
Submitted:
18 July 2024
Posted:
18 July 2024
You are already at the latest version
Abstract
B-cell lymphoid malignancies is a heterogeneous group of hematologic cancers, where Bruton's tyrosine kinase (BTK) inhibitors have received FDA approval for several subtypes. The first-in-class covalent BTK inhibitor, ibrutinib, binds to the C481 amino acid residue to block the BTK enzyme and prevent the downstream signaling. Resistance to covalent BTK inhibitors can occur through mutations at the BTK binding site (C481S) but also other BTK sites and the phospholipase C gamma 2 (PLCγ2) resulting in downstream signaling. To bypass the C481S mutation, non-covalent BTK inhibitors, such as pirtobrutinib, were developed and are active against both wild-type and the C481S mutation. In this review, we discuss the molecular and genetic mechanisms which contribute to acquisition of resistance to covalent and non-covalent BTK inhibitors. In addition, we discuss the new emerging class of BTK degraders, which utilize the evolution of proteolysis-targeting chimeras (PROTACs) to degrade the BTK protein and constitute an important avenue of overcoming resistance. The moving landscape of resistance to BTK inhibitors and the development of new therapeutic strategies highlight the ongoing advances, that are being made towards the pursue of cure of B-cell lymphoid malignancies.
Keywords:
lymphoma
; B‐cell
; hematologic neoplasms
; bruton tyrosine kinase
; protein kinase inhibitors
; drug resistance
; neoplasm
; ibrutinib
; pirtobrutinib
; molecular targeted therapy
; chronic lymphocytic leukemia
; hematopoietic stem cell transplantation
; covalent inhibitors
; protein degradation
Introduction
Non-Hodgkin’s lymphomas are a diverse group of lymphoid malignancies that represent the fourth most common cancer and the sixth leading cause of cancer death in the United States [1]. Treatment has traditionally required the use of chemotherapy, which caused a wide variety of adverse events, but the paradigm has changed since the introduction of targeted therapies. Bruton's tyrosine kinase (BTK), a key enzyme of the TEC family of kinases, is crucial for the normal function and development of B-cells. Because of the reliance of the B-lymphocytes to the BTK, its inhibition also constitutes an important approach in the treatment of B-cell malignancies including the chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL) and Waldenström's macroglobulinemia (WM)[2].
The introduction of covalent BTK inhibitors marked a significant shift in treating these conditions. Ibrutinib, the first of these inhibitors, targets the BTK enzyme by binding to the C481 residue site, blocking the ATP-binding pocket and therefore inhibiting its catalytic activity [3]. This interaction prevents BTK from participating in the B-cell receptor signaling process downstream of the B-cell receptor (BCR). Early on, in the first clinical trials of ibrutinib, it was described that resistance to the covalent BTK inhibitors can occur, primarily through mutations at the BTK C481 binding site and the PLCγ2, a downstream substrate of BTK [4].
To overcome these resistance mechanisms, noncovalent (reversible) BTK inhibitors were subsequently developed. These newer agents do not rely on binding to the C481 residue and can inhibit both wild-type and the C481S mutant, resulting in favorable clinical outcomes as shown in the BRUIN Phase 1–2 trial [5,6]. Herein, we describe the genetic and molecular mechanisms of acquisition of resistance to covalent and non-covalent BTK inhibitors. Additionally, we present a new category of emerging BTK directed therapies, such as the BTK degraders, which result in proteolysis of the BTK protein, offering another novel mechanism to overcome resistance to BTK inhibitors [7].
Bruton Tyrosine Kinase Protein
History of BTK Discovery
BTK enzyme is a non-receptor member of the Tec family of non-receptor tyrosine kinases. BTK is mutated in the X-linked agammaglobulinemia (XLA), a rare genetic disorder which was initially described in 1952 [8]. In 1993, two groups identified that the BTK protein is mutated in patients with XLA, leading to a compromise of B cell development in the bone marrow with subsequent depletion of serum immunoglobulins and humoral immunodeficiency [9,10]. This was further confirmed by Khan et al, showing that introduction of specific mutations to BTK in mouse models, can lead to diminished BTK expression, leading to reduced numbers of mature conventional B cells and IgM and IgG3 deficiency [11].
Molecular Family, Biochemistry and Activation of BTK
The TEC family is the second largest non-receptor tyrosine kinase family and is comprised by 5 proteins: the bone-marrow expressed kinase (BMX), the interleukin-2-inducible T-cell kinase (ITK), the tyrosine kinase expressed in hepatocellular carcinoma (TEC), the tyrosine kinase protein (TXK) and the BTK [2]. The BTK protein has 5 main domains: (i) The N-terminal pleckstrin domain, (ii) a TEC homology domain, (iii/iv) two SRC homology domains (SH3 followed by SH2) and (v) the kinase domain, which harbors the enzymatic activity and transfers the phosphate group from ATP to tyrosine residues on substrate proteins [12]. BTK becomes active after cell membrane association and subsequent phosphorylation of the Y551 in the kinase domain by the upstream proteins of the SRC family kinase or by the spleen tyrosine kinase (SYK) [13]. These events activate the catalytic activity of BTK and result in autophosphorylation of Y223 in the SH3 domain [14]. While phosphorylated Y223 has shown to mirror the catalytic activity of the BTK, it's not clear whether it influences the biological function of BTK [15].
