
Submitted:
15 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
Every individual at some point encounters the progressive biological process of aging which is considered one of the major risk factors for common diseases. The main drivers of aging are oxi-dative stress, senescence, and Reactive oxygen species (ROS). The renin-angiotensin-aldosterone system (RAAS) includes several systematic processes for the regulation of blood pressure which is caused by an imbalance of electrolytes. During activation of RAAS, binding of angiotensin II (ANG II) to angiotensin II type I receptor (AGTR1) activates intracellular NADPH oxidase to generate superoxide anions and promote uncoupling of endothelial NO synthase, which in turn decreases NO availability and increases ROS production. Promoting oxidative stress and DNA damage mediated by ANG II is tightly regulated. Individuals with sodium deficiency-associated diseases such as Gitelman syndrome (GS) and Bartter syndrome (BS) show down-regulation of inflammation-related processes and have reduced oxidative stress and ROS. Additionally, the histone deacetylase-Sirtuin-1(SIRT1) has a significant impact on the aging process with reduced activity with age. However, GS/BS patients generally sustain higher levels of SIRT1 activity than aged-matched healthy individuals.
In this review, we highlight the importance of the hallmarks of aging, inflammation, and the RAAS system in GS/BS patients and how this might impact healthy aging. We further propose fu-ture research directions for studying the etiology of GS/BS at the molecular level using pa-tient-derived renal stem cells and induced pluripotent stem cells.
Keywords:
Sodium deficiency diseases
; distal convoluted tubules
; Renin-angiotensin-aldosterone system
; ageing
; sodium accumulation
1. Introduction
The kidney is a paired organ associated with the urinary system and performs numerous tasks such as filtration of urine and regulation of homeostasis of electrolytes such as sodium, potassium, calcium, magnesium, and phosphate [1].
The processes involved in the regulation of sodium and water balance, including blood pressure and the glomerular filtration rate, have a decisive influence on sodium and water transport in the renal tubule. An imbalance is accompanied by undesirable consequences. Several diseases are associated with sodium deficiency, these include Gitelman syndrome (GS) and Bartter syndrome (BS) [2,3,4,5,6].
Gitelman syndrome is an autosomal recessive tubular disorder caused by biallelic mutations in a gene encoding the protein which is a sodium-chloride co-transporter. These are responsible for approximately 7 - 10% of tubular absorption of electrolytes. GS is characterized by an increased excretion of magnesium (hypermagnesiuria) and a reduced excretion of calcium (hypocalciuria) in the urine. In addition, GS patients have hypokalemic alkalosis, i.e., a disorder of the acid-base balance and salt loss. Other signs of illness include muscle weakness, fatigue, cramps, and low blood pressure caused by an electrolyte imbalance due to distinct mutations within the SLC12A3 encoding gene [7,8,9].
Each individual at some point encounters the progressive biological process of aging associated with oxidative stress, senescence, and reactive oxygen species. GS/BS patients show down-regulation of inflammation-related processes and have reduced oxidative stress and ROS. In this context, GS, and BS patients in comparison to healthy individuals sustain higher levels of SIRT1 activity, which seem to have a significant impact on hypertension and the aging process.
We highlight the importance of the hallmarks of aging and the renin-angiotensin-aldosterone system in Gitelman syndrome patients and how this might impact on healthy aging.
2. Gitelman Syndrome
The nephrologist Hillel Jonathan Gitelman first observed patients with various conditions associated with metabolic alkalosis, electrolyte imbalance, and salt wasting in the late 1960s [8]. Gitelman syndrome (GS) (OMIM 263800), is one of the most common renal tubulopathy and is known as familial hypokalemia-hypomagnesemia. It is an autosomal recessive inherited disorder caused by a disturbance in the function of the thiazide-sensitive sodium chloride co-transporter (NCC) in the kidney. In this process, blood levels show magnesium and potassium deficiency, while the urine has high levels of magnesium excretion and decreased excretion of calcium. GS is characterized by metabolic alkalosis, hypomagnesemia, and hypercalciuria. The etiology is a transport defect of the distal convoluted tubule (DCT). It is manifested in adolescence or adulthood and has an estimated prevalence of 1:40.000 Caucasian individuals [10]. It is more prevalent in the Asian population and is estimated at around 1.7 per 1000 people [10]. The actual frequency of GS is difficult to determine in the general population due to its inconspicuousness or misdiagnosis.
The disease is caused by biallelic inactivating mutations within the SLC12A3 encoding gene, which encodes the thiazide-sensitive sodium chloride co-transporter-NCC. Its genomic locus is chromosome 16q13 and consists of 26 exons that code for 1002 amino acids [9,11,12].
NCC is expressed by cells of the apical membrane lining the distal convoluted tubule [9]. Studies have identified more than 350 distinct mutations within the SLC12A3 encoding gene in GS patients [13,14,15]. In addition to this, mutations in the cation channels subfamily 6 of the protein Claudin 16 gene (TRPM6) which is involved in magnesium transport in distal tubules [16,17].
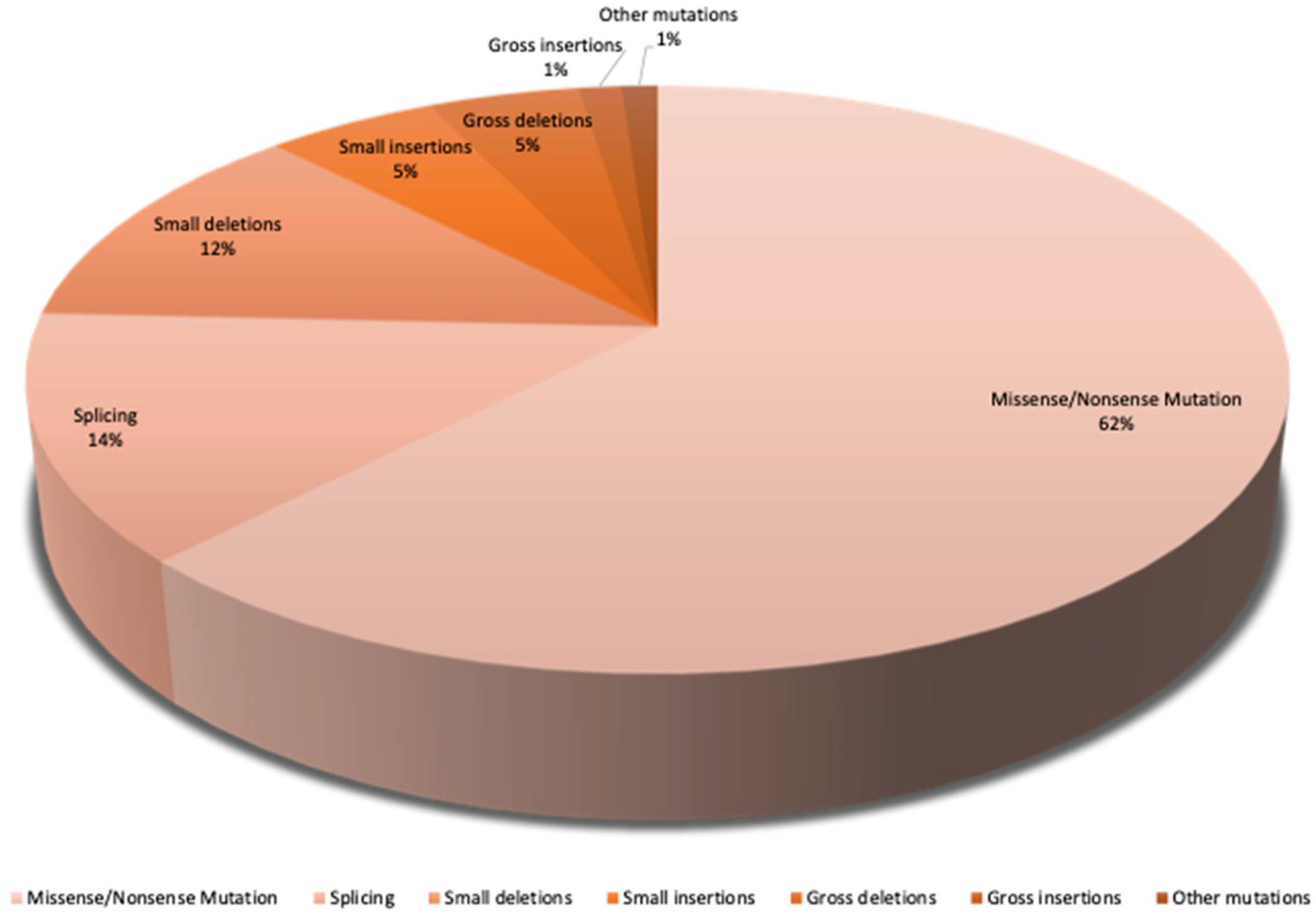
The largest proportion of mutations in SLC12A3 is 62,1% missense and nonsense mutations. A missense mutation is when the error in spelling a single base pair creates a different amino acid. If the mutation is meaningless (nonsense mutation), a so-called stop codon results, and the synthesis of the protein is terminated. Furthermore, the splice mutations are present at around 13,5% and small deletions at around 12%. Finally, there are the major deletions and small insertions at 5%. 7,4% account for other mutations (Figure.1) [15].
The most common mutation is heterozygous, which has a prevalence of 1% in Europe [18]. Studies have shown that homozygous mutations have also been identified in a Spanish cohort [19].
An example of multiple mutations was found in patients in Taiwan. Here, triple mutations in GS patients are presented with a prevalence of approximately 12%, which is certainly due to the higher frequency of heterozygous carriers within the Asian population [20].
Figure 1.
SLC12A3 mutation variants. Gitelman syndrome is caused by biallelic inactivating mutations in the SLC12A3 gene. It consists of 26 exons that code for 1002 amino acids. There are more than >350 different mutations that are caused by missense and nonsense variants, frameshift, splice, and deep intronic variants, as well as large genomic rearrangements. The pie chart represents all variants of mutations in the SLC12A3 gene. The largest population of mutations are missense and nonsense mutations with a prevalence of approximately 62,1%. The splice mutations are present at approximately 14% and small deletions at around 12%. Major deletions and small insertions make up approximately 5%, whereas the remaining mutation types account for 7,4% [15].
Figure 1.
SLC12A3 mutation variants. Gitelman syndrome is caused by biallelic inactivating mutations in the SLC12A3 gene. It consists of 26 exons that code for 1002 amino acids. There are more than >350 different mutations that are caused by missense and nonsense variants, frameshift, splice, and deep intronic variants, as well as large genomic rearrangements. The pie chart represents all variants of mutations in the SLC12A3 gene. The largest population of mutations are missense and nonsense mutations with a prevalence of approximately 62,1%. The splice mutations are present at approximately 14% and small deletions at around 12%. Major deletions and small insertions make up approximately 5%, whereas the remaining mutation types account for 7,4% [15].
The mutations lead to sodium efflux and activation of adaptive mechanisms such as the renin-angiotensin-aldosterone system (RAAS) [21]. The inactivation of NCC leads to the activation of RAAS, which results in reduced tubular re-absorption of sodium and chloride. This deficiency leads to dehydration, which triggers RAAS as a compensatory mechanism. Additionally, low levels of potassium in blood serum have been described, as well as an increase in the levels of renin and aldosterone.
