
Submitted:
15 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
Pancreatic ductal adenocarcinoma (PDAC) represents an extremely aggressive type of cancer due in part to early invasion and metastasis, which in turn involves epithelial-mesenchymal transition (EMT) of the tumor cells. Prompted by the discovery that two PDAC-derived tumor cell lines of the quasi-mesenchymal subtype (PANC-1, MIA PaCa-2) exhibit neuroendocrine differentiation (NED), we asked whether NED is associated with EMT. Using quantitative real-time PCR and immunoblot analysis, we initially verified endogenous expression of various NED markers, i.e., chromogranin A (CHGA), synaptophysin (SYP), somatostatin receptor 2 (SSTR2) and SSTR5 in PANC-1 and MIA PaCa-2 cells. In HPDE6c7 normal pancreatic duct epithelial cells and in BxPC-3, another PDAC cell line with an epithelial phenotype, the expression of CHGA, SYP and neuron-specific enolase 2 (NSE) was either undetectable or much lower than in PANC-1 and MIA PaCa-2 cells. Parental cultures of PANC-1 cells exhibit EM plasticity (EMP) and harbor clonal subpopulations with different EMT phenotypes [Ungefroren et al 2022]. Monitoring the NED state of mesenchymal and epithelial PANC-1 subclones showed that the NE phenotype is more pronounced in M-type clones when compared to E-type ones. Inducing EMT in parental cultures of PANC-1 cells by treatment with transforming growth factor (TGF)-β1 repressed epithelial genes and co-induced mesenchymal and NED genes, except for SSTR5. Surprisingly, treatment with bone morphogenetic protein (BMP)-7 differentially affected gene expression in PANC-1 and MIA PaCa-2 cells. It synergised with TGF-β1 in the induction of vimentin, SNAIL, SSTR2 and NSE but antagonised it in the regulation of CHGA and SSTR5. From these data, we conclude that in EMT of PDAC cells mesenchymal and NED markers are co-regulated, and that mesenchymal-epithelial transition (MET) is associated with a loss of both the mesenchymal and NED phenotypes. Analysing NED in another tumor type, small cell carcinoma of the ovary hypercalcemic type (SCCOHT), revealed that two model cell lines of this disease (SCCOHT-1, BIN-67) do express CDH1, SNAI1, VIM, CHGA, SYP, ENO2 and SSTR2, but that in contrast to BMP-7 none of these genes was transcriptionally regulated by TGF-β1. We conclude that in PDAC-derived tumor cells, NED is closely linked to EMT and TGF-β signaling, which may have implications for the therapeutic use of TGF-β inhibitors in PDAC management.
Keywords:
pancreatic ductal adenocarcinoma
; epithelial-mesenchymal transition
; mesenchymal-epithelial transition
; neuroendocrine differentiation
; TGF-β
1. Introduction
Pancreatic cancer is one of the most common causes of cancer-related deaths worldwide. Its two major histological subtypes are pancreatic ductal adenocarcinoma (PDAC), accounting for 90% of all cases, and pancreatic neuroendocrine neoplasm (panNEN), which makes up 3-5% of all cases. PanNEN is classified into well-differentiated pancreatic NE tumor and poorly-differentiated pancreatic NE carcinoma (panNEC). Although PDAC and panNEN are commonly thought to be different diseases with distinct biology, cell of origin, and genomic abnormalities, the idea that PDAC and panNEC share common cells of origin has been gaining support. This is supported by molecular profiling data suggesting that panNEC is genetically and phenotypically related to PDAC [1]. PanNENs represent a heterogeneous group of epithelial tumors with NE differentiation (NED) that are classified into well-differentiated pancreatic NE tumors (panNETs), including G1, G2, and G3 tumors, and poorly differentiated panNECs [2]. PanNETs can be regarded as a unique category, where G1-G2 tumors may progress to G3 tumors mainly driven by DAXX/ATRX mutations [2]. Conversely, panNECs display totally different histomolecular features more closely related to PDAC, including TP53 and Rb alterations [2] was therefore not surprising that two PDAC-derived tumor cell lines of the quasi-mesenchymal subtype, PANC-1 and MIA PaCa-2, were demonstrated to harbor a NED phenotype [3]. PANC-1 expresses CK5.6, MNF-116, vimentin (VIM), chromogranin A (CHGA), neural cell adhesion molecule (NCAM/CD56) and somatostatin receptor-2 (SSTR2) but not E-cadherin (ECAD), synaptophysin (SYP) or neurotrophin receptor-1 (NTR1), while MIA PaCa-2 expresses CK5.6, AE1/AE3, ECAD, VIM, CHGA, SYP, SSTR2 and NTR1 but not NCAM [3]. In addition to the NED markers, we [4] and others [5] demonstrated that PANC-1 and MIA PaCa-2 cells express genes associated with endocrine/neuroendocrine differentiation such as MAFA, NEURODI, PDX1, and NEUROG3. Of note, the PANC-1 cell line has been shown to exhibit epithelial-mesenchymal plasticity (EMP). Parental cultures comprise several clonal subpopulations with different EM transdifferentiation (EMT) phenotypes of which some are more epithelial and others more mesenchymal in nature [6]. The importance of EMT is also considered in other gastrointestinal (GI) cancers such as colorectal NEC (coloNEC) and a distinct subtype of ovarian cancer (OC), e.g., the heterogeneous small cell carcinoma of the ovary hypercalcemic type (SCCOHT). The SCCOHT represents a rare form of an aggressive ovarian tumor, which is predominantly observed in young women. This malignant neoplasia is associated with paraneoplastic hypercalcemia and affected patients often have a lethal outcome already within a few months after diagnosis [7,8]. Cellular models for this tumor entity were established and characterized as the SCCOHT-1 [9] and BIN-67 cell lines [10,11]. Indeed, the characterization of SCCOHT-1 cells and chemotherapeutic responses has revealed a constitutive expression of NED markers such as NCAM in the original patient tumor and derived mouse xenograft tumors [9,12]. Thus, the heterogeneity and plasticity of SCCOHT-1 cells may also involve EMP and maturation along a NED phenotype [13].
A series of studies suggests that NED is closely associated with EMT in different tumor types, mainly prostate, lung and colon/pancreas. In prostate carcinoma (PCa), androgen deprivation has been shown to activate both EMT and NE transdifferentiation programs [14]. Many factors have been associated with the onset and progression of NED in clinically typical prostate adenocarcinomas including loss of androgen receptor (AR) expression and/or signaling, conventional therapy, and dysregulated cytokine function. The AR is a critical driver of tumor progression as well as therapeutic response in patients with metastatic castrate-resistant PCa (CRPC) [15]. Emerging evidence suggests that the acquisition of EMT and a cancer stem cell (CSC) phenotype are associated with the development of NED in PCa [16]. EMT and NED may also be induced by androgen-targeted therapy [14] and is considered a resistance mechanism to treatments in PCa [17]. The resulting NE PCa (NEPC) is highly aggressive exhibiting reactivation of developmental programs associated with EMT induction and stem cell-like characteristics.
Interestingly, an inverse relationship between NED and EMT has also been described in some tumors, i.e., small cell lung carcinoma (SCLC), Merkel cell carcinoma and gastroenteropancreatic (GEP)-NET [18]. In SCLC, an association was revealed between the loss of NED and EMT induction [19] as inferred from the observation that the low NED subtype had undergone EMT and had activated - amongst others - the TGF-β pathway. However, differential effects of TGF-β on both programs were observed in SCLC in that TGF-β seems to be required for promoting EMT but not NED. In the panNET cell lines, BON-1 (BON), QGP-1 and NT-3 previous work has shown that the ECAD and VIM expression profiles are indicative of a well-differentiated epithelial phenotype [20]. In BON cells, TGF-β1 has been shown to control proliferation and NED through the SST/SSTR system [21,22]. Of note, disrupting either the TGF-β or SST signaling pathway resulted in NED-mesenchymal transition, which is characterized by the loss of NED markers, decreased ECAD and elevated VIM expression. This inverse correlation of TGF-β sigaling activity and EMT was surprising since TGF-β is known as one of the most potent inducers of EMT. In PCa, aberrant TGF-β signaling accelerates progression in a transgenic mouse model via effects on EMT and NED driving tumor progression to CRPC [23].
Given the concept of a positive association between EMT and NED mainly arising from the PCa model, an important issue remains whether the reverse process, mesenchymal-epithelial transition (MET) affects NED. We have recently shown that PDAC-derived tumor cells can be forced to undergo MET in response to a transdifferentiation culture through exposure to a combination of three cytokines, IL-1β, IFN-γ and TNF-α (TDC-IIT) [6}. Moreover, a member of the TGF-β superfamily of growth and differentiation factors, bone morphogenetic protein-7 (BMP-7), has been reported to be able to induce MET in adult renal fibroblasts of the injured kidney [24], hepatic stellate cells [25] and melanoma cells [26], generating functional epithelial cells [27]. Interestingly, BMP-7 has been shown to induce MET through downregulation of the EMT-related transcription factor SNAIL [28], SNAIL-induced α-smooth muscle actin, and concomitant upregulation of ECAD. BMP-7 also acts as an inhibitor of fibrotic progression in many organs through activation of the SMAD1/5 arm of TGF-β signaling and inhibition of TGF-β–mediated EMT [29,30] via suppression of canonical TGF-β/SMAD2/3 signaling [31].
Given the above-mentioned findings, particularly the inverse relationship between EMT and NED in panNET, we decided to study the effects of EMT and MET inducers on EMP and NED in pancreatic cells (normal duct cells and PDAC-derived tumor cells of the epithelial and quasi-mesenchymal subtype), and in cell lines of SCCOHT. Specifically, we asked whether forced conversion from epithelial to mesenchymal (via treatment with TGF-β1) or vice versa (via stimulation with TDC-IIT or BMP-7) will affect NED markers in the same way as EMT markers.
2. Material and Methods
2.1. Cells
PANC-1, MIA PaCa-2 and BxPC-3 human PDAC cells were originally obtained from ATCC (Manassas, VA) and propagated in RPMI 1640 supplemented with fetal bovine serum (FBS, 10%), Penicillin-Streptomycin-Glutamine (1%, Life Technologies/Thermo Fisher Scientific, Darmstadt, Germany) and sodium pyruvate (1%, Merck Millipore). HPDEH6c7 were purchased from AddexBio (#T0018001), BON cells were established from a functional human pancreatic carcinoid tumor and was originally provided by C.M. Townsend (University of Texas, Galveston, TX, USA). The NT-3 cell line has been established and characterized by our group in 2018 [32]. Maintenance of BON and NT-3 cells has been described in detail previously [20,22]. The generation of individual PANC-1 cell clones by limited dilution has been described in detail earlier [6]. Cell counting of cells was performed with a Neubauer chamber. For determination of basal mRNA levels several independent RNA isolates (3-6) were retrieved from continuous cultures of cells and subjected to qPCR analysis.
