Submitted:
15 July 2024
Posted:
15 July 2024
You are already at the latest version
Abstract
Nicotinamide is an important functional compound and, in the form of nicotinamide adenine dinucleotide (NAD), is used as a cofactor by protein-based enzymes to catalyze redox reactions. In the context of the RNA world hypothesis, it is therefore reasonable to assume that ancestral ribozymes could have used cofactors such as NAD or its simpler analog nicotinamide riboside (NAR) to catalyze redox reactions. The only described example of such an engineered ribozyme uses a nicotinamide moiety bound to the ribozyme through non-covalent interactions. Covalent attachment of NAR to RNA could be advantageous, but the demonstration of such scenarios to date has suffered from the chemical instability of both NAR and its reduced form NARH, making their use in oligonucleotide synthesis less straightforward. Here, we review the literature describing the chemical properties of the oxidized and reduced species of NAR, their synthesis, and previous attempts to incorporate either species into RNA. We discuss how to overcome the stability problem and succeed in generating RNA structures incorporating NAR.
Keywords:
Co-factor
; Nicotinamide ribonucleotide
; Redox reaction
; Ribozyme
; RNA
1. Introduction
In the early stages of life, RNA is thought to have been the sole actor responsible for all catalytic activities and the maintenance of genetic information. Hypothetically, a set of catalysts consisting only of RNA supported all the reactions required for the synthesis of single ribonucleotides, polynucleotides and other biomolecules [1,2]. At some point, regulation of these ribozyme activities was needed to adapt to fluctuations in, for example, metabolite concentrations or external signals [3,4,5]. This would require the binding of external regulators, which could be amino acids, small peptides or other molecules, for allosteric control of activity [1]. In addition, ribozymes may have required cofactors for function, as is known for many protein enzymes today. Many cofactors that occur in the modern cell are derivatives of ribonucleotides and are therefore considered remnants of the RNA world [6]. Interestingly, RNA is able to support the synthesis of the cofactors coenzyme A (CoA), nicotinamide dinucleotide (NAD) and flavine adenine dinucleotide (FAD). However, this occurs only at the final stage of their respective biosynthetic pathways, when the basic structure of the cofactor is already established [7]. It is therefore reasonable to postulate that these cofactors originated separately from RNA in early life through prebiotic chemistry and may have supported RNA function. Initially, ribozymes and cofactors may have formed a complex through non-covalent interactions or through a covalent linkage at the 5' or 3' terminus of the ribozyme, possibly giving the ribozyme an evolutionary advantage through extended or simply better functional performance [8,9].
A number of laboratory-developed artificial ribozymes have been created that require a cofactor for activity. One such example is a ribozyme that utilizes covalently bound β-nicotinamide mononucleotide for RNA ligation, a cofactor that has been shown to arise under prebiotic conditions [10,11,12,13]. In modern biological systems, nicotinamide remains an important functional compound, and in the form of NAD, is used as a cofactor by protein-based enzymes for catalyzing redox reactions [14]. Furthermore, the cofactors pyrrolochinolinchinone (PQQ), NAD, and FAD have been demonstrated to exhibit redox activity when complexed with a DNA aptamer [15]. It is therefore reasonable to hypothesize that primordial ribozymes could have utilized cofactors such as NAD to catalyze redox reactions. Indeed, through in vitro selection, a ribozyme, designated Ribox02, was successfully generated with an activity analogous to that of an alcohol dehydrogenase (ADH) [16,17].
Given their role as a crucial mechanism in prebiotic catalysis, redox reactions supported by ribozymes in conjunction with co-factors such as NAD appear to be a highly plausible avenue of investigation. There are numerous possibilities for the design of redox-active ribozymes. In addition to the non-covalent interaction of the co-factor with the ribozyme, the covalent attachment of the redox cofactor to the ribozyme as a prosthetic group represents a compelling proposition. This could result in the creation of more functional ribozymes that are capable of mimicking their respective enzyme counterparts in modern biochemistry. The gold standard for synthesizing RNA incorporating modified nucleotides remains the phosphoramidite method. Consequently, it is of great interest to ascertain whether the particular cofactor is compatible with this chemistry. This review will focus on nicotinamide riboside (NAR) as a redox cofactor and address the question of whether it is possible to chemically incorporate it into RNA structures (Figure 1). Attaining this goal would provide further insight into the potential functions of RNA redox catalysis during the early stages of the RNA world. Furthermore, it would facilitate the development of redox ribozymes.
2. Stability of Nicotinamide Riboside
The thermal stability of the oxidized and reduced species, namely NAR and NARH, at elevated temperatures rarely presents a challenge during chemical synthesis. Nevertheless, both forms are susceptible to degradation by nucleophiles in either acidic or alkaline environments, whereas they exhibit tolerable stability under neutral or physiological conditions.
2.1. Nucleophilic Attack On The Anomeric Carbon And The Pyridinium Moiety Leads To Decomposition Of Nicotinamide Riboside Under Alkaline Conditions
Nicotinamide riboside (NAR) exhibits remarkable stability in acidic media [18], but due to its intrinsically cationic nature, it is labile under neutral and alkaline conditions by various degradation routes [19], which for simplicity have been divided into two major pathways (Figure 2).
The first pathway entails the cleavage of the glycosidic bond, which occurs because the pyridinium moiety functions as an excellent leaving group in the presence of suitable nucleophiles, such as hydroxide anions. This leads to the formation of nicotinamide and D-ribose. The glycosidic bond cleavage is influenced by temperature, with an increase of approximately 10 °C resulting in a doubling of the rate. The reaction is facilitated by the assistance of the sugar moiety, which forms an oxocarbenium intermediate [19]. It can be reasonably deduced that this process could be effectively suppressed by replacing the N-glycosidic linkage with a more stable C-C linkage. This could be achieved by employing a C-nucleoside analogue of NAR. Nevertheless, it may be a synthetically rather challenging process. In addition, the inductive effect of substituents in 2'-substituted nicotinamide arabinosides (Figure 3; 21) [20] was analyzed, supporting a dissociative mechanism involving a cationic intermediate and suggesting that electron withdrawing substituents retard the rate of anomeric displacement by destabilizing the oxocarbenium ion. The other degradation pathway is based on the fact that the pyridinium ring is susceptible to degradation by the attack of various C-, N-, O- and S-nucleophiles (for example cyanide, alkoxide/hydroxide ions, hydroxylamines, sulfides etc.) on carbon 2/6 and 4 [21]. It has been reported that polarizable ions, such as cyanide, iodide, sulfide, and dithionite preferentially give 1,4-addition products, whereas less polarizable ions, such as hydroxide, alkoxide, or hydride give 1,2- and 1,6-addition products [22]. This is consistent with the observed regioselectivity of dithionite-induced reduction, which mainly yields 1,4-dihydropyridine, whereas borohydride preferentially yields 1,6- and 1,2- dihydropyridine [23]. Subsequent reactions may follow the nucleophilic attack, which typically involves the opening and re-closure of the ring, to generate various degradation products [24,25]. However, this is not a cause for concern in general, given that these reactions are reversible.
It is noteworthy that under strong alkaline conditions (above pH 12.5), the nucleophilic attack of hydroxide ions on the pyridinium ring dominates over the above-mentioned glycosidic hydrolysis. The process is initiated by a sophisticated mechanism, which ultimately results in the formation of 2-pyridone-3-carbaldehyde as the primary product. This may subsequently degrade into various species [19,26]. It is noteworthy that stable adducts of NAR have also been described, which can be reconverted under appropriate conditions. Consequently, stable NAR adducts may prove a valuable alternative to the challenging cationic NAR or NARH in chemical synthesis strategies. For instance, the cyanide adduct of NAR monophosphate (NAR-CN) was employed for the enzymatic synthesis of NAR-CN-containing oligonucleotides, which were subsequently transformed into poly-NAR oligonucleotides by incubation in aqueous buffer. In contrast, the utilization of the parent nucleotide, NARH monophosphate, yielded markedly inferior results. Nevertheless, from a modern perspective, the analytical characterization appears to be insufficient and at times less convincing [27].
2.2 . Dihydronicotinamide Riboside (NARH) Is Prone To Degradation By Acid-Catalyzed Addition And Nucleophilic Displacement Of The Dihydropyridine Moiety In Alkaline Media
As anticipated, 1,4-dihydronicotinamide riboside is readily oxidized by a plethora of oxidants, including iodine, oxygen, selenium dioxide, permanganate and various nitrates, amongst others [28,29,30,31,32]. In contrast to the well-known fact that NARH will slowly re-oxidize to NAR if exposed to humid air, its tendency for isomerization to 1,2- and 1,6-dihydronicotinamide riboside under these conditions is rather unknown. Nevertheless, following an extended period of incubation, an initially pure sample of NARH may exhibit a minimal presence of these isomers, up to 4%. These differ widely in stability compared to the parent compound and are potent inhibitors for NAD(P)H-dependent oxidoreductases, indicating their antagonistic role in maintaining cellular redox homeostasis [23]. Both the 1,2-isomer and the 1,6-isomer decompose rapidly, as previously noted [29]. Similarly, NARH is also susceptible to dissociative degradation under alkaline conditions, as previously discussed in the context of NAR. However, the latter is undoubtedly more susceptible to hydrolysis in terms of the leaving group quality. Nevertheless, NARH also undergoes significant degradation under aqueous-alkaline conditions, forming dihydronicotinamide and D-ribose by nucleophilic attack of hydroxide on the anomeric carbon [33] (Figure 4). It is essential to consider this side reaction when adjusting the pH in an experiment. It is notable that this presents a challenge for the chemical reduction of NAR with dithionite, a standard procedure for the synthesis of 1,4-NARH. Even a slight increase in pH (from 8.1 to 8.5) has been observed to increase dihydronicotinamide dissociation, a finding that is evident from a mechanistic perspective. During the course of the reaction, the sulfinate intermediate is relatively stable under alkaline conditions. However, it eventually requires protonation in order to form NARH. Consequently, an increase in pH hinders the nucleophilic replacement at the anomeric carbon by simultaneously formed anions (i.e. thiosulfate, bisulfite), which are more nucleophilic than hydroxide. Consequently, the reduction with dithionite indirectly promotes the hydrolysis of the glycosidic bond, prolongs the reaction time and leads to the accumulation of the 1,6-isomer [33]. In particular, the vinylamine structure of the dihydropyridine ring defines its stability and reactivity. Consequently, the enamine carbon atom is susceptible to electrophilic attack, whereas the carbonyl-analogous position undergoes nucleophilic addition [34]. The presence of electron-donating substituents attached to the enamine nitrogen or enamine carbon, respectively, has been demonstrated to facilitate this reaction [35]. It is well established that 1,4-dihydropyridines degrade under aqueous conditions by hydration of the 2,3-double bond. In contrast, the 5,6-double bond appears to be either unaffected or less reactive. This may be due to the presence of the electron-withdrawing carbamoyl group, which disfavors the initial protonation of the 3 position. NARH, as the representative structural moiety of the genuine cofactors NAD(P)H, is rapidly hydrated to form 2-hydroxy-1,4,5,6-tetrahydronicotinamide riboside (2OH-THNAR), in acidic and neutral media (Figure 4). The acid-catalyzed nucleophilic addition is typically accompanied by a characteristic hypochromic shift of the absorption maximum, from 340 nm to 290 nm [23,36]. This ultimately restricts the range of viable reaction conditions, with implications for biochemical applications, particularly in the context of buffer composition. In practice, however, this is not a significant concern because under physiological conditions, this side reaction is relatively slow and negligible during measurements. Indeed, stability studies have demonstrated that complete degradation occurs immediately at low pH, but is tolerable under physiological or alkaline conditions. Moreover, the integrity of NARH remains intact even after prolonged incubation [33]. Although previously known, the 2'-hydroxyl group of the sugar moiety has been overlooked in recent years. This group is also capable of undergoing acid-catalyzed enamine addition, which leads to the formation of cTHNAR. Due to its ribo configuration, this process is only possible following ring opening, passing through an imine intermediate and subsequent formation of the alpha anomer. The cyclisation process may compete with the hydration of the beta anomer (formation of 2OH-THNAR), depending on the buffer substance, concentration and pH. At low pH (pH < 3), anomerization occurs at a faster rate than hydration, resulting in the irreversible formation of cTHNAR [37]. In contrast, in neutral or alkaline media, hydration is the predominant process. It is noteworthy that hydration occurs at comparable rates in the majority of solvents and buffer systems, with the exception of the presence of several ions, such as acetate [33] or phosphate, where hydration/addition of the 2’-hydroxyl group is significantly faster [38]. Although the synergistic buffer effect of ions on the hydration of NARH is well established, the underlying mechanism has not yet been definitively elucidated. Furthermore, the formation of additional degradation products under acidic conditions has been described in the literature [39,40]. Nevertheless, these equilibrium reactions in buffers and solvents appear to be insignificant in comparison to the primary acid modification (i.e. the formation of 2OH-THNAR and cTHNAR) (Figure 4).