Studies based on GS rodent models revealed that Ca2+ deficiency has significant consequences on DCT function as compromised NCC function leads to structural remodeling of DCTs [22]. The disease can be asymptomatic or associated with mild symptoms, such as weakness, fatigue, craving for salt, thirst, or nocturia. GS patients often have transient episodes of muscle weakness and tetany, sometimes combined with abdominal pain, vomiting, and fever [23,24]. The presence of hypocalciuria and hypomagnesemia has high predictive value for the clinical diagnosis of GS [25,26].
If GS is suspected, there are not only clinical complaints but also biochemical criteria which include: Chronic hypokalemia (<3.5 mmol/L) and renal potassium loss as a spot urine sample with a potassium/creatinine ratio >2.0 mmol/mol (>18 mmol/g)[27].
Hypocalciuric as a spontaneous urine calcium/creatinine ratio <0.2 mmol/mmol (0.07 mg/mg). GS patients have low urinary calcium excretion [28,29], high fractional magnesium excretion, and hypomagnesemia [8,28].
Metabolic alkalosis; hypomagnesemia <0.7 mmol/L (<1.70 mg/dl) [30] and renal magnesium loss (fractional magnesium excretion >4%) [31]. Gitelman syndrome occurs in both males and females alike.
In adults, additional criteria include fractional chloride excretion >0.5%, normal or low blood pressure, and the absence of morphologic and functional renal abnormalities [27,32]. Including increased renin activity and aldosterone expression [33].
GS is caused by a variety of mutations within the genes encoding SLC12A3 (Thiazide-sensitive sodium chloride cotransporter- NCC). HNF1B (Hepatocyte nuclear factor 1β), FXYD2 (Sodium/potassium-transporting ATPase gamma chain - a member of the FXYD family of transmembrane proteins), KCNJ10 (ATP-sensitive inward rectifier potassium channel 10, a member of the inward rectifier-type potassium channel family [13,34,35,36,37]. EAST or SeSAME syndrome closely resembles Gitelman syndrome. This is an autosomal recessive disorder caused by mutations within the KCNJ10 gene. EAST is an acronym for Epilepsy, Ataxia, Sensorineural deafness, and (a renal salt-wasting) tubulopathy where SeSAME stands for Seizures, Sensorineural deafness, Ataxia, Mental retardation, and Electrolyte imbalance [35,36,38].
Mitochondria are essential components of eukaryotic cells. Mitochondrial DNA (mtDNA)mutations can occur during mitosis, which then results in changes in the degree of heteroplasmy. If the proportion of mtDNA exceeds the threshold value of 60-80% [39], this results in a considerable drop in energy production leading to the development of metabolic disorders. Pathogenic mitochondrial DNA (mtDNA) variants of genes encoding the transfer RNAs for phenylalanine (MT-TF) and isoleucine (MT-TI) cause a Gitelman-like syndrome [40].
Genetic counseling has an important role in this autosomal recessive disease. The risk of recurrence is 25% for parents with a child diagnosed with GS. Since the clinical symptoms may appear later, DNA analysis can be used to determine whether GS is present. For adult patients with GS, the risk of having children with GS is low (~1 in 400) unless the patient and their partner are blood related.
There is currently no specific therapy, but potassium and magnesium supplementation and a high-salt diet are recommended for all GS patients. To counteract disturbed hormone levels, fluid and electrolyte balance, spironolactone or triamterene are prescribed to GS patients to reduce potassium and magnesium losses [41]. In GS patients, a high dose of potassium chloride is used to combat hypokalemia. Salts with poor absorbable anions such as aspartan should not be administered to patients, as these do not lead to any improvement and may even exacerbate alkalosis.
3. Bartter Syndrome
In 1962, Bartter et al. identified a new syndrome and named it Bartter syndrome (BS) [44]. BS is one of the rare hereditary renal tubular disorders caused by impaired salt re-absorption in the thick ascending limb (TAL) of the loop of Henle. It is associated with several electrolyte abnormalities including low potassium and chloride, salt wasting, hypokalemia, and metabolic alkalosis with hyperaldosteronism with normal blood pressure and hyperplasia of the juxtaglomerular apparatus (JGA) [44,45].
In contrast to GS, Bartter syndrome is a more severe congenital kidney injury that often manifests itself earlier, usually within the first year of life [46]. BS is a polygenic disease caused by homozygous or mixed heterozygous mutations in one of the following genes: SLC12A1, KCNJ1, CLCNKB, BSND or CASR (Figure. 2).
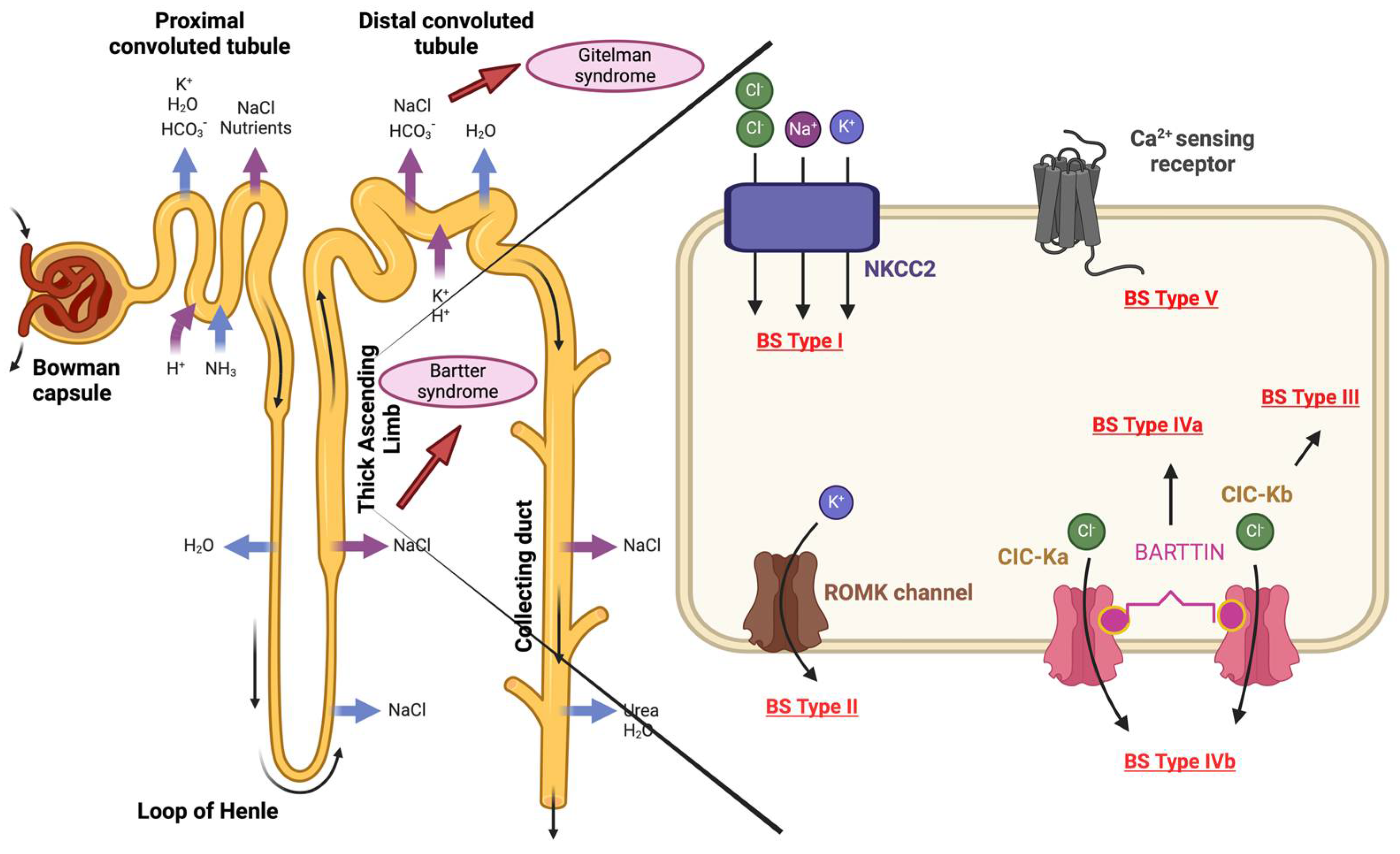
Depending on the mutated gene and thus the affected transporter protein, the BS can be divided into at least five subtypes, which are expressed in the tubular epithelial cells of the TAL of the Henle loop. Type I BS is characterized by loss-of-function mutations within the SLC12A1 gene and is known as neonatal Bartter syndrome (hyperprostaglandin E syndrome). This codes for the apical sodium-potassium-chloride co-transporter NKCC2. Type II of BS, also known as neonatal Bartter syndrome, is caused by loss-of-function mutations in the KCNJ1 gene, which codes for the apical inwardly rectifying potassium channel ROMK. BS type III, also known as classic Bartter syndrome, is caused by mutations in the CLCNKB gene, which codes for the basolateral chloride channel- ClC-Kb. Distinct variants of chloride channels are present in most organs but CLCNKB affected here is found exclusively in the kidney, so the clinical manifestation of a mutation in this channel is only seen in the kidney [47].
Classical Bartter syndrome with sensorineural hearing loss (hyperprostaglandin E syndrome), or BS type IVa, is caused by loss-of-function mutations in the BSND gene, which encodes Barttin, a β-subunit for ClC-Ka and ClC-Kb. Parallel to BS type IVa, simultaneous mutations in CIC-Ka and CIC-Kb are present in type IVb.Bartter syndrome type V with hypocalcemia is also known as autosomal dominant hypoparathyroidism. This is characterized by gain-of-function mutations in the Ca2+-sensing receptor-CASR, which codes for the basolateral calcium receptor CASR.
Figure 2.
Mutations associated with Bartter syndrome. This is caused by homozygous or mixed heterozygous mutations in one of the following genes: SLC12A1, KCNJ1, CLCNKB, BSND or CASR. Depending on the mutated gene, BS can be divided into five subtypes. BS Type I: SLC12A1 codes for the sodium-potassium-chloride co-transporter NKCC2. BS Type II: KCNJ1 codes for the potassium channel- ROMK. BS Type III: CLCNKB codes for chloride channel- ClC-Kb. BS Type IVa: BSND codes for BARTTIN which is a subunit of the chloride channels ClC-Ka and ClC-Kb. BS Type IVb- simultaneous mutations in CIC-Ka and CIC-Kb. BS Type V: CASR, which encodes the basolateral calcium receptor- CASR. Figure 2 was created with BioRender.com.
Figure 2.
Mutations associated with Bartter syndrome. This is caused by homozygous or mixed heterozygous mutations in one of the following genes: SLC12A1, KCNJ1, CLCNKB, BSND or CASR. Depending on the mutated gene, BS can be divided into five subtypes. BS Type I: SLC12A1 codes for the sodium-potassium-chloride co-transporter NKCC2. BS Type II: KCNJ1 codes for the potassium channel- ROMK. BS Type III: CLCNKB codes for chloride channel- ClC-Kb. BS Type IVa: BSND codes for BARTTIN which is a subunit of the chloride channels ClC-Ka and ClC-Kb. BS Type IVb- simultaneous mutations in CIC-Ka and CIC-Kb. BS Type V: CASR, which encodes the basolateral calcium receptor- CASR. Figure 2 was created with BioRender.com.
