
Submitted:
15 July 2024
Posted:
16 July 2024
You are already at the latest version
Abstract
Lung cancer (LC) continues to be an important public health problem being the most common form of cancer and a major cause of cancer deaths worldwide. Despite the great bulk of research to identify genetic susceptibility genes by genome-wide association studies, only few loci associated to nicotine dependence have been consistently replicated. Our previously published study in few phenotypically discordant sib-pairs identified a combination of germline truncating mutations in known cancer susceptibility genes in never smoker early-onset LC patient does not present in their healthy sib. These results firstly demonstrated the presence of an oligogenic combination of disrupted cancer predisposing genes in non-smokers patients, giving experimental support to a model of a “private genetic epidemiology”. Here, we used a combination of whole-exome and RNA sequencing coupled with a discordant sib’s model in a novel cohort of pairs of never-smokers early-onset LC patients and in their healthy sibs used as controls. We selected rare germline variants predicted as deleterious by CADD and SVM bio-informatics tools and absent in the healthy sib. Overall, we identified an average of 200 variants per patient, about 10 of which in cancer predisposing genes. In most of them, RNA sequencing data reinforced the pathogenic role of the identified variants showing: i) downregulation in LC tissue (indicating “second hit” in tumor suppressor genes); ii) upregulation in cancer tissue (likely oncogene); iii) downregulation in both normal and cancer tissue (indicating transcript instability). The combination of the two techniques demonstrates that each patient has an average of six (with a range from four to eight) private mutations with functional effect in tumor predisposing genes. The presence of a unique combination of disrupting events in the affected subjects may explain the absence of familial clustering of non-small cell lung cancer. In conclusion, these findings indicate that each patient has his/her own “predisposing signature” to cancer development and suggest the use of personalized therapeutic strategies in lung cancer.
Keywords:
whole-exome sequencing
; lung cancer susceptibility
; germline variants
; next-generation sequencing
; oligogenic model
1. Introduction
Lung cancer (LC) is an important public health problem and a major cause of cancer death worldwide [1]. Despite the strong relationship to tobacco smoking, less than 1 out of 5 heavy smokers develop lung cancer [2], whereas 25% of patients with lung cancer did never smoke [3], suggesting a role for genetic factors in individual susceptibility to lung cancer. So far, no specific environmental or genetic risk factors have been detected in these individuals.
Previous genome-wide association studies (GWAS) have identified up to 45 susceptibility loci for lung cancer in population-based series, mainly including smokers [4,5,6,7,8,9,10,11,12,13,14,15]. On the other hand, several studies found the rare germline T790M mutation in EGFR correlated with familial clustering of lung cancer [16,17,18,19,20,21,22,23]. The EGFR is a major cancer predisposition gene with an estimated 31% risk of lung cancer development in non-smoking carriers [19]. However, the EGFR T790M mutation is weakly oncogenic and became significantly enhanced when associated with a second activating mutation [18] more frequently with female sex and non-smoking status.
Still today, there is a need to better understanding of the genetic factors associated with lung cancer in never-smokers’ patients in non-predisposed families.
In this frame, using the whole-exome sequencing approach, in a previous study analyzing discordant sib-pairs, we identified a combination of germline disruptive mutations in known cancer susceptibility genes in never smoker with lung adenocarcinoma (ADCA) of early onset, that were not present in their healthy control sibs [24]. These results demonstrated that each affected subject had an oligogenic combination of disrupted cancer predisposing genes. This evidence gives experimental support to a model of “private genetic epidemiology” for lung cancer susceptibility that has previously only been hypothesized. The oligogenic nature of the model may therefore explain the non-heritability of the condition.
In the present study, we used a combination of genetic technical tools (whole-exome sequencing analysis and RNA sequencing) coupled with a pedigree model in discordant pairs of non-smokers with lung cancer of early onset. Healthy sibs were used as controls. We identified in affected subjects a unique combination of private “predisposing signatures”, that further confirms and exploits the oligogenic model of the disease.
2. Materials and Methods
2.1. Study design and samples
This study analyzed four non-smoker lung cancer patients with lung cancer (cases), who underwent lung lobectomy in the Thoracic Surgery Unit at the Azienda Ospedaliero-Universitaria Senese (AOUS, Siena, Italy), in comparison with their healthy sibs (controls). The genetic comparison of discordant sibs, that share 50% of the genome, facilitates the identification of variants associated with lung cancer susceptibility. For each patient, formalin-fixed paraffin-embedded (FFPE) samples of tumoral and non-tumoral lung tissues were obtained from the Pathology Unit of the recruiting hospital and analyzed through whole-exome sequencing and RNA sequencing (RNA-seq).
Each patient and sib signed the informed consent declaration for the use of their biological samples and clinical data for research purposes. The study protocol was approved by the Institutional Ethical Committees. Information on histological diagnoses (by the Pathology Unit) was retrieved from the clinical records.
2.2. DNA and RNA extraction
Genomic DNA of cases and controls was isolated from EDTA peripheral blood using QIAamp DNA Blood Kit (Qiagen®, Hilden, Germany) according to the manufacturer’s protocol. DNA was extracted from FFPE lung tumoral and non-tumoral tissue samples using MagCore® Genomic DNA FFPE One-Step Kit for MagCore® System (Diatech Pharmacogenetics srl, Jesi, Ancona, Italy). RNA was extracted from FFPE lung tumoral and non-tumoral tissue samples using High Pure FFPE-Tissue RNA Isolation Kit (Roche, Basel, Switzerland) following the manufacturer’s instructions. RNA samples were processed to remove ribosomal RNA (rRNA) using Ribo-Zero rRNA Removal Kit for Human samples (Illumina, Grand Island, USA) following the manufacturer’s instructions. RNA integrity was verified using the Agilent Eukaryote Total RNA Nano Kit (Agilent Technologies, Palo Alto, CA) on Agilent2100 Bioanalyzer (Agilent Technologies). Both DNA and RNA were quantified by spectrophotometry (ND-2000c; NanoDrop Products, Wilmington, DE) and Qubit® Fluorometer with Qubit® dsDNA HS Assay and Qubit® RNA HS Assay Kits (LifeTechnologies), respectively.
2.3. Whole Exome Sequencing and data analysis
Whole exome sequencing was performed using the Illumina Nextseq 550 on genomic DNA samples of cases and controls and tumor tissues as previously described [25]. After variant annotation against external datasets, including 1000 genomes (http://www.1000genomes.org/) and dbSNP, in order to identify susceptibility variants, we selected for rare variants (minor allele frequency – MAF ≤ 0.01) with a potentially damaging effect and with a functional link to cancer development. Additional filtering procedures were thus implemented for: i) retrieving exonic rare variants with a potential detrimental impact on protein function, i.e., truncating frameshift insertion and deletion and nonsense variants predicted as deleterious that were present in affected but not in unaffected sibs and vice versa; ii) identifying somatic mutations present in tumor tissues.
2.4. RNA sequencing and data analysis
RNA sequencing was performed using Illumina HiSeq2500 platform (Illumina), in a 2x100bp paired-end (PE) configuration in High Output mode (V4 chemistry), with a total of at least 250 million reads per lane. After quality check, RNA (50 ng) was used to prepare sample libraries. Sequencing library construction included these steps: library construction using Illumina TruSeq RNA Sample Pre Kit (Illumina), library purification using Beckman AMPure XP beads (Beckman Coulter s.r.l., Milan, Italy), insert fragments test using Agilent High Sensitivity DNA Kit on Agilent 2100 Bioanalyzer (Agilent Technologies), quantitative analysis of library (ABI 7500 real time PCR instrument; KAPA SYBR green fast universal 2 9 qPCR master mix. GRN), and cBOT automatic cluster (TruSeq PE Cluster Kit v3-cBotHS). Post-library quality controls were performed using the Agilent RNA 6000 Nano kit (Agilent Technologies) on Agilent2100 Bioanalyzer (Agilent Technologies) and Qubit® Fluorometer (Life Technologies). Libraries were then loaded on HiSeq2500 sequencing platform (Illumina) and sequenced using 2x100bp pair-end High Output Mode (V4 chemistry) per lane. The reads generated on the HiSeq2500 were provided under FASTQ format.