Established and characterized cellular models of SCCOHT are represented by the two cell lines BIN-67 (kindly provided by Dr. Barbara Vanderhyden, University of Ottawa, Canada) and SCCOHT-1. BIN-67 were cultured with DMEM/F12:DMEM medium (1:1, v/v) (Sigma Aldrich, St. Louis, MO) supplemented with fetal calf serum (FCS, 20%) (PAN-Biotech GmbH, 94501 Aidenbach, Germany), L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 µg/ml) (all from Capricorn Scientific GmbH, Ebsdorfergrund, Germany) [11].
The SCCOHT-1 cells were isolated, processed, and cultured as a spontaneously growing primary culture derived from a tumor biopsy of a 31-year-old patient with recurrent SCCOHT [9]. Studies with these cells have been approved by the Ethics Committee of Hannover Medical School, Project #3916 on June 15th, 2005, and prior informed written consent was obtained from the patient for the use of this material. SCCOHT-1 cells were cultured in RPMI 1640 (Sigma) supplemented with 10% FCS (PAN-Biotech GmbH), 100 U/ml L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (all from Capricorn Scientific GmbH). The cell culture was performed at 37°C in a humidified atmosphere of 5% CO2 and the culture medium was changed at intervals of 3 to 4 d. For subculture, SCCOHT-1 cells were detached by repeated pipetting, centrifuged (320 g/6 min), and resuspended in the appropriate growth medium.
In some experiments, cells were treated with recombinant human TGF-β1 (#300-023, ReliaTech, Wolfenbüttel, Germany) (PANC-1: 5 ng/ml; MIA PaCa-2, BIN-67, SCCOHT-1: 10 ng/ml) or human BMP-7 (#120-03P, Preprotech, Hamburg, Germany) (200 ng/ml).
2.2. QPCR Analysis
Total RNA was extracted and purified from cells using affinity chromatography on columns (innuPREP RNA Mini Kit 2.0, IST Innuscreen GmbH, Berlin, Germany) according to manufacturer’s instructions. An amount of 2.5 μg RNA per sample was subjected to reverse transcription in a total volume of 20 μl at 37 °C for 1 h using M-MLV Reverse Transcriptase (200 U) and random hexamers (2.5 μM) (Thermo Fisher Scientific). The relative expression of the genes of interest was quantified by quantitative realtime PCR on an I-Cycler (BioRad, Munich, Germany) using Maxima SYBR Green Mastermix (Thermo Fisher Scientific). Following the generation of Ct values for the target genes, these were normalized with those for either TATA-box-binding protein (TBP) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The sequences of PCR amplification primers are given in Table S1.
2.3. Cell Lysis and Immunoblotting
Subconfluent cultures of ells were washed once with ice-cold PBS and lysed with 1x PhosphoSafe lysis buffer (Merck Millipore). Following sonication and clearing total protein concentration of the supernatants was determined with the BioRad DC Protein Assay. Samples were fractionated on mini-PROTEAN TGX any-kD precast gels (BioRad) and blotted to 0.45 μm PVDF membranes. Following blocking of the membranes with nonfat dry milk or BSA their incubation with primary antibodies proceeded overnight at 4°C. HRP-linked secondary antibodies and Amersham ECL Prime Detection Reagent (GE Healthcare, Munich, Germany) were used for chemoluminescent detection of proteins on a BioRad ChemiDoc XRS imaging system. We employed the following primary antibodies: anti-HSP90 (F-8), #sc-13119, and anti-vimentin, sc-6260 (Santa Cruz Biotechnology), anti-RAC1b, #09-271 (Merck Millipore, Darmstadt, Germany), anti-E-cadherin, #3195, anti-GAPDH (14C10), #2118, and anti-Snail, #3895 (Cell Signaling Technology), anti-Synaptophysin and anti-human Chromogranin A, clone DAK-A3 (both from Dako, Glostrup, Denmark), and anti-Somatostatin Receptor-2 antibody [UMB1]-C-terminal, #ab134152 (Abcam, Cambridge, UK). HRP-linked anti-rabbit, #7074, and anti-mouse, #7076, secondary antibodies were from Cell Signaling Technology.
2.4. Statistical Analysis
Statistical significance was calculated using the unpaired Student’s t test. Results were considered significant at p < 0.05 (∗).
3. Results
3.1. PANC-1 and MIA PaCa-2 Cells, but Not Normal Pancreatic Duct Epithelial Cells or Epithelial-Subtype Pancreatic Tumor Cells, Constitutively Express Various NED Markers
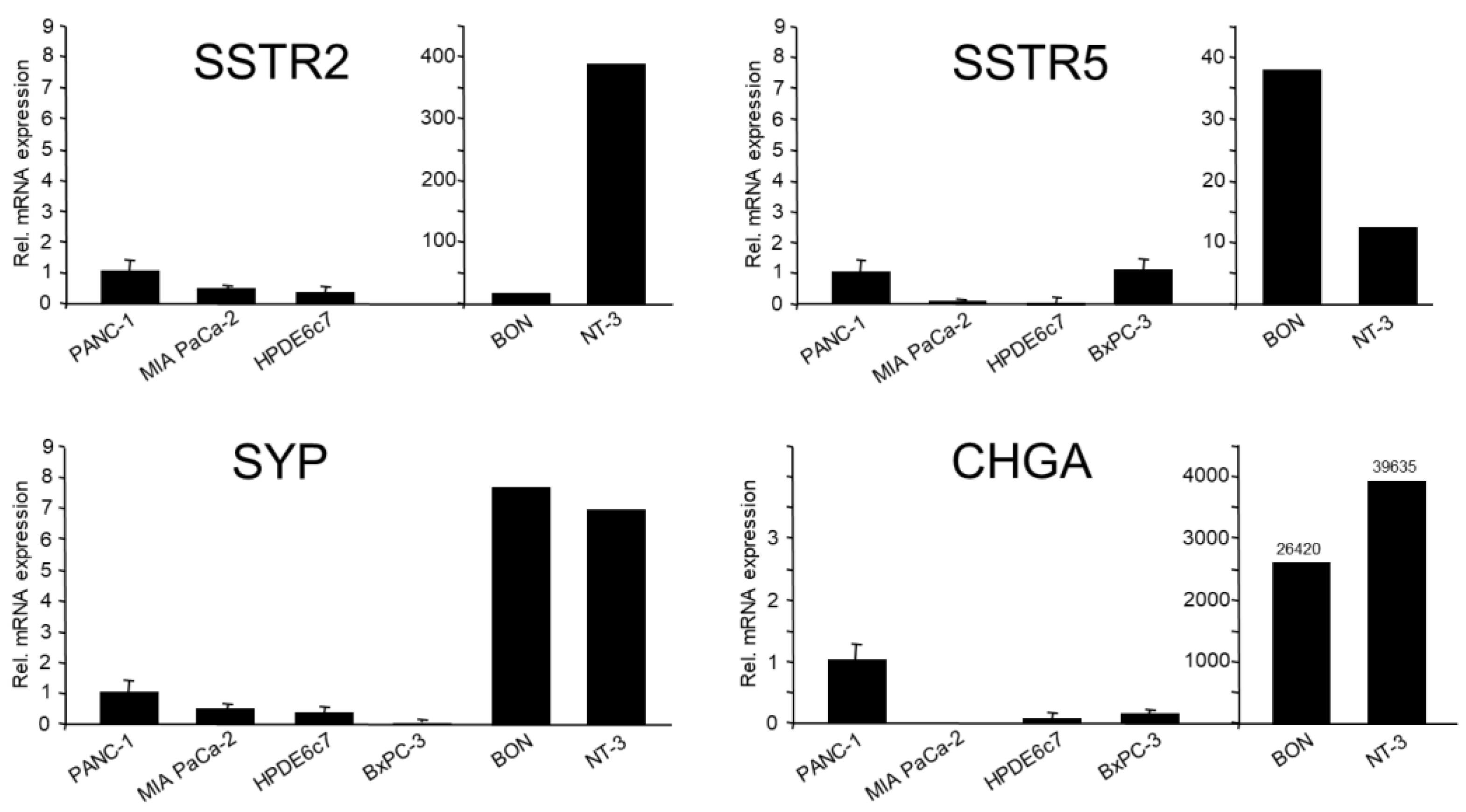
As mentioned in the Introduction, the quasi-mesenchymal cell lines PANC-1 and MIA PaCa-2 have been shown earlier to express various NED markers [3,4,5]. Here, we confirmed endogenous expression of the SYP, SSTR2 and SSTR5 genes in both cell lines by qPCR analysis, which was clearly higher in PANC-1 cells (Figure 1). This was most apparent for another prominent NED marker, CHGA, the mRNA of which was not detectable in MIA PaCa-2 cells. As positive controls, we employed the panNET cell lines, BON-1 and NT-3 [22] (Figure 1).
If NED segregates with a transformed/tumor cell phenotype then HPDE6c7, a normal (immortalised) cell line established from pancreatic ductal epithelial cells, should be devoid of expression of NED markers. Indeed, in qPCR analysis HPDE6c7 cells were either negative or only weakly positive for all NED markers tested except SSTR5 (Figure 1), and this was confirmed for CHGA, SYP and SSTR2 by immunoblot analysis (data not shown). The PDAC-derived moderately differentiated cell line BxPC-3 was shown earlier to be strongly positive for ECAD, but negative for VIM [6]. This suggests that these cells despite their transformed state have retained an epithelial phenotype, hence representing the classical/epithelial histomorphological subtype of PDAC [6]. Based on the postulated assumption that NED is associated with a mesenchymal phenotype, we reasoned that BxPC-3 cells should exhibit lower expression of SYP and CHGA. When compared to PANC-1, this was indeed the case (CHGA: 8-fold lower), SYP (400-fold lower) (Figure 1), and NSE (10-fold lower, not shown). However, SSTR2 and SSTR5 were expressed by BxPC-3 at a higher or the same level, respectively, compared to PANC-1 (Figure 1) and, in contrast to CHGA and SYP, SSTR2 was also detectable by immunoblot analysis (Supplementary Figure 1). For protein expression of SYP and SSTR2 in PANC-1 cells, see Figure 3, Figure 4 and Figure 5. We conclude that poorly differentiated PDAC cell lines, in particular PANC-1, but not their presumed nontransformed precursor cells, express moderate amounts of SYP and CHGA. Moderately differentiated PDAC cells - as exemplified by BxPC-3 - exhibit lower mRNA levels of SYP and CHGA (but not of SSTR2 and 5) when compared to PANC-1 cells.