BTK in the B-Cell Receptor Signaling Pathway
The IgM B-cell receptor pathway (BCR) is essential for the survival of the peripheral B-lymphocytes (Figure 1) [16]. Because of its short cytoplasmic domain, the IgM cannot signal directly but it associates with the transmembrane proteins CD79a/CD79b. This heterodimer contains the immunoreceptor tyrosine-based activation motifs (ITAMs) in their cytoplasmic domain, which can be phosphorylated by the Src-family protein tyrosine kinases such as LYN upon BCR engagement by an antigen, with subsequent creation of docking sites for the spleen tyrosine kinase (SYK) [17]. Upon BTK activation by SYK, BTK leads to phosphorylation of PLCγ2 at Y753 and Y759 which are important for its catalytic activity [18]. The active PLCγ2 cleaves the phosphatidylinositol bisphosphate (PIP2) into two second messengers: diacylglycerol (DAG) and inositol triphosphate (IP3). The IP3 regulates the intracellular calcium and leads to activation of the activated T cells (NFAT) transcription, through calcineurin and calmodulin. The DAG activates protein kinase Cβ (PKCβ), which oversees the activation of IκB kinase (IKK) proteins and the downstream NF-κB pathway. In summary BTK, links the BCR pathway to the activation of the NF-κB, a vital pathway for the B-cell proliferation, maturation, and differentiation [19].
The Role of BTK in B-Cell Malignancies
The central role of BTK in B-cell malignancies was first discovered by Davis et al, through RNA interference genetic screen in human lymphoma cell lines, demonstrating that BTK is essential for the survival of the activated B-cell-like (ABC) a subtype of diffuse large B-cell lymphoma (DLBCL) addicted to the NF-κΒ pathway [20]. These experiments were also recapitulated through pharmacological inhibition of BTK with the ibrutinib (formerly known as PCI-32765), a first in class BTK inhibitor. Ibrutinib forms an irreversible covalent bond with the cysteine residue on C481, resulting in the inhibition of the BTK enzymatic activity, abrogation of the autophosphorylation at Y223 and subsequent halting of the downstream BCR signaling [3,21]. Besides the ABC-DLBCL, ibrutinib demonstrated activity in preclinical models of CLL, where it showed to abrogate downstream survival pathway including ERK1/2, PI3K and NF-κB, providing a solid and significant support for the development of ibrutinib as a therapeutic agent in a variety of B-cell malignancies [22].
Covalent BTK Inhibitors in Clinical Practice
The first clinical data demonstrating the fundamental therapeutic role of BTK inhibitors in patients with relapsed/refractory B-cell lymphoma and CLL were described in a phase I trial by Advani et al, where ibrutinib (imbruvica) showed an objective response rate (ORR) of 60% [23]. The first FDA approval of ibrutinib was obtained in 2013 for the treatment of relapsed/refractory (R/R) mantle cell lymphoma (MCL) based on a phase II trial (PCYC-1104), which showed an ORR of 67%, a complete response (CR) of 23%, median progression-free survival (mPFS) of 13 months and median overall survival (OS) of 22.5 months [24,25]. In 2014, ibrutinib was approved for patients with R/R CLL, who had received at least one prior therapy, based on the RESONATE study, a multicenter open-label phase 3 study, where Ibrutinib was compared to Ofatumumab, an anti-CD20 monoclonal antibody. The ibrutinib showed an ORR of 42.6% vs. 4% with Ofatumumab, mPFS of 44.1m vs 8.1m and mOS of 67.7m vs 65.1m with the latter irrespective of the extensive (68%) crossover to ibrutinib [26,27]. The role of BTK inhibitors in Waldenström's Macroglobulinemia (WM) which lead to the first FDA approval in the R/R setting was published in 2015, based on phase II showing an overall response rate (ORR) of 90.5% and a major response rate of 73% [28]. Following the FDA approvals of ibrutinib in the R/R setting for CLL and WM, subsequent studies demonstrated benefit in the front line, gaining new indications, which transformed the management of various B-cell malignancies [29]. Notably, a common observation among the clinical trials and real-world experience of ibrutinib, was the high rates of cardiovascular adverse events including hypertension, atrial fibrillation and bleeding, which led to FDA to withdraw approval of the indications for MCL and marginal zone lymphoma (MZL). This clinical challenge was addressed subsequently with the development of the newer generation of covalent-BTK inhibitors of acalabrutinib and Zanubrutinib.
Acalabrutinib (Calquence) was the first of the two second-generation covalent-BTK inhibitors to obtain FDA approval. In 2017, the phase study ACE-LY-004 enrolled patients with R/R MCL and demonstrated an ORR of 81%, with 48% CR and a median PFS of 22 months [30,31]. Subsequently, in 2019, acalabrutinib was approved for adults with CLL based on the ELEVATE-TN and the ASCEND trial for treatment naive patient (TN) and patients with R/R disease, respectively. The ELEVATE-TN, compared acalabrutinib alone or in combination with obinutuzumab vs. chlorambucil plus obinutuzumab and demonstrated a superior median PFS at a median follow up of 28.3 months for acalabrutinib-obinutuzumab and acalabrutinib monotherapy (mPFS not reached), compared with 22.6 months for obinutuzumab-chlorambucil [32]. The ASCEND trial compared acalabrutinib to idelalisib or bendamustine plus rituximab, and after a median follow-up of 16.1 months, the mPFS was significantly longer for the acalabrutinib (not reached) compared to the investigator's choice of 16.5 months [33].