Sequence reads in FASTQ format were processing using the Fastqc software (http:// www.bioinformatics.babraham.ac.uk/projects/fastqc/) for data quality check and removing excess adaptors to get high-quality and clean reads. The high-quality reads were aligned to the GRCh38/hg38 reference human genome (ftp://jgi-psf.org/pub/compgen/phyto zome/v9.0/Ptrichocarpa/assembly/Ptrichocarpa_210.fa.gz) using the TopHat software (version 2.0.9) [26]. Transcript assembling and expression quantification were carried out using Cufflinks (version 2) [27]. Gene expression was expressed as fragments per kilo-base transcript per million mapped reads (FPKM) value [28]. This normalized value was used for visualization on a genome browser (http://genome.ucsc.edu/) [29], as well as to compare read coverage between and throughout different genes. Statistical analysis was performed to compute the mean FPKM level with the associated P-value for lung normal tissues together with the mean FPKM level with the associated P-value for lung cancer tissues. Cuffdiff tool from Cufflinks was used to identify differentially expressed genes [30]. Potential gene fusion events were detected by TopHat-fusion [31] with spanning reads ≥ 10 in cancer and normal tissue. The cancer-specific fusion genes were obtained by excluding the fusion genes that were also identified in distant normal tissue. Gene Ontology (GO) and pathways analyses were performed using the Database for Annotation, Visualization and Integrated Discovery functional annotation tool (DAVID Bioinformatics Resources 6.7, https://david.ncifcrf.gov).
3. Results
3.1. Exome Analysis of Constitutive DNA
We carried out whole-exome sequencing of DNA from blood tissue in four discordant sib pairs (Table 1). Patients had early onset lung cancer in the absence of smoking history, and we used as controls their unaffected sibling. Exome sequencing generated a mean of 28,960,442 reads for sample with a mean read length of 160bp.
Overall, we identified a total of 349,411 genetic variants in eight samples. After removing variants with low coverage and filtering for exonic mutations with MAF≤0.01 or not reported, we obtained 6,086 total variants (Figure 1). We then filtered excluding variants with clinical significance as “benign” or “likely benign” and present in an in-house database of variants identifying 3,235 variants. Among them, we selected deleterious variants applied the Combined Annotation Dependent Depletion (CADD) and the MetaSVM (Support Vector Machine) bioinformatics tools. In this way, we obtained 961 potential deleterious variants of which 370 were non-synonymous and 591 were truncating variants (insertions and deletions (indels) causing exonic frameshifts, and nonsense mutations leading to truncated proteins).
Out of these, 40 variants mapped in genes that have been found previously associated with cancer of lung or other tissues and, interestingly, all of these were present exclusively in the affected sib (Table 2). Validation of these variants was carried out using custom NGS panel for Ion PGM sequencer (Life Technologies) and led to the confirmation of 40 variants probably responsible of lung cancer susceptibility in our cases. All the 40 sequence variants identified were in heterozygous state. No common variants to all four cases were found. However, three variants were common to two out of four patients (ANGPLT4, CARS, ESRRA). The two variants in ANGPLT4 (Angiopoietin Like 4) and ESRRA (Estrogen Related Receptor Alpha) genes were common to case 1 and case 4, while the same variant in CARS (Cysteinyl-TRNA Synthetase) gene was found in case 3 and case 4. Each of the 40 variants were also present in the relative lung tumor tissue in heterozygous state.
3.2. Somatic Mutations in Tumor Lung Tissues
In the 4 tumor lung samples, we identified an average of 2.568 somatic mutations per tumor (ranging from 2.169 to 3.348). When we limited the mutations in the coding regions, the average number of mutations in each tumor was 1.510 (range 1.172-1.865), among which an average of 1092 (range 797-1542) were missense, 374 (range 85-735) were frameshift ins/del and 43 (range 32-56) were nonsense mutations (Figure 2). The number of mutations was not associated with clinical and pathological variables (stage and age at diagnosis). No recurrent mutations, such as mutations in EGFR, KRAS or AKT genes, were present in our tumor samples.
3.3. RNA-seq Analysis of Tumors and Non-Involved Lung Tissues
To detect differences in the expression level, splicing pattern and/or polyadenylation sites that could both help the understanding of the functional role of germline variants in the development of lung cancer and shade light on the pathogenic mechanisms, we performed RNA sequencing (RNA-seq) analysis of RNA extracted from both FFPE tumor and non-involved lung tissues of the four lung cancer patients. RNA-seq generated a mean of 73,865,049 reads per sample and 91.07% of bases sequenced above Q30 quality score (Supplementary Table 1).
To assess the potential functional role of the 40 germline variants identified by WES (whole-exome sequencing) and predicted as deleterious by bioinformatics tools, we combined results from WES experiments with the respective expression profile from RNA-seq. In 16 variants mapping in 16 genes, RNA sequencing data reinforced the pathogenic role of the identified variants showing three different effects (Figure 3). Firstly, 8 genes (ACACA, ANGPTL4, BUB1B, FBN2, MEN1, MMP14, TP73, WWTR1) showed a downregulation in lung cancer tissue indicating a possible “second hit” in tumor suppressor genes responsible of gene inactivation (Figure 3, panel A). Secondly, 3 genes (ACAP2, ENO3, PSCA) showed an upregulation in lung cancer tissue suggesting their role as oncogene (Figure 3, panel B). Lastly, 5 remaining genes (AMIGO3, CARS, DEPTOR, IQGAP2, RNASEL) resulted as downregulated in both normal and cancer tissue indicating a possible transcript instability (Figure 3, panel C). In addition, a nonsense variant that was predicted as deleterious in the putative tumor suppressor URI1 gene, although it was not associated with changes in mRNA levels, was also retained in the panel of candidate genes.
Data obtained from tumor tissues were then compared with those from normal tissues in order to identify differences in the expression level using well-established and accepted analysis tools such as Cufflink-Cuffdiff [30]. Cuffdiff differential expression analysis identified 315 genes significantly down- regulated (fold≤-2 change, P-value ≤0.05) and 347 genes significantly up-regulated (fold change≥2, P-value ≤0.05) in lung tumor tissues compared to normal tissues. We then interrogated these data both for the enrichment of genes involved in peculiar cell functions and for the involvement of specific pathways. GO analysis of upregulated differentially expressed genes revealed a statistically significant enrichment for genes mainly involved in cellular process (p-value=1.29x10-4), cellular component organization (p-value=1.78x10-4) and developmental process (p-value=0.002) (Figure 4A). GO terms of downregulated differentially expressed genes were mainly related to acute inflammatory response (p-value=0.015) and cell adhesion (p-value=0.028) (Figure 4B).
Pathways analysis individually performed on the differentially expressed genes between each normal-tumor tissue pair, showed the ECM-receptor interaction pathway as the common involved pathway in all four lung tumor tissues (Table 3).
3.4. Analysis of Tumor Loss of Heterozygosity
We performed whole-exome sequencing of DNA from the four tumor tissues in order to identify potential driver genes. Analysis of exome sequencing in DNA from blood compared to exome sequencing in DNA from tumor tissues allowed to identify the presence of 9 variants in 9 genes that were in heterozygous state in DNA from patient’s blood and in homozygous state in tumor tissue (Figure 5), thus representing a possible second hit responsible of gene inactivation in this tissue.
3.5. Protein-Protein Interaction
To explore possible pathogenic mechanisms in lung cancer, we further investigated the existence of possible interactions among the involved cancer genes that had germline mutations. We found three networks involving mutated genes belonging to different cases (Figure 5). The main network connecting EPHB6 gene (mutated in case 1) with ACACA and ENO3 genes (mutated in case 3), CARS gene (mutated in case 3 and 4) and ACAP2 gene (mutated in case 2). Overall, each patients had deleterious germline variants in genes belonging to this network.
Expanding the analysis of this network with a threshold of 20 interactors, we identified associations between the mutated genes in our patients with many genes involved in cancer development. In particular, the network clustered in two relevant groups, the former involving PRKA interactors, in which are comprised our mutated genes, and the latter involving RAD51 interactors. (Figure 6).
4. Discussion
In this study, we used an integrative approach of next-generation sequencing technologies to dissect genetic susceptibility to lung cancer in nonsmoker patients for the identification of a genetic profile that could be predictive of the individual risk for lung cancer. Whole-exome sequencing technique, coupled with a model of cases and controls deriving from the same kindred, demonstrated that each patient has a combination of an average of 10 (range 7-14) deleterious “private” germline variants in tumor predisposing genes. These mutations were absent in unaffected sibs.