Bartter syndrome is an inherited disorder of the renal tubules caused by impaired salt reabsorption. It leads to several electrolyte abnormalities including low potassium and chloride, salt wasting, hypokalemia, and metabolic alkalosis [6,48].
Previously, GS and BS patients were classified based on age, the presence of hypercalciuria, polyhydramnios or growth retardation, and severity. Based on differences in the metabolism of divalent cations, BS and GS can be distinguished by laboratory measurements which include urinary Ca2+- excretion, Ca2+/Creatinine, and serum Mg2+. BS patients tend to have a less severe manifestation compared to GS patients. BS patients have increased urinary calcium excretion and fractional excretion rates, whereas serum magnesium levels are within the normal range [47]. To differentiate between BS and GS, a thiazide test was proposed using a cohort of genetically proven GS and BS patients given oral administration of hydrochlorothiazide (1 mg/kg to 50 mg) [49]. In patients with GS, fractional chloride excretion showed very little change (<2.3%) as the defect was in the thiazide-sensitive Na/Cl cotransporter (NCCT). In contrast, patients with BS showed an increased response to HCO3 administration, which led to a more pronounced excretion. Despite the successful differentiation between GS and BS, the test is not routinely recommended due to the increased risk of volume depletion [49].
Due to the rare nature of the syndrome, there is only limited clinical information on treatment. Therapeutically, BS patients urgently need sufficient fluid and electrolyte replacement. For newborns, it is important to control the risk of pre-renal failure due to imbalances in fluid homeostasis. The classic pharmacological therapy for BS patients include the administration of potassium chloride, prostaglandin inhibitors (indomethacin), and aldosterone antagonists (spironolactone) [50,51].
The symptoms of BS patients are initially treated with the help of potassium supplementation. Also, potassium-sparing diuretics such as spironolactone, eplerenone, or amiloride are often prescribed. According to the literature, these diuretics lead to reduced aldosterone levels, which subsequently increases serum potassium and hence counteracting metabolic alkalosis. In cases of persistent hypokalemia and or other side-effects of medications, potassium-sparing diuretics, renin-angiotensin system blockers, or NSAIDs are also prescribed. Combinations of these medications are also recommended in certain cases [42,45,52]. Angiotensin-converting enzyme inhibitors (ACE inhibitors) are used to correct low K+ levels or to counteract proteinuria [52,53].
Table 1 Comparisons between Gitelman and Bartter syndrome. Gitelman syndrome (GS) and Bartter syndrome (BS) have distinct genetic mutations. BS is divided into five subtypes depending on the affected gene (I, II, III, IVa, IVb, V). NKCC2: Furosemide-sensitive sodium-potassium-2-chloride cotransporter; ROMK: Renal outer medullary potassium channel; CLC-Kb: Chloride channel Kb; CLC-Ka: Chloride channel Ka; CaSR: Calcium-sensing receptor; Gitelman syndrome is caused by biallelic inactivating mutations in the SLC12A3 gene. NCC: Thiazide-sensitive sodium chloride cotransporter. MT-TF: Mitochondria encoded tRNA-Phe (UUU/C)); KCNJ1: Potassium Inwardly Rectifying Channel Subfamily J Member 10; FXYD2: FXYD Domain Containing Ion Transport Regulator 2; HNF1B: hepatocyte nuclear factor 1-beta. Modified version from Fulchiero et al. [54].
4. Aging and RAAS
Aging is a biological process that affects all organizational levels of an organism. The aging process is considered one of the major risk factors for the most prevalent diseases worldwide [55]. The hallmarks of aging contribute to processes leading to the aging phenotype which include epigenetic alterations, chronic inflammation, dysbiosis, deregulated nutrient sensing, mitochondrial dysfunction, genomic instability, telomere attrition, loss of proteostasis, deregulated macro-autophagy, stem cell exhaustion, altered intercellular communication and cellular senescence [56,57,58]. Cellular aging is the result of the accumulation of damaged macromolecules, which are chemically altered by reactive oxygen species (ROS). ROS impairs the integrity and function of mitochondria, resulting in, for example, reduced ATP generation [59].
The renin-angiotensin-aldosterone system (RAAS) includes several systematic processes for the regulation of blood pressure. This is a consequence of increasing peripheral resistance or vascular tone and cardiac contractility, as well as enhanced water and sodium reabsorption, increased aldosterone secretion, and facilitated catecholamine release from sympathetic nerve terminals [1]. Regulation of hypertension and extracellular volume is regulated by Angiotensin II (ANG II) as the biological effector of RAAS [60,61,62]. ANG II preferentially binds two subtypes of Angiotensin II receptors- AT1 (AGTR1) and AT2 (AGTR2). AGTRs are expressed in various segments of the nephron in the distal tubule, collecting duct, and renal vasculature [63,64,65,66,67,68]. The known physiological and pathological effects are induced via the AT1 receptor. In humans, AGTR1 receptors are expressed in a wide variety of organs. They are present in glomerular mesangial cells, proximal and distal tubular epithelial cells, medullary interstitial cells, and renal vasculature [69,70,71]. Binding of ANG II to AGTR1 activates four classical signaling cascades, including phospholipase A2, phospholipase C, phospholipase D, and L-type calcium channels, and normally blocks the adenylate cycle.
ANG II binding to renal AGTR1 initiates vasoconstriction, sodium reabsorption, protein synthesis, and cellular growth [71,72].
In comparison, the activation of AGTR2 mediates K+ channel activity via the protein-coupled receptors and activates protein tyrosine phosphatase (PTP), which leads to a reduction in the activities of MAPK and ERK1 signaling pathways [73,74]. Activation of AGTR2 leads to increased bradykinin production, which induces vasodilation via the nitric oxide (NO)/cyclic guanosine monophosphate (GMP) pathway [71]. ANG II binding to AGTR2 initiates vasodilation, differentiation, apoptosis, anti-proliferation, and sodium transport imbalance of membrane ionic effects [75].
Both types of receptors are involved in the regulation of renal hemodynamics, tubular function, renal cellular growth, and matrix formation [71].
During activation of RAAS, binding of ANG II to AGTR1 leads intracellular NADPH oxidase to generate superoxide anions and promotes uncoupling of endothelial NO synthase, which in turn decreases NO availability, and increases ROS production. The promotion of oxidative stress and DNA damage mediated by ANG II is tightly regulated. However, if uncontrolled, ANGII-mediated ROS production occurs, this leads to cellular senescence associated with mitochondrial dysfunction, hypertrophy, inflammation, and fibrosis in the kidneys, heart, and brain [76,77]. ANG II-mediated signaling via AGTR1 additionally leads to shortening of telomeres and ultimately glomerular mesangial cell senescence but this can be counteracted by losartan [78].
The mammalian ortholog of the silent information regulator (Sir2) of yeast, sirtuin-1 (SIRT1) is an NAD+-dependent class III histone deacetylase involved in many aging-associated processes such as apoptosis, cell differentiation, development, stress response, metabolism, and tumorigenesis [39,57,79]. SIRT1 expression is reduced in senescent mesenchymal stem cells, whereas its over-expression slows the onset of senescence and loss of differentiation capacity [80]. SIRT1 is associated with longevity, and in a hypertensive patient, low SIRT1 levels lead to an acceleration of the aging process [81,82]. The same is true for endothelial progenitor cells induced by ANG II, which presumably have reduced SIRT1 expression [83,84]. To counteract this, studies have been conducted investigating the role of polyphenol resveratrol in relation to hypertension [85,86]. Resveratrol (3,5,4′-trihydroxystilbene) is a polyphenol found in red wine that has several beneficial effects on cardiovascular diseases [87,88]. In mammalian cells, resveratrol can promote various cellular functions and signaling pathways including longevity, cell cycle regulation, apoptosis, DNA damage repair, and muscle differentiation through the activation of SIRT1 [89,90]. In a study carried out by Miyazaki et. al., resveratrol intake was shown to reduce AGTR1 expression in vascular smooth muscle cells [85]. Furthermore, Resveratrol has been shown to downregulate AGTR1 expression via SIRT1 and has beneficial effects on hypertension[85]. Currently, several approaches can be employed to counteract premature aging such as caloric restriction, strength training, or nutrition [91,92,93,94,95]. In DNA repair mutant mice, caloric restriction reduced ROS and other reactive compounds, resulting in lower levels of DNA damage [96].
According to a study by the Robert Koch Institute, around every third person in Germany and therefore around 20 - 30 million German citizens are afflicted with high blood pressure [97]. Worldwide, it is about a fourth of the world's population. The prevalence of high blood pressure increases with age, with almost two-thirds of people aged 65 and over having a diagnosis of high blood pressure [97]. Globally, hypertension is responsible for ten million deaths each year and is a critical risk factor for cardiovascular disease.
Multiple key mechanisms including inflammation, oxidative stress as well endothelial dysfunction lead to accelerated aging and hypertension. They can occur either independently or together [98,99,100]. Hypertension is an age-associated disease as illustrated in Figure 3.
The hallmarks of aging in the etiology of sodium deficiency-associated diseases.
The aging process is one of the main causes of cancer, metabolic disorders, heart and kidney diseases. Aged tissue is accompanied by a progressive loss of physiological integrity, which leads to dysfunction and is prone to death. The accumulation of Reactive Oxygen and Nitrogen Species (RONS)-induced cellular and organ damage is often associated with aging. RONS are generated by various endogenous and exogenous processes, which are normally neutralized by antioxidant defenses, e.g., lipoic, and uric acid, coenzyme Q. When an imbalance occurs between the attack and defense system, oxidative stress ensues. Oxidative stress is part and parcel associated with various age-related diseases. The formation of RONS can be attributed to endogenous as well as exogenous sources.
Exogenous sources of RONS include tobacco, alcohol, heavy metals, air and water pollution, radiation, etc., which are subsequently converted into free radicals [101]. Endogenous sources include, for example, myeloperoxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and Angiotensin II [102]. NADPH is an enzyme complex consisting of several subunits that uses nicotinamide adenine dinucleotide phosphate to produce superoxide anions (O2 •–) [103]. Here, most of the di-oxygen is dismutated into hydrogen peroxide[101]. This can form the highly reactive ROS hydroxyl ion (OH-) by a Fenton or Haber-Weiss reaction. In addition, nitric oxide, which is formed from L-arginine by nitric oxide synthase (NOS), is also one of the endogenous sources. This can be divided into three main isoforms: epithelial NOS, neuronal NOS, and inducible NOS. Epithelial NOS is associated with vasodilation and vascular regulation and has a fundamental link with RAAS as well as with tubular disorder diseases such as Gitelman syndrome and Bartter syndrome (Figure.4) [104].
The cortical segments of the nephron have a high density of mitochondria [105,106]. Mitochondria not only play an essential role in the metabolism of the kidney but also signal transmission, and they facilitate stress reactions of kidney cells. Mitochondrial defects such as mutations in nuclear or mitochondrial DNA (mtDNA) are common causes of mitochondrial dysfunction leading ultimately to the development of multiple kidney diseases [107,108,109]. In detail, permanent mitochondrial dysfunction can lead to inflammation, oxidative stress, loss in cellular functions and structure of renal cells, as well as reduced adenosine triphosphate (ATP) production [105,110,111]. These consequences are also linked to the aging process.
