Submitted:
17 July 2024
Posted:
18 July 2024
You are already at the latest version
Abstract
The use of chimeric antigen receptors (CAR-T cells) for the treatment of patients with malignant haematological diseases has become a well-established application, for conditions such as refractory or relapsed B cell acute lymphoblastic leukaemia (B-ALL), B cell lymphomas (BCL), and multiple myeloma (MM). Nearly 35,000 patients have received autologous CAR-T cells for the treatment of these conditions only in the USA. Since their approval by the Food and Drug Administration (FDA) in 2017, over 1200 clinical trials have been initiated globally and there are at least 10 different CAR-T cells with approval by different regulatory agencies around the globe. In the USA the Food and Drug Administration (FDA) has approved 6 commercial CAR-T cells that are widely distributed worldwide. At the time of this writing, several clinical trials have been performed in patients with solid tumours such as glioblastoma, renal and pancreatic cancer, as well as in patients with autoimmune conditions such as systemic lupus erythematosus (SLE), idiopathic inflammatory myositis (IIM), and systemic sclerosis (SS). There are also several studies showing the potential benefit of CAR-T cells for other non-malignant diseases such as asthma and even fungal infections. In this review, without pretending to cover all the current areas of treatments with CAR-T cells, we offer a brief summary of some of the most relevant aspects of the use of CAR-T cells for some of these conditions.
Keywords:
advanced therapy medicinal products (ATMPs)
; chimeric antigen receptors (CAR-T cells)
; B acute lymphoblastic leukaemia (B-ALL)
; food and drug administration (FDA)
; systemic lupus erythematosus (SLE)
; idiopathic inflammatory myositis (IIM)
; systemic sclerosis (SS)
; memorial sloan kettering cancer centre (MSKCC)
; hematopoietic stem cell transplantation [HSCT]
; non-Hodgkin’s cell lymphoma (NHCL)
; multiple myeloma (MM)
; follicular lymphoma
; (FL)
; large B cell-lymphoma (LBCL)
; mantle cell lymphoma (MCL)
; myeloid-derived suppressor cells (MDSCs)
; rheumatoid arthritis (RA)
; immune effector cell-associated neurotoxicity syndrome (ICANS)
; natural killer group member D ligands (NKG2DLs)
; acute myelogenous leukaemia (AML)
Introduction
In recent years, Advanced Therapy Medicinal Products (ATMPs) have revolutionized medicine with the promise of new treatments for previously incurable diseases. Chimeric antigen receptor (CAR) T cell therapies, in particular, have emerged as a highly active area of research and entrepreneurship [1].
By now, something that started very promising in the first decade of this century has become a reality and CAR T cell therapy has been extraordinarily successful for the treatment of patients with haematological malignancies. More than 10,000 patients receive CAR T cells each year and by early 2024, more than 34,000 cases have been treated with these therapies just in the United States. CAR T cells are successful against B acute lymphoblastic leukaemia (B-ALL), non-Hodgkin’s lymphoma (NHCL), multiple myeloma (MM), follicular lymphoma, (FL), large B cell-lymphoma (LBCL), and mantle cell lymphoma (MCL) [8,9,10] and encouraging results in solid tumours and autoimmune disorders have been obtained [2].