Regarding Zanubrutinib (Brukinsa), the first approval was granted in 2019 for R/R MCL based on 2 single arm clinical trials which assessed ORR as primary endpoint and collectively demonstrated an ORR of 84% [34,35]. In 2021, Zanubrutinib was granted its second approval for the treatment of WM based on ASPEN trial, a phase 3 study, comparing zanubrutinib and ibrutinib in patients with WM [36]. In the first cohort of the ASPEN with the MYD88 L265P mutation, the response rates of CR + very good partial response (VGPR) at 44.4 month median follow- up was 36.3% for Zanubrutinib vs 25.3% for ibrutinib, whereas in the second cohort with the MYD88 wildtype or unknown mutational status, the response rates were 30.8% with one CR [37]. Subsequently, the approval for R/R MZL was granted in 2021 based on the efficacy results from MAGNOLIA and the BGB-3111-AU-003 trial [38]. The MAGNOLIA phase 2 study demonstrated an ORR of 68.2% with a CR of 25.8% CR, whereas the BGB-3111-AU-003, a phase 1/2 study of zanubrutinib in B-cell malignancies showed an ORR of 80.0% with CR of 20% in MZL [35,39]. Regarding the approval of zanubrutinib in CLL, it was granted based on two phase 3 randomized trials. The SEQUOIA study compared Zanubrutinib versus bendamustine and rituximab in TN-CLL and met the prespecified criteria for superiority during the median follow up of 26.2 months with HR for PFS of 0.42 (P<0.0001) [40]. The ALPINE trial compared Zanubrutinib vs Ibrutinib in the R/R setting and demonstrated a higher ORR 83.5% vs 74.2% and also a superior PFS with HR 0.65 at the median follow up of 29.6 months [41]. The most recent approval of Zanubrutinib was granted in 2024 in follicular lymphomas based on the ROSEWOOD trial which compared zanubrutinib plus obinutuzumab vs single agent obinutuzumab. The study showed a ORR of 69% for the combination vs. 46% for the obinutuzumab monotherapy with a durable response of 69% during the 18-month DoR [42].
Collectively, these clinical data, highlight the importance of the covalent BTK inhibitors in the treatment of various B-cell malignancies, but even more importantly underscores the fact, that the duration of response can be limited due to acquisition of resistance.
Resistance Mechanisms to Covalent BTK Inhibitors
Despite the remarkable efficiency of the covalent BTK inhibitors in the treatment of B-cell malignancies, resistance can develop. From clinical standpoint, resistance is classified as primary and secondary. Primary resistance is defined as resistance that occur in patients who fail to respond to the BTK inhibitors upfront, whereas secondary resistance is defined as resistance which develops in patients who initially responded to the BTK inhibitors and then developed relapse. From biological standpoint, the mechanisms of resistance can be broken down to 1. Intrinsic mechanisms involving mutations of the cancer cells and upregulation of survival signaling pathways and 2. Extrinsic mechanisms involving the tumor microenvironment. Herein, we primary focus on the secondary mechanisms of resistance which are more frequently observed in the clinical setting of CLL with more emphasis in the acquisition of BTK mutations.
The mPFS of ibrutinib in treatment naive patients with CLL is 8.9 years as shown in the phase 3 Resonate-2 study, whereas the mPFS is shorter 3.6 years in the R/R setting [27,43]. Patients with primary refractory disease or early progression within 15 months for the initiation of therapy often present with Richter’s syndrome, a histologic transformation of CLL to DLBCL [44,45]. In contrast, progression due to CLL occurs later with acquisition of mutations to BTK C481S and PLCΓ2 (within the autoinhibitory domain) in approximately 80% of patients with late progression [46].
While the BTK C481S is the most commonly observed mutation described in all the 3 FDA approved covalent BTKi, mutations to other amino acid residues have been described as well (Figure 1) [47,48,49,50]. With functional studies based on enzymatic activity and a differential interactome, two district classes of BTK mutations of the kinase domain have been defined including the Kinase Proficient drug resistance mutations (C481S and T474I) and the Kinase Deficient/Dead drug resistance mutations (L528W) [7]. Similarly to the C481S mutation, the T474I mutation can increase the autophosphorylation of BTK-Y223 in the absence of BCR stimulation and activate the downstream signaling. While T474I has been described both in R/R CLL cases on ibrutinib and acalabrutinib, it has not been described in Zanubrutinib yet, highlighting that the acquisition of resistance to the covalent BTK inhibitors does not have a universal mechanism that can be applied for every BKTi, but there can specific mechanisms for each individual BTKi [51,52].
Figure 2.
BTK and PLCγ2 mutations described in patients treated with the FDA approved BTK inhibitors ibrutinib, acalabrutinib, zanubrutinib and pirtobrutinib. The domains of the BTK and PLCγ2 are illustrated. BTK E41*: E41 K/V, BTK C481*: C481 S/F/T/Y/R/G, BTK T474*: T474I/F. PLCG2 S707*: S707 Y/F/P, L845*: L845 F/L, D993* : D993 G/H/Y, D1139*: D1139G/del, D1140*:D1140 E/G/N, M1141*: M1141 K/R.
Figure 2.
BTK and PLCγ2 mutations described in patients treated with the FDA approved BTK inhibitors ibrutinib, acalabrutinib, zanubrutinib and pirtobrutinib. The domains of the BTK and PLCγ2 are illustrated. BTK E41*: E41 K/V, BTK C481*: C481 S/F/T/Y/R/G, BTK T474*: T474I/F. PLCG2 S707*: S707 Y/F/P, L845*: L845 F/L, D993* : D993 G/H/Y, D1139*: D1139G/del, D1140*:D1140 E/G/N, M1141*: M1141 K/R.
