
Submitted:
12 July 2024
Posted:
15 July 2024
You are already at the latest version
Abstract
AUTAC-Biguanide is a hybrid compound designed to target mitochondria inducing their degradation by mitophagy. This study unveils the potential of biguanides as cancer cell-targeting agents, emphasizing AUTAC-Biguanide's superior antiproliferative properties compared to metformin and its selectivity for cancer cells. The mechanism behind this heightened effect includes the ability of AUTAC-Biguanide to triggers mitophagy. By providing a comprehensive analysis of these findings, the study adds valuable insights to the field of mitochondrial-targeting anticancer agents.
Keywords:
AUTAC
; biguanide
; pancreatic cancer
1. Introduction
The influence of biguanide derivatives on the mitochondrial respiratory chain has become a well-established area of research. Their antitumor properties, to some extent, stem from their ability to inhibit oxidative phosphorylation.1 While numerous studies have demonstrated the inhibition of complex I in response to metformin and phenformin treatments2, the precise molecular target of biguanides within the mitochondria remains unclear.3, 4 Furthermore, some studies indicate a lack of biguanide activity on isolated mitochondria, suggesting that these compounds may only exert their effects within intact cells.5 One plausible explanation is that these compounds do not directly interact with mitochondria.4 Another hypothesis suggests that the action of biguanides is, indeed, the result of a direct interaction but is triggered by their accumulation within these organelles.4-6 This raises an intriguing question regarding whether biguanides primarily target mitochondria themselves or if their target is actually in the cytoplasm, the inhibition of oxidative phosphorylation being an indirect consequence of this interaction.
The propensity of monosubstituted biguanides to localize within mitochondria primarily hinges on their protonation status at physiological pH. Positively charged biguanides exhibit an affinity for the negatively charged mitochondrial inner membrane.7, 8 Since the inception of research on biguanide derivatives in the 1920s-1930s, limited attention has been given to exploring their interactions within mitochondria.9 A notable breakthrough occurred in 2018 when a study demonstrated the co-localization of these compounds with cytochrome c.10 In pursuit of this understanding, the researchers conducted an in vitro experiment where they conjugated a metformin derivative with a fluorescent marker using a copper-catalyzed azide-alkyne cycloaddition method applicable within intracellular environments. Their observations revealed the accumulation of the metformin derivative in mitochondria.
Nevertheless, several crucial questions persist. Does the observed accumulation in mitochondria suffice to designate mitochondria as the primary target of biguanides? Furthermore, does incorporating a biguanide function into a compound redirect its mechanisms of action toward mitochondria? To delve into the interaction between biguanidium salts and mitochondria, and with the aspiration of formulating a potentially more potent anticancer agent than metformin, we fused metformin with a ligand capable of activating the cell's autophagy system. The effects of this hybrid compound on mitochondrial structure may offer compelling evidence supporting the notion that biguanides indeed target mitochondria.
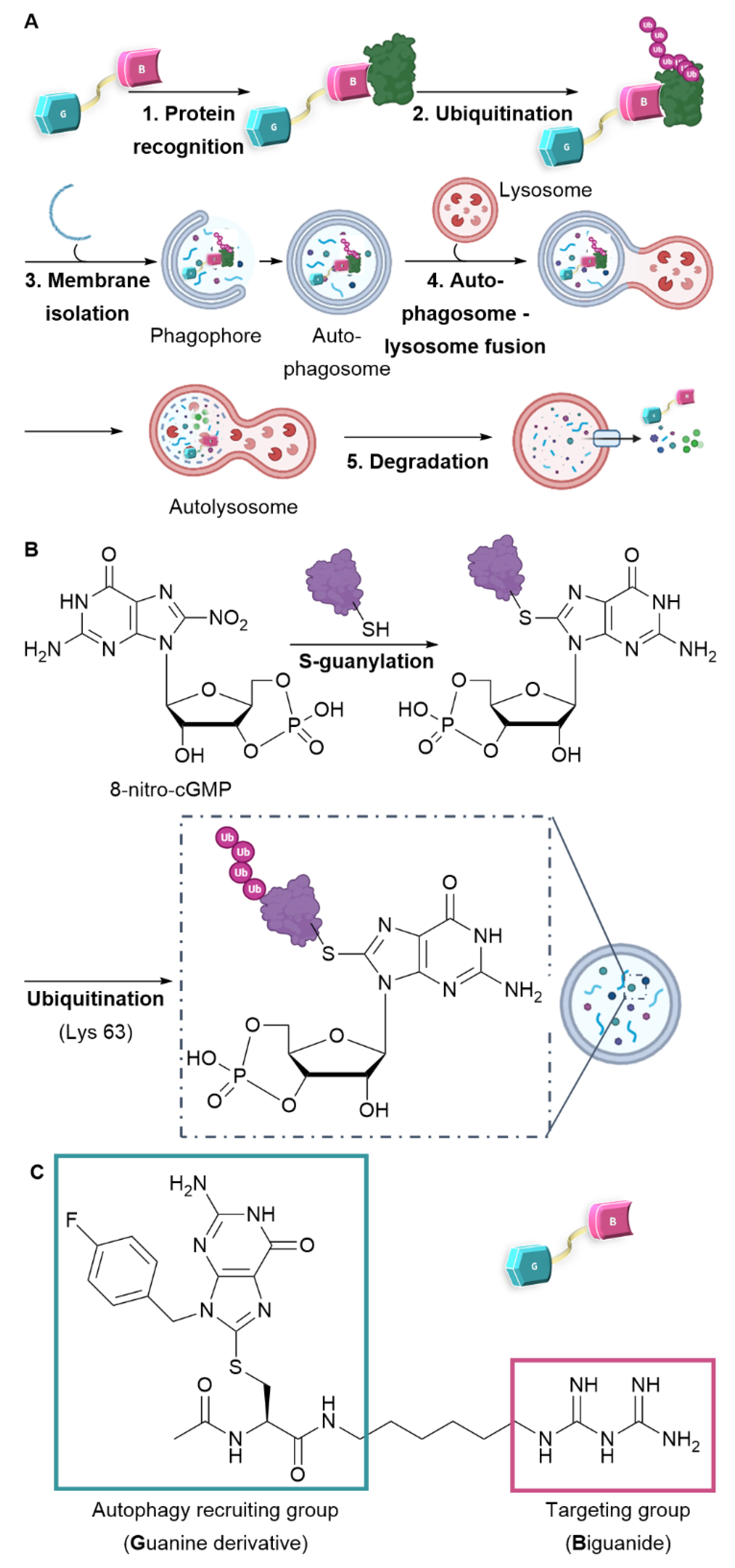
An emerging strategy involves promoting the degradation of a target molecule by attaching it to a ligand that can harness the cell's intrinsic degradation pathways.11-13 Autophagy is a cellular mechanism that facilitates the degradation of a broad spectrum of substrates, ranging from individual proteins to entire organelles. This process operates through the selective tagging of proteins and their subsequent incorporation into autophagosomes. Upon fusion with lysosomes, these autophagosomes allow the enzymatic degradation of their contents by hydrolases.14 To specifically target proteins or organelles of interest using this mechanism, recent developments have led to the creation of bifunctional molecules, including AUTACs (Autophagy Targeting Chimeras) and ATTECs (Autophagosome Tethering Compounds)11, 12, 15. These molecules consist of two essential components: one ligand targeting the protein to be degraded, represented by the pink motif (Biguanide in our case), and another ligand that recruits the degradation system, denoted by the blue motif (Guanine-derivative) (see Figure 1A). Notably, the creation of an ATTEC compound with mitochondria-targeting capabilities has already been successfully realized through the incorporation of a triphenylphosphonium (TPP) targeting group.16 Liu et al. have successfully showcased the capacity of ATTEC-TPP derivatives to trigger mitophagy.17 Among the diverse approaches to activate autophagy, the creation of AUTAC compounds draws inspiration from protein S-guanylation as a triggering mechanism. In fact, a study conducted by Arimoto et al. in 2013 emphasized the significance of S-guanylation as a marker that induces bacterial autophagy within the cytoplasm of mammalian cells.18, 19 This process is characterized by the interaction between the compound 8-nitro-cGMP (Cyclic Guanosine Monophosphate) and a protein's cysteine residue. The creation of a thioether bond facilitates the subsequent attachment of ubiquitin through lysine 63 (K63), thereby marking the protein for degradation by the lysosome.20 As a result, the ubiquitin chain is subsequently identified by autophagy receptors and integrated into the autophagosome (as illustrated in Figure 1B). Taking inspiration from this recognition mechanism, Arimoto et al. achieved the synthesis of the initial AUTAC compound. This one comprises a guanine derivative and a mitochondria-targeting ligand and has demonstrated the ability to induce mitophagy.15
