
Submitted:
16 July 2024
Posted:
17 July 2024
You are already at the latest version
Abstract
Polyethylene terephthalate (PET) is one of the most produced plastic materials in the world. The emer-gence of microplastics and nanoplastics (MPs/NPs) as a significant environmental contaminant has become a matter of increasing concern. While the toxicological effects of PET NPs have been widely re-searched, there is a lack of methodologies for studying their accumulation. The present study introduces a novel method to monitor the distribution of PET NPs in germinating wheat (Triticum aestivum L.) seeds. This involves the functionalization of superparamagnetic iron oxide nanoparticles (SPIONs) with PET NPs (PET-fSPION) coupled with magnetic resonance microimaging (µMRI) to provide insight into their distribution within the seed. The present study has demonstrated that PET-fSPION accumulates in specific regions of germinating wheat seeds, including the shoot apical meristem, the radicle, the cole-optile, the plumule, and the scutellum. Furthermore, the accumulation of PET-fSPION has been shown to exert a discernible effect on spin-spin relaxation, as observed via MRI and quantitative T2 relaxation time analysis. The accumulation of PET NPs in embryo regions was also confirmed by SEM. Diffu-sion-weighted magnetic resonance imaging (DW-MRI) and non-invasive chemical shift imaging analyses demonstrated that PET NPs resulted in restricted diffusion within the highlighted areas, as well as an impact on lipid content. Our study reveals that using µMRI with fSPION provides a non-invasive method to monitor the biodistribution of PET nanoparticles in wheat seeds. Additionally, it offers valuable in-sights into the microstructural interactions of PET.
Keywords:
Polyethylene terephthalate
; nanoplastics
; µMRI
; SPION
; wheat
1. Introduction
Plastics are persistent pollutants that circulate in the primary food chain and can pose a threat to our environment. It is estimated that 390.7 million tons of plastic products are produced each year and 55 percent of global plastic waste end up in landfill [1,2]. More than 690 marine species have been affected by plastic pollution and are reported to ingest plastics, with adverse effects postulated through physical impairment, direct metabolic toxicity or physiological effects from additives and adsorbed pollutants [3]. The major concerns regarding plastics in terrestrial systems are their observed prevalence, their transport through the soil, and the further translocation of these particles through the food chain. According to the estimations, humans exposed to 39,000 to 211,000 micro- and nanoplastics (MP/NPs) particles per year via drinking water bottles, air, and food [4]. Polyethylene terephthalate (PET) is one of the most widely used polymeric packaging materials in the world. PET plastic particles have been found in groundwater, drinking water, soil, sediments, and air [5,6,7,8].The hazardous effects of PET NPs in various organisms (e.g. zebrafish, copepods) and humans have been studied [5,8,9,10]. These particles can alter cell physiology and affect cellular metabolism, leading to hepatoxicity associated with membrane dysfunction and alterations in pathways involved in energy metabolism [11]. Although there are many toxicological studies on the influence of PET NPs in various biological systems, a suitable method for tracking and internalization of these particles is still lacking.
Previous studies have attempted to track the accumulation of these particles using fluorescently labelled NPs [12,13,14]. However, the use of fluorescence techniques is challenging due to numerous factors such as background effect, scattering and bleaching, which further affect the sensitivity and accuracy of detection. To enhance the understanding of uptake and biodistribution of NPs some studies focused on with radiolabeled NPs [15,16,17]. For example, radiolabeled zirconium-89 (t1/2 ~ 3.3 days) and 14C-labelled (t1/2 ~ 5,730 years) PS NPs have been used to study the biodistribution of these particles in vivo using positron emission tomography [18]. In addition, a quantitative imaging approach, radionuclide imaging tracking, has been used to follow the spatial and temporal biodistribution of various emerging contaminants, including NPs, in vivo [19]. However, it has limited applicability due to its hazardous nature, cost, and high stability [20]. A previous study has focused on tracking PET NPs in vivo using inductively coupled plasma mass spectroscopy with an iridium-containing organic molecular agent [21]. Although this method provides quantitative information on the spatial and temporal distribution of NPs, it requires time-consuming sample preparation, which can also lead to sample loss.
Advances in magnetic resonance imaging (MRI) provide the possibility to assess the structure and metabolism of living organisms non-invasively for diagnostic and research purposes [22,23,24]. MRI has been used to study dynamic processes in a plant biology using contrast agents (e.g. Gd-based) [25,26]. This approach relies on the immediate impact of paramagnetic contrast agents on tissue-specific magnetic properties, predominantly altering the longitudinal relaxation times (T1). Despite the complex tissue architecture of plants, the adaptability of MRI techniques allows for effective imaging, mitigating many of the challenges associated with plant versus animal tissue differences [24]. Recently, Sarcletti et al. proposed a method for the quantification of polystyrene (PS) and polymethylmethacrylate (PMMA) NPs/MPs in water samples using iron-based contrast agents (e.g. superparamagnetic iron oxide NPs (SPION) with different iron cores (Fe3O4 or Fe2O3) with corresponding phosphonic acid monolayers) [25]. These contrast agents have previously been shown to be non-toxic to leukemia cells [27]. SPION are single-domain iron oxide nanoparticles with a core diameter between 10 nm and 100 nm [28,29]. Due to the unique distinct characteristics, SPION have been used in a variety of biomedical applications, including diagnostics and therapeutic applications [7,30,31]. These nanoparticles are widely used as magnetic-based T2 contrast agents due to their high chemical stability, lack of toxicity, and biodegradability [11,32]. It has a higher molar ratio with a darkening effect on MRI.
The unique chemical and relaxation properties of PET NPs pose significant challenges for their detection via traditional MRI methods, underscoring the need for innovative approaches. The use of MRI contrast agents attached to PET NPs can be a feasible approach to track the localization of these NPs in biological organisms, as they can influence the relaxation of the nearby protons. Attachment of another type of NPs namely PS with functionalized SPION has been shown in a previous study for determination of NPs in water sample [33].
In this proof-of-concept study, we attached PET NPs with functionalized SPION (PET-fSPION) and present a non-invasive method to monitor accumulation of PET NPs in germinating wheat (Triticum aestivum L.) seeds using µMRI. Wheat is a widely grown crop, accounting for 30% of global crop consumption. Given the widespread presence of PET NPs, comprehending their accumulation and effects on metabolism is essential. Our results show that PET NPs bound with fSPION can be non-invasively monitored using µMRI. Furthermore, we show that PET NPs significantly influence the microstructural changes in germinating wheat seeds. Our findings demonstrate the potential of µMRI in advancing our understanding of nanoparticle interactions and provides a valuable insight for future research in this field.
2. Results and Discussion
In this study, we aimed to develop a biocompatible method using fSPION as a contrast agent to track PET NPs in wheat seeds non-invasively using µMRI. The PET NPs and fSPION were synthesized, and their successful attachment was verified through comprehensive characterization. We assessed the relaxivity of PET-fSPION to ensure their effectiveness as MRI contrast agents. µMRI was then employed to visualize distribution of PET-fSPION in germinating wheat seeds, providing detailed insights into their localization and interaction.
2.1. Characterization and Analysis of PET-fSPION
At first, the stable PET NPs and fSPION were prepared as described previously [35,36]. Subsequently, PET NPs were attached with fSPION to generate PET-fSPION. To assess interaction between PET NPs and fSPION characterization was performed using transmission electron microscopy (TEM), dynamic light scattering (DLS), and Fourier transform infrared spectroscopy (FTIR).
Detailed insights into the morphology of both PET NPs and PET-fSPION assessed via TEM is shown in Figure 1. TEM imaging revealed that the PET NPs were spherical and there was an interaction between fSPION within the PET NPs (Figure 1A). This interaction is observed as the fSPION consistently agglomerate around PET NPs spheres. The PET NPs have an average size of ~ 44 nm, while the fSPION particles have an average size of 8.8 nm. To investigate details of the distribution of fSPION around PET NPs, material contrast imaging was performed by high-angular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDX). The HAADF-STEM imaging shows an overview of fSPION around PET NPs (Figure 1B). The STEM-EDX map illustrates the elemental distribution with net intensity EDX signals. The yellow signals correspond to iron, indicating the presence and distribution of the fSPION within the PET NPs (Figure 1C). Additionally, carbon signals, depicted in purple, emphasized the carbon-rich nature of the PET NPs as well as the carbon support film of the TEM grid (Figure 1D). The size distribution of PET NPs and fSPIONs is shown in Supplementary Figure S1.
DLS analysis was employed to assess the hydrodynamic size distribution and zeta po-tential of PET NPs, fSPION, and PET-fSPION, as shown in Figure 2A and B. This analysis revealed that the hydrodynamic sizes of PET NPs, fSPION, and PET-fSPION were 67 ± 3 nm, 185 ± 15 nm, and 245 ± 20 nm, respectively (Figure 2B). Zeta potential measurements demonstrated that PET NPs exhibit a negative charge (-20 ±5 mV), whereas fSPION displayed a positive charge (+20 ± 5 mV). The combination of PET NPs with fSPION in PET-fSPION yielded a composite with a surface charge of +15 ± 4 mV nearly as positive as that of pure fSPION. The relatively larger size of the PET-fSPION composite compared to the individual components could be due to the formation of a stable and integrated nanostructure, potentially due to strong electrostatic interactions and physical entanglement between the particles. Furthermore, hydrodynamic size of PET-fSPION (245 ± 20 nm) indicates that these particles are within an effective range for plant tissue penetration [34]. The positive zeta potential of PET-fSPION suggests that the fSPION’s surface properties predominantly influence the overall charge of the composite. The reduced absolute value of the zeta potential in the composite compared to pure fSPION (+15 mV vs. +20 mV) might be due to partial neutralization of charges, indicating electrostatic interaction between the components. The polydispersity index (PDI) values suggest that both the PET-NPs and PET-fSPION are homogeneous in solution and show low tendencies towards aggregation and remaining stable throughout the measurements. For detailed characteristics of each particle group, refer to Figure 2B.
FTIR spectrum of amorphous PET film, PET-fSPION, fSPION and SPION is shown in Figure 2C. The FTIR analysis revealed specific vibrational modes associated with the chemical bonds present in the samples. The PET film exhibited strong stretching vibrations of the C--O bond ν(C-O) at 1715 cm–1 and vibrations associated with ν(C–O) (esters and ethers) around 1240 cm–1. Weak absorption bands corresponding to alkyl ν(C-H) and aryl groups, as well as characteristic bending vibrations of aromatic compounds, were observed at 2960 cm–1 and 1407 cm–1, respectively.