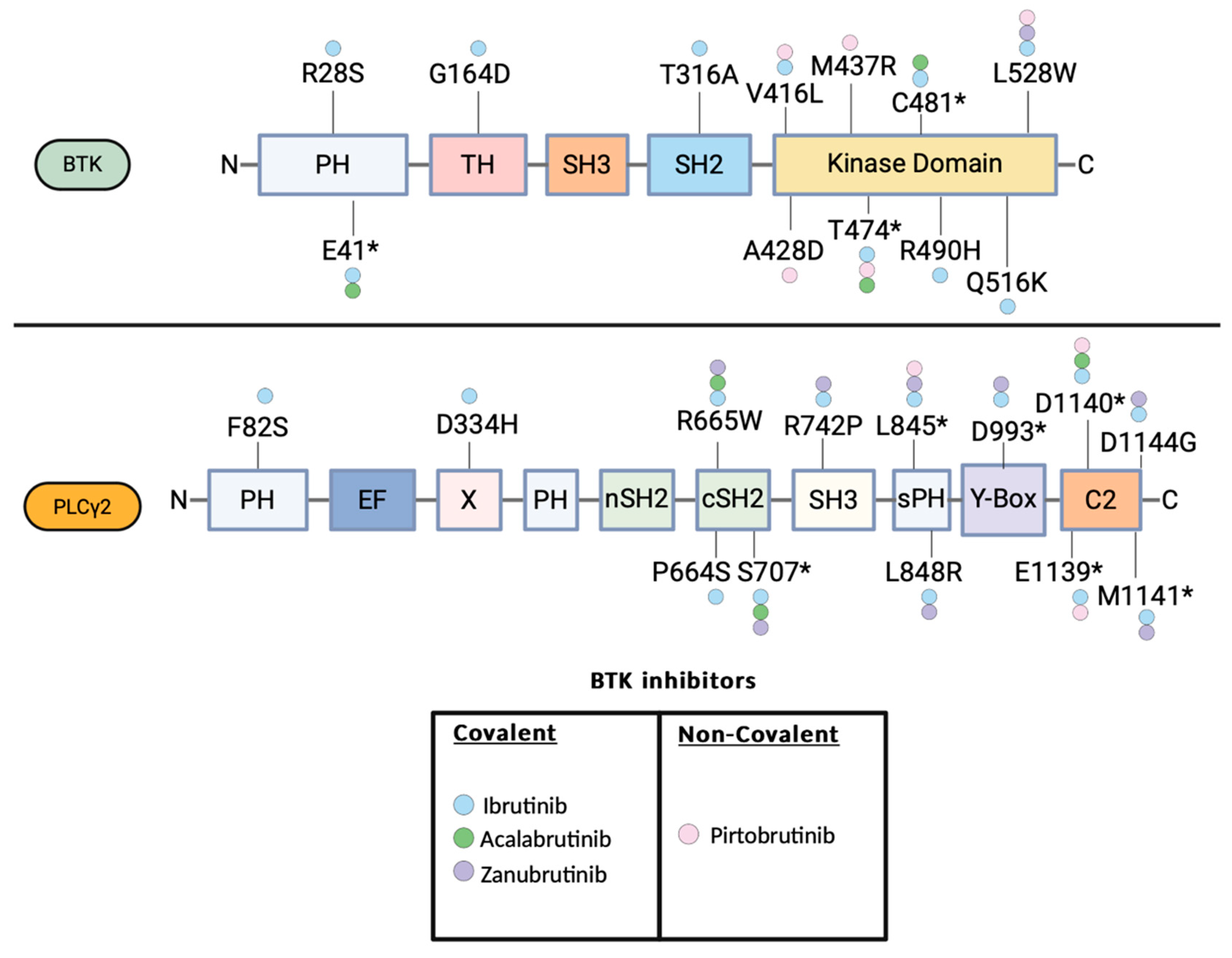
Regarding the Kinase Deficient/Dead drug resistance mutations, the L528W mutation has been described in patients with ibrutinib and zanubrutinib [48,53]. L528W mutation is associated with reduced autophosphorylation of the BTK-Y223 and reduced BTK enzymatic activity. Functional experiments with immunoprecipitation mass spectrometry have revealed a scaffold function for the BKT L528W with downstream cell signaling, which skews the survival dependence on surrogate kinases that can find to kinase-impaired BTK proteofolds.
Directly downstream of the BTK, mutations in phospholipase C gamma 2 (PLCγ2) can also lead to resistance. Several nonsynonymous mutations in PLCΓ2 have been described in ibrutinib resistant CLL, with the majority of them occurring in the SH2 domain including the P664S, R665W and S707[46,50,51,54,55,56,57,58]. Notably mutations of the PCLG2 SH2 domain has been shown to activate the PCLG2 independently of the BTK, and reconstitute the BCR signaling, which can explain their acquisition in the context BTK inhibition [59,60]. Besides the mutations of BTK and PCLG2, studies with whole exome and deep-targeted sequencing studies have also described the clonal evolution of CLL developing resistance to BTK inhibition with expansion of clones with deletion of 8p and additional driver mutations of EP300, MLL2, EIF2A [55,61].
In MCL, in contrast to the CLL, primary resistance to ibrutinib is more common, occurring in 32%, which could stem from the fact, that MCL has a higher rate of high-risk mutations such as mutations in the ATM and TP53 gene in 44% and 27% accordingly [24] [62]. Primary resistance is mainly mediated though genetic lesions in the alternative NF-κΒ pathway as shown in human cell lines and MCL samples with mutations in TRAF2 and BIRC3 [63] and also, through activation of compensatory signaling pathways such as the PI3K/AKT/mTOR, MEK/ERK and canonical NF-κB pathway. Unlike CLL, mutations in the BTK are rarely detected at disease progression. Similarly to the primary resistance, secondary resistance, is mainly mediated through the alternative compensatory signaling pathways, albeit mutations have been described in D1 (CCND1) gene and CDKN2A/MTAP gene [64].
Noncovalent BTK Inhibitors: Clinical Data and Emergence of Resistance
The resistance mechanisms discussed above pose a major limitation to the use of covalent BTK inhibitors, which lead to the development of noncovalent or reversible BTK inhibitors. This class of therapies was designed to have the original properties of covalent BTK inhibitors but with the additional capacity to bind to C481 mutant BTK [54]. Preclinical studies assessing pirtobrutinib (LOXO-305), demonstrated inhibition of the BCR pathway in the presence of the C481S mutation, which moved this agent to the clinic [65].
