
Submitted:
17 July 2024
Posted:
18 July 2024
You are already at the latest version
Abstract
Chronic granulomatous disease (CGD) is a group of rare primary inborn errors of immunity characterised by a defect in the phagocyte respiratory burst, which leads to severe and life-threatening infective and inflammatory complications. Despite recent advances in our understanding of the genetic and molecular pathophysiology of X-linked and autosomal recessive CGD, and growth in the availability of functional and genetic testing, there remain significant barriers to early and accurate diagnosis. In the current review, we provide an up-to-date summary of CGD pathophysiology, which underpins methods of diagnostic testing for CGD and closely related disorders. We present an overview of the benefits of early diagnosis and when to suspect and test for CGD. We discuss current and historical methods for functional testing of NADPH oxidase activity, as well as assays for measuring protein expression of NADPH oxidase subunits. Lastly, we focus on genetic and genomic methods employed to diagnose CGD, including gene-targeted panels, comprehensive genomic testing and ancillary methods. Throughout, we highlight general limitations of testing, and caveats specific to interpretation of results in the context of CGD and related disorders, and provide an outlook for newborn screening and the future.
Keywords:
Inborn errors of immunity (IEI)
; Chronic granulomatous disease (CGD)
; NADPH oxidase
; Nitroblue tetrazolium (NBT)
; Dihydrorhodamine (DHR)
; Genetic testing
; Genomic testing
; Screening
1. Introduction
Chronic granulomatous disease (CGD) represents a group of rare and complex primary inborn errors of immunity, whose common pathology is a significant deficit in the respiratory burst pathway in phagocytes, recently reviewed in [1,2,3,4,5]. Our understanding of CGD pathogenesis, diagnosis and management has improved significantly since early studies, which identified CGD as a functional disorder of phagocytes [6,7,8,9,10,11]. Despite these advances, it remains challenging to achieve a timely and accurate clinical diagnosis of CGD. The current review, informed by our modern understanding of CGD in its molecular pathophysiology, aims to provide a comprehensive overview of the diagnosis of CGD and associated challenges, with an up-to-date focus on functional assays, molecular and genetic testing.
2. Clinical Features and Inheritance
CGD is felt overall to be a rare disease, with varied reported estimates of prevalence, which differ geographically and by population rate of consanguinity [11,12,13,14]. In Western Europe and the US, prevalence is reported as 1 in 200,000-250,000 [15,16]. The clinical spectrum of CGD varies in terms of disease manifestations, severity and age at presentation, which presents a challenge to recognition and diagnosis. Severe cases (predominantly X-linked recessive in inheritance), tend to present early at 9-14 months of age, and are diagnosed early at 2.1-4.9 years [13,15,17,18]. Milder cases (predominantly autosomal recessive) tend to present later at 2.5-3.4 years, and are also diagnosed later at 5.8-8.8 years [13,15,17,18]. Diagnoses in early infancy, adolescence and adulthood are unusual, but occur increasingly due to increased awareness and uptake of testing [11,19,20,21,22,23].
CGD is characterised by four major clinical features, reviewed in [1,2,11,24,25,26,27,28,29]: (1) predisposition to infection by a classically described subset of catalase-positive bacteria and fungi, (2) development of tissue granulomas in respiratory, gastrointestinal and genitourinary tracts, which may lead to local complications, (3) predisposition to inflammatory bowel disease (IBD), which may be very early in onset and severe, and (4) other autoinflammatory or autoimmune complications. The overall incidence and severity of infective complications is highest in the first decade of life, and lower thereafter [30]. Most pathogens that cause infection in CGD patients also cause sporadic infection in immunocompetent hosts (e.g., Staphylococcus aureus, Serratia marcescens, and Burkholderia cepacia complex), but infections in CGD patients are more likely to be severe and frequent [30,31]. Virulence determinants other than catalase are also likely to be essential to virulence in CGD, as reviewed in [32], and evidenced by mouse models and clinical data [33,34,35,36]. Children with CGD are more likely to experience failure to thrive, organ dysfunction, poor quality of life, and increased risk of secondary inflammatory complications, such as haemophagocytic lymphohistiocytosis (HLH) [37,38,39,40]. Prior to advent of modern therapies, most children with CGD died before 10 years of age [8], and infection, particularly invasive fungal infection, remains the leading cause of death [17,30,41,42]. Mortality risk is influenced by organ dysfunction [43], and the degree of detectable residual respiratory burst activity [44].
CGD can occur sporadically, but also demonstrates inheritance in X-linked recessive (XR) and autosomal recessive (AR) patterns. XR disease is most common worldwide, predominantly affecting males, and rarely females with very skewed X-chromosome inactivation (XCI) [12,17,40,45,46]. AR CGD has equal sex distribution, and is more common in consanguineous populations, surpassing XR CGD in prevalence in some countries [47,48,49]. There is also a well-described carrier phenotype for haploinsufficient female carriers of the XR mutation, characterised by predisposition to autoinflammatory and autoimmune phenomena, and milder infective and IBD-like symptoms [50,51]. Indeed, some argue that a subset of haploinsufficient carriers should be managed akin to bona fide CGD cases [45,50,52].
3. Molecular and Cellular Pathophysiology
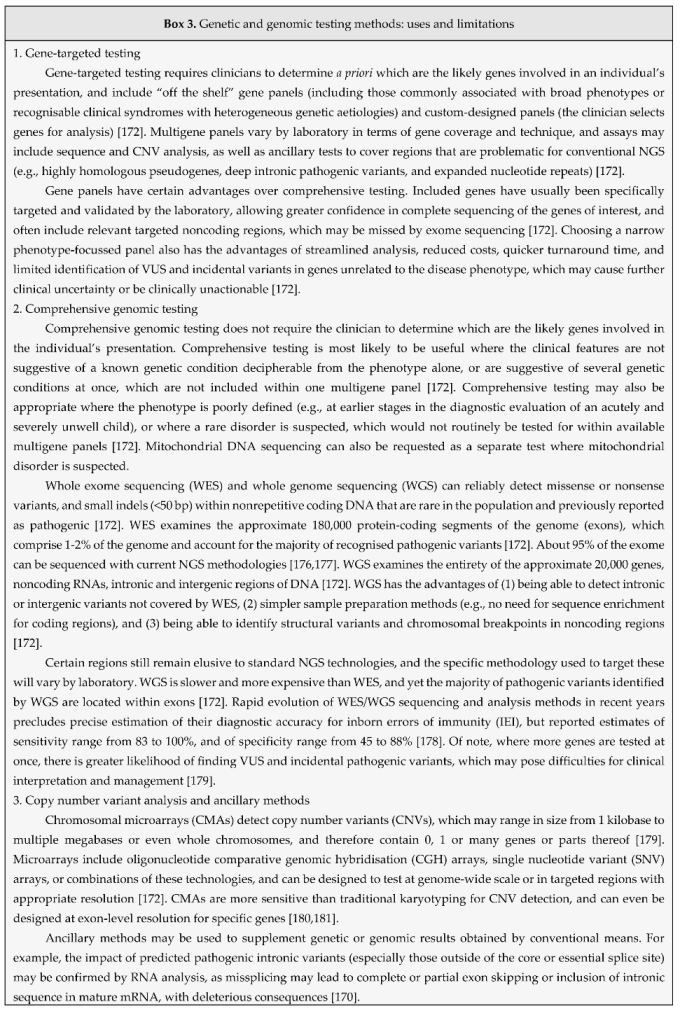
All forms of CGD share a common pathology: failure of phagocytes (primarily neutrophils, monocytes and tissue macrophages), to produce reactive oxygen species (ROS) in the respiratory burst, reviewed in [53]. Central to this pathway is the 5-subunit enzyme complex nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Figure 1). Deficiency of any one subunit of NADPH oxidase (gp91phox, p22phox, p47phox, p67phox, p40phox; Table 1), or of other factors that contribute to the respiratory burst pathway (CYBC1/Eros, Rac2-GTPase, G6PD, MPO, GSS; Table 2) may culminate in CGD or a CGD-like phagocyte disorder, reviewed in [54].
CYBB (cytochrome b558 beta component) encodes the gp91phox component of NADPH oxidase on the X chromosome, and mutation of this gene accounts for XR disease and 65-70% of all CGD [56]. Most cases of CYBB-related CGD are hemizygous males, although haploinsufficient females with very skewed XCI are also well-recognised, and show predominantly autoimmune phenotypes [45,59,60]. gp91phox is a glycosylated integral membrane protein and the central enzymatic subunit of the NADPH oxidase complex. CYBA (cytochrome b558 alpha component) encodes the p22phox subunit, and mutation of this gene is responsible for about 5% of CGD, heritable in an AR manner and equal in severity to XR disease [55,61]. p22phox forms a heterodimer with gp91phox, and stable expression of gp91phox-p22phox depends on the chaperone protein CYBC1/Eros. More recently described rare mutations of this gene are responsible for a small subset of AR CGD [62,63,64,65,66].
The p47phox, p67phox and p40phox subunits form a distinct heterotrimeric complex in the cytosol in resting phagocytes, and together activate NADPH oxidase when the respiratory burst pathway is triggered, e.g., by microorganisms engaged via membrane receptors. NCF1 (neutrophil cytosolic factor 1) encodes p47phox, and mutation of this gene accounts for about 20% of all CGD [67,68,69]. NCF1-related CGD is generally mild in severity [55]. NCF2 encodes p67phox, and mutation of this gene causes AR disease that is as severe as XR CGD [67,69]. NCF4 encodes p40phox, and mutation of this gene causes a rare, mild and atypical form of CGD with more inflammatory clinical manifestations [19,70,71]. Upon encountering phagocytic or inflammatory stimuli, signalling pathways converge on NADPH oxidase to activate the respiratory burst [70,72,73,74]. The complex is also engaged by activated Rac2-GTPase, which enhances its enzymatic activity [72,74]. Mutation of RAC2 therefore disrupts signalling to the NADPH oxidase complex [75], resulting in defects in ROS production and other neutrophil and lymphocyte functions, which manifest as an autosomal dominant (AD) CGD-like syndrome [4,76,77].
Severe G6PD deficiency can compromise supply of NADPH to the respiratory burst, and manifest clinically as mild CGD [4,73,78]. Because of its relatively high prevalence and XR inheritance, G6PD deficiency is an important differential diagnosis for CGD, especially where other clinical features are suggestive (e.g., haemolytic anaemia) [54,56]. Deficiencies of glutathione synthetase, glutathione peroxidase and glutathione reductase are also associated with inflammatory complications akin to CGD, through the roles of these enzymes in oxidative balance within phagocytes [79]. Of note, deficiencies of other ROS-producing systems, such as the mitochondrial respiratory chain, xanthine oxidases, lipoxygenases, nitric oxide synthases or cyclooxygenases do not typically confer susceptibility to infection [2].
NADPH oxidase transfers electrons to apical molecular oxygen to form superoxide radical, which is then converted to other ROS (hydrogen peroxide [H2O2] and hydroxyl radicals) at the apical membrane, reviewed in [73]. These ROS serve as substrates for peroxidase enzymes (e.g., myeloperoxidase [MPO]), to produce other reactive molecules, e.g., hypochlorous acid and secondary amines. NADPH oxidase activation furthermore leads to potassium ion influx into the phagosome lumen, which activates microbicidal proteases from the granule matrix [72,74,80]. Within phagocytes, these activities are both directly microbicidal and stimulate inflammatory signalling [81]. MPO deficiency is more common than CGD, with estimated prevalence of 1 in 2,000-4,000, but is usually asymptomatic or causes a limited immunodeficiency with susceptibility to Candida infections [54,82], though not true CGD.
Overall, neutrophils deficient in the respiratory burst show defective killing of microorganisms by phagocytosis, neutrophil extracellular traps, and other forms of regulated cell death [83,84,85,86]. However, defects of ROS-related inflammatory regulation and other innate immune pathways are thought to be responsible for the hyperinflammatory manifestations of CGD [81,87,88,89,90]. Furthermore, granulomas are believed to arise where microbes fail to be eliminated, or there is persistent dysregulated local cytokine production following microbial sterilisation. The resulting chronic inflammatory infiltrate may organise into a structured granuloma containing lymphocytes and histiocytes [28,91].
4. Benefits of Early Diagnosis of CGD
Like many inborn errors of immunity (IEI), early diagnosis of CGD is important for several reasons, which together support optimal clinical outcomes (Figure 2): (1) early instigation of effective preventative care, (2) appropriate and tailored management of acute infections and complications, (3) early definitive treatment, and (4) early identification of affected family members. Preventative care includes prophylactic antimicrobials (e.g., co-trimoxazole and azole antifungals), active surveillance, immunomodulation (some centres advocate use of interferon-gamma), immunisation advice, and avoidance of risks (e.g., decaying organic matter and high-risk water exposures) [19,24]. Definitive treatment in the form of allogeneic haematopoietic stem cell transplantation (HSCT) is indicated for severe CGD, and is ideally performed early in life, prior to episodes of severe infection, inflammatory complications, alloimmunisation, or organ scarring and dysfunction, which increase the risk of complications associated with HSCT [3,92]. The psychosocial and economic burden of unexplained significant ill-health and its implications for quality of life are also likely to be substantial for patients with undiagnosed severe CGD [22,93,94,95]. Furthermore, survey data highlighting the impact of CGD on psychosocial and functional outcomes for patients and carriers further emphasise the potential benefits of early diagnosis and definitive treatment [96,97,98,99]. For these reasons, prompt recognition and testing are essential.