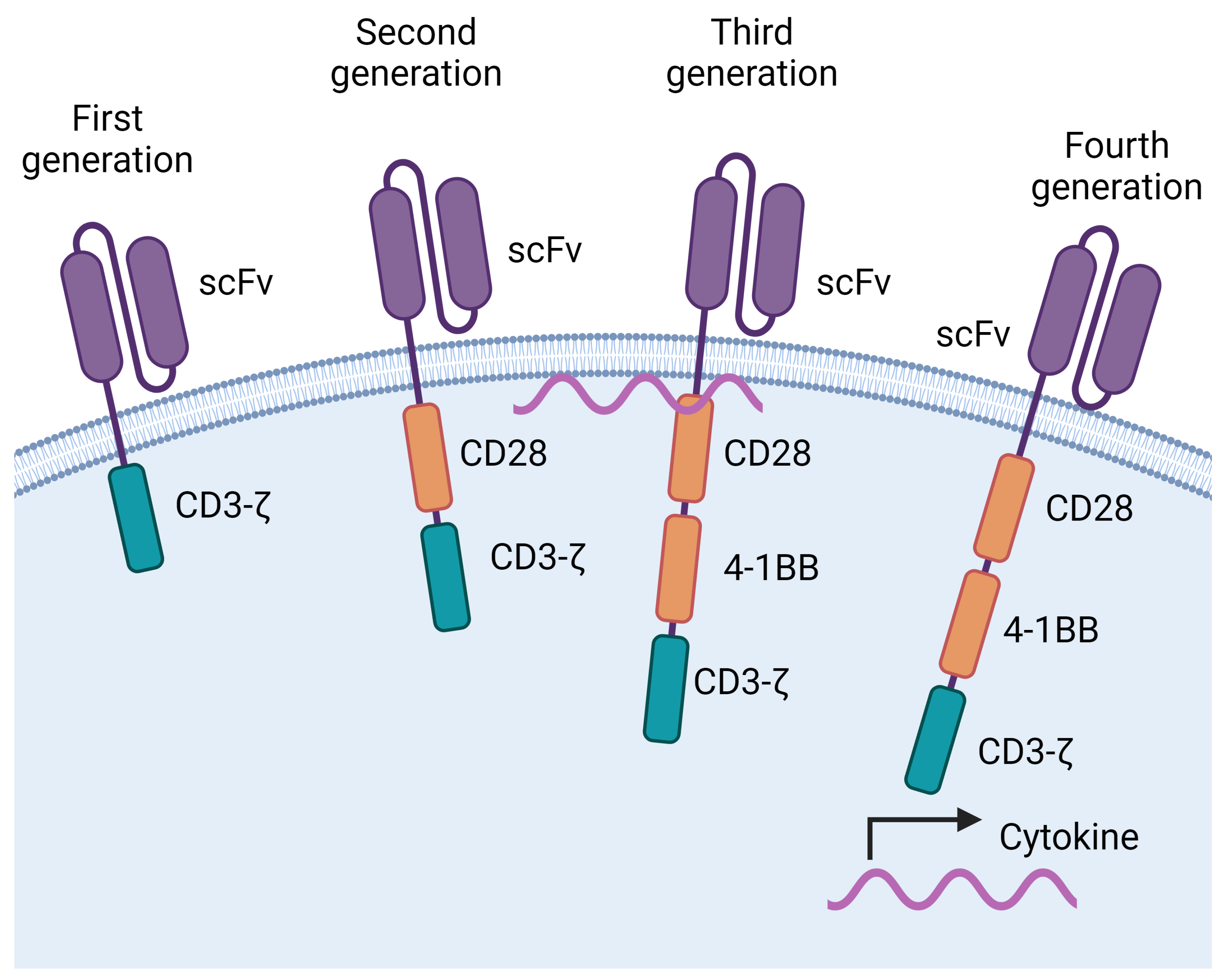
CAR-T cells are generated by introducing into T cells a transgene that encodes a chimeric receptor (CAR). The CAR confers the T cells the capacity to recognise a molecule expressed by the tumour in a non-HLA-dependent manner. This is achieved because the CAR has an antigen binding site similar to an antibody that can bind to molecules in their native conformation. In the CARs that have been approved for clinical use, the antigen binding site is a single chain variable fragment (scFv). The scFv is linked to the cell by a hinge and a transmembrane domain. Additionally, the CAR has intracytoplasmic domains that allow it to deliver an activating signal to the cell. These are usually derived from CD3z and from costimulatory molecules, typically 4-1BB, CD28, or both (Figure 1). Thus, the specificity of the CAR is mediated by the scFv whereas the other elements transduce signals into the T cell that lead to its activation and proliferation [1,2,3,4,5,6].
