The potential of metabolic and lipid profiling in inflammatory bowel diseases: a pilot study, Bosn J of Basic Med Sci 2019. [CrossRef]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review, Journal of Crohn’s and Colitis 2021, 15, 813–826. [CrossRef]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Lin, E.; Borody, T.J.; Wilkins, M.R.; Colombel, J.-F.; Mitchell, H.M.; Kaakoush, N.O. Specific Bacteria and Metabolites Associated With Response to Fecal Microbiota Transplantation in Patients With Ulcerative Colitis, Gastroenterology 2019, 156, 1440–1454.e2. [CrossRef]
- Magnusson, M.K.; Isaksson, S.; Öhman, L. The Anti-inflammatory Immune Regulation Induced by Butyrate Is Impaired in Inflamed Intestinal Mucosa from Patients with Ulcerative Colitis, Inflammation 2020, 43, 507–517. [CrossRef]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile Acid Signaling in Inflammatory Bowel Diseases, Dig Dis Sci 2021, 66, 674–693. [CrossRef]
- Bares, M.; Cantero, J.M.B.; Flores, E.I.; Alcalde, B.G.; Ortega, E.M.; Muret, F.R.M.; Asenjo, E.C.; Sánchez, V.G.; Casas, J.A.V. Bile acid malabsorption in patients with chronic diarrhea and Crohn�s disease, Rev Esp Enferm Dig 2018, 111,. [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; Bridonneau, C.; Dumetz, F.; Grill, J.-P.; Masliah, J.; Beaugerie, L.; Cosnes, J.; Chazouillères, O.; Poupon, R.; Wolf, C.; Mallet, J.-M.; Langella, P.; Trugnan, G.; Sokol, H.; Seksik, P. ; Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases, Gut 2013, 62, 531–539. [CrossRef]
- Washio, J.; Sato, T.; Koseki, T.; Takahashi, N. Hydrogen sulfide-producing bacteria in tongue biofilm and their relationship with oral malodour, Journal of Medical Microbiology 2005, 54, 889–895. [CrossRef]
- Metwaly, A.; Dunkel, A.; Waldschmitt, N.; Raj, A.C.D.; Lagkouvardos, I.; Corraliza, A.M.; Mayorgas, A.; Martinez-Medina, M.; Reiter, S.; Schloter, M.; Hofmann, T.; Allez, M.; Panes, J.; Salas, A.; Haller, D. Integrated microbiota and metabolite profiles link Crohn’s disease to sulfur metabolism, Nat Commun 2020, 11, 4322. [CrossRef]
- Leschelle, X.; Goubern, M.; Andriamihaja, M.; Blottière, H.M.; Couplan, E.; Gonzalez-Barroso, M.-M.; Petit, C.; Pagniez, A.; Chaumontet, C.; Mignotte, B.; Bouillaud, F.; Blachier, F. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide, Biochimica et Biophysica Acta (BBA) - General Subjects 2005, 1725, 201–212. [CrossRef]
- Smith, F.M.; Coffey, J.C.; Kell, M.R.; O’Sullivan, M.; Redmond, H.P.; Kirwan, W.O. A characterization of anaerobic colonization and associated mucosal adaptations in the undiseased ileal pouch, Colorect Dis 2005, 7, 563–570. [CrossRef]
- Li, X.; Zhang, Z.; Zabed, H.M.; Yun, J.; Zhang, G.; Qi, X. An Insight into the Roles of Dietary Tryptophan and Its Metabolites in Intestinal Inflammation and Inflammatory Bowel Disease, Mol. Nutr. Food Res. 2021, 65, 2000461. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis, Cell 2015, 161, 264–276. [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; Colgan, S.P. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor, The American Journal of Pathology 2018, 188, 1183–1194. [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; Corr, S.C.; McManus, G.; Ryan, D.; Jacobs, H.T.; Szibor, M.; Xavier, R.J.; Braun, T.; Frezza, C.; Murphy, M.P.; O’Neill, L.A. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages, Cell 2016, 167, 457–470.e13. [CrossRef]