Mutations within mtDNA can result in Gitelman and Bartter syndrome [40,112,113]. Active mitochondria increase ROS levels thus activating the renin-angiotensin-aldosterone system (RAAS). This is associated with increased NCC activity and thus with the regulation of DCT-specific NaCl re-absorption [114].
In a healthy person, kidney function often declines, leading to an accumulation of metabolic waste products and excessive electrolytes. Low potassium concentration associated with excessive salt concentration appears to be a highly prevalent cause of hypertension. High sodium intake induces intra-renal and vascular RAAS activation, which ultimately produces pro-inflammatory and pro-fibrotic conclusions [115,116,117].
GS and BS patients have salt wasting and are protected from the well-documented increase in sodium re-absorption and retention promoted by Angiotensin II. Excessive sodium consumption leads to a significant increase in blood pressure linked to hypertension and cardiovascular diseases [118,119].
GS patients have reduced serum phosphate [120], Ca2+ reduction [121] and PKC activation in contrast to a healthy individual [119,121,122]. PKC is a negative regulator of endothelial nitric oxide synthase (eNOS) and has a positive effect on vasodilation in GS and BS patients as evidenced by increased urinary eNOS expression. Thus, it can be concluded that increased eNOS and NO-mediated vasodilation in GS/BS patients is crucial for the response of endothelial cells to increased flow in the brachial artery [32,123,124].
ANG II induces a reduction in the number of endothelial progenitor cells (EPCs) which are crucial for regulating heart diseases such as hypertension [124]. ANG II binding to AGTR1 leads to induced senescence of EPCs in mice and the development of oxidative stress [125].
In GS/BS patients, senescence as well as the development of oxidative stress remains under control at non-deleterious levels. It appears that in GS/BS patients, there is drastically reduced hypertension, cardiac remodeling, and the reduction of oxidative stress and its associated proteins despite constitutive RAAS stimulation secondary to salt wasting [123,125]. In hypertensive patients, ACE inhibitors (e.g., ramipril, benazapril, and captopril) or AGTR1 antagonists (e.g., losartan, candesartan, and irbesartan) are used to counteract AGTR1-modulated effects [61,62,72,126]. Mammalian aging is characterized by somatic mutations and other forms of DNA damage such as chromosomal abnormalities [127]. When these changes occur, cell cycle is arrested in the G1 phase which is triggered by TP53, TP22 as well as TP16 [57,128].
Four distinct processes can be initiated depending on the cell type, transient cell cycle arrest associated with DNA repair, apoptosis, senescence or cell differentiation [129]. A connection between TP53 and Gitelman syndrome is currently not established. According to the literature, p22/PRG1 is a novel target gene of p53 target [130]. Caló et al. reported that GS/BS patients have less oxidative stress and low levels of p22 expression [131]. In relation to p53, this may imply that GS/BS patients have lower p53 activity and consequently the activation of p22 is lower [83,127,132].
5. Conclusions
Salt intake is a determinant of high blood pressure and as such has an important role in the sensitivity of hypertensive responses. Salt-sensitive individuals show strong hypertension responses compared to those who are salt-resistant [134,135]. Several studies related to hypertension and salt intake have also been carried out in animal and human models [136]. Several diseases are associated with magnesium deficiency such as Gitelman and Bartter syndromes. The binding of Angiotensin II to AGTR1 leads to ROS production and subsequently to cellular senescence associated with mitochondrial dysfunction, hypertrophy, inflammation, and renal fibrosis [76,77,137]. However, none of these hypertensive consequences have been observed to date in GS/BS patients despite their high salt medication [138,139]. GS and BS patients show down-regulation of inflammation-related processes and have reduced oxidative stress and ROS production. Explanations for this may include reduced release of messenger substances by AGTR1 [140], reduced Ca2+ release and PKC activation [140], or higher SIRT1 expression [133]. As these reports are sparse, more studies are needed to confirm these observations.
Another link can be made to immune deficiency in patients with salt-wasting tubulopathies, including GS [141]. Here, the effects of salt wasting may provide a further link between GS/BS and protection against some effects of aging by analyzing the signaling pathways and excreted proteins. The addition of salt as a therapeutic intervention, which is already used as therapy today, could play a crucial role in this process.
These findings should not only apply to GS/BS but could also provide general links to salt-wasting tubulopathies.
6. The Future Direction of Research
We previously demonstrated that urine can serve as a non-invasive source of bipotential SIX2-positive renal progenitor cells [142], which can be differentiated into renal tubular cells and podocytes [60,61,142]. These cells can be employed for studying kidney-associated aging processes [57], ANGII-mediated RAAS activation [60,61], and reprogrammed into induced pluripotent stem cells to enable studying the effects of a mutation on other cell types [143].
We are currently isolating SIX2-positive urine derived progenitor cells from GS and BS patients with the aim of differentiating these into either tubular cells or podocytes which will be ideal for studying the effect of Angiotensin II and the RAAS pathway in these patients.
More recently, iPSCs have been generated from GS patients harboring the common Asian mutations [144,145]. GS and BS patient-derived iPSCs will enable far-reaching investigations using revolutionized gene editing methods such as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) to correct the mutation(s) in the GS/BS patient-derived iPSC lines to provide isogenic disease and healthy lines which can be differentiated either to tubular cells or podocytes to study disease mechanisms and also toxicology and drug screening. There are GS and BS knockout mice which can be used to investigate the link between hypertension, oxidative stress and aging [146,147,148,149,150,151]. With these human cellular and mouse models the path is now paved for increased efforts in uncovering the underlying etiology leading to Gitelman and Bartter syndrome. These cellular models will enable drug screening/development and toxicology studies thus leading to better and more targeted treatment options in the near future.
Author Contributions
Conceptualization, C.T. and J.A. Original draft preparation, C.T. Editing, C.T. and J.A.; Figures, C.T.
Funding
This research was funded by the Medical faculty-Heinrich-Heine University, Duesseldorf.
Institutional Review Board Statement
Not applicable.
Acknowledgments
James Adjaye acknowledges funding from the medical faculty of Heinrich Heine University, Duesseldorf, Germany.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fountain, J.H.; Kaur, J.; Lappin, S.L. Physiology, Renin Angiotensin System. StatPearls Publishing LLC.; 2023.
- Walsh, S.B.; Unwin, R.J. Renal tubular disorders. Clin Med (Lond). 2012, 12, 476–9. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat Rev Dis Primers. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; Benigni, A.; Remuzzi, A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. JCI. 2006, 288–96. [Google Scholar] [CrossRef] [PubMed]
- Currie, G.; Delles, C. Proteinuria and its relation to cardiovascular disease. Int J Nephrol Renovasc Dis. 2014, 13–24. [Google Scholar]
- da Silva Cunha, T.; Pfefermann Heilberg, I. Bartter syndrome: causes, diagnosis, and treatment. Int J Nephrol Renovasc Dis. 2018, 11, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Shaer, A.J.; Bia, M.J.; Lifton, R.P.; Simon, D.B. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney International. 2001, 59, 710–7. [Google Scholar] [CrossRef] [PubMed]
- Gitelman, H.J.; Graham, J.B.; Welt, L.G. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966, 79, 221–35. [Google Scholar] [PubMed]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; Gainza, F.J.; Gitleman, H.J.; Lifton, R.P. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nature Genetics. 1996, 12, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Nagano, C.; Ishiko, S.; Omori, T.; Aoto, Y.; Rossanti, R.; Sakakibara, N.; Horinouchi, T.; Yamamura, T.; Nagai, S.; Okada, E.; Shima, Y.; Nakanishi, K.; et al. Examination of the predicted prevalence of Gitelman syndrome by ethnicity based on genome databases. Scientific Reports. 2021, 11. [Google Scholar]
- San-Cristobal, P.; de los Heros, P.; Ponce-Coria, J.; Moreno, E.; Gamba, G. WNK Kinases, Renal Ion Transport and Hypertension. Am J Nephrol. 2008, 28, 860–70. [Google Scholar] [CrossRef]
- Gamba, G. Role of WNK kinases in regulating tubular salt and potassium transport and in the development of hypertension. American Journal of Physiology - Renal Physiology. 2005, 288, 245–52. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Mishima, E.; Shima, H.; Akiyama, Y.; Suzuki, C.; Suzuki, T.; Kobayashi, T.; Suzuki, Y.; Nakayama, T.; Takeshima, Y.; Vazquez, N.; Ito, S.; Gamba, G.; et al. Exonic Mutations in the SLC12A3 Gene Cause Exon Skipping and Premature Termination in Gitelman Syndrome. J Am Soc Nephrol. 2015, 26, 271–9. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Poussou, R.; Dahan, K.; Kahila, D.; Venisse, A.; Riveira-Munoz, E.; Debaix, H.; Grisart, B.; Bridoux, F.; Unwin, R.; Moulin, B.; Haymann, J.-P.; Vantyghem, M.-C.; Rigothier, C.; et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011, 22, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003, 21, 577–81. [Google Scholar] [CrossRef]
- Hsu, Y.-J.; Hoenderop, J.G.; Bindels, R.J.M. TRP channels in kidney disease. Biochim Biophys Acta. 2007, 1772, 928–36. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N.V.A. Inherited forms of renal hypomagnesemia: an update. Pediatr Nephrol. 2009, 24, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Chen, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nature Genetics volume. 2008, 40, 592–9. [Google Scholar] [CrossRef]
- Herrero-Morín, J.D.; Rodríguez, J.; Coto, E.; Gil-Peña, H.; Alvarez, V.; Espinosa, L.; Loris, C.; Gil-Calvo, M.; Santos, F. Gitelman syndrome in Gypsy paediatric patients carrying the same intron 9 + 1 G>T mutation. Clinical features and impact on quality of life. Nephrol Dial Transplant. 2011, 26, 151–5. [Google Scholar] [CrossRef]
- Tseng, M.-H.; Yang, S.-S.; Hsu, Y.-J.; Fang, Y.-W.; Wu, C.-J.; Tsai, J.-D.; Hwang, D.-Y.; Lin, S.-H. Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation. J Clin Endocrinol Metab. 2012, 97, 1478–82. [Google Scholar] [CrossRef]
- Wang, L.; Dong, C.; Xi, Y.-G.; Su, X. Thiazide-sensitive Na+-Cl- cotransporter: genetic polymorphisms and human diseases. Acta Biochim Biophys Sin (Shanghai). 2015, 47, 325–34. [Google Scholar] [CrossRef]
- Loffing, J.; Loffing-Cueni, D.; Valderrabano, V.; Kläusli, L.; Hebert, S.C.; Rossier, B.C.; Hoenderop, J.G.; Bindels, R.J.; Kaissling, B. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. American Journal of Physiology - Renal Physiology. 2001, 281, F1021–7. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N.V.A.M.; Levtchenko, E. Gitelman syndrome. Orphanet J Rare Dis. 2008, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Fedeli, C.; Moroni, L.; Cosmai, L.; Badalamenti, S.; Ponticelli, C. Gitelman syndrome: pathophysiological and clinical aspects*. QJM: An International Journal of Medicine. 2010, 103, 741–8. [Google Scholar] [CrossRef] [PubMed]
- Bettinelli, A.; Bianchetti, M.G.; Girardin, E.; Caringella, A.; Cecconi, M.; Appiani, A.C.; Pavanello, L.; Gastaldi, R.; Isimbaldi, C.; Lama, G.; Marchesoni, C.; Matteucci, C.; Patriarca, P.; et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. 1992, 120, 38–43.