Over the years, various attempts have been made to overcome this problem through the synthesis of derivatives and mimetics. For example, the hydration of the dihydropyridine ring can be effectively reduced by substituents. Electron donating substituents generally increase acid lability, while electron withdrawing groups suppress acid catalyzed degradation, but may result in base labile compounds. Substituents on ring carbons or annulated aromatic rings that act as a permanent protective group of the 2,3-double bond are usually accompanied by drastic changes in the redox potential and may lead to loss of function, as observed in enzymatic studies [41,42]. Furthermore, the modification of the 5-substituent, which affects the reactivity of the dihydronicotinamide moiety through mesomeric effects, will have an impact on the overall stability (Figure 5). Consequently, N-benzyl dihydronicotinamide derivatives with variations of the carbonyl group were synthesized and the rate of the primary reaction with acid was determined. In comparison to nicotinamide, any substituent that decreases the thermodynamic stability of the conjugated acid intermediate retards the rate of acid catalyzed degradation and thus increases the stability against hydration [43].
It was also shown that, similar to the adenosyl residue in NAD(P)H, an aromatic substituent attached to the ring nitrogen can improve stability through π-π stacking interactions. It has been reported that biomimetic cofactors of NAD containing a phenyl ring with alkyl chains of varying lengths exhibit differences in pH and temperature stability. The mimetic containing an ethyl chain demonstrated stability comparable to NADH at elevated temperatures and low pH. In contrast, derivatives with shorter or longer alkyl chains were less stable due to unfavorable molecular geometry or non-ideal stacking distance [44]. Recently, the carbocyclic derivative carbo NADH, which has a cyclopentane ring instead of ribofuranose, was shown to be almost unaffected by hydration under acidic, neutral and alkaline conditions (pH 3 - 10.5). It also has increased thermal stability compared to NADH and is not susceptible to buffer-assisted hydration [45]. The increased stability against nucleophilic substitution of the heterocycle is intuitively understandable for both the oxidized and reduced forms, since, unlike the glycosidic acetal bond, the dissociation of the heterocycle is not supported by neighboring group effects. Nevertheless, the improved hydration stability of the reduced species is truly remarkable. However, the underlying structure-reactivity relationship that gives rise to this extraordinary stability is still under investigation.
3. Synthesis of Nicotinamide Riboside
3.1. Synthetic Paths To Nicotinamide Riboside
Due to the stability issues of NAR discussed above, incorporation of the reduced form, NARH, into RNA followed by oxidation may be a possible way to provide the redox cofactor within and covalently bound to an RNA structure. To do this, NARH needs to be synthesized followed by decoration with the typical functionalities to convert it into a phosphoramidite building block. One way to synthesize the nucleoside and its derivatives is to glycosylate nicotinamide with a suitable ribose derivative. Early methods include the use of O-acyl-protected halosugars (Figure 6a). These mostly tri-O-benzoyl or -acetyl halo ribofuranosides can then be condensed with nicotinamide to give the corresponding tri-acyl nicotinamide nucleoside, which can be deprotected using methanolic ammonia, whether the reduced or oxidized species [46,47]. The selectivity of glycosylation tends to yield the desired β-anomer with approximately 90% yield, while yields of up to 5% of the α-anomer have been reported [48,49]. The reason for the stereoselectivity has been attributed to the formation of an intermediate 1,2-acyloxonium sugar, which is formed by anchimeric assistance of the 2-O-acyl group [50]. Without the acyl groups, the resulting nucleoside would be racemic.
Another strategy is to use a Lewis acid such as trimethylsilyltrifluoromethane sulfonate (TMSOTf) to catalyze glycosylation (Figure 6b). Here, TMSOTf reacts with the pearcetylated ribose to form the 1,2-acyloxonium sugar mentioned above. This facilitates the condensation of the sugar with nicotinamide to give triacetyl nicotinamide riboside. As with the previous strategy, the β-anomer is the preferred product, but it is reported that approximately 13% of the α-anomer is formed [51]. In order to suppress the appearance of this unwanted by-product, an optimized version of this reaction has been developed. The key element of this procedure is the silylation of nicotinamide with trimethylsilyl chloride and the use of catalytic amounts of TMSOTf in the subsequent coupling with peracetylated ribose. Under these controlled conditions, the beta-anomer appears as the sole product [52].
Instead of using nicotinamide, ethyl or methyl nicotinate can be condensed with tetraacetyl ribose in the presence of TMSOTf. The resulting cationic nicotinate ester can then be treated with methanolic ammonia, resulting in simultaneous deacetylation of the hydroxy groups and ester to amide conversion to give beta-nicotinamide riboside [53,54].
The Zincke reaction is another possibility for the chemical synthesis of nicotinamide riboside. Nicotinamide, a pyridine derivative, can react with an electron-poor aromatic compound such as 2,4-dinitrochlorobenzene to form the Zincke salt (Figure 6c). This salt can then be condensed with 1-amino sugars to form the corresponding nicotinamide nucleoside. It is noteworthy that the absolute stereoconfiguration of the anomeric carbon remains the same, since the nucleobase is generated from the sugar moiety having a defined stereoconfiguration [55].
Reduction of the pyridinium moiety can be carried out with or without protection of the hydroxyl groups. However, if the reaction is carried out in an aqueous system, NARH can be extracted with an organic solvent as long as the hydroxyl groups are acylated. This greatly facilitates purification after reduction. The general procedure is to dissolve triacetylated nicotinamide riboside in dichloromethane and mix it with an aqueous solution of NaHCO3 and Na2S2O4. After a few hours the organic layer containing the reduced nucleoside can be separated. After removing the acetyl groups with methanol and K2CO3, NARH is obtained [56]. As mentioned above the choice of the reducing agent is crucial for this reaction, as three different isomers of NARH can be formed under different conditions. The only stable and biologically relevant isomer, 1,4-dihydronicotinamide, is the main product when sodium dithionite is used as the reducing agent [57]. This regioselectivity is due to the particular orientation of the dithionite anion to the pyridinium salt, which determines the position of attack as carbon 4. It is supported by the distance between the two nucleophilic oxygen atoms of dithionite, which is very similar to the interatomic distance between the ring nitrogen and the electrophilic C4 atom [58]. Interestingly, when performing the dithionite-mediated reduction with NAR mimetics this regioselectivity disappears [59]. Moreover, if a stronger reducing agent such as NaBH4 is used, a mixture of the 1,2-, 1,4-, and 1,6-isomers was obtained in a ratio of 1:0.44:1 [57]. The hydride anion has the potential to attack any of the three carbon atoms in the pyridinium ring. However, selectivity is observed towards the 1,2- and 1,6-isomers due to their weak polarizability, making them more favorable targets for the hydride anion compared to carbon 4.
3.2. Synthetic Modification of the Structure of the Ribose Moiety
As discussed in Chapter 2, the limited stability of nicotinamide riboside is a consequence of its readily cleavable glycosidic bond. This must be taken into account when attempting to convert the nucleoside derivative into a phosphoramidite building block for incorporation into RNA by solid phase synthesis. Functional group protection and phosphoramidite synthesis involve multiple chemical reaction steps that are prone to degradation of NAR. To circumvent this problem, several NAD mimetics with different properties have been developed.
It has already been shown that the alternative nucleoside-structured carbocyclic NAR has greatly increased stability compared to natural NAR at various pH values. Therefore, carbocyclic nicotinamide (Figure 3; 19) [60] can be used to maintain the general idea of incorporating nicotinamide as a nucleoside into RNA via solid phase synthesis. The carbocycles of the canonical bases have previously been shown to be functionalized as phosphoramidites and incorporated into RNA via solid phase synthesis [61]. Carbocyclic sugar analoges (Figure 3; 16, 17) [62,63] are synthesized from (R)-Vince lactam, which is first converted to amino carba-ribose. It can then react with the corresponding nicotinamide Zincke salt to form nicotinamide carba-riboside (Figure 7a) [60].
The same lactam can also be used to prepare carbocyclic deoxyribonucleosides [62]. The reaction pathway slightly differs from that of the ribonucleosides resulting in a different precursor. However, the final step also involves the aforementioned Zincke reaction. Recently, the chemoenzymatic replacement of the endocyclic ribose oxygen with a less electronegative sulfur has been achieved, resulting in nicotinamide 4'-thioriboside (Figure 3; 18) [64], with reduced reactivity of the N-glycosidic linkage while maintaining the riboside geometry.
Another modification that can be made to the ribose structure is to change from the typical ring sugar to an acyclic sugar, accessible by simple nucleophilic substitution from the corresponding halide (Figure 3; 1-8) [65,66,67,68,69,70,71,72]. The absence of the 2’- and 3’-hydroxy groups facilitates the functionalization of the nucleoside for use in chemical RNA synthesis. For example, a prototypical synthesis of an acyclic nucleoside involves the reaction of a linear halide with nicotinamide. The acyclic sugar analog can be synthesized by acetolysis of 1,3-dioxolane with acetyl bromide. The resulting alkyl halide can then be reacted with nicotinamide to give acetylated pyridinium bromide. Subsequent deacetylation in methanolic ammonia yields the acyclic nicotinamide riboside (Figure 7b; Figure 3; 4) [68,73].
3.3. Synthetic Modifications Of The Nicotinamide Base
In addition to structural modifications of the ribose sugar, the glycosidic linkage can also be strengthened by altering the nicotinamide moiety. One promising option is to replace the N-glycosidic bond with a C-glycosidic bond, which significantly increases its stability. There are a number of ways to make such a carbon link, and pyridine C-nucleosides have already been synthesized. One way is to use a fully protected ribonolactone which reacts with lithiopyridine to give the corresponding hemiacetal (Figure 8). This intermediate can then be reduced to the corresponding C-ribonucleoside [74].
Other methods include Friedel-Crafts glycosylation with aromatic compounds and acetylfuranoses, photoredox coupling of furanosyl acids with aryl bromides, and nucleophilic additions [75,76,77,78,79]. For the synthesis of the nicotinamide C-nucleoside, it is important to choose a pyridine derivative with a functional group in position 3 that can be converted to a carbamoyl group, such as bromine or nitrile [74,80,81]. In addition, the ring nitrogen of the C-nucleoside must be alkylated, for example with methyl iodide, to exhibit redox activity similar to that of N-glycosidic NAR [80].