The research presented below intends to confirm the mitochondria-targeting potential of biguanides and enhance their efficacy as anticancer agents. We designed an AUTAC-Biguanide compound by incorporating a biguanide functional group as a targeting moiety to the guanine motif, known for its ability to recruit autophagy, as originally reported by Arimoto et al.15 The two ligands connected by a carbon chain (Figure 1C) were assessed for their capacity to target and modify mitochondria and inhibit the proliferation of cancer cells in vitro.
2. Results and Discussion
The synthesis of the AUTAC-Biguanide was the result of the conjugation of two building blocks. N-Boc-1,6-hexanediamine was introduced in stoichiometric quantities with dicyandiamide and TMSCl to form compound 1 in salt form, by in situ deprotection of the tert-Butoxycarbonyl (Boc) group (Scheme 1A). To form the guanine ligand, the synthetic route previously described by Arimoto et al.15 has been revisited with the aim of reducing the number of reaction steps. By substituting 4-fluorobenzyl alcohol with fluorobenzyl bromide, it was possible to streamline the synthesis process by eliminating three steps, reducing it to just four steps. The initial step involved the formation of compound 2 through an SN2 reaction involving the purine imidazole and fluorobenzyl bromide, yielding the desired isomer with an 89% yield. Subsequent addition of formic acid led to the complete formation of intermediate 3, followed by bromination to yield compound 4. This one then reacted with acetylcysteine thiol to yield guanine derivative ligand 5, incorporating an essential carboxylic acid function to enable coupling with the previously synthesized biguanide derivative 1 (see S1-S14). After HPLC-prep purification, compound 6 was successfully obtained with a 12% yield, as depicted in Scheme 1B. A control derivative was also synthesized without the biguanide function. In this case, compound 5 was coupled with hexylamine to retain only the guanine ligand and the spacer, following the synthetic route depicted in Scheme 1C. This approach offers the advantage of simplicity, requiring only a few steps.
To evaluate its potential as an anticancer agent, AUTAC-Biguanide 6 underwent testing on two pancreatic cancer cell lines, namely KP4 and PANC1. Its effectiveness was subsequently compared with that of metformin, as well as guanine derivatives AUTAC-COOH 5 and AUTAC-Hexyl 7. The results showed that controls 5 and 7 had no discernible impact on the growth of cancer cells, while metformin exhibited toxic activity at concentrations ranging from 1 to 4 mM. Notably, the combination of the AUTAC ligand with the biguanide proved significantly more potent, demonstrating a median effective concentration of approximately 0.1 mM. This concentration is ten times lower than that of metformin, highlighting the compound's potential antitumor properties (see Table 1). The compounds were also tested on two healthy cell lines, hTERT-HPNE (immortalized pancreatic cells) and IMR90 (lung fibroblasts), to assess their toxicity. Metformin had no noticeable effect on either cell line at the concentrations examined. Conversely, the controls, AUTAC-COOH 5 and AUTAC-Hexyl 7, appeared to affect hTERT-HPNE without exhibiting toxicity toward IMR90. AUTAC-Biguanide, much like metformin, had no adverse impact on any of the healthy cell lines. However, it's important to note that determining the EC50 (effective concentration reducing cell viability to 50%) for these compounds became challenging as the solubility of the compound exceeded 1 mM in the culture medium (see Table 1). Both AUTAC-Biguanide 6 and metformin exhibited a remarkable selectivity for cancer cells over healthy cells, indicating the potential of the biguanide function as a cancer cell-targeting mechanism. These derivatives demonstrated a selectivity index greater than 10, establishing them as promising anti-cancer agents.
Notably, compound 6 outperformed metformin, as its anticancer effects were observed at lower concentrations, as illustrated in Figure 2. Evidently, AUTAC-Biguanide stands out from metformin due to its significantly improved antiproliferative properties in cancer cells. To gain insights into the underlying reasons for this enhanced effect, a comprehensive investigation of the compound's mechanism of action was undertaken and subsequently compared with its biguanide and guanine controls.