- Jeck, N.; Schlingmann, K.P.; Reinalter, S.C.; Kömhoff, M.; Peters, M.; Waldegger, S.; Seyberth, H.W. Salt handling in the distal nephron: lessons learned from inherited human disorders. Am J Physiol Regul Integr Comp Physiol. 2005, 288, 782–95. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calò, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.A.; Konrad, M.; Lin, S.-H.; Vargas-Poussou, R. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International. 2017, 91, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Colussi, G.; Macaluso, M.; Brunati, C.; Minetti, L. Calcium metabolism and calciotropic hormone levels in Gitelman’s syndrome. Miner Electrolyte Metab. 1994, 20, 294–301. [Google Scholar]
- Rudin, A.; Aurell, M.; Wilske, J. Low urinary calcium excretion in Bartter’s syndrome. Scand J Urol Nephrol. 1988, 22, 35–9. [Google Scholar] [CrossRef] [PubMed]
- Agus, Z.S. Hypomagnesemia. J Am Soc Nephrol. 1999, 10, 1616–22. [Google Scholar] [CrossRef]
- Elisaf, M.; Panteli, K.; Theodorou, J.; Siamopoulos, K.C. Fractional excretion of magnesium in normal subjects and in patients with hypomagnesemia. Magnes Res 1. 1997, 10, 315–20. [Google Scholar]
- Ravarotto, V.; Bertoldi, G.; Stefanelli, L.F.; Nalesso, F.; Calò, L.A. Gitelman’s and Bartter’s Syndromes: From Genetics to the Molecular Basis of Hypertension and More. Kidney Blood Press Res. 2022, 47, 556–64. [Google Scholar] [CrossRef]
- Fuhimura, J.; Nozu, K.; Yamamura, T.; Minamikawa, S.; Nakanishi, K.; Horinouchi, T.; Nagano, C.; Sakakibara, N.; Nakanishi, K.; Shima, Y.; Miyako, K.; Nozu, Y.; Morisada, N.; et al. Clinical and Genetic Characteristics in Patients With Gitelman Syndrome. Kidney Int Rep. 2019, 4, 119–25. [Google Scholar] [CrossRef]
- Adalat, S.; Hayes, W.N.; Bryant, W.; Booth, J.; Woolf, A.S.; Kleta, R.; Subtil, S.; Clissold, R.; Colclough, K.; Ellard, S.; Bockenhauer, D. HNF1B Mutations Are Associated With a Gitelman-like Tubulopathy That Develops During Childhood. Kidney Int Rep. 2019, 4, 1304–11. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G.; Arora, R.; Sirimanna, T.; Thompson, D.; et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009, 360, 1960–70. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009, 106, 5842–7. [Google Scholar] [CrossRef] [PubMed]
- Mayan, H.; Farfel, Z.; Karlish, S.J.D. Renal Mg handling, FXYD2 and the central role of the Na,K-ATPase. Physiol Rep. 2018, 6, e13843. [Google Scholar] [CrossRef] [PubMed]
- Suzumoto, Y.; Columbano, V.; Gervasi, L.; Giunta, R.; Mattina, T.; Trimarchi, G.; Capolongo, G.; Simeoni, M.; Perna, A.F.; Zacchia, M.; Toriello, G.; Pollastro, R.M.; Rapisarda, F.; et al. A case series of adult patients affected by EAST/SeSAME syndrome suggests more severe disease in subjects bearing KCNJ10 truncating mutations. Intractable Rare Dis Res. 2021, 10, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature. 2009, 460, 587–91. [Google Scholar] [CrossRef] [PubMed]
- Viering, D.; Schlingmann, K.P.; Hureaux, M.; Nijenhuis, T.; Mallett, A.; Chan, M.M.Y.; van Beek, A.; van Eerde, A.; Coulibaly, J.-M.; Vallet, M. ; et. al. Gitelman-Like Syndrome Caused by Pathogenic Variants in mtDNA. J Am Soc Nephrol. 2022, 33, 305–25. [Google Scholar]
- Yang, L.; Fan, J.; Liu, Y.; Ren, Y.; Liu, Z.; Fu, H.; Qi, H.; Yang, J. Case report: Gitelman syndrome with diabetes: Confirmed by both hydrochlorothiazide test and genetic testing. Medicine (Baltimore). 2023, 102, e33959. [Google Scholar] [CrossRef]
- Nuñez-Gonzale, L.; Carrera, N.; Garcia-Gonzale, M.A. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Bartter and Gitelman Syndromes: A Primer for Clinicians. Int J Mol Sci. 2021, 22, 11414. [Google Scholar] [CrossRef]
- Liaw, L.C.; Banerjee, K.; Coulthard, M.G. Dose related growth response to indometacin in Gitelman syndrome. Arch Dis Child. 1999, 81, 508–10. [Google Scholar] [CrossRef] [PubMed]
- Bartter, F.C.; Pronove, P.; Gill, J.R.; Maccardle, R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med. 1962, 33, 811–28. [Google Scholar] [CrossRef]
- Lee, B.H.; Cho, H.Y.; Lee, H.; Han, K.H.; Kang, H.G.; Ha, I.S.; Lee, J.H.; Park, Y.S.; Shin, J.I.; Lee, D.-Y.; Kim, S.-Y.; Choi, Y.; Cheong, H.I. Genetic basis of Bartter syndrome in Korea. ndt. 2012, 27, 1516–21. [Google Scholar] [CrossRef]
- Konrad, M.; Vollmer, M.; Lemmink, H.H.; Van Den Heuvel, L.P.W.J.; Jeck, N.; Vargas-Poussou, R.; Lakings, A.; Ruf, R.; Deschênes, G.; Antignac, C.; Guay-Woodford, L.; Knoers, N.V.A.M.; Seyberth, H.W.; et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000, 11, 1449–59. [Google Scholar] [CrossRef]
- Kurtz, I. Molecular pathogenesis of Bartter’s and Gitelman’s syndromes. Kidney Int. 1998, 54, 1396–410. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, S.R.A.; Zulfiqar, H.; Mansur, A. Bartter Syndrome. StatPearls Publishing LLC.; 2023.
- Colussi, G.; Bettinelli, A.; Tedeschi, S.; De Ferrari, M.E.; Syrèn, M.L.; Borsa, N.; Mattiello, C.; Casari, G.; Bianchetti, M.G. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007, 2, 454–60. [Google Scholar] [CrossRef] [PubMed]
- Verberckmoes, R.; van Damme, B.; Clement, J.; Amery, A.; Michielsen, P. Bartter’s syndrome with hyperplasia of renomedullary cells: successful treatment with indomethacin. Kidney Int. 1976, 9, 302–7. [Google Scholar] [CrossRef]
- Nascimento, C.L.P.; Garcia, C.L.; Schvartsman, B.G.S.; Vaisbich, M.H. Treatment of Bartter syndrome. Unsolved issue. J Pediatr (Rio J). 2014, 90, 512–7. [Google Scholar] [CrossRef]
- Morales, J.M.; Ruilope, L.M.; Coto, A.; Alcazar, J.M.; Prieto, C.; Nieto, J.; Rodicio, J.L. Long-term enalapril therapy in Bartter’s syndrome. Nephron. 1988, 48, 327. [Google Scholar] [CrossRef]
- Hené, R.J.; Koomans, H.A.; Dorhout Mees, E.J.; vd Stolpe, A.; Verhoef, G.E.; Boer, P. Correction of Hypokalemia in Bartter’s Syndrome by Enalapril. Am J Kidney Dis. 1987, 9, 200–5. [Google Scholar] [CrossRef]
- Fulchiero, R.; Seo-Mayer, P. Bartter Syndrome and Gitelman Syndrome. Pediatr Clin North Am. 2019, 66, 121–34. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.; de Cabo, R.; Mannick, J.; Partridge, L.; van Deursen, J.; Villeda, S. Translational strategies in aging and age-related disease. Nat Med. 2015, 21, 1395–9. [Google Scholar] [CrossRef] [PubMed]
- Lòpez-Otìn, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell. 2013, 153, 1194–217. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, L.; Adjaye, J. Crosstalk between age accumulated DNA-damage and the SIRT1-AKT-GSK3ß axis in urine derived renal progenitor cells. Aging. 2022, 14, 8179–204. [Google Scholar] [CrossRef] [PubMed]
- López-Otìn, C.; Blasco, M.A.; Patridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell. 2023, 186, 243–78. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardò, F.V.; Viña, J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life. 2000, 49, 427–35. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, L.; Thimm, C.; Bohndorf, M.; Rahman, M.S.; Wruck, W.; Adjaye, J. Activation of the Renin–Angiotensin System Disrupts the Cytoskeletal Architecture of Human Urine-Derived Podocytes. Cells. 2022, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Thimm, C.; Erichsen, L.; Wruck, W.; Adjaye, J. Unveiling Angiotensin II and Losartan-Induced Gene Regulatory Networks Using Human Urine-Derived Podocytes. Int J Mol Sci. 2023, 24, 10551. [Google Scholar] [CrossRef]
- Erichsen, L.; Kloss, L.D.F.; Thimm, C.; Bohndorf, M.; Schichel, K.; Wruck, W.; Adjaye, J. Derivation of the immortalized cell line-UM51-PrePodo-hTERT and its responsiveness to Angiotensin II and activation of RAAS. Cells. 2023, 12. [Google Scholar] [CrossRef]
- Barreto-Chaves, M.L.; Mello-Aires, M. Effect of luminal angiotensin II and ANP on early and late cortical distal tubule HCO3- reabsorption. American Journal of Physiology. 1996, 271, F977–84. [Google Scholar] [CrossRef]
- Kwon, T.-H.; Nielsen, J.; Kim, Y.-H.; Knepper, M.A.; Frøkiaer, J.; Nielsen, S. Regulation of sodium transporters in the thick ascending limb of rat kidney: response to angiotensin, I. I. American Journal of Physiology. 2003, 285, F152–65. [Google Scholar] [CrossRef] [PubMed]
- Rocque, B.L.; Babayeva, S.; Li, J.; Leung, V.; Nezvitsky, L.; Cybulsky, A.V.; Gros, P.; Torban, E. Deficiency of the Planar Cell Polarity Protein Vangl2 in Podocytes Affects Glomerular Morphogenesis and Increases Susceptibility to Injury. Journal of the American Society of Nephrology. 2015, 26, 576–86. [Google Scholar] [CrossRef] [PubMed]
- Tojo, A.; Tisher, C.C.; Madsen, K.M. Angiotensin II regulates H(+)-ATPase activity in rat cortical collecting duct. American Journal of Physiology. 1994, 267, F1045–51. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Xu, C.; Zhang, M.; Wang, M.; Min, L.; Qian, C.; Wang, Q.; Ni, Z.; Mou, S.; Dai, H.; Pang, H.; Gu, L. Yes-associated protein regulates podocyte cell cycle re-entry and dedifferentiation in adriamycin-induced nephropathy. Cell Death & Disease. 2019, 10, 915. [Google Scholar]
- Wang, T.; Giebisch, G. Effects of angiotensin II on electrolyte transport in the early and late distal tubule in rat kidney. American Journal of Physiology. 1996, 271, F143–9. [Google Scholar] [CrossRef]
- Zhuo, J.; Maric, C.; Harris, P.J.; Alcorn, D.; Mendelsohn, F. Localization and Functional Properties of Angiotensin II AT1 Receptors in the Kidney: Focus on Renomedullary Interstitial Cells. Hypertension Research. 1997, 20, 233–50. [Google Scholar] [CrossRef] [PubMed]
- Gasparo, M.; Unger, T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacological Reviews [Internet]. 2000 [cited 2021 Aug 22]. Available from: https://www.researchgate. 1234. [Google Scholar]
- Siragy, H.M. AT1 and AT2 Receptor in the Kidney: Role in Health and Disease. Seminars in Nephrology. 2004, 24, 93–100. [Google Scholar] [CrossRef]
- Ames, M.; Atkins, C.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. Journal of Veterinary Internal Medicine. 2019, 33, 363–82. [Google Scholar] [CrossRef]
- Berry, C.; Touyz, A.F.; Dominiczak, R.; Webb, C.; Johns, D.G. Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. AM J Physiol Heart Circ Physiol. 2001, 2337–65. [Google Scholar] [CrossRef]
- Widdop, R.; Jones, E.; Hannan, R.; Gaspari, T. Angiotensin AT2 receptors: cardiovascular hope or hype? British Journal of Pharmacology. 2003, 809–24. [Google Scholar] [CrossRef]
- Nouet, S.; Nahmias, C. Signal Transduction from the Angiotensin II AT2 Receptor. 2000, 1–6.