In addition to perform a genome-wide analysis in the search of oligogenic signatures that could differentiate affected from non-affected siblings, we also analyzed, in the same patients RNA-seq data in tumor specimens, comparing tumoral vs non-tumoral tissue. Using this approach, we confirmed the potential functional effect of most of the identified variants with an average of 6 (range 4-8) variants per patients. In particular, we distinguished three class of alterations: 1) variants associated with downregulation of the gene in lung tumoral tissue compared to non-tumoral tissue indicating the presence of a “second hit” in a putative tumor suppressor gene; 2) variants associated with upregulation in tumoral compared with non-tumoral tissue of the gene that could represent a likely oncogene; 3) variants associated with downregulation in both tumoral and non-tumoral tissue indicating possible mechanisms for transcript instability.
Concerning oncogenic genes, the comparison of RNA-seq data in the tumoral vs non-tumoral tissue showed the involvement of genes belonging to molecular localization, cell movement and cellular component assembly pathways among the upregulated genes while, among the downregulated genes, the mainly involved pathways were cell localization, transport, negative regulation of cellular processes and response to stress. These results seem to suggest that cancer cells miss proper intracellular molecular localization and are more resistant to stress. Comparing differentially expressed genes in matched tissue pairs, we showed involvement of the ECM-receptor pathway in all of four pairs of siblings. Similar findings have already been reported in prostate cancer [32].
Network analysis concerning the identified cancer susceptibility genes showed three networks that were shared by different patients, suggesting a possible common path for private oligogenic signatures. The main network involving genes mutated in all four patients (ACACA, ACAP2, ENO3, EPHB6, CARS) connected the candidate mutated genes with the group of RAD51 (RAD51 recombinase) and PRKA interactors. RAD51 encodes for a key recombinase that seems to be essential for homologous recombination and DNA repair [33]. It interacts with a large number of proteins involved in double-strand DNA breaks and in cell cycle with important implications in tumorigenesis, such as BRCA1, and TP53 (as reviewed in 34). PRKA encodes for the catalytic subunit of AMP-activated protein kinase (AMPK) that is a cellular energy sensor that maintains energetic homeostasis [35] and has been suggested as a novel target for anticancer therapy since its activation determines a reduction of mRNA translation and protein synthesis [36]. Figure 5 and Figure 6 show the possible network (Figure 5) and “expanded” network (Figure 6) of proposed cooperation among the main mutated genes in the occurrence of “increased susceptibility” to non small cell lung cancer. We have previously discussed the putative role of each gene in the occurrence of lung cancer.
Our study has a number of strengths and limitations. One of the main strengths is that we used an integrative next-generation sequencing approach combine whole-exome sequencing technique (germline and tumor DNA) with transcriptome sequencing (tumor and matched normal tissue). In addition, the original selection strategy of discordant sibs has been used. Among the limitation, variants in noncoding regions, copy number variations, genome rearrangements and common susceptibility variants have been missed due to the study design. Secondly, the number of samples analyzed is relatively small. However, the private nature of the oligogenic susceptibility reduces the potential contribution of additional discordant sib-pairs.
Anyway, the “novelty” that present results strongly suggest is the following. Opposite to what occurs in inherited multitumoral syndromes, such as Familial Adenomatous Polyposis (FAP), where germline mutations of a single gene (i.e. APC gene) determines colon cancer in 100% of affected patients, in the present model, that is likely to occur in most frequent sporadic cancers, variable oligogenic combinations of germline mutations, that change from an individual to another, all together are responsible for the occurrence of a “susceptible” or “resistant” phenotype towards a given cancer.
Overall, our findings showed that private oligogenic signatures could be part of an individual way to cancer. We suggest that every patient has his/her peculiar signature of germline mutations, governing personal cancer susceptibility. This signature may play a major role in the development and growth of lung cancer, namely in nonsmoker patients and may therefore explain the non-heritability of the condition. In fact, lung cancer in non-smokers rarely shows familial aggregation but rather seems sporadic or occurs in phratries. These two possibilities resulted perfectly explained by our private oligogenic model of inheritance (Figure 7). The proposed model may have important implications in the evaluation of individual risk for lung cancer and may lead to a “germline genetic signature”, which, in the modern era of personalized medicine, could be of benefit for early detection of cancer.
5. Conclusions
In conclusion further study is mandatory to confirm present findings in larger studies. Anyway, our “focused analysis” in a small number of patients could contribute to a deeper insight into the complexity of the various subtypes of lung cancer and the variable interplay among gene programs involving biological processes, which could seem apparently distinct and/or distant from each other.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table 1.
Author Contributions
“Conceptualization, M.P. and E.F.; methodology, V.B.S., D.M., A.R.; software, D.R., O.S.; validation, M.G., C.B.; F.M.; data curation, M.P., E.F., M.G.; writing E.F., M.P., B.V.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NextGenerationEU of PNRR “Piano Nazionale di Ripresa e Resilienza” Missione 4 Componente 2 – M4C2a - Project THE – Tuscany Health Ecosystem – SPOKE 6 – CUP B63C22000680007 (MILESTONE n. 6.3.2: Innovative tools for target genes, disease pathways and therapeutic strategies discovery). The funders had no role in the design or conduct of the study, in the collection, analysis or interpretation of the data, or in the preparation, review or approval of the manuscript.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors thank the NextGenerationEU of PNRR “Piano Nazionale di Ripresa e Resilienza” Missione 4 Componente 2 – M4C2a - Project THE – Tuscany Health Ecosystem – SPOKE 6 – CUP B63C22000680007 (MILESTONE n. 6.3.2: Innovative tools for target genes, disease pathways and therapeutic strategies discovery); the EU funding within the NextGenerationEU with Italian Ministry of University and Research (MUR) for “Fondo per il Programma Nazionale di Ricerca e Progetti di Rilevante Interesse Nazionale (PRIN)” - PRIN 2022 – Project 2022ARXHR2_002 - CUP B53D23007910006 (E.F.); the EU funding within the “NextGenerationEU of Piano Nazionale di Ripresa e Resilienza (PNRR) - Missione 4 “Istruzione e Ricerca” - Componente 2 – M4C2a Investimento 1.1, “Fondo per il Programma Nazionale di Ricerca e Progetti di Rilevante Interesse Nazionale (PRIN)” - PRIN 2022 PNRR – Project P2022NLEBP - CUP B53D23025110001 (E.F.); the “INAIL (BRiC - 2022) Piano Attività di Ricerca 2022-2024” (E.F); Italian Ministry of University and Research for PNRR-National Center for Gene Therapy and Drugs based on RNA Technology - CN00000041 (F.M.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- W.CP.; W.E. et al. World Cancer Report: Cancer Research for Cancer Prevention 2020. International Agency for Research on Cancer, 2020.
- P.R.; D.S. et al. Smoking, smoking cessation, and lung cancer in the UK since 1950: Combination of national statistics with two case-control studies. Br Med J 2000, 9, 321-323. [CrossRef]
- L.E.; P.H. et al. Functional studies of lung cancer GWAS beyond association. Hum Mol Genet. 2022, R1, R22-R36. [CrossRef]
- W.Y.; B.P. et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 2008, 40, 1407–1409. [CrossRef]
- Mc.J.D.; H.R.J. et al. Lung cancer susceptibility locus at 5p15.33. Nat. Genet, 2008, 40, 1404–1406. [CrossRef]
- L.M.T.; C.N. et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet., 2009, 85, 679–691. [CrossRef]
- W.Y.; McJ.D. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet., 2014, 46, 736–741. [CrossRef]
- Mc.J.D.; HR.J. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet., 2017, 49, 1126–1132. [CrossRef]
- W.Y.; B.P.et al. Variation in TP63 is associated with lung adenocarcinoma in the UK population. Cancer Epidemiol. Biomark. Prev., 2014, 20, 1453–1462. [CrossRef]
- H.Z.; W.C. et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet., 2011, 43, 792–796.
- D.J.; H.Z. et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population. Nat. Genet., 2012, 44, 895–899. [CrossRef]
- S.K.; K.H. et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat. Genet., 2012, 44, 900–903. [CrossRef]
- L.Y.; S.C.C. et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol., 2010, 11, 321–330. [CrossRef]
- L.Q.; H.C.A. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet., 2012, 44, 1330–1335. [CrossRef]
- D.J.; Lv.J. et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Respir. Med., 2019, 7, 881–891. 10.1016/S2213-2600(19)30144-4.