As the impact of biguanide compounds is primarily linked to mitochondria, a study was conducted to investigate whether the guanine motif had the potential to induce autophagy in these organelles. In healthy cells, mitochondria form a dynamic network that evolves throughout the cell cycle. This network presents itself as a collection of mobile filaments, which tend to undergo fragmentation at the onset of apoptosis, resulting in a disorganized network with a punctate appearance.21, 22 The morphology of mitochondria can be influenced by various stimuli, which may have a more or less direct impact on these organelles. These alterations can be visualized using confocal microscopy. Depending on their morphology, mitochondria can be categorized as filamentous, fragmented (representing an intermediate state with both filaments and dots), or punctate.21,22 A fluorochrome that specifically targets mitochondria (MitoTracker Deep Red FM or MTDR) was employed to examine the mitochondrial morphology in KP4 cells.23 In the presence of metformin and AUTAC-Hexyl 7, mitochondria predominantly formed a filamentous network. However, upon treatment with AUTAC-Biguanide, they exhibited a punctate configuration. Notably, the extent of this effect was concentration-dependent and became evident only when concentrations exceeded the EC50 threshold (100 µM). At this concentration, more than 50% of cells displayed a fragmented or punctate mitochondrial network within just 24 hours of treatment. Doubling this concentration resulted in fewer than 10% of cells maintaining a filamentous network. Moreover, there was a decrease in fluorescence intensity, indicating potential mitochondrial degradation, which aligns with previous findings (Figure 3A). To enhance image resolution and validate the disparity in the effects of metformin and AUTAC-Biguanide, immunostaining was performed. In this context, an immunofluorescent label was applied to TOMM20, a mitochondrial outer membrane protein. The results were consistent with MTDR results, showing that the majority of cells assumed a punctate appearance after treatment with 200 µM compound 6, while 2 mM metformin induced a mild fragmentation of the network, characterized by the presence of filaments (indicated by white arrows) and dots (indicated by red arrows) (Figure 3B). Furthermore, mitochondria seem to adopt the same morphology with both AUTAC-Biguanide and ATTEC-TPP treatments as reported by Liu et al.17, suggesting that both compounds have the same mode of action of inducing mitophagy.
To further assess mitochondrial mass reduction induced by the action of compound 6, its effect on the levels of proteins belonging to the mitochondrial respiratory chain was studied (Figure 4). To this end, protein transfer was performed following 24 h treatment of compound 6 and metformin in KP4. A cocktail of antibodies was used to reveal and quantify various mitochondrial proteins involved in oxidative phosphorylation (OXPHOS). This mixture enabled the detection of one protein from each of the respiratory chain complexes: vATP5A (complex V), UQCR2 (complex III), SDHB (complex II), COXII (complex IV) and NDUFB8 (complex I). While metformin did not appear to affect the levels of the proteins studied, a decrease was visible upon treatment with compound 6 for the majority of proteins, with the exception of vATP5A and SDHB.
To delve deeper into the mechanism of action of AUTAC-Biguanide 6, investigations were conducted to ascertain whether autophagy played a role in inducing mitochondrial degradation. The autophagic process hinges on the formation of autophagosomes, which encapsulate proteins or organelles. These autophagosomes subsequently fuse with lysosomes, leading to the degradation of their contents.24 One of the most widely used markers of autophagy is the LC3-II protein, associated with the membranes of autophagosomes and autophagolysosomes.25 The cytosolic protein LC3-I (microtubule-associated protein 1A/1B-light chain 3) is recruited during autophagy and conjugated with a phosphatidylethanolamine (PE) molecule to form LC3-II. This can then be incorporated into the phagophore membrane, and is degraded only after fusion to the lysosome by hydrolases.25 Its expression is therefore closely correlated with autophagy, and its detection by immunoblot has proved an effective method for monitoring this process.26 In addition, LC3-II interacts with the p62 protein, capable of binding to ubiquitinated proteins or organelles.27 This interaction ensures selective degradation of components incorporated into the autophagosome. Autophagy is a naturally observed degradation process, which contributes to cell homeostasis by targeting damaged or overexpressed proteins or organelles.24
There is therefore a basal level of LC3-II, which may differ depending on the cell line studied or the experimental conditions. To assess the impact of compound 6 on the induction of the autophagy process, LC3-I and LC3-II levels were therefore determined in KP4 after 24 h of different treatments and expressed as a relative quantity compared to the protein level in control-treated cells (DMSO) (Figure 5). According to previous experiments, AUTAC-Biguanide was most effective after a 24 h treatment at 200 µM in KP4, with major disruption of the mitochondrial network. Cells were then treated during the same time with 100 µM (EC50) and 200 µM (2 x EC50) of compound 6. To evaluate the impact of the biguanide component on autophagy levels, two different concentrations of metformin were examined: 200 µM, to assess the effect of the biguanide ligand at a concentration equivalent to that of compound 6, and 2 mM, the concentration at which metformin demonstrates efficacy as an anticancer agent in these cells. For each of these metformin concentrations, it was observed that the level of LC3-II did not increase, indicating that the anticancer properties of metformin are not tied to its ability to induce autophagy (Figure 5A). At a concentration of 200 µM, AUTAC-Hexyl 7 demonstrated an ability to elevate LC3-II levels, underscoring the capacity of the AUTAC ligand to induce autophagy. However, it's important to note that AUTAC lacks anticancer properties and doesn't affect mitochondrial morphology at higher concentrations. Its participation in the autophagy process does not inherently lead to cytotoxicity, and it doesn't specifically target mitochondria. On the other hand, AUTAC-Biguanide showed a mild increase in LC3-II levels after treatment at 100 µM and 200 µM. This suggests that it retains the autophagy-inducing properties of the AUTAC ligand, but its conjugation with the biguanide function directs its effect towards mitochondria, potentially leading to mitophagy.
However, when studying the effects of a chemical agent on autophagy, the increase in LC3-II protein levels can result from either enhanced conversion of LC3-I to LC3-II (indicative of autophagy activation) or reduced elimination by the lysosome (indicative of lysosomal degradation inhibition). To determine whether compound 6 serves as an autophagy activator or an inhibitor of lysosomal degradation, the same experiment as described above was conducted in the presence of the autophagosome-lysosome fusion inhibitor, chloroquine (Figure 5B). The introduction of chloroquine to the medium led to an increase in LC3-II levels by inhibiting its degradation. A comparison between the medium without chloroquine and with chloroquine helped determine whether AUTAC-Biguanide exhibited similar properties to this inhibitor. If this were the case, the addition of chloroquine in its presence would have had little impact on LC3-II levels. Immunoblot results revealed a significant overexpression of LC3-II in the presence of chloroquine under all conditions, including when AUTAC-Biguanide was present. Therefore, it can be concluded that AUTAC-Biguanide does not affect the degradation of LC3B. These experiments align with the original hypothesis behind the design of AUTAC-Biguanide. AUTAC is specifically engineered to induce autophagy in cells through S-guanylation, thereby promoting the degradation of its target. The examination of LC3-II levels indeed suggests the activation of upstream autophagy. The incorporation of the biguanide component allows this degradation process to be selectively directed towards mitochondria, consequently inducing mitophagy.