4. Attempts to Incorporate Nicotinamide Riboside in RNA
4.1. Synthesis of NAR/NARH Building Blocks
Obviously, the site-specific incorporation of nicotinamide riboside into RNA is by no means trivial. To date, attachment to the 5'- and 3'-terminus has been reported, but solid-phase synthesis (SPS) approaches for the synthesis of nicotinamide-containing oligoribonucleotides (NCRs) have not yet been demonstrated. Efforts to modify the 5'-terminus have been driven primarily by studies of the so-called 5'-NAD cap. These studies provide access to ligation of nicotinamide mononucleotide (NMN) to the 5'-terminus of oligonucleotides via a pyrophosphate linkage instead of the natural phosphodiester bond, using a large excess of activated intermediates such as NMN-imidazolide (Figure 3; 11, 12) [82,83]. Among these NMN amidates, conjugation via the activated nicotinamide carbamoyl group is feasible (Figure 3; 9, 10) [84] and has been used several times as a straightforward approach for the construction of quite intriguing redox relays [85]. In addition, several azido- or alkyne-modified nicotinamide derivatives (Figure 3; 13, 14, 15) [86,87,88] are described, suggesting conjugation via alkyne-azide cycloaddition or Staudinger ligation, respectively. However, both approaches, active ester and click chemistry, are of little avail for NCR synthesis, when structural similarity to canonical incorporated nucleotides is demanded. Transcriptional priming with excess NAD or tethering of NMN by an in vitro selected variant of the hairpin ribozyme catalyzing the addition of NAD-2′,3′-cyclic phosphate has also been described [89]. Göckel and Richert reported primer extension with NMN-imidazolide, which is attached to the 3'-terminus via a phosphoramidate linkage. This linkage is isoelectronic to the natural phosphodiester bond, although it exhibits slightly altered stereoelectronic properties [83,90]. Decades ago, the enzymatic de-novo polymerisation of NMN and primer extension using the di- and triphosphates of reduced NMN (NARH-5'-monophosphate) and NMN-CN were reported to be catalyzed by polynucleotide phosphorylase [91]. While the reduced species and the cyanide adduct were found to be suitable substrates, the oxidized form was less efficient, but instead was successfully used to extend a DNA primer by one or two nucleotides in the presence of terminal deoxyribonucleotide transferase [91].
Although it has been claimed that the lability of NAR and NARH towards bases or acids precludes the SPS of NCRs, there is as yet no convincing experimental evidence to support this claim. Instead, we argue the opposite, as preliminary results from our laboratory suggest that the synthesis of NCRs using the phosphoramidite approach could be feasible through sophisticated processing. This is further supported by the fact that key compounds such as 5'-O-trityl protected NAR (Figure 3; 22) [92] and NARH phosphoramidite (Figure 3; 23) [93] have been described previously. Nevertheless, we would like to point out that the attempts to incorporate NAR or NARH into oligonucleotides present several synthetic challenges. Based on our own observations, we can confirm that the synthesis of phosphoramidite building blocks starting from commercially available NAR or NARH is associated with serious problems, as the conditions required for the incorporation of protective groups lead to side reactions and decomposition of NAR or NARH due to their inherent instability. The key to the synthesis of such building blocks is the formation of the glycosidic bond by the Zincke reaction using an amino sugar with the required 5'-O-trityl and 2'-O-protective groups [92]. For this approach, the use of a base-stable 2'-O-protecting group, such as the triisopropylysilyloxymethyl (TOM) group, rather than the commonly used tert-butyl dimethylsilyl (tBDMS) group, is mandatory, as silyl group migration is certainly inevitable under these strong basic conditions. However, in view of the above-mentioned degradation pathways of NARH, it would be reasonable to carry out such a synthesis with a 2'-deoxyamino sugar or the corresponding carbo cycle (Figure 3; 16, 20) [20,62], starting from the Vince lactam (Figure 10) [63]. Finally, the phosphoramidite group (i.e. the CED group) is introduced. This approach should also be applicable to the synthesis of building blocks for the H-phosphonate or phosphotriester method.
4.2. Application of a NAR/NARH-Phosphoramidite
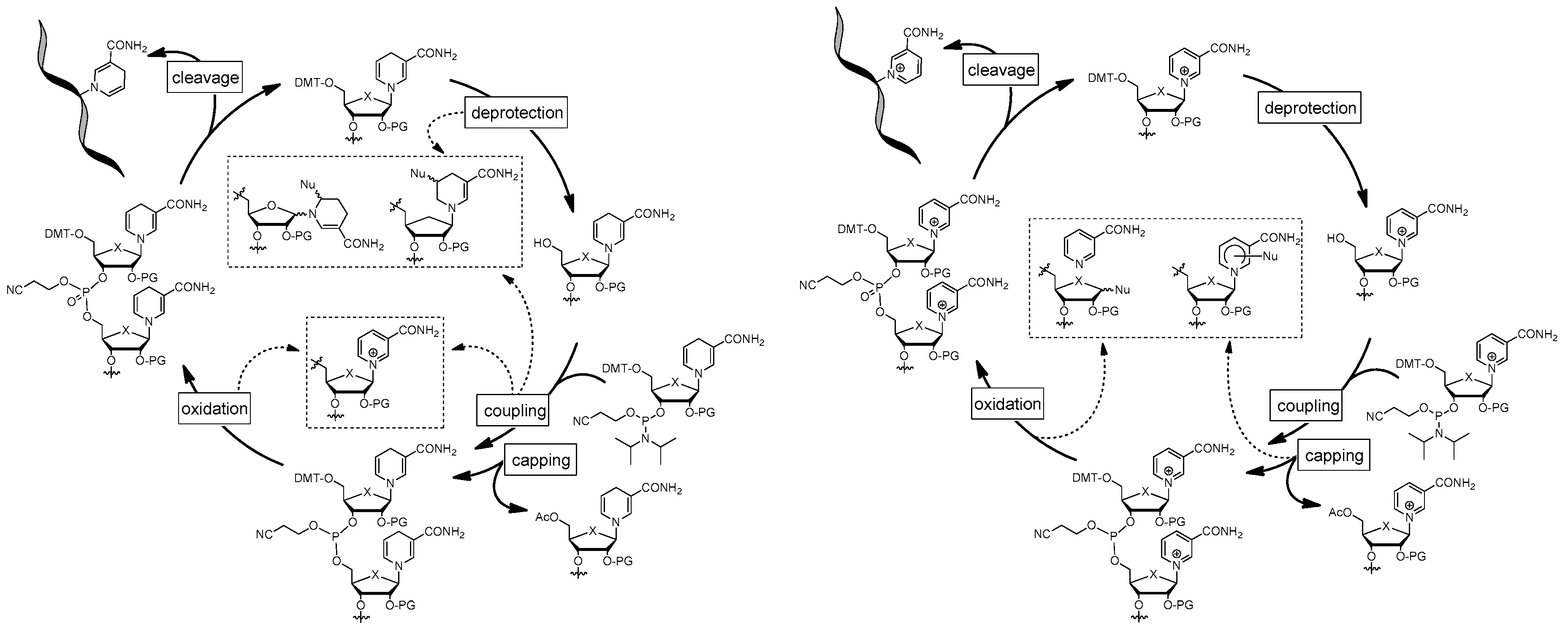
Hypothetically, when using a NAR phosphoramidite in oligonucleotide synthesis, we predict that the heterocycle will be unaffected during detritylation and coupling, as it is stable under acidic conditions. In contrast, reagents that require basic processing (i.e. capping and oxidation) could lead to degradation. However, stability studies have shown that even after prolonged incubation of NAR with the oxidation reagent (i.e. iodine), no reaction or degradation was observed in the solvents used (ACN, trimethylpyridine, water) (unpublished data). Decomposition as a result of capping is not to be expected either, since although the process is carried out under basic conditions, only poor nucleophiles are present (Figure 11). Therefore, the use of a NAR phosphoramidite seems to be feasible. However, an effect on the solubility of the phosphoramidite is to be expected due to the cationic nature of the nicotinamide moiety. This may be an issue during SPS, but should be easily overcome by the non-polar protecting groups present in a fully functionalized phosphoramidite. Nevertheless, the extent to which the cationic pyridinium moiety influences subsequent coupling after its incorporation and partial deprotection is still unexplored. However, it is known that the nucleophilicity of the 5'-hydroxyl group is reduced compared to canonical nucleosides due to electrostatic interactions with the pyridinium moiety [94]. This would adversely affect the coupling efficiency of the subsequent building blocks, but could possibly be compensated by prolonged coupling times and/or multiple coupling cycles. However, the fact that 5’-silylation of NAR with sterically hindered triisopropylsilyl chloride (Figure 3; 24) [95] and subsequent deprotection was achieved suggests a promising outcome during the coupling step and also indicates compatibility of NAR with silicon-based protection group chemistry.
When using NARH phosphoramidites, it is expected that the dihydropyridine moiety will be immediately reconverted to the pyridinium species (i.e. NAR) during the oxidation step. Interestingly, we observed that NARH is indeed oxidized in quantitative yield, but no further degradation of the reduced or oxidized species was observed (unpublished data). Again, the oxidation of the dihydropyridine moiety is likely to be negligible as long as the coupling of the next building block is not affected. Alternatively, chemoselective oxidation of the phosphite could be carried out with hydrogen peroxide in aqueous methanolic solution, leaving the dihydropyridine moiety virtually intact [93]. Interestingly, oxidation of the dihydropyridine moiety was subsequently mediated by cobalt acetate under the same conditions [93]. Exploratory experiments also showed that NARH is inert towards the capping reagent. A more serious issue is the expected degradation of the dihydropyridine moiety during acidic processing (i.e. detritylation and coupling/activation). As expected, we observed the formation of cTHNAR from NARH upon exposure to the detritylation reagent (dichloroacetic acid in dichloroethane) (unpublished data). Hypothetically, this cyclisation is blocked in a NARH phosphoramidite by the 2'-O-protecting group. Nevertheless, anomerization and formation of tetrahydropyridine derivatives by addition of nucleophiles is likely to occur. However, detritylation using Lewis acids such as zinc halides [96], offers a simple way to overcome this hurdle, as no degradation was observed after prolonged incubation of NARH in zinc chloride/dichloroethane (unpublished data). As the activation step is also acidic, degradation of the dihydropyridine might also be expected. However, this was not observed. To our surprise, incubation of NARH with benzylmercaptotetrazole (BMT), currently the most commonly used activator for phosphoramidite coupling, leads to oxidation (i.e. formation of NAR) (unpublished data). In principle, this is in line with widely used colorimetric assays for the characterization of NAD(P)H-dependent dehydrogenases by the formation of formazan derivatives from tetrazolium salts (e.g. MTT assay) [97]. Surprisingly, tetrazole has been successfully used for phosphoramidite coupling with 2',3'-protected NARH, suggesting the use of alternative activators, although tetrazole is certainly a less efficient variant [93].
5. Conclusions
In the context of the RNA world hypothesis, ribozymes played the key role as catalysts to support chemical reactions. On the evolutionary path to modern biochemistry, the initial rather simple catalytic activity must have evolved into more complex scenarios, including regulation of RNA catalytic activity and recruitment of co-factors for function, before proteins evolved and took over. The regulation of activity and the use of co-factors would require other molecules (ligands) to bind to the RNA, possibly covalently. Several examples of such allosterically regulated ribozymes have been described in the literature [98,99,100], and cofactor-dependent ribozyme activity has been demonstrated with redox-active ligands such as FMN or NAD [101]. For the further development of redox-active ribozymes and aptamers, the incorporation of electron donors and/or acceptors into RNA is a highly desirable goal, as it would pave the way for ribozyme-catalyzed enantioselective and regioselective redox reactions and provide the basis for novel biotechnological platforms. Promising initial results have been obtained with pyrrolochinolinchinone (PQQ), NAD and FAD, which exhibit redox activity when complexed with a DNA aptamer [15]. With regard to the RNA world hypothesis, precursors of today's known cofactors, with simpler structures and eventually covalently bound to RNA, may have played a key role. Thus, much effort has been devoted to the incorporation of NAR or NARH into RNA, which has proven to be a challenging task. The limited stability of the oxidized species (NAR) in the presence of bases and the reduced species (NARH) in the presence of acids is a problem, as is the susceptibility of C1' and the pyridinium moiety to be attacked by a nucleophile, leading to decomposition. Similarly, NARH is susceptible to degradation by acid-catalyzed nucleophile addition. The synthesis of NAR(H) is possible by various glycosylation reactions. However, care must be taken because of the stability problems described above. The use of carba-ribose and/or C-linked nucleosides may be a way to circumvent the stability problems, but has not been described in this specific case.