Achieving a genetic diagnosis of CGD confers several specific additional benefits. Genetic diagnosis can guide prognosis for affected individuals, genetic testing and counselling for family members, and family planning, which may take into account prenatal or preimplantation testing [19]. Genetic diagnosis can also guide appropriate selection of noncarrier HSCT donors among HLA-matched family members, and is essential for directing efforts towards effective gene therapies, which at present remain aspirational and a very active field of research [100,101,102]. Indeed, the potential benefits of genetic diagnosis warrant consideration of DNA banking for affected deceased probands who lack a defined genetic cause for their CGD, as testing methods continue to improve [103]. Prenatal and preimplantation detection of CGD for at-risk pregnancies is now common practice, and methods are reliable for both XR and AR forms of the disorder, particularly where a precise familial mutation is known [19,104,105]. Of note, family and prenatal testing are still possible where a genetic diagnosis is lacking, as conventional functional assays can still be performed on peripheral blood or umbilical vein samples [19,106].
5. When to Suspect and Test for CGD
CGD testing is indicated where there is history of repeated invasive bacterial or fungal infections typical for this disorder, tissue granulomas, inflammatory manifestations or complications, especially in the context of a suggestive family history or characteristic histologic or radiologic findings (although the latter are not essential) [107,108] (Box 1). The European Society for Immunodeficiencies (ESID) registry provides working definitions for definite and probable cases of CGD, which reflect the clinical features described above, and require laboratory testing of both the respiratory burst pathway and genetics [109].

Typically, an immunological workup in CGD reflects the central deficit in NADPH oxidase [107]. The differential white cell count and immunoglobulin levels are likely to be normal, or nonspecifically altered in the context of intercurrent infection or complication. Complement studies, lymphocyte subsets, vaccine responses and phagocyte morphology are typically normal. Expression of neutrophil cell surface adhesion molecules and tests of chemotaxis are expected to be normal, whereas direct tests of NADPH oxidase function and phagocytic killing are abnormal, as below. Genetic testing is informed by the clinical details and family history, particularly if the familial mutation is already known [108]. Functional and genetic carriership testing should also be offered to adult female relatives of confirmed XR cases, particularly if there are infective or inflammatory manifestations of CGD [45,50,108].
The main differential diagnoses for CGD are disorders of recurrent severe or atypical infection, granulomas and hyperinflammation [107], including cystic fibrosis, severe G6PD deficiency [54], glutathione synthetase deficiency [58], protein kinase C delta deficiency [123,124], leukocyte adhesion deficiency [54], combined or common variable immunodeficiency [91], MyD88/IRAK4 deficiency, hyper-IgE syndrome [125], allergic bronchopulmonary aspergillosis, sarcoidosis, IBD, MPO deficiency [54] and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis). Distinctive phenotypic features may point clinical suspicion in these directions, and prompt alternative methods of diagnostic testing.
6. Testing of NADPH Oxidase Function
Defective ROS production mediates the effect of genotype on phenotype in CGD, and diagnosis requires demonstration of a significantly impaired respiratory burst [126]. Most CGD cases exhibit a total lack of NADPH oxidase activity [127], however functional assays, which provide quantitative information on residual enzymatic activity, are important for guiding prognosis, because of the well-recognised survival benefit conferred by residual oxidase function [44].
Tests of NADPH oxidase activity can be performed on anticoagulated whole blood or purified leukocytes or neutrophils, reviewed in [108,126,127,128]. Misleading assay results may occur due to pyrogen contamination, sample mishandling and delay in processing, due in part to the short ex vivo lifespan of functional neutrophils, and therefore strict quality controls are required (e.g., a healthy comparator specimen) [108,126,129,130]. Of note, in certain circumstances, the polymorphonuclear fraction of circulating leukocytes may contain significant numbers of non-neutrophils (e.g., eosinophilia due to atopy or parasitic infection) [126].
Because NADPH oxidase is inactive in resting neutrophils, exogenous stimulation of the pathway is required for functional testing. Serum-opsonised particulate stimuli, such as zymosan (a yeast membrane derivative), E. coli, or S. aureus provoke the respiratory burst by engagement of Fc-gamma receptor (FCγR) and complement receptor 3 (CR3) on the phagocyte cell surface [108]. Fluid-phase stimuli such as phorbol myristate acetate (PMA) or formyl-methionyl-leucyl-phenylalanine (fMLP) require priming by platelet-activating factor (PAF), and also provoke the respiratory burst [108]. Comprehensive testing of neutrophil function thus requires assays that test multiple different stimuli.
Tests of NADPH function include measurement of oxygen consumption, plate-based assays (ferricytochrome c reduction, lucigenin/isoluminol, Amplex® Red), nitroblue tetrazolium, dihydrorhodamine and dichlorofluorescein assays. Tests of phagocytosis and neutrophil microbicidal function may be carried out in addition as part of a wider workup of a suspected disorder of neutrophil function, but are outside the scope of the current review [127].
6.1. Measurement of Oxygen Consumption
6.2. Plate-Based Assays
Ferricytochrome c reduction is a colorimetric assay of superoxide production. Ferricytochrome c is added extracellularly and does not permeate cell membranes [126]. Superoxide produced by phagocytes and released to the extracellular space converts ferricytochrome c to the reduced product ferrocytochrome c, which can be measured by spectrophotometry [108,126,132]. The result gives a pooled reading for the whole population of cells tested, and cannot distinguish mixed populations [126]. Having been superseded by other tests, the ferricytochrome c assay is no longer widely used for CGD diagnosis.
Reagents that release energy in the form of light upon reaction with ROS can also be used to measure NADPH oxidase activity by chemiluminescence, e.g., lucigenin or isoluminol [133]. These assays are sensitive, and can be carried out relatively quickly on few available cells [126,132,134]. They detect both intra- and extracellular H2O2, and rely on cellular peroxidases, so may misdiagnose MPO deficiency as CGD [132,134].
Amplex® Red is an alternative chemical probe for H2O2. The colourless, nonfluorescent dihydroresorufin derivative Amplex® Red is oxidised by H2O2 to resorufin, which is detectable as red fluorescence, and can be measured using a plate reader [135]. The Amplex® Red assay is also sensitive to small numbers of cells, reliable and relatively easy to perform, but is not truly quantitative, and again is not routinely used in practice for diagnosis of CGD [124].
6.3. Nitroblue Tetrazolium (NBT)
The nitroblue tetrazolium (NBT) assay measures superoxide production, and has been in clinical use for decades for diagnosis of CGD [136]. In this assay, superoxide produced by activated NADPH oxidase converts the pale yellow dye NBT to its dark blue insoluble formazan product, which precipitates within activated phagocytes [126,137] (Figure 3). Quantification can be done by counting formazan-positive and -negative neutrophils by light microscopy of peripheral blood in a chamber slide, or by spectrophotometric measurement of the solubilised formazan product in neutrophil lysate [108,132]. The assay is theoretically semi-quantitative when carried out by an experienced technician, but is most sensitive for severe CGD, where there is very little to no formazan deposition in any cell [138]. It also has low sensitivity for detecting mixed populations of cells in female carriers haploinsufficient for pathogenic CYBB mutations, and for detecting hypomorphic variants with residual NADPH oxidase function (both milder AR and protein-positive XR CGD) [126]. High-confidence current estimates of sensitivity and specificity of the NBT assay for diagnosing CGD are lacking in published literature. However, because it is quick to perform using nonspecialist materials and equipment, it is particularly useful in low-resource settings, or where a rapid result is required, and local expertise is available [139,140,141].
6.4. Dihydrorhodamine (DHR)
The dihydrorhodamine-1,2,3 (DHR) assay is considered the current gold standard test of NADPH oxidase activity [126]. In the presence of a peroxidase enzyme (e.g., MPO or eosinophil peroxidase) and stimulated conditions, it allows quantitative measurement of H2O2 production [142]. DHR freely diffuses into cells and reacts with H2O2, becoming reduced to rhodamine, which is trapped intracellularly and fluoresces green [142]. The fluorescent signal can be measured in individual cells by flow cytometry [142] (Figure 4). Extracellular addition of catalase minimises background rhodamine signal [126]. Three classic patterns of abnormal DHR result are seen: (1) complete or severe deficiency (rhodamine-positive cells comprise <5% of total neutrophils or <5% of control), (2) hypomorphic or partial deficiency (where the rhodamine signal is uniformly intermediate between unstimulated and stimulated control neutrophils), and (3) mosaic patterns, where a proportion of neutrophils show reduced NADPH oxidase function and the remainder are normal [126,143] (Figure 4).
The DHR assay is highly sensitive, quantitative and relatively easy to perform in facilities with standard flow cytometry capability [126], though this has obvious implications for test availability in low-resource settings [139,140,141]. The DHR assay is especially useful because of its ability to quantify residual NADPH oxidase function, and to discriminate carrier status and quantify donor chimerism post-HSCT [44,50,144,145]. Notably, within XR CGD carriers, the proportion of DHR-positive neutrophils is reported to vary over time [143,146].
False positive DHR results can arise in severe MPO deficiency, which mimics CGD, although superoxide production, and hence ferricytochrome c reduction and the NBT assay, are normal [147,148]. Other false positives can occur in severe G6PD deficiency [78] and SAPHO syndrome [149], where superoxide production is abnormal, and in conditions of acquired neutrophil dysfunction, such as granulocytic ehrlichiosis [150,151], and reportedly after exposure to anti-inflammatory drugs [152,153,154]. The DHR assay cannot detect carriers of AR CGD, because a single functional allele of an autosomal gene is usually sufficient for cellular function. Some residual H2O2 (but not superoxide) production is reported to persist in NCF1-deficient neutrophils [127,155]. Routine DHR testing is also reportedly not sensitive for the rare cases of CGD caused by mutation in NCF4 [19]. In the latter case, the PMA/fMLP-stimulated DHR response is normal or only mildly reduced, but oxidative responses to particulate stimuli are defective, and killing of certain microorganisms is also impaired [71,127]. In Rac2-GTPase deficiency, PMA-stimulated superoxide production is normal or increased compared to control, whereas fMLP-stimulated chemotaxis and superoxide production are impaired [75,76], along with other non-phagocyte immune defects [2,156]. In CYBC1/Eros deficiency, as expected there is demonstrable loss of PMA-stimulated respiratory burst measured by the DHR assay, due to the lack of expressed gp91phox-p22phox [62].
6.5. Dichlorofluorescein (DCF)
2’7’-dichlorofluorescein (DCF) and 5,6-carboxy-2’7’-dichlorofluorescein diacetate (CDCF) are alternative fluorescent probes, which can be used in flow cytometric assays of NADPH oxidase function, but are used less commonly in practice than DHR [157].
7. Molecular Diagnostics: Protein Assays
Where NADPH oxidase activity is confirmed to be absent by functional assays, protein expression of NADPH oxidase components can be measured to narrow the differential diagnosis of genetic causes of CGD [127]. These assays largely rely on antibodies specific for protein components of NADPH oxidase, and can be carried out alongside other protein assays for suspected disorders of neutrophil function, using anticoagulated blood [108,126]. Protein assays include western blot of neutrophil lysates [108] and flow cytometry-based assays that use surface- or intracellular antibody stains [108,158,159,160,161]. Additionally, antibody-based immunofluorescence and immunohistochemistry for NADPH oxidase subunits have been reported in formalin-fixed, paraffin-embedded tissues, but are not routinely used for CGD diagnosis [162]. It is also possible to detect NADPH oxidase proteins by mass spectrometry, but this is not widely used outside the research space of high-throughput screening for IEI [163,164].
Although western blot is sensitive to detect even small traces of protein, which can be relatively quantified by densitometry, certain caveats complicate the use of this method for diagnostic testing, and require strict quality control and validation [108,126]. Antibody binding and specificity may be sensitive to degradation of blood or tissue samples following collection [162]. Antibodies may be nonspecific to individual NADPH oxidase subunits, but rather recognise the complex as a whole or individual hemi-complexes (gp91phox-p22phox or p47phox-p67phox-p40phox) [126,165,166]. As described, gp91phox-p22phox expresses as a stable complex integral to the membrane, so isolated deficiency of either subunit alone will not be distinguished from the other solely by protein-based assays [102]. Another caveat is that positive antibody staining does not guarantee that functional protein is present, as intact expression of enzymatically inactive protein will still result in respiratory burst deficiency and CGD [167]. Protein assays may not therefore reliably discriminate between genetic diagnoses, although the result can be suggestive when considered in the context of the inheritance pattern and clinical features [165]. Of note, flow cytometry-based assays using antibody probes may distinguish mixed populations in XR carrier or mosaic states, and be useful in these cases [127].
Cell-free NADPH oxidase assays have also been developed to distinguish biochemical deficits in cytosolic and membrane-bound NADPH oxidase components [168]. Purified proband neutrophil membranes are mixed with healthy neutrophil cytosol (or vice-versa), and incubated with NADPH oxidase substrates and activators. Oxidase activity is then measured by superoxide production or oxygen consumption [168]. Furthermore, the nonfunctioning CGD gene may be identified by restoration of the biochemical deficit in EBV-transformed B cells from the proband, by transducing a retroviral vector to express wild-type cDNA for each of the NADPH oxidase components in turn [169]. Again, however, these assays are not routinely used in practice for diagnosis of CGD.