- L.Y.; P.C.V. et al. Case report: germline mutation of T790M and dual/multiple EGFR mutations in patients with lung adenocarcinoma. Clin Lung Cancer. 2016, 17(2):1–13. [CrossRef]
- T.A.; X.L. et al. Concurrent molecular alterations in tumors with germ line epidermal growth factor receptor T790M mutations. Clin Lung Cancer. 2013, 14(4):452–456. [CrossRef]
- G.A.; R.L. et al. Hereditary lung cancer syndrome targets never smokers with germline egfr gene T790M mutations. J Thorac Oncol. 2014, 9(4):456–463. [CrossRef]
- Y.HA.; A.ME. et al. Germline EGFR T790M mutation found in multiple members of a familial cohort. J Thorac Oncol. 2014, 9(4):554–558. [CrossRef]
- O.GR.; M.VA. et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol. 2012, 7(6):1049–1052. [CrossRef]
- L.C.; P.C. Inherited germline T790M mutation and somatic epidermal growth factor receptor mutations in non-small cell lung cancer patients. J Thorac Oncol. 2011, 6(2):395–396. [CrossRef]
- B.DW.; Gore I. et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005, 37(12):1315–1316. [CrossRef]
- R.L.; B.MN. et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One. 2007, 2(8):1–10. [CrossRef]
- R.A.; M.AM. et al. Oligogenic germline mutations identified in early non-smokers lung adenocarcinoma patients. Lung Cancer. 2014, 85(2):168-74. [CrossRef]
- I.V.; M.MA. Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress. Int J Mol Sci 2016, 17(3):306. [CrossRef]
- T.C.; P.L. et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25:1105-11. [CrossRef]
- T.C.; R.A. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7:562-78. [CrossRef]
- M.A.; B.AW. et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5:621-8. [CrossRef]
- K.WJ.; C.WS. et al. The human genome browser at UCSC. Genome Res. 2002, 12:996-1006. [CrossRef]
- T.C.; W.BA. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010, 28:511-515. [CrossRef]
- D.K.; S.LS. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol., 2011, 12:R72. [CrossRef]
- S.DA.; C.CR. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol Endocrinol. 2004, 2:2. [CrossRef]
- T.T.; F.Y. et al. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. 1993, USA 93, 6236–6240. [CrossRef]
- R.C. RAD51, genomic stability, and tumorigenesis. Cancer Lett. 2005, 218: 127-139. [CrossRef]
- K.S.; R.DR. et al. Gene of the month. AMP kinase (PRKAA1). J Clin Pathol 2014, 67:758–63. [CrossRef]
- C.S.; Z.X. et al. Combined cancer therapy with non-conventional drugs: all roads lead to AMPK. Mini Rev Med Chem. 2014, 14(8):642-54.
Figure 1.
Flowchart illustrating filtering process and variants selection.
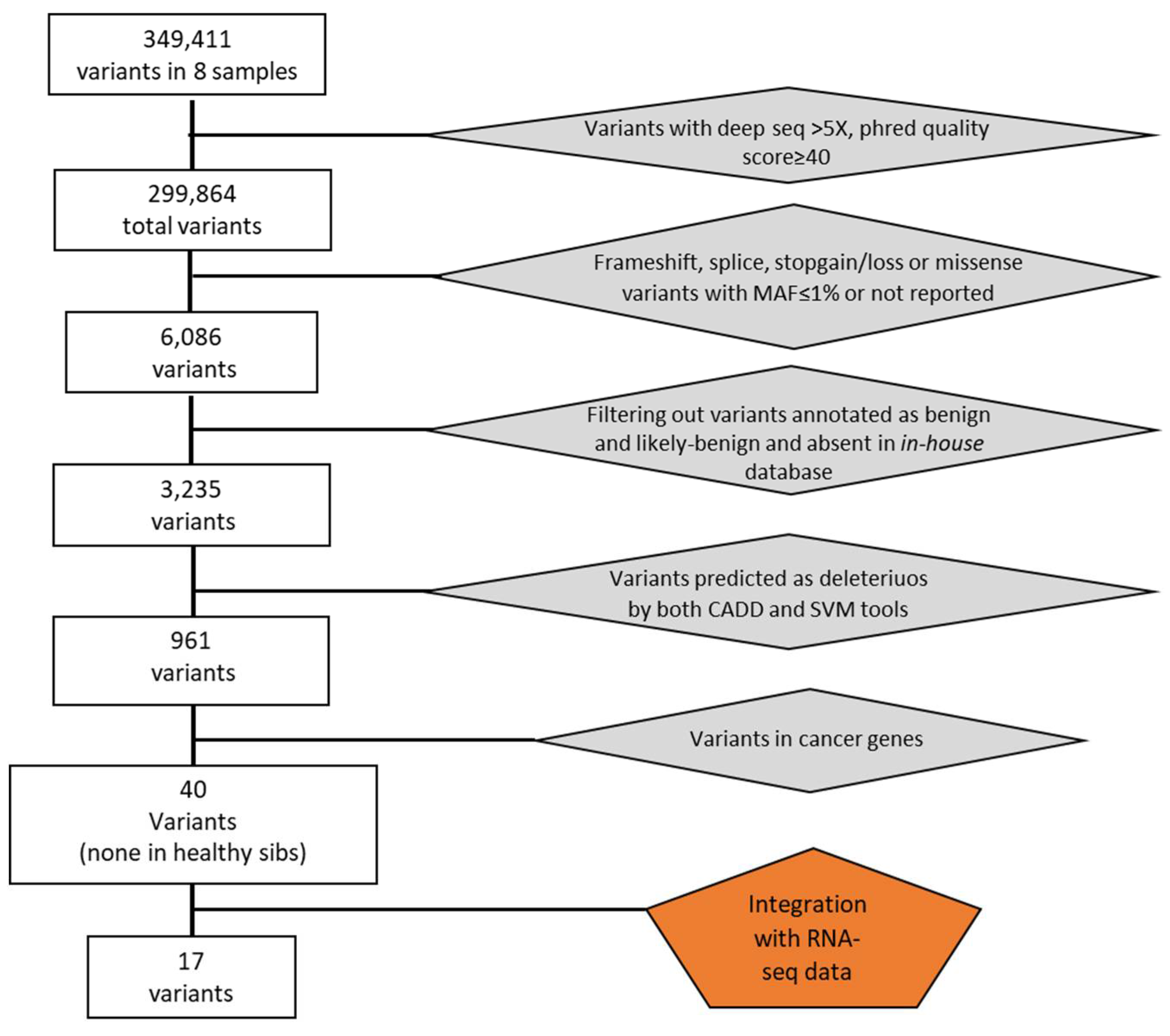
Figure 2.
Average number of mutations. In white the missense mutation with a numerical range from 797 to 1542. In gray the indel mutations ranging in number from 85 to 735 and in white the nonsense mutation with a range from 32 to 56.
Figure 2.
Average number of mutations. In white the missense mutation with a numerical range from 797 to 1542. In gray the indel mutations ranging in number from 85 to 735 and in white the nonsense mutation with a range from 32 to 56.