The efficacy of pirtobrutinib was assessed in the BRUIN study, an open-label, single arm, multicohort, phase 1/2 trial in patients with B-cell malignancies. The CLL cohort included 317 patients, who had previously treated with at least 2 prior lines of therapy including a BTK inhibitor and a BCL-2 inhibitor. Among the patients who had previously treated with a covalent BTK inhibitor, the ORR was 73.3%, whereas in the subgroup of patients previously received both a covalent BTK and BCL2 inhibitor, the ORR was 70% [66]. Notably in an earlier report of the BRUIN study the ORR was noted to be similar among patients with WT-BTK with ORR 66% and those with C481 mutation with ORR 71% [6]. Regarding the MCL cohort which included 90 patients with R/R MCL, the ORR 57.8%, including 20% CR. These data lead in 2023 to the accelerated approval of pirtobrutinib for R/R MCL after at least two lines of systemic therapy, including a BTK inhibitor and for R/R CLL/SLL after at least two lines of systemic therapy, including a BTK inhibitor and a BCL2 inhibitor. Notably, pirtobrutinib was well tolerated in the BRUIN study with low rates of discontinuation due to treatment related adverse events around 3% [66].
Besides pirtobrutinib, other noncovalent BTKis are currently under investigation. Nemtabrutinib is being tested in the BELLWAVE-001 (NCT03930953) phase 1/2 trial for R/R CLL/SLL and other B-cell malignancies. Early results for CLL showed an ORR of 75% and grade 3/4 adverse events are neutropenia (23.4%), febrile neutropenia (14.9%), and pneumonia (14.9%) [67]. Its use is also being investigated in the upfront setting in the phase 3 randomized trial, BELLWAVE-011, comparing nemtabrutinib vs ibrutinib or acalabrutinib in patients with untreated CLL/SLL, which is actively enrolling [68].
Despite, the relative short experience with the noncovalent BTKi pirtobrutinib, progressions and relapses have been described. A recent genomic analysis of pretreatment samples as well samples obtained at that time of progression in R/R CLL patients treated with pirtobrutinib, identified on-target BTK mutations and also downstream PLCγ2 mutations that allowed escape from the BTK inhibition [69]. Specifically, a key set of BTK mutations were identified in 9 out of 55 patients with CLL refractory to pirtobrutinib including V416L, A428D, M437R, T474I, and L528W [69]. All these mutations are clustered in the kinase domain of BTK and can confer resistance to both covalent and noncovalent BTK inhibitors [69]. Enzymatic analysis demonstrated that the V416L, A428D, M437R, and L528W resulted in diminished BTK enzymatic function, while the T474I resulted in enhanced BTK enzymatic function. Notably, the T474I and the L528W have also been described in the context of covalent BTK inhibition, highlighting the fact that common mechanism and resistance can occur regardless, the covalent or not-covalent nature of the BTK inhibition. Besides the BTK mutations, genetic studies revealed that PLCγ2 mutations can also confer resistance to pirtobrutinib. 2 patients with stable disease to pirtobrutinib had preexisting PLCG2 mutations E1139 and D1140E accordingly whereas one patient with preexisting D1144G mutation who initially had a stable response to pirtobrutinib, developed Richter transformation to DLBCL after 5 months of treatment. The BTK and PLCγ2 mutations that have been described in patients with CLL that progressed on pirtobrutinib are illustrated in Figure 1 (PMID: 37706363).
These mechanisms of resistance are important in designing the next-generation therapies by targeting the scaffold functions of BTK rather than its kinase activity. This could then disrupt crucial protein–protein interactions that would be necessary for BCR signaling to take place, annulling resistance that is driven by mutations in the kinase domain of BTK. Moreover, combination therapies involving non-covalent inhibitors with other agents may be able to overcome acquired resistant clones and enhance therapy efficacy [70].
BTK Degraders: The Future of BTK Targeted Therapies
BTK degraders represent an emerging alternative in the context of growing resistance to both covalent and noncovalent BTK inhibitors. These molecules utilize proteolysis-targeting chimeras (PROTACs) technology. PROTACs against BTK are bifunctional molecules with one end binding to the target protein, BTK, and the other end binding to an E3 ubiquitin ligase. This binding induces ubiquitination and subsequent proteasomal degradation of the BTK by effectively reducing its levels and inhibiting its signaling pathway [71]. Several BTK degraders are currently in various stages of research and development. Key studies and trials have demonstrated the potential of these compounds to overcome resistance and improve outcomes in patients with relapsed or refractory B-cell malignancies.
NX-2127 is one of the first molecules to exhibit pre-clinical and clinical efficacy. To synthesize a BTK degrader, a set of numerous BTK binders linked to the CRBN ligand with flexible polyethylene glycol and alkyl linkers of variable length were evaluated for their ability to selectively degrade BTK [72]. Further optimization of the initial compounds was done through medical chemistry, structure-based drug design, and empirical testing in both the laboratory and on animal model led to the design of NX-2127, an orally bioavailable BTK degrader [72]. NX-2127 was shown to bind to all classes of drug-resistant BTK mutant proteins including BTK proficient and BTK dead/deficient and inducing a stable complex formation with CRBN-DDB1. Additionally, NX-2127 was shown to degrade the BTK drug resistant mutant proteins with subsequent abrogation of the downstream BCR signaling. As a proof of concept, NX-2127 was evaluated in a phase 1 trial for R/R B-cell malignancies which included 29 patients with CLL/SLL among a total of 47 patients [73]. Among the efficacy evaluable patient with CLL, there were 9 PRs/PR with rebound lymphocytosis, 11 patients with Stable disease (SD) and 4 with PD. Notable, rapid and robust BTK degradation was observed in all patients regardless the tumor type. NX-5948 is another BTK degrader, which in contrast to the NX-2127 which degrades both BTK and IKZF3, it selectively degrades BTK. NX-5948 can induces degradation of wild-type and mutant forms of BTK in B-cells at a sub-nanomolar levels. NX-5948 is currently being tested in a phase 1a trial (NCT05131022) and preliminary data from 14 patients with R/R CLL or NHL show well tolerability and no discontinuation due to adverse events. All patients had a rapid and sustained reduction in BTK protein due to degradation, regardless of tumor type, drug dose or baseline BTK level [74].