8. Molecular Diagnostics: Genetic Testing
Confirmation of a CGD diagnosis following positive functional testing is established by identifying a pathogenic variant in one of the CGD-related genes that encode or permit assembly or function of NADPH oxidase [19]. Algorithms that incorporate different genetic and genomic techniques into a standardised workflow for the investigation of CGD have been proposed [170], and reporting of genetic results generally involves classifying identified variants according to the likelihood that they are responsible for the clinical phenotype (Box 2). Genetic testing methods, reviewed in [19], generally include: (1) gene-targeted testing in single- or multi-gene panels performed by Sanger or next-generation sequencing (NGS) techniques; (2) comprehensive genomic testing, including whole exome (WES) and whole genome sequencing (WGS); (3) copy number variant (CNV) analysis, e.g., by multiplex ligation-dependent probe amplification (MLPA) or comparative genomic hybridisation (CGH) arrays; and (4) ancillary methods, e.g., single nucleotide variant (SNV) arrays, mRNA size and sequence analysis, deep amplicon sequencing, and optical gene mapping [171] (Box 3). The choice of genetic techniques applied depends on the phenotype and clinical suspicion (clinical details, family history, and functional assay results), and confirmatory testing should be applied as standard within genetic testing pipelines. Family history is essential to guide analysis and interpretation of sequencing data (e.g., by application of bioinformatic filters based on inferred modes of inheritance), and testing is far easier where the familial mutation is already known [172]. For genetic testing, EDTA blood is preferred, and genomic DNA or total RNA can be extracted from peripheral blood mononuclear cells (PBMCs) [108].

9. Mutations in CGD
The heterogeneity of clinical phenotypes observed in CGD is reflected in the variety of mutations identified in responsible genes. Multigene panels for suspected IEI routinely include the core genes responsible for CGD (CYBB, CYBA, NCF1, NCF2, NCF4 and CYBC1), and should also include other genes of interest responsible for important differential diagnoses (e.g., CFTR, G6PD, RAC2 and GSS). These genes should all be captured by WES/WGS methods, but it is important to confirm that they are included within the analysis when CGD is suspected.
CYBB mutations range from single nucleotide variants (missense, nonsense) and small insertions and deletions (indels), to deletions of several megabases, and may involve exons or splice sites [56]. Large insertions are relatively less common, and are usually caused by duplications or transposable elements [56,127,182]. Some CYBB mutations result in complete absence of gp91phox protein expression, and complete deficiency of NADPH oxidase function, termed the “Xb0 phenotype” [127]. Some CYBB mutations result in a degree of residual functional protein expression (e.g., amino acid substitutions or small in-frame indels, and splice site mutations that leave some residual normal mRNA), termed the “Xb- phenotype” [127]. These comprise mostly pathogenic missense variants in the region encoding the N-terminal 309 amino acids, and tend to confer relatively better prognosis [44]. Variants affecting the C-terminal portion affect cofactor and substrate binding domains, and render the expressed protein largely nonfunctional with relatively poorer prognosis [44]. Yet other CYBB mutations result in expression of normal levels of enzymatically inactive protein, termed the “Xb+ phenotype” [127]. Of note, a distinct spectrum of hypomorphic missense mutations in CYBB, which specifically impair NADPH oxidase function in macrophages and B cells (rather than granulocytes), leads to XR Mendelian susceptibility to mycobacterial disease (MSMD) [183,184].
About 15% of CYBB mutations arise de novo, where maternal leukocyte DNA does not contain the mutant allele, but this does not exclude the possibility of maternal somatic mosaicism, where some or all of the maternal gonadal or germ cells harbour the mutation. As such, if maternal testing is negative for the mutation, there may still be a risk that further offspring will be affected [13]. As described earlier, skewed XCI is also responsible for clinical features of overt CGD in a subset of female carriers of pathogenic CYBB variants [11,59]. Where this is suspected, the standard HUMARA assay for XCI may be used, which assesses epigenetic silencing of X-chromosome genes [185,186]. Novel methods, which use NGS to directly assess differential transcription of X-chromosome genes have also been developed more recently to assess for skewed XCI [170]. Individuals with suspected XR CGD and clinical manifestations of McLeod syndrome (neuromuscular abnormalities and haemolytic anaemia) should undergo CMA to detect larger deletions within the X chromosome [187,188]. CMA with adequate resolution to identify the deletion boundary can define whether there is loss of implicated genes contiguous to CYBB in these cases, e.g., XK (Kx blood group antigen), RPGR (retinitis pigmentosa), DMD (Duchenne muscular dystrophy) and OTC (ornithine transcarbamoylase) [187,188].
NCF1 has two neighbouring paralogous pseudogenes >99% identical in sequence to NCF1, which pose a particular challenge to genetic diagnosis by conventional sequencing [189]. Both pseudogenes harbour a two-nucleotide GT deletion at the beginning of exon 2, which leads to frameshift and a premature stop codon, resulting in absent expression of active p47phox protein [127,190]. Loss of the functional NCF1 gene may occur due to unequal crossover between the intact gene and either pseudogene during meiosis [189,191]. NCF1-ΔGT accounts for about 90% of all p47phox-deficient CGD, and compound heterozygotes with other NCF1 mutations have also been reported [55]. To detect these variants, PCR and sequencing may rely on primers that discriminate between NCF1 and its pseudogenes [108], or on methods that can demonstrate a 2 bp length discrepancy between amplified fragments of the NCF1 gene or pseudogene [192,193]. mRNA and protein assays may also have a role in confirming pathogenicity of detected NCF1 variants [194]. Of note, another distinct group of NCF1 variants is reported to be associated with autoimmune and autoinflammatory disorders without the other features of CGD [195].
Mutations in CYBA and NCF2 include indels, missense, nonsense and splice site variants [55]. Most lead to complete absence of protein expression, but a few reported mutations lead to decreased expression of functional protein, and thus reduced NADPH oxidase activity [196,197]. There are few reported cases of NCF4 deficiency, but described variants include missense and nonsense mutations affecting exons, introns or essential splice sites, as well as in-frame deletions [70,71,167]. Mutations of CYBC1 result in loss of expression of the gp91phox-p22phox protein complex, but levels of CYBB and CYBA mRNA are preserved [63,64]. Reported mutations of RAC2 include missense variants, which encode an inactive protein unable to bind GTP [4,75,76,156]. Only severe forms of G6PD deficiency result in such scarcity of NADPH as to phenocopy the infection susceptibility of milder forms of CGD [54]. As such, these mutations (mostly missense) comprise only a small subset of all recognised G6PD mutations [56]. Although a theoretical possibility, very skewed XCI in haploinsufficient carriers of G6PD deficiency may also result in CGD features in females.
10. Limitations of Genetic Testing for CGD
Broadly, challenges in genetic diagnosis of CGD reflect both technical and biological factors common to other genetic disorders. Where sequence variants are not detected by genetic or genomic methods, there are several potential explanations.
Specimen type and handling can impact the yield and quality of nucleic acid, and thus sequencing performance, e.g., NGS read depth and accuracy of base calling, although standard quality controls should identify these issues early [108]. NGS technologies are subject to technical variables that affect diagnostic sensitivity, including the sequence enrichment method used for exome sequencing, use of paired- versus single-end read methods, and use of short- versus long-read technologies [172]. It is worth noting that standard NGS techniques cannot detect variants that influence gene expression without changing DNA sequence (e.g., epigenetic or imprinting errors, and uniparental disomy), and do not reliably detect variants in highly repetitive regions of DNA that are difficult to sequence (e.g., nucleotide repeat expansions or contractions), variants involving a gene that is highly homologous to other gene family members or pseudogenes (e.g., NCF1), and variants that are somatically acquired or mosaic (i.e., only present in a small percentage of cells, such that base calls do not pass quality thresholds).
Gene panels or WES may not detect pathogenic variants in regions (e.g., introns or regulatory regions) not covered by the sequencing method used. Conventional sequence analysis alone is also poorly sensitive for structural variants and large whole-exon or whole-gene deletions or duplications (e.g., McLeod syndrome), and these types of mutation require alternative methods of CNV analysis or high-resolution karyotyping [19]. However, creative new methods based on NGS and Sanger sequencing have the potential to address some of these historic challenges in diagnosing CGD and investigating skewed XCI and mosaicism [170].
It is worth emphasising that genetic and genomic testing workflows depend upon the quality of the clinical information and family history provided by the requesting clinician, and diagnostic sensitivity may be affected if these are inadequate. Interpretation of the significance of detected variants is also subject to the limitations of existing literature, databases and bioinformatic prediction tools [173] (Box 2). Notwithstanding technical factors, the proband may also truly not have a pathogenic variant in the tested genes, and an alternative explanation for their clinical syndrome should be sought. Where clinical suspicion for CGD remains high, this warrants close inspection of the methods employed, and periodic re-analysis of existing data to take account of updated bioinformatic methods and reference databases. In some cases, following discussion with genomic service providers, repeat testing may be warranted in due course to account for technical improvements, which might improve diagnostic yield.
Because CGD is currently understood to be a rare diagnosis, clinical awareness and suspicion may not be sufficient to prompt testing for CGD in the first place, and awareness of the need to test presents a significant challenge to timely diagnosis of this disorder. Availability of genetic and genomic testing is also restricted in resource-limited settings, which remains one of the ongoing challenges to identifying and helping those CGD cases whose need is greatest [198,199,200]. Even in high-resource settings such as the UK, significant reporting backlogs in centralised genomic testing services have the potential to contribute to significant delays in diagnosis and treatment [201].
11. Screening
Routine newborn population screening for CGD is not commonplace, despite the potential benefits of early diagnosis [163]. This is due at least in part to the infeasibility of a CGD-relevant screening assay, which can be run in parallel with other tests, although potential assays have been proposed using mass spectrometry for protein detection in newborn dried blood spot samples [164]. Screening of asymptomatic individuals is thus usually restricted to family members of known CGD probands. However, there is growing interest in the use of genomic tests for newborn screening [202,203], and it is timely that five CGD-related genes are included in those to be tested in the Generation Study of newborn genomic screening currently being piloted in sites across the UK [204]. This furthermore emphasises the need for reliable and accurate confirmatory diagnostic testing for screen-positive individuals identified prior to onset of any infective or other complications.
12. Outlook
Within the realm of testing for CGD and other genetic disorders, gene panels, sequencing technologies, bioinformatic tools, reference databases and diagnostic sensitivity are likely to vary between laboratories and over time. As services evolve to keep pace with novel developments, we are heartened that functional and genetic testing for CGD continues to improve. We hope that these improvements will help to overcome the diagnostic challenges in CGD discussed here, and facilitate wider access and uptake of testing. Particularly with the expansion of screening into younger age groups using genomic testing in infancy, the future for early diagnosis and definitive treatment of CGD looks much brighter than it did before.
Author Contributions
Conceptualization, MA, COD, JB; literature review, COD, MA, LT; original draft preparation, COD, MA, LT; review and editing, COD, JB, AG, MR; supervision, JB, AG. All authors have read and agreed to the published version of the manuscript.
Funding
This review received no specific funding. COD is funded by Academic Clinical Fellowship ACF-2022-25-003 from the National Institute for Health and Care Research (NIHR), UK. The views expressed are those of the author(s) and not necessarily those of the NIHR or the UK Department of Health and Social Care.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to gratefully acknowledge the careful reading and feedback of our anonymous reviewers, which has enhanced the content of this review article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [CrossRef]
- Nunoi, H.; Nakamura, H.; Nishimura, T.; Matsukura, M. Recent topics and advanced therapies in chronic granulomatous disease. Hum. Cell 2022, 36, 515–527. [CrossRef]
- Yu, H.-H.; Yang, Y.-H.; Chiang, B.-L. Chronic Granulomatous Disease: a Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 61, 101–113. [CrossRef]
- Roos D, Holland SM, Kuijpers TW. Chronic granulomatous disease. In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases, a molecular and genetic approach. 3rd ed. New York: Oxford University Press; 2014. p. 689–722. [CrossRef]
- Justiz-Vaillant, A.A.; Williams-Persad, A.F.-A.; Arozarena-Fundora, R.; Gopaul, D.; Soodeen, S.; Asin-Milan, O.; Thompson, R.; Unakal, C.; Akpaka, P.E. Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment. Microorganisms 2023, 11, 2233. [CrossRef]
- Janeway CA, Craig J, Davidson M, Downey W, Gitlin D, Sullivan JC. Hypergammaglobulinemia associated with severe, recurrent and chronic non-specific infection. Am J Dis Child. 1954;88:388-92.
- Landing, B.H.; Shirkey, H.S. A syndrome of recurrent infection and infiltration of viscera by pigmented lipid histiocytes. Pediatrics 1957, 20, 431–438. [CrossRef]
- A Bridges, R.; Berendes, H.; A Good, R. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. Am J Dis Child. 1959, 97, 387–408. [CrossRef]
- Holmes B, Quie PG, Windhorst DB, Good RA. Fatal granulomatous disease of childhood.: an inborn abnormality of phagocytic function. Lancet. 1966;287(7449):1225-8.
- Babior BM. The respiratory burst oxidase. Hematol Oncol Clin North Am. 1988;2(2):201-12.
- Gennery, A. Recent advances in understanding and treating chronic granulomatous disease. F1000Research 2017, 6, 1427. [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic Granulomatous Disease: Report on a National Registry of 368 Patients. Medicine 2000, 79, 155–169. [CrossRef]
- Wolach, B.; Gavrieli, R.; de Boer, M.; Gottesman, G.; Ben-Ari, J.; Rottem, M.; Schlesinger, Y.; Grisaru-Soen, G.; Etzioni, A.; Roos, D. Chronic granulomatous disease in Israel: Clinical, functional and molecular studies of 38 patients. Clin. Immunol. 2008, 129, 103–114. [CrossRef]
- Fattahi, F.; Badalzadeh, M.; Sedighipour, L.; Movahedi, M.; Fazlollahi, M.R.; Mansouri, S.D.; Khotaei, G.T.; Bemanian, M.H.; Behmanesh, F.; Hamidieh, A.A.; et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2011, 31, 792–801. [CrossRef]
- Jones, L.B.K.R.; McGrogan, P.; Flood, T.J.; Gennery, A.R.; Morton, L.; Thrasher, A.; Goldblatt, D.; Parker, L.; Cant, A.J. Special Article: Chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin. Exp. Immunol. 2008, 152, 211–218. [CrossRef]
- Rider, N.L.; Jameson, M.B.; Creech, C.B. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S2–S5. [CrossRef]
- Berg, J.M.v.D.; van Koppen, E.; Åhlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Español, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic Granulomatous Disease: The European Experience. PLOS ONE 2009, 4, e5234. [CrossRef]
- Martire, B.; Rondelli, R.; Soresina, A.; Pignata, C.; Broccoletti, T.; Finocchi, A.; Rossi, P.; Gattorno, M.; Rabusin, M.; Azzari, C.; et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: An Italian multicenter study. Clin. Immunol. 2008, 126, 155–164. [CrossRef]
- Leiding JW, Holland SM. Chronic Granulomatous Disease. 2022 2012 Aug 9 [updated 2022 Apr 21]. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle.