CAR-T cells that recognise CD19 have had impressive results in patients with B-lymphocyte cancers, particularly B-ALL and some types of lymphomas [6]. Because the tumour cell of B-ALL and B-cell lymphomas expresses CD19, the same CAR can be used to treat both types of cancer [7]. There are four CARs directed against CD19 that have been approved by the USA FDA [8,9,10]. These second generation anti-CD19 CAR-T cells (tisagenlecleucel/tisa-cel {Kymriah}; brexucabtagene autoleucel/brexu-cel {Tecartus}; axicabtagene ciloleucel/axi-cel {Yescartar}; lisocabtagene maraleucel/liso-cel {Breyanzi}) have demonstrated in several clinical trials to induce long term (at least 10 years now) remission in a large number of patients with B-ALL, follicular lymphoma, (FL), large B cell-lymphoma (LBCL), and mantle cell lymphoma (MCL) NHCL [8,9,10]. There is another type of cancer, multiple myeloma, where the tumour cell is a plasma cell. There are two CARs directed against an antigen present on myeloma cells (BCMA) that have been approved by the FDA (Idecabtagene vicleucel {Abecma}, Ciltacabtagene autoleucel {Carvykte}) [10,11]. The six FDA-approved CARs represent hope for a cure for patients with life-threatening diseases, and globally up to 10 distinct CAR-T cell products have received regulatory approval by different countries including China such as Yikaida’s (Axicabtagene ciloleucel) and Relma-cel’s (Relmacabtagene) approval from the NMPA in China, Actaly-cel’s (Actalycabtagene autoleucel) in India, Fucaso’s (Equecabtagene autoleucel) approval in China) and Ebvallo’s (tabelecleucel) approval from the European Commission [11,12,13].
The result of multiple trials using these CAR-T cells has revolutionised the treatment of patients with terminal conditions, in particular for patients with relapsed B-ALL where the media overall survival is only between 4 and 9 months and no longer that 8 months in patients treated with Blinatumomab and inotuzumab ozogamicin [13,14,15]. For instance, Park et al reported a long term follow up of 53 patients with BALL treated with CAR-T CD19 cells (19-28ζ) in a phase 1 clinical trial at Memorial Sloan Kettering Cancer Centre (MSKCC). Eighty-three percent of patients reached complete remission [14]. Interestingly, although all 53 patients were heavily pretreated, they found that the disease characteristics did not influence the long-term outcome (68% of them received CAR-T cells as a third or later salvage treatment; 23% had primary refractory disease; 19 of them had previous hematopoietic stem cell transplantation [HSCT]; 25% were treated previously with Blinotumomab). They concluded that their CAR-T cell (19-28ζ) treatment of 53 adult patients with relapsed B-ALL heavy treated, achieved 83% complete remission with an impressive median overall survival of more than 20 months and low incidence of severe cytokine release syndrome (26%) [14].
Figure 1.
Evolution of chimeric antigen receptors (CARs). CARs have an antigen binding domain that recognizes molecules in their native conformation, usually a single chain fragment variable (scFV). This domain is tethered to the membrane through a hinge and a transmembrane domain, that connect to intracellular signalling domains. First generation CARs had a domain derived from the CD3-z chain. A costimulatory domain, derived from CD28, was added to second generation CARs. Third generation CARs have 2 costimulatory domains, usually derived from CD28 and 4-1BB. Fourth generation CARs encode additional genes that are produced by the CAR T cell. For example, genes that encode cytokines. Created with BioRender.com.
Figure 1.
Evolution of chimeric antigen receptors (CARs). CARs have an antigen binding domain that recognizes molecules in their native conformation, usually a single chain fragment variable (scFV). This domain is tethered to the membrane through a hinge and a transmembrane domain, that connect to intracellular signalling domains. First generation CARs had a domain derived from the CD3-z chain. A costimulatory domain, derived from CD28, was added to second generation CARs. Third generation CARs have 2 costimulatory domains, usually derived from CD28 and 4-1BB. Fourth generation CARs encode additional genes that are produced by the CAR T cell. For example, genes that encode cytokines. Created with BioRender.com.
Although CAR T cells have shown promise, accumulating data shows that multiple factors influence their efficacy. For example, the stage of the disease, their persistence in patients after infusion, the conditioning regimes, and the immunosuppressive drugs that the patients receive. Based on these observations, there are now several clinical trials meant to analyse these and other factor in patients that receive CAR T cells for MM, B-cell lymphomas, and B-ALL. Additionally, their use as second line of treatment is being tested with very encouraging results.