To the best of our knowledge, NAR(H) has not been successfully transferred into a posphoramidite building block and used in solid phase oligoribonucleotide synthesis. The only described examples of nicotinamide species covalently attached to RNA are a 5'-5'-linked NAD-cap at the 5'-terminus [82] and a NAR moiety attached to the 3'-terminus through a phosphoramidate linkage [83]. The incorporation of NAR(H) into oligonucleotides should be possible in principle, although it requires careful optimization of the synthesis conditions. For example, benzylmercaptotetrazole (BMT) is the most suitable activator for coupling phosphoramidites, but at the same time it can act as an oxidant, which can be problematic when incorporating NARH. NAR is the more challenging derivative for oligonucleotide synthesis because it is poorly soluble in acetonitrile and has a less nucleophilic 5'-OH group, which would make the coupling reaction less efficient. Nevertheless, key intermediates such as NARH-5'-phosphoramidite or 5'-O-DMT protected NAR have already been synthesized [92,93], and it is likely that the incorporation of NAR(H) into oligonucleotides will be successful in the near future.
References
- Panchapakesan, Shanker S. S., and Ronald R. Breaker. "The Case of the Missing Allosteric Ribozymes." Nat Chem Biol 17, no. 4 (2021): 375-382. [CrossRef]
- Robertson, M. P., and G. F. Joyce. "The Origins of the RNA World." Cold Spring Harbor Perspectives in Biology 4, no. 5 (2012). [CrossRef]
- Vitreschak, A. "Riboswitches: The Oldest Mechanism for the Regulation of Gene Expression?" Trends Genet. 20, no. 1 (2004): 44-50. [CrossRef]
- Breaker, R. R. "Riboswitches and the RNA World." Cold Spring Harbor Perspectives in Biology 4, no. 2 (2012). [CrossRef]
- Breaker, Ronald R. "Riboswitches: From Ancient Gene-Control Systems to Modern Drug Targets." Future Microbiol 4, no. 7 (2009): 771-773. [CrossRef]
- Goldman, Aaron D., and Betul Kacar. "Cofactors Are Remnants of Life’s Origin and Early Evolution." J Mol Evol 89, no. 3 (2021): 127-133. [CrossRef]
- Huang, Faqing, Charles Walter Bugg, and Michael Yarus. "RNA-Catalyzed Coa, NAD, and FAD Synthesis from Phosphopantetheine, NMN, and FMN." Biochemistry 39, no. 50 (2000): 15548-15555. [CrossRef]
- Kirschning, Andreas. "Coenzymes and Their Role in the Evolution of Life." Angew Chem Int Ed 60, no. 12 (2021): 6242-6269. [CrossRef]
- Breaker, Ronald R., and Gerald F. Joyce. "Self-Incorporation of Coenzymes by Ribozymes." J Mol Evol 40, no. 6 (1995): 551-558. [CrossRef]
- Fujita, Yuki, Hiroyuki Furuta, and Yoshiya Ikawa. "Construction of an Artificial Ribozyme Which Ligates an RNA Fragment Activated by Nicotinamide Mononucleotide." Nucleic Acids Symp Ser 50, no. 1 (2006): 231-232. [CrossRef]
- Fujita, Yuki, Hiroyuki Furuta, and Yoshiya Ikawa. "Tailoring RNA Modular Units on a Common Scaffold: A Modular Ribozyme with a Catalytic Unit for -Nicotinamide Mononucleotide-Activated RNA Ligation." RNA 15, no. 5 (2009): 877-888. [CrossRef]
- Cleaves, H. James, and Stanley L. Miller. "The Nicotinamide Biosynthetic Pathway Is a By-Product of the RNA World." J Mol Evol 52, no. 1 (2001): 73-77. [CrossRef]
- Kim, Hyo-Joong, and Steven A. Benner. "A Direct Prebiotic Synthesis of Nicotinamide Nucleotide." Chem Eur J 24, no. 3 (2018): 581-584. [CrossRef]
- Griffiths, Hollie B. S., Courtney Williams, Sarah J. King, and Simon J. Allison. "Nicotinamide Adenine Dinucleotide (NAD+): Essential Redox Metabolite, Co-Substrate and an Anti-Cancer and Anti-Ageing Therapeutic Target." Biochem Soc Trans 48, no. 3 (2020): 733-744. [CrossRef]
- Emahi, Ismaila, Paige R. Gruenke, and Dana A. Baum. "Effect of Aptamer Binding on the Electron-Transfer Properties of Redox Cofactors." J Mol Evol 81, no. 5-6 (2015): 186-193. [CrossRef]
- Tsukiji, S., K. Ramaswamy, and H. Suga. "Ribozymes That Use Redox Cofactors." Pure Appl Chem 76, no. 7-8 (2004): 1525-1536. [CrossRef]
- Tsukiji, Shinya, Swetansu B. Pattnaik, and Hiroaki Suga. "An Alcohol Dehydrogenase Ribozyme." Nat Struct Mol Biol 10, no. 9 (2003): 713-717. [CrossRef]
- Campbell, M. T. D., D. S. Jones, G. P. Andrews, and S. Li. "Understanding the Physicochemical Properties and Degradation Kinetics of Nicotinamide Riboside, a Promising Vitamin B(3)Nutritional Supplement." Food Nutr Res 63 (2019). [CrossRef]
- Oppenheimer, N. J. "NAD Hydrolysis: Chemical and Enzymatic Mechanisms." Mol Cell Biochem 138, no. 1-2 (1994): 245-251. [CrossRef]
- Handlon, Anthony L., and Norman J. Oppenheimer. "Substituent Effects on the pH-Independent Hydrolysis of 2'-Substituted Nicotinamide Arabinosides." J Org Chem 56, no. 17 (1991): 5009-5010. [CrossRef]
- Minato, H., E. Yamazaki, and M. Kobayashi. "Reactions of Several Nucleophiles with Quaternary Salts of Nicotinamide." Chem. Lett. 5, no. 5 (1976): 525-530. [CrossRef]
- Engbersen, J. F. J., A. Koudijs, H. M. Sleiderink, and M. C. R. Franssen. "Addition of Cyanide Ion to Nicotinamide Cations in Acetonitrile. Formation of Non-Productive Charge-Transfer Complexes." J. Chem. Soc., Perkin Trans. 2, no. 1 (1990): 79-83. [CrossRef]
- Makarov, M. V., F. Hayat, B. Graves, M. Sonavane, E. A. Salter, A. Wierzbicki, N. R. Gassman, and M. E. Migaud. "Chemical and Biochemical Reactivity of the Reduced Forms of Nicotinamide Riboside." ACS Chem Biol 16, no. 4 (2021): 604-614. [CrossRef]
- You, W., K. M. Hugar, R. C. Selhorst, M. Treichel, C. R. Peltier, K. J. T. Noonan, and G. W. Coates. "Degradation of Organic Cations under Alkaline Conditions." J Org Chem 86, no. 1 (2021): 254-263. [CrossRef]
- Johnson, S. L., and D. L. Morrison. "The Alkaline Reaction of Nicotinamide Adenine Dinucleotide, a New Transient Intermediate." J Biol Chem 245, no. 17 (1970): 4519-4524. [CrossRef]
- Guilbert, C. C., and S. L. Johnson. "Isolation and Characterization of the Fluorescent Alkali Product from Diphosphopyridine Nucleotide." Biochemistry 10, no. 12 (1971): 2313-2316. [CrossRef]
- Liu, R., and L. E. Orgel. "Enzymatic Synthesis of Polymers Containing Nicotinamide Mononucleotide." Nucleic Acids Res 23, no. 18 (1995): 3742-3749. [CrossRef]
- Zeynizadeh, Behzad, Karim Akbari Dilmaghani, and Asli Roozijoy. "Aromatization of Hantzsch Ester 1,4-Dihydropyridines with Iodine under Normal Conditions and Ultrasound Irradiation." J Chin Chem Soc 52, no. 5 (2005): 1001-1004. [CrossRef]
- Cai, Xiao-hua, Hai-jun Yang, and Guo-lin Zhang. "Aromatization of 1,4-Dihydropyridines with Selenium Dioxide." Can J Chem 83, no. 3 (2005): 273-275. [CrossRef]
- Nakamichi, Natsuki, Yuka Kawashita, and Masahiko Hayashi. "Activated Carbon-Promoted Oxidative Aromatization of Hantzsch 1,4-Dihydropyridines and 1,3,5-Trisubstituted Pyrazolines Using Molecular Oxygen." Synthesis, no. 7 (2004): 1015-1020. [CrossRef]
- Abdoli-Senejani, M., and K. Karami. "Ultrasound-Assisted Heterogeneous Oxidation of 1,4-Dihydropyridines." Org Prep Proced Int 52, no. 4 (2020): 274-281. [CrossRef]
- Boecker, Ronald H., and F. Peter Guengerich. "Oxidation of 4-Aryl- and 4-Alkyl-Substituted 2,6-Dimethyl-3,5-Bis(Alkoxycarbonyl)-1,4-Dihydropyridines by Human Liver Microsomes and Immunochemical Evidence for the Involvement of a Form of Cytochrome P-450." J Med Chem 29, no. 9 (1986): 1596-1603. [CrossRef]
- Zarei, A., L. Khazdooz, M. Enayati, S. Madarshahian, T. J. Wooster, G. Ufheil, and A. Abbaspourrad. "Dihydronicotinamide Riboside: Synthesis from Nicotinamide Riboside Chloride, Purification and Stability Studies." RSC Adv 11, no. 34 (2021): 21036-21047. [CrossRef]