BGB-16673 is also a novel BTK degrader, which is currently tested in patients with R/R B-Cell Malignancies. Preliminary data from a phase 1 trial (NCT05006716) of 26 patients, with at least 2 previous lines of treatment, showed substantial reductions in BTK protein levels in peripheral blood and tumor tissue, even at the lowest dose. Among the 18 response-evaluable patients, 5/6 patients with CLL, 2/2 with MZL and 3/4 with WM demonstrated a PR, whereas 1/3 with MCL had a CR [75].
Conclusions
The introduction of BTK inhibitors has revolutionized the treatment of B-cell malignancies, leading to improvement of the survival rates. In this review, we summarized the clinical data of the landmark clinical trials that led to the FDA approval of the various covalent and noncovalent BTK inhibitors. We reviewed the mechanisms of resistance to the BTK inhibitors by focusing to the BTK mutations and described how the new class of BTK degraders can overcome the BTK proficient and BTK deficient/dead mutations.
Currently, studies assessing novel BTK degraders are underway, whereas trials with combinational programs of BTK inhibitors and other molecular targeted therapies are aiming to improve the survival rates of B-cell malignancies. Ultimately, genetic interrogation of patient samples, will be vital in understanding the patterns of resistance as we design new therapies to move the field forward.
Author Contributions
Conceptualization G.P, Investigation, A.B, M.A, D.W, J.N and G.P, writing original draft preparation A.B, M.A, J.N, D.W and GP, writing-review and editing M.A, D.W, and G.P., Supervision G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Conflicts of Interest
A.B, M.A, D.W, J.N. declare no conflict of interest. G.P. is shareholder of Amgen, Eli Lilly, Crispr Therapeutics and equity holder of Mevox ltd.
References
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US Lymphoid Malignancy Statistics by World Health Organization Subtypes. CA Cancer J Clin 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The Role of Bruton’s Tyrosine Kinase in the Immune System and Disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Quinquenel, A.; Fornecker, L.-M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.-S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 Mutations in a Real-Life CLL Cohort Still on Ibrutinib after 3 Years: A FILO Group Study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef]
- Wang, M.; Shah, N.N.; Alencar, A.J.; Gerson, J.N.; Patel, M.R.; Fakhri, B.; Jurczak, W.; Tan, X.N.; Lewis, K.; Fenske, T.S.; et al. Pirtobrutinib, A Next Generation, Highly Selective, Non-Covalent BTK Inhibitor in Previously Treated Mantle Cell Lymphoma: Updated Results from the Phase 1/2 BRUIN Study. Blood 2021, 138, 381. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in Relapsed or Refractory B-Cell Malignancies (BRUIN): A Phase 1/2 Study. The Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.M.; et al. Kinase-Impaired BTK Mutations Are Susceptible to Clinical-Stage BTK and IKZF1/3 Degrader NX-2127. Science 2024, 383, eadi5798. [Google Scholar] [CrossRef] [PubMed]
- Bruton, O.C. Agammaglobulinemia. Pediatrics 1952, 9, 722–728. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovský, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarström, L.; Kinnon, C.; Levinsky, R.; Bobrow, M. The Gene Involved in X-Linked Agammaglobulinaemia Is a Member of the Src Family of Protein-Tyrosine Kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S. Deficient Expression of a B Cell Cytoplasmic Tyrosine Kinase in Human X-Linked Agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Khan, W.N.; Alt, F.W.; Gerstein, R.M.; Malynn, B.A.; Larsson, I.; Rathbun, G.; Davidson, L.; Müller, S.; Kantor, A.B.; Herzenberg, L.A. Defective B Cell Development and Function in Btk-Deficient Mice. Immunity 1995, 3, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.-M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s Tyrosine Kinase (Btk): Function, Regulation, and Transformation with Special Emphasis on the PH Domain. Immunol Rev 2009, 228, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Scharenberg, A.M.; Park, H.; Wahl, M.I.; Lin, S.; Kato, R.M.; Fluckiger, A.C.; Witte, O.N.; Kinet, J.P. Activation of BTK by a Phosphorylation Mechanism Initiated by SRC Family Kinases. Science 1996, 271, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Wahl, M.I.; Afar, D.E.; Turck, C.W.; Rawlings, D.J.; Tam, C.; Scharenberg, A.M.; Kinet, J.P.; Witte, O.N. Regulation of Btk Function by a Major Autophosphorylation Site within the SH3 Domain. Immunity 1996, 4, 515–525. [Google Scholar] [CrossRef]
- Estupiñán, H.Y.; Bouderlique, T.; He, C.; Berglöf, A.; Cappelleri, A.; Frengen, N.; Zain, R.; Karlsson, M.C.I.; Månsson, R.; Smith, C.I.E. In BTK, Phosphorylated Y223 in the SH3 Domain Mirrors Catalytic Activity, but Does Not Influence Biological Function. Blood Adv 2024, 8, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.P.; Kühn, R.; Rajewsky, K. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell 1997, 90, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Rolli, V.; Gallwitz, M.; Wossning, T.; Flemming, A.; Schamel, W.W.A.; Zürn, C.; Reth, M. Amplification of B Cell Antigen Receptor Signaling by a Syk/ITAM Positive Feedback Loop. Mol Cell 2002, 10, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Sekiya, F.; Poulin, B.; Bae, Y.S.; Rhee, S.G. Mechanism of B-Cell Receptor-Induced Phosphorylation and Activation of Phospholipase C-Gamma2. Mol Cell Biol 2004, 24, 9986–9999. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, U.D.; Zhang, K.; Teutsch, M.; Sen, R.; Wortis, H.H. Bruton’s Tyrosine Kinase Links the B Cell Receptor to Nuclear Factor kappaB Activation. J Exp Med 2000, 191, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc Natl Acad Sci U S A 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.M.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton Tyrosine Kinase Represents a Promising Therapeutic Target for Treatment of Chronic Lymphocytic Leukemia and Is Effectively Targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Has Significant Activity in Patients with Relapsed/Refractory B-Cell Malignancies. J Clin Oncol 2013, 31, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Blum, K.A.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Long-Term Follow-up of MCL Patients Treated with Single-Agent Ibrutinib: Updated Safety and Efficacy Results. Blood 2015, 126, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Byrd John, C.; Brown Jennifer, R.; O’Brien Susan; Barrientos Jacqueline C. ; Kay Neil E.; Reddy Nishitha M.; Coutre Steven; Tam Constantine S.; Mulligan Stephen P.; Jaeger Ulrich; et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. New England Journal of Medicine 2014, 371, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final Analysis from RESONATE: Up to Six Years of Follow-up on Ibrutinib in Patients with Previously Treated Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Am J Hematol 2019, 94, 1353–1363. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N Engl J Med 2015, 372, 1430–1440. [Google Scholar] [CrossRef]
- IMBRUVICA® (Ibrutinib) Official Patient Website. Available online: https://www.imbruvica.com/ (accessed on 8 July 2024).