3. Conclusions
AUTAC-Biguanide 6 demonstrates significant antiproliferative properties against cancer cells, leading to mitochondrial perturbations leading to a punctate mitochondrial network following treatment at the median effective concentration. Notably, the EC50 for AUTAC-Biguanide is significantly lower than that of metformin, underscoring its heightened antiproliferative activity against cancer cells.
Furthermore, AUTAC-Biguanide demonstrates notable selectivity for cancer cells compared to healthy cells. Consequently, this bifunctional molecule proves to be a superior anticancer agent compared to metformin. Moreover, the observed perturbations of mitochondrial network following treatment with AUTAC-Biguanide serves as compelling evidence that biguanidium salts effectively target these organelles and suggests that the biguanide function could be considered a mitochondria-targeting agent, akin to TPP group. While TPP is commonly employed to direct molecules towards mitochondria, it has the drawback of being highly apolar, making conjugation with excessively hydrophobic molecules a challenge due to resulting insolubility in biological media. The biguanide function, being highly polar, offers an alternative to TPP. Conjugation with hydrophobic molecules strikes a balance between hydrophilicity and hydrophobicity, ensuring both the diffusion of molecules across cell membranes and their solubility.
In contrast to the mito-AUTAC previously reported that improved cell viability,15 we present new biguanide-based AUTACs that are toxic to cancer cells and thus hold potential for anticancer therapies. The action of AUTACs depends on the presence of 8-nitro-cGMP, a metabolite produced from cGMP by the action of nitric oxide (NO) and ROS. Pancreatic cancer cells bearing KRAS mutations exhibit high levels of ROS and NO, suggesting why biguanide AUTACs can be toxic to these cells, which likely have elevated levels of 8-nitro-cGMP. It is also conceivable that the toxicity relies on the ability of biguanides to specifically target cancer mitochondria, but there is currently no data available to support this hypothesis.
The synthesis of this biguanide derivative, capable of inducing autophagy, has, therefore, paved the way for the development of a more potent anti-cancer agent.
Author Contributions
A.R.S and G.F. conceived the project, wrote funding grants and received funding for the project; J.V. performed all the experiments; V.G. supervised the biological experiments; J.V. wrote and V.B., G.F. and A.R.S edited the manuscript.
Conflicts of interest
There are no conflicts to declare.
Notes and references
Electronic Supplementary Information (ESI) available: General information for the synthetic procedures, synthetic procedures and characterization of the compounds, materials and methods for the in vitro experiments. See DOI: 10.1039/x0xx00000x.
References
- Bridges, H.R.; Sirviö, V.A.; Agip, A.N.; Hirst, J. BMC Biol. 2016, 14, 65–75.
- Parisotto, M.; Vuong-Robillard, N.; Kalegari, P.; Meharwade, T.; Joumier, L.; Igelmann, S.; Bourdeau, V.; Rowell, M.C.; Pollak, M.; Malleshaiah, M.; Schmitzer, A.R.; Ferbeyre, G. Cancers 2022, 14, 5597–5615.
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; Chandel, N.S. Elife 2014, 3, 02242– 02259.
- Fontaine, E. Front . Endocrinol. 2018, 9, 753–762. [Google Scholar]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. J. J . Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Cancer Metab. 2014, 2, 12–26.
- Murphy, M.P. , Biochim . Biophys. Acta., 2008, 1777, 1028–1031. [Google Scholar]
- Finichiu, P.G.; James, A.M.; Larsen, L.; Smith, R.A.; Murphy, M.P. J. J . Bioenerg. Biomembr. 2013, 45, 165–173. [Google Scholar] [CrossRef]
- Grytsai, O.; Myrgorodska, I.; Rocchi, S.; Ronco, C.; Benhida, R. Eur . J. Med. Chem. 2021, 224, 113726–113751. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Versini, A.; Sindikubwabo, F.; Belthier, G.; Niyomchon, S.; Pannequin, J.; Grimaud, L.; Cañeque, T.; Rodriguez, R. PLoS ONE 2018, 13, 0206764–0206780.
- Garber, K. Nat . Biotechnol. 2022, 40, 1709–1713. [Google Scholar]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. , Signal Transduction Targeted Ther . 2022, 7, 113–126. [Google Scholar] [PubMed]
- Ding, Y.; Fei, Y.; Lu, B. Trends Pharmacol . Sci. 2020, 41, 464–474. [Google Scholar]
- Whitmarsh-Everiss, T.; Laraia, L. Nat . Chem. Biol. 2021, 17, 653–664. [Google Scholar]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. Mol . Cell 2019, 76, 797–810. [Google Scholar]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. J. J . Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qin, G.; Yang, J.; Wang, W.; Zhang, W.; Lu, B.; Ren, J.; Qu, X. Chem . Sci. 2023, 14, 11192–11202. [Google Scholar]
- Arimoto, H. BMC Pharmacol . Toxicol. 2015, 16, 14–16. [Google Scholar]
- Ito, C.; Saito, Y.; Nozawa, T.; Fujii, S.; Sawa, T.; Inoue, H.; Matsunaga, T.; Khan, S.; Akashi, S.; Hashimoto, R.; Aikawa, C.; Takahashi, E.; Sagara, H.; Komatsu, M.; Tanaka, K.; Akaike, T.; Nakagawa, I.; Arimoto, H. Mol . Cell 2013, 52, 794–804. [Google Scholar]
- Richard, T.J.C.; Herzog, L.K.; Vornberger, J.; Rahmanto, A.S.; Sangfelt, O.; Salomons, F.A.; Dantuma, N.P. Sci . Rep. 2020, 10, 22334–22346. [Google Scholar]
- Ahmad, T.; Aggarwal, K.; Pattnaik, B.; Mukherjee, S.; Sethi, T.; Tiwari, B.K.; Kumar, M.; Micheal, A.; Mabalirajan, U.; Ghosh, B.; Roy, S.S.; Agrawal, A. Cell Death Dis. 2013, 4, 461–461.
- Karbowski, M.; Youle, R.J. Cell Death Differ. 2003, 10, 870–880.
- Chazotte, B. Cold Spring Harb . Protoc. 2011, 2011, 990–992. [Google Scholar]
- Zhang, J. Redox Biol. 2015, 4, 242–259.