The process of CAR T cell generation entails the in vitro activation and expansion of a heterogeneous pool of T cells. Each individual patient may have a different composition of the peripheral T cell pool, including varying proportions of CD4 and CD8 T cells and different amounts of naïve and memory T cells, including cells with suppressive or exhausted phenotypes. Depending on the relative proportion of these T cell subsets, the resulting CAR T cells may have different capacities to expand, persist, and lyse target cells. Several studies have shown that T cells from patients with an increased proportion of naïve cells have a greater potential of expansion and efficacy. Because of this, attempts are being developed to enrich the cells to be transduced with a higher proportion of cells that maintain proliferative capacity. In particular, naïve-like or central memory T cells [16,17].
An important limiting factor in the use of CAR-T cells is their high cost compared with other treatments. An interesting cost evaluation analysis was performed at the Vanderbilt University Medical Center. Schulthess et al. compared the internal cost for the management and treatment of patients with ALL with CAR-T with the total cost of treating them with HSCT. When the cost of the CAR-T product was excluded, CAR-T associated management cost was 61% of the cost of HSCT (US$118.795 vs $303,065). When the cost of the CAR-T product was included ($475,000) the total treatment cost with CAR-T cell was in the order of nearly 5,800,000 dollars. Interestingly in this retrospective study they compared the outcome of treatment with CAR-T versus HSCT as the next-best optimal treatment option. The analysis, performed retrospectively, compared 29 patients treated with HSCT versus 14 treated with CAR-T cells, revealed a high rate of relapse with CAR-T treatment with a 3-year relapse free survival probability of 46% versus 68% with HSCT [18].
One way to improve the outcome of CAR-T cells is arming them with extra pro-inflammatory cytokines such as IL-18. In a recent presentation at ASCO 2024, Carl June described a phase I clinical trial in 20 patients with NHCL that had relapsed or had no longer responded to treatment with commercially available CAR-T cells. The patients received an “armoured” IL-18-producing CAR-T developed at the University of Pennsylvania. Their intention was to test whether the presence of IL-18 could protect the CAR-T and enhance their capacity to destroy the lymphoma cells. In this report they also described the capacity to reduce the manufacturing time for the CAR-T cells from the current time of 9 to 14 days to only three days, which represents a critical factor during treatment of patients with aggressively growing tumour cells [19].
Another critical factor that determines the outcome of CAR-T treatment is their capacity to expand and persist in vivo. In this regard, attempts to modify the existent commercially approved CAR-T cells, that express murine-derived scFv, to CARs with a fully human scFv (hscFv) have been published [20]. Absence of murine-derived sequences could prolong the lifespan of CAR-T cells by diminishing anti-CAR immune responses. Another element that influences the CAR-T lifespan is the quality of the signal that the receptor conveys to the T cell. The choice of costimulatory motifs included in the intracellular part of the receptor can affect the magnitude of the T cell activation and the acquisition of memory-like properties. So-called second-generation CARs contain, in addition to the CD3z motif, a fused CD28 or 4-1BB costimulatory domain. Third-generation CAR-T cells include both 4-1BB and CD28 (CD28-41BB) domains. This is based on the observations that the presence of CD28 domains elicit higher “anti-cancer” activity than 4-1BB domains, whereas the latter confer longer persistence [20,21]. The addition of the intracellular domain of CD27 to the CAR-T construct (4SCAR19) showed long term high efficiency and safety against B-ALL, however, longer and larger clinical trials are needed to demonstrate the superiority of these constructs over other constructs and commercially available CAR-T cells [21].