3.2. NED Marker Expression Varies in Single Cell-Derived Clones of PANC-1 Cells with Different EMT Phenotypes
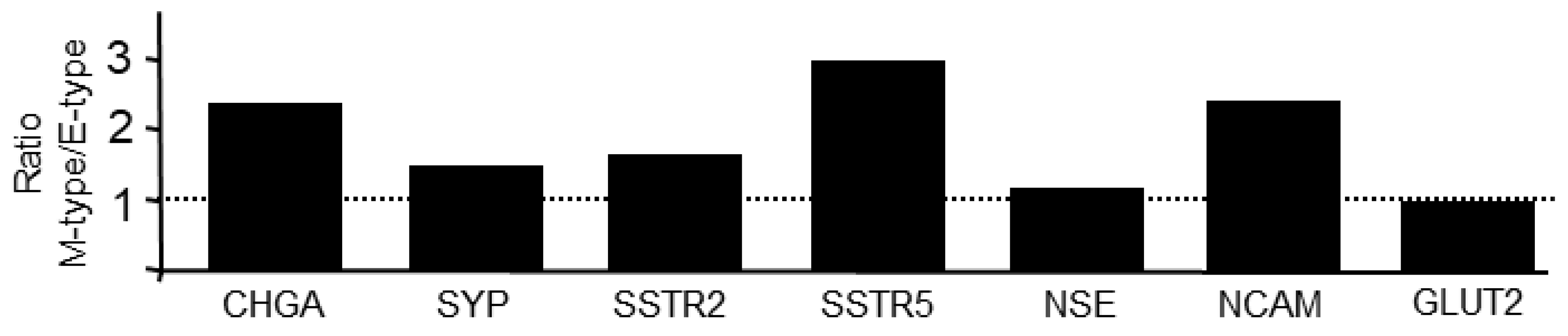
We have shown recently that the PANC-1 cell line displays EMP; parental cultures consist of clonal subpopulations with different EMT phenotypes as evidenced by different ratios of ECAD:VIM expression in single cell-derived clones [6]. This unique feature allowed us to study the association between EMT and NED within a genetically identical background. Here, we analyzed seven clones, exhibiting either a more epithelial (E) or mesenchymal (M) phenotype, by qPCR analysis for expression of CHGA, SYP, NCAM, NSE, GLUT2, SSTR2, and SSTR5. Results indicate that clones with a low ECAD/VIM ratio (M-type: P1C3, P3D10, P4B9, P2E8) when compared to clones with a high ECAD/VIM ratio (E-type: P4B11, P3D2, P1G7) present with higher levels (mean±SD) of CHGA (5.21±2.1 vs. 2.26±1.12), SYP (1.18±0.44 vs. 0.811±0.16), NSE (1.28±0.35 vs. 1.16±0.14), NCAM (2.04±0.92 vs. 0.86±0.16), SSTR2 (1.49±0.42 vs. 0.92±0.10) and SSTR5 (2.90±1.34 vs. 0.98±0.40) (Figure 2). Yet another marker, GLUT2, was not different between M and E-type cells (0.86±0.42 vs. 0.90±0.19) (Figure 2). We conclude that NED is more pronounced in clonal subcultures with an M-phenotype when compared to those with an E-phenotype.
Figure 2.
NED markers are enriched in PANC-1 single cell-derived clones with an M-phenotype over those with an E-phenotype. Seven individual clones previously grouped according to their EMT state (three E-type and four M-type clones) [6] were subjected to qPCR analysis for the indicated NED markers. Data represent the ratio of mean values between M and E-type clones for each marker. At 1.0, expression is equally high in M and E-type clones (indicated by the stippled line).
Figure 2.
NED markers are enriched in PANC-1 single cell-derived clones with an M-phenotype over those with an E-phenotype. Seven individual clones previously grouped according to their EMT state (three E-type and four M-type clones) [6] were subjected to qPCR analysis for the indicated NED markers. Data represent the ratio of mean values between M and E-type clones for each marker. At 1.0, expression is equally high in M and E-type clones (indicated by the stippled line).
3.3. Treatment of PANC-1 Cells with TGF-β1 or BMP-7 Alters EMT and NED-Associated Gene Expression
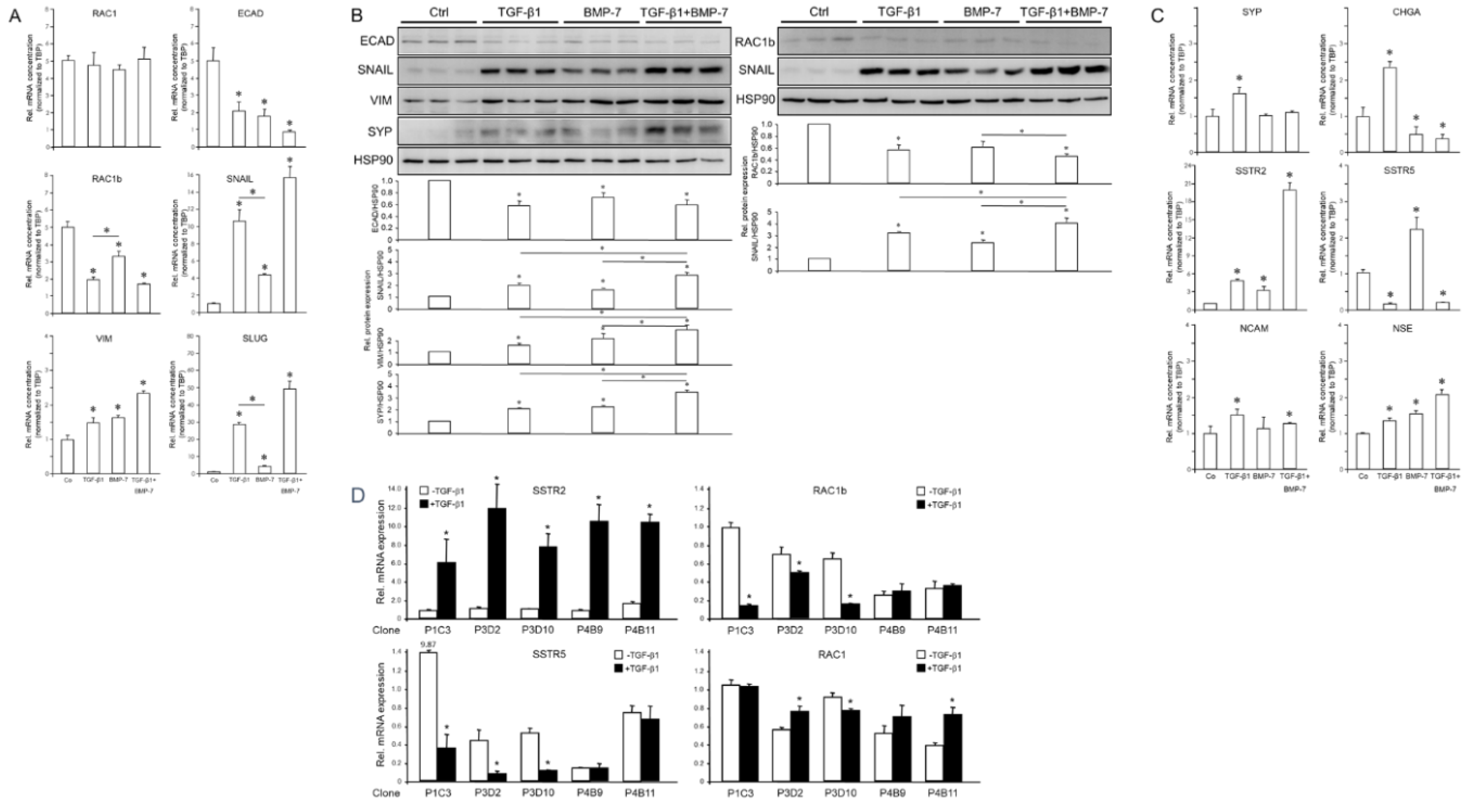
Prompted by the crucial role of TGF-β in pancreatic NE tumors [21] and the known existence of a PDAC subtype exhibiting NED, we sought to study the response of PANC-1 cells to TGF-β1 and BMP-7. The former growth factor is a strong inducer of EMT, while the latter is an inhibitor of EMT, promoter of MET [22,23,24,25] and a presumed suppressor of canonical TGF-β/SMAD2/3 signaling [29]. In PANC-1 cells, treatment with TGF-β1 or BMP-7 downregulated ECAD and another epithelial marker, RAC1b (a tumor-associated splice isoform of human RAC1) [33]. At the same time, both growth factors upregulated the mesenchymal markers SNAIL1, SNAIL2/SLUG, and VIM, while another mesenchymal marker, RAC1, remained unaltered (Figure 3A). The effect of TGF-β1 was more potent than that of BMP-7 for RAC1b, SNAIL and SLUG, while no significant differences between both growth factors were seen for RAC1, ECAD and VIM (Figure 3A). The simultaneous cotreatment with TGF-β1 and BMP-7 acted in an additive manner to suppress ECAD and to enhance SNAIL, SLUG and VIM (Figure 3A).
The regulatory effects of TGF-β1 and BMP-7 on EMT markers at the mRNA level were reproduced for the ECAD, RAC1b, SNAIL and VIM proteins as shown by immunoblotting (Figure 3B). This suggested that both TGF-β1 and, unexpectedly also BMP-7, induce EMT in PANC-1 cells and that BMP-7 can synergize with TGF-β1 in EMT induction.
Next, we monitored in TGF-β1 or BMP-7-treated parental cultures of PANC-1 cells the expression of NED-associated genes. Immunoblotting revealed that SYP protein levels were induced by both TGF-β1 and BMP-7 (Figure 3B). Moreover, combined treatment of PANC-1 cells with TGF-β1 and BMP-7 had a synergistic effect on SYP protein abundance (Figure 3B). However, unlike TGF-β1, BMP-7 failed to induce SYP mRNA (Figure 3C), suggesting that regulation of SYP by BMP-7 involves alterations in protein stability/half-life rather than de novo transcription. In contrast to SYP, SSTR2 mRNA (Figure 3C) was induced by both TGF-β1 and BMP-7 with the induction by TGF-β1 being greater than that by BMP-7 (Figure 3C, p=0.023). Intriguingly, the combined treatment of PANC-1 cells with TGF-β1 and BMP-7 had a synergistic effect on SSTR2 but not on SYP mRNA levels (Figure 3C). In contrast to SSTR2, SSTR5 was strongly downregulated by TGF-β1 and TGF-β1+BMP-7 but, surprisingly, was upregulated by BMP-7 alone (Figure 3C). In addition, TGF-β1 treatment induced CHGA, NCAM, and NSE (Figure 3C). In contrast, BMP-7 suppressed CHGA (Figure 3C) but like TGF-β1 induced NSE (Figure 3C). For a selected panel of genes, which have been shown above to be either upregulated (SSTR2) or downregulated (RAC1b, SSTR5) by TGF-β1 in parental cultures of PANC-1 cells, we also evaluated whether the TGF-β1 effects vary among individual clones. Analysis of five clones indeed showed that these responded differently with respect to induction of SSTR2 and suppression of RAC1b and SSTR5, but not RAC1 (Figure 3D).