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Doorduijn, J.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib in Relapsed or Refractory Mantle Cell Lymphoma (ACE-LY-004): A Single-Arm, Multicentre, Phase 2 Trial. Lancet 2018, 391, 659–667. [Google Scholar] [CrossRef]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.H.; Casasnovas, R.-O.; Smith, S.D.; Damaj, G.L.; Doorduijn, J.K.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib Monotherapy in Patients with Relapsed/Refractory Mantle Cell Lymphoma: Long-Term Efficacy and Safety Results from a Phase 2 Study. Blood 2020, 136, 38–39. [Google Scholar] [CrossRef]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without Obinutuzumab versus Chlorambucil and Obinutuzmab for Treatment-Naive Chronic Lymphocytic Leukaemia (ELEVATE TN): A Randomised, Controlled, Phase 3 Trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol 2020, 38, 2849–2861. [Google Scholar] [CrossRef]
- Song, Y.; Zhou, K.; Zou, D.; Zhou, J.; Hu, J.; Yang, H.; Zhang, H.; Ji, J.; Xu, W.; Jin, J.; et al. Zanubrutinib in Relapsed/Refractory Mantle Cell Lymphoma: Long-Term Efficacy and Safety Results from a Phase 2 Study. Blood 2022, 139, 3148–3158. [Google Scholar] [CrossRef]
- Phillips, T.; Chan, H.; Tam, C.S.; Tedeschi, A.; Johnston, P.; Oh, S.Y.; Opat, S.; Eom, H.-S.; Allewelt, H.; Stern, J.C.; et al. Zanubrutinib Monotherapy in Relapsed/Refractory Indolent Non-Hodgkin Lymphoma. Blood Adv 2022, 6, 3472–3479. [Google Scholar] [CrossRef]
- Research, C. for D.E. and FDA Approves Zanubrutinib for Waldenström’s Macroglobulinemia. FDA 2021.
- Dimopoulos, M.A.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Garcia-Sanz, R.; et al. Zanubrutinib Versus Ibrutinib in Symptomatic Waldenström Macroglobulinemia: Final Analysis From the Randomized Phase III ASPEN Study. J Clin Oncol 2023, 41, 5099–5106. [Google Scholar] [CrossRef]
- Research, C. for D.E. and FDA Grants Accelerated Approval to Zanubrutinib for Marginal Zone Lymphoma. FDA 2022. [Google Scholar]
- Opat, S.; Tedeschi, A.; Linton, K.; McKay, P.; Hu, B.; Chan, H.; Jin, J.; Sobieraj-Teague, M.; Zinzani, P.L.; Coleman, M.; et al. The MAGNOLIA Trial: Zanubrutinib, a Next-Generation Bruton Tyrosine Kinase Inhibitor, Demonstrates Safety and Efficacy in Relapsed/Refractory Marginal Zone Lymphoma. Clin Cancer Res 2021, 27, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Brown, J.R.; Kahl, B.S.; Ghia, P.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; et al. Zanubrutinib versus Bendamustine and Rituximab in Untreated Chronic Lymphocytic Leukaemia and Small Lymphocytic Lymphoma (SEQUOIA): A Randomised, Controlled, Phase 3 Trial. Lancet Oncol 2022, 23, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Eichhorst, B.; Hillmen, P.; Jurczak, W.; Kaźmierczak, M.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Zhou, K.; et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 2023, 388, 319–332. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Mayer, J.; Flowers, C.R.; Bijou, F.; De Oliveira, A.C.; Song, Y.; Zhang, Q.; Merli, M.; Bouabdallah, K.; Ganly, P.; et al. ROSEWOOD: A Phase II Randomized Study of Zanubrutinib Plus Obinutuzumab Versus Obinutuzumab Monotherapy in Patients With Relapsed or Refractory Follicular Lymphoma. J Clin Oncol 2023, 41, 5107–5117. [Google Scholar] [CrossRef]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- M, A.; Sr, J.; G, P. Richter’s Syndrome (RS): Emerging Therapeutic Horizons. Journal of Oncology Research and Therapy 2024. [Google Scholar]
- Woyach, J.A. Patterns of Resistance to B Cell-Receptor Pathway Antagonists in Chronic Lymphocytic Leukemia and Strategies for Management. Hematology Am Soc Hematol Educ Program 2015, 2015, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.M.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal Evolution Leading to Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef]
- Tsushima, T.; Sato, N.; Guo, Y.-M.; Nakamura, H.; Kunisada, K.; Chi, S.-G.; Akie, K.; Takahashi, Y.; Nakamura, S.; Shimada, K.; et al. Richter Transformation Acquiring PLCG2 Mutation during Bruton Tyrosine Kinase Inhibitors Treatment. EJHaem 2024, 5, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.R.; Lew, T.E.; Tiong, I.S.; Bennett, R.; Cheah, C.Y.; Lewis, K.L.; Handunnetti, S.M.; Tang, C.P.S.; Roberts, A.; et al. Enrichment of BTK Leu528Trp Mutations in Patients with CLL on Zanubrutinib: Potential for Pirtobrutinib Cross-Resistance. Blood Adv 2022, 6, 5589–5592. [Google Scholar] [CrossRef]