Accessibility, treatment rate, and reasons for non-treatment is a critical factor and varies among countries. According to a recent report from IQVIA [22], in the USA, the country with more patients treated with CAR-Ts, a large majority of the patients received the treatment in the context of clinical trials, in around 800 clinical trial sites. In contrast, only 198 centres are approved and validated to treat patients with commercially available CAR-T cells. In their report, the average time to start treatment after referral varies between 1.3 to 2.2 months. Disease progression (79%) and patient ineligibility (55%) were the main reasons for refusal of treatment after referral in the nine countries they evaluated. The number of patients with access to CAR-T cell therapy also varies among different countries, in Brazil is as low as 25%, in Italy 70%, Germany 31% and surprisingly in USA only 38%. Even though time for treatment after leukapheresis is expected to be shorter with new technologies that reduce the time to produce CAR-T cells, the production time is still in average 13 to 26 days, and the waiting time for it to be performed around 4-6 months [22].
Durable remission after CAR-T cell treatment has been shown to be the result of several intercorrelated factors, as described by Cappell and Kochenderfer [23]. These include depth of response and malignancy type. Substantial differences between the rate of remission between patients with B cell lymphomas and B-ALL versus MM, that achieve less sustained remissions. Another factor negatively associated sustained remission is tumour burden and distribution (e.g. extramedullary location). On the other hand, use of lymphodepleting chemotherapy and high peak blood CAR-T cell numbers are associated with higher durable responses and higher rates of remission [23]. Other potential mechanisms of tumour resistance are the release of immunosuppressive cytokines by the tumour, the presence of tumour-associated macrophages, the abundance of T-regs or myeloid-derived suppressor cells (MDSCs), or a combination of all of them.
The excellent results obtained with CAR-T cells in haematological malignancies has raised the hope that they can be applied for the treatment of solid tumours, however several challenges have been identified that make their use for many tumours more difficult. For example, target tumour toxicity, tumour target heterogeneity, antigen escape, T cell persistence and efficacy decline, to mention some few [24]. Del Bufalo et al. has reported the use of CAR-T cell targeting the disialoganglioside-GD2 glycolipid, which is highly expressed in multiple tumour times including neuroblastoma. Their CAR-T cells (CART01) were modified to express two costimulatory domains plus an inducible caspase 9 gene, in order to induce the death of the CAR T cells in case of associated toxic effects. In a phase 1-2 clinical trial for patients with refractory or relapsed high-risk neuroblastoma, they showed extremely encouraging results, with a complete response in 33% of the 27 patients that they treated with a 3-year event-free survival in 36% patients [24,25].
Another emerging and highly promising application for the use of CAR-T cells is the area of autoimmune disorders. Studies in animal models and in humans in disorders such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), type 1 diabetes, pemphigus, and multiple sclerosis have yielded encouraging results. In these diseases, the B- and T-cell-mediated pathophysiology leads to autoreactive B-cell clonal activity with autoantibodies targeting self-antigens [27]. In 2021, CAR T cells were used to treat a 20-year-old patient with severe treatment-refractory SLE, and complete clinical remission was obtained [28]. In this initial case, a single infusion of autologous CAR-T cells targeting CD19, led to remarkable improvement and remission of all the clinical associated manifestation of SLE. Proteinuria disappeared, renal function improved, and most auto-antibodies, including anti-double stranded DNA (dsDNA) became negative. Remarkably, there was very little toxicity associated to the use of CAR-Ts and immunosuppressive medication could be discontinued. The patient remains in complete remission after 18 months of the initial treatment [28]. The impressive response of lupus nephritis to CAR T cell treatment has been confirmed in a recent report by Krickau et al., who administered a single infusion of CD19 CAR-T cells into a 15-year-old female with severe rapidly progressive and refractory lupus nephritis. Before treatment she had an estimated glomerular filtration rate of 8 mL/min and blood creatinine concentration of 4.96 mg/dL and required haemodialysis every 2 to 3 days. Three months after infusion of CD19 CAR-T cells, the creatinine concentration was reduced to 1.2 mg/dL, the estimated glomerular filtration rate increased to 43 mL/min, and she was haemodialysis free. In addition, there was a dramatic improvement of all the SLE clinical activity with the resolution of arthritis, and normalisation of C3 and C4 within 6 weeks, with the disappearance of anti-nuclear antibodies, anti-dsDNA, and other autoantibodies. As the authors conclude, this case shows the potential benefit of early use of CAR-T cells in patients with juvenile aggressive SLE [29].