- Tanida, I.; Ueno, T.; Kominami, E. In Autophagosome and Phagosome, ed. V. Deretic, Humana Press, Totowa, NJ, 2008; pp. 77–88. [CrossRef]
- Mizushima, N.; Yoshimori, T. Autophagy 2007, 3, 542–545.
- Tanida, I.; Waguri, S. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols, eds. P. Bross and N. Gregersen, Humana Press, Totowa, NJ, 2010; pp. 193–214. [CrossRef]
Figure 1.
Design and mechanism of AUTAC-Biguanide. (A) Mode of action of the AUTAC compound and the mechanism of autophagy. (B) S-guanylation and ubiquitination protein recognition mode for selective degradation by autophagy. (C) Design and structure of AUTAC-Biguanide.
Figure 1.
Design and mechanism of AUTAC-Biguanide. (A) Mode of action of the AUTAC compound and the mechanism of autophagy. (B) S-guanylation and ubiquitination protein recognition mode for selective degradation by autophagy. (C) Design and structure of AUTAC-Biguanide.
Scheme 1.
Synthesis of AUTAC-Biguanide and its control AUTAC-Hexyl. (A) Synthesis of intermediate 1. (B) Synthesis of AUTAC-Biguanide 6. (C) Synthesis of AUTAC-Hexyl 7.
Scheme 1.
Synthesis of AUTAC-Biguanide and its control AUTAC-Hexyl. (A) Synthesis of intermediate 1. (B) Synthesis of AUTAC-Biguanide 6. (C) Synthesis of AUTAC-Hexyl 7.
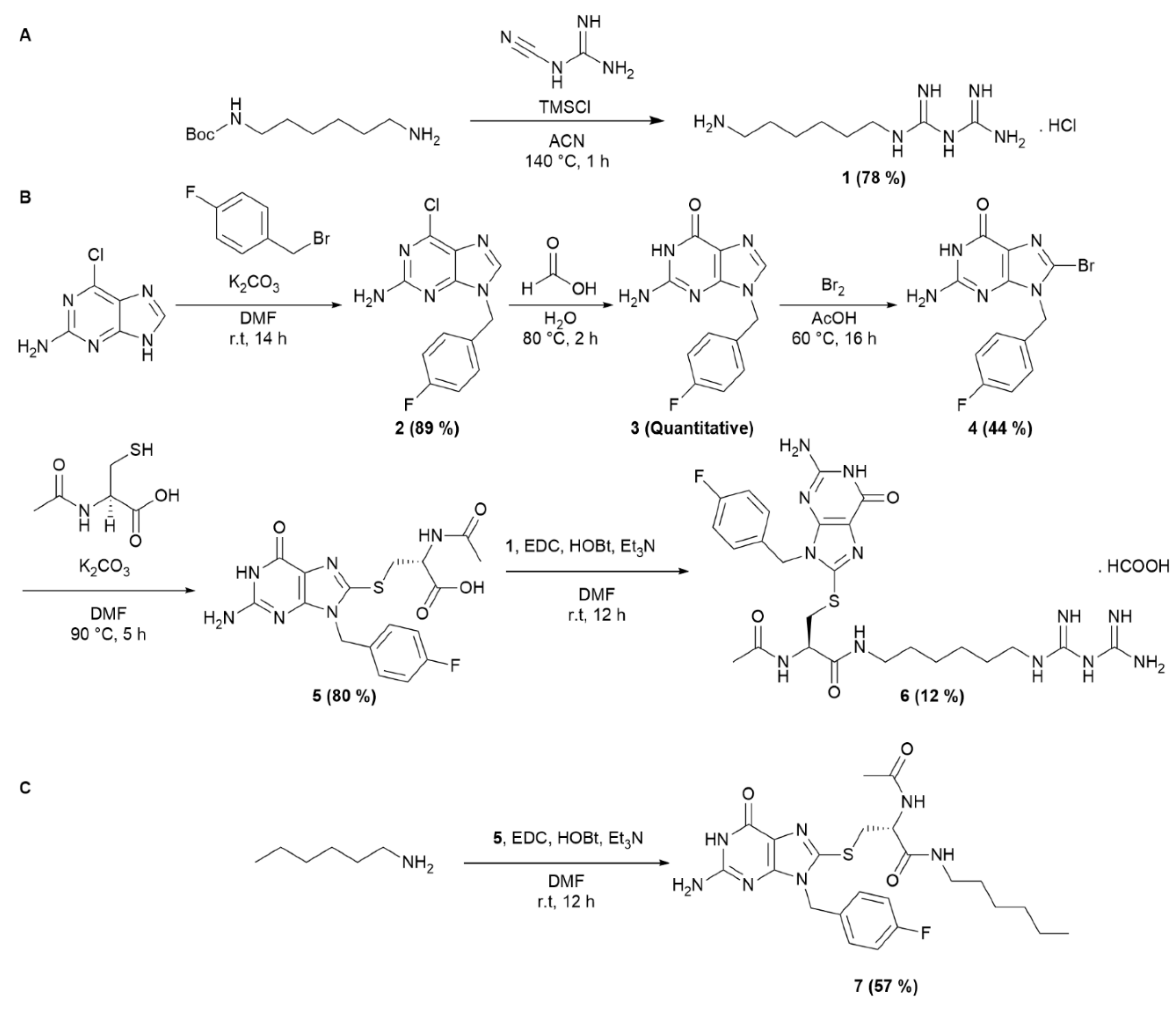
Figure 2.
Study of the effect of AUTAC-Biguanide 6 on different cell lines. (A) Effect of 6 on pancreatic cancer cells KP4 compared to metformin (B) Effect of 6 on the proliferation and growth of KP4 and PANC1 (cancer cells) and hTERT-HPNE and IMR90 (healthy cells), to assess its selectivity for cancer cells versus healthy cells.
Figure 2.
Study of the effect of AUTAC-Biguanide 6 on different cell lines. (A) Effect of 6 on pancreatic cancer cells KP4 compared to metformin (B) Effect of 6 on the proliferation and growth of KP4 and PANC1 (cancer cells) and hTERT-HPNE and IMR90 (healthy cells), to assess its selectivity for cancer cells versus healthy cells.