- Sollenberger, P. Y., and R. B. Martin. "Mechanism of Enamine Hydrolysis." J. Am. Chem. Soc. 92, no. 14 (1970): 4261–4270. [CrossRef]
- Maas, W., M. J. Janssen, E. J. Stamhuis, and H. Wynberg. "Mechanism of Enamine Reactions. IV. The Hydrolysis of Tertiary Enamines in Acidic Medium." J. Org. Chem. 32, no. 4 (1967): 1111–1115. [CrossRef]
- Johnston, C. C., D. E. Metzler, and J. L. Gardner. "Acid-Catalyzed Addition of Water to 1 4-Dihydronicotinamide Derivatives." Biochemistry 2, no. 4 (1963): 689-696. [CrossRef]
- Oppenheimer, N. J., and N. O. Kaplan. "The Alpha Beta Epimerization of Reduced Nicotinamide Adenine Dinucleotide." Arch Biochem Biophys 166, no. 2 (1975): 526-535. [CrossRef]
- Johnson, S. L., and P. T. Tuazon. "Acid-Catalyzed Hydration of Reduced Nicotinamide Adenine Dinucleotide and Its Analogues." Biochemistry 16, no. 6 (1977): 1175-1183. [CrossRef]
- Branlant, G., B. Eiler, and J. F. Biellmann. "A Word of Caution: 1,4,5,6-Tetrahydronicotinamide Adenine Dinucleotide (Phosphate) Should Be Used with Care in Acidic and Neutral Media." Anal Biochem 125, no. 2 (1982): 264-268. [CrossRef]
- Ammon, H. L., and L. H. Jensen. "The "Dimeric Acid Product" of 1-Methyl-1,4-Dihydronicotinamide." J Am Chem Soc 88, no. 3 (1966): 613-614. [CrossRef]
- Ilic, S., U. Pandey Kadel, Y. Basdogan, J. A. Keith, and K. D. Glusac. "Thermodynamic Hydricities of Biomimetic Organic Hydride Donors." J Am Chem Soc 140, no. 13 (2018): 4569-4579. [CrossRef]
- Zhu, X. Q., Y. Tan, and C. T. Cao. "Thermodynamic Diagnosis of the Properties and Mechanism of Dihydropyridine-Type Compounds as Hydride Source in Acetonitrile with "Molecule ID Card"." J Phys Chem B 114, no. 5 (2010): 2058-2075. [CrossRef]
- Anderson Jr., A. G., and G. Berkelhammer. "A Study of the Primary Acid Reaction on Model Compounds of Reduced Diphosphopyridine Nucleotide." J. Am. Chem. Soc. 80, no. 4 (1958): 992–999. [CrossRef]
- Nowak, C., A. Pick, L. I. Csepei, and V. Sieber. "Characterization of Biomimetic Cofactors According to Stability, Redox Potentials, and Enzymatic Conversion by Nadh Oxidase from Lactobacillus Pentosus." ChemBioChem 18, no. 19 (2017): 1944-1949. [CrossRef]
- Zachos, I., M. Doring, G. Tafertshofer, R. C. Simon, and V. Sieber. "Carba Nicotinamide Adenine Dinucleotide Phosphate: Robust Cofactor for Redox Biocatalysis." Angew Chem Int Ed 60, no. 26 (2021): 14701-14706. [CrossRef]
- Haynes, L. J., and A. R. Todd. "66. Codehydrogenases. Part I. The Synthesis of Dihydronicotinamide-D-Ribofuranoside [N-D-Ribofuranosidyl-1 : 2(or 6)-Dihydronicotinamide]." J Chem Soc (Resumed) (1950): 303. [CrossRef]
- Haykes, L. J., N. A. Hughes, G. W. Kenner, and Alexander Todd. "734. Codehydrogenases. Part II. A Synthesis of Nicotinamide Nucleotide." J Chem Soc (Resumed) (1957): 3727. [CrossRef]
- Mikhailopulo, I. A., and A. I. Miroshnikov. "New Trends in Nucleoside Biotechnology." Acta Naturae 2, no. 2 (2010): 36-58. [CrossRef]
- Lee, Jaemoon, Hywyn Churchil, Woo-Baeg Choi, Joseph E. Lynch, F. E. Roberts, R. P. Volante, and Paul J. Reider. "A Chemical Synthesis of Nicotinamide Adenine Dinucleotide (NAD+)." Chem Commun, no. 8 (1999): 729-730. [CrossRef]
- Jarman, M., and W. C. J. Ross. "4-Substituted Nicotinic Acids and Nicotinamides. Part II. The Preparation of 4-Methylnicotinamide Riboside." J Chem Soc C: Organic, no. 2 (1969): 199. [CrossRef]
- Tanimori, Shinji, Takeshi Ohta, and Mitsunori Kirihata. "An Efficient Chemical Synthesis of Nicotinamide Riboside (NAR) and Analogues." Bioorg Med Chem Lett 12, no. 8 (2002): 1135-1137. [CrossRef]
- Franchetti, Palmarisa, Michela Pasqualini, Riccardo Petrelli, Massimo Ricciutelli, Patrizia Vita, and Loredana Cappellacci. "Stereoselective Synthesis of Nicotinamide β-Riboside and Nucleoside Analogs." Bioorg Med Chem Lett 14, no. 18 (2004): 4655-4658. [CrossRef]
- Yang, Tianle, Noel Yan-Ki Chan, and Anthony A. Sauve. "Syntheses of Nicotinamide Riboside and Derivatives: Effective Agents for Increasing Nicotinamide Adenine Dinucleotide Concentrations in Mammalian Cells." J Med Chem 50, no. 26 (2007): 6458-6461. [CrossRef]
- Zhang, Ning, and Anthony A. Sauve. "Synthesis of β-Nicotinamide Riboside Using an Efficient Two-Step Methodology." Curr Prot Nucleic Acid Chem 71, no. 1 (2017). [CrossRef]
- Atkinson, M. R., R. K. Morton, and R. Naylor. "98. Synthesis of Glycosylpyridinium Compounds from Glycosylamines and from Glycosyl Halides." J Chem Soc (Resumed) (1965): 610. [CrossRef]
- Makarov, M. V., N. W. Harris, M. Rodrigues, and M. E. Migaud. "Scalable Syntheses of Traceable Ribosylated NAD+ Precursors." Org Biomol Chem 17, no. 38 (2019): 8716-8720. [CrossRef]
- Makarov, Mikhail V., Faisal Hayat, Briley Graves, Manoj Sonavane, Edward A. Salter, Andrzej Wierzbicki, Natalie R. Gassman, and Marie E. Migaud. "Chemical and Biochemical Reactivity of the Reduced Forms of Nicotinamide Riboside." ACS Chem Biol16, no. 4 (2021): 604-614. [CrossRef]
- Carelli, Vincenzo, Felice Liberatore, Luigi Scipione, Barbara Di Rienzo, and Silvano Tortorella. "Dithionite Adducts of Pyridinium Salts: Regioselectivity of Formation and Mechanisms of Decomposition." Tetrahedron 61, no. 43 (2005): 10331-10337. [CrossRef]
- Paul, Caroline E., Isabel W. C. E. Arends, and Frank Hollmann. "Is Simpler Better? Synthetic Nicotinamide Cofactor Analogues for Redox Chemistry." ACS Catalysis 4, no. 3 (2014): 788-797. [CrossRef]
- Szczepankiewicz, Bruce G., Han Dai, Karsten J. Koppetsch, Dongming Qian, Fan Jiang, Cheney Mao, and Robert B. Perni. "Synthesis of Carba-NAD and the Structures of Its Ternary Complexes with Sirt3 and Sirt5." J Org Chem 77, no. 17 (2012): 7319-7329. [CrossRef]
- Akabane-Nakata, Masaaki, Tyler Chickering, Joel M. Harp, Mark K. Schlegel, Shigeo Matsuda, Martin Egli, and Muthiah Manoharan. "RNAs Containing Carbocyclic Ribonucleotides." Org Lett 24, no. 2 (2022): 525-530. [CrossRef]
- Largy, E., W. Liu, A. Hasan, and D. M. Perrin. "Base-Pairing Behavior of a Carbocyclic Janus-AT Nucleoside Analogue Capable of Recognizing A and T within a DNA Duplex." ChemBioChem 14, no. 16 (2013): 2199-2208. [CrossRef]
- Rydzik, Anna M., Regina Balk, Martin Koegler, Tobias Steinle, Doris Riether, and Dirk Gottschling. "Access to 1′-Amino Carbocyclic Phosphoramidite to Enable Postsynthetic Functionalization of Oligonucleotides." Org Lett 23, no. 17 (2021): 6735-6739. [CrossRef]
- Dai, Zhefu, Xiao-Nan Zhang, Fariborz Nasertorabi, Qinqin Cheng, Hua Pei, Stan G. Louie, Raymond C. Stevens, and Yong Zhang. "Facile Chemoenzymatic Synthesis of a Novel Stable Mimic of NAD+." Chem Sci 9, no. 44 (2018): 8337-8342. [CrossRef]
- Zang, Hongjun, Jing Lou, Shuolei Jiao, Huanxin Li, Yannan Du, and Jiao Wang. "Valorization of Chitin Derived N-Acetyl-D-Glucosamine into High Valuable N-Containing 3-Acetamido-5-Acetylfuran Using Pyridinium-Based Ionic Liquids." J Mol Liq 330 (2021): 115667. [CrossRef]
- Craig, John H., Ping C. Huang, T. Gordon Scott, and Nelson J. Leonard. "Synthetic Spectroscopic Models Related to Coenzymes and Base Pairs. X. Quaternized Bisnicotinamides." J Am Chem Soc 94, no. 16 (1972): 5872-5879. [CrossRef]
- Knox, Richard J., Terence C. Jenkins, Stephen M. Hobbs, Shiuan Chen, Roger G. Melton, and Philip J. Burke. "Bioactivation of 5-(Aziridin-1-Yl)-2,4-Dinitrobenzamide (CB 1954) by Human NAD(P)H Quinone Oxidoreductase 2: A Novel Co-Substrate-Mediated Antitumor Prodrug Therapy1." Canc Res 60, no. 15 (2000): 4179-4186.