In a larger number of patients with different autoimmune diseases (15 in total), Müller et al., recently reported the outcome of the use of CD19 CAR-T cells. Eight patients had severe SLE, 3 patients idiopathic inflammatory myositis (IIM), and 4 patients systemic sclerosis (SS). All these patients have severe progressive diseases and were resistant to at least two courses of immunosuppressive standard-care treatments. The reported results are dramatic. In all 8 patients with SLE, after only 6 months there was a resolution of all the major disease categories scores and they remain in remission after a long-term follow-up of up to 29 months, including absence of detectable anti-dsDNA antibodies and proteinuria, as well as normalisation of complement factor C3. Similarly, in all 3 patients with IIM and in the 4 patients with SS, there was a remarkable response according to the respective disease scores. In general, there was very little toxicity or associated cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome (ICANS), but 6 patients had to be treated with Tocilizumab and one patient received steroids for the treatment of possible mild immune effector-associated neurotoxicity, and one patient required hospitalisation due to pneumonia, seven weeks after the CAR-T cell infusion. Interestingly, even though all the patients had full B-cell reconstitution for up to 2 years, all of them could successfully stop all the immunosuppressive treatment without relapse or disease worsening or antibody activity. Based on that, the authors suggest that the CAR-T treatment may have induced a reset of the B cell population responsible of all these autoimmune conditions. It will be important to have a long term follow up of these patients to be sure that the treatment with CD19 CAR-T cells can induce long-term remission without any further immunosuppressive treatment [30].
Thus, the use of CD19 CAR-T cell to treat autoimmune disorders is proving to be a robust resource to rescue patients with refractory immune mediated conditions and these and other publications are starting to prove the relevance of their use even in disorders such as severe multidrug-resistant dermatomyositis (anti-synthetase syndrome) [31] and type 1 diabetes [32].
Although out of the scope of this review, it is wort to mention there are several promising animal model studies where the use of CAR-T cells has demonstrated its potential value in conditions such as asthma, aging, fungal infections, etc. For instance, Yang et al showed that targeting the natural killer group member D ligands (NKG2DLs) using an NKG2D-CAR-T can eliminate senescent cells with substantial improvement of aging and age-associated diseases driven by senescence in aged nonhuman primates [33]. Similarly, Jin et al showed that multifunctional CAR T cell targeting IL-15/IL-4/IL-13 simultaneously induce sustained remission of type 2-high asthma in mice [34]. Finally, the group of Löffler [35] has demonstrated that CAR-T cells can be used to treat effectively invasive pulmonary aspergillosis targeting Aspergillus fumigatus in preclinical animal models. All these preclinical studies exemplify the enormous potential of CAR-T cells for the treatment of a broad variety of clinical life-threatening conditions.
Future Challenges for the Use of CAR-T Cells
Although autologous CAR-T cell therapy has proven to be a potential cure for many B-cell neoplasms, for relapsed or refractory MM, and for autoimmune disorders, its high associated costs, as well as laborious-time of production has led to the development of allogenic CAR cells, including NK CAR cells or genetically modified T cells, in order to generate of off-the-shelf cellular products [36] that will allow the targeting of multiple antigens and their wider use for other leukaemias such as acute myelogenous leukaemia (AML) that has proven to be a complex condition to treat with autologous CAR-T cells [37,38].
Another important emerging issue that must be addressed is the potential generation of second cancers associated with the use of CAR- T cells. There have been over 20 reported cases of T-cell cancer, potentially due to the viral vector integration, but other potential explanations may be antigen driven proliferation of T cells, and potential genetic or epigenetic changes during the T cell excessive expansion [39,40,41].
Finally, there are several acute adverse clinical complications associated with the use of CAR-T cells, such as cytokine release syndrome, cytopenias, neurotoxicity, and infection. All these possible adverse effects must be considered, when the use of CAR-T therapy is proposed to treat non-life-threatening conditions. Nevertheless, current clinical studies have shown that, in general, CAR-T cells can be employed safely, with relatively low adverse effects that can be manageable for instance with the use of Tocilizumab [42].