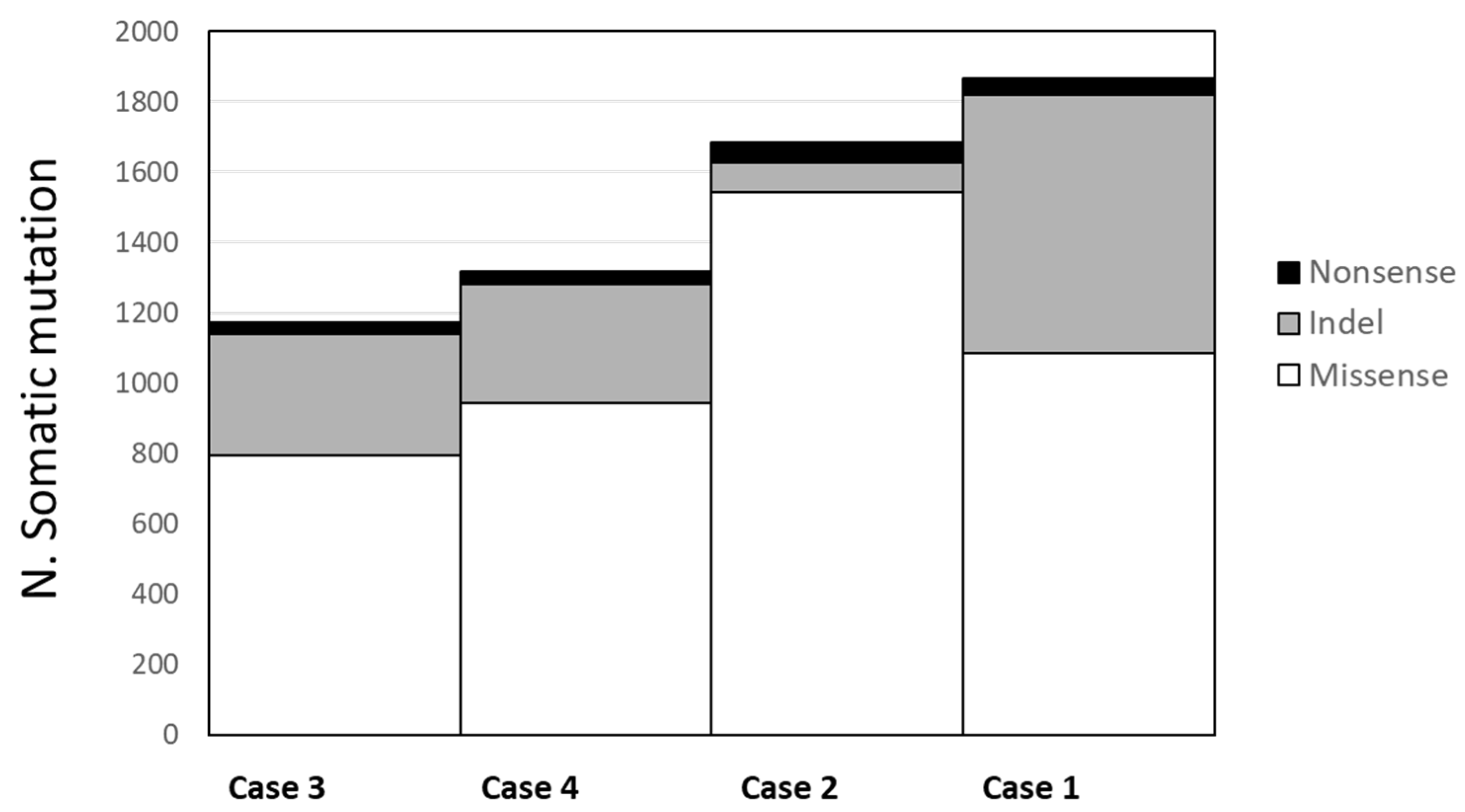
Figure 3.
RNA transcript levels in normal and tumor tissue pairs of 16 candidate germline mutated genes. RNA levels were expressed as FPKM value in tumor tissue (T), in the normal counterpart (N) and in the group of normal tissues (controls).
Figure 3.
RNA transcript levels in normal and tumor tissue pairs of 16 candidate germline mutated genes. RNA levels were expressed as FPKM value in tumor tissue (T), in the normal counterpart (N) and in the group of normal tissues (controls).
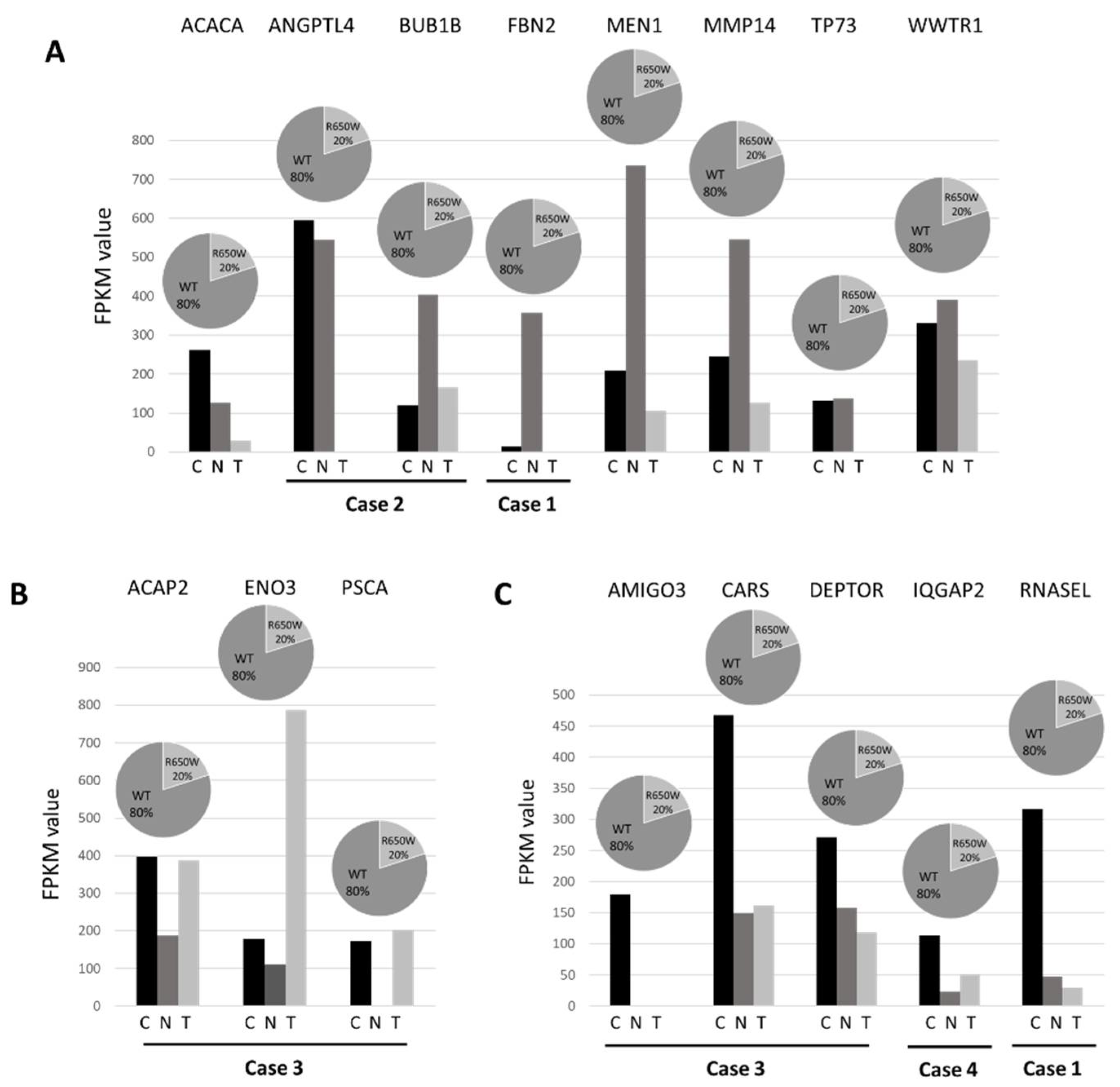
Figure 4.
Results of GO analysis of upregulated (panel A) and downregulated (panel B) transcripts in lung tumor tissues compared with lung normal tissues (all components with >10% and p-value<0.05).
Figure 4.
Results of GO analysis of upregulated (panel A) and downregulated (panel B) transcripts in lung tumor tissues compared with lung normal tissues (all components with >10% and p-value<0.05).