These findings are consistent with the molecular structure of PET [35]. For both fSPION and commercially purchased SPION (γ-Fe2O3), the stretching bands appeared in the low wavenumber range of 500-750 cm–1. Additional weak signals were detected at 1430 cm–1 (associated with CHn vibrations), 1630 cm–1 (related to C-C vibrations), and around 3300 cm–1 (typical of ν(O-H) vibrations). The fSPION exhibited prominent signals corresponding to alkyl chains at 2920 cm–1 ν(C-H) and 2850 cm–1 ν(N-C), as expected based on the structural characteristics [36]. A broader peak around 1000 cm–1, attributed to the phosphate group, was also observed. The absorption bands characteristic of SPION were still present in fSPION but exhibited different transmittance properties. When mixing PET NPs with fSPION and analyzing PET-fSPION the same characteristic bands of PET, as well as the fSPION, were observed. These results suggest that coupling of PET NPs with fSPION in PET-fSPION did not alter the properties of PET as well as fSPION and it remained intact during the mixing process. Thus, FTIR analysis indicates that the attachment of PET NPs with fSPION was successful. The adsorption of fSPION to PET NPs may have been facilitated by both electrostatic and van der Waals interactions, as evidenced in previous studies where similar processes have been observed in the use of other types of NPs, including PS and PMMA. [33].
2.2. Relaxivity of PET-fSPION and fSPION
To investigate whether the attachment of PET NPs with fSPION can influence their relaxation properties, we analysed the T2 relaxivity of fSPION and PET-fSPION. The standard curve using various concentrations of fSPION and PET-fSPION was made to obtain an R2 value of 0.9933 for r2 SPION and an R2 value of 0.9970 for r2 PET-fSPION (Figure 3A and 3B). R2 values shows the fit quality of the data to the regression model. The standard curve equation was as follows: y = 254.63x + 5.6377 (PET-fSPION) and y = 289.4480x + 3.5588 (fSPION), where x represents the concentration of Fe2+ ions (mM) and y are the relaxation rate (s-1). The results indicate a slight variation in r2 values between fSPION (289 mM-1s-1) and PET-fSPION (254 mM-1s-1) respectively. The plotted transverse relaxation rate, R2 (1/T2), exhibited a linear correlation with fSPION and PET-fSPION concentration (mM). The relaxivity (r2) value of our fSPION showed a relaxation property-altering effective T2 value with an r2 of 289 mM-1s-1. This finding is consistent with previous measurements of SPION integrated with a chitosan-based polymer, which exhibited a relaxivity (r2) value of 294.8 mM-1s-1 at the same Fe2+ ion concentration used in our study. This consistency extends across various cluster sizes in other studies with 150 nm clusters of 8 nm particles, 100 nm clusters of 7 nm particles, and 50 nm clusters of 8 nm particles exhibiting 294 mM-1s-1, 294 mM-1s-1, and 227 mM-1s-1 in relaxivity, respectively [37-39]. The slight variation in magnetic relaxivity between PET-fSPION and fSPION might be related to the changes in local magnetic field within the solution resulting from the introduction of non-magnetic PET NPs. Thus, even non-magnetic materials like plastics, can affect the local magnetic field to some extent due to their physical presence [40]. This is a phenomenon known as the magnetic susceptibility of materials [41]. Although the effect of PET NPs on the magnetic field is minimal, it can still alter the local magnetic environment, resulting in slightly different relaxivity values for PET-fSPION compared to fSPION. This has been explained in earlier studies concerning other non-magnetic materials [44].
2.3. Localization of PET NPs in Wheat: Insights from SEM
Scanning electron microscopy (SEM) was employed to track the distribution and morphology of the PET-fSPION particles within wheat seeds (Figure 4). The SEM analysis revealed the presence of PET NPs as aggregates, exhibiting a distinct chain-like formation. This morphological evidence indicates translocation of PET NPs and their specific localization within the embryo region of the wheat seed. Moreover, these particles were also seen in the endosperm and crease regions of wheat seed (Figure S2). These results suggest that PET-fSPION were absorbed by seeds during imbibition and effectively penetrated through the seed coats and enter seed tissues. These results are in line with earlier studies where other types of nanoparticles have been shown to be absorbed by the seeds during imbibition [43]. Direct absorption of nanoparticles in seeds is known to occur by entering the coat via parenchymatic intercellular spaces, accompanied by diffusion in the cotyledon [42].
2.4. Impact of PET-fSPION on Spin-Spin Relaxation in Wheat Seeds
Representative T2-weighted MRI images of germinating wheat seed treated with only water (control) or PET-fSPION, are shown in Figure 5. The embryo region is shown in magnification. As PET-fSPION are magnetic and have a stronger effect on the local magnetic field, they shorten T2 relaxation times of neighbouring protons [43]. Thus, the presence of PET-fSPION at various locations in the seed is clearly visible as darker areas in the images (indicated by arrows in the magnified area). A marked reduction in signal intensity was also observed in the region around the root cap and root sheath.
These observations are further supported by quantitative T2 relaxation analysis (Figure 6). The shortning of T2 due to penetration of PET-fSPION was observed in different areas of the embryos, including the shoot apical meristem (ROI 1), radicle (ROI 2), coleoptile (ROI 3), plumule (ROI 4) and scutellum (ROI 5), as illustrated in Figure 6A. A colour T2 map as shown in Figure 6B, also clearly indicate the reduction in T2, due to the penetration of PET-fSPION in various locations within the embryo. The T2 relaxation time values of all five selected regions of interest (ROIs) in the embryo were quantitatively analysed (Figure 6C). Figure 6D represents the results of the one-way ANOVA statistical analysis performed on the T2 values from selected ROIs. The results indicated that there were significant changes in the T2 values in radicle and scutellum (p < 0.05), with more significant changes in coleoptile and plumule regions (p < 0.01) when exposed to PET-fSPION compared to the control. In contrast, shoot apical meristem exhibited a less pronounced effect on T2 relaxation time.
These results indicate that, regions such as the coleoptile and plumule, which play an active role in early seedling development, showed higher accumulation of PET-fSPION. Coleoptile is a protective sheath covering the young shoot of the embryo that helps in guiding the shoot through the soil, and plumule is the part of the embryo that develops into the shoot system, including the first true leaves of the plant. These areas are highly metabolically active. The enhanced permeability and retention effect of other nanoparticles in these regions with high metabolic activity and permeability has been documented [44,45].
2.5. Diffusion MRI Measurements
The regions which show high accumulation of PET NPs were further analysed by diffusion-weighted MRI (DW-MRI), a technique that enables the visualization of both the diffusion restriction and microstructural alterations resulting from PET NPs accumulation.
The apparent diffusion coefficient (ADC), a well-established marker of cellular integrity and tissue structure [46]. It was used to assess the intracellular diffusion barrier. DW-MRI measurements were performed in selected ROIs which show PET-fSPION accumulation. Figure 7A shows the diffusion image of wheat seed and indicate selected ROIs. The ADC map generated through the ’dtraceb’ algorithm illustrated the diffusion behavior across the seed (Figure 7B). A normalized mono-exponential fitting curve for each selected ROIs, in embryo regions, is shown in Figure 7C. In all ROIs, PET NPs treated seeds show attenuated signal decay, indicating restricted diffusion compared to control wheat. ADC values were significantly lower in PET NPs treated seeds across all ROIs (Figure 7D). The coleoptile (ROI 3), plumule (ROI 4) and scutellum (ROI 5) show significant reduction in ADC values. These results indicate that accumulation of PET NPs in these areas cause diffusion restriction, either directly or due to their interaction with the microstructure of the wheat seed embryo.
2.6. Chemical Shift Imaging
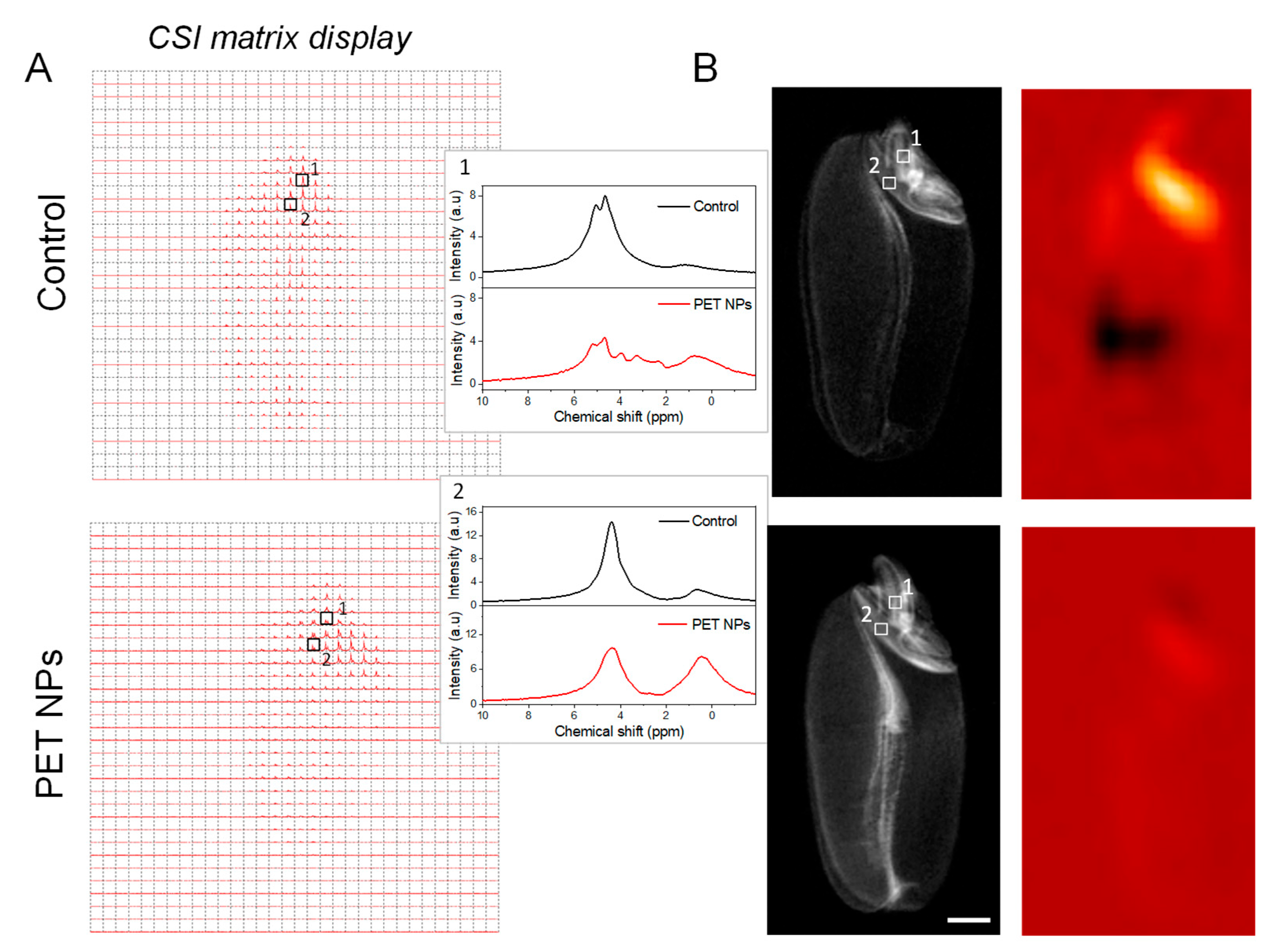
Non-invasive chemical shift imaging (CSI) was used to analyze the content and distribution of metabolites in wheat seed at the spectroscopic level. CSI is particularly adept at mapping the spatial distribution of hydrogen nuclei, which are typically associated with either water or lipid molecules (Figure 8). In Figure 8A, the CSI data from scutellum and radicle regions is presented as a matrix array of spectra. The peak between ~0.5 and 3 ppm corresponds to the aliphatic protons -(CH2)n- of carbon atoms (Figure 8A). This suggests that this region contains lipids that might have been mobilized from stored triacylglycerol during the early stages of seed germination. [47–49]. The peak at 4.7 ppm is from water signal.