Figure 3.
Effect of treatment with TGF-β1 or BMP-7 on EMT and NED markers in PANC-1 cells. Cells were treated with TGF-β1 (5 ng/ml), BMP-7 (200 ng/ml), or vehicle (Ctrl), either singly or in combination, for 24 h and subsequently subjected to qPCR (A) or immunoblot (B) analysis of the indicated EMT and NED-associated genes. (C) As in (A), except that only NED-associated markers were detected. (D) Five single cell-derived clones were treated with TGF-β1 (5 ng/ml) for 24 h and subjected to qPCR analysis for the indicated markers. The graphs below the blots in (B) display the results of densitometry-based quantification of band intensities. Data shown (mean ± SD of three parallel wells) are each from a representative experiment out of at least three experiments performed in total. The asterisks (∗) denote significant differences (two-tailed unpaired Student’s t-test).
Figure 3.
Effect of treatment with TGF-β1 or BMP-7 on EMT and NED markers in PANC-1 cells. Cells were treated with TGF-β1 (5 ng/ml), BMP-7 (200 ng/ml), or vehicle (Ctrl), either singly or in combination, for 24 h and subsequently subjected to qPCR (A) or immunoblot (B) analysis of the indicated EMT and NED-associated genes. (C) As in (A), except that only NED-associated markers were detected. (D) Five single cell-derived clones were treated with TGF-β1 (5 ng/ml) for 24 h and subjected to qPCR analysis for the indicated markers. The graphs below the blots in (B) display the results of densitometry-based quantification of band intensities. Data shown (mean ± SD of three parallel wells) are each from a representative experiment out of at least three experiments performed in total. The asterisks (∗) denote significant differences (two-tailed unpaired Student’s t-test).
3.4. Treatment of MIA PaCa-2 Cells with BMP-7 but Not TGF-β1 Alters EMT and NED-Associated Gene Expression
MIA PaCa-2 cells have been reported to be insensitive to TGF-β stimulation due to a lack of expression of the TGF-β type II receptor [34]. Accordingly, MIA PaCa-2 cells failed to respond with any changes in EMT or NED marker expression to treatment with TGF-β1 alone (Figure 4A), but whether these cells can respond to BMP-7 was unknown. As in PANC-1 cells, treatment with BMP-7 (200 ng/ml) induced the mRNA expression of SNAIL (x1.31), SLUG (x2.91), VIM (x1.41), and SSTR5 (x2.72) (Figure 4A), while no significant changes were seen for RAC1, RAC1b, SYP and SSTR2 (data not shown). Both ECAD and CHGA were undetectable in MIA PaCa-2 even by qPCR.
Expression of SNAIL and VIM in MIA PaCa-2 cells was also analysed by immunoblotting. SNAIL, but not VIM, was induced by BMP-7 alone, however, when combined with TGF-β1, BMP-7 was able to also increase VIM levels (Figure 4B). As expected from the high Ct values in qPCR analysis of MIA PaCa-2 cells, SYP and SSTR2 were below detection limits in immunoblots. The data presented in Figure 3 and Figure 4 show that in PANC-1 cells, TGF-β1 repressed epithelial genes and induced mesenchymal and NED genes, except for SSTR5. In contrast, BMP-7 differentially impacted gene expression in both PANC-1 and MIA PaCa-2; it synergised with TGF-β1 in the induction of VIM, SNAI1, SSTR2 and ENO2 (encoding NSE), but antagonised it in the regulation of SSTR5 and CHGA (if expressed).
Figure 4.
Effect of treatment with TGF-β1 or BMP-7 on EMT and NED markers in MIA PaCa-2 cells. Cells were treated with 5 ng/ml TGF-β1, 200 ng/ml BMP-7 or vehicle (Ctrl) either singly, or with both growth factors simultaneously, for 24 h and subjected to qPCR (A) or immunoblot (B) analysis of the indicated EMT and NED-associated markers. The graphs below the blots in (B) display the results of densitometric band quantification. Data shown (mean ± SD of three parallel wells) are from a representative experiment out of at least three experiments performed in total. The asterisks (∗) denote significant differences (on-tailed unpaired Student’s t-test).
Figure 4.
Effect of treatment with TGF-β1 or BMP-7 on EMT and NED markers in MIA PaCa-2 cells. Cells were treated with 5 ng/ml TGF-β1, 200 ng/ml BMP-7 or vehicle (Ctrl) either singly, or with both growth factors simultaneously, for 24 h and subjected to qPCR (A) or immunoblot (B) analysis of the indicated EMT and NED-associated markers. The graphs below the blots in (B) display the results of densitometric band quantification. Data shown (mean ± SD of three parallel wells) are from a representative experiment out of at least three experiments performed in total. The asterisks (∗) denote significant differences (on-tailed unpaired Student’s t-test).
3.5. The Expression of NED Markers is Altered During MET and Positively Correlates with that of Mesenchymal Markers
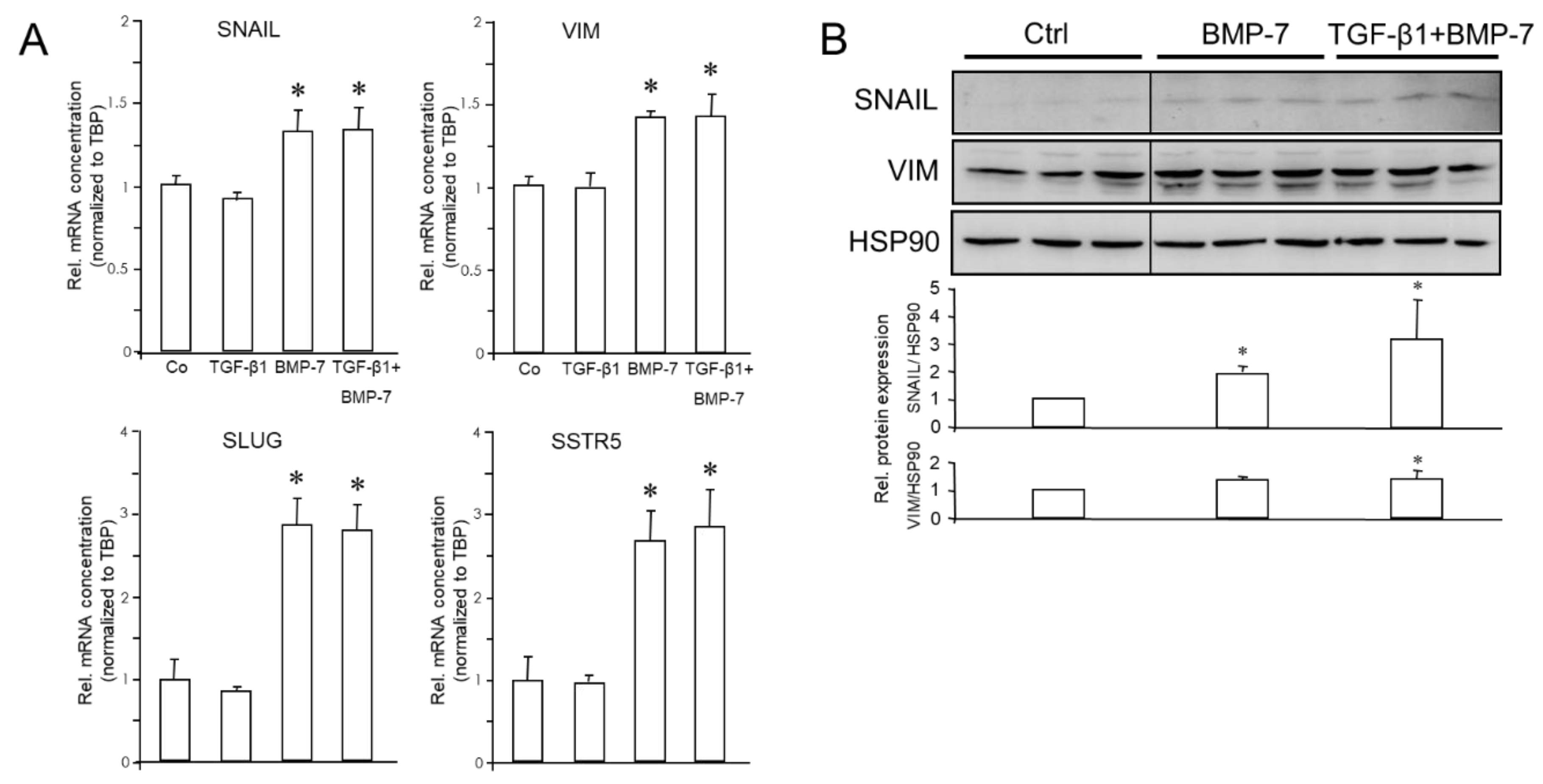
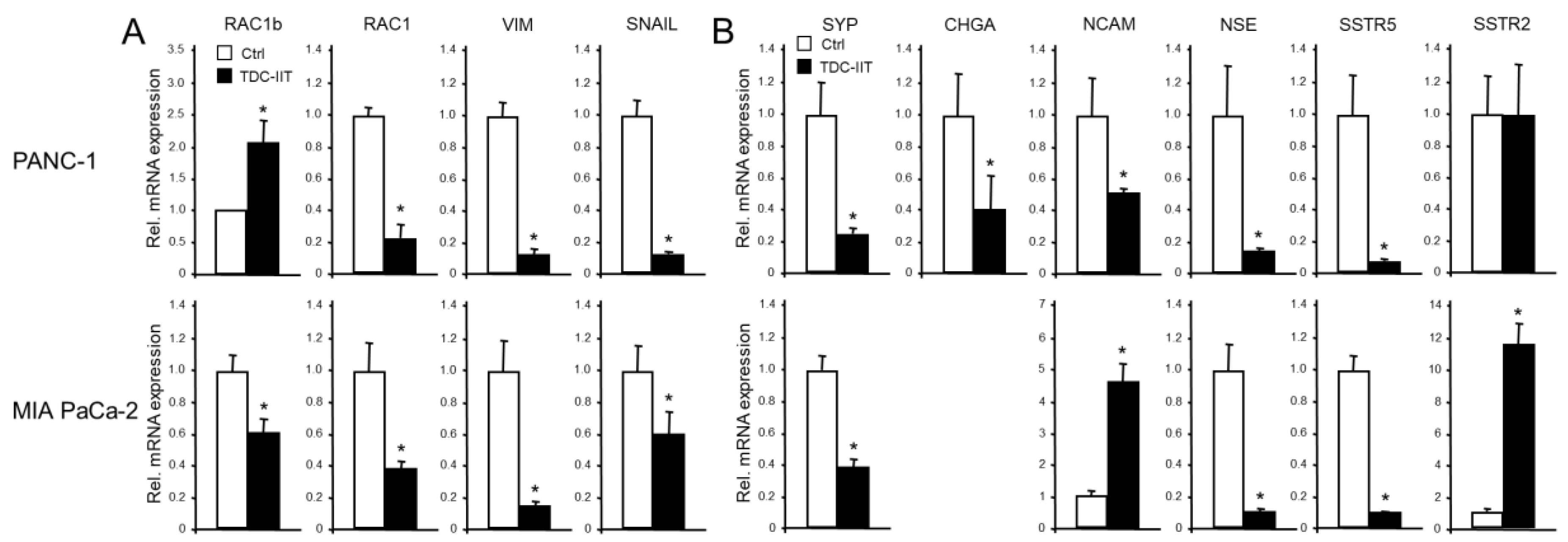
Our previous studies have shown that poorly differentiated quasi-mesenchymal PANC-1 and MIA PaCa-2 cells can be induced to undergo MET by a 72-h treatment with IL1-β, IFN-γ and TNF-α (IIT) [6]. This transdifferentiation culture (TDC) of both cell lines with IIT (TDC-IIT) was associated with reversal of EMT, as evidenced by upregulation of epithelial markers ECAD, CLDN4, CK19, GRHL2 and OVOL2 [6]. Here, we noted, in addition, an induction of RAC1b mRNA in PANC-1 (but repression in MIA PaCa-2, Figure 5A), and a decline in RAC1, VIM and SNAIL mRNAs in both cell lines (Figure 5A).