- Baxter, J.; Smith, D.; Webb, C. Adult-diagnosed Chronic Granulomatous Disease: The Need to Increase Awareness. Mil. Med. 2021, 188, e410–e411. [CrossRef]
- Di Matteo, G.; Finocchi, A. Late diagnosis and advances in genetics of chronic granulomatous disease. Clin. Exp. Immunol. 2020, 203, 244–246. [CrossRef]
- Barkai, T.; Somech, R.; Broides, A.; Gavrieli, R.; Wolach, B.; Marcus, N.; Hagin, D.; Stauber, T. Late diagnosis of chronic granulomatous disease. Clin. Exp. Immunol. 2020, 201, 297–305. [CrossRef]
- Lee, E.; Bosi, I.; Peacock, K.; Lau, C.; Ford, L.S.; Wong, M.; Hsu, P. Novel infantile presentations of chronic granulomatous disease. Pediatr. Allergy Immunol. 2024, 35, e14190. [CrossRef]
- Magnani, A.; Mahlaoui, N. Managing Inflammatory Manifestations in Patients with Chronic Granulomatous Disease. Pediatr. Drugs 2016, 18, 335–345. [CrossRef]
- Mouy, R.; Fischer, A.; Vilmer, E.; Seger, R.; Griscelli, C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J. Pediatr. 1989, 114, 555–560. [CrossRef]
- Falcone EL, Holland SM. Gastrointestinal Complications in Chronic Granulomatous Disease. In: Knaus UG, Leto TL, editors. NADPH Oxidases: Methods and Protocols. 1982: Humana Press; 2019. p. 573-86. [CrossRef]
- Grammatikos, A.; Gennery, A.R. Inflammatory Complications in Chronic Granulomatous Disease. J. Clin. Med. 2024, 13, 1092. [CrossRef]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current Concepts of Hyperinflammation in Chronic Granulomatous Disease. J. Immunol. Res. 2011, 2012, 1–6. [CrossRef]
- Staudacher O, von Bernuth H. Clinical presentation, diagnosis, and treatment of chronic granulomatous disease. Front Pediatr. 2024;12:1384550. [CrossRef]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common Severe Infections in Chronic Granulomatous Disease. Clin. Infect. Dis. 2014, 60, 1176–1183. [CrossRef]
- Prince, B.T.; Thielen, B.K.; Williams, K.W.; Kellner, E.S.; E Arnold, D.; Cosme-Blanco, W.; Redmond, M.T.; Hartog, N.L.; Chong, H.J.; Holland, S.M. Geographic Variability and Pathogen-Specific Considerations in the Diagnosis and Management of Chronic Granulomatous Disease. Pediatr. Heal. Med. Ther. 2020, ume 11, 257–268. [CrossRef]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.-H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2016, 41, 139–157. [CrossRef]
- Chang, Y.C.; Segal, B.H.; Holland, S.M.; Miller, G.F.; Kwon-Chung, K.J. Virulence of catalase-deficient aspergillus nidulans in p47(phox)-/- mice. Implications for fungal pathogenicity and host defense in chronic granulomatous disease.. J. Clin. Investig. 1998, 101, 1843–1850. [CrossRef]
- Messina, C.G.; Reeves, E.P.; Roes, J.; Segal, A.W. Catalase negative Staphylococcus aureus retain virulence in mouse model of chronic granulomatous disease. FEBS Lett. 2002, 518, 107–110. [CrossRef]
- Kottilil, S.; Malech, H.; Gill, V.; Holland, S. Infections with Haemophilus species in chronic granulomatous disease: insights into the interaction of bacterial catalase and H2O2 production. Clin. Immunol. 2003, 106, 226–230. [CrossRef]
- Reichenbach J, Lopatin U, Mahlaoui N, Beovic B, Siler U, Zbinden R, et al. Actinomyces in chronic granulomatous disease: an emerging and unanticipated pathogen. Clin Infect Dis. 2009;49(11):1703-10. [CrossRef]
- Rawat, A.; Singh, S.; Suri, D.; Gupta, A.; Saikia, B.; Minz, R.W.; Sehgal, S.; Vaiphei, K.; Kamae, C.; Honma, K.; et al. Chronic Granulomatous Disease: Two Decades of Experience From a Tertiary Care Centre in North West India. J. Clin. Immunol. 2013, 34, 58–67. [CrossRef]
- van Montfrans, J.M.; Rudd, E.; van de Corput, L.; Henter, J.; Nikkels, P.; Wulffraat, N.; Boelens, J.J. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr. Blood Cancer 2008, 52, 527–529. [CrossRef]
- Parekh, C.; Hofstra, T.; Church, J.A.; Coates, T.D. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr. Blood Cancer 2010, 56, 460–462. [CrossRef]
- Bortoletto P, Lyman K, Camacho A, Fricchione M, Khanolkar A, Katz BZ. Chronic Granulomatous Disease: A Large, Single-center US Experience. Pediatr Infect Dis J. 2015;34(10):1110-4. [CrossRef]
- Falcone EL, Holland SM. Invasive fungal infection in chronic granulomatous disease: insights into pathogenesis and management. Curr Opin Infect Dis. 2012;25(6):658-69. [CrossRef]
- Beaute J, Obenga G, Le Mignot L, Mahlaoui N, Bougnoux ME, Mouy R, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J. 2011;30(1):57-62. [CrossRef]
- Feld, J.J.; Hussain, N.; Wright, E.C.; Kleiner, D.E.; Hoofnagle, J.H.; Ahlawat, S.; Anderson, V.; Hilligoss, D.; Gallin, J.I.; Liang, T.J.; et al. Hepatic Involvement and Portal Hypertension Predict Mortality in Chronic Granulomatous Disease. Gastroenterology 2008, 134, 1917–1926. [CrossRef]
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.L.; Soule, B.P.; et al. Residual NADPH Oxidase and Survival in Chronic Granulomatous Disease. New Engl. J. Med. 2010, 363, 2600–2610. [CrossRef]
- Marciano, B.E.; Zerbe, C.S.; Falcone, E.L.; Ding, L.; DeRavin, S.S.; Daub, J.; Kreuzburg, S.; Yockey, L.; Hunsberger, S.; Foruraghi, L.; et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 2018, 141, 365–371. [CrossRef]
- de Oliveira-Junior, E.B.; Zurro, N.B.; Prando, C.; Cabral-Marques, O.; Pereira, P.V.S.; Schimke, L.-F.; Klaver, S.; Buzolin, M.; Blancas-Galicia, L.; Santos-Argumedo, L.; et al. Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report from the LASID Registry. Pediatr. Blood Cancer 2015, 62, 2101–2107. [CrossRef]
- Köker, M.Y.; Camcıoğlu, Y.; van Leeuwen, K.; Kılıç, S..; Barlan, I.; Yılmaz, M.; Metin, A.; de Boer, M.; Avcılar, H.; Patıroğlu, T.; et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J. Allergy Clin. Immunol. 2013, 132, 1156–1163.e5. [CrossRef]
- El Kares, R.; Barbouche, M.R.; Elloumi-Zghal, H.; Bejaoui, M.; Chemli, J.; Mellouli, F.; Tebib, N.; Abdelmoula, M.S.; Boukthir, S.; Fitouri, Z.; et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J. Hum. Genet. 2006, 51, 887–895. [CrossRef]
- Wolach, B.; Gavrieli, R.; Wolach, O.; Salamon, P.; De Boer, M.; van Leeuwen, K.; Abuzaitoun, O.; Broides, A.; Gottesman, G.; Grisaru-Soen, G.; et al. Genotype-Phenotype Correlations in Chronic Granulomatous Disease: Insights From a Large National Cohort. Blood J. 2024. [CrossRef]
- Battersby, A.C.; Cale, C.M.; Goldblatt, D.; Gennery, A.R. Clinical Manifestations of Disease in X-Linked Carriers of Chronic Granulomatous Disease. J. Clin. Immunol. 2013, 33, 1276–1284. [CrossRef]
- Hauck, F.; Koletzko, S.; Walz, C.; von Bernuth, H.; Klenk, A.; Schmid, I.; Belohradsky, B.H.; Klein, C.; Bufler, P.; Albert, M.H. Diagnostic and Treatment Options for Severe IBD in Female X-CGD Carriers with Non-random X-inactivation. J. Crohn’s Colitis 2015, 10, 112–115. [CrossRef]
- Roesler, J. Carriers of X-linked chronic granulomatous disease at risk. Clin. Immunol. 2009, 130, 233. [CrossRef]
- Stasia, M.J.; Li, X.J. Genetics and immunopathology of chronic granulomatous disease. Semin. Immunopathol. 2008, 30, 209–235. [CrossRef]
- Zerbe, C.S.; Holland, S.M. Functional neutrophil disorders: Chronic granulomatous disease and beyond. Immunol. Rev. 2024, 322, 71–80. [CrossRef]
- Roos, D.; van Leeuwen, K.; Hsu, A.P.; Priel, D.L.; Begtrup, A.; Brandon, R.; Rawat, A.; Vignesh, P.; Madkaikar, M.; Stasia, M.J.; et al. Hematologically important mutations: The autosomal forms of chronic granulomatous disease (third update). Blood Cells, Mol. Dis. 2021, 92, 102596. [CrossRef]
- Roos, D.; van Leeuwen, K.; Hsu, A.P.; Priel, D.L.; Begtrup, A.; Brandon, R.; Stasia, M.J.; Bakri, F.G.; Köker, N.; Köker, M.Y.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (fourth update). Blood Cells, Mol. Dis. 2021, 90, 102587. [CrossRef]
- Marchetti, C.; Patriarca, P.; Solero, G.P.; Baralle, F.E.; Romano, M. Genetic characterization of myeloperoxidase deficiency in Italy. Hum. Mutat. 2004, 23, 496–505. [CrossRef]
- Njålsson, R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. 2005, 62, 1938–1945. [CrossRef]
- Battersby, A.C.; Braggins, H.; Pearce, M.S.; Cale, C.M.; Burns, S.O.; Hackett, S.; Hughes, S.; Barge, D.; Goldblatt, D.; Gennery, A.R. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J. Allergy Clin. Immunol. 2017, 140, 628–630.e6. [CrossRef]
- Cale, C.M.; Morton, L.; Goldblatt, D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin. Exp. Immunol. 2007, 148, 79–84. [CrossRef]
- Dinauer, M.C.; A Pierce, E.; A Bruns, G.; Curnutte, J.T.; Orkin, S.H. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J. Clin. Investig. 1990, 86, 1729–1737. [CrossRef]
- Arnadottir, G.A.; Norddahl, G.L.; Gudmundsdottir, S.; Agustsdottir, A.B.; Sigurdsson, S.; Jensson, B.O.; Bjarnadottir, K.; Theodors, F.; Benonisdottir, S.; Ivarsdottir, E.V.; et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. 2018, 9, 1–9. [CrossRef]
- Thomas, D.; Charbonnier, L.-M.; Schejtman, A.; Aldhekri, H.; Coomber, E.L.; Dufficy, E.R.; E Beenken, A.; Lee, J.; Clare, S.; O Speak, A.; et al. EROS/CYBC1 mutations: Decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. 2019;143(2):782-5 e1. [CrossRef]
- Thomas, D.; Clare, S.; Sowerby, J.; Pardo, M.; Juss, J.; Goulding, D.; Van Der Weyden, L.; Storisteanu, D.; Prakash, A.; Espéli, M.; et al. Eros is a novel transmembrane protein that controls the phagocyte respiratory burst and is essential for innate immunity. J Exp Med. 2017;214(4):1111-28. [CrossRef]
- Mortimer, P.M.; Nichols, E.; Thomas, J.; Shanbhag, R.; Singh, N.; Coomber, E.L.; Malik, T.H.; Pickering, M.C.; Randzavola, L.; Rae, W.; et al. A novel mutation in EROS (CYBC1) causes chronic granulomatous disease. Clin. Immunol. 2023, 255, 109761. [CrossRef]
- Chiriaco, M.; De Matteis, A.; Cifaldi, C.; Di Matteo, G.; Rivalta, B.; Passarelli, C.; Perrone, C.; Novelli, A.; De Benedetti, F.; Insalaco, A.; et al. Characterization of AR-CGD female patient with a novel homozygous deletion in CYBC1 gene presenting with unusual clinical phenotype. Clin. Immunol. 2023, 251, 109316. [CrossRef]
- Nunoi, H.; Rotrosen, D.; Gallin, J.I.; Malech, H.L. Two Forms of Autosomal Chronic Granulomatous Disease Lack Distinct Neutrophil Cytosol Factors. Science 1988, 242, 1298–1301. [CrossRef]
- Curnutte, J.; Kuver, R.; Scott, P. Activation of neutrophil NADPH oxidase in a cell-free system. Partial purification of components and characterization of the activation process.. J. Biol. Chem. 1987, 262, 5563–5569. [CrossRef]
- Umei, T.; Takeshige, K.; Minakami, S. NADPH-binding component of the superoxide-generating oxidase in unstimulated neutrophils and the neutrophils from the patients with chronic granulomatous disease. Biochem. J. 1987, 243, 467–472. [CrossRef]
- Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114(15):3309-15. [CrossRef]
- van de Geer, A.; Nieto-Patlán, A.; Kuhns, D.B.; Tool, A.T.; Arias, A.A.; Bouaziz, M.; de Boer, M.; Franco, J.L.; Gazendam, R.P.; van Hamme, J.L.; et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J. Clin. Investig. 2018, 128, 3957–3975. [CrossRef]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH Oxidase and Associated Ion Fluxes During Phagocytosis. Traffic 2013, 14, 1118–1131. [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48, e12951. [CrossRef]
- Bréchard, S.; Plançon, S.; Tschirhart, E.J. New Insights into the Regulation of Neutrophil NADPH Oxidase Activity in the Phagosome: A Focus on the Role of Lipid and Ca2+Signaling. Antioxidants Redox Signal. 2013, 18, 661–676. [CrossRef]
- A Williams, D.; Tao, W.; Yang, F.; Kim, C.; Gu, Y.; Mansfield, P.; E Levine, J.; Petryniak, B.; Derrow, C.W.; Harris, C.; et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96(5):1646-54.