Wheat seeds treated with PET NPs showed an increase in lipid content and a decrease in water content in the scutellum as well as in radicle regions during the early stages of germination (Figure 8). While some lipid content is necessary for seed viability, excessively high levels might impede other critical functions. For example, high lipid accumulation may affect water uptake or oxygen diffusion, which could have a negative impact on germination rates and overall seedling vigour [50–52]. The observed increase in lipid content may be indicative of a stress response to PET NPs exposure, as has been previously documented in plant studies with other NPs [53]. The underlying mechanisms by which exposure to PET NPs leads to enhanced lipid accumulation remain unclear and necessitate further investigation. To visualize the spatial distribution of lipids, a color map is reconstructed from the lipid signal and displayed and overlaid on an MR image of the same slice (Figure 8B).
The second noticeable peak in radicle region, occurring in the range of ~3 to 5 ppm, is characteristic of protons found in overlapping carbohydrate molecules [54–56]. These are likely present due to the degradation of starches and other polysaccharides as the seed activates its metabolism. To contextualize these findings, we refer to literature on the effects of another nanoplastics namely PS NPs on starch granules during wheat seed germination [57]. It has been observed that PS NPs can induce higher α-amylase activity, leading to significant degradation of starch granules [57]. α-amylase typically initiates the hydrolysis of starch by pitting the surface of starch granule and then penetrating into the interior, hydrolyzing the granule from the inside out. They observed that starch granules have rougher and eroded appearance in PS NPs treated seeds, implying increased α-amylase penetration and higher starch decomposition [57]. By analogy, we hypothesize that in our study, PET NPs may have similar effects leading to high degradation of starch and accumulation of sugars as seen in our CSI data (Figure 8). Despite the elevated level of sugars observed in the radicle of PET NPs-treated seeds, it was not utilized for boosting radicle growth, as we observed inhibitory growth of the radicle in PET NPs treated seeds compared to non-treated controls. It appears that these sugars may have been accumulated as a means of coping with the stress generated by PET NPs treatment.
3. Materials and Methods
3.1. Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. 12-dodecylphosphonic acid-N, N-dimethyl-N-octadecylammonium bromide (PAC12NC18) was purchased from Sikemia, France. SPION (used as a hypernym for Fe2O3) with a size below 45 nm were provided by COMAR Chemicals, South Africa.
3.2. Preparation of PET-fSPION
Initially, PET NPs were prepared according to the previous method with slight modifications [58,59]. Briefly, 100 mg of amorphous PET (Goodfellow, Germany) was dissolved in 10 mL of hexafluoroisopropanol (1% v/v) at room temperature for 12 h. The PET suspension was then transferred to the buret and the solution was added dropwise to ice-cooled deionized (DI) water (200 mL) while stirring continuously. To eliminate larger particles, the suspension was filtered (type 12, cellulose membrane, 125 mm diameter, Roth, Germany). The organic solvent was then removed from the solution using a rotary evaporator (Heidolph Instruments, USA) at elevated temperature and reduced pressure (50 °C, ~ 250 mbar). The NPs were then allowed to settle in the cylinder for 2 h and the upper 40 mL suspension was collected. The concentration of NPs was determined gravimetrically by drying 2 mL of the suspension in a pre-dried polymer pellet (3x) at 50 °C for 24 h and then weighing to quantify the residue. The stock concentration of PET NPs used was 0.35 mg/ml. The prepared solution was stored at room temperature, and it was stable for several weeks without any sedimentation.
To attach PET NPs to SPION, the SPION were first functionalized. Functionalized SPION (fSPION) were prepared using gamma-Fe2O3 core labeled with PAC12NC18 according to the previous following established procedure [33,60,61]. Briefly, PAC12NC18 (20 mM) as well as SPION (3 mg/mL) were dispersed in methanol. PAC12NC18 and SPION were mixed in a ratio of 3: 10 and sonicated in a SONOCOOL 255 (Bandelin, Germany) for 30 min. The dispersion was then centrifuged to collect fSPION. The collected SPION were washed twice in methanol and the supernatant of the second centrifugation step was transferred into DI water using a Hei-Vap Advantage rotary evaporator (Heidolph Instruments, Germany) at 60 °C and 337 mbar. The final fSPION stock concentration was 0.35 mg/ml.
Subsequently, PET NPs were mixed with fSPION in a concentration ratio of 2: 1 to prepare PET-fSPION.
3.3. Dynamic Light Scattering (DLS)
DLS (ZetaPALS, Brookhaven, US) was used to determine the size of the fSPION, PET NPs and PET-fSPION in water. For analysis of PET-fSPION the solution was first diluted tenfold with water before the DLS measurement. Samples were allowed to equilibrate in the sample compartment at 25 °C for at least 2 min and measured in acrylic cuvettes (10 × 10 × 45 mm, Sarstedt, Germany) at 25 °C. Each measurement was carried out three times and zeta potential measurement conducted in 10 successive runs. The hydrodynamic diameter and zeta potential (according to the Smoluchowski equation) of PET NPs were evaluated [58,62]
3.4. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
ATR-FTIR was performed on a Bruker Alpha II FTIR spectrometer (Bruker Optik GmbH, Germany) equipped with a Diamond Crystal ATR attachment. The spectrum of the diamond crystal was obtained as background. Analyzed samples included fSPION, SPION, PET-fSPION, and PET film. For analysis, 20 µL of each sample (fSPION and PET-fSPION) solution was applied to the ATR crystal and then dried at 60 °C for 15 minutes. Solid samples, such as SPION and PET film, were directly placed on the ATR crystal Absorbance spectra were recorded between 4000 and 399 cm-1 with a resolution of 2 cm-1 and 128 averaged scans. Data acquisition was performed using Opus 7.8 software (Bruker Optik GmbH, Germany) and the acquired data were further plotted using OriginPro v.8 (OriginLab, Northampton, MA, USA).
3.5. Scanning Electron Microscopy (SEM)
Wheat seeds were soaked in DI water or PET-fSPION for 24 h. Subsequently, the samples were fixed with 4% formaldehyde (FA) for 24 h and rinsed with PBS buffer. After fixation, the samples were manually cut in half with a scalpel and dried at 50°C for 6 h. Subsequently, they were mounted onto sample holders using silver DAG (PLANO) and platinum coating, approximately 5 nm thick, was applied using a BAL-TEC SCD 050 sputter coater for 40 seconds at 40 mA, while maintaining a working distance of 50 mm. SEM images were then taken using a Thermo Fisher (formerly FEI) NOVA NANO® SEM 200 equipped with a Schott field emitter. The imaging conditions were set to a high voltage of 10 kV with magnifications of 700x and 10000x, and a working distance of 5.5 mm for each image.
3.6. Transmission Electron Microscopy (TEM)
TEM was employed for characterization of PET-fSPION. The sample preparation was performed by transferring a droplet (10 μl) of PET-fSPION solution onto a holey carbon TEM-grid, which was allowed to settle and dried at room temperature. TEM analysis was performed using a JEM2100Plus microscope (Jeol, Japan). The microscope is equipped with a LaB6 filament and high-resolution pole piece and was operated at an accelerating voltage of 200 keV. Elemental analysis was performed by energy dispersive X-ray (EDX) analysis using a windowless Optima T-30 detector (EDAX, UK). Furthermore, images were acquired with a 4K ultrafast CMOS camera system (TVIPS, Germany) and analyzed with the associated EMMeasure software (TVIPS, Germany). High-angle annular darkfield (HAADF) scanning transmission electron microscopy (STEM) was employed to obtain material contrast imaging of the heavy Fe2O3 and the lighter PET NPs. The acquisition of HAADF STEM images was performed by the TEM center software (Jeol, Japan) and evaluation of the images and size of the particles evaluated by SightX-Viewer (Jeol, Japan).
3.7. Determination of Iron Content in fSPION
Prior to evaluating the relaxivities of fSPION and PET-fSPION, the iron concentration was determined using the 1, 10-phenanthroline method. [63]. Briefly, a standard solution of iron was prepared by dissolving iron ammonium sulfate hexahydrate in water to achieve a concentration of 70 mg/L (equivalent to 0.178 mmol/L), with a volume of 200 mL. To this, 2.5 mL of concentrated sulfuric acid was added before diluting to 1 L with water in a volumetric flask. Calibration standards were created at concentrations of 0, 0.7, 3.5, 7, 14, and 24.5 mg/L from the iron standard, with DI water serving as the blank. The appropriate volumes, as indicated in Table S1, were transferred into volumetric flasks. To each flask, including the blank, the following were added: 1 mL of a 100 g/L hydroxylamine solution, 10 mL of a 1 g/L 1, 10-phenanthroline solution, and 8 mL of a 98.4 g/L sodium acetate solution. The fSPION sample was prepared in the same manner as the calibration standards, with an iron concentration of approximately 7 mg/L. After allowing all solutions to stand for 10 minutes for color development, the absorbance was measured thrice at 510 nm using a Shimadzu 1900i UV/VIS spectrometer equipped with a TCC240A temperature control system. The iron concentration in the fSPION sample was then calculated from the calibration curve established by the absorbance readings of the standard solutions.
3.8. Relaxivity Measurement of fSPION and PET-fSPION
The performance of fSPION as a contrast agent was assessed by evaluating the relaxivities of fSPION and PET-fSPION using a 200 MHz (4.7 T) MRI system. Samples were linearly diluted either in DI water or PET NPs. T2 values were recorded using a spin-echo pulse sequence. The imaging parameters for the experiments were: number of averages (NA) = 64, number of slices = 3 with a slice thickness of 0.5 mm, number of echoes = 40 with echo spacing = 4.28, repetition time (RT) = 2 s, effective spectral bandwidth = 100 kHz, filed of view (FOV) = 20 × 20 cm², matrix size = 128 × 128, providing an effective in-plane resolution of 0.078 × 0.078 mm² and a voxel resolution of 10 × 10 × 3 mm³. The total acquisition time for the experiment was 4 minutes.
3.9. MRI Experiments
The MRI experiments were conducted using a Bruker 300 MHz (7T) or 200 MHz (4.7T) vertical bore system (Bruker Biospin GmbH, Germany). A birdcage transmit/receive radiofrequency (RF) coil with an internal diameter of 10 mm or 5 mm was utilized at magnets. The workstation was connected to a Linux operating system running Paravision 5.1 (at 7T) or Paravision 360 v2.1 (at 4.7T) imaging software (Bruker Biospin GmbH, Germany). All measurements conducted at room temperature.