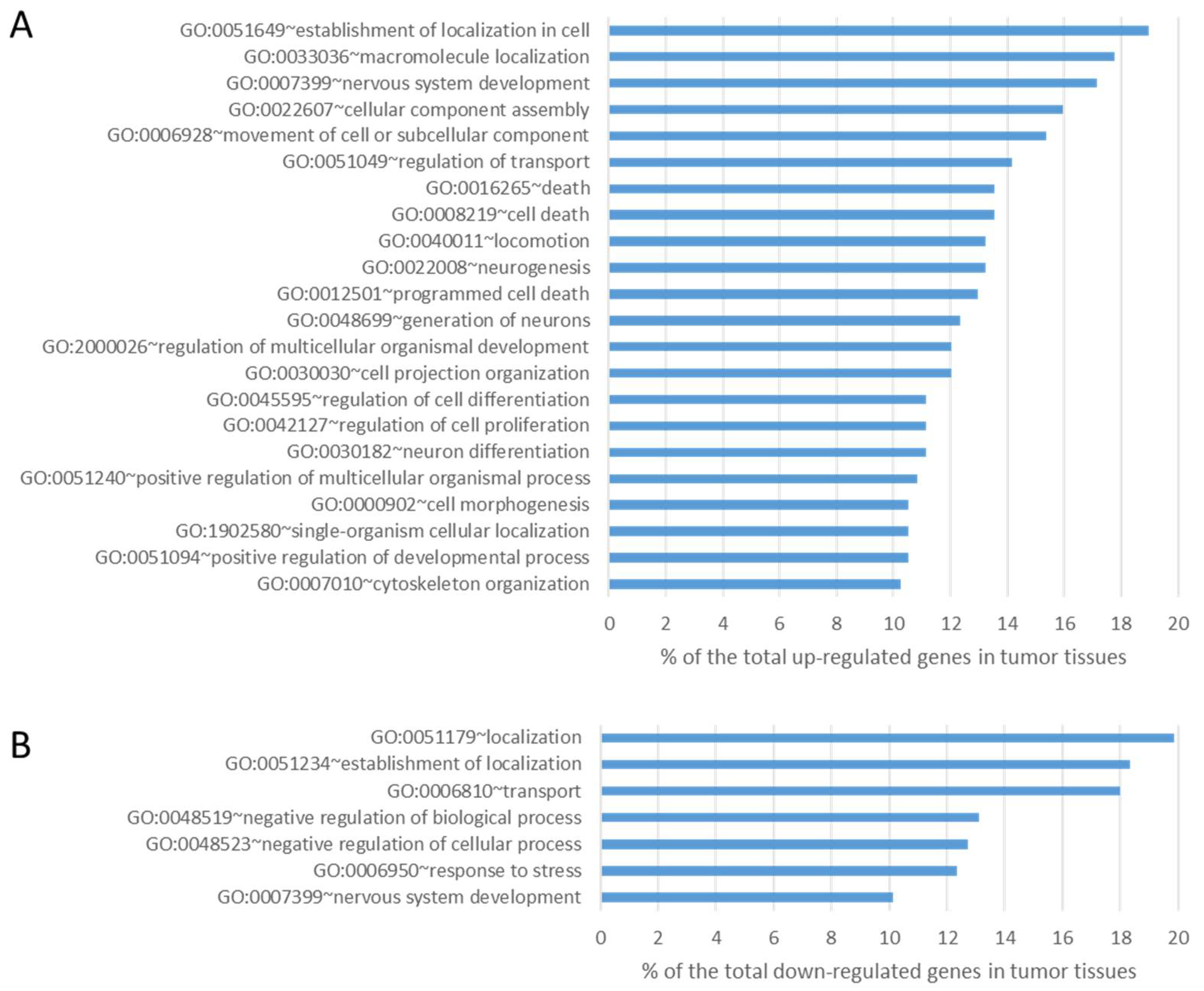
Figure 5.
Networks among germline mutated genes. .
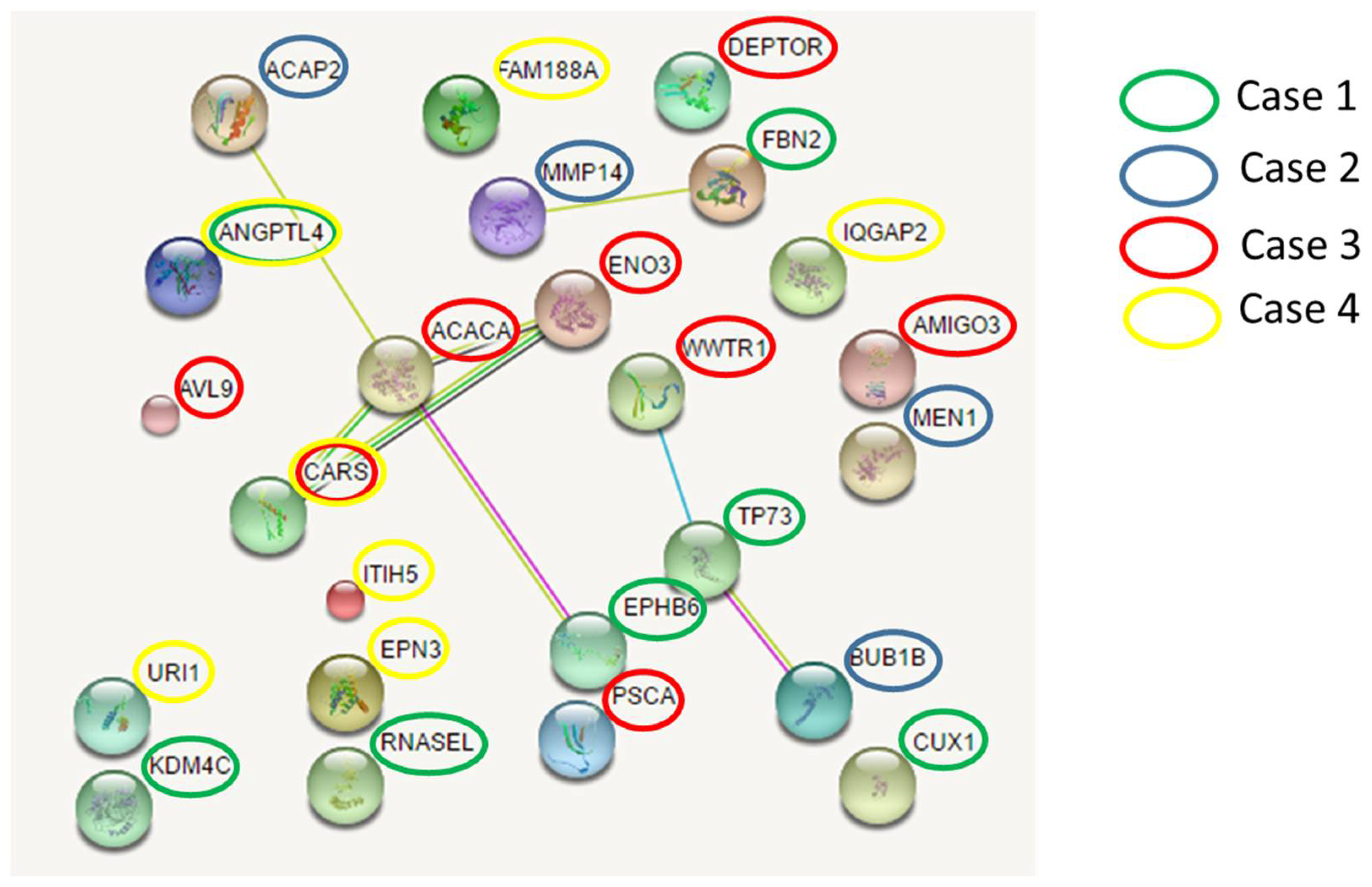
Figure 6.
Expanded network among EPHB6, ACACA, ENO3, CARS and ACAP2 genes. .