- Gilliam-Davis, S.; Payne, V.S.; Kasper, S.O.; Tommasi, E.N.; Robbins, M.E.; Diz, D.I. Long-term AT1 receptor blockade improves metabolic function and provides renoprotection in Fischer-344 rats. Am J Physiol Heart Circ Physiol. 2007, 293, H1327–33. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ Res. 2008, 102, 201–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, L.; Li, Y. Change of telomere length in angiotensin II-induced human glomerular mesangial cell senescence and the protective role of losartan. Mol Med Rep. 2011, 4, 255–60. [Google Scholar] [PubMed]
- Brooks, C.; Gu, W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009, 9, 123–8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.-F.; Zhai, C.; Yan, X.-L.; Zhao, D.-D.; Wang, J.-X.; Zeng, Q.; Chen, L.; Nan, X.; He, L.-J.; Li, S.-T.; Yue, W.; Pei, X.-T. SIRT1 is required for long-term growth of human mesenchymal stem cells. J Mol Med (Berl). 2012, 90, 389–400. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Clifton, P.; Wozniak, D.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and, L. H. Cell Metab. 2013, 18, 416–30. [Google Scholar] [CrossRef] [PubMed]
- Jun, P.; Chang-ping, H.; Yuan-Jian, L. Reply to ‘Number and function of circulating endothelial progenitor cells and calcitonin gene-related peptide in hypertension: support from and opportunities in Bartter’s and Gitelman’s syndromes patients. ’ Journal of Hypertension. 2010, 28, 2171. [Google Scholar]
- Liu, T.; Yang, Q.; Zhang, X.; Qin, R.; Shan, W.; Zhang, H.; Chen, X. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. 2020, 257, 118116.
- Miyazaki, R.; Ichiki, T.; Hashimoto, T.; Inanaga, K.; Imayama, I.; Sadoshima, J.; Sunagawa, K. SIRT1, a Longevity Gene, Downregulates Angiotensin II Type 1 Receptor Expression in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2008, 28, 1263–9. [Google Scholar] [CrossRef]
- Jang, I.-A.; Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Effects of Resveratrol on the Renin-Angiotensin System in the Aging Kidney. Nutrients. 2018, 10, 1741. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; Moon, R.C.; Pezzuto, J.M. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997, 275, 218–20. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–47. [Google Scholar] [CrossRef] [PubMed]
- Dryden, S.C.; Nahhas, F.; Nowak, J.E.; Goustin, A.-S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 2003, 23, 3173–85. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004, 73, 417–35. [Google Scholar] [CrossRef] [PubMed]
- Nishikori, S.; Yasuda, J.; Murata, K.; Takegaki, J.; Harada, Y.; Shirai, Y.; Fujita, S. Resistance training rejuvenates aging skin by reducing circulating inflammatory factors and enhancing dermal extracellular matrices. Sci Rep. 2023, 13, 10214. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; White, T.A.; Heeren, A.A.; Hulshizer, C.A.; Saul, D.; Zhang, X.; Molina, A.J.A.; Redman, L.M.; Martin, C.K. ; et. al. Calorie restriction reduces biomarkers of cellular senescence in humans. Aging Cell. 2023, 23. [Google Scholar]
- Dorling, J.L.; Martin, C.K.; Redman, L.M. Calorie restriction for enhanced longevity: The role of novel dietary strategies in the present obesogenic environment. Ageing Res Rev. 2020, 64, 101038. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.W.; Most, J.; Mey, J.T.; Redman, L.M. Calorie Restriction and Aging in Humans. Annu Rev Nutr. 2020, 40, 105–33. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.F.; Hanson, E.D.; Sheaff, A.K. Strength Training as a Countermeasure to Aging Muscle and Chronic Disease. Sports Med. 2012, 41, 289–306. [Google Scholar] [CrossRef]
- Vermeji, W.P.; Dollé, M.E.T.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C.R.; Wu, H.; Roks, A.J.M.; Botter, S.M.; van der Eerden, B.C.; Youssef, S.A.; Kuiper, R.V.; Nagarajah, B.; et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016, 537, 427–31. [Google Scholar] [CrossRef]
- Neuhauser, H.; Kuhnert, R.; Born, S. 12-Monats-Prävalenz von Bluthochdruck in Deutschland. Journal of Health Monitoring. 2017, 2, 57–63. [Google Scholar]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertension Research volume. 2012, 35, 1039–47. [Google Scholar] [CrossRef] [PubMed]
- Buford, T.W. Hypertension and aging. Ageing Research Reviews. 2016, 26, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Zhongjie, S. Aging, Arterial Stiffness, and Hypertensio. Hypertension. 2015, 65, 252–6. [Google Scholar]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal. 2007, 19, 1807–19. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, D.; Bronas, U. Reactive oxygen and nitrogen species: impact on endothelial dysfunction. Nurs Res. 2015, 64, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Rastogi, R.; Geng, X.; Ding, Y. Nicotinamide adenine dinucleotide phosphate oxidase activation and neuronal death after ischemic stroke. Neural Regen Res. 2019, 14, 948–53. [Google Scholar]
- Guo, D.; Liang, S.; Wang, S.; Tang, C.; Yao, B.; Wan, W.; Zhang, H.; Jiang, H.; Ahmed, A.; Zhang, Z.; Gu, Y. Role of epithelial Na+ channels in endothelial function. J Cell Sci. 2016, 129, 290–7. [Google Scholar] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017, 13, 629–46. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010, 92, 1369–77. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: potential therapeutic approaches. Kidney Res Clin Pract. 2020, 39, 244–58. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016, 12, 267–80. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney International. 2017, 92, 1051–7. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res Clin Pract. 2019, 38, 414–26. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Dong, Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–81. [Google Scholar] [CrossRef]
- Goto, Y.; Itami, N.; Kajii, N.; Tochimaru, H.; Endo, M.; Horai, s. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990, 116, 904–10. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; Pizzini, C.; Tessa, A.; Di Giandomenico, S.; Onetti-Muda, A.; Santorelli, F.M.; Bertini, E.; Rizzoni, G. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. 2006, 21, 355–60. [Google Scholar] [CrossRef]
- Gonzalez-Vicente, A.; Hong, N.; Garvin, J.L. Effects of reactive oxygen species on renal tubular transport. Am J Physiol Renal Physiol. 2019, 317, F444–55. [Google Scholar] [CrossRef] [PubMed]
- Kwakernaak, A.J.; Waanders, F.; Slagman, M.C.J.; Dokter, M.M.; Laverman, G.D.; de Boer, R.A.; Navis, G. Sodium restriction on top of renin–angiotensin–aldosterone system blockade increases circulating levels of N-acetyl-seryl-aspartyl-lysyl-proline in chronic kidney disease patients. J Hypertens. 2013, 31, 2425–32. [Google Scholar] [CrossRef]
- Kobori, H.; Nishiyama, A.; Abe, Y.; Navar, L.G. Enhancement of Intrarenal Angiotensinogen in Dahl Salt-Sensitive Rats on High Salt Diet. Hypertension. 2003, 41, 592–7. [Google Scholar] [CrossRef]
- Kocks, M.; Buikema, H.; Gschwend, S.; Boomsma, F.; de Zeeuw, D.; Navis, G. High Dietary Sodium Blunts Effects of Angiotensin-converting Enzyme Inhibition on Vascular Angiotensin I–to–Angiotensin II Conversion in Rats. Journal of Cardiovascular Pharmacology. 2003, 42, 601–6. [Google Scholar] [CrossRef] [PubMed]
- Felder, R.A.; Gildea, J.J.; Xu, P.; Yue, W.; Armando, I.; Carey, R.M.; Jose, P.A. Inverse Salt Sensitivity of Blood Pressure: Mechanisms and Potential Relevance for Prevention of Cardiovascular Disease. Curr Hypertens Rep. 2022, 24, 361–74. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, D.; Calò, L.A.; Favaro, S.; Borsatti, A. Resting and stimulated cytosolic free calcium levels in neutrophils from patients with Bartter’s syndrome. Clin Sci (Lond). 1987, 72, 483–8. [Google Scholar] [CrossRef] [PubMed]
- Verploegen, M.; Vargas-Poussou, R.; Walsh, S.B.; Alpay, H.; Amouzegar, A.; Ariceta, G.; Atmis, B.; Francesco, E.; Nijenhuis, T. Parathyroid hormone and phosphate homeostasis in patients with Bartter and Gitelman syndrome: an international cross-sectional study. Nephrol Dial Transplant. 2022, 37, 2474–86. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; Ceolotto, G.; Milani, M.; Pagnin, E.; van den Heuvel, L.P.; Sartori, M.; Davis, P.A.; Costa, R.; Semplicini, A. Abnormalities of Gq-mediated cell signaling in Bartter and Gitelman syndromes. Kidney Int. 2001, 60, 882–9. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; D’Angelo, A.; Cantaro, S.; Rizzolo, M.; Favaro, S.; Antonello, A.; Borsatti, A. Intracellular calcium signaling and vascular reactivity in Bartter’s syndrome. Nephron. 1996, 72, 570–3. [Google Scholar] [CrossRef]
- Calò, L.A.; Puato, M.; Schiavo, S.; Zanardo, M.; Tirrito, C.; Pagnin, E.; Balbi, G.; Davis, P.A.; Palatini, P.; Pauletto, P. Absence of vascular remodelling in a high angiotensin-II state (Bartter’s and Gitelman’s syndromes): implications for angiotensin II signalling pathways. Nephrology Dialysis Transplantation. 2008, 23, 2804–9. [Google Scholar] [CrossRef]
- Tura, O.; Mills, N.L.; Hadoke, P.W. Does Bartter’s syndrome/Gitelman’s syndrome provide a clinical model for investigating the association between calcitonin gene-related peptide and angiotensin II-mediated senescence of endothelial progenitor cells? J Hypertens. 2010, 28, 2170–1. [Google Scholar] [CrossRef]
- Ravarotto, V.; Simioni, F.; Sabbadin, C.; Pagnin, E.; Maiolino, G.; Armanini, D.; Caló, L.A. Proinflammatory/profibrotic effects of aldosterone in Gitelman’s syndrome, a human model opposite to hypertension. J Endocrinol Invest. 2019, 42, 521–6. [Google Scholar] [CrossRef]
- Atlas, S. The Renin-Angiotensin Aldosterone System: Pathophysiological Role and Pharmacologic Inhibition. J Manag Care Pharm. 2007, 9–20. [Google Scholar] [CrossRef]
- Calò, L.A.; Davis, P.A. Number and function of circulating endothelial progenitor cells and calcitonin gene-related peptide in hypertension: support from and opportunities in Bartter’s and Gitelman’s syndromes patients. J Hypertens. 2012, 28, 2169–70. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Weinberg, R.A. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004, 113, 8–13. [Google Scholar] [CrossRef]
- Sherman, M.H.; Bassing, C.H.; Teitell, M.A. Regulation of cell differentiation by the DNA damage response. Trends Cell Biol. 2011, 21, 312–9. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, H.; Trauzold, A.; Sebens, T.; Deppert, W.; Fölsch, U.R.; Schmidt, W.E. The proliferation-associated early response gene p22/PRG1 is a novel p53 target gene. oncogene. 1998, 16, 2479–87. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; Pagnin, E.; Davis, P.A.; Sartori, M.; Semplicini, A. Oxidative stress-related factors in Bartter’s and Gitelman’s syndromes: relevance for angiotensin II signalling. 2003, 18, 1518–25.