Conclusions
After only 7 years of FDA approval of the first CAR-T cell, at least 35,000 patients have been treated with CAR-T cell therapy, just in the USA for haematological malignancies, using commercial CAR-T cells. In addition, over 10 different CARs have been approved globally by different agencies. The field of immunotherapy and cell therapy with the use of CAR-T cell is expanding to other clinical conditions such as solid tumour malignancies and autoimmune disorders but also with the development of allogenic genetically modify T-cells the potential expand its application in many clinical disorders is enormous.
Contributors
All authors wrote the first draft of the paper and revised the text. AM did the literature search for haematologic diseases. JCC did the literature search for autoimmune diseases.
Acknowledgments
We are grateful for the review and comments from Dr. Luis Alonzo Herrera from the Escuela de Medicina y Ciencias de la Salud, Tecnológico de Monterrey, Monterrey, México, for his editing recommendations.
Declaration of interests
AM has received speaker honoraria and consulting fees from Celgene in the past.
References
- Irvine D, Maus M, Mooney D, et al.: The future of engineered immune cell therapies, Science 2022;378:853–858. [CrossRef]
- Passweg J, Baldomero H, Ciceri F, et al.: Hematopoietic cell transplantation and cellular therapies in Europe 2022. CAR-T activity continues to grow; transplant activity has slowed: a report from the EBMT. Bone Marrow Transplant 2024 March-4 (Epub ahead of print). [CrossRef]
- June C, & Sadelain M.: Chimeric antigen receptor therapy. N Engl J Med 2018; 379:64–73.
- Sadelain M, Riviere I, Riddell S.: Therapeutic T cell engineering. Nature 2017; 545:423–31. [CrossRef]
- van der Stegen S, Hamieh M, Sadelain M.: The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 2015;14:499–509. [CrossRef]
- Imai C, Mihara K, Andreansky M, et al.: Chimeric receptors with 4–1BB signalling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004;18:676–84. [CrossRef]
- Maher J, Brentjens R, Gunset G, Riviere I, Sadelain M.: Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 2002;20:70–5.
- Sheykhhasan M, Manoochehri H, Dama P.: Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: a review study. Cancer Gene Therapy 2021 29:1080–1096. [CrossRef]
- Sterner R & Sterner R.: CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal 2021:11:1–11.
- Munshi N, Anderson L, Shah N, et al.: Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med 2021:384:705–716. [CrossRef]
- Berdeja J, Madduri D, Usmani S, et al.: Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 2021;398:314–324. [CrossRef]
- https://hillman.upmc.com/mario-lemieux-center/treatment/car-t-cell-therapy/fda-approved-therapieshttps://www.frontiersin.org/articles/10.3389/fphar.2022.915342/full.
- Hu Y, Feng J, Gu T, et al.: CAR T-cell therapies in China: rapid evolution and a bright future. Lancet Haematol 2022; 9(12):e930-e94. [CrossRef]
- Park J, Rivière I, Gonen M, et al.: Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 2018;378:449-59. [CrossRef]
- Kantarjian H, DeAngelo D, Stelljes M, et al.: Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med 2016; 375: 740-53. [CrossRef]
- Kantarjian H, Stein A, kbuget N, et al: Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 2017; 376:836-47. [CrossRef]
- Ghassemi S, Joseph S, Durgin J, et al.: Rapid manufacturing of non-activated potent CAR T cells. Nature Biomedical engineering. 2022;6:118–128. [CrossRef]
- Crompton J, Sukumar M, Roychoudhuri R, et al.: Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75:296–305. [CrossRef]
- Schulthess D, Gassull D, Makady A, et al.: Are CAR-T therapies living up to their hype? A study using real-world data in two cohorts to determine how well they are actually working in practice compared with bone marrow transplantsBMJ Evidence-Based Medicine Epub. [CrossRef]
- Svobada J et al ASCO, New ‘Armored’ CAR produces significant responses in patients whose cancers don't respond to current CAR T cell therapies. abstract 7004, June 2024. CT https://clinicaltrials.gov/study/NCT05989204.