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; Deboer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [CrossRef]
- Alkhairy, O.K.; Rezaei, N.; Graham, R.R.; Abolhassani, H.; Borte, S.; Hultenby, K.; Wu, C.; Aghamohammadi, A.; Williams, D.A.; Behrens, T.W.; et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 135, 1380–1384.e5. [CrossRef]
- van Bruggen R, Bautista JM, Petropoulou T, de Boer M, van Zwieten R, Gómez-Gallego F, et al. Deletion of leucine 61 in glucose-6-phosphate dehydrogenase leads to chronic nonspherocytic anemia, granulocyte dysfunction, and increased susceptibility to infections. Blood. 2002;100(3):1026-30. [CrossRef]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16–16. [CrossRef]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.M.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [CrossRef]
- Trevelin, S.C.; Shah, A.M.; Lombardi, G. Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol. Lett. 2020, 221, 39–48. [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [CrossRef]
- Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6. [CrossRef]
- Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619-22. [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [CrossRef]
- Roxo-Junior P, Simao HM. Chronic granulomatous disease: why an inflammatory disease? Braz J Med Biol Res. 2014;47(11):924-8. [CrossRef]
- Hartl, D.; Lehmann, N.; Hoffmann, F.; Jansson, A.; Hector, A.; Notheis, G.; Roos, D.; Belohradsky, B.H.; Wintergerst, U. Dysregulation of innate immune receptors on neutrophils in chronic granulomatous disease. J. Allergy Clin. Immunol. 2008, 121, 375–382.e9. [CrossRef]
- Garay, J.A.; Silva, J.E.; Di Genaro, M.S.; Davicino, R.C. The Multiple Faces of Nitric Oxide in Chronic Granulomatous Disease: A Comprehensive Update. Biomedicines 2022, 10, 2570. [CrossRef]
- Segal, B.H.; Leto, T.L.; Gallin, J.I.; Malech, H.L.; Holland, S.M. Genetic, Biochemical, and Clinical Features of Chronic Granulomatous Disease. Medicine 2000, 79, 170–200. [CrossRef]
- Sacco, K.A.; Gazzin, A.; Notarangelo, L.D.; Delmonte, O.M. Granulomatous inflammation in inborn errors of immunity. Front. Pediatr. 2023, 11, 1110115. [CrossRef]
- A Connelly, J.; Marsh, R.; Parikh, S.; Talano, J.-A. Allogeneic Hematopoietic Cell Transplantation for Chronic Granulomatous Disease: Controversies and State of the Art. J. Pediatr. Infect. Dis. Soc. 2018, 7, S31–S39. [CrossRef]
- Xiao, N.; Huang, X.; Zang, W.; Kiselev, S.; Bolkov, M.A.; Tuzankina, I.A.; Chereshnev, V.A. Health-related quality of life in patients with inborn errors of immunity: a bibliometric analysis. Front. Immunol. 2024, 15, 1371124. [CrossRef]
- Nicholson, B.; Goodman, R.; Day, J.; Worth, A.; Carpenter, B.; Sandford, K.; Morris, E.C.; Burns, S.O.; Ridout, D.; Titman, P.; et al. Quality of Life and Social and Psychological Outcomes in Adulthood Following Allogeneic HSCT in Childhood for Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1451–1460. [CrossRef]
- Bai, G.; Herten, M.H.; Landgraf, J.M.; Korfage, I.J.; Raat, H. Childhood chronic conditions and health-related quality of life: Findings from a large population-based study. PLOS ONE 2017, 12, e0178539. [CrossRef]
- Cole, T.; McKendrick, F.; Titman, P.; Cant, A.J.; Pearce, M.S.; Cale, C.M.; Goldblatt, D.; Gennery, A.R. Health Related Quality of Life and Emotional Health in Children with Chronic Granulomatous Disease: A Comparison of Those Managed Conservatively with Those That Have Undergone Haematopoietic Stem Cell Transplant. J. Clin. Immunol. 2012, 33, 8–13. [CrossRef]
- Cole, T.S.; Jones, L.K.R.; McGrogan, P.; Pearce, M.S.; Flood, T.J.; Cant, A.J.; Goldblatt, D.; Thrasher, A.J.; Gennery, A.R.; McKendrick, F.; et al. Emotional and behavioural difficulties in chronic granulomatous disease. Arch. Dis. Child. 2011, 97, 87–87. [CrossRef]
- Battersby, A.C.; Braggins, H.; Pearce, M.S.; McKendrick, F.; Campbell, M.; Burns, S.; Cale, C.M.; Goldblatt, D.; Gennery, A.R. Health-Related Quality of Life and Emotional Health in X-Linked Carriers of Chronic Granulomatous Disease in the United Kingdom. J. Clin. Immunol. 2019, 39, 195–199. [CrossRef]
- Pulvirenti, F.; Sangerardi, M.; Plebani, A.; Soresina, A.; Finocchi, A.; Pignata, C.; Cirillo, E.; Trizzino, A.; Aiuti, A.; Migliavacca, M.; et al. Health-Related Quality of Life and Emotional Difficulties in Chronic Granulomatous Disease: Data on Adult and Pediatric Patients from Italian Network for Primary Immunodeficiency (IPINet). J. Clin. Immunol. 2019, 40, 289–298. [CrossRef]
- Ghanim, H.Y.; Porteus, M.H. Gene regulation in inborn errors of immunity: Implications for gene therapy design and efficacy. Immunol. Rev. 2024, 322, 157–177. [CrossRef]
- Fischer, A. Gene therapy for inborn errors of immunity: past, present and future. Nat. Rev. Immunol. 2022, 23, 397–408. [CrossRef]
- Gennery, A.R. Progress in treating chronic granulomatous disease. Br. J. Haematol. 2020, 192, 251–264. [CrossRef]
- Overwater, E.; Smulders, Y.; van der Burg, M.; Lombardi, M.P.; Meijers-Heijboer, H.E.; Kuijpers, T.W.; Houweling, A.C. The value of DNA storage and pedigree analysis in rare diseases: a 17-year-old boy with X-linked lymphoproliferative disease (XLP) caused by a de novo SH2D1A mutation. Eur. J. Pediatr. 2014, 173, 1695–1698. [CrossRef]
- Chen, X.; Peng, C.; Chen, H.; Zhou, F.; Keqie, Y.B.; Li, Y.B.; Liu, S.; Ren, J. Preimplantation genetic testing for X-linked chronic granulomatous disease induced by a CYBB gene variant: A case report. Medicine 2024, 103, e37198. [CrossRef]
- Modarresi, S.Z.; Sabetkish, N.; Badalzadeh, M.; Tajik, S.; Esmaeili, B.; Fazlollahi, M.R.; Houshmand, M.; Gharehdaghi, J.; Niroomanesh, S.; Sherbaf, F.R.; et al. The Critical Role of Prenatal Genetic Study in Prevention of Primary Immunodeficiency in High-risk Families: The Largest Report of 107 Cases. Iran. J. Allergy, Asthma Immunol. 2020, 19, 478–483. [CrossRef]
- Kulkarni, M.; Gupta, M.; Madkaikar, M. Phenotypic Prenatal Diagnosis of Chronic Granulomatous Disease: A Useful Tool in The Absence Of Molecular Diagnosis. Scand. J. Immunol. 2017, 86, 486–490. [CrossRef]
- Yu JE, Azar AE, Chong HJ, Jongco AM, 3rd, Prince BT. Considerations in the Diagnosis of Chronic Granulomatous Disease. J Pediatric Infect Dis Soc. 2018;7(suppl_1):S6-S11.
- Roos, D.; Boer, M. Molecular diagnosis of chronic granulomatous disease. Clin. Exp. Immunol. 2014, 175, 139–149. [CrossRef]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; van Montfrans, J.; Scheible, R.; Rusch, S.; Gasteiger, L.M.; Grimbacher, B.; et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pr. 2019, 7, 1763–1770. [CrossRef]
- Rosen, Y. Pathology of Granulomatous Pulmonary Diseases. Arch. Pathol. Lab. Med. 2021, 146, 233–251. [CrossRef]
- Moskaluk CA, Pogrebniak HW, Pass HI, Gallin JI, Travis WD. Surgical pathology of the lung in chronic granulomatous disease American Journal of Clinical Pathology. 1994;102(5):684–91.
- Marks, D.J.B.; Miyagi, K.; Rahman, F.Z.; Novelli, M.; Bloom, S.L.; Segal, A.W. Inflammatory Bowel Disease in CGD Reproduces the Clinicopathological Features of Crohn’s Disease. Am. J. Gastroenterol. 2008, 104, 117–124. [CrossRef]
- Brown, I.; Kumarasinghe, M.P. Granulomas in the gastrointestinal tract: deciphering the Pandora’s box. Virchows Arch. 2017, 472, 3–14. [CrossRef]
- Khangura, S.K.; Kamal, N.; Ho, N.; Quezado, M.; Zhao, X.; Marciano, B.; Simpson, J.; Zerbe, C.; Uzel, G.; Yao, M.D.; et al. Gastrointestinal Features of Chronic Granulomatous Disease Found During Endoscopy. Clin. Gastroenterol. Hepatol. 2015, 14, 395–402.e5. [CrossRef]
- Marciano, B.E.; Rosenzweig, S.D.; Kleiner, D.E.; Anderson, V.L.; Darnell, D.N.; Anaya-O’Brien, S.; Hilligoss, D.M.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Gastrointestinal Involvement in Chronic Granulomatous Disease. Pediatrics 2004, 114, 462–468. [CrossRef]
- Alimchandani M, Lai JP, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal Histopathology in Chronic Granulomatous Disease: A Study of 87 Patients. The American Journal of Surgical Pathology. 2013;37(9):1365-72.
- Levine S, Smith VV, Malone M, Sebire NJ. Histopathological features of chronic granulomatous disease (CGD) in childhood. Histopathology. 2005;47(5):508-16.
- Grenier, P.A.; Brun, A.L.; Longchampt, E.; Lipski, M.; Mellot, F.; Catherinot, E. Primary immunodeficiency diseases of adults: a review of pulmonary complication imaging findings. Eur. Radiol. 2023, 34, 4142–4154. [CrossRef]
- Salvator, H.; Mahlaoui, N.; Catherinot, E.; Rivaud, E.; Pilmis, B.; Borie, R.; Crestani, B.; Tcherakian, C.; Suarez, F.; Dunogue, B.; et al. Pulmonary manifestations in adult patients with chronic granulomatous disease. Eur. Respir. J. 2015, 45, 1613–1623. [CrossRef]
- Bhattacharya, S.; Koethe, Y.; Ling, A.; Kamal, N.; Khangura, S.; Alimchandani, M.; Quezado, M.M.; Zerbe, C.S.; Malech, H.L.; Gallin, J.I.; et al. Gastrointestinal Computed Tomography Findings in Chronic Granulomatous Disease with Subgroup Clinicopathologic Analysis. Dig. Dis. Sci. 2021, 67, 1831–1842. [CrossRef]
- Lee, M.; Lee, M.S.; Lee, J.S.; Ko, S.Y.; Jeong, S.Y. Spectrum of imaging findings of chronic granulomatous disease: a single center experience. Diagn. Interv. Radiol. 2017, 23, 472–477. [CrossRef]
- Towbin AJ, Chaves I. Chronic granulomatous disease. Pediatr Radiol. 2010;40(5):657-68; quiz 792-3.
- Jefferson, L.; Ramanan, A.V.; Jolles, S.; Bernatoniene, J.; Mathieu, A.-L.; Belot, A.; Roderick, M.R. Phenotypic Variability in PRKCD: a Review of the Literature. J. Clin. Immunol. 2023, 43, 1692–1705. [CrossRef]
- Neehus AL, Tuano K, Le Voyer T, Nandiwada SL, Murthy K, Puel A, et al. Chronic Granulomatous Disease-Like Presentation of a Child with Autosomal Recessive PKCdelta Deficiency. J Clin Immunol. 2022;42(6):1244-53.
- Hafsi W, Yarrarapu SNS. Job Syndrome Treasure Island (FL): StatPearls Publishing; 2023 [.
- Kuhns DB. Diagnostic Testing for Chronic Granulomatous Disease. In: Knaus UG, Leto TL, editors. Methods Mol Biol. 1982. New York: Humana Press; 2019. p. 543-71.
- Roos D. Chronic Granulomatous Disease. In: Knaus UG, Leto TL, editors. NADPH Oxidases: Methods and Protocols. 1982: Humana Press; 2019. p. 531-42.