Given the reciprocal pattern of regulation of epithelial and mesenchymal markers in MET compared to EMT, it was of interest to analyse if exposing parental cultures of PANC-1 or MIA PaCa-2 cells to TDC-IIT also alters NED gene expression. Based on the positive association of NED and mesenchymal genes (see above), we reasoned that the expression of NED genes should decrease in response to the MET-inducing conditions. Intriguingly, using qPCR analysis, we observed in PANC-1 cells strong downregulation of CHGA, SYP, NCAM, NSE and SSTR5 but no effect on SSTR2 (Figure 5B, upper graphs). However, immunoblots indicated a decline in protein levels of SSTR2 (Supplementary Figure 2), which was not unexpected due to its upregulation by the EMT inducers TGF-β1 and BMP-7 (see Figure 3). In MIA PaCa-2 cells, we noted downregulation of SYP, NSE and SSTR5 and upregulation of NCAM, SSTR2 (Figure 5B, lower graphs), and GLUT2 (not shown). This clearly shows that the major EMT and NED markers are co-regulated and that MET is associated with a loss of both the mesenchymal and NED phenotypes.
Figure 5.
Expression of EMT and NED markers in response to MET induction via TDC-IIT. PANC-1 (upper graphs) and MIA PaCa-2 (lower graphs) cells were subjected to TDC-IIT or control culture (Ctrl) for 72 h followed by lysis and qPCR analysis of markers of EMT (A) or NED (B). The data represent the mean ± SD of quadruplicate wells of a representative assay out of at least three assays performed in total. The asterisks (∗) denote significant differences relative to Ctrl.
Figure 5.
Expression of EMT and NED markers in response to MET induction via TDC-IIT. PANC-1 (upper graphs) and MIA PaCa-2 (lower graphs) cells were subjected to TDC-IIT or control culture (Ctrl) for 72 h followed by lysis and qPCR analysis of markers of EMT (A) or NED (B). The data represent the mean ± SD of quadruplicate wells of a representative assay out of at least three assays performed in total. The asterisks (∗) denote significant differences relative to Ctrl.
3.6. Cell Lines Derived from SCCOHT Differentially Respond to TGF-β1 or BMP-7 Treatment with Upregulation of EMT and NED Markers
Two different cell lines, BIN-67 and SCCOHT-1, representing cellular models for SCCOHT [9,10,11], were employed to test possible effects of TGF-β1 and BMP-7 on EMT and NED-associated genes. An intial qPCR-based screening indicated that BIN-67 and SCCOHT-1 cells only express ECAD, SNAIL, VIM, CHGA, SYP and SSTR2 to a significant extent. Other genes (SLUG, NCAM, GLUT2, SSTR5) were either negative or only weakly positive, while NSE was expressed only in SCCOHT-1 but not in BIN-67 cells (data not shown).
Both cell lines were then treated for 24 h with either TGF-β1, BMP-7, or a combination of both, and subjected to qPCR analysis of the above EMT and NED markers. Results show that BIN-67 cells were refractory to stimulation with either growth factor for the above-mentioned genes, except ECAD, which was induced by BMP-7 by a factor of 1.6 (data not shown).
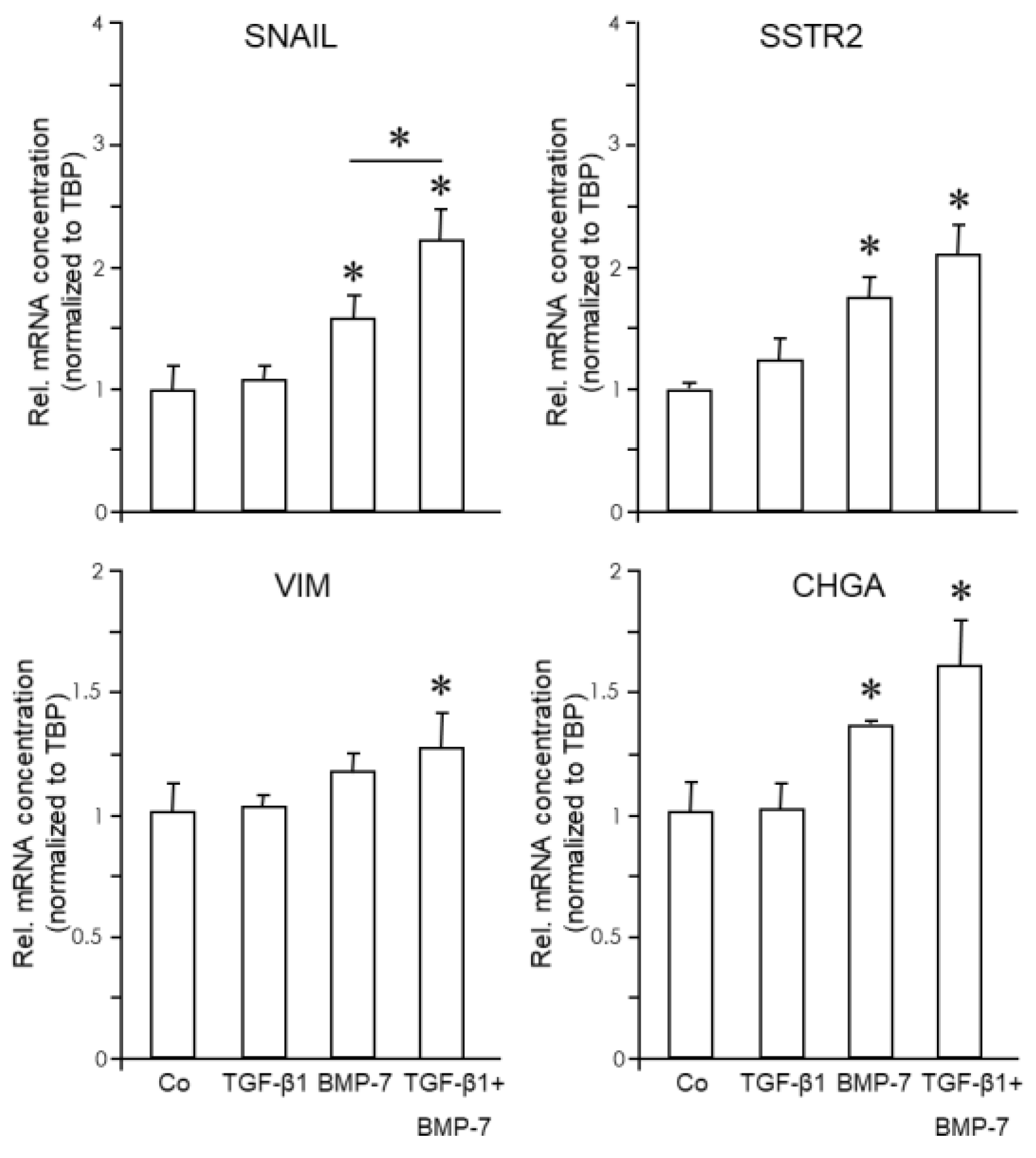
In SCCOHT-1 cells, both growth factors were unable to alter the expression of ECAD, SYP or NSE. However, SNAI1 failed to respond to TGF-β1 treatment (not shown) but was induced 1.82-fold (p=0.02) by BMP-7 and 2.33-fold (p=0.002) by TGF-β1+BMP-7 (Figure 6). The enhancing effect of the combination of TGF-β1+BMP-7 over BMP-7 alone was statistically significant (p=0.045) (Figure 6). Likewise, although VIM was upregulated 1.2-fold (p=0.065) by BMP-7, only the combined treatment generated a statistically significant induction (1.3-fold, p=0.004, Figure 6). The only NED markers that were responsive to BMP-7 were CHGA (1.35-fold, p=0.017), and 1.59-fold (p=0.031) by TGF-β1+BMP-7, and SSTR2 (1.78-fold, p=0.007) by BMP-7 and 2.13-fold (p=0.021) by TGF-β1+BMP-7 (Figure 6). We conclude that both cell lines were unresponsive to TGF-β1 (even in response to a high concentration such as 10 ng/ml), while SCCOHT-1 but not BIN-67 cells have retained sensitivity to BMP-7. Intriguingly, although resistant to single treatment, TGF-β1 when used in combination with BMP-7, was able to enhance the BMP-7 effect on SNAIL (Figure 6). For VIM, CHGA and SSTR2, we observed the same trend but the differences between BMP-7 and TGF-β1+BMP-7 missed statistical significance (not shown).
Figure 6.
Expression of EMT and NED markers in response to TGF-β1 and BMP-7 in SCCOHT. SCCOHT-1 cells were treated with TGF-β1 (10 ng/ml) or BMP-7 (200 ng/ml) followed by lysis and qPCR analysis of markers of EMT (A) or NED (B). The graphs represent the mean ± SD of quadruplicate wells of a representative assay out of at least three independent assays performed in total. The asterisks (∗) denote a significant difference.