Prior to imaging, wheat samples were imbibed for 24 h in DI water (control) and PET NPs with and without fSPION in a round 96-well microplate (Greiner Bio-One, Germany). For microimaging, the imbibed seeds were carefully surface-dried with a paper towel and inserted into NMR tube and the ends of the glass cylinder were sealed with parafilm (American Can Comp., Greenwich, CT) to avoid possible drying during the MRI experiment-The seeds were imaged with multi-slice multi-echo (MSME), rapid acquisition with relaxation enhancement sequences (RARE), diffusion-weighted imaging (DWI) and chemical shift imaging (CSI).
Each measurement started with a gradient-echo sequence to determine the position and to select the desired region for the further experiments. For anatomical imaging, RARE sequence was used. RARE, a rapid imaging sequence, utilizes an RF excitation pulse succeeded by a series of refocusing pulses [64]. This technique generates multiple RF spin echoes, enabling faster image acquisition by capturing more than one k-space line during each repetition. The imaging parameters for the RARE sequence were as follows: echo time = 20 ms, RT = 2 s, NA = 64, rare factor = 4, number of slices = 10 with a slice thickness of 0.2 mm, FOV = 10 × 10 cm², effective in-plane resolution of 0.078 × 0.078 mm², and a voxel resolution of 10 × 10 × 3 mm³.
T2 values were measured using the MSME sequence, based on the Carr-Purcell-Meiboom-Gill (CPMG) sequence. In this method, transverse magnetization from a 90-degree pulse is refocused by a train of 180-degree pulses, generating a series of echoes [65,66]. The imaging parameters for in vivo experiments were: NA = 64, number of slices = 3 with a slice thickness of 0.5 mm, number of echoes = 16 with echo spacing = 4.28, TR = 2 s, effective spectral bandwidth = 100 kHz, FOV = 8 × 8 cm², matrix size = 256 × 256, providing an effective in-plane resolution of 0.078 × 0.078 mm² and a voxel resolution of 10 × 10 × 3 mm³. The total acquisition time for the experiment was 2 h and 16 minutes.
Diffusion measurements were performed using a spin-echo pulse sequence with a pair of mono-polar diffusion-sensitizing gradients [67]. Gradient orientations were evenly distributed in one direction. Effective B-values range: 100, 500, 1000, 1500, 2000, 2200 s/mm2. A diffusion gradient duration (δ) of 2 ms was utilized in conjunction with an 8 ms diffusion gradient separation (Δ), resulting in a total TR and TE of 2000 and 14 ms, respectively. In order to achieve sufficient signal-to-noise ratio, 12 averages were acquired, resulting in an overall acquisition time of 12 h. The FOV was 8 × 8 mm2, matrix size 256 × 256 and the slice thickness was set to 0.3 mm, resulting in a spatial resolution of 0.031 x 0.031 mm2.
Multivoxel spectroscopic information was acquired using CSI in the spin-echo slab-selective mode. CSI employs two orthogonal phase encoding steps with pulsed gradients, capturing a pure spectroscopic echo during acquisition, distinct from the conventional readout gradient used in imaging. The simultaneous achievement of spatial and spectral resolution allows for the generation of multi-voxel spectra, resulting in spatial distribution maps of individual metabolites [68,69]. To improve the spatial response function, a Hanning function-weighted k-space acquisition scheme was employed, following the Bruker ’weighted’ measurement method. The fundamental measurement parameters included TE = 15 ms, TR = 2000 ms, matrix size = 16 × 16, FOV = 10 × 10 mm², slice thickness = 3 mm, resolution = 0.625 × 0.625 mm2, and the number of scans = 4096. Data underwent reconstruction into a 32 × 32 matrix size with linear smoothing for display purposes. Excitation and refocusing utilized Sinc3 pulses with a bandwidth of 8012 Hz. Echoes were acquired in 2048 points over 204.80 ms, yielding a spectral resolution of 2.4 Hz per point, and a spectral width of 10 kHz (13.3 ppm). Magnetic field homogeneity in the selected volume was optimized through water resonance shimming. VAPOR suppression, with a duration of 625 ms, efficiently saturated the water signal. Interpulse RF delays between seven hermite shaped CSI modules were set at 150, 80, 160, 80, 100, 37.2 and 15 ms. The RF bandwidth was 900 Hz, and the excitation offset was -75 Hz (-0.1 ppm).
3.10. Data Processing
To calculate T2, ROIs were drawn on images using an image sequence analysis package (Paravision 2.1, Bruker) using a fit function [y = A + C × exp (-t/ T2)], where A=absolute bias, C = signal intensity, and T2 = transverse relaxation time. ROIs were defined manually for germ (e.g., the shoot apical meristem, the radicle, the coleoptile, the plumule, and the scutellum). For all samples, T2 was calculated from ROIs that were drawn on the sagittal sides of the wheat. For the phantoms, a ROI in the form of a cylinder was drawn on the axial images. The transverse relaxation rate (R2) was obtained from the equation: R2 = 1/T2 (s-1). Relaxivity (r2) was calculated as the slope of the linear regression line of a plot of R2 versus MRI contrast agent concentration [70].
Diffusion data was processed with a Paravision 360 v2.1. The results were consequently analysed using the Bruker image sequence analysis tool. Signal intensity and standard deviation were derived from the internal fitting function ‘dtraceb’. Mono-exponential signal attenuation: I = A + C × exp (-b × ADC) where A in this equation represents absolute bias, C is the contrast term reflecting diffusion-related signal attenuation, b is the diffusion weighting factor, and ADC is apparent diffusion coefficient. Five ROIs were selected from embryo region for further calculations.
For maping of lipids in CSI images, the image slices were exported and analyzed in Bruker CSI Visualization Tool. To reconstruct the CSI images, a plug-in was used to select the area corresponding to lipid signal. The hyperintense signal of the ROI was then calculated. The data were then exported to Origin Pro v. 8 software for further analysis.
The statistical analysis was carried out using one-way analysis of variance (ANOVA) with OriginPro v.8 (OriginLab, Northampton, MA, USA). This analysis aimed to compare relaxation times across five different ROIs (including the shoot apical meristem, the radicle, the coleoptile, the plumule, and the scutellum) in the embryo region along with their corresponding relaxation times. For statistically significant results obtained from the one-way ANOVA, a Tukey post hoc analysis was performed as a multiple comparison of means test to evaluate the differences between individual groups. A significance level of p < 0.05 was used to determine statistical significance. To ensure the reliability of the results, the homogeneity of variance was evaluated using the Levene test. Groups with a p-value greater than 0.05 were considered to exhibit equal variance between them.
4. Conclusions
In this study, we introduced a biocompatible and non-invasive method for tracking the PET NPs in wheat seeds using fSPION as MRI contrast agents. The PET-fSPION was effectively visualised within wheat seeds, particularly in key developmental regions such as the shoot apical meristem, radicle, coleoptile, plumule, and scutellum. The successful attachment of PET NPs to fSPION was confirmed by comprehensive characterisation, including TEM, DLS, and FTIR. The relaxivity measurements demonstrated that PET-fSPION are effective as MRI contrast agents, indicating their capacity to enhance image contrast in wheat seeds. Furthermore, SEM confirmed the presence and specific localization of PET NPs within the wheat embryo, thereby providing additional validation of the µMRI findings. DW-MRI and CSI revealed significant microstructural and biochemical alterations in the seeds treated with PET. The presence of PET-fSPION resulted in restricted diffusion and an increased lipid content due to exposure to PET NPs.
In conclusion, this study provides a valuable tool for future research into the environmental impact of plastic pollution and offers new perspectives on the interactions between nanoplastics and biological organisms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
N.B, AA, J.M conception & design of the work; N.B., D.D., H.G., M.H., E.B., L.A., D.P. conducted experiments; N.B., D.P., E.B., L.A. AA analyzed & interpret the data, N.B., A.A wrote the manuscript; J.M., A.A. and CT supervised the work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the SFB TRR-386 HYP*MOL grant.
Data Availability Statement
All relevant data are within the paper and its Supporting Information
files.
Acknowledgments
The authors thank to Rico Singer for help in diffusion MRI measurements. We also thank Ronny Frank for useful discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- (PEMRG), P., Annual production of plastics worldwide from 1950 to 2021 (in million metric tons). In Statista, December 2, 2022.
- Geyer, R., J.R. Jambeck, and K.L. Law, Production, use, and fate of all plastics ever made. Sci Adv, 2017. 3(7): p. e1700782.
- Gall, S.C. and R.C. Thompson, The impact of debris on marine life. Mar Pollut Bull, 2015. 92(1-2): p. 170-179. [CrossRef]
- Mason, S.A., V.G. Welch, and J. Neratko, Synthetic Polymer Contamination in Bottled Water. Front Chem, 2018. 6: p. 407. [CrossRef]
- Zhang, H., et al., Pulmonary toxicology assessment of polyethylene terephthalate nanoplastic particles in vitro. Environ Int, 2022. 162: p. 107177. [CrossRef]
- Lin, S., et al., Metabolomics Reveal Nanoplastic-Induced Mitochondrial Damage in Human Liver and Lung Cells. Environ Sci Technol, 2022. 56(17): p. 12483-12493. [CrossRef]
- Jiang, Q., et al., Effects of Acute Exposure to Polystyrene Nanoplastics on the Channel Catfish Larvae: Insights From Energy Metabolism and Transcriptomic Analysis. Front Physiol, 2022. 13: p. 923278. [CrossRef]
- Dhaka, V., et al., Occurrence, toxicity and remediation of polyethylene terephthalate plastics. A review. Environ Chem Lett, 2022. 20(3): p. 1777-1800. [CrossRef]
- Heinder, F.M., et al., Toxic effects of polyethylene terephthalate microparticles and Di(2-ethylhexyl)phthalate on the calanoid copepod,. Ecotoxicology and Environmental Safety, 2017. 141: p. 298-305.
- Magrì, D., et al., Laser Ablation as a Versatile Tool To Mimic Polyethylene Terephthalate Nanoplastic Pollutants: Characterization and Toxicology Assessment. ACS Nano, 2018. 12(8): p. 7690-7700. [CrossRef]
- Li, L., et al., Superparamagnetic Iron Oxide Nanoparticles as MRI contrast agents for Non-invasive Stem Cell Labeling and Tracking. Theranostics, 2013. 3: p. 595-615. [CrossRef]
- Pitt, J.A., et al., Uptake, tissue distribution, and toxicity of polystyrene nanoparticles in developing zebrafish (Danio rerio). Aquatic Toxicology, 2018. 194: p. 185-194. [CrossRef]
- van Pomeren, M., et al., Exploring uptake and biodistribution of polystyrene (nano)particles in zebrafish embryos at different developmental stages. Aquatic Toxicology, 2017. 190: p. 40-45. [CrossRef]
- Catarino, A.I., A. Frutos, and T.B. Henry, Use of fluorescent-labelled nanoplastics (NPs) to demonstrate NP absorption is inconclusive without adequate controls. Science of The Total Environment, 2019. 670: p. 915-920. [CrossRef]
- Varani, M., et al., Methods for Radiolabelling Nanoparticles: SPECT Use (Part 1). Biomolecules, 2022. 12(10): p. 1522. [CrossRef]
- Stricker, A., et al., Radiolabeling of Micro-/Nanoplastics via In-Diffusion. Nanomaterials, 2023. 13(19): p. 2687.