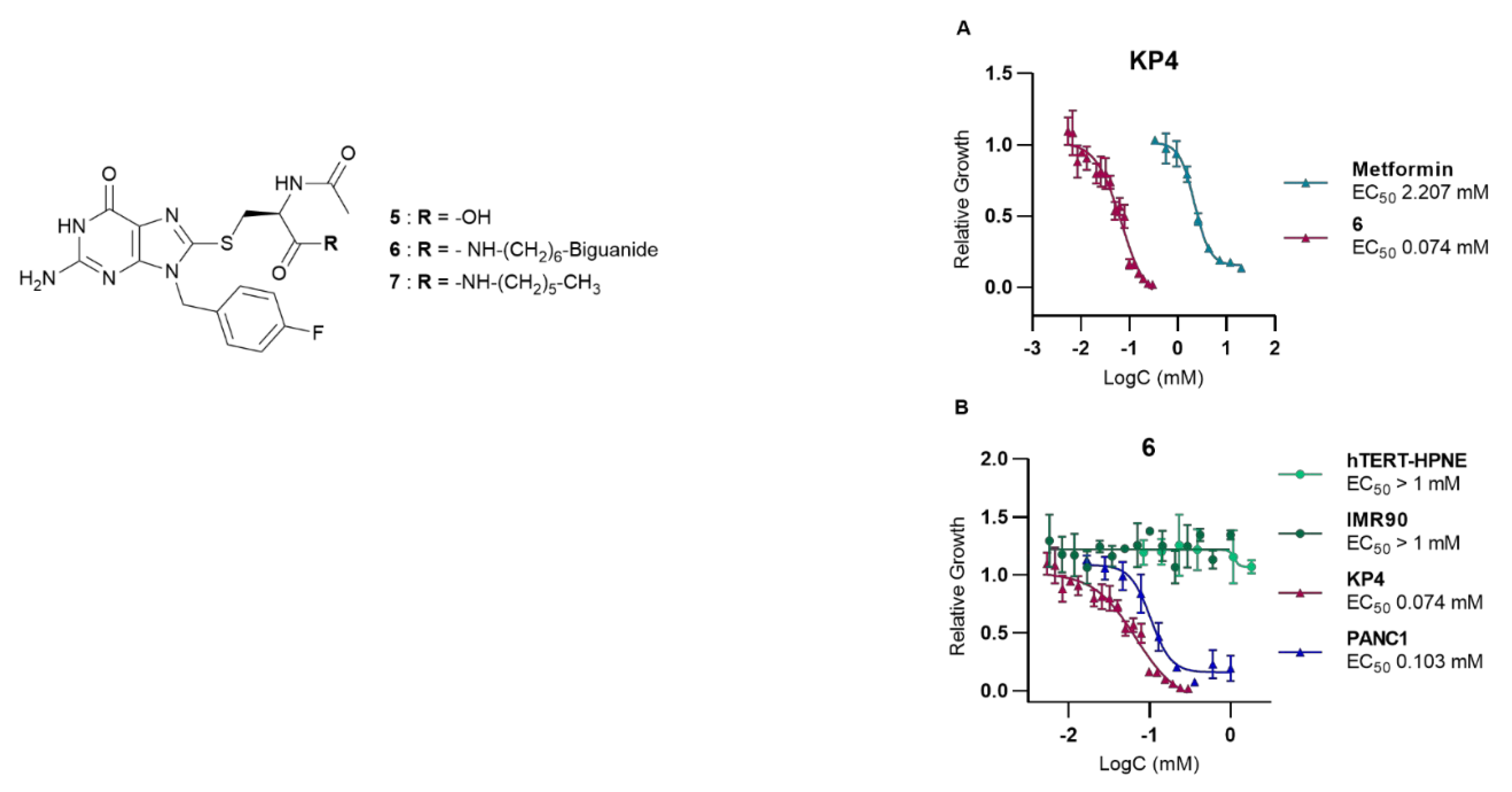
Figure 3.
Confocal microscopy images of KP4 pancreatic cancer cells following treatment with AUTAC-Biguanide 6 or controls. (A) Morphology of KP4 cells after 24 h of treatment with DMSO, metformin (2 mM), compounds 7 (1 mM) or 6 (50 μM, 100 μM and 200 μM) and staining of mitochondria with the Mitotracker® Deep Red FM (MTDR) fluorescent probe and the nuclei with the fluorescent dye DAPI; Cell morphology counts performed on a minimum of 100 cells per treatment (n > 2). (B) Observation of KP4 cells after 24 h of treatment with DMSO, metformin (2 mM) or compound 6 (200 μM) by immunofluorescence of the TOMM20 protein performed with the combination of antibodies against TOMM20 and an anti-mouse Alexa Fluor 488. The nuclei are stained with the fluorescent dye DAPI. Scale bars : 10 µm.
Figure 3.
Confocal microscopy images of KP4 pancreatic cancer cells following treatment with AUTAC-Biguanide 6 or controls. (A) Morphology of KP4 cells after 24 h of treatment with DMSO, metformin (2 mM), compounds 7 (1 mM) or 6 (50 μM, 100 μM and 200 μM) and staining of mitochondria with the Mitotracker® Deep Red FM (MTDR) fluorescent probe and the nuclei with the fluorescent dye DAPI; Cell morphology counts performed on a minimum of 100 cells per treatment (n > 2). (B) Observation of KP4 cells after 24 h of treatment with DMSO, metformin (2 mM) or compound 6 (200 μM) by immunofluorescence of the TOMM20 protein performed with the combination of antibodies against TOMM20 and an anti-mouse Alexa Fluor 488. The nuclei are stained with the fluorescent dye DAPI. Scale bars : 10 µm.