Figure 6.
Expression of EMT and NED markers in response to TGF-β1 and BMP-7 in SCCOHT. SCCOHT-1 cells were treated with TGF-β1 (10 ng/ml) or BMP-7 (200 ng/ml) followed by lysis and qPCR analysis of markers of EMT (A) or NED (B). The graphs represent the mean ± SD of quadruplicate wells of a representative assay out of at least three independent assays performed in total. The asterisks (∗) denote a significant difference.
4. Discussion
The relationship between EMT and NED, and the role of TGF-β signaling in controlling these differentiation programs appears to vary among different tumor entities, and even between cancers affecting the same organ, such as the pancreas. While these associations have been studied in detail in a panNET cell line [21], only little information was available for PDAC in this respect. For other tumor entities such as SCCOHT no such data, whatsoever, were available. We thus initially focussed on the pancreatic model comparing poorly differentiated tumor cells of the quasi-mesenchymal subtype (PANC-1, MIA PaCa-2) with moderately differentated ones of the epithelial subtype (BxPC-3) and with the presumed progenitor cells, non-transformed pancreatic ductal epithelial cells (HPDE6c7). Based on our assumption that NED is associated with EMT and a mesenchymal phenotype, we hypothesised that HPDE6c7 and BxPC-3 cells should not exhibit a NED phenotype. Indeed, HPDE6c7 expressed much lower levels of NED-associated markers than PANC-1 and MIA PaCa-2. The same was true for BxPC-3 with respect to CHGA, SYP and NSE although, unlike HPDE6c7, not for SSTR2 and SSTR5. Hence, we have shown in PANC-1 and MIA PaCa-2 cells endogenous expression NED markers, some of which had previously been detected by flow cytometry [3].
PANC-1 cells exhibit EMP, meaning that parental cultures of this cell line consist of a mixture of subclones each displaying a different EMT phenotype despite being genetically identical. A previous histomorphological subtyping with a panel of epithelial and mesenchymal markers has shown that these clones can be grossly classified as epithelial (E-type), mesenchymal (M-type), or mixed [6]. We, therefore, considered it appropriate to test whether NED markers are enriched in either the E or the M-type clones. Monitoring seven (three E-type and four M-type) single cell-derived clones for expression of SYP, CHGA, NCAM, NSE, GLUT2, SSTR2, and SSTR5 showed that M-type clones (P1C3, P3D10, P4B9, P2E8) present with higher levels of these NED markers, except for GLUT2, than E-type clones (P4B11, P3D2, P1G7). This led us to conclude that NED is preferentially associated with a mesenchymal phenotype.
Next, we sought to know if TGF-β1, a powerful promoter of EMT, and BMP-7, another member of the TGF-β superfamily of growth and differentiation factors and promoter of MET, impact EMT and NED-associated gene expression in pancreatic tumor cells. While PANC-1 cells are highly sensitive to this growth factor, MIA PaCa-2 cells are refractory due to a defective type II receptor. These cells could thus only be employed to study the effects of BMP-7. Treatment of PANC-1 cells with TGF-β1 downregulated ECAD and RAC1b and upregulated VIM and SNAIL. However, both PANC-1 and MIA PaCa-2 cells responded to treatment with BMP-7 with induction of SNAIL and VIM, while RAC1b was only suppressed in PANC-1 and RAC1 remained unaltered in both cell lines. The effect of TGF-β1 on VIM and SNAIL was more potent than that of BMP-7. Cotreatment with both growth factors acted in either an additive or synergistic manner to suppress ECAD (only in PANC-1 since MIA PaCa-2 are ECAD-null) and RAC1b, and to enhance SNAIL and VIM expression at both the RNA and protein level. We thus concluded that TGF-β1 and BMP-7 in PANC1, and BMP-7 in MIA PaCa-2 cells, can induce EMT, and that BMP-7 can synergize with TGF-β1 in EMT induction. The observation that BMP-7 promoted EMT was surprising since this growth factor has been identified in other cellular models as either an inhibtor of EMT or even a promoter of MET [24,25,26,27,28].
Both TGF-β1 and BMP-7 were capable of inducing SYP protein in PANC-1, however, only TGF-β1 appears to accomplish this by a transcriptional mechanism. Intriguingly, a very strong inductive effect of TGF-β1 or BMP-7 was observed on SSTR2 in PANC-1 cells that could be further enhanced synergistically by combined treatment. In MIA PaCa-2 cells, however, SSTR2 mRNA levels remained unchanged in response to TGF-β1 or BMP-7, while the related SSTR5 was strongly downregulated by TGF-β1, but upregulated by BMP-7 in both cell lines. In addition, TGF-β1 treatment of PANC-1 induced CHGA, NCAM, and NSE, while BMP-7 treatment only upregulated NSE, but downregulated CHGA. These results clearly indicate that at least in control of SSTR5 and CHGA, BMP-7 can also act in an antagonistic fashion to TGF-β1.
SSTR2 and 5 represent not only established markers of NED but also possess tumor-relevant functions. SSTR2 is an inhibitory G protein-coupled receptor, the expression of which is lost in most human pancreatic cancers [35]. Of note, murine Sstr2 has been identified as a transcriptional target of TGF-β [35], which suggested the possibility that loss of SMAD4 accounts for the loss of SSTR2 expression in human PDAC. This event may contribute to a growth advantage of tumor cells [35] and is consistent with findings that SSTR2 exhibits anti-tumor properties. Here, we have confirmed - for the first time - in human PDAC-derived tumor cells a strong positive regulation of SSTR2 by TGF-β1. Also, for the first time, SSTR5 was identified here as a negative transcriptional target gene of TGF-β1 as evidenced by the dramatic downregulation of its mRNA. This mode of regulation suggests the possibility that SSTR5 normally antagonises TGF-β-dependent EMT or even other cellular responses to TGF-β, such as growth arrest. Hence, while SSTR2 qualifies as a gene involved in growth arrest in accordance with the proposed anti-tumor function, the reverse may be true for SSTR5. Moreover, while SSTR2 appears to be involved in mesenchymal conversion, SSTR5 may have a role in promoting MET or an epithelial phenotype. An interesting finding in this context came from a study with a highly invasive paclitaxel-resistant OC cell line. This cell line expresses CD105/endoglin, a stem cell marker and TGF-β co-receptor that may promote EMT and metastasis of OC by inhibiting expression of ECAD. Of note, after CD105 knockdown, the expression of both SSTR5 and ECAD (amongst others) was markedly upregulated [36]. Conversely, coactivation of SSTR2 in PDAC cells led to increased expression of mesenchymal markers and decreased expression of an epithelial marker [37]. Moreover, the expression of SSTR2 (along with those of SNAIL, SLUG and VIM) was associated with invasive non-functioning NETs of the pituitary [38].
Treatment of PANC-1 or MIA PaCa-2 cells with BMP-7 downregulated ECAD and upregulated VIM and SNAIL. This pro-EMT effect was quite surprising as BMP-7 has been identified previously as a MET-inducing (and thus anti-EMT) factor in a range of different cell types such as alveolar type II cells, adult renal epithelial tubular cells and fibroblasts, hepatic stellate cells, and melanoma cells [24,25,26,27,28]. Moreover, BMP-7 stimulation of PANC-1 or MIA PaCa-2 cells also altered the expression of some NED genes, i.e., SSTR2, the same way as the EMT-associated genes. The SSTR2 mRNA was induced by BMP-7, although the extent of induction was not as great as that with TGF-β1. In contrast to the suppressive effect of TGF-β1, BMP-7 upregulated SSTR5 mRNA, which would be consistent with a pro-epithelial effect based on the above proposed function for SSTR5. However, the observation that BMP-7 induced only a few NED markers, while others were either inhibited or remained unaffected questions its role as a general promoter of NED. Surprisingly, upon combined stimulation of PANC-1 cells with TGF-β1 and BMP-7, additive effects on induction of SSTR2 were noted. Together, this clearly suggests an association of EMT and NED through TGF-β signaling, while BMP-7 only partially shares this ability in common with TGF-β.
Prompted by the newly discovered positive association of EMT and TGF-β signaling with NED, we evaluated in another set of experiments the possibility that, conversely, MET in PANC-1 cells is associated with a loss of NED. In agreement with this hypothesis, we observed that during IIT-induced MET most NED markers, except SSTR2, were downregulated along with VIM, RAC1 and SNAIL at the mRNA (Figure 5) and protein [6] level, while concomitantly, the epithelial markers ECAD, CLDN4, GRHL2, OVOL2, CK19 and RAC1b were all upregulated [6]. This clearly shows a simultaneous loss of NED and mesenchymal markers during MET and the acquisition of an epithelial phenotype. Mechanistically, this may – at least in part - be mediated through inhibition of the SMAD2/3 arm of TGF-β signaling, since all markers that are responsive to TGF-β1 treatment were also affected by TDC-IIT but in an antagonistic fashion.
As control for cells with a strong NED phenotype, we employed the panNET cell lines, BON and NT-3, which are both epithelial in nature [19]. This is in sharp contrast to PDAC cells with NED, which are poorly differentiated/quasi-mesenchymal. In this study, we have, therefore, revealed fundamental differences between two major types of pancreatic cancer, PDAC and panNET, with respect to the association of NED with EMT and TGF-β signaling. In panNET, NED is associated with a well-differentiated epithelial phenotype and functional TGF-β and SST signaling, and defective TGF-β/SST signaling causes loss of NED with mesenchymal conversion [21]. However, in the present study the reverse situation is operating; NED occurs in poorly differentiated mesenchymal cells and can still be further enhanced by activation of TGF-β or BMP-7 signaling provided the cells have retained responsiveness to these growth factors. Thus, the role of SST/SSTR in TGF-β signaling may differ between panNET (BON, NT-3) and PDAC, which is also supported by the antagonistic regulation of SSTR5 by TGF-β1. We have thus identified a newly distinguishing feature between panNET and PDAC. Consequently, it will be highly intriguing to test these properties in panNEC, a pancreatic cancer entity that combines features of both panNET and PDAC. Intriguingly, we have shown recently that not only treatment with TGF-β but also the stimulation with SST, or the SST analogs octreotide and lanreotide, was able to regulate a set of NED genes and alter the NED state [22].