- Malver, Olaf, Mina J. Sebastian, and Norman J. Oppenheimer. "Alteration in Substrate Specificity of Horse Liver Alcohol Dehydrogenase by an Acyclic Nicotinamide Analog of NAD+." DNA Repair 23 (2014): 95-100. [CrossRef]
- Bušić, Valentina, Karolina Vrandečić, Tamara Siber, Sunčica Roca, Vice Tomičić, and Dajana Gašo Sokač. "Novel Synthetic Routes to Quaternary Pyridinium Salts and Their Antifungal Activity." Croat Chem Acta 95, no. 1 (2022): 31-38. [CrossRef]
- Chennamaneni, Srinivas Rao, Venkateswarlu Vobalaboina, and Achaiah Garlapati. "Quaternary Salts of 4,3′ and 4,4′ Bis-Pyridinium Monooximes: Synthesis and Biological Activity." Bioorg Med Chem Lett 15, no. 12 (2005): 3076-3080. [CrossRef]
- Friedman, Orrie M., Kurt Pollak, and Ezra Khedouri. "1-(β-Chloroethyl)-3-Carbamylpyridinium Chloride. Prototype of a New Class of Latently Cytotoxic Potential Antitumor Agents." J Med Chem 6, no. 4 (1963): 462-463. [CrossRef]
- Plapp, Bryce V., Christoph Woenckhaus, and Gerhard Pfleiderer. "Evaluation of N1-(Ω-Bromoacetamidoalkyl)Nicotinamides as Inhibitors of Dehydrogenases." Arch Biochem Biophys 128, no. 2 (1968): 360-368. [CrossRef]
- Robins, Morris J., and Peter W. Hatfield. "Nucleic Acid Related Compounds. 37. Convenient and High-Yield Syntheses of N-[(2-Hydroxyethoxy)Methyl] Heterocycles as "Acyclic Nucleoside" Analogues." Can J Chem 60, no. 5 (1982): 547-553. [CrossRef]
- Štefko, Martin, Lenka Slavětínská, Blanka Klepetářová, and Michal Hocek. "General and Modular Synthesis of Isomeric 5-Substituted Pyridin-2-yl and 6-Substituted Pyridin-3-yl C-Ribonucleosides Bearing Diverse Alkyl, Aryl, Hetaryl, Amino, Carbamoyl, and Hydroxy Groups." J Org Chem 76, no. 16 (2011): 6619-6635. [CrossRef]
- Ma, Y., S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang, and Y. Zhang. "Highly Stereoselective Synthesis of Aryl/Heteroaryl-C-Nucleosides Via the Merger of Photoredox and Nickel Catalysis." Chem Commun 55, no. 97 (2019): 14657-14660. [CrossRef]
- Maeba, Isamu, Katsumi Iwata, Fumitaka Usami, and Hiroshi Furukawa. "C-Nucleosides. 1. Synthesis of 3-(β-D-Ribofuranosyl)Pyridazines." J Org Chem 48, no. 18 (1983): 2998-3002. [CrossRef]
- Singh, Ishwar, and Oliver Seitz. "Diastereoselective Synthesis of -Aryl-C-Nucleosides from 1,2-Anhydrosugars." Org Lett 8, no. 19 (2006): 4319-4322. [CrossRef]
- Brotschi, Christine, Adrian Häberli, and Christian J. Leumann. "A Stable DNA Duplex Containing a Non-Hydrogen-Bonding and Non-Shape-Complementary Base Couple: Interstrand Stacking as the Stability Determining Factor." Angew Chem Int Ed 40, no. 16 (2001): 3012-3014. [CrossRef]
- Franchetti, Palmarisa, Loredana Cappellacci, Mario Grifantini, Anna Barzi, Giuseppe Nocentini, Hongyoan Yang, Ayrn O'Connor, Hiremagalur N. Jayaram, Christopher Carrell, and Barry M. Goldstein. "Furanfurin and Thiophenfurin: Two Novel Tiazofurin Analogs. Synthesis, Structure, Antitumor Activity, and Interactions with Inosine Monophosphate Dehydrogenase." J Med Chem 38, no. 19 (1995): 3829-3837. [CrossRef]
- Kabat, Marek M., Krzysztof W. Pankiewicz, and Kyoichi A. Watanabe. "Nucleosides. 141. Synthesis of 5-β-D-Ribofuranosylnicotinamide and Its N-Methyl Derivative. The Isosteric and Isoelectronic Analogs of Nicotinamide Nucleoside." J Med Chem 30, no. 5 (1987): 924-927. [CrossRef]
- Pankiewicz, Krzysztof, Kyoichi Watanabe, Krystyna Lesiak-Watanabe, Barry Goldstein, and Hiremagalur Jayaram. "The Chemistry of Nicotinamide Adenine Dinucleotide (NAD) Analogues Containing C-Nucleosides Related to Nicotinamide Riboside." Curr Med Chem 9, no. 7 (2002): 733-741. [CrossRef]
- Höfer, Katharina, Florian Abele, Jasmin Schlotthauer, and Andres Jäschke. "Synthesis of 5′-NAD-Capped RNA." Bioconjugate Chem 27, no. 4 (2016): 874-877. [CrossRef]
- Göckel, A., and C. Richert. "Synthesis of an Oligonucleotide with a Nicotinamide Mononucleotide Residue and Its Molecular Recognition in DNA Helices." Org Biomol Chem 13, no. 41 (2015): 10303-10309. [CrossRef]
- Kjellstrand, Martin, Klas Broo, Linda Andersson, Cecilia Farre, Åke Nilsson, and Lars Baltzer. "The Site-Selective Incorporation of a NAD+ Cofactor Mimic into a Folded Helix–Loop–Helix Polypeptide Motif." J Chem Soc, Perkin Trans 2, no. 12 (1997): 2745-2750. [CrossRef]
- Zhang, B., X. Q. Zhu, J. Y. Lu, J. He, P. G. Wang, and J. P. Cheng. "Polysiloxane-Supported NAD(P)H Model 1-Benzyl-1,4-Dihydronicotinamide: Synthesis and Application in the Reduction of Activated Olefins." J Org Chem 68, no. 8 (2003): 3295-3298. [CrossRef]
- Chen, Zhe, Anna Ka Yee Kwong, Zhenjun Yang, Liangren Zhang, Hon Cheung Lee, and Lihe Zhang. "Cheminform Abstract: Studies of the Synthesis of Nicotinamide Nucleoside and Nucleotide Analogues and Their Inhibitions Towards CD38 NADase." ChemInform 43, no. 15 (2012). [CrossRef]
- Kovarik, Zrinka, Jarosław Kalisiak, Nikolina Maček Hrvat, Maja Katalinić, Tamara Zorbaz, Suzana Žunec, Carol Green, Zoran Radić, Valery V. Fokin, K. Barry Sharpless, and Palmer Taylor. "Reversal of Tabun Toxicity Enabled by a Triazole-Annulated Oxime Library—Reactivators of Acetylcholinesterase." Chem Eur J 25, no. 16 (2019): 4100-4114. [CrossRef]
- Brunner, Heiko, Stefanie Ackermann, Sandra Lucks, Jun Wu, and James Adolf. "Aqueous Composition for Depositing a Cobalt Deposit and Method for Electrolytically Depositing Such a Deposit." Atotech Deutschland Gmbh. WO Patent 2019/013762 A1, January 17, 2019.
- Mutschler, Hannes, and Philipp Holliger. "Non-Canonical 3′-5′ Extension of RNA with Prebiotically Plausible Ribonucleoside 2′,3′-Cyclic Phosphates." J Am Chem Soc 136, no. 14 (2014): 5193-5196. [CrossRef]
- Tereshko, Valentina, Sergei Gryaznov, and Martin Egli. "Consequences of Replacing the DNA 3‘-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3‘ → P5‘ Phosphoramidate DNA Dodecamer Duplex." J Am Chem Soc 120, no. 2 (1998): 269-283. [CrossRef]
- Liu, Rihe, and Leslie E. Orgel. "Enzymatic Synthesis of Polymers Containing Nicotinamide Mononucleotide." Nucleic Acids Res 23, no. 18 (1995): 3742-3749. [CrossRef]
- Kam, Bernard L., and Norman J. Oppenheimer. "Synthesis of a New Class of D-Aldopentofuranosylamines, the 5-O-Trityl-D-Aldopentofuranosylamines." Carbohyd Res 77, no. 1 (1979): 275-280. [CrossRef]
- Livingston, D., J. M. McKearin, B. Szczepankiewicz, and J. N. Kremsky. 2017. "Nicotinamide Mononucleotide Derivatives and Their Uses for Treating Diseases and Improving Cell and Tissue Survival." WO Patent 2017/024255 A1, February 09, 2017.
- Hayat, F., and M. E. Migaud. "Nicotinamide Riboside-Amino Acid Conjugates That Are Stable to Purine Nucleoside Phosphorylase." Org Biomol Chem 18, no. 15 (2020): 2877-2885. [CrossRef]
- Xu, Hui, and Fangjun Chen. "Method for Preparing Beta-Nicotinamide Mononucleotide." Changsha Innovative Drug Industry Tech Research Institute Limited Company. CN Patent 114685582 A, January 07, 2022.
- Matteucci, M. D., and M. H. Caruthers. "The Use of Zinc Bromide for Removal of Dimethoxytrityl Ethers from Deoxynucleosides." Tetrahedron Lett 21, no. 34 (1980): 3243-3246. [CrossRef]
- Berridge, M. V., and A. S. Tan. "Characterization of the Cellular Reduction of 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT): Subcellular Localization, Substrate Dependence, and Involvement of Mitochondrial Electron Transport in MTT Reduction." Arch Biochem Biophys 303, no. 2 (1993): 474-482. [CrossRef]
- Araki, M., Y. Okuno, Y. Hara, and Y. Sugiura. "Allosteric Regulation of a Ribozyme Activity through Ligand-Induced Conformational Change." Nucleic Acids Res 26, no. 14 (1998): 3379-3384. [CrossRef]
- Strohbach, D., N. Novak, and S. Müller. "Redox-Active Riboswitching: Allosteric Regulation of Ribozyme Activity by Ligand-Shape Control." Angew Chem Int Ed 45, no. 13 (2006): 2127-2129. [CrossRef]
- Chen, A. G., N. Sudarsan, and R. R. Breaker. "Mechanism for Gene Control by a Natural Allosteric Group I Ribozyme." RNA 17, no. 11 (2011): 1967-1972. [CrossRef]
- Strohbach, Denise, Nina Novak, and Sabine Müller. "Redox-Active Riboswitching: Allosteric Regulation of Ribozyme Activity by Ligand-Shape Control." Angew Chem Int Ed 45, no. 13 (2006): 2127-2129. [CrossRef]
Figure 1.
Scheme of the redox cycle of an artificial nicotinamide-containing RNA structure with oxidoreductase activity.
Figure 1.
Scheme of the redox cycle of an artificial nicotinamide-containing RNA structure with oxidoreductase activity.
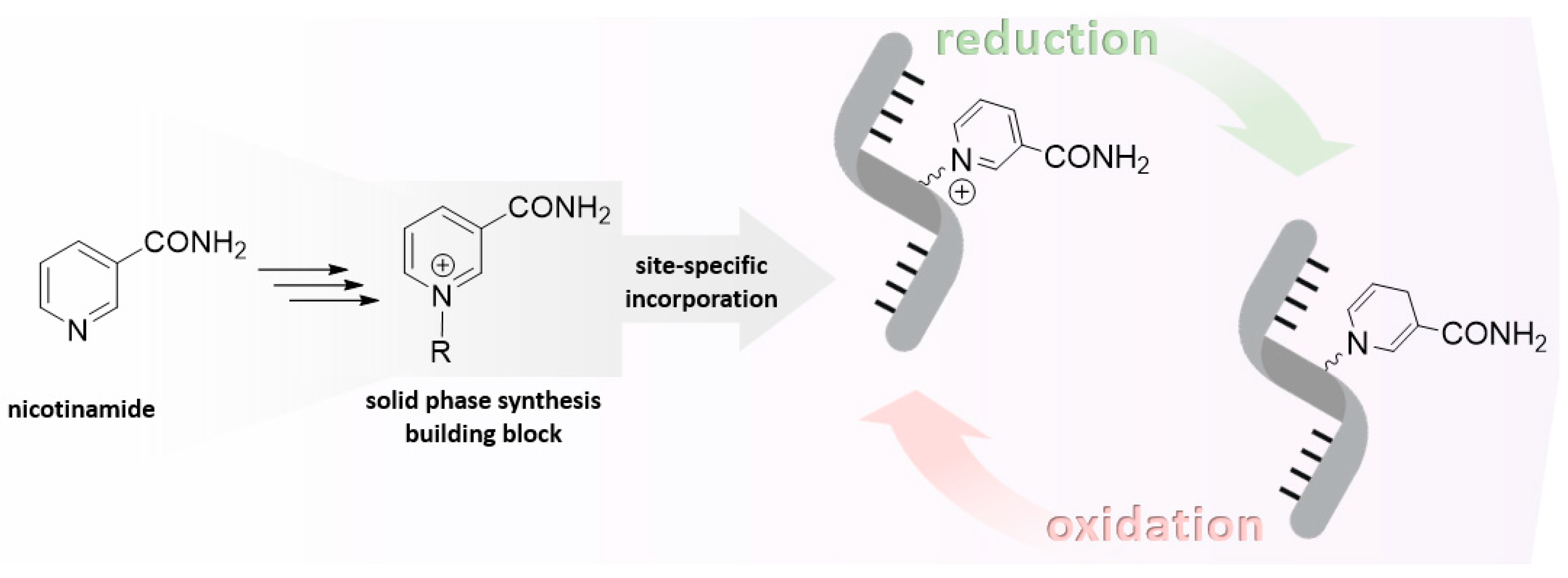
Figure 2.
Nucleophilic degradation of NAR by either attack onto the heterocycle or the anomeric carbon.
Figure 2.
Nucleophilic degradation of NAR by either attack onto the heterocycle or the anomeric carbon.