- Macias-Ceja, D.C.; Ortiz-Masiá, D.; Salvador, P.; Gisbert-Ferrándiz, L.; Hernández, C.; Hausmann, M.; Rogler, G.; Esplugues, J.V.; Hinojosa, J.; Alós, R.; Navarro, F.; Cosin-Roger, J.; Calatayud, S.; Barrachina, M.D. Succinate receptor mediates intestinal inflammation and fibrosis, Mucosal Immunol 2019, 12, 178–187. [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; Bousvaros, A.; Korzenik, J.; Sands, B.E.; Xavier, R.J.; Huttenhower, C. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment, Genome Biol 2012, 13, R79. [CrossRef]
- Zhang, Y.; Bhosle, A.; Bae, S.; McIver, L.J.; Pishchany, G.; Accorsi, E.K.; Thompson, K.N.; Arze, C.; Wang, Y.; Subramanian, A.; Kearney, S.M.; Pawluk, A.; Plichta, D.R.; Rahnavard, A.; Shafquat, A.; Xavier, R.J.; Vlamakis, H.; Garrett, W.S.; Krueger, A.; Huttenhower, C.; Franzosa, E.A. Discovery of bioactive microbial gene products in inflammatory bowel disease, Nature 2022, 606, 754–760. [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; Binder, V.; Finkel, Y.; Cortot, A.; Modigliani, R.; Laurent-Puig, P.; Gower-Rousseau, C.; Macry, J.; Colombel, J.F.; Sahbatou, M.; Thomas, G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease, Nature 2001, 411, 599–603. [CrossRef]
- Krela-Kaźmierczak, I.; Zakerska-Banaszak, O.; Skrzypczak-Zielińska, M.; Łykowska-Szuber, L.; Szymczak-Tomczak, A.; Zawada, A.; Rychter, A.M.; Ratajczak, A.E.; Skoracka, K.; Skrzypczak, D.; Marcinkowska, E.; Słomski, R.; Dobrowolska, A. Where Do We Stand in the Behavioral Pathogenesis of Inflammatory Bowel Disease? The Western Dietary Pattern and Microbiota-A Narrative Review, Nutrients 2022, 14, 2520. [Google Scholar] [CrossRef]
- Arabyan, N.; Park, D.; Foutouhi, S.; Weis, A.M.; Huang, B.C.; Williams, C.C.; Desai, P.; Shah, J.; Jeannotte, R.; Kong, N.; Lebrilla, C.B.; Weimer, B.C. Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling, Sci Rep 2016, 6, 29525. [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD, Autophagy 2020, 16, 38–51. [CrossRef]
- Pickard, J.M.; Chervonsky, A.V. Intestinal fucose as a mediator of host-microbe symbiosis, J Immunol 2015, 194, 5588–5593. [CrossRef]
- Tang, X.; Wang, W.; Hong, G.; Duan, C.; Zhu, S.; Tian, Y.; Han, C.; Qian, W.; Lin, R.; Hou, X. Gut microbiota-mediated lysophosphatidylcholine generation promotes colitis in intestinal epithelium-specific Fut2 deficiency, J Biomed Sci 2021, 28, 20. 28. [CrossRef]
- Cheng, S.; Hu, J.; Wu, X.; Pan, J.-A.; Jiao, N.; Li, Y.; Huang, Y.; Lin, X.; Zou, Y.; Chen, Y.; Zhu, L.; Zhi, M.; Lan, P. Altered gut microbiome in FUT2 loss-of-function mutants in support of personalized medicine for inflammatory bowel diseases, J Genet Genomics 2021, 48, 771–780. [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: regulators of inflammation in health and disease, Nat Rev Immunol 2014, 14, 9–23. [CrossRef]
- Kudelka, M.R.; Hinrichs, B.H.; Darby, T.; Moreno, C.S.; Nishio, H.; Cutler, C.E.; Wang, J.; Wu, H.; Zeng, J.; Wang, Y.; Ju, T.; Stowell, S.R.; Nusrat, A.; Jones, R.M.; Neish, A.S.; Cummings, R.D. Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk, Proc Natl Acad Sci U S A 2016, 113, 14787–14792. [CrossRef]
- Nighot, P.K.; Hu, C.-A.A.; Ma, T.Y. Autophagy Enhances Intestinal Epithelial Tight Junction Barrier Function by Targeting Claudin-2 Protein Degradation, Journal of Biological Chemistry 2015, 290, 7234–7246. [CrossRef]