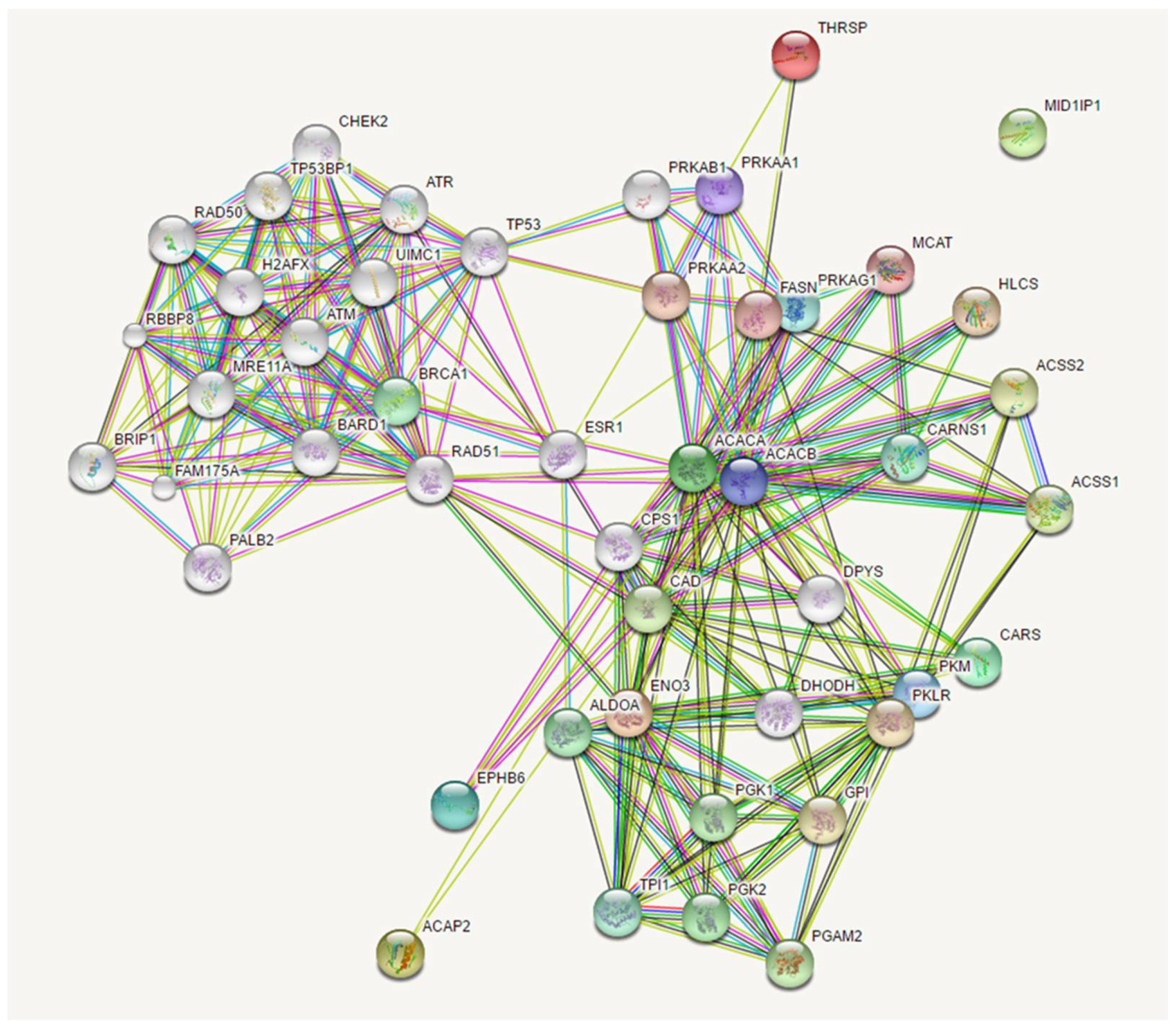
Figure 7.
Model of private oligogenic inheritance of lung cancer in non-smokers.
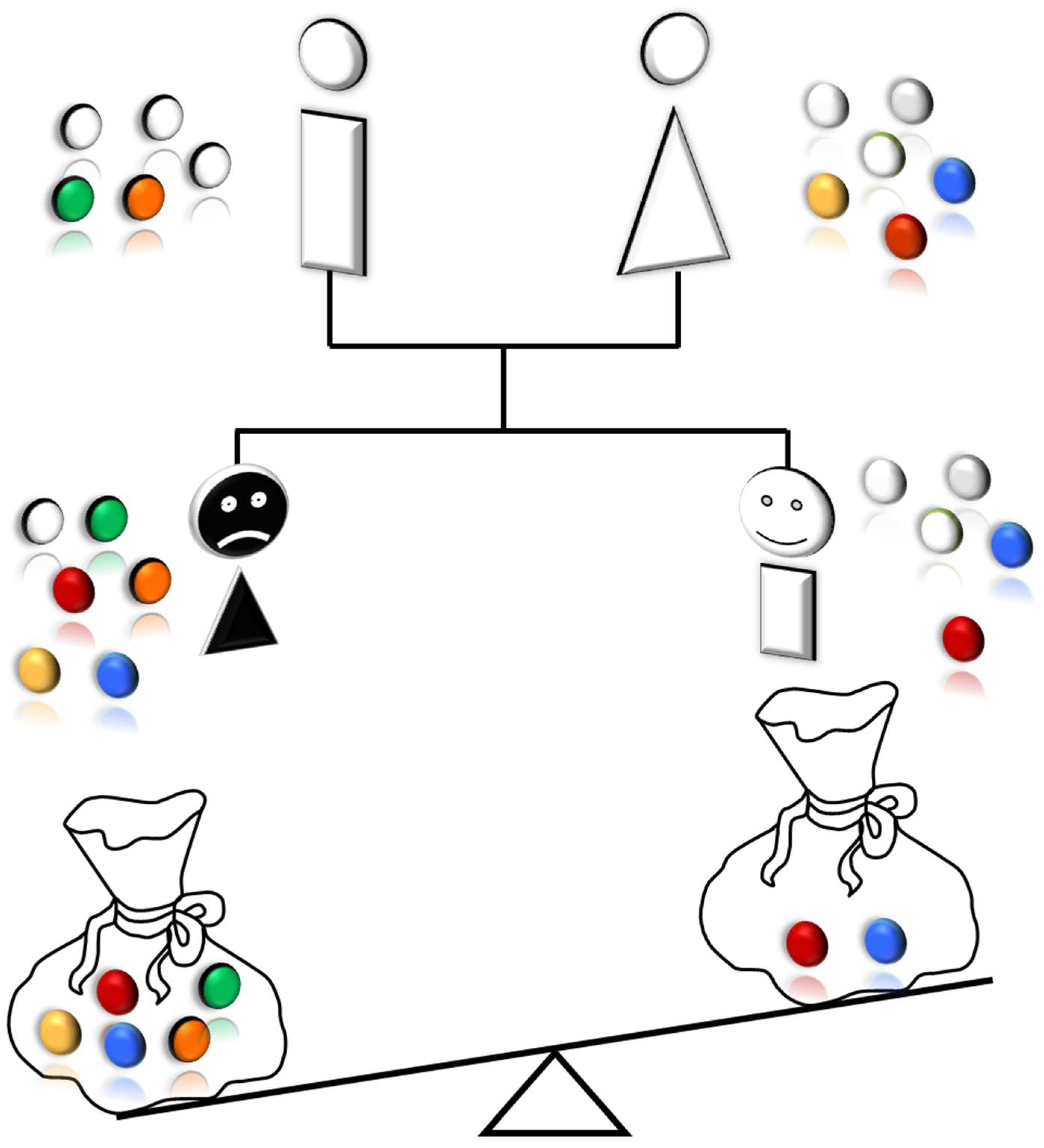

Table 1.
Clinical characteristics and sequencing data for lung cancer cases and their healthy sibling.
Table 1.
Clinical characteristics and sequencing data for lung cancer cases and their healthy sibling.
| Case 1 | Sib 1 | Case 2 | Sib 2 | Case 3 | Sib 3 | Case 4 | Sib 4 | |
| Gender | F | M | F | F | F | F | F | M |
| Age at diagnosis, years | 52 | NA | 65 | NA | 69 | NA | 55 | NA |
| Age at sampling, years | NA | 44 | NA | 64 | NA | 75 | NA | 54 |
| Smoker status | Never | Never | Never | Never | Never | Never | Never | Never |
| Histologic type | ADCA | NA | ADCA | NA | ADCA | NA | ADCA | NA |
| Follow-up status | Alive | Alive | Alive | Alive | Alive | Alive | Alive | Alive |
| Age at follow-up | 53 | 45 a | 66 | 65 a | 71 | 75 | 57 | 54 |
| Exome sequence data, Mbp | 5,63 | 5,48 | 9,06 | 6,72 | 3,11 | 11,8 | 12,8 | 5,3 |
| Number of Reads, Mil | 38,3 | 37,5 | 49,9 | 36,8 | 17,1 | 65,1 | 69,0 | 28,3 |
NA, not applicable; a cancer-free.
Table 2.