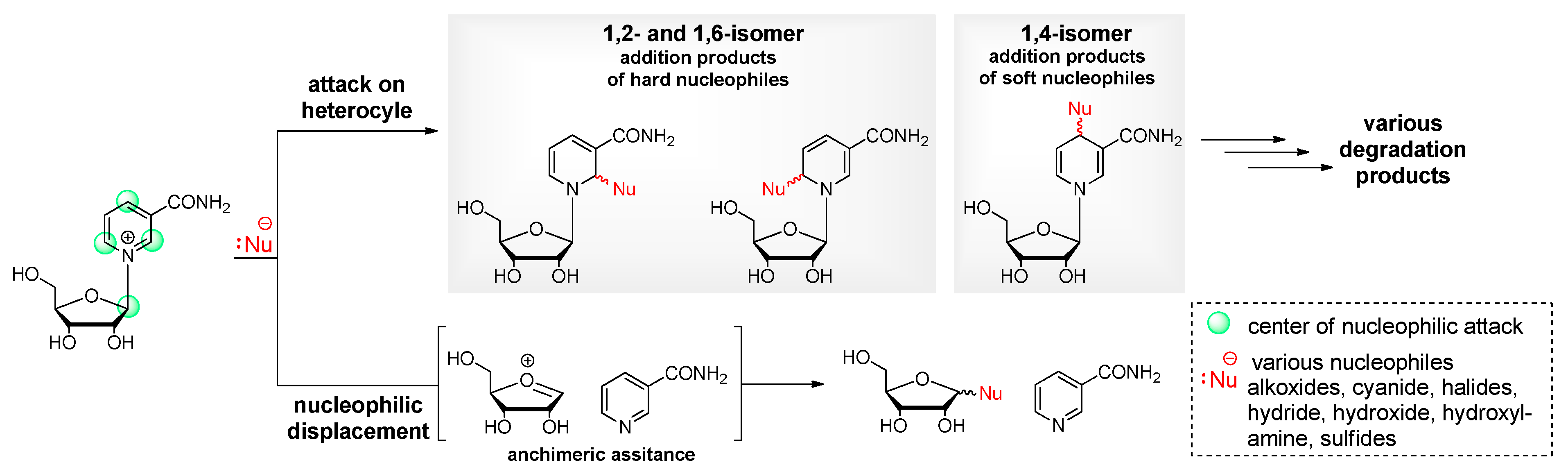
Figure 3.
Selection of key nicotinamide-based compounds important for incorporation into RNA structures.
Figure 3.
Selection of key nicotinamide-based compounds important for incorporation into RNA structures.
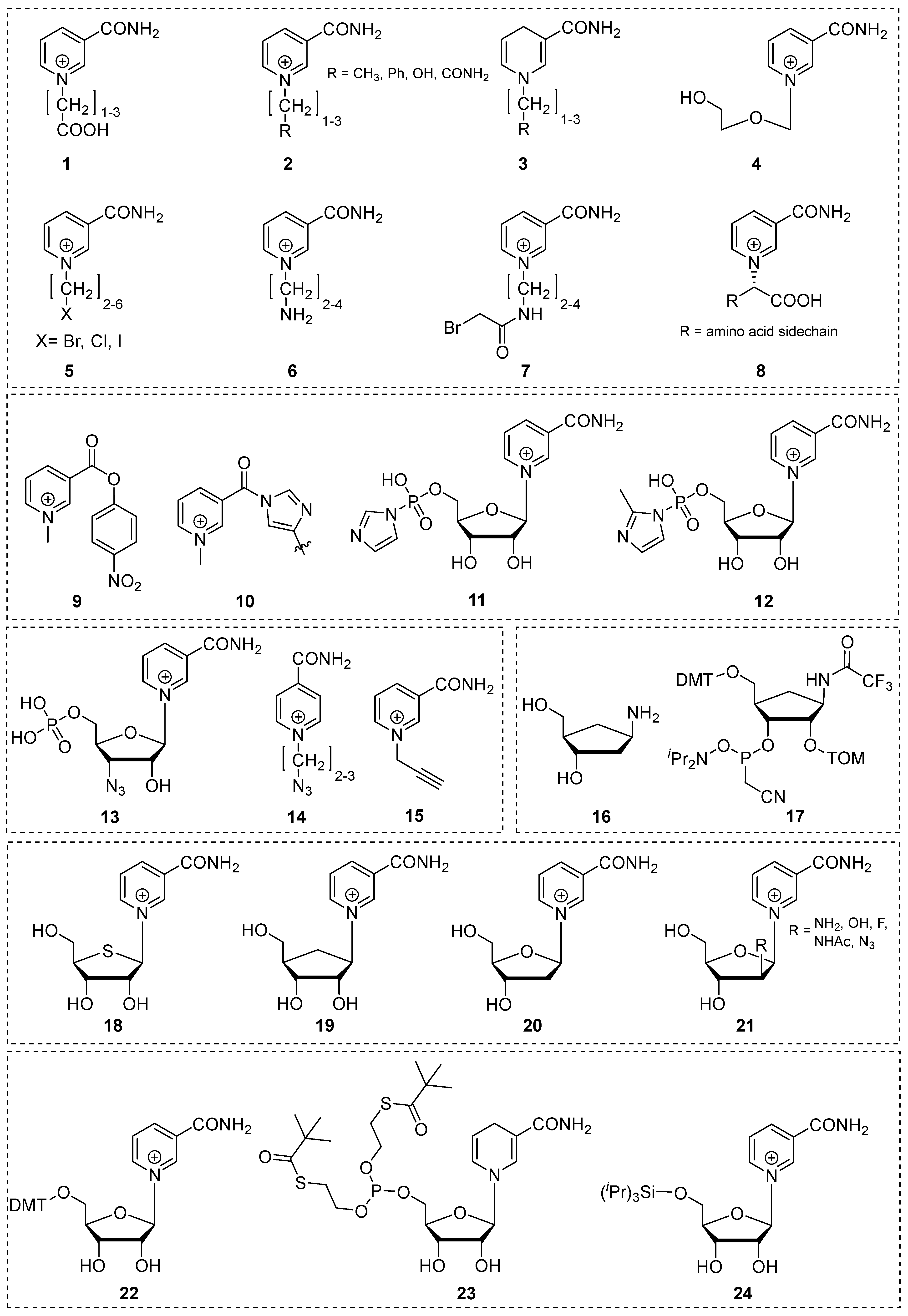
Figure 4.
Acid-catalyzed vinylamine addition, anomeric displacement and isomerization leading to deagradation of NARH.
Figure 4.
Acid-catalyzed vinylamine addition, anomeric displacement and isomerization leading to deagradation of NARH.
Figure 5.
Alteration in resonance contribution of the carbonyl substituent of NARH affecting the rate of initial protonation. The stabilization induced by an additional resonance structure facilitates protonation of the nicotinamide moiety.
Figure 5.
Alteration in resonance contribution of the carbonyl substituent of NARH affecting the rate of initial protonation. The stabilization induced by an additional resonance structure facilitates protonation of the nicotinamide moiety.
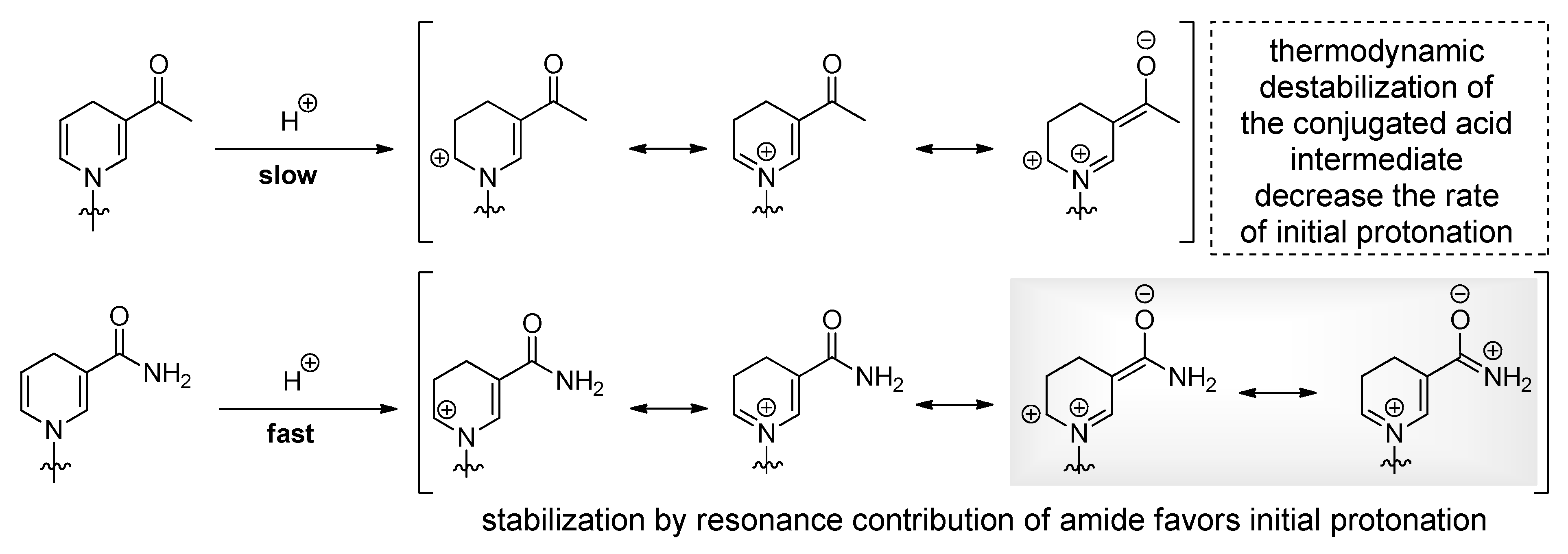
Figure 6.
Access to NAR(H) derivatives by (a) nucleophilic substitution of halo sugars; (b) Lewis acid catalyzed glycosylation of peracetylated ribose and (c) formation of the nicotinamide moiety through Zincke reaction.
Figure 6.
Access to NAR(H) derivatives by (a) nucleophilic substitution of halo sugars; (b) Lewis acid catalyzed glycosylation of peracetylated ribose and (c) formation of the nicotinamide moiety through Zincke reaction.
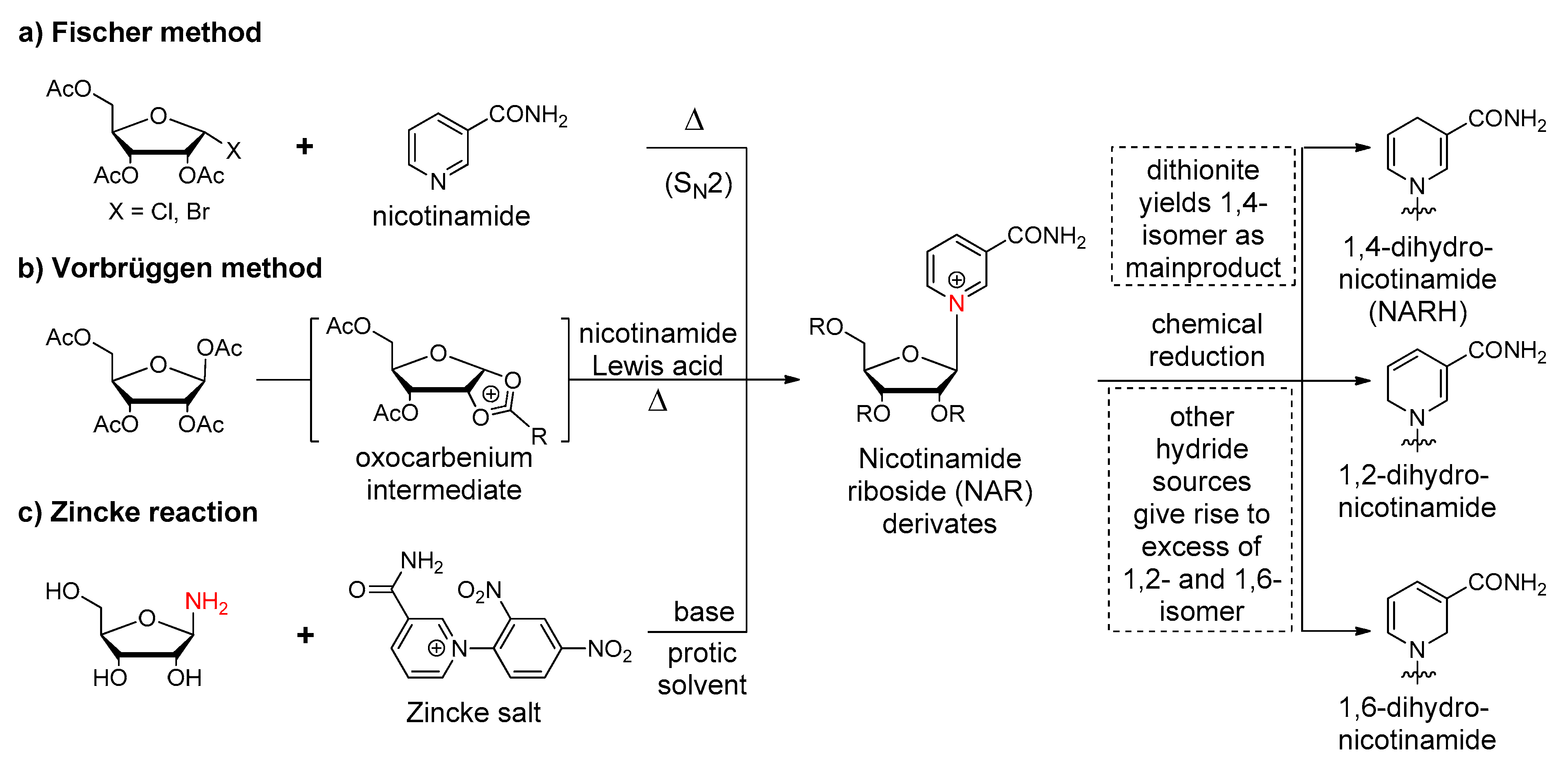
Figure 7.