- Battistini, C.; Ballan, R.; Herkenhoff, M.E.; Saad, S.M.I.; Sun, J. Vitamin D Modulates Intestinal Microbiota in Inflammatory Bowel Diseases, Int J Mol Sci 2020, 22, E362. [CrossRef]
- Sidiq, T.; Yoshihama, S.; Downs, I.; Kobayashi, K.S. Nod2: A Critical Regulator of Ileal Microbiota and Crohn’s Disease, Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Turpin, W.; Bedrani, L.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Smith, M.I.; Garay, J.A.R.; Lee, S.-H.; Guttman, D.S.; Griffiths, A.; Moayyedi, P.; Panaccione, R.; Huynh, H.; Steinhart, H.A.; Aumais, G.; Dieleman, L.A.; Turner, D. CCC IBD GEM Project research team, A. D. Paterson, K. Croitoru, Associations of NOD2 polymorphisms with Erysipelotrichaceae in stool of in healthy first degree relatives of Crohn’s disease subjects, BMC Med Genet 2020, 21, 204. [Google Scholar] [CrossRef]
- Seiderer, J.; Brand, S.; Herrmann, K.A.; Schnitzler, F.; Hatz, R.; Crispin, A.; Pfennig, S.; Schoenberg, S.O.; Göke, B.; Lohse, P.; Ochsenkuhn, T. Predictive value of the CARD15 variant 1007fs for the diagnosis of intestinal stenoses and the need for surgery in Crohn’s disease in clinical practice: results of a prospective study, Inflamm Bowel Dis 2006, 12, 1114–1121. [CrossRef]
- Frank, D.N.; Robertson, C.E.; Hamm, C.M.; Kpadeh, Z.; Zhang, T.; Chen, H.; Zhu, W.; Sartor, R.B.; Boedeker, E.C.; Harpaz, N.; Pace, N.R.; Li, E. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases, Inflamm Bowel Dis 2011, 17, 179–184. [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; Steege, R.W.F.T.; Huttenhower, C.; Dijkstra, G.; Xavier, R.J.; Festen, E.A.M.; Wijmenga, C.; Zhernakova, A.; Weersma, R.K. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease, Gut 2018, 67, 108–119. [CrossRef]
- Liu, H.; Gao, P.; Jia, B.; Lu, N.; Zhu, B.; Zhang, F. IBD-Associated Atg16L1T300A Polymorphism Regulates Commensal Microbiota of the Intestine, Front. Immunol. 2022, 12, 772189. [Google Scholar] [CrossRef]
- Rokhsefat, S.; Lin, A.; Comelli, E.M. Mucin-Microbiota Interaction During Postnatal Maturation of the Intestinal Ecosystem: Clinical Implications, Dig Dis Sci 2016, 61, 1473–1486. [CrossRef]
- Colquhoun, C.; Duncan, M.; Grant, G. Inflammatory Bowel Diseases: Host-Microbial-Environmental Interactions in Dysbiosis, Diseases 2020, 8, 13. 8. [CrossRef]
- Taman, H.; Fenton, C.G.; Hensel, I.V.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. Transcriptomic Landscape of Treatment-Naïve Ulcerative Colitis, J Crohns Colitis 2018, 12, 327–336. [CrossRef]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies, Front. Immunol. 2020, 11, 2054. [Google Scholar] [CrossRef] [PubMed]
- Padra, M.; Adamczyk, B.; Flahou, B.; Erhardsson, M.; Chahal, G.; Smet, A.; Jin, C.; Thorell, A.; Ducatelle, R.; Haesebrouck, F.; Karlsson, N.G.; Lindén, S.K. Helicobacter suis infection alters glycosylation and decreases the pathogen growth inhibiting effect and binding avidity of gastric mucins, Mucosal Immunol 2019, 12, 784–794. [CrossRef]
- Farooq, S.M.; Stillie, R.; Svensson, M.; Svanborg, C.; Strieter, R.M.; Stadnyk, A.W. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis, J Pharmacol Exp Ther 2009, 329, 123–129. [CrossRef]