- Wutti-in Y, Sujjitjoon J, Sawasdee N, et al.: Development of a Novel Anti-CD19 CAR Containing a Fully Human scFv and Three Costimulatory Domains.Front. Oncol.2022; 11:802876. [CrossRef]
- Chang L, Dong L, Liu Y, et al.: Safety and Efficacy Evaluation of 4SCAR19 Chimeric Antigen Receptor-Modified T Cells Targeting B Cell Acute Lymphoblastic Leukemia - Three-Year Follow-Up of a Multicenter Phase I/II Study. Blood 2016; 128:587–7. [CrossRef]
- IQVIA Institute for Human Data Science. Strengthening Pathways for Cell and Gene Therapies: Current State and Future Scenarios. March 2024. Available from www.iqviainstitute.org.
- Cappell, K & Kochenderfer J.: Long term outcome following CAR T cell therapy: what we know so far.: Nature Reviews Clinical Oncology 2023;20:359-371.
- Chen N, Li X, Chintala N, et al. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol 2018;51:103–10. [CrossRef]
- Del Bufalo F, De Angelis B, Caruana I, et al.: GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma N Engl J Med 2023;388:1284-95. [CrossRef]
- Yeku O & Longo D,: CAR T cells for neuroblastoma. N. Eng. J Med 388;14:1328-1331.
- Schett G, Mackensen A, Mougiakakos D.: CAR T-cell therapy in autoimmune diseases. The Lancet 2023,. [CrossRef]
- Mougiakakos D, Kroenke G, Voelkl S, et al.: CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med 2021:385:567-69.
- Krickau T, Naumann-Bartsch M, Kharboutli S, et al.; CAR T-cell therapy rescues adolescent with rapidly progressive lupus nephritis from hemodialysis. The Lancet 2024:403:1627-1630.
- Müller F, Taubmann J, Bucci, L.: CD19 CAR T-Cell Therapy in Autoimmune Disease —A Case Series with Follow-up. N Engl J Med 2024;390:687-700. [CrossRef]
- Müller F, Boeltz S, Knitza J et al.: CD19 targeted CAR-T cells in refractory antisynthetase syndrome. Lancet 2023; 401:815-18. [CrossRef]
- Orvain C, Boulch M, Bousso P et al.: Is there a place for chimeric antigen receptor-T cells in the treatment of chronic autoimmune rheumatic diseases. Arthritis Rheymatol 2021:73:1954-65. [CrossRef]
- Yang D, Sun B, Li S, et al: NKG2D-CAR T cells eliminate senescent cells in aged mice and non-human primates. Science Translational Medicine 2023;15,709. [CrossRef]
- Jin G, Liu Y, Wang L et al. A single infusion of engineered long-lived and multifunctional T cells confers durable remission of asthma in mice. Nature Immunology.2024;25:1059-1072. [CrossRef]
- Seif M, kakoschke T, Ebel F, et al.: CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci Transl Med. 2022: 28;14(664),1-15. Epub 2022 Sep 28. [CrossRef] [PubMed]
- Liu E, Tong Y, Dotti G, et al: Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018;32:520-531. Epub 2017, 20. [CrossRef]
- Chiesa R, Georgiadis, G, Syed, F et al.: Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N Engl J Med 2023;389:899-910. [CrossRef]
- Longo, D.: Engineering CAR T Cells for Off-the-Shelf Use. N Engl J Med, 2023;389:953-957. [CrossRef]
- Hamilton M, Sugio T, Noordenbos T, et al.: Risk of second tumors and T-cell lymphoma after CAR T-cell therapy. N Engl J Med 2024;390:2047-60. [CrossRef]
- Ozdemirli M, Loughney T, Deniz E, et al.: Indolent CD4+ CAR T-cell lymphoma after cilta-cel CAR T-cell therapy. N Engl J Med 2024;390:2074-82. [CrossRef]
- Mitchell E & Vassiliou G.: T-Cell Cancer after CAR T-Cell Therapy. N Engl J Med 2024;390:2120-2121. [CrossRef]
- Brudno J, & Kochenderfer, J.; Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev 2019;34:45–55. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