- Pagnin, E.; Paul, D.; Sartori, M.; Semplicini, A.; Pessina, A.C.; Calò, L.A. Rho kinase and PAI-1 in Bartter’s/Gitelman’s syndromes relationship to angiotensin II signaling. Journal of Hypertension. 2004, 22, 1963–9. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.A.; Pagnin, E.; Dal Maso, L.; Caielli, P.; Maiolino, G.; Fusaro, M.; Rossi, G.P.; Calò, L.A. SIRT1, heme oxygenase-1 and NO-mediated vasodilation in a human model of endogenous angiotensin II type 1 receptor antagonism: implications for hypertension. Hypertension Research. 2013, 36, 873–8. [Google Scholar] [CrossRef]
- Morimoto, A.; Uzu, T.; Fujii, T.; Nishimura, M.; Kuroda, S.; Nakamura, S.; Inenaga, T.; Kimura, G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997, 350, 1734–7. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.H.; Fineberg, N.S.; Fineberg, S.E.; Weinberger, M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001, 37, 429–32. [Google Scholar] [CrossRef]
- Joe, B.; Shapiro, J.I. Molecular mechanisms of experimental salt-sensitive hypertension. J Am Heart Assoc. 2012, 1, e002121. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, T.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996, 97, 1916–23. [Google Scholar]
- Sartori, M.; Parotto, E.; Bonso, E.; Semplicini, A.; Palatini, P.; Pessina, A.C.; Calò, L.A. Autonomic nervous system function in chronic hypotension associated with Bartter and Gitelman syndromes. Am J Kidney Dis. 49: 330–5.
- Harris, P.J.; Young, J.A. Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Arch. 1977, 367, 295–7. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; Davis, P.A.; Rossi, G.P. Understanding the mechanisms of angiotensin II signaling involved in hypertension and its long-term sequelae: insights from Bartter’s and Gitelman’s syndromes, human models of endogenous angiotensin II signaling antagonism. J Hypertens. 2014, 32, 2109–19. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.D.R.; Antonelou, M.; Sathiananthamoorthy, S.; Rega, M.; Henderson, S.; Ceron-Gutierrez, L.; Barcenas-Morales, G.; Müller, C.A.; Doffinger, R.; Walsh, S.B.; Salama, A.D. Inherited salt-losing tubulopathies are associated with immunodeficiency due to impaired IL-17 responses. Nat Commun. 2020, 11, 4368. [Google Scholar] [CrossRef]
- Rahman, M.S.; Wruck, W.; Spitzhorn, L.-S.; Nguyen, L.; Bohndorf, M.; Martins, S.; Asar, F.; Ncube, A.; Erichsen, L.; Graffmann, N.; Adjaye, J. The FGF, TGFβ and WNT axis Modulate Self-renewal of Human SIX2 + Urine Derived Renal Progenitor Cells. Scientific Reports. 2020, 10, 739. [Google Scholar] [CrossRef] [PubMed]
- Bohndorf, M.; Ncube, A.; Spitzhorn, L.-S.; Enczmann, J.; Wruck, W.; Adjaye, J. Derivation and characterization of integration-free iPSC line ISRM-UM51 derived from SIX2-positive renal cells isolated from urine of an African male expressing the CYP2D6 *4/*17 variant which confers intermediate drug metabolizing activity. Stem Cell Res. 2017, 25, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qian, R.; Shan, X.; Yang, L.; Chen, H.; Ding, Y.; Chen, C.; Chu, M.; Lin, J.; Wang, D. Generation of a human induced pluripotent stem cell line (WMUi021-A) from a Gitelman syndrome patient carrying a SLC12A3 gene mutation (c.179C > T). Stem Cell Research. 2021, 53.
- Lim, S.W.; Shin, Y.J.; Cui, S.; Ko, E.J.; Lee, K.I.; Lee, J.Y.; Chung, B.H.; Yang, C.W. Generation of a human induced pluripotent stem cell line (CMCi002-A) from a patient with Gitelman’s syndrome. Stem Cell Research. 2020, 49. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-S.; Lo, Y.-F.; Wu, C.-C.; Lin, S.-W.; Yeh, C.-J.; Chu, P.; Sytwu, H.-K.; Uchida, S.; Sasaki, S.; Lin, S.-H. SPAK-Knockout Mice Manifest Gitelman Syndrome and Impaired Vasoconstriction. J Am Soc Nephrol. 2010, 21, 1868–77. [Google Scholar] [CrossRef] [PubMed]
- Nicolet-Barousse, L.; Blanchard, A.; Roux, C.; Pietri, L.; Bloch-Faure, M.; Kolta, S.; Chappard, C.; Geoffroy, V.; Morieux, C.; Jeunemaitre, X.; Shull, G.E.; Meneton, P.; Paillard, M.; et al. Inactivation of the Na-Cl Co-Transporter ( NCC ) Gene Is Associated With High BMD Through Both Renal and Bone Mechanisms: Analysis of Patients With Gitelman Syndrome and Ncc Null Mice. J Bone Miner Res. 2005, 20, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Tajima, M.; Sugawara, N.; Morimoto, T.; Kondo, Y.; Ohno, M.; Uchida, K.; Mutig, K.; Bachmann, S.; Soleimani, M.; Ohta, E.; Ohta, A.; Sohara, E.; et al. Generation and analyses of R8L barttin knockin mouse. Am J Physiol-Ren Physiol. 2011, 301, F297–307. [Google Scholar] [CrossRef]
- Matsumura, Y.; Uchida, S.; Kondo, Y.; Miyazaki, H.; Ko, S.B.H.; Hayama, A.; Morimoto, T.; Liu, W.; Arisawa, M.; Sasaki, S.; Marumo, F. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat Genet. 1999, 21, 95–8. [Google Scholar] [CrossRef]
- Dong, K.; Yan, Q.; Lu, M.; Wan, L.; Hu, H.; Guo, J.; Boulpaep, E.; Wang, W.; Giebisch, G.; Hebert, S.C.; Wang, T. Romk1 Knockout Mice Do Not Produce Bartter Phenotype but Exhibit Impaired K Excretion. J Biol Chem. 2016, 291, 5259–69. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-H.; Yu, I.-S.; Jiang, S.-T.; Lin, S.-W.; Chu, P.; Chen, A.; Sytwu, H.-K.; Sohara, E.; Uchida, S.; Sasaki, S.; Yang, S.-S. Impaired phosphorylation of Na + -K + -2Cl − cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci. 2011, 108, 17538–43. [Google Scholar] [CrossRef] [PubMed]
Figure 3.
The Role of the RAAS in the aging process. Cellular aging and stimulation of RAAS are closely intertwined. Sustained stimulation of angiotensin II (ANGII) receptor type 1 (AGTR1) causes intracellular NADPH oxidase to generate superoxide anions and promote uncoupling of endothelial NO synthase, which in turn decreases NO availability and increases reactive oxygen species (ROS) production. Uncontrolled ANGII-induced ROS production ultimately results in cellular senescence, mitochondrial dysfunction, hypertrophy, inflammation, and fibrosis in the kidney. Sirtuin-1 (SIRT1) involved in many age-associated processes can counteract the uncontrolled ANGII-induced effects, by regulating the expression of AGTR1. Resveratrol is a natural compound capable of promoting SIRT1 activity and has been shown to have beneficial effects on hypertension. Adapted from “Activation of Protein Kinase C (PKC)”, by BioRender.com (2023). Figure 3 was generated from https://app.biorender.com/biorender-templates.
Figure 3.
The Role of the RAAS in the aging process. Cellular aging and stimulation of RAAS are closely intertwined. Sustained stimulation of angiotensin II (ANGII) receptor type 1 (AGTR1) causes intracellular NADPH oxidase to generate superoxide anions and promote uncoupling of endothelial NO synthase, which in turn decreases NO availability and increases reactive oxygen species (ROS) production. Uncontrolled ANGII-induced ROS production ultimately results in cellular senescence, mitochondrial dysfunction, hypertrophy, inflammation, and fibrosis in the kidney. Sirtuin-1 (SIRT1) involved in many age-associated processes can counteract the uncontrolled ANGII-induced effects, by regulating the expression of AGTR1. Resveratrol is a natural compound capable of promoting SIRT1 activity and has been shown to have beneficial effects on hypertension. Adapted from “Activation of Protein Kinase C (PKC)”, by BioRender.com (2023). Figure 3 was generated from https://app.biorender.com/biorender-templates.