- Melhem, H.; Regan-Komito, D.; Niess, J.H. Mucins Dynamics in Physiological and Pathological Conditions, IJMS 2021, 22, 13642. [CrossRef]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions, Proc Natl Acad Sci U S A 108 Suppl 2011, 1, 4659–4665. [CrossRef]
- Van der Sluis, M.; De Koning, B.A.E.; De Bruijn, A.C.J.M.; Velcich, A.; Meijerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; Einerhand, A.W.C. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection, Gastroenterology 2006, 131, 117–129. [CrossRef]
- McCole, D.F. IBD candidate genes and intestinal barrier regulation, Inflamm Bowel Dis 2014, 20, 1829–1849. [CrossRef]
- Yamamoto-Furusho, J.K.; Ascaño-Gutiérrez, I.; Furuzawa-Carballeda, J.; Fonseca-Camarillo, G. Differential Expression of MUC12, MUC16, and MUC20 in Patients with Active and Remission Ulcerative Colitis, Mediators of Inflammation 2015, 2015, 1–8. [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease, Nature 2020, 578, 527–539. [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host–microbiota maladaptation in colorectal cancer, Nature 2020, 585, 509–517. [CrossRef]
- Read, E.; Curtis, M.A.; Neves, J.F. The role of oral bacteria in inflammatory bowel disease, Nat Rev Gastroenterol Hepatol 2021, 18, 731–742. [CrossRef]
- Jergens, A.E.; Parvinroo, S.; Kopper, J.; Wannemuehler, M.J. Rules of Engagement: Epithelial-Microbe Interactions and Inflammatory Bowel Disease, Front. Med. 2021, 8, 669913. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Horst, R.T.; Jansen, T.; Jacobs, L.; Bonder, M.J.; Kurilshikov, A.; Fu, J.; Joosten, L.A.B.; Zhernakova, A.; Huttenhower, C.; Wijmenga, C.; Netea, M.G.; Xavier, R.J. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity, Cell 2016, 167, 1125–1136.e8. [CrossRef]
- M. Sadaghian, Interaction between the gut and its microbiota in inflammatory bowel disease. (n.d.).
- Pugazhendhi, S.; Baskaran, K.; Santhanam, S.; Ramakrishna, B.S. Association of ATG16L1 gene haplotype with inflammatory bowel disease in Indians, PLoS One 2017, 12, e0178291. [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome, Pathogens 2019, 8, 126. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Dalmasso, G.; Müller, S.; Carrière, J.; Seibold, F.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy, Gastroenterology 2014, 146, 508–519. [CrossRef]
- Kee, B.P.; Ng, J.G.; Ng, C.C.; Hilmi, I.; Goh, K.L.; Chua, K.H. Genetic polymorphisms of ATG16L1 and IRGM genes in Malaysian patients with Crohn’s disease, J Dig Dis 2020, 21, 29–37. [CrossRef]
- Negroni, A.; Pierdomenico, M.; Cucchiara, S.; Stronati, L. NOD2 inflammation current insights. J Inflamm Res 2018, 11, 49–60. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; Drummond, H.; Lees, C.W.; Khawaja, S.A.; Bagnall, R.; Burke, D.A.; Todhunter, C.E.; Ahmad, T.; Onnie, C.M.; McArdle, W.; Strachan, D.; Bethel, G.; Bryan, C.; Lewis, C.M.; Deloukas, P.; Forbes, A.; Sanderson, J.; Jewell, D.P.; Satsangi, J.; Mansfield, J.C. Wellcome Trust Case Control Consortium, L. Cardon, C.G. Mathew, Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility, Nat Genet 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; Drummond, H.; Lees, C.W.; Khawaja, S.A.; Bagnall, R.; Burke, D.A.; Todhunter, C.E.; Ahmad, T.; Onnie, C.M.; McArdle, W.; Strachan, D.; Bethel, G.; Bryan, C.; Lewis, C.M.; Deloukas, P.; Forbes, A.; Sanderson, J.; Jewell, D.P.; Satsangi, J.; Mansfield, J.C. Wellcome Trust Case Control Consortium, L. Cardon, C.G. Mathew, Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility, Nat Genet 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Moehle, C.; Ackermann, N.; Langmann, T.; Aslanidis, C.; Kel, A.; Kel-Margoulis, O.; Schmitz-Madry, A.; Zahn, A.; Stremmel, W.; Schmitz, G. Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease, J Mol Med 2006, 84, 1055–1066. [CrossRef]