- Munir, M., et al., Iodine-131 radiolabeled polyvinylchloride: A potential radiotracer for micro and nanoplastics bioaccumulation and biodistribution study in organisms. Marine Pollution Bulletin, 2023. 188: p. 114627. [CrossRef]
- Keinänen, O.; et al. Harnessing PET to track micro- and nanoplastics in vivo. Scientific Reports, 2021. 11(1): p. 11463. [CrossRef]
- Fan, Y.; et al. Radiolabelling and in vivo radionuclide imaging tracking of emerging pollutants in environmental toxicology: A review. Science of The Total Environment, 2023. 866: p. 161412. [CrossRef]
- Feiner, I.V.J.; et al. The Race for Hydroxamate-Based Zirconium-89 Chelators. Cancers (Basel), 2021. 13(17).
- Gao, Q.; et al. Tracking of realistic nanoplastics in complicated matrices by iridium element labeling and inductively coupled plasma mass spectroscopy. Journal of Hazardous Materials, 2022. 424: p. 127628. [CrossRef]
- Hwang, J.H. and C.S. Choi, Use of in vivo magnetic resonance spectroscopy for studying metabolic diseases. Exp Mol Med, 2015. 47(2): p. e139. [CrossRef]
- Mansfield, P. and P.K. Grannell, Nmr Diffraction in Solids. Journal of Physics C-Solid State Physics, 1973. 6(22): p. L422-L426.
- Kockenberger, W. Functional imaging of plants by magnetic resonance experiments. Trends Plant Sci, 2001. 6(7): p. 286-92. [CrossRef]
- Lux, J. and A.D. Sherry, Advances in gadolinium-based MRI contrast agent designs for monitoring biological processes in vivo. Curr Opin Chem Biol, 2018. 45: p. 121-130. [CrossRef]
- Liu, Z.; et al. Effects of the magnetic resonance imaging contrast agent Gd-DTPA on plant growth and root imaging in rice. PLoS One, 2014. 9(6): p. e100246. [CrossRef]
- Lauterbur, P.C. Image-Formation by Induced Local Interactions - Examples Employing Nuclear Magnetic-Resonance. Clinical Orthopaedics and Related Research, 1989(244): p. 3-6.
- Wahajuddin and, S. Arora, Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine, 2012. 7: p. 3445-71. [CrossRef]
- Frey, N.A.; et al. Magnetic nanoparticles: synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem Soc Rev, 2009. 38(9): p. 2532-42. [CrossRef]
- Xiao, Y. and J. Du, Superparamagnetic nanoparticles for biomedical applications. J Mater CChem B, 2020. 8(3): p. 354-367. [CrossRef]
- Vangijzegem, T.; et al. Superparamagnetic Iron Oxide Nanoparticles (SPION): From Fundamentals to State-of-the-Art Innovative Applications for Cancer Therapy. Pharmaceutics, 2023. 15(1). [CrossRef]
- Wu, X.; et al. SPIONs Magnetophoresis and Separation via Permanent Magnets: Biomedical and Environmental Applications. Processes, 2023. 11(12): p. 3316. [CrossRef]
- Sarcletti, M.; et al. The remediation of nano-/microplastics from water. Materials Today, 2021. 48: p. 38-46.
- Wang, X.; et al. Nanoparticles in Plants: Uptake, Transport and Physiological Activity in Leaf and Root. Materials (Basel), 2023. 16(8). [CrossRef]
- Pereira, A.P.D.; et al. Processing and Characterization of PET Composites Reinforced With Geopolymer Concrete Waste. Materials Research-Ibero-American Journal of Materials, 2017. 20: p. 411-420. [CrossRef]
- Vidal-Vidal, J., J. Rivas, and M.A. López-Quintela, Synthesis of monodisperse maghemite nanoparticles by the microemulsion method. Colloids and Surfaces a-Physicochemical and Engineering Aspects, 2006. 288(1-3): p. 44-51. [CrossRef]
- Hobson, N.J.; et al. Facile aqueous, room temperature preparation of high transverse relaxivity clustered iron oxide nanoparticles. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2019. 570: p. 165-171. [CrossRef]
- Ragheb, R.R.T.; et al. Induced Clustered Nanoconfinement of Superparamagnetic Iron Oxide in Biodegradable Nanoparticles Enhances Transverse Relaxivity for Targeted Theranostics. Magnetic Resonance in Medicine, 2013. 70(6): p. 1748-1760. [CrossRef]
- Wang, D.; et al. Control of the interparticle spacing in superparamagnetic iron oxide nanoparticle clusters by surface ligand engineering. Chinese Physics B, 2016. 25(7). [CrossRef]
- Vo, T. and G. Pollack, Surprising attraction of non-magnetic materials to magnets. JOURNAL OF ADVANCES IN PHYSICS, 2018. 14: p. 5520-5525. [CrossRef]
- Gaeta, M.; et al. Magnetism of materials: theory and practice in magnetic resonance imaging. Insights Imaging, 2021. 12(1): p. 179. [CrossRef]
- Tripathi, D.K.; et al. Silicon nanoparticles more effectively alleviated UV-B stress than silicon in wheat (Triticum aestivum) seedlings. Plant Physiology and Biochemistry, 2017. 110: p. 70-81. [CrossRef]
- Zhou, Z.; et al. Artificial local magnetic field inhomogeneity enhances T2 relaxivity. Nature Communications, 2017. 8(1): p. 15468. [CrossRef]
- Pérez-de-Luque, A. Interaction of Nanomaterials with Plants: What Do We Need for Real Applications in Agriculture? Frontiers in Environmental Science, 2017. 5.
- Munir, N.; et al. Plant–Nanoparticle Interactions: Transcriptomic and Proteomic Insights. Agronomy, 2023. 13(8): p. 2112. [CrossRef]
- Gruwel, M.L.H.; et al. On the Diffusion Constant of Water in Wheat. Journal of Agricultural and Food Chemistry, 2008. 56(1): p. 59-62.
- Quettier, A.L. and P.J. Eastmond, Storage oil hydrolysis during early seedling growth. Plant Physiol Biochem, 2009. 47(6): p. 485-90. [CrossRef]
- Theodoulou, F.L. and P.J. Eastmond, Seed storage oil catabolism: a story of give and take. Current Opinion in Plant Biology, 2012. 15(3): p. 322-328. [CrossRef]
- Park, S.H. and N. Morita, Changes of Bound Lipids and Composition of Fatty Acids in Germination of Quinoa Seeds. Food Science and Technology Research, 2004. 10(3): p. 303-306. [CrossRef]
- Xu, K.; et al. Lipid Droplets from Plants and Microalgae: Characteristics, Extractions, and Applications. Biology, 2023. 12(4): p. 594. [CrossRef]
- Cai Feng, C.F.; et al. Lipid peroxidation and antioxidant responses during seed germination of Jatropha curcas. International Journal of Agriculture and Biology. 13(1): p. 25–30.
- Farooq, M.A.; et al. Roles of Reactive Oxygen Species and Mitochondria in Seed Germination. Front Plant Sci, 2021. 12: p. 781734. [CrossRef]
- Ekner-Grzyb, A.; et al. Plants oxidative response to nanoplastic. Front Plant Sci, 2022. 13: p. 1027608. [CrossRef]
- Aoki, N.; et al. Pathway of sugar transport in germinating wheat seeds. Plant Physiol, 2006. 141(4): p. 1255-63. [CrossRef]
- Jackowiak, H.; et al. Scanning electron microscopy of mature wheat kernels infected with Fusarium culmorum. Journal of applied genetics, 2002. 43A: p. 167-176.
- Hung, P.V., D. W. Hatcher, and W. Barker, Phenolic acid composition of sprouted wheats by ultra-performance liquid chromatography (UPLC) and their antioxidant activities. Food Chem, 2011. 126(4): p. 1896-901. [CrossRef]
- Lian, J.; et al. Impact of polystyrene nanoplastics (PSNPs) on seed germination and seedling growth of wheat (Triticum aestivum L.). Journal of Hazardous Materials, 2020. 385: p. 121620. [CrossRef]
- Bashirova, N.; et al. A mechanistic understanding of the effects of polyethylene terephthalate nanoplastics in the zebrafish (Danio rerio) embryo. Sci Rep, 2023. 13(1): p. 1891. [CrossRef]
- Welzel, K., R. -J. Müller, and W.-D. Deckwer, Enzymatischer Abbau von Polyester-Nanopartikeln. Chemie Ingenieur Technik, 2002. 74(10): p. 1496-1500. [CrossRef]
- Sarcletti, M.; et al. Superoleophilic Magnetic Iron Oxide Nanoparticles for Effective Hydrocarbon Removal from Water. Advanced Functional Materials, 2019. 29(15): p. 1805742.
- Portilla, L. and M. Halik, Smoothly Tunable Surface Properties of Aluminum Oxide Core–Shell Nanoparticles By A Mixed-Ligand Approach. ACS Applied Materials & Interfaces, 2014. 6(8): p. 5977-5982. [CrossRef]
- Smoluchowski, M.V. Drei Vortrage uber Diffusion, Brownsche Bewegung und Koagulation von Kolloidteilchen. Zeitschrift fur Physik, 1916. 17: p. 557-585.
- Nisah, K.; et al. Study of linearity and stability of Pb(II)-1,10-phenanthroline complex with the presence of Fe (II) dan Mg (II) matrix ions using UV-Vis spectrophotometry. IOP Conference Series: Materials Science and Engineering, 2021. 1087(1): p. 012052. [CrossRef]
- Hennig, J., A. Nauerth, and H. Friedburg, Rare Imaging - a Fast Imaging Method for Clinical Mr. Magnetic Resonance in Medicine, 1986. 3(6): p. 823-833. [CrossRef]
- Carr, H.Y. and E.M. Purcell, Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Physical Review, 1954. 94(3): p. 630-638. [CrossRef]
- Meiboom, S. and D. Gill, Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Review of Scientific Instruments, 1958. 29(8): p. 688-691. [CrossRef]
- Schadewijk, R.V.; et al. Non-invasive magnetic resonance imaging of oils in Botryococcus braunii green algae: Chemical shift selective and diffusion-weighted imaging. PLoS One, 2018. 13(8): p. e0203217.
- Luyten, P.R.; et al. Metabolic imaging of patients with intracranial tumors: H-1 MR spectroscopic imaging and PET. Radiology, 1990. 176(3): p. 791-9. [CrossRef]
- Keevil, S.F. Spatial localization in nuclear magnetic resonance spectroscopy. Phys Med Biol, 2006. 51(16): p. R579-636. [CrossRef]
- Rohrer, M.; et al. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Investigative Radiology, 2005. 40(11): p. 715-724. [CrossRef]
Figure 1.