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 1–32. [CrossRef]
- Roesler, J. Remarks on the article Genetics and immunopathology of chronic granulomatous disease by Marie José Stasia and Xing Jun Li. Semin. Immunopathol. 2008, 30, 365–365. [CrossRef]
- Quach, A.; Glowik, S.; Putty, T.; Ferrante, A. Delayed Blood Processing Leads to Rapid Deterioration in the Measurement of the Neutrophil Respiratory Burst by the Dihydrorhodamine-123 Reduction Assay. Cytom. Part B: Clin. Cytom. 2019, 96, 389–396. [CrossRef]
- Weening, R.S.; Roos, D.; A Loos, J. Oxygen consumption of phagocytizing cells in human leukocyte and granulocyte preparations: a comparative study. J Lab Clin Med. 1974, 83, 570–7.
- Elloumi HZ, Holland SM. Diagnostic assays for chronic granulomatous disease and other neutrophil disorders. Methods Mol Biol. 2007;412:505-23.
- Ríos N, Prolo C, Álvarez MN, Piacenza L, Radi R. Peroxynitrite Formation and Detection in Living Cells. Nitric Oxide2017. p. 271-88.
- Dahlgren C, Karlsson A, Bylund J. Measurement of respiratory burst products generated by professional phagocytes. Methods Mol Biol. 2007;412:349-63.
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A Stable Nonfluorescent Derivative of Resorufin for the Fluorometric Determination of Trace Hydrogen Peroxide: Applications in Detecting the Activity of Phagocyte NADPH Oxidase and Other Oxidases. Anal. Biochem. 1997, 253, 162–168. [CrossRef]
- Baehner, R.L.; Nathan, D.G. Quantitative Nitroblue Tetrazolium Test in Chronic Granulomatous Disease. New Engl. J. Med. 1968, 278, 971–976. [CrossRef]
- Ochs, H.D.; Igo, R.P. The NBT slide test: A simple screening method for detecting chronic granulomatous disease and female carriers. J. Pediatr. 1973, 83, 77–82. [CrossRef]
- Roesler J, Emmendorffer A. Diagnosis of chronic granulomatous disease [letter; comment]. Blood. 1991;78(5):1387-9.
- Israeli, S.; Golden, A.; Atalig, M.; Mekki, N.; Rais, A.; Storey, H.; Barbouche, M.-R.; Peck, R. A Novel Point-of-Care Rapid Diagnostic Test for Screening Individuals for Antibody Deficiencies. J. Clin. Immunol. 2021, 42, 394–403. [CrossRef]
- Alemayehu, T.; Deribessa, S.J. A First Case Report of DiGeorge Syndrome from Ethiopia Highlights Challenges in Identifying and Treating Children with Primary T-Cell Deficiencies in Low Resource Settings. Case Rep. Immunol. 2020, 2020, 1–3. [CrossRef]
- Kosack, C.S. Experience of Médecins Sans Frontières in laboratory medicine in resource-limited settings. Clin Chem Lab Med. 2012, 50, 1221–1227. [CrossRef]
- Emmendörffer, A.; Hecht, M.; Lohmann-Matthes, M.-L.; Roesler, J. A fast and easy method to determine the production of reactive oxygen intermediates by human and murine phagocytes using dihydrorhodamine 123. J. Immunol. Methods 1990, 131, 269–275. [CrossRef]
- Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):89-99, viii.
- Jirapongsananuruk, O.; Malech, H.L.; Kuhns, D.B.; Niemela, J.E.; Brown, M.R.; Anderson-Cohen, M.; Fleisher, T.A. Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. J. Allergy Clin. Immunol. 2003, 111, 374–379. [CrossRef]
- Kim, H.-Y.; Kim, H.-J.; Ki, C.-S.; Kim, D.W.; Yoo, K.H.; Kang, E.-S. Rapid Determination of Chimerism Status Using Dihydrorhodamine Assay in a Patient with X-linked Chronic Granulomatous Disease Following Hematopoietic Stem Cell Transplantation. Ann. Lab. Med. 2013, 33, 288–292. [CrossRef]
- Rösen-Wolff, A.; Soldan, W.; Heyne, K.; Bickhardt, J.; Gahr, M.; Roesler, J. Increased susceptibility of a carrier of X-linked chronic granulomatous disease (CGD) to Aspergillus fumigatus infection associated with age-related skewing of lyonization. Ann. Hematol. 2001, 80, 113–115. [CrossRef]
- Mauch, L.; Lun, A.; O’gorman, M.R.; Harris, J.S.; Schulze, I.; Zychlinsky, A.; Fuchs, T.; Oelschlägel, U.; Brenner, S.; Kutter, D.; et al. Chronic Granulomatous Disease (CGD) and Complete Myeloperoxidase Deficiency Both Yield Strongly Reduced Dihydrorhodamine 123 Test Signals but Can Be Easily Discerned in Routine Testing for CGD. Clin. Chem. 2007, 53, 890–896. [CrossRef]
- Milligan KL, Mann D, Rump A, Anderson VL, Hsu AP, Kuhns DB, et al. Complete Myeloperoxidase Deficiency: Beware the “False-Positive” Dihydrorhodamine Oxidation. J Pediatr. 2016;176:204-6. [CrossRef]
- Ferguson, P.J.; Lokuta, M.A.; El-Shanti, H.I.; Muhle, L.; Bing, X.; Huttenlocher, A. Neutrophil dysfunction in a family with a SAPHO syndrome–like phenotype. Arthritis Rheum. 2008, 58, 3264–3269. [CrossRef]
- Mott, J.; Rikihisa, Y. Human Granulocytic Ehrlichiosis Agent Inhibits Superoxide Anion Generation by Human Neutrophils. Infect. Immun. 2000, 68, 6697–6703. [CrossRef]
- Banerjee R, Anguita J, Roos D, Fikrig E. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J Immunol. 2000;164(8):3946-9.
- Almutairi, A.; Zaman, F.; Day-Lewis, M.; Tsitsikov, E.; Reiter, A.; Xue, K.; Geha, R.S.; Chou, J.; Yee, C.S. Acetaminophen Inhibits the Neutrophil Oxidative Burst: Implications for Diagnostic Testing. J. Allergy Clin. Immunol. Pr. 2020, 8, 3543–3548. [CrossRef]
- Suematsu, M.; Suzuki, M.; Miura, S.; Nagata, H.; Oshio, C.; Asakura, H.; Watanabe, M.; Tsuchiya, M. Sulfasalazine and its metabolites attenuate respiratory burst of leukocytes--a possible mechanism of anti-inflammatory effects. J Clin Lab Immunol. 1987;23(1):31-3.
- Costa, D.; Marques, A.P.; Reis, R.L.; Lima, J.L.; Fernandes, E. Inhibition of human neutrophil oxidative burst by pyrazolone derivatives. Free. Radic. Biol. Med. 2005, 40, 632–640. [CrossRef]
- Cross, A.R.; Curnutte, J.T. The Cytosolic Activating Factors p47phox and p67phox Have Distinct Roles in the Regulation of Electron Flow in NADPH Oxidase. J Biol Chem. 1995, 270, 6543–6548. [CrossRef]
- Accetta, D.; Syverson, G.; Bonacci, B.; Reddy, S.; Bengtson, C.; Surfus, J.; Harbeck, R.; Huttenlocher, A.; Grossman, W.; Routes, J. Human phagocyte defect caused by a Rac2 mutation detected by means of neonatal screening for T-cell lymphopenia. J. Allergy Clin. Immunol. 2011, 127, 535–538.e2. [CrossRef]
- Vowells, S.; Sekhsaria, S.; Malech, H.; Shalit, M.; Fleisher, T. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J. Immunol. Methods 1995, 178, 89–97. [CrossRef]
- Yamauchi A, Yu L, Potgens AJ, Kuribayashi F, Nunoi H, Kanegasaki S, et al. Location of the epitope for 7D5, a monoclonal antibody raised against human flavocytochrome b558, to the extracellular peptide portion of primate gp91phox. Microbiol Immunol. 2001;45(3):249-57. [CrossRef]
- Köker, M.Y.; Sanal, .; Van Leeuwen, K.; De Boer, M.; Metin, A.; Patıroğlu, T.; Özgür, T.T.; Tezcan, I.; Roos, D. Four different NCF2 mutations in six families from Turkey and an overview of NCF2 gene mutations. Eur. J. Clin. Investig. 2009, 39, 942–951. [CrossRef]
- Nakamura M, Murakami M, Koga T, Tanaka Y, Minakami S. Monoclonal antibody 7D5 raised to cytochrome b558 of human neutrophils: immunocytochemical detection of the antigen in peripheral phagocytes of normal subjects, patients with chronic granulomatous disease, and their carrier mothers. Blood. 1987;69(5):1404-8. [CrossRef]
- Wada, T.; Muraoka, M.; Toma, T.; Imai, T.; Shigemura, T.; Agematsu, K.; Haraguchi, K.; Moriuchi, H.; Oh-Ishi, T.; Kitoh, T.; et al. Rapid Detection of Intracellular p47phox and p67phox by Flow Cytometry; Useful Screening Tests for Chronic Granulomatous Disease. J. Clin. Immunol. 2013, 33, 857–864. [CrossRef]
- Sekar, A.; Gupta, K.; Rawat, A.; Jindal, A.; Pandiarajan, V.; Suri, D.; Gupta, A.; Kaur, G.; Kumar, I.; Gummadi, A.; et al. Utility of Immunohistochemistry and Immunofluorescence in Determining the Pathogenic Variants of Chronic Granulomatous Disease. J. Clin. Immunol. 2021, 42, 85–93. [CrossRef]
- Blom, M.; Bredius, R.G.M.; van der Burg, M. Future Perspectives of Newborn Screening for Inborn Errors of Immunity. Int. J. Neonatal Screen. 2021, 7, 74. [CrossRef]
- Collins, C.J.; Yi, F.; Dayuha, R.; Whiteaker, J.R.; Ochs, H.D.; Freeman, A.; Su, H.C.; Paulovich, A.G.; Segundo, G.R.S.; Torgerson, T.; et al. Multiplexed Proteomic Analysis for Diagnosis and Screening of Five Primary Immunodeficiency Disorders From Dried Blood Spots. Front. Immunol. 2020, 11, 464. [CrossRef]
- Rawat A, Bhattad S, Singh S. Chronic Granulomatous Disease. Indian J Pediatr. 2016;83(4):345-53.
- Tsunawaki, S.; Mizunari, H.; Nagata, M.; Tatsuzawa, O.; Kuratsuji, T. A Novel Cytosolic Component, p40phox, of Respiratory Burst Oxidase Associates with p67phox and Is Absent in Patients with Chronic Granulomatous Disease Who Lack p67phox. Biochem. Biophys. Res. Commun. 1994, 199, 1378–1387. [CrossRef]
- Anjani, G.; Vignesh, P.; Joshi, V.; Shandilya, J.K.; Bhattarai, D.; Sharma, J.; Rawat, A. Recent advances in chronic granulomatous disease. Genes Dis. 2020, 7, 84–92. [CrossRef]
- Molshanski-Mor S, Mizrahi A, Ugolev Y, Dahan I, Berdichevsky Y, Pick E. Cell-free assays: the reductionist approach to the study of NADPH oxidase assembly, or “all you wanted to know about cell-free assays but did not dare to ask”. In: Quinn MT, DeLeo FR, Bokoch GM, editors. Neutrophil Methods and Protocols. 412. New Jersey: Humana Press; 2007. p. 385-428.
- Chanock, S.J.; Faust, L.R.; Barrett, D.; Bizal, C.; Maly, F.E.; Newburger, P.E.; Ruedi, J.M.; Smith, R.M.; Babior, B.M. O2- production by B lymphocytes lacking the respiratory burst oxidase subunit p47phox after transfection with an expression vector containing a p47phox cDNA. Proc. Natl. Acad. Sci. USA 1992, 89, 10174–10177. [CrossRef]
- Batlle-Masó, L.; Rivière, J.G.; Franco-Jarava, C.; Martín-Nalda, A.; Garcia-Prat, M.; Parra-Martínez, A.; Aguiló-Cucurull, A.; Castells, N.; Martinez-Gallo, M.; Soler-Palacín, P.; et al. Molecular Challenges in the Diagnosis of X-Linked Chronic Granulomatous Disease: CNVs, Intronic Variants, Skewed X-Chromosome Inactivation, and Gonosomal Mosaicism. J. Clin. Immunol. 2023, 43, 1953–1963. [CrossRef]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. [CrossRef]
- Wallace SE, Bean LJH. Educational Materials — Genetic Testing: Current Approaches. 2020. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle.
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [CrossRef]
- Kelly P, Mahon S, Friend P. When Germline Genetic Testing Results Are Unclear: Highlighting Variants of Uncertain Significance. J Adv Pract Oncol. 2023;14(7):631-8. [CrossRef]
- East K, Chung W, Foreman K, Gilmore M, Gornick M, Hindorff L, et al. Guide to interpreting genomic reports: A genomics toolkit. In: Research CSE, editor. 2017. p. 2020-04.