- McAuley, J.L.; Linden, S.K.; Png, C.W.; King, R.M.; Pennington, H.L.; Gendler, S.J.; Florin, T.H.; Hill, G.R.; Korolik, V.; McGuckin, M.A. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection, J. Clin. Invest. 2007, 117, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xin, Y.; Zhou, J.; Tian, Z.; Liu, C.; Yu, X.; Meng, X.; Jiang, W.; Zhao, S.; Dong, Q. Gastric Mucosa-Associated Microbial Signatures of Early Gastric Cancer, Front Microbiol 2020, 11, 1548. [CrossRef]
- Visschedijk, M.C.; Alberts, R.; Mucha, S.; Deelen, P.; de Jong, D.J.; Pierik, M.; Spekhorst, L.M.; Imhann, F.; van der Meulen-de Jong, A.E.; van der Woude, C.J.; van Bodegraven, A.A.; Oldenburg, B.; Löwenberg, M.; Dijkstra, G.; Ellinghaus, D.; Schreiber, S.; Wijmenga, C. The Initiative on Crohn and Colitis, Parelsnoer Institute, M. A. Rivas, A. Franke, C.C. van Diemen, R.K. Weersma, Pooled Resequencing of 122 Ulcerative Colitis Genes in a Large Dutch Cohort Suggests Population-Specific Associations of Rare Variants in MUC2, PLoS ONE 2016, 11, e0159609. [Google Scholar] [CrossRef]
- Padra, M.; Adamczyk, B.; Flahou, B.; Erhardsson, M.; Chahal, G.; Smet, A.; Jin, C.; Thorell, A.; Ducatelle, R.; Haesebrouck, F.; Karlsson, N.G.; Lindén, S.K. Helicobacter suis infection alters glycosylation and decreases the pathogen growth inhibiting effect and binding avidity of gastric mucins, Mucosal Immunol 2019, 12, 784–794. [CrossRef]
- Luo, P.; Yang, Z.; Chen, B.; Zhong, X. The multifaceted role of CARD9 in inflammatory bowel disease, J Cell Mol Med 2020, 24, 34–39. [CrossRef]
- Zhernakova, A.; Festen, E.M.; Franke, L.; Trynka, G.; van Diemen, C.C.; Monsuur, A.J.; Bevova, M.; Nijmeijer, R.M.; van ’t Slot, R.; Heijmans, R.; Boezen, H.M.; van Heel, D.A.; van Bodegraven, A.A.; Stokkers, P.C.F.; Wijmenga, C.; Crusius, J.B.A.; Weersma, R.K. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP, Am J Hum Genet 2008, 82, 1202–1210. [CrossRef]
- Wu, S.; Zhang, Y.; Ma, J.; Liu, Y.; Li, W.; Wang, T.; Xu, X.; Wang, Y.; Cheng, K.; Zhuang, R. Interleukin-6 absence triggers intestinal microbiota dysbiosis and mucosal immunity in mice, Cytokine 2022, 153, 155841. [CrossRef]
- Borecki, K.; Zawada, I.; Salkić, N.N.; Karakiewicz, B.; Adler, G. Relationship between the IL23R SNPs and Crohn’s Disease Susceptibility and Phenotype in the Polish and Bosnian Populations: A Case-Control Study, IJERPH 2019, 16, 1551. [CrossRef]
- Alharbi, R.S.; Shaik, N.A.; Almahdi, H.; ElSokary, H.A.; Jamalalail, B.A.; Mosli, M.H.; Alsufyani, H.A.; Al-Aama, J.Y.; Elango, R.; Saadah, O.I.; Banaganapalli, B. Genetic association study of NOD2 and IL23R amino acid substitution polymorphisms in Saudi Inflammatory Bowel Disease patients, Journal of King Saud University - Science 2022, 34, 101726. [CrossRef]
- Peng, L.-L.; Wang, Y.; Zhu, F.-L.; Xu, W.-D.; Ji, X.-L.; Ni, J. IL-23R mutation is associated with ulcerative colitis: A systemic review and meta-analysis, Oncotarget 2017, 8, 4849–4863. [CrossRef]
Figure 1.