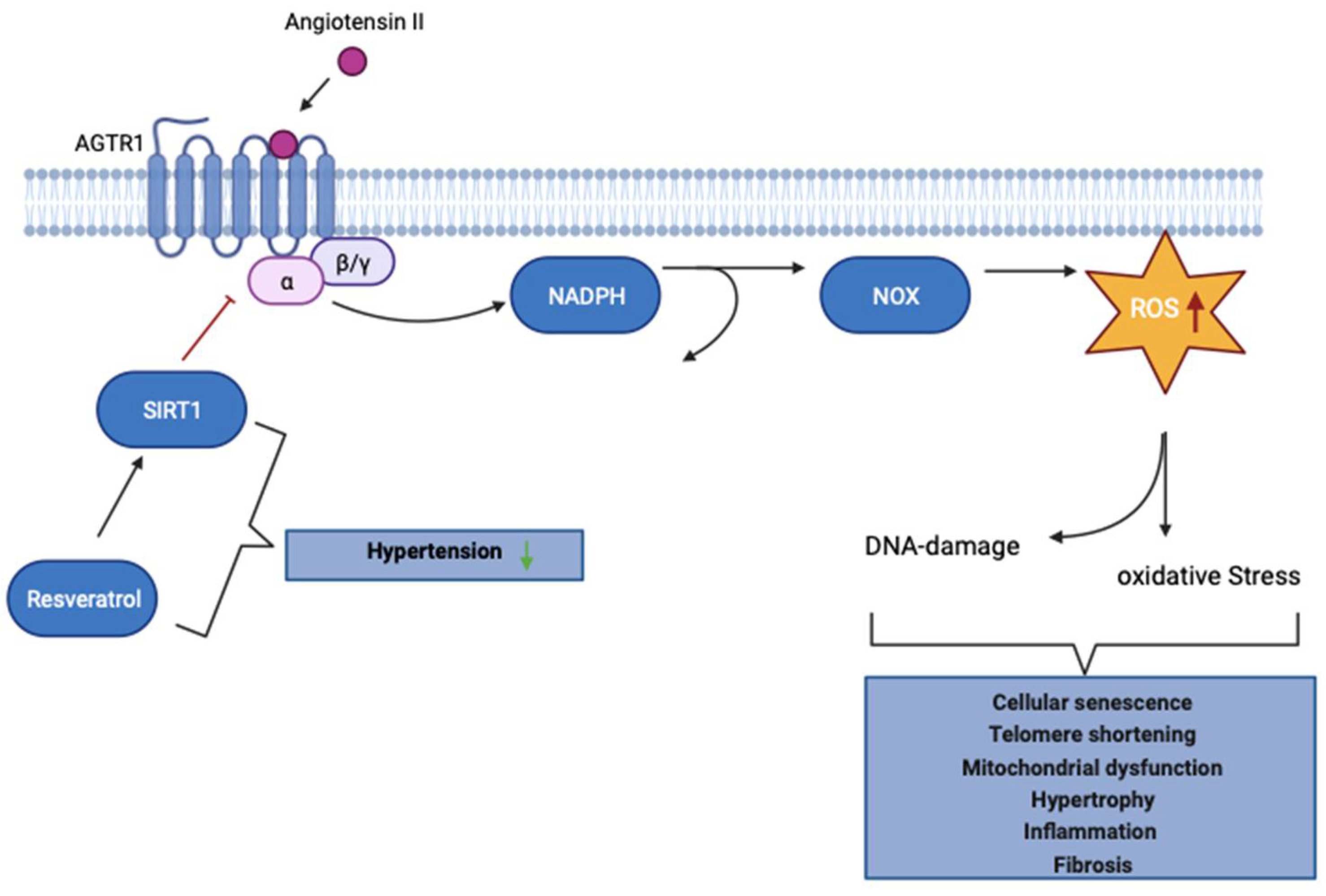
Figure 4.
A proposed mechanism of how ROS, SIRT1, and RAAS impinge on Gitelman and Bartter syndrome. Regulation of the renin-angiotensin-aldosterone system (RAAS) is regulated by the octapeptide Angiotensin II (ANGII). In a young individual independent in healthy or in Bartter syndrome (BS) /Gitelman syndrome (GS) patients ANGII binds to either of the two subtypes of Angiotensin II receptors, AT1 (AGTR1) initiates vasoconstriction and proliferation, and AT2 (AGTR2) initiates vasodilation and differentiation. In a healthy but aged individual, such a stimulatory response of AGTR1 leads to increased arteriolar vasoconstriction, proliferation, and reactive oxygen species (ROS) activation which then induces DNA damage, cellular senescence, and acceleration of the aging process. This process can be reversed/minimized by high levels of sirtuin-1 (SIRT1). It is assumed that GS and BS patients have comparable levels of SIRT1 as seen in young individuals. The figure was created with BioRender.com.
Figure 4.
A proposed mechanism of how ROS, SIRT1, and RAAS impinge on Gitelman and Bartter syndrome. Regulation of the renin-angiotensin-aldosterone system (RAAS) is regulated by the octapeptide Angiotensin II (ANGII). In a young individual independent in healthy or in Bartter syndrome (BS) /Gitelman syndrome (GS) patients ANGII binds to either of the two subtypes of Angiotensin II receptors, AT1 (AGTR1) initiates vasoconstriction and proliferation, and AT2 (AGTR2) initiates vasodilation and differentiation. In a healthy but aged individual, such a stimulatory response of AGTR1 leads to increased arteriolar vasoconstriction, proliferation, and reactive oxygen species (ROS) activation which then induces DNA damage, cellular senescence, and acceleration of the aging process. This process can be reversed/minimized by high levels of sirtuin-1 (SIRT1). It is assumed that GS and BS patients have comparable levels of SIRT1 as seen in young individuals. The figure was created with BioRender.com.
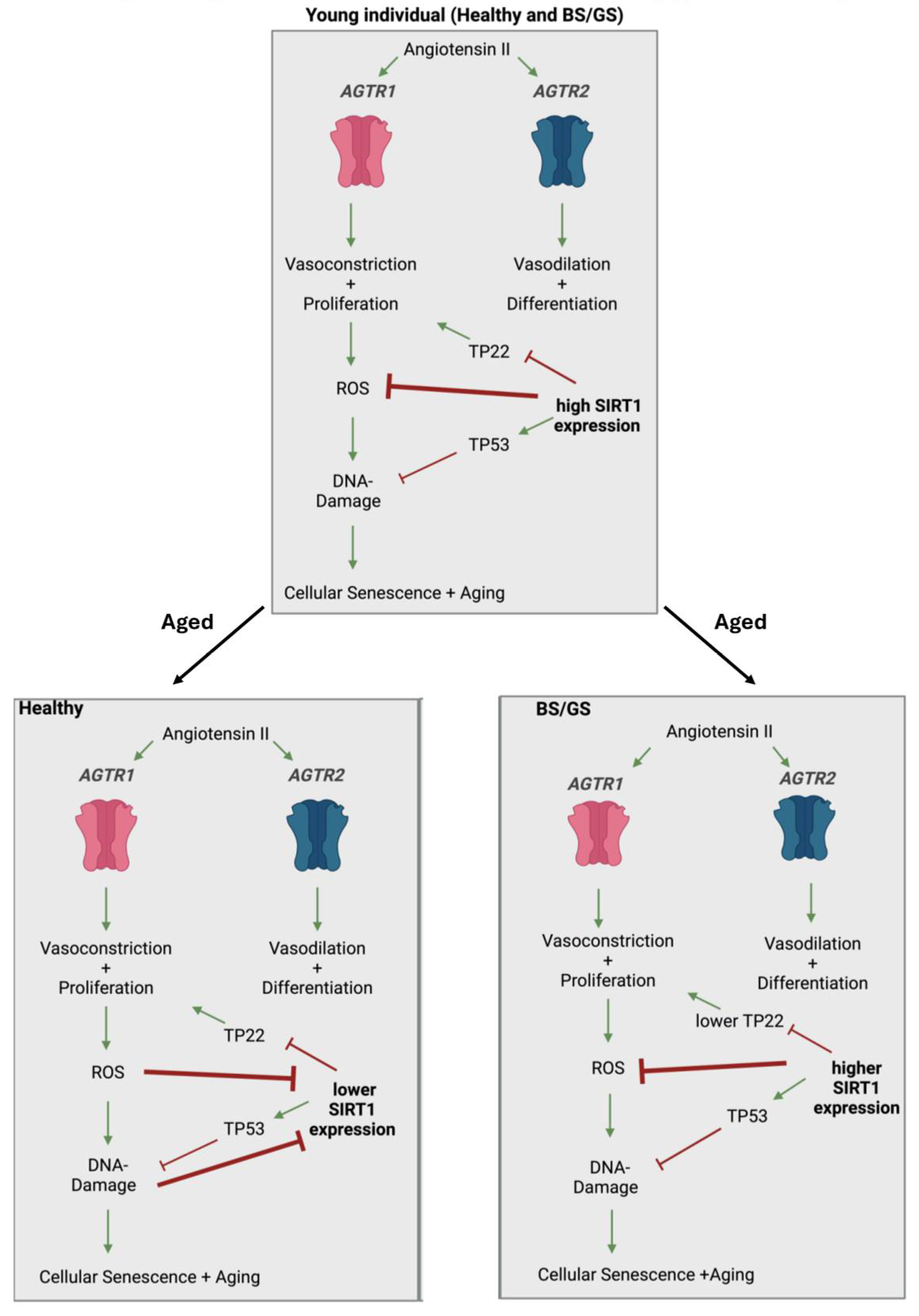

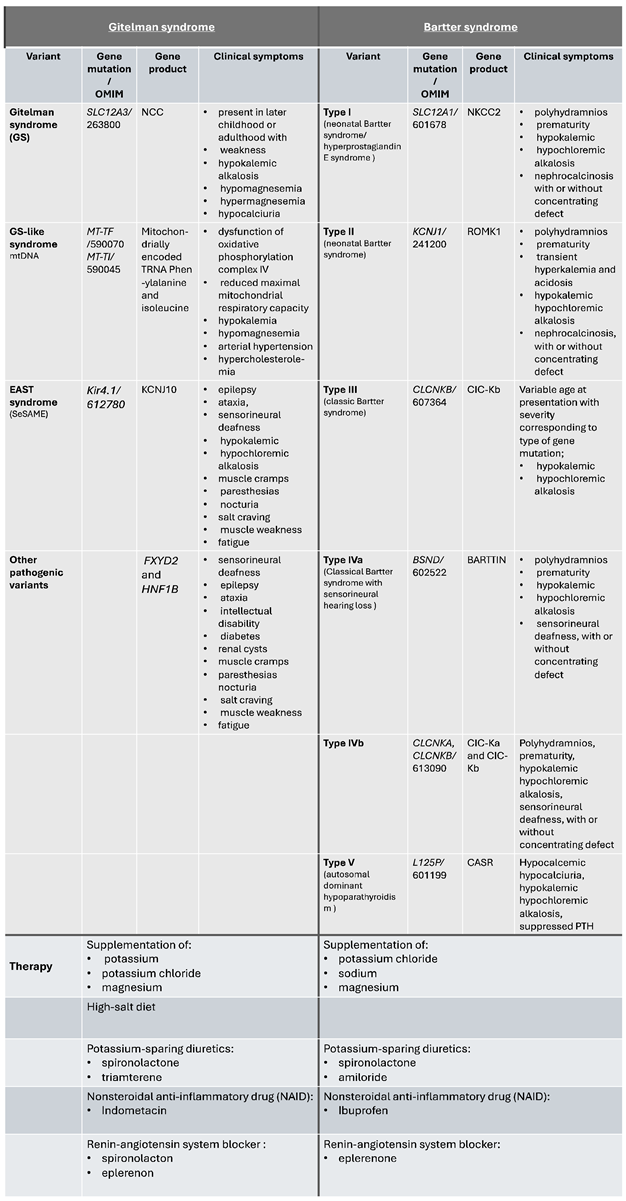
Table 1.
provides an overview of the distinct gene variants, clinical symptoms, and treatment options for GS/BS patients.
Table 1.
provides an overview of the distinct gene variants, clinical symptoms, and treatment options for GS/BS patients.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