- Gahl, W.A.; Markello, T.C.; Toro, C.; Fajardo, K.F.; Sincan, M.; Gill, F.; Carlson-Donohoe, H.; Gropman, A.; Pierson, T.M.; Golas, G.; et al. The National Institutes of Health Undiagnosed Diseases Program: Insights into rare diseases. Genet Med. 2012;14(1):51-9. [CrossRef]
- Lazaridis KN, Schahl KA, Cousin MA, Babovic-Vuksanovic D, Riegert-Johnson DL, Gavrilova RH, et al. Outcome of Whole Exome Sequencing for Diagnostic Odyssey Cases of an Individualized Medicine Clinic: The Mayo Clinic Experience. Mayo Clin Proc. 2016;91(3):297-307. [CrossRef]
- Yska, H.A.F.; Elsink, K.; Kuijpers, T.W.; Frederix, G.W.J.; van Gijn, M.E.; van Montfrans, J.M. Diagnostic Yield of Next Generation Sequencing in Genetically Undiagnosed Patients with Primary Immunodeficiencies: a Systematic Review. J. Clin. Immunol. 2019, 39, 577–591. [CrossRef]
- Taylor, J.C.; Martin, H.C.; Lise, S.; Broxholme, J.; Cazier, J.-B.; Rimmer, A.; Kanapin, A.; Lunter, G.; Fiddy, S.; Allan, C.; et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 2015, 47, 717–726. [CrossRef]
- Manning M, Hudgins L, Professional P, Guidelines C. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):742-5. [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [CrossRef]
- de Boer M, van Leeuwen K, Geissler J, Weemaes CM, van den Berg TK, Kuijpers TW, et al. Primary immunodeficiency caused by an exonized retroposed gene copy inserted in the CYBB gene. Hum Mutat. 2014;35(4):486-96. [CrossRef]
- Bustamante, J.; A Arias, A.; Vogt, G.; Picard, C.; Galicia, L.B.; Prando, C.; Grant, A.V.; Marchal, C.C.; Hubeau, M.; Chapgier, A.; et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat. Immunol. 2011, 12, 213–221. [CrossRef]
- Bustamante, J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum. Genet. 2020, 139, 993–1000. [CrossRef]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992, 51, 1229–39.
- Sauteraud, R.; Stahl, J.M.; James, J.; Englebright, M.; Chen, F.; Zhan, X.; Carrel, L.; Liu, D.J. Inferring genes that escape X-Chromosome inactivation reveals important contribution of variable escape genes to sex-biased diseases. Genome Res. 2021, 31, 1629–1637. [CrossRef]
- Peng, J.; Redman, C.M.; Wu, X.; Song, X.; Walker, R.H.; Westhoff, C.M.; Lee, S. Insights into extensive deletions around the XK locus associated with McLeod phenotype and characterization of two novel cases. Gene 2007, 392, 142–150. [CrossRef]
- E Watkins, C.; Litchfield, J.; Song, E.; Jaishankar, G.B.; Misra, N.; Holla, N.; Duffourc, M.; Krishnaswamy, G. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clin. Mol. Allergy 2011, 9, 1–6. [CrossRef]
- Görlach, A.; Lee, P.L.; Roesler, J.; Hopkins, P.J.; Christensen, B.; Green, E.D.; Chanock, S.J.; Curnutte, J.T. A p47-phox pseudogene carries the most common mutation causing p47-phox- deficient chronic granulomatous disease.. J. Clin. Investig. 1997, 100, 1907–1918. [CrossRef]
- Tarazona-Santos, E.; Machado, M.; Magalhães, W.C.; Chen, R.; Lyon, F.; Burdett, L.; Crenshaw, A.; Fabbri, C.; Pereira, L.; Pinto, L.; et al. Evolutionary Dynamics of the Human NADPH Oxidase Genes CYBB, CYBA, NCF2, and NCF4: Functional Implications. Mol. Biol. Evol. 2013, 30, 2157–2167. [CrossRef]
- Hayrapetyan, A.; Dencher, P.C.; van Leeuwen, K.; de Boer, M.; Roos, D. Different unequal cross-over events between NCF1 and its pseudogenes in autosomal p47phox-deficient chronic granulomatous disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2013, 1832, 1662–1672. [CrossRef]
- Dekker, J.; de Boer, M.; Roos, D. Gene-scan method for the recognition of carriers and patients with p47phox-deficient autosomal recessive chronic granulomatous disease. Exp. Hematol. 2001, 29, 1319–1325. [CrossRef]
- Roos D, de Boer M, Koker MY, Dekker J, Singh-Gupta V, Ahlin A, et al. Chronic granulomatous disease caused by mutations other than the common GT deletion in NCF1, the gene encoding the p47phox component of the phagocyte NADPH oxidase. Hum Mutat. 2006;27(12):1218-29. [CrossRef]
- Kuhns, D.B.; Hsu, A.P.; Sun, D.; Lau, K.; Fink, D.; Griffith, P.; Huang, D.W.; Priel, D.A.L.; Mendez, L.; Kreuzburg, S.; et al. NCF1 (p47phox)–deficient chronic granulomatous disease: comprehensive genetic and flow cytometric analysis. Blood Adv. 2019, 3, 136–147. [CrossRef]
- Zhao, J.; Ma, J.; Deng, Y.; A Kelly, J.; Kim, K.; Bang, S.-Y.; Lee, H.-S.; Li, Q.-Z.; Wakeland, E.K.; Qiu, R.; et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 2017, 49, 433–437. [CrossRef]
- Leusen, J.H.; Bolscher, B.G.; Hilarius, P.M.; Weening, R.S.; Kaulfersch, W.; A Seger, R.; Roos, D.; Verhoeven, A.J. 156Pro-->Gln substitution in the light chain of cytochrome b558 of the human NADPH oxidase (p22-phox) leads to defective translocation of the cytosolic proteins p47-phox and p67-phox.. J. Exp. Med. 1994, 180, 2329–2334. [CrossRef]
- Roos, D.; van Buul, J.D.; Tool, A.T.; Matute, J.D.; Marchal, C.M.; Hayee, B.; Köker, M.Y.; de Boer, M.; van Leeuwen, K.; Segal, A.W.; et al. Two CGD Families with a Hypomorphic Mutation in the Activation Domain of p67phox. J Clin Cell Immunol. 2014;5(3):1000231.
- Lavelle TA, Feng X, Keisler M, Cohen JT, Neumann PJ, Prichard D, et al. Cost-effectiveness of exome and genome sequencing for children with rare and undiagnosed conditions. Genet Med. 2022;24(6):1349-61. [CrossRef]
- Owolabi, M.O.; Suwanwela, N.C.; Yaria, J. Barriers to implementation of evidence into clinical practice in low-resource settings. Nat. Rev. Neurol. 2022, 18, 451–452. [CrossRef]
- Barbouche, M.; Galal, N.; Ben-Mustapha, I.; Jeddane, L.; Mellouli, F.; Ailal, F.; Bejaoui, M.; Boutros, J.; Marsafy, A.; Bousfiha, A.A. Primary immunodeficiencies in highly consanguineous North African populations. Ann. New York Acad. Sci. 2011, 1238, 42–52. [CrossRef]
- North Thames Genomic Medicine Service. Genetic test turnaround times. 2023 [Available from: https://norththamesgenomics.nhs.uk/genetic-test-ordering/turn-around-times/] Last accessed 2024-06-25.
- Horton, R.; Lucassen, A. Ethical issues raised by new genomic technologies: the case study of newborn genome screening. Camb. Prism. Precis. Med. 2022, 1, 1–16. [CrossRef]
- Genomics England. Newborn Genomes Programme: Vision Document. 2021 [Available at https://files.genomicsengland.co.uk/documents/Newborns-Vision-Final_SEP_2021-11-02-122418_jjne.pdf] Last accessed 2024-06-25.
- Genomics England. Newborn Genomes Programme Generation Study: List of conditions and genes. 2024 [Available from: https://www.genomicsengland.co.uk/initiatives/newborns/choosing-conditions/conditions-list-generation-study] Last accessed 2024-06-25.
Figure 1.
Factors required for expression, activation and function of NADPH oxidase. CYBC1/EROS cytochrome b558 chaperone 1, G6PD glucose 6-phosphate dehydrogenase, GDP guanosine diphosphate, gp91phox/p22phox/p67phox/p40phox/p47phox NADPH oxidase core enzyme components, GTP guanosine triphosphate, H+ hydrogen ion, H2O2 hydrogen peroxide, HOCl hypochlorous acid, K+ potassium ion, MPO myeloperoxidase, NADPH/NADP nicotinamide adenine dinucleotide diphosphate, O2 molecular oxygen, ·O2 superoxide radical, ·OH hydroxyl radical, RAC2 Rac family small GTPase 2, SOD superoxide dismutase. Figure made with Biorender®.
Figure 1.
Factors required for expression, activation and function of NADPH oxidase. CYBC1/EROS cytochrome b558 chaperone 1, G6PD glucose 6-phosphate dehydrogenase, GDP guanosine diphosphate, gp91phox/p22phox/p67phox/p40phox/p47phox NADPH oxidase core enzyme components, GTP guanosine triphosphate, H+ hydrogen ion, H2O2 hydrogen peroxide, HOCl hypochlorous acid, K+ potassium ion, MPO myeloperoxidase, NADPH/NADP nicotinamide adenine dinucleotide diphosphate, O2 molecular oxygen, ·O2 superoxide radical, ·OH hydroxyl radical, RAC2 Rac family small GTPase 2, SOD superoxide dismutase. Figure made with Biorender®.
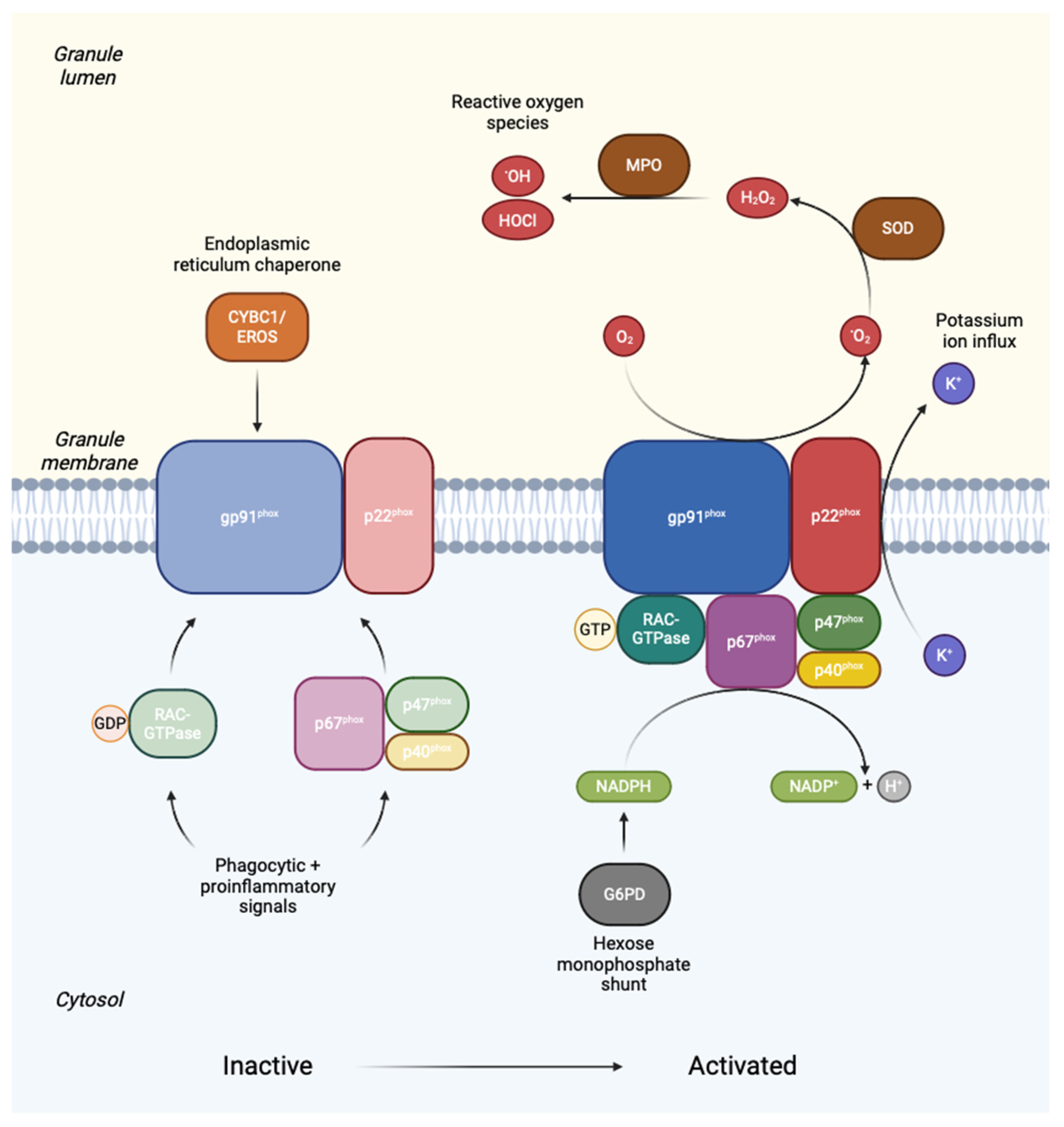
Figure 2.
Benefits of early diagnosis in chronic granulomatous disease. HSCT haematopoietic stem cell transplant. Figure made with Biorender®.
Figure 2.
Benefits of early diagnosis in chronic granulomatous disease. HSCT haematopoietic stem cell transplant. Figure made with Biorender®.