TEM, STEM and EDX characterization of PET-fSPION. (A) TEM images of PET-fSPION. (B) HAADF STEM image of PET-fSPION showing that fSPION are well dispersed around PET NPs spheres. (C&D) STEM-EDX map of PET-fSPION where net intensity EDX signals of iron in yellow (C) and of carbon in purple (D) is shown. Scale bars: 90 nm (in A) and 200 nm (in B-D).
Figure 1.
TEM, STEM and EDX characterization of PET-fSPION. (A) TEM images of PET-fSPION. (B) HAADF STEM image of PET-fSPION showing that fSPION are well dispersed around PET NPs spheres. (C&D) STEM-EDX map of PET-fSPION where net intensity EDX signals of iron in yellow (C) and of carbon in purple (D) is shown. Scale bars: 90 nm (in A) and 200 nm (in B-D).
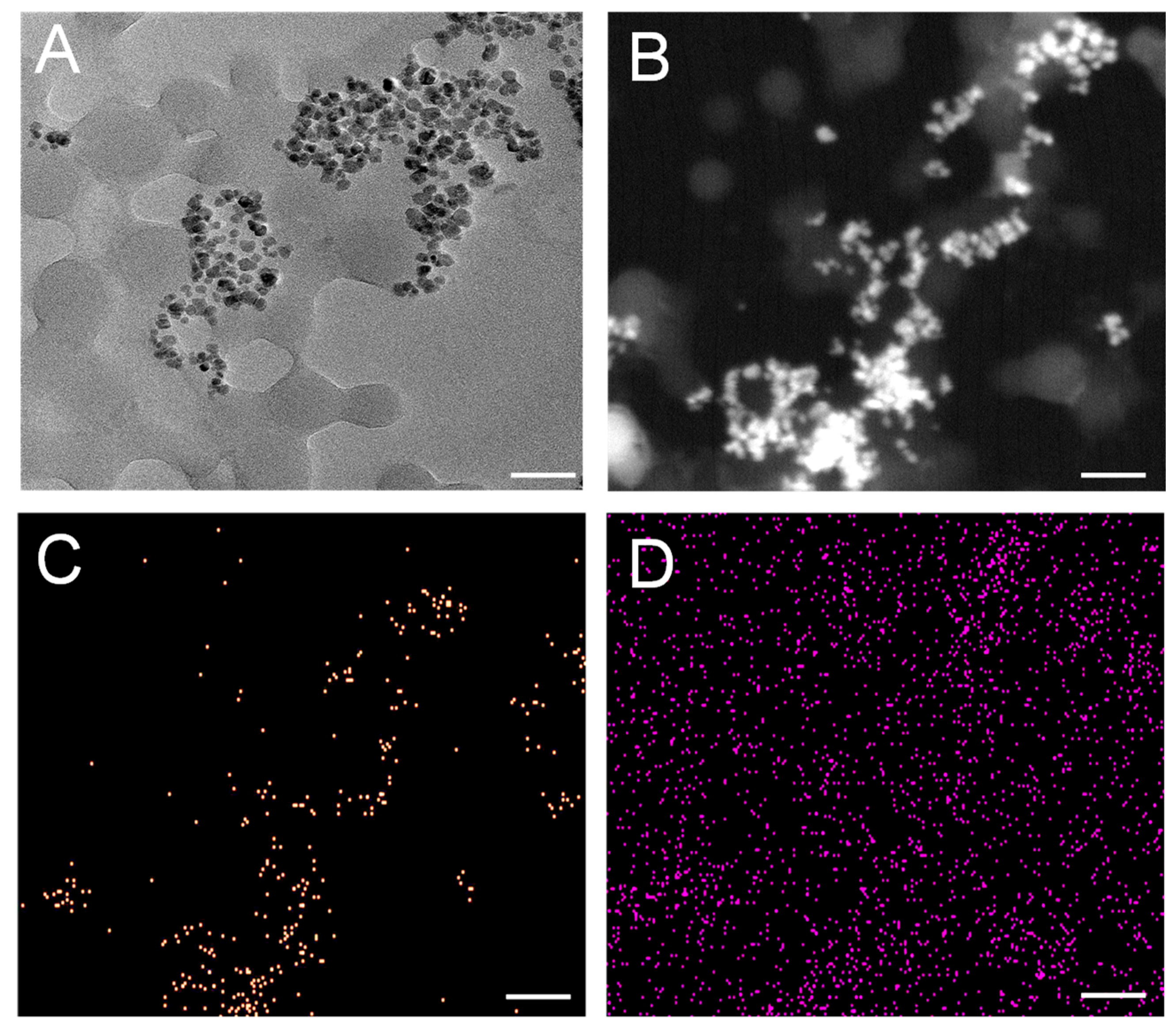
Figure 2.
Characterization of PET NPs with and without functionalized fSPION. (A) Hydrody-namic diameter distribution of PET-fSPION. (B) Particle size, PDI and zeta potential of PET, fSPION and PET-fSPION determined by DLS. (C) ATR-FTIR spectra of PET film, PET-fSPION, fSPION and SPION.
Figure 2.
Characterization of PET NPs with and without functionalized fSPION. (A) Hydrody-namic diameter distribution of PET-fSPION. (B) Particle size, PDI and zeta potential of PET, fSPION and PET-fSPION determined by DLS. (C) ATR-FTIR spectra of PET film, PET-fSPION, fSPION and SPION.
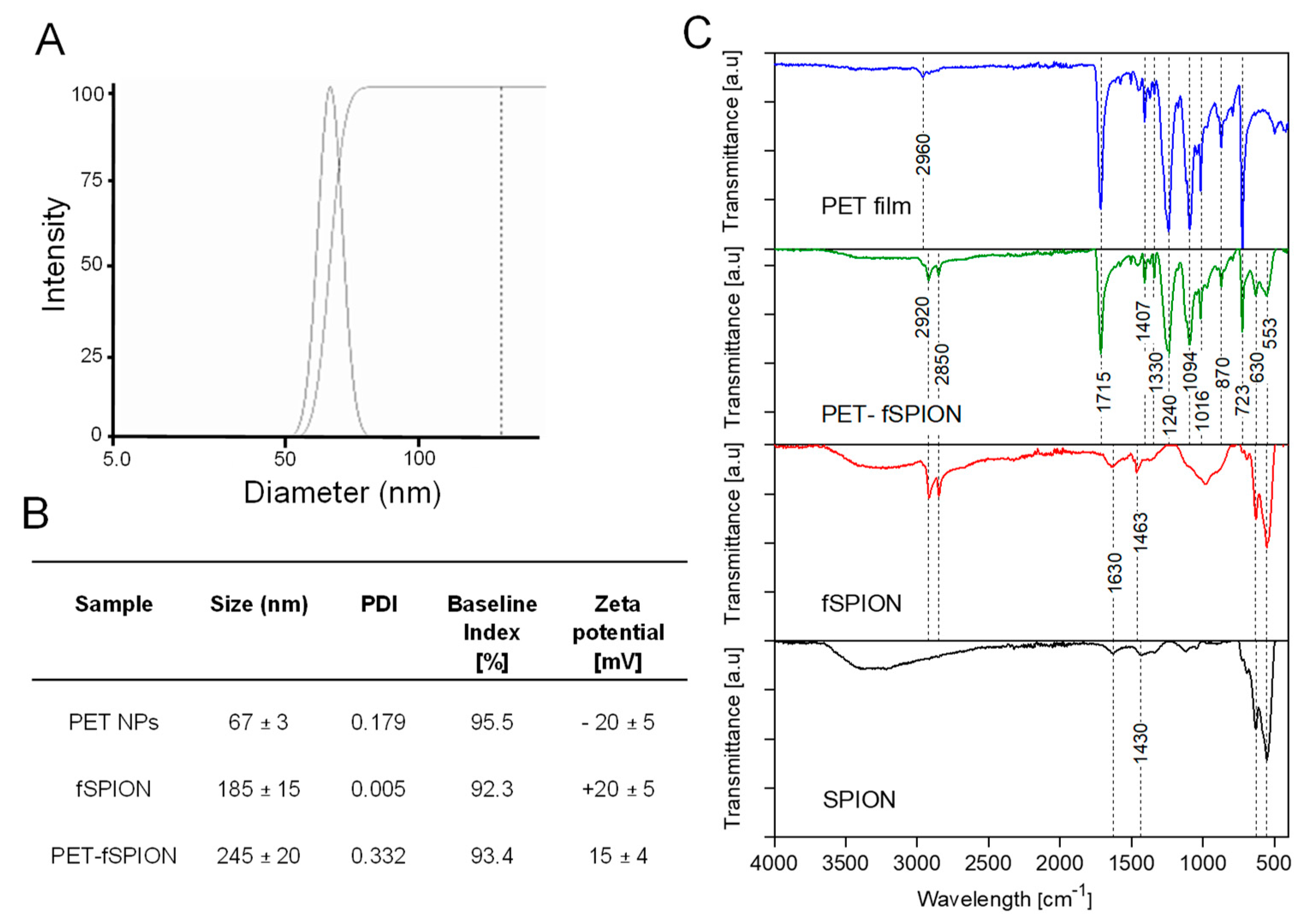
Figure 3.
The relaxivity of fSPION and PET-fSPION was determined at different concentrations using µMRI. The relaxation rates 1/T2 were plotted against the Fe concentration. The relaxivities (r2) were calculated from the linear regression slopes. The r2 for fSPION was 289 mM-1s-1 with an R² of 0.9933, while the r2 for PET-fSPION was 254 mM-1s-1 with an R² of 0.9970.
Figure 3.
The relaxivity of fSPION and PET-fSPION was determined at different concentrations using µMRI. The relaxation rates 1/T2 were plotted against the Fe concentration. The relaxivities (r2) were calculated from the linear regression slopes. The r2 for fSPION was 289 mM-1s-1 with an R² of 0.9933, while the r2 for PET-fSPION was 254 mM-1s-1 with an R² of 0.9970.