While the phenotypic association between EMT and NED seems to be well established in some cancers, this is not the case for the underlying molecular mechanism(s). Initial insights came from the PCa model [39] with the identification of microRNA-147b as an inducer of NED through targeting the ribosomal protein PRS15A [40]. More recently, activation of NFκB-STAT3 signaling by tumor protein D52, isoform 3 (TPD52) has been found to induce distinct NED features (as measured by CHGA and NSE) through EMT under androgen-depleted conditions [39]. Moreover, the authors were able to show that TPD52 also positively regulates EMT of PCa cells towards NED (as revealed by induction of N-cadherin, VIM and ZEB1, another EMT-associated transcription factor) via activation of NFκB-STAT3. These changes were orchestrated by SNAIL, since silencing of SNAI1 in TPD52-positive cells blocked the progression of NED [39]. SNAIL may thus promote tumor aggressiveness in PCa cells through multiple processes; induction of EMT to promote migration, while, in turn, induction of NED promotes tumor proliferation through a paracrine mechanism [41]. In addition, ZEB1 has been shown to promote NED in PCa [42]. Liu and colleagues investigated the molecular mechanisms by which androgen deprivation therapy (ADT) induces NED in advanced Pca and found transmembrane protein 1 (MCTP1) to be abundantly expressed in samples from patients with advanced PCa. Of note, after ADT, MCTP1 through SNAIL promoted EMT, NED and cell migration of PC3 and C4-2 PCa cells [43].
Therapeutic targeting of SNAIL or ZEB1 may thus prove beneficial in abrogating not only EMT but also NED [41]. Apart from NFκB-STAT3 [39], other signaling pathways are involved in acquiring NED characteristic features of PCa cells, such as AMPK/SIRT1-p38MAPK-IL6 [44] and ERK [45]. Of note, MEK-ERK signaling is also activated by TGF-β and is critically involved in driving TGF-β-dependent EMT, migration, invasion and metastasis [46,47,48], and TGF-β1-induced downregulation of CDH1 and upregulation of SNAI1 [33]. We are currently carrying out ERK immunoblot analysis of PANC-1 subclones to reveal whether the extent of ERK1/2 activation corresponds with NED marker expression. Interestingly, treatment of MIA PaCa-2 and PANC-1 cells with grape seed proanthocyanidins (GSPs) resulted in decreased phosphorylation of ERK1/2, inactivation of NFκB, reversal of EMT, upregulation of ECAD, downregulation of NCAD and VIM, and reduced cell migration [49]. It is thus conceivable that GSP-induced inhibition of ERK activation also reduces NED in these cells.
The importance of EMT is also considered in other GI cancers such as coloNEC. Current efforts are therefore underway to reveal an association between EMT and NED in model cell lines of this disease, e.g., the coloNEC-derived cell lines, SS-2 and LCC-18. Here, we have studied a non-GI cancer, namely small cell hypercalcemic ovarian cancer, represented by the cell lines SCCOHT-1 [9] and BIN-67 [10,11]. SCCOHT-1 cells constitutively express NED markers such as NCAM in the original patient tumor and derived mouse xenograft tumors [9,12]. Moreover, both cell lines developed biallelic deleterious SMARC A4 gene mutations whereby phenotypic and genetic similarities were observed between SCCOHT and highly malignant childhood-onset atypical teratoid/rhabdoid tumors (AT/RTs) of the central nervous system [50,51]. This heterogeneity and plasticity of SCCOHT-1 indicates the potential for transdifferentiation involving EMT and maturation along a NED phenotype [13]. We found both cell lines to be refractory to stimulation with TGF-β1 for any of the above-mentioned EMT/NED genes but have partially retained sensitivity to BMP-7 as evidenced by induction of ECAD in BIN-67 and SNAIL, VIM, CHGA and SSTR2 in SCCOHT-1 cells.
Since both PANC-1 and MIA PaCa-2 cells are highly invasive and metastatic [52], the NED phenotype besides the mesenchymal subtype may contribute to this property. In addition, the TGF-β1 effects on various NED markers may have therapeutic significance as it was shown that PANC-1 and MIA PaCa-2 cell lines when subjected to (fractionated) radiation upregulate not only the expression of NED genes [5] but also induce the synthesis and secretion of TGF-β [53]. Both radiotherapy and chemotherapy induce TGF-β activity, possibly promoting metastatic progression, and high levels of TGF-β are associated with resistance to anticancer treatments [54]. Therefore, irradiation-induced secretion of TGF-β1 by tumor cells may account for changes in NED marker expression. Given that the resulting induction of EMT and NED may enhance tumor invasion and metastasis in PDAC, the concomitant application of TGF-β inhibitors between radiation cycles should be considered to prevent an unwanted increase in tumor aggressiveness [54].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Immunoblot analysis of the indicated NED markers in BxPC-3 cells; Figure S2: Immunoblot analysis of SSTR2 in PANC-1 cells induced to undergo MET.
Author Contributions
H.U., J.v.d.O., R.B. and R.H. performed the experiments and analysed the data; H.U. and J.S. provided cells and essential reagents; H.U., R.H. and B.K. conceived and designed the experiments; H.U. wrote the paper; R.B., J.S., H.L., J.U.M., R.H. and B.K. critically read and edited the manuscript.
Funding
This work was supported by a grant from the Niedersächsische Krebsgesellschaft e.V. with respect to the NDR charity campaign ‘Hand in Hand für Norddeutschland 2019’ to Ralf Hass.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of the study are available from the corresponding author upon reasonable request.
Acknowledgments
We are indebted to H. Albrecht for excellent technical assistance.
Conflict of Interest
The authors declare no conflicts of interest.
References
- Gao, H.L.; Wang, W.Q.; Yu, X.J.; Liu, L. Molecular drivers and cells of origin in pancreatic ductal adenocarcinoma and pancreatic neuroendocrine carcinoma. Exp. Hematol. Oncol. 2020, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Scarpa, A. Neoplastic Progression in Neuroendocrine Neoplasms of the Pancreas. Arch. Pathol. Lab. Med. 2023, Mar 7. [CrossRef]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1 - pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef] [PubMed]
- Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.U.; Ungefroren, H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers (Basel). 2021, 13, 4663. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Stone, B.; Thibodeau, B.J.; Baschnagel, A.M.; Galoforo, S.; Fortier, L.E.; Ketelsen, B.; Ahmed, S.; Kelley, Z.; Hana, A.; et al. The significance of Trk receptors in pancreatic cancer. Tumour Biol. 2017, 39, 1010428317692256. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1-A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers (Basel) 2022, 14, 2057. [Google Scholar] [CrossRef] [PubMed]
- Dickersin, G.R.; Kline, I.; Scully, R.E. Small cell carcinoma of the ovary with hypercalcemia: a report of eleven cases. Cancer. 1982, 49, 188–197. [Google Scholar] [CrossRef]
- Young, R.H.; Oliva, E.; Scully, R.E. Small cell carcinoma of the hypercalcemic type in the ovary. Gynecol. Oncol. 1995, 57, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Göhring, G.; Steinemann, D.; Schlegelberger, B.; Groos, S.; Länger, F.; Kreipe, H.H.; Schambach, A.; Neumann, T.; Hillemanns, P.; et al. A tumor-derived population (SCCOHT-1) as cellular model for a small cell ovarian carcinoma of the hypercalcemic type. Int. J. Oncol. 2012, 41, 765–775. [Google Scholar] [CrossRef]
- Upchurch, K.S.; Parker, L.M.; Scully, R.E.; Krane, S.M. Differential cyclic AMP responses to calcitonen among human ovarian carcinoma cell lines: a clacitonin-responsive line derived from a rare tumor type. J. Bone Miner. Res. 1986, 1, 299–304. [Google Scholar] [CrossRef]
- Gamwell, L.F.; Gambaro, K.; Merziotis, M. , Crane, C.; Arcand, S.L.; Bourada, V.; Davis, C.; Squire, J.A.; Huntsman, D.G.; Tonin, P.N.; et al. Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics. Orphanet J. Rare Dis. 2013, 8, 33. [Google Scholar] [CrossRef]
- Otte, A.; Rauprich, F.; Hillemanns, P.; Park-Simon, T.W.; von der Ohe, J.; Hass, R. In vitro and in vivo therapeutic approach for a small cell carcinoma of the ovary hypercalcaemic type using a SCCOHT-1 cellular model. Orphanet J. Rare Dis. 2014, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Hass, R. von der Ohe, J. Ungefroren, H. The intimate relationship among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to be exploited for therapeutic purposes. Cancers (Basel). 2020, 12, 3674. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Ratther, E.; Stylianou, N.; Nelson, C.C.; Hollier, B.G.; Williams, E.D. Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: an opportunity for intervention. Front. Oncol. 2014, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. EMT, stemness and tumor plasticity in aggressive variant neuroendocrine prostate cancers. Biochim- Biophys. Acta Rev. Cancer. 2018, 1870, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Aieta, M.; Amadori, D.; De Giorgi, U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Crit. Rev. Oncol. Hematol. 2014, 92, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.D.; Choo, R.; Huang, J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Gravemeyer, J.; Lange, A.; Ritter, C.; Spassova, I.; Song, L.; Picard, D.; Remke, M.; Horny, K.; Sriram, A.; Gambichler, T.; et al. Classical and Variant Merkel Cell Carcinoma Cell Lines Display Different Degrees of Neuroendocrine Differentiation and Epithelial-Mesenchymal Transition. J. Invest. Dermatol. 2021, 141, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Groves, S.M.; Panchy, N.; Tyson, D.R.; Harris, L.A.; Quaranta, V.; Hong, T. Involvement of Epithelial-Mesenchymal Transition Genes in Small Cell Lung Cancer Phenotypic Plasticity. Cancers (Basel). 2023, 15, 1477. [Google Scholar] [CrossRef] [PubMed]
- Luley, K.B.; Biedermann, S.B.; Künstner, A.; Busch, H.; Franzenburg, S.; Schrader, J.; Grabowski, P.; Wellner, U.F.; Keck, T.; Brabant, G.; et al. A Comprehensive Molecular Characterization of the Pancreatic Neuroendocrine Tumor Cell Lines BON-1 and QGP-1. Cancers (Basel). 2020, 12, 691. [Google Scholar] [CrossRef]