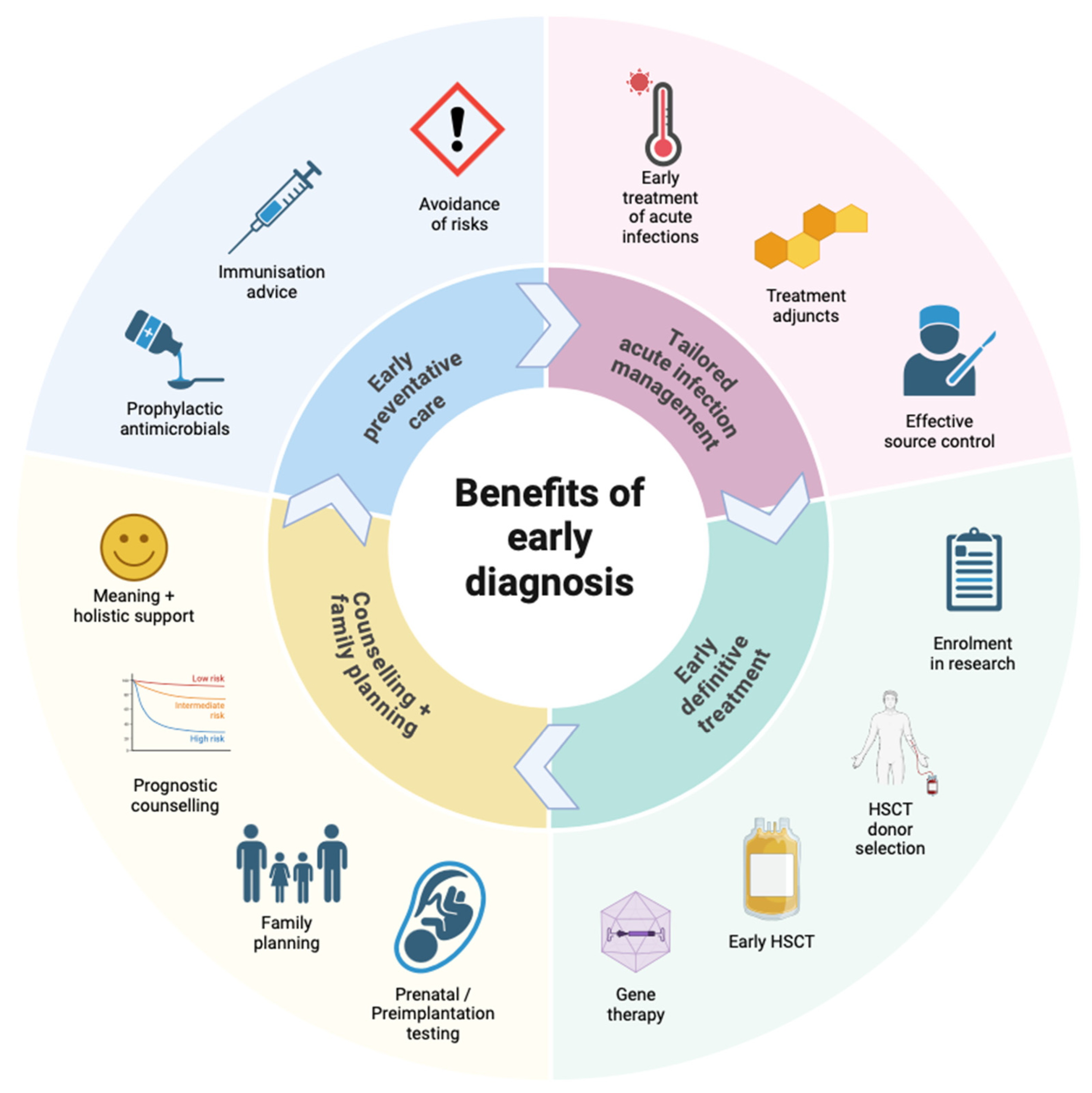
Figure 3.
Nitroblue tetrazolium (NBT) test. Figure made with Biorender®.
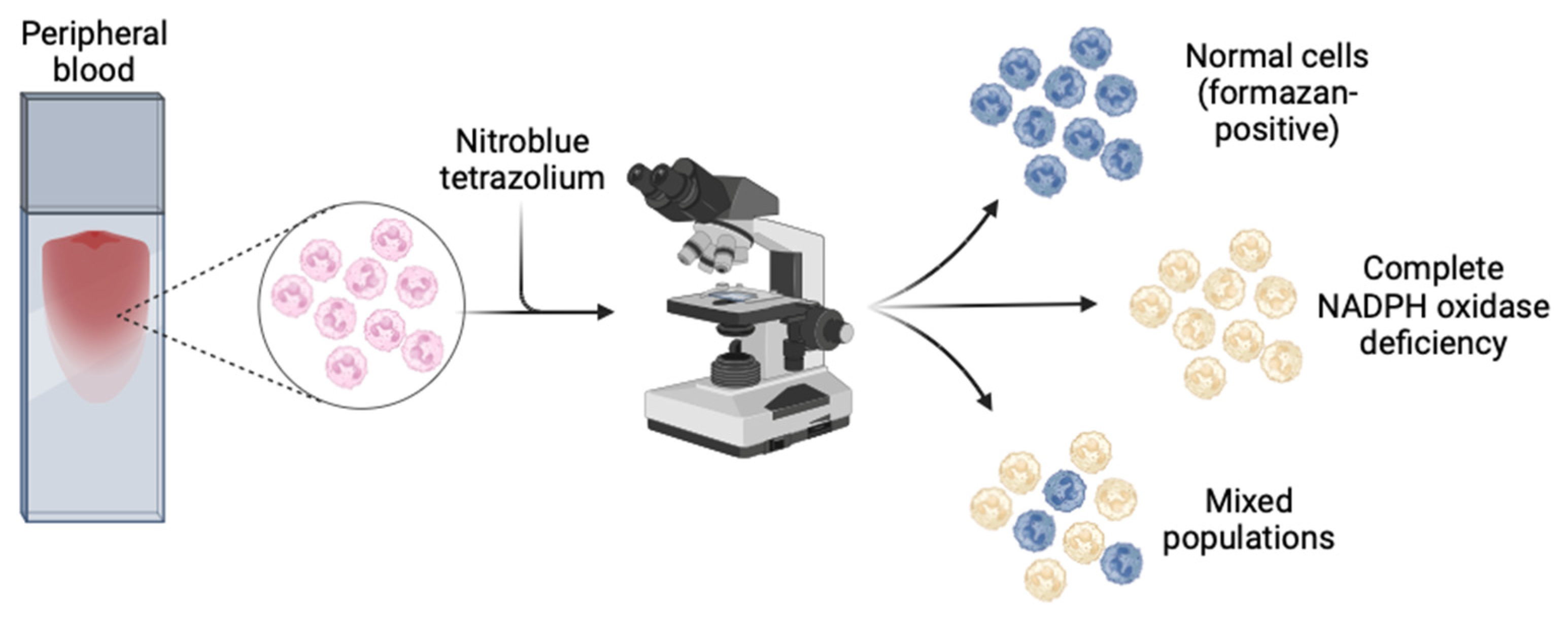
Figure 4.
Dihydrorhodamine (DHR) test. Figure made with Biorender®.
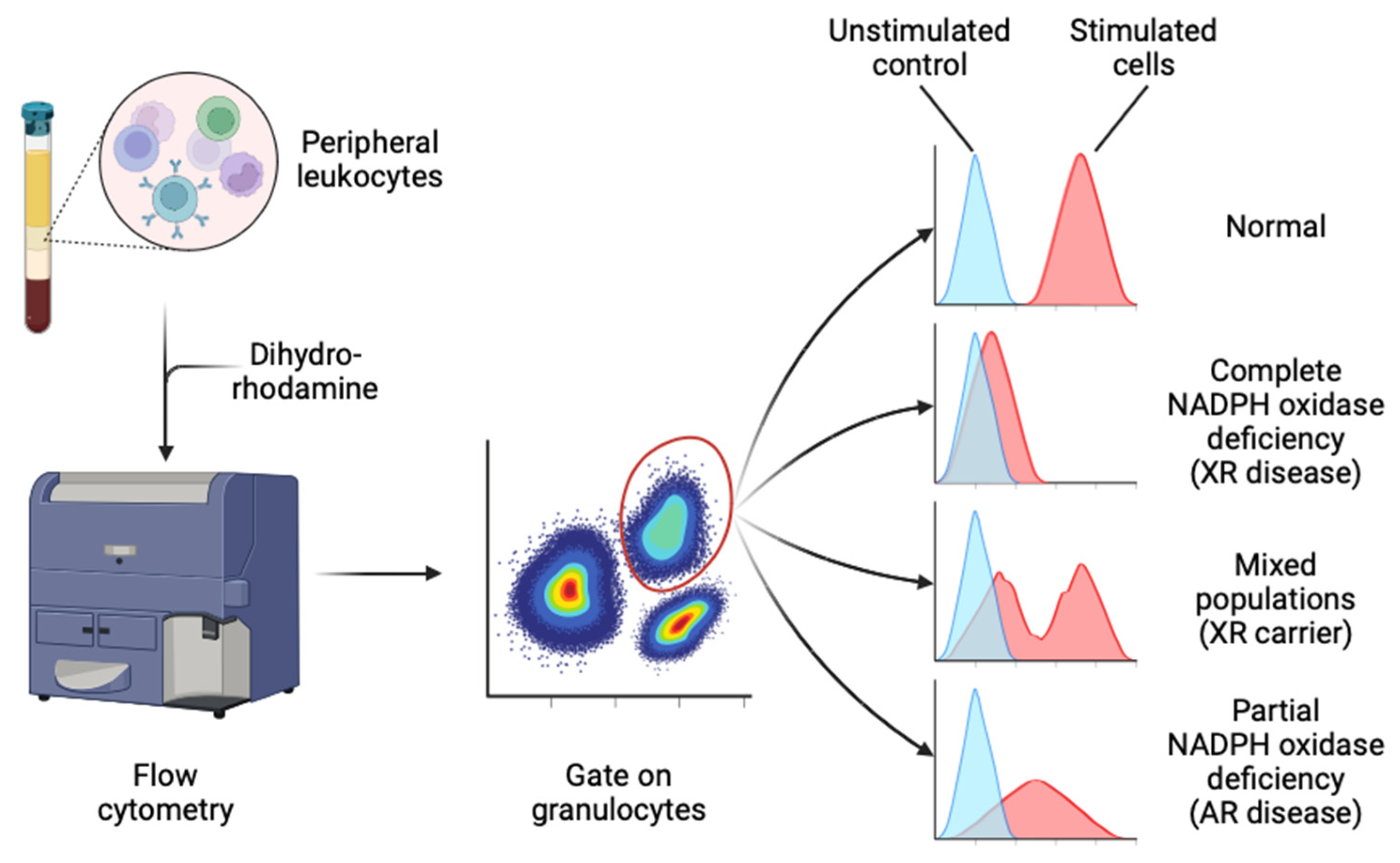

Table 1.
Genetic causes of chronic granulomatous disease (CGD): NADPH oxidase components.
| Gene symbol | CYBB | CYBA | NCF1 | NCF2 | NCF4 |
| NCBI gene ID | 1536 | 1535 | 653361 | 4688 | 4689 |
| Gene name | Cytochrome b558 beta | Cytochrome b558 alpha | Neutrophil cytosolic factor 1 | Neutrophil cytosolic factor 2 | Neutrophil cytosolic factor 4 |
| Gene location | Xp21.1-p11.4 | 16q24.2 | 7q11.23 | 1q25.3 | 22q12.3 |
| Exon count | 14 | 6 | 11 | 20 | 12 |
| Protein | gp91phox / NOX2 | p22phox | p47phox | p67phox | p40phox |
| Protein location | Specific granule membrane, plasma membrane | Specific granule membrane, plasma membrane | Cytosol, cytoskeleton | Cytosol, cytoskeleton | Cytosol, cytoskeleton |
| Frequency | 65-70% | 5% | 20% | 5% | Rare |
| Inheritance | XR | AR | AR | AR | AR |
| Clinical severity | Severe$Early onset | Severe$Early onset | Mild$Late onset | Severe$Early onset | Mild$Atypical |
| Mutations | Missense$Nonsense$Missplicing$Large or small deletions$Insertions$Duplications$Transposable elements | Insertions$Deletions$Missense$Nonsense$Missplicing | Unequal meiotic crossover events with neighbouring pseudogenes$Missense$Nonsense$Insertions$Deletions$Missplicing | Insertions$Deletions$Missense$Nonsense$Missplicing | Insertions$Deletions$Missense$Nonsense$Missplicing |
| Pickup on sequence analysis | 85% | 85% | 75% | 85% | 90% |
| Pickup on deletion/duplication analysis | 15% | 15% | 25% | 15% | 10% |
Table 2.
Rare genetic causes of chronic granulomatous disease (CGD) or related disorders: non-NADPH oxidase causes.
Table 2.
Rare genetic causes of chronic granulomatous disease (CGD) or related disorders: non-NADPH oxidase causes.
| Gene symbol | CYBC1 | RAC2 | G6PD | MPO | GSS |
| NCBI gene ID | 79415 | 5880 | 2539 | 4353 | 2937 |
| Gene name | Cytochrome b558 chaperone 1 | Rac family small GTPase 2 | Glucose-6-phosphate dehydrogenase | Myeloperoxidase | Glutathione synthetase |
| Gene location | 17q25.3 | 22q13.1 | Xq28 | 17q22 | 20q11.22 |
| Exon count | 8 | 8 | 14 | 12 | 15 |
| Protein | CYBC1/Eros | Rac2-GTPase | G6PD | MPO | GSS |
| Protein location | Endoplasmic reticulum | Cytosol | Cytosol | Granule lumen | Cytosol, nucleus, mitochondria |
| Frequency | Rare | Rare | Severe deficiency is rare | Common but unlikely to cause CGD-like features | Rare |
| Inheritance | AR | AD | XR | AR | AR |
| Clinical severity | Few cases described | Few cases described | Spectrum from mild to severe | Asymptomatic or susceptible to Candida | Spectrum from mild to severe |
| Mutations | Nonsense$Missense$Missplicing | Nonsense$Missense | Deletion$Missense | Nonsense$Missense$Missplicing$Deletions | Indels$Nonsense$Missense$Missplicing |
| Notes | Also regulates expression of proteins other than gp91phox-p22phox, e.g., P2X7 | Also associated with abnormalities of neutrophil chemotaxis and lymphocyte function | Mild cases: relatively common but lack CGD-like features$Severe cases: generally mild CGD features | Rarely solely responsible for immunodeficiency | Mild cases: haemolytic anaemia only$Moderate: metabolic acidosis$Severe: progressive neurological dysfunction and infection susceptibility |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