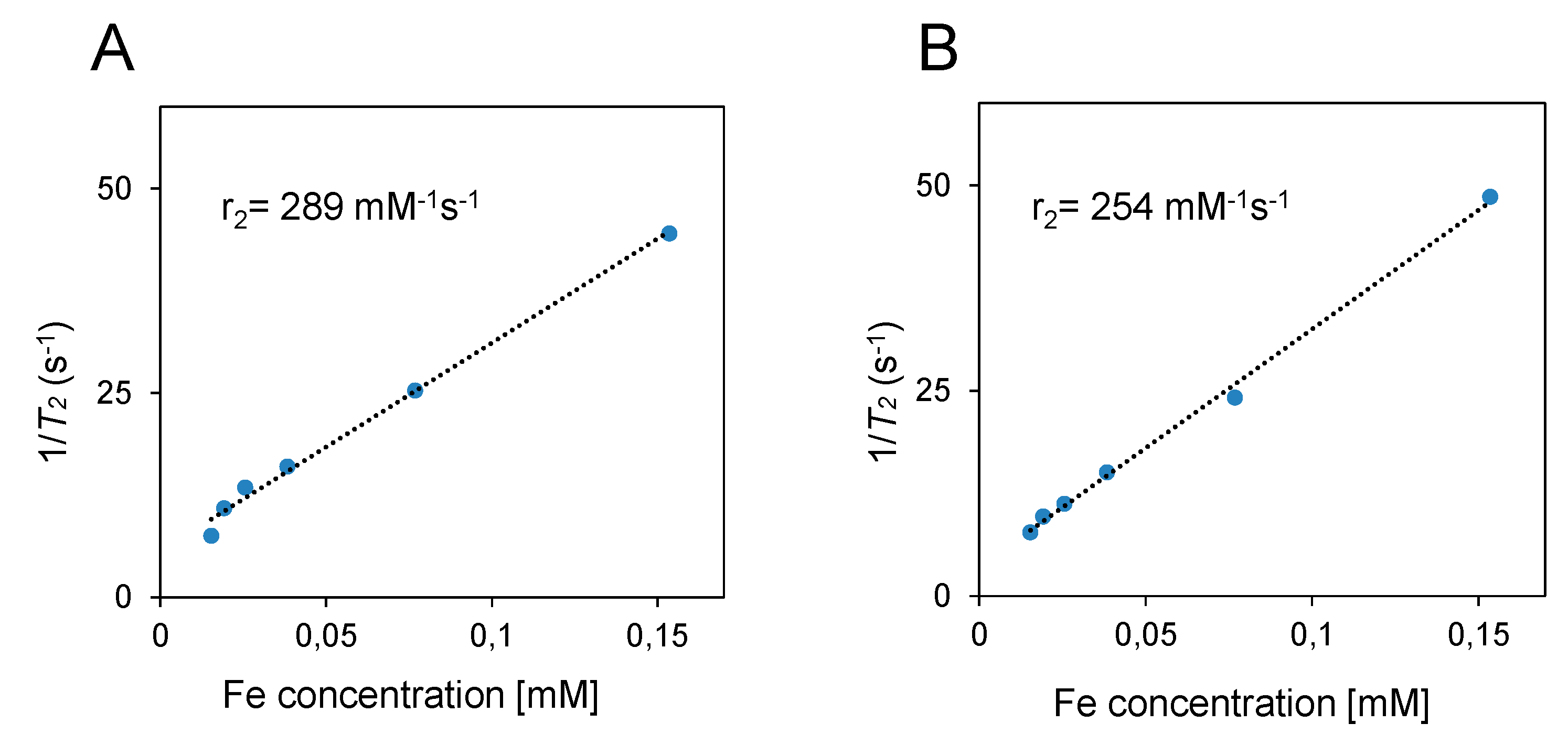
Figure 4.
SEM images showing the localization of PET-fSPION in wheat seeds. Seeds were treated with a PET-fSPION. Aggregations of PET-fSPION NPs are visible. The area in the white squares is magnified to the right, highlighting the detailed interaction between the nanoparticles and embryo surface.
Figure 4.
SEM images showing the localization of PET-fSPION in wheat seeds. Seeds were treated with a PET-fSPION. Aggregations of PET-fSPION NPs are visible. The area in the white squares is magnified to the right, highlighting the detailed interaction between the nanoparticles and embryo surface.

Figure 5.
Representative MRI images of wheat seeds: (A) shows the control group with no treatment (e.g., DI water), while (B) shows seeds treated with PET-fSPION. Arrows indicate the presence of PET-fSPION. Insets below each image provide magnified views of the regions highlighted by the dashed boxes, illustrating the internal structure of the wheat seeds. Measurements performed using a RARE sequence with the following parameters: TR = 2000 ms, TE = 9.15 ms, slice thickness = 0.3 mm, RARE factor = 4, FOV = 8 × 8 cm, number of scans = 256, in-plane spatial resolution = 0.31 × 0.31 mm², total acquisition time ≈ 9h 6 min. Scale bar: 1 mm.
Figure 5.
Representative MRI images of wheat seeds: (A) shows the control group with no treatment (e.g., DI water), while (B) shows seeds treated with PET-fSPION. Arrows indicate the presence of PET-fSPION. Insets below each image provide magnified views of the regions highlighted by the dashed boxes, illustrating the internal structure of the wheat seeds. Measurements performed using a RARE sequence with the following parameters: TR = 2000 ms, TE = 9.15 ms, slice thickness = 0.3 mm, RARE factor = 4, FOV = 8 × 8 cm, number of scans = 256, in-plane spatial resolution = 0.31 × 0.31 mm², total acquisition time ≈ 9h 6 min. Scale bar: 1 mm.
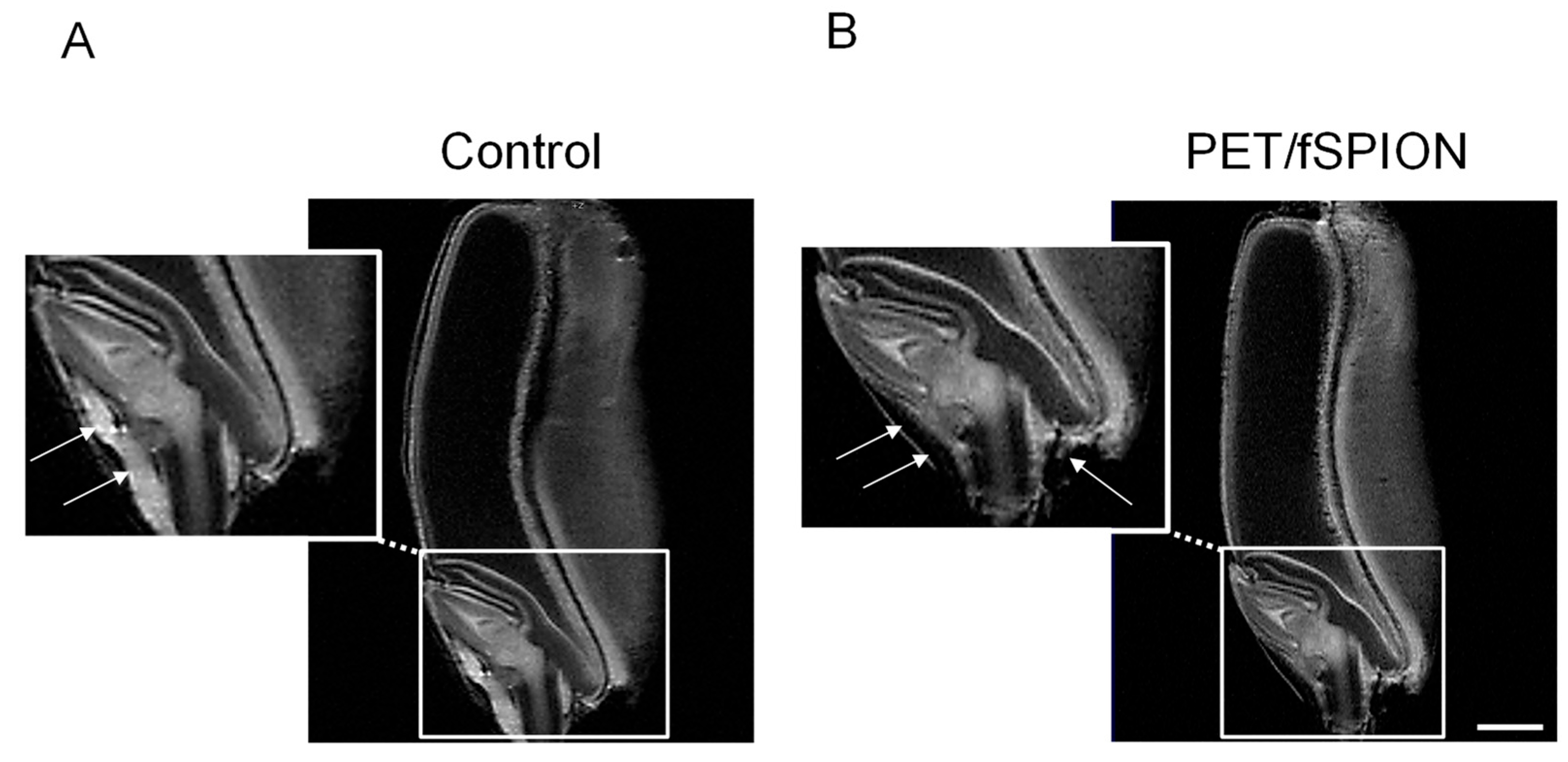
Figure 6.
T2-weighted MRI analysis of control and PET-fSPION treated wheat seeds (A) T2 –weighted MRI image of wheat seed, showing various regions used for T2 measurements. (B) Representative color-coded T2 maps of control and PET-fSPION treated wheat seed. The color scale indicates T2 relaxation times ranging from 0 to 30 ms. Short T2 values are observed in the PET-fSPION treated wheat in comparison to the control. (C) Quantitative T2 relaxation time values in different ROIs. Data are presented as mean ± standard deviation for each ROI. (D) Reliability test of T2 measurements was performed using ANOVA to accomplish pairwise comparisons of the data. At the 0.05 level, the T2 population means of control and PET-fSPION exposed wheat are significantly different (*p<0.05, **p<0.01). Data represents the mean T2 in ms ± SD with error bars. T2 measurements performed using a 2D MSME sequence with the following parameters: TR = 2000 ms; sixteen TEs ranging from 4.3 ms to 68.8 ms; slice thickness = 0.3 mm; FOV = 8 × 8 cm; in-plane spatial resolution = 0.31 × 0.31 mm²; acquisition time = 2 h 16 minutes. Scale bar: 1 mm.
Figure 6.
T2-weighted MRI analysis of control and PET-fSPION treated wheat seeds (A) T2 –weighted MRI image of wheat seed, showing various regions used for T2 measurements. (B) Representative color-coded T2 maps of control and PET-fSPION treated wheat seed. The color scale indicates T2 relaxation times ranging from 0 to 30 ms. Short T2 values are observed in the PET-fSPION treated wheat in comparison to the control. (C) Quantitative T2 relaxation time values in different ROIs. Data are presented as mean ± standard deviation for each ROI. (D) Reliability test of T2 measurements was performed using ANOVA to accomplish pairwise comparisons of the data. At the 0.05 level, the T2 population means of control and PET-fSPION exposed wheat are significantly different (*p<0.05, **p<0.01). Data represents the mean T2 in ms ± SD with error bars. T2 measurements performed using a 2D MSME sequence with the following parameters: TR = 2000 ms; sixteen TEs ranging from 4.3 ms to 68.8 ms; slice thickness = 0.3 mm; FOV = 8 × 8 cm; in-plane spatial resolution = 0.31 × 0.31 mm²; acquisition time = 2 h 16 minutes. Scale bar: 1 mm.