Synthetic strategies towards modified sugar moieties. (a) Preparation of carbocyclic nicotinamide nucleosides. (b) Prototypic synthesis of an acyclic NAR analog.
Figure 7.
Synthetic strategies towards modified sugar moieties. (a) Preparation of carbocyclic nicotinamide nucleosides. (b) Prototypic synthesis of an acyclic NAR analog.
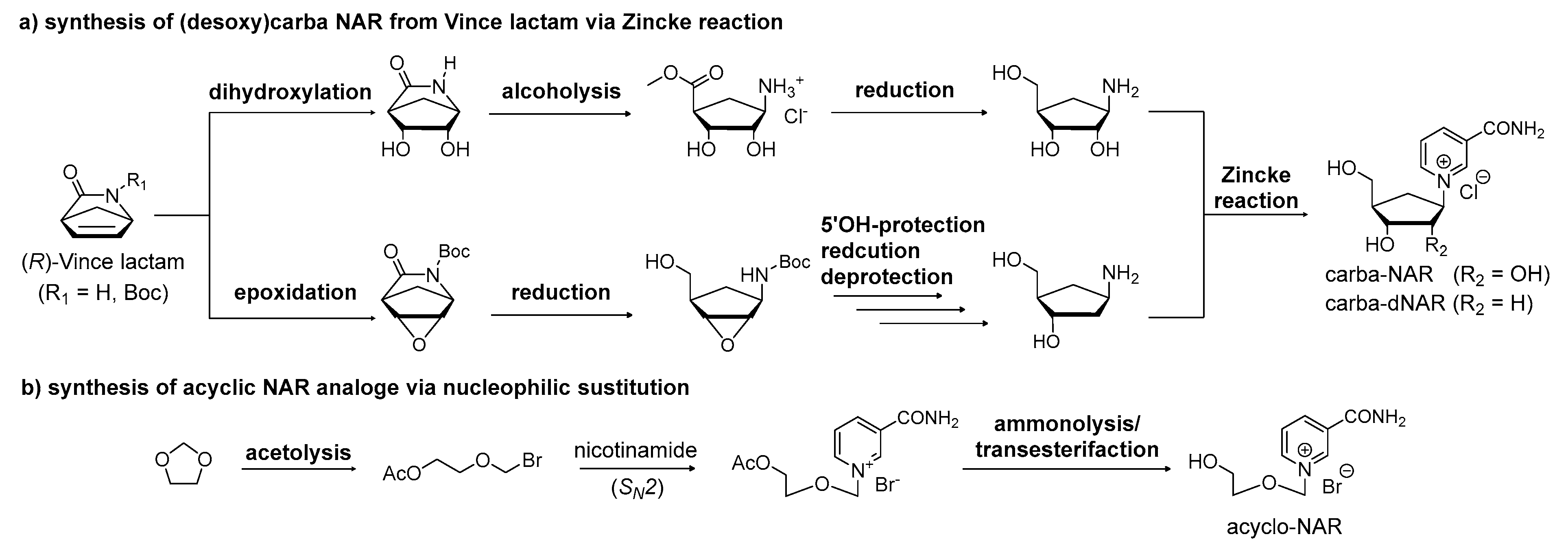
Figure 8.
General strategies for the synthesis of nicotinamide derived C-ribonucleosides. After formation of the nucleosidic bond, an alkylation of the ring nitrogen is required for redox activity.
Figure 8.
General strategies for the synthesis of nicotinamide derived C-ribonucleosides. After formation of the nucleosidic bond, an alkylation of the ring nitrogen is required for redox activity.
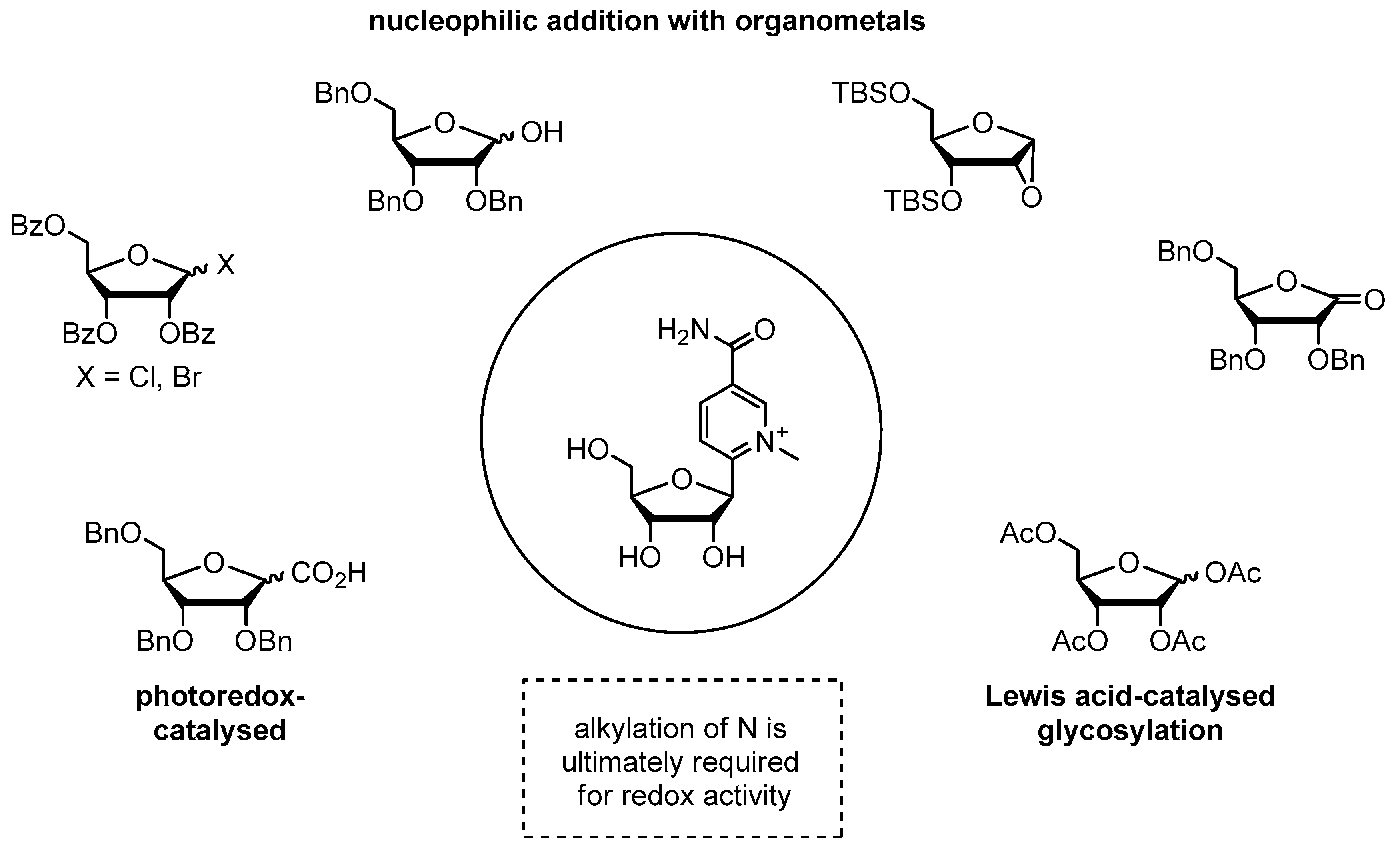
Figure 10.
Proposed strategy for synthesis of NCRs by the phosphoramidite approach. The Zincke reaction enables access to partially protected nucleosides from pre-functionalized sugar moieties.
Figure 10.
Proposed strategy for synthesis of NCRs by the phosphoramidite approach. The Zincke reaction enables access to partially protected nucleosides from pre-functionalized sugar moieties.
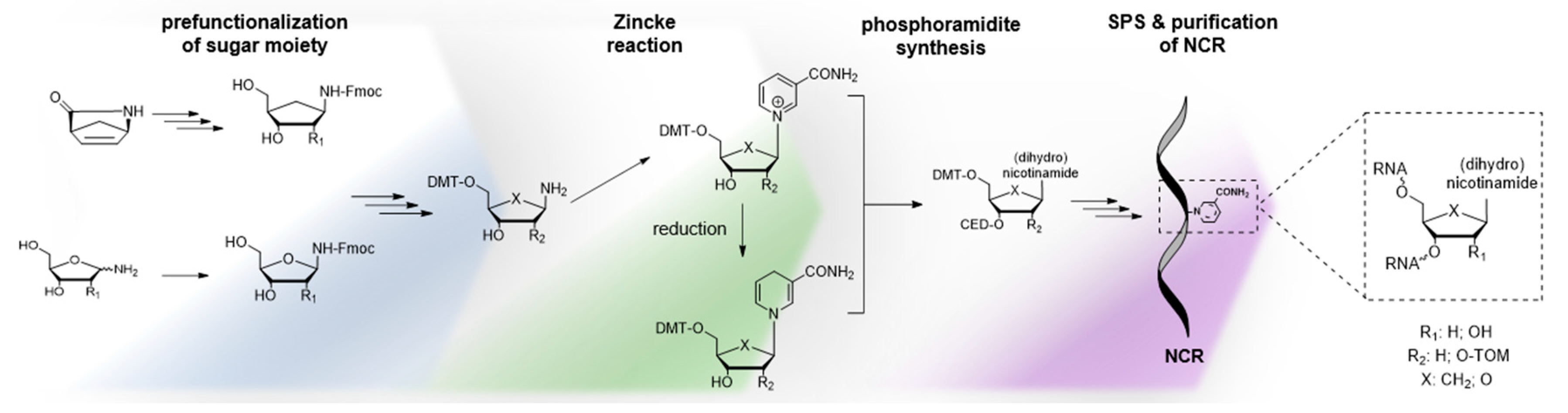
Figure 11.
Hypothetic outcome of chemical solid phase synthesis using NARH (left) or NAR (right) phosphoramidite.
Figure 11.
Hypothetic outcome of chemical solid phase synthesis using NARH (left) or NAR (right) phosphoramidite.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