Genes having an impact on gut microbiota.
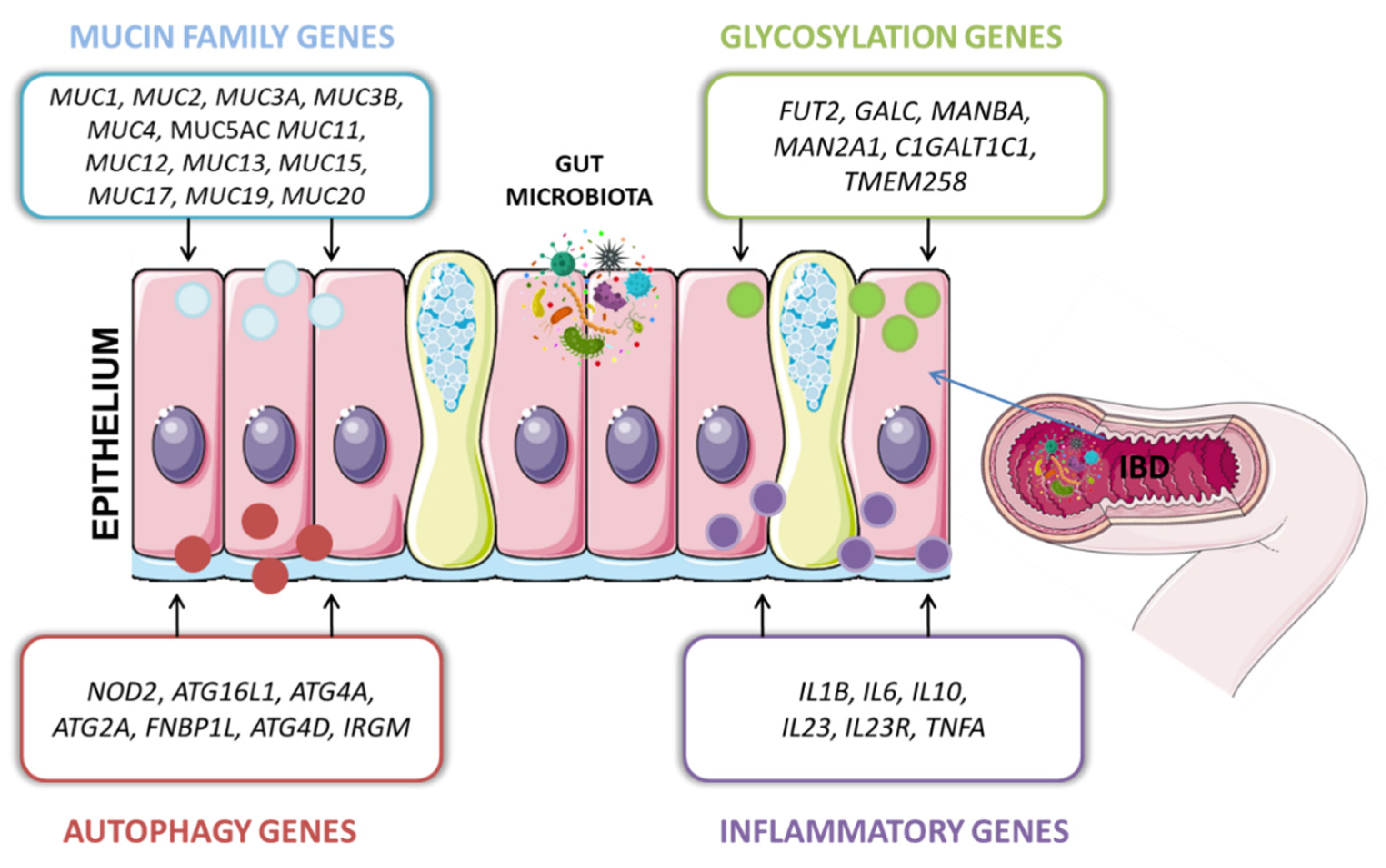
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