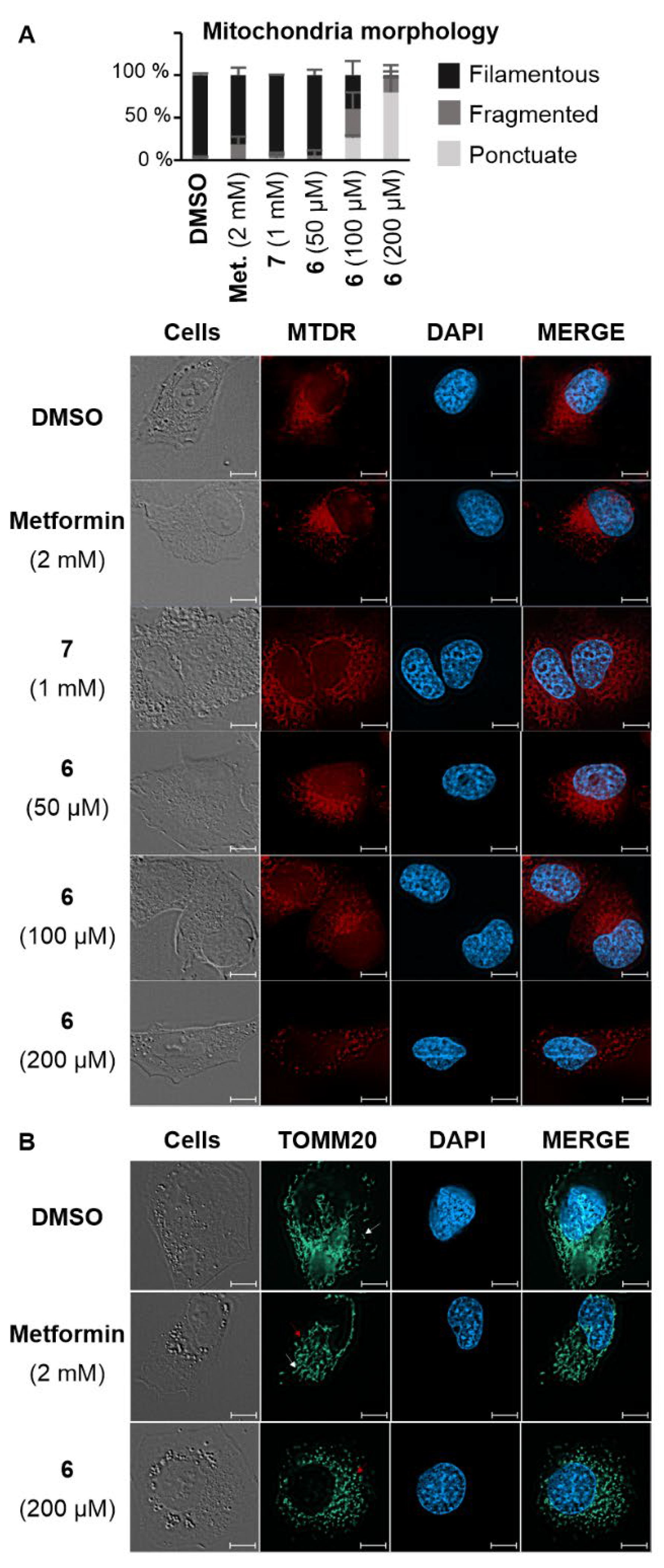
Figure 4.
Effect of AUTAC-Biguanide 6 and its controls on OXPHOS proteins levels in KP4 after 24 h of treatment and relative quantification of proteins levels for metformin and AUTAC-Biguanide (6); *p ≤ 0,05 ; **p ≤ 0.01 (ANOVA).
Figure 4.
Effect of AUTAC-Biguanide 6 and its controls on OXPHOS proteins levels in KP4 after 24 h of treatment and relative quantification of proteins levels for metformin and AUTAC-Biguanide (6); *p ≤ 0,05 ; **p ≤ 0.01 (ANOVA).
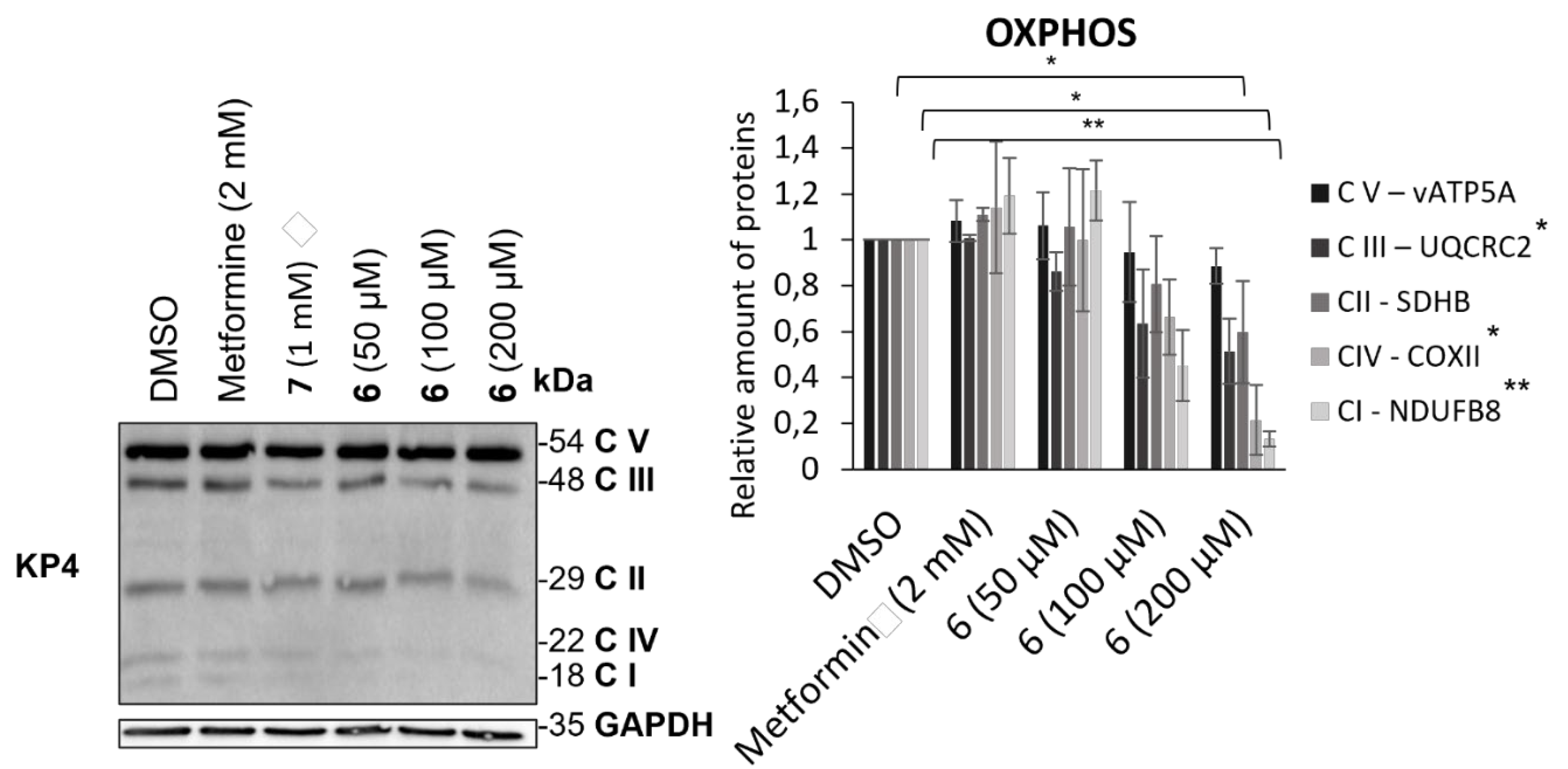
Figure 5.