- Leu, F.P.; Nandi, M.; Niu, C. The effect of transforming growth factor beta on human neuroendocrine tumor BON cell proliferation and differentiation is mediated through somatostatin signaling. Mol. Cancer Res. 2008, 6, 1029–1042. [Google Scholar] [CrossRef]
- Ungefroren, H.; Künstner, A.; Busch, H.; Franzenburg, S.; Luley, K.; Viol, F.; Schrader, J.; Konukiewitz, B.; Wellner, U.F.; Meyhöfer, S.M.; et al. Differential Effects of Somatostatin, Octreotide, and Lanreotide on Neuroendocrine Differentiation and Proliferation in Established and Primary NET Cell Lines: Possible Crosstalk with TGF-β Signaling. Int. J. Mol. Sci. 2022, 23, 15868. [Google Scholar] [CrossRef] [PubMed]
- Dicken, H.; Hensley, P.J.; Kyprianou, N. Prostate tumor neuroendocrine differentiation via EMT: The road less traveled. Asian J. Urol. 2019, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Li, R. X.; Yiu, W.H.; Tang, S.C. Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 2015, 6, 114. [Google Scholar] [CrossRef]
- Pan, Q.; Wang, Y.Q.; Li, G.M.; Duan, X.Y.; Fan, J.G. Fuzheng Huayu Recipe Ameliorates Liver Fibrosis by Restoring Balance between Epithelial-to-Mesenchymal Transition and Mesenchymal-to-Epithelial Transition in Hepatic Stellate Cells. Biomed. Res. Int. 2015, 2015, 935903. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Seok, S.H.; Kim, D.J.; Han, J.H.; Kim, T.H.; Jung, H.; Lee, B.H.; Park, J.H. Bone morphogenetic protein 7 induces mesenchymal-to-epithelial transition in melanoma cells, leading to inhibition of metastasis. Cancer Sci. 2009, 100, 2218–2225. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef]
- Bi, W.R.; Jin, C.X.; Xu, G.T.; Yang, C.Q. Bone morphogenetic protein-7 regulates Snail signaling in carbon tetrachloride-induced fibrosis in the rat liver. Exp. Ther. Med. 2012, 4, 1022–1026. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, C.H.; Song, S.H.; Um, J.E.; Kim, H.S.; Yun, J.S.; Han, D.; Cho, E.S.; Nam, B.Y.; Yook, J. I,.; et al. Micellized Protein Transduction Domain-Bone Morphogenetic Protein-7 Efficiently Blocks Renal Fibrosis Via Inhibition of Transforming Growth Factor-Beta-Mediated Epithelial-Mesenchymal Transition. Front. Pharmacol. 2020, 11, 591275. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, D.; Zhu, Z.; Li, X.; An, G.; Niu, P.; Chen, L.; Tian, L. Bone morphogenetic protein-7 prevented epithelial-mesenchymal transition in RLE-6TN cells. Toxicol. Res. (Camb). 2016, 5, 931–937. [Google Scholar] [CrossRef]
- Cho, K.; Kim, N.H.; Seo, S.H.; Song, S.H.; Jeong, C.H.; Kim, H.S.; Um, J.E.; Ku, M.; Yang, J.; Park, J.Y.; et al. A micellized bone morphogenetic protein-7 prodrug ameliorates liver fibrosis by suppressing transforming growth factor-β signaling. Am. J. Cancer Res. 2022, 12, 763–778. eCollection 2022. [Google Scholar]
- Benten D, Behrang Y, Unrau L, Weissmann V, Wolters-Eisfeld G, Burdak-Rothkamm S, Stahl FR, Anlauf M, Grabowski P, Möbs M. ; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A Guardian of the Epithelial Phenotype and Protector Against Epithelial-Mesenchymal Transition. Cells. 2019, 8, 1569. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.W.; Mattingly, C.A.; Strodel, W.E. Increased tumorigenicity in the human pancreatic cell line MIA PaCa-2 is associated with an aberrant regulation of an IGF-1 autocrine loop and lack of expression of the TGF-beta type RII receptor. J. Cell. Physiol. 1995, 165, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Puente, E.; Saint-Laurent, N.; Torrisani, J.; Furet, C.; Schally, A.V.; Vaysse, N.; Buscail, L.; Susini, C. Transcriptional activation of mouse sst2 somatostatin receptor promoter by transforming growth factor-beta. Involvement of Smad4. J. Biol. Chem. 2001, 276, 13461–13468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sang, X.; Zhang, R.; Chi, J.; Bai, W. CD105 expression is associated with invasive capacity in ovarian cancer and promotes invasiveness by inhibiting NDRG1 and regulating the epithelial-mesenchymal transition. Am. J. Transl. Res. 2021, 13, 12461–12479. eCollection 2021. [Google Scholar] [PubMed]
- Jorand, R.; Biswas, S.; Wakefield, D.L.; Tobin, S.J.; Golfetto, O.; Hilton, K.; Ko, M.; Ramos, J.W.; Small, A.R.; Chu, P.; et al. Molecular signatures of mu opioid receptor and somatostatin receptor 2 in pancreatic cancer. Mol. Biol. Cell. 2016, 27, 3659–3672. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Marques-Pamies, M.; Valassi, E.; Serra, G.; Salinas, I.; Xifra, G.; Casano-Sancho, P.; Carrato, C.; Biagetti, B.; Sesmilo, G.; et al. Molecular characterization of epithelial-mesenchymal transition and medical treatment related-genes in non-functioning pituitary neuroendocrine tumors. Front. Endocrinol. (Lausanne). 2023, 14, 1129213. [Google Scholar] [CrossRef] [PubMed]
- Sruthi, K.K.; Natani, S.; Ummanni, R. Tumor protein D52 (isoform 3) induces NF-κB - STAT3 mediated EMT driving neuroendocrine differentiation of prostate cancer cells. Int. J. Biochem. Cell Biol. 2024, 166, 106493. [Google Scholar] [CrossRef] [PubMed]
- Natani, S.; Ramakrishna, M.; Nallavolu, T.; Ummanni, R. MicroRNA-147b induces neuroendocrine differentiation of prostate cancer cells by targeting ribosomal protein RPS15A. Prostate. 2023, 83, 936–949. [Google Scholar] [CrossRef] [PubMed]
- McKeithen, D.; Graham, T.; Chung, L.W.; Odero-Marah, V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate. 2010, 70, 982–992. [Google Scholar] [CrossRef]
- Bery, F.; Cancel, M.; Guéguinou, M.; Potier-Cartereau, M.; Vandier, C.; Chantôme, A.; Guibon, R.; Bruyère, F.; Fromont, G.; Mahéo, K. Zeb1 and SK3 Channel Are Up-Regulated in Castration-Resistant Prostate Cancer and Promote Neuroendocrine Differentiation. Cancers (Basel)., 2021, 13, 2947. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Chen, W.Y.; Yeh, H.L.; Chen, W.H.; Jiang, K.C.; Li, H.R.; Dung, P.V.T.; Chen, Z.Q.; Lee, W.J.; Hsiao, M.; et al. MCTP1 increases the malignancy of androgen-deprived prostate cancer cells by inducing neuroendocrine differentiation and EMT. Sci. Signal. 2024, 17, eadc9142. [Google Scholar] [CrossRef]
- Natani, S.; Dhople, V.M.; Parveen, A.; Sruthi, K.K.; Khilar, P.; Bhukya, S.; Ummanni, R. AMPK/SIRT1 signaling through p38MAPK mediates Interleukin-6 induced neuroendocrine differentiation of LNCaP prostate cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119085. [Google Scholar] [CrossRef]
- Bosutti, A.; Zanconati, F.; Grassi, G.; Dapas, B.; Passamonti, S.; Scaggiante, B. Epigenetic and miRNAs Dysregulation in Prostate Cancer: The role of Nutraceuticals. Anticancer Agents Med. Chem. 2016, 16, 1385–1402. [Google Scholar] [CrossRef]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar]
- Ungefroren, H.; Konukiewitz, B.; Braun, R.; Wellner, U.F.; Lehnert, H.; Marquardt, J.U. TAp73 Inhibits EMT and Cell Migration in Pancreatic Cancer Cells through Promoting SMAD4 Expression and SMAD4-Dependent Inhibition of ERK Activation. Cancers (Basel). 2023, 15, 3791. [Google Scholar] [CrossRef]
- Thakur, A.K.; Nigri, J.; Lac, S.; Leca, J.; Bressy, C.; Berthezene, P.; Bartholin, L.; Chan, P.; Calvo, E.; Iovanna, J.L.; et al. TAp73 loss favors Smad-independent TGF-β signaling that drives EMT in pancreatic ductal adenocarcinoma. Cell Death Differ. 2016, 23, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Katiyar, S.K. Grape seed proanthocyanidins inhibit migration potential of pancreatic cancer cells by promoting mesenchymal-to-epithelial transition and targeting NF-κB. Cancer Lett. 2013, 334, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Meehan, B.; Macdonald, E.; Venneti, S.; Wang, X.Q.D.; Witkowski, L.; Jelinic, P.; Kong, T.; Martinez, D.; Morin, G.; et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat. Commun. 2019, 10, 558. [Google Scholar] [CrossRef]
- Otte, A.; Rauprich, F.; von der Ohe, J.; Yang, Y.; Kommoss, F.; Feuerhake, F.; Hillemanns, P.; Hass, R. c-Met inhibitors attenuate tumor growth of small cell hypercalcemic ovarian carcinoma (SCCOHT) populations. Oncotarget. 2015, 6, 31640–31658. [Google Scholar] [CrossRef]
- Takada, M.; Hirata, K.; Ajiki, T.; Suzuki, Y.; Kuroda, Y. Expression of receptor for advanced glycation end products (RAGE) and MMP-9 in human pancreatic cancer cells. Hepatogastroenterology. 2004, 51, 928–930. [Google Scholar] [PubMed]
- Carl, C.; Flindt, A.; Hartmann, J.; Dahlke, M.; Rades, D.; Dunst, J.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell. Mol. Life Sci. 2016, 73, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; de Gramont, A.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. Perspectives of TGF-beta inhibition in pancreatic and hepatocellular carcinomas. Oncotarget. 2014, 5, 78–94. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Relative expression of NED markers in PDAC-derived cell lines, PANC-1, MIA PaCa-2 and BxPC-3, and in the normal pancreatic duct epithelial cell line, HPDE6c7. The panNET-derived cell lines, BON and NT-3 were employed here as control. Cells were lysed at different times during continuous culture and subjected to RNA isolation, reverse transcription and real-time PCR analysis. Expression levels for the indicated genes are displayed relative to those in PANC-1 cells set arbitrarily at 1.0. Data represent the means ± SD of 3-6 independent preparations. Please note the extra scales for BON and NT-3 cells in the SSTR2, 5 and CHGA graphs, which indicate the orders of magnitude higher expression.
Figure 1.
Relative expression of NED markers in PDAC-derived cell lines, PANC-1, MIA PaCa-2 and BxPC-3, and in the normal pancreatic duct epithelial cell line, HPDE6c7. The panNET-derived cell lines, BON and NT-3 were employed here as control. Cells were lysed at different times during continuous culture and subjected to RNA isolation, reverse transcription and real-time PCR analysis. Expression levels for the indicated genes are displayed relative to those in PANC-1 cells set arbitrarily at 1.0. Data represent the means ± SD of 3-6 independent preparations. Please note the extra scales for BON and NT-3 cells in the SSTR2, 5 and CHGA graphs, which indicate the orders of magnitude higher expression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