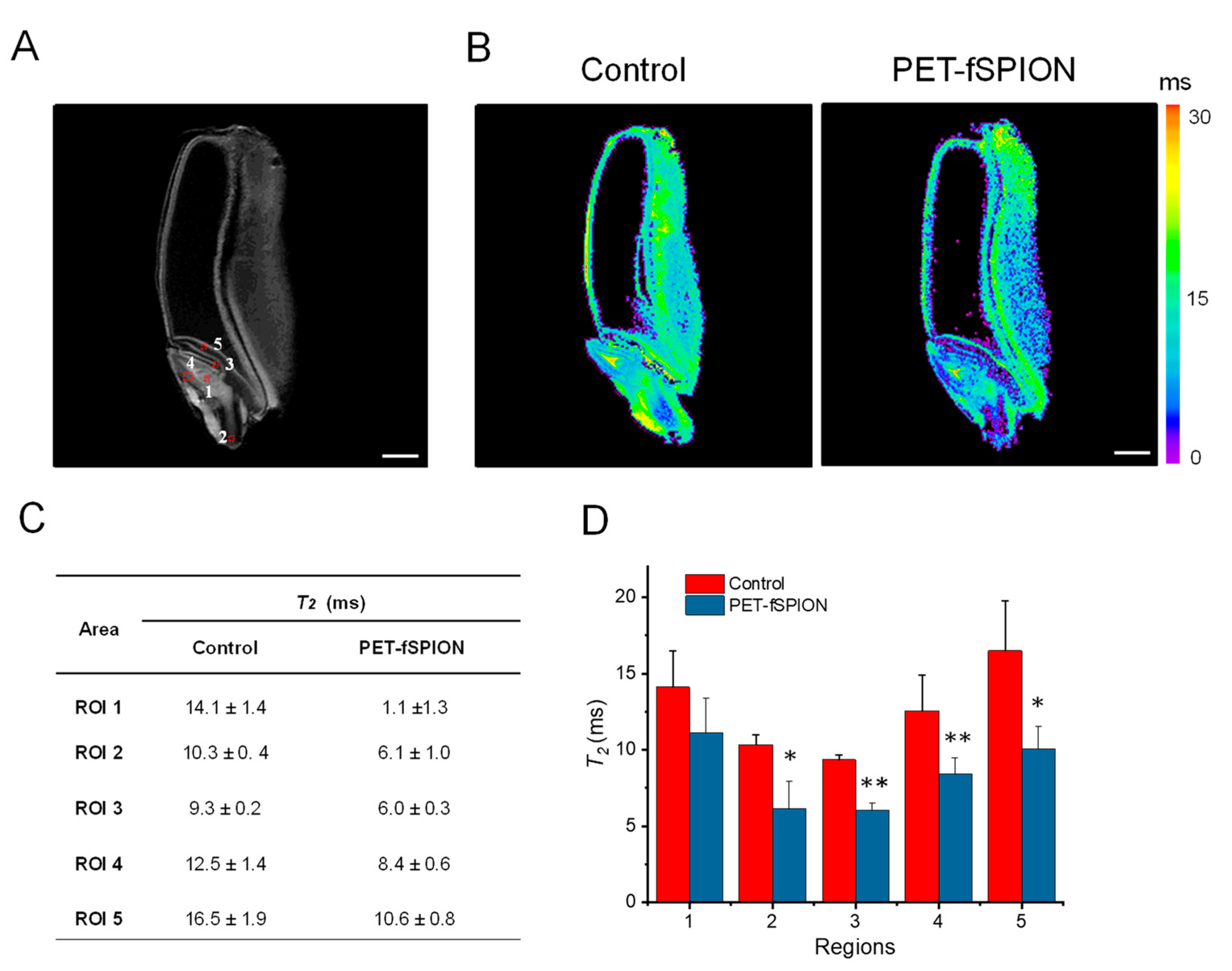
Figure 7.
Diffusion-weighted MRI in embryo regions of control and PET NPs treated wheat seed. Diffusion measurements were performed using a spin-echo pulse sequence containing a pair of mono-polar diffusion-sensitizing gradients (TR = 2000 ms; TE = 14 ms; diffusion gradient duration = 2 ms and gradient separation = 8 s; effective B-values range: 100, 500, 1000, 1500, 2000, 2200 s/mm2). (A) A diffusion image of control wheat showing regions of interest (ROI) located in various embryo regions of wheat. The ROIs are located in the shoot apical meristem (ROI 1), in the radicle of wheat (ROI 2), in coleoptile (ROI 3), in plumule (ROI 4) and in scutellum (ROI 5). (B) Representative ADC map image of control wheat generated through Bruker internal ‘dtraceb’ algorithm. It shows the distribution of ADC values where higher ADC appears bright and lower ADC appears darker. (C) Signal decay curve in respective ROIs as shown in (A). (D) Table of calculated ADC values from reinterest is shown in (A). Scale bar: 1 mm.
Figure 7.
Diffusion-weighted MRI in embryo regions of control and PET NPs treated wheat seed. Diffusion measurements were performed using a spin-echo pulse sequence containing a pair of mono-polar diffusion-sensitizing gradients (TR = 2000 ms; TE = 14 ms; diffusion gradient duration = 2 ms and gradient separation = 8 s; effective B-values range: 100, 500, 1000, 1500, 2000, 2200 s/mm2). (A) A diffusion image of control wheat showing regions of interest (ROI) located in various embryo regions of wheat. The ROIs are located in the shoot apical meristem (ROI 1), in the radicle of wheat (ROI 2), in coleoptile (ROI 3), in plumule (ROI 4) and in scutellum (ROI 5). (B) Representative ADC map image of control wheat generated through Bruker internal ‘dtraceb’ algorithm. It shows the distribution of ADC values where higher ADC appears bright and lower ADC appears darker. (C) Signal decay curve in respective ROIs as shown in (A). (D) Table of calculated ADC values from reinterest is shown in (A). Scale bar: 1 mm.
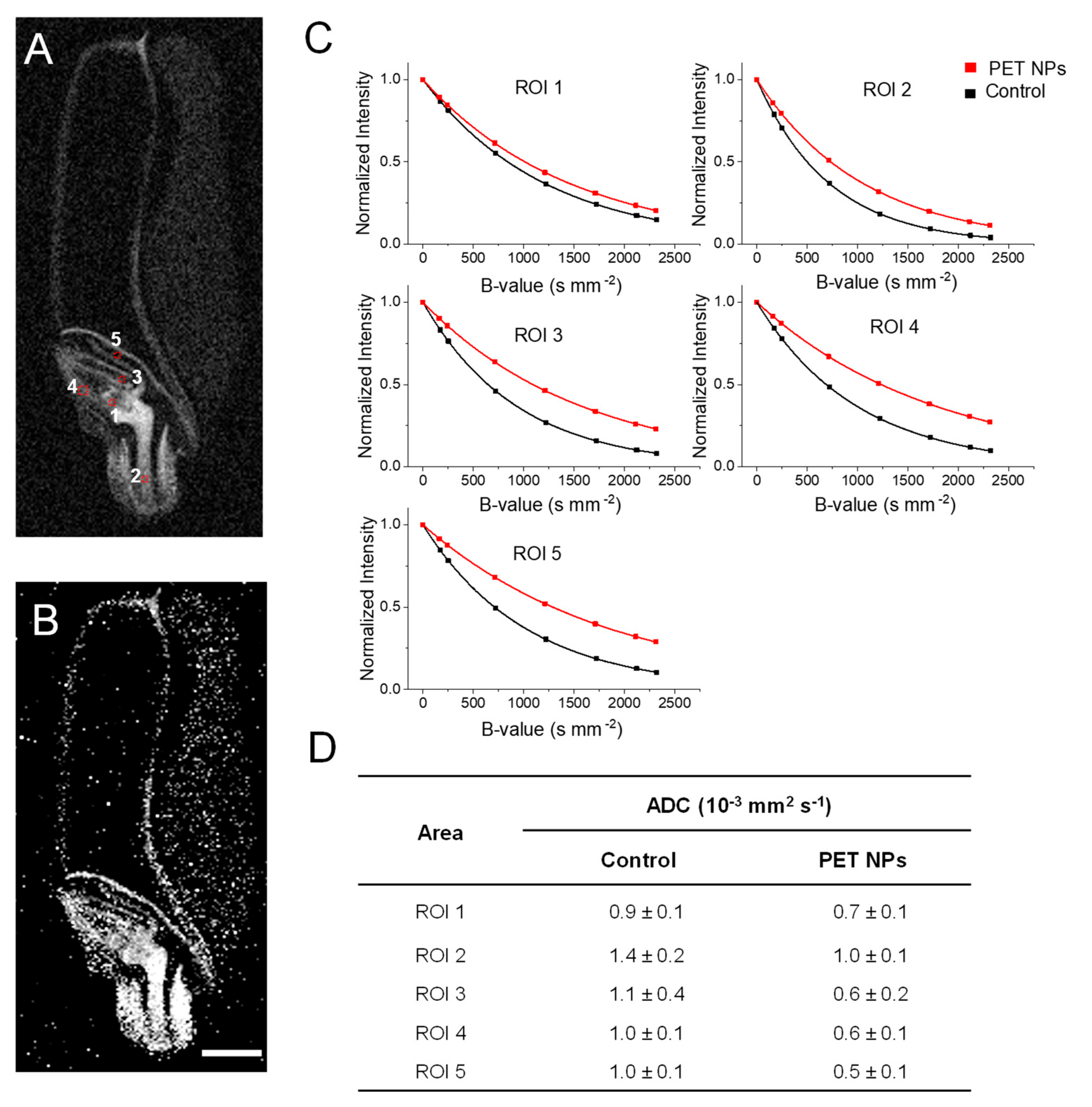
Figure 8.
Chemical shift imaging. (A) Matrix display of chemical shift imaging spectra of control and PET NPs treated wheat seeds. Spectra from radicle (1) and scutellum (2) regions are shown in magnification. Signals between ~0.5 to 3 ppm are from lipids and 4.7 ppm from water. Overlapping signals from carbohydrate (glucose, fructose, or other sugars) between ~3 to 5 ppm are present in radicle region in PET NPs-treated samples. (B) Corresponding CSI voxel intensity thresholding of lipid signals which was reconstruct as color map and overlaid with corresponding T2-weighted RARE images using the Bruker CSI Visualisation Tool. CSI data was recorded with a TR = 2000 ms; TE = 15 ms and slice thick-ness was 3 mm. Total averages were 256. Resolution obtained was 0.625 × 0.625 mm2. Spectral width used was 10 kHz (13.33 ppm) and 16x16 matrix was reconstructed into 32 × 32 voxels. Scale bar: 1 mm.
Figure 8.
Chemical shift imaging. (A) Matrix display of chemical shift imaging spectra of control and PET NPs treated wheat seeds. Spectra from radicle (1) and scutellum (2) regions are shown in magnification. Signals between ~0.5 to 3 ppm are from lipids and 4.7 ppm from water. Overlapping signals from carbohydrate (glucose, fructose, or other sugars) between ~3 to 5 ppm are present in radicle region in PET NPs-treated samples. (B) Corresponding CSI voxel intensity thresholding of lipid signals which was reconstruct as color map and overlaid with corresponding T2-weighted RARE images using the Bruker CSI Visualisation Tool. CSI data was recorded with a TR = 2000 ms; TE = 15 ms and slice thick-ness was 3 mm. Total averages were 256. Resolution obtained was 0.625 × 0.625 mm2. Spectral width used was 10 kHz (13.33 ppm) and 16x16 matrix was reconstructed into 32 × 32 voxels. Scale bar: 1 mm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