Effect of AUTAC-Biguanide 6 and its controls on LC3-I and LC3-II protein levels. (A) Immunoblot for the study of LC3-I and LC3-II protein levels in KP4 after 24 h of treatment and relative quantification of protein levels for metformin (Met.), AUTAC-Hexyl (7) and AUTAC-Biguanide (6); *p ≤ 0.05; p ≤ 0.001 (ANOVA). (B) Effect of AUTAC-Biguanide 6 and its controls on LC3-I and LC3-II protein levels in the absence and presence of chloroquine; Immunoblot for the study of LC3-I and LC3-II protein levels in KP4, with and without chloroquine, after 24 h of treatment and relative quantification of protein levels for metformin, 7 and 6; **p ≤ 0.01; p ≤ 0.001 (ANOVA).
Figure 5.
Effect of AUTAC-Biguanide 6 and its controls on LC3-I and LC3-II protein levels. (A) Immunoblot for the study of LC3-I and LC3-II protein levels in KP4 after 24 h of treatment and relative quantification of protein levels for metformin (Met.), AUTAC-Hexyl (7) and AUTAC-Biguanide (6); *p ≤ 0.05; p ≤ 0.001 (ANOVA). (B) Effect of AUTAC-Biguanide 6 and its controls on LC3-I and LC3-II protein levels in the absence and presence of chloroquine; Immunoblot for the study of LC3-I and LC3-II protein levels in KP4, with and without chloroquine, after 24 h of treatment and relative quantification of protein levels for metformin, 7 and 6; **p ≤ 0.01; p ≤ 0.001 (ANOVA).
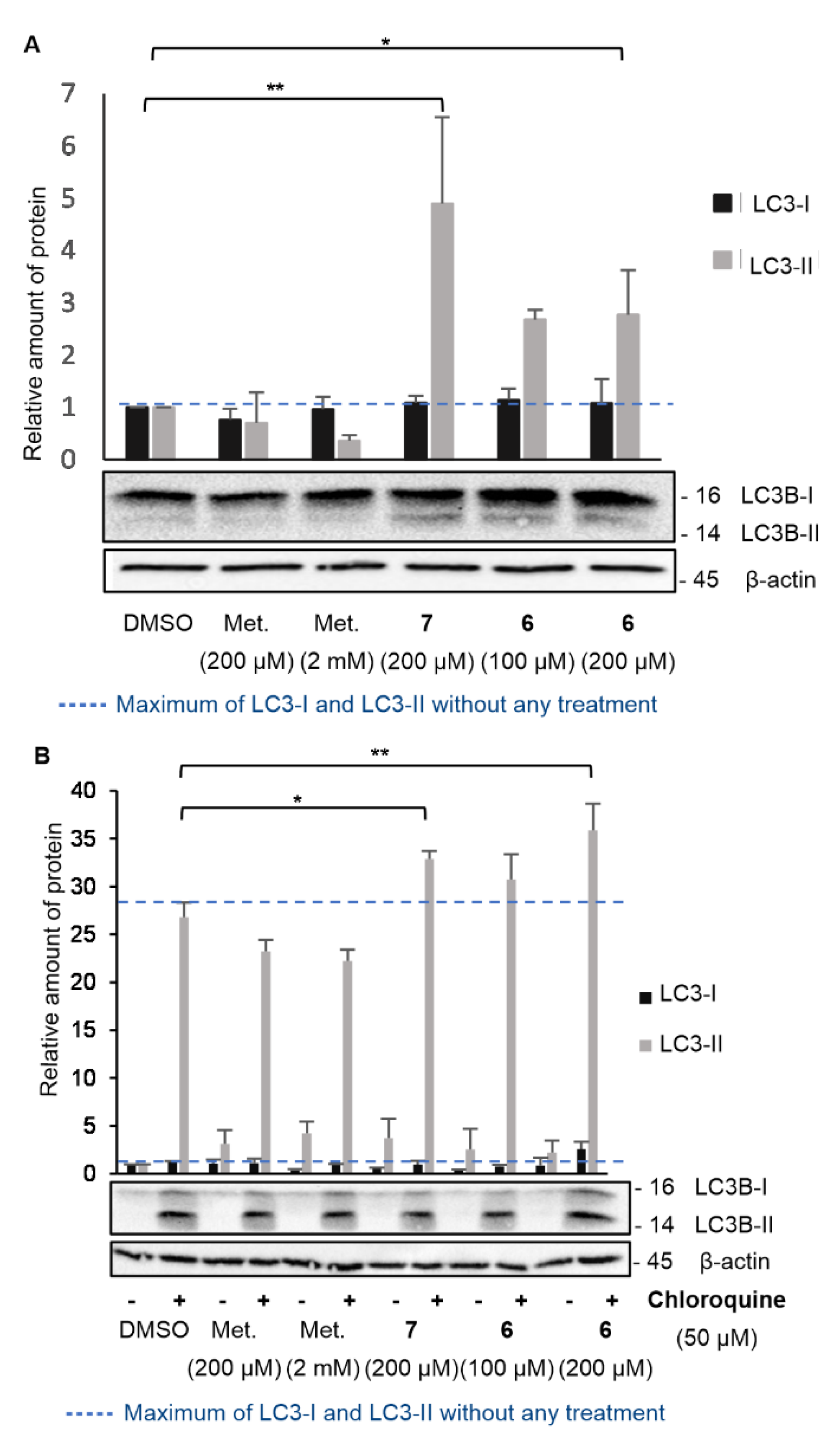

Table 1.
EC50 and selectivity index (SI) of AUTAC-Biguanide and its controls, determined after 72 h of treatment in different cell lines (mean ± SD).
Table 1.
EC50 and selectivity index (SI) of AUTAC-Biguanide and its controls, determined after 72 h of treatment in different cell lines (mean ± SD).
| Entry | EC50 (µM) | |||||
|---|---|---|---|---|---|---|
| KP4 | PANC1 | IMR90 | hTERT-HPNE | SI | ||
| Met.a | 1.49 ± 0.55 | 3.13 ± 1.41 | >20 | > 20 | >13 | |
| 5 | >5* | >5* | >5* | 3.51 ± 1.45 | - | |
| 6 | 0.10 ± 0.03 | 0.15 ± 0.03 | >1* | > 1 * | >10 | |
| 7 | >1* | >1* | >1* | 0.11 ± 0.02 | - | |
See section “Material and Methods” in Supporting Information. * Could not be determined due to solubility issues. a Met. = Metformin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