Sequence variants in cancer-related genes identified by whole-exome sequencing.
| Gene symbol: mutation | Case 1 | Case 2 | Case 3 | Case 4 | Gene name | KEGG Pathway / Function |
|---|---|---|---|---|---|---|
| ACAP2:c.C976T:p.R326X | ✓ | ArfGAP with coiled-coil, ankyrin repeat and PH domains 2 | Endocytosis / Arf6 signaling events | |||
| ACTL6A: c.T673A:p.S225T | ✓ | Actin like 6A | Chromatin organization / DNA Double-Strand Break Repair | |||
| BUB1B:c.T2609C:p.V870A | ✓ | Budding uninhibited by benzimidazoles 1 homolog beta | Cell cycle / Mitotic function (TS) | |||
| DDX11: c.G814A:p.V272M | ✓ | DEAD/H-box helicase 11 | NA / (TS) | |||
| EPB41::c.G1264A:p.E422K | ✓ | Erythrocyte membrane protein band 4.1 | Tight junction / Sertoli-Sertoli Cell Junction dynamics | |||
| MEN1: c.G301A:p.V101I | ✓ | Menin 1 | Transcriptional misregulation in cancer / Putative TS associated with a syndrome known as multiple endocrine neoplasia type 1 | |||
| ACACA:c.C1948T:p.R650W | ✓ | Acetyl-Coenzyme A carboxylase alpha | Fatty acid biosynthesis, Pyruvate metabolism, Propanoate metabolism, Insulin signaling pathway | |||
| AMIGO3: c.C669A:p.C223X | ✓ | Adhesion molecule with Ig like domain 3 | NA | |||
| AVL9: c.37_38del:p.R13fs | ✓ | AVL9 cell migration associated | NA / Late secretory pathway protein AVL9 homolog | |||
| CTBP2: c.C2149T:p.R717C | ✓ | C-terminal binding protein 2 | Wnt signaling, Notch signaling, Pathways in cancer | |||
| CTSZ:c.G358A:p.V120M | ✓ | Cathepsin Z | Lysosome / Apoptosis (candidate O) | |||
| DEPTOR: c.A631T:p.R211X | ✓ | DEP domain containing MTOR-interacting protein | mTOR signaling pathway / | |||
| ENO3: c.C642G:p.Y214X | ✓ | Enolase 3 | Glycolysis, Gluconeogenesis, RNA degradation / Possible TS in lung cancer (17p13.3) | |||
| GRM1:c.C2185A:p.P729T | ✓ | Glutamate receptor, metabotropic 1 | Calcium signaling pathway, Neuroactive ligand-receptor interaction, Gap junction | |||
| MYO10: c.C5690T:p.S1897F | ✓ | Myosin X | Fc gamma R-mediated phagocytosis / Epithelial Adherens Junctions , Innate Immune System , RhoGDI (Putative O) | |||
| PFKP:c.G311A:p.R104Q | ✓ | Phosphofructokinase, platelet | Glycolysis, Gluconeogenesis, Pentose phosphate pathway, Fructose and mannose metabolism, Galactose metabolism | |||
| PSCA:c.G326A:p.W109X | ✓ | Prostate stem cell antigen | NA / Overexpressed in prostate cancer | |||
| ROCK1:c.C727T:p.P243S | ✓ | Rho-associated, coiled-coil containing protein kinase 1 | Chemokine signaling pathway, Vascular smooth muscle contraction, Wnt signaling pathway, TGF-beta signaling pathway, Axon guidance, Focal adhesion, Leukocyte transendothelial migration, Regulation of actin cytoskeleton / Cytoskeleton remodeling | |||
| WWTR1:c.1199_1200insTTTA:p.L400_X401delinsLX | ✓ | WW domain containing transcription regulator 1 | Hippo signaling pathway / Gene Expression (TS) | |||
| CARS:c.G775A:p.G259S | ✓ | ✓ | Cysteinyl-tRNA synthetase | Aminoacyl-tRNA biosynthesis / Localized in an important tumor-suppressor gene region (11p15.5) | ||
| ANGPTL4:c.637delC:p.P213fs | ✓ | Angiopoietin-Like 4 | PPAR signaling pathway. Also known as Peroxisome proliferator-activated receptor (PPAR). PPAR activates gene expression. | |||
| CUX1: c.2413dupC:p.G804fs | ✓ | Cut like homeobox 1 | NA / FGFR1 mutant receptor activation (TS) | |||
| EPHB6: c.840delC:p.S280fs | ✓ | EPH receptor B6 | Axon guidance / (TS) | |||
| FBN2:c.G3883A:p.D1295N | ✓ | Fibrillin 2 | NA/ ERK Signaling, Degradation of the extracellular matrix (TS) | |||
| GANAB: c.C583T:p.R195C | ✓ | Glucosidase II alpha subunit | N-Glycan biosynthesis / Metabolism (TS) | |||
| KDM4C:c.3110delG:p.S1037fs | ✓ | Lysine demethylase 4C | NA / Involved in Signal Transduction, Signaling by Rho GTPases and Chromatin organization (Putative O) | |||
| MMP14:c.C609A:p.Y203X | ✓ | Matrix metallopeptidase 14 | GnRH signaling pathway / Cell adhesion_ECM remodeling | |||
| PTPN23:c.G4189T:p.G1397C | ✓ | Protein tyrosine phosphatase non-receptor type 23 | Involved in the regulation of small nuclear ribonucleoprotein assembly and pre-mRNA splicing (within a putative tumor suppressor region) | |||
| RNASEL: c.G793T:p.E265X | ✓ | Ribonuclease L | Immune System / Mutations in this gene have been associated with predisposition to prostate cancer | |||
| TP73:c.G749T:p.G250V | ✓ | Tumor protein p73 | p53 signaling pathway, Neurotrophin signaling pathway / Cell cycle (TS) | |||
| ESRRA: c.C1162T:p.L388F | ✓ | ✓ | Estrogen related receptor alpha | NA / Nuclear Receptor transcription pathway (O) | ||
| ESRRA: c.C1165T:p.R389C | ✓ | ✓ | Estrogen related receptor alpha | NA / Nuclear Receptor transcription pathway (O) | ||
| ABHD5:c.G341T:p.R114L | ✓ | Abhydrolase domain containing 5 | Regulation of lipolysis in adipocytes / Metabolism of lipids and lipoproteins | |||
| ACAD9:c.G976A:p.A326T | ✓ | Acyl-CoA dehydrogenase family member 9 | Mitochondrial biogenesis / Respiratory electron transport | |||
| EPHA7: c.A2009C:p.Q670P | ✓ | EPH receptor A7 | Axon guidance | |||
| EPN3:c.879delA:p.L293fs | ✓ | Epsin 3 | Endocytosis / Promoting senescence (O) | |||
| FAM188A:c.1107delT:p.F369fs | ✓ | Family with sequence similarity 188 member A | NA (Novel TS in NSCLC) | |||
| IQGAP2:c.G1135C:p.E379Q | ✓ | IQ motif containing GTPase activating protein 2 | Regulation of actin cytoskeleton, | |||
| ITIH5: c.1063delG:p.D355fs | ✓ | Inter-alpha-trypsin inhibitor heavy chain family member 5 | NA / RHO GTPase Effectors (TS) | |||
| PSAT1:c.G511C:p.A171P | ✓ | Phosphoserine aminotransferase 1 | Glycine, serine and threonine metabolism, Vitamin B6 metabolism / Metabolism of amino acids and derivatives | |||
| URI1:c.G1303T:p.E435X | ✓ | URI1, prefoldin like chaperone | NA / scaffolding protein with roles in ubiquitination and transcription (Putative TS) |
In bold: mutations associated with an effect at RNA level.
Table 3.
Results of pathways analysis on deregulated genes in tumor tissues compared with normal tissues.
Table 3.
Results of pathways analysis on deregulated genes in tumor tissues compared with normal tissues.
| Case 1 | Case 2 | Case 3 | Case 4 | |||||
|---|---|---|---|---|---|---|---|---|
| KEGG pathway | p-value | Count | p-value | Count | p-value | Count | p-value | Count |
| ECM-receptor interaction | 9,50E-08 | 22 | 1,40E-06 | 17 | 5,20E-05 | 20 | 2,60E-03 | 13 |
| Focal adesion | 4,20E-03 | 25 | 1,80E-04 | 23 | 3,10E-02 | 26 | ||
| ABC transporters | 8,30E-03 | 9 | ||||||
| Integrin signaling pathway | 3,70E-02 | 26 | ||||||
| Tight junction | 2,40E-02 | 13 | ||||||
| Cell adhesion molecules (CAMs) | 4,70E-02 | 12 | ||||||
| Calcium signaling pathway | 3,90E-02 | 23 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