- Jones, D.; Woyach, J.A.; Zhao, W.; Caruthers, S.; Tu, H.; Coleman, J.; Byrd, J.C.; Johnson, A.J.; Lozanski, G. PLCG2 C2 Domain Mutations Co-Occur with BTK and PLCG2 Resistance Mutations in Chronic Lymphocytic Leukemia Undergoing Ibrutinib Treatment. Leukemia 2017, 31, 1645–1647. [Google Scholar] [CrossRef]
- Sedlarikova, L.; Petrackova, A.; Papajik, T.; Turcsanyi, P.; Kriegova, E. Resistance-Associated Mutations in Chronic Lymphocytic Leukemia Patients Treated With Novel Agents. Front Oncol 2020, 10, 894. [Google Scholar] [CrossRef]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol 2015, 1, 80–87. [Google Scholar] [CrossRef]
- Stephens, D.M.; Byrd, J.C. Resistance to Bruton Tyrosine Kinase Inhibitors: The Achilles Heel of Their Success Story in Lymphoid Malignancies. Blood 2021, 138, 1099–1109. [Google Scholar] [CrossRef]
- Brown, J.; Mashima, K.; Fernandes, S.; Naeem, A.; Shupe, S.; Fardoun, R.; Davids, M. Mutations Detected in Real World Clinical Sequencing during BTK Inhibitor Treatment in CLL. Res Sq 2024. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N Engl J Med 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal Evolution in Patients with Chronic Lymphocytic Leukaemia Developing Resistance to BTK Inhibition. Nat Commun 2016, 7, 11589. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Jones, D.; Jurczak, W.; Robak, T.; Illes, A.; Kater, A.P.; Ghia, P.; Byrd, J.C.; Seymour, J.F.; Long, S.; et al. Mutational Profile of Previously Treated Chronic Lymphocytic Leukemia Patients Progressing on Acalabrutinib or Ibrutinib. Blood, 2024. [Google Scholar] [CrossRef]
- Tatarczuch, M.; Waltham, M.; Shortt, J.; Polekhina, G.; Hawkes, E.A.; Ho, S.-J.; Trotman, J.; Brasacchio, D.; Co, M.; Li, J.; et al. Molecular Associations of Response to the New-Generation BTK Inhibitor Zanubrutinib in Marginal Zone Lymphoma. Blood Adv 2023, 7, 3531–3539. [Google Scholar] [CrossRef]
- Woolston, D.W.; Lee, N.D.; Shadman, M.; Latorre-Esteves, E.; Tee, X.R.; Fredrickson, J.; Kohrn, B.F.; Ujjani, C.; Eckel, A.; Till, B.; et al. Ultra-Deep Mutational Landscape in Chronic Lymphocytic Leukemia Uncovers Dynamics of Resistance to Targeted Therapies. Haematologica 2024, 109, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic Mutation of Phospholipase C, Γ2 Acquired in Ibrutinib-Resistant CLL Confers BTK Independency upon B-Cell Receptor Activation. Blood 2015, 126, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Walliser, C.; Hermkes, E.; Schade, A.; Wiese, S.; Deinzer, J.; Zapatka, M.; Désiré, L.; Mertens, D.; Stilgenbauer, S.; Gierschik, P. The Phospholipase Cγ2 Mutants R665W and L845F Identified in Ibrutinib-Resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J Biol Chem 2016, 291, 22136–22148. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Sun, C.; Rosebrock, D.; Herman, S.E.M.; Fein, J.; Sivina, M.; Underbayev, C.; Liu, D.; Hoellenriegel, J.; Ravichandran, S.; et al. The Evolutionary Landscape of Chronic Lymphocytic Leukemia Treated with Ibrutinib Targeted Therapy. Nat Commun 2017, 8, 2185. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic Mutations and Features of Mantle Cell Lymphoma: A Systematic Review and Meta-Analysis. Blood Adv 2020, 4, 2927–2938. [Google Scholar] [CrossRef]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chan, F.C.; Meissner, B.; Bhang, H.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and Genomic Profiling Identifies NF-κB-Targeted Treatment Strategies for Mantle Cell Lymphoma. Nat Med 2014, 20, 87–92. [Google Scholar] [CrossRef]
- Nakhoda, S.; Vistarop, A.; Wang, Y.L. Resistance to Bruton Tyrosine Kinase Inhibition in Chronic Lymphocytic Leukaemia and Non-Hodgkin Lymphoma. Br J Haematol 2023, 200, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.T.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.K.; Rogers, K.A.; et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discov 2018, 8, 1300–1315. [Google Scholar] [CrossRef]
- Mato, A.R.; Woyach, J.A.; Brown, J.R.; Ghia, P.; Patel, K.; Eyre, T.A.; Munir, T.; Lech-Maranda, E.; Lamanna, N.; Tam, C.S.; et al. Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N Engl J Med 2023, 389, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Stephens, D.M.; Flinn, I.W.; Bhat, S.A.; Savage, R.E.; Chai, F.; Eathiraj, S.; Reiff, S.D.; Muhowski, E.M.; Granlund, L.; et al. First-in-Human Study of the Reversible BTK Inhibitor Nemtabrutinib in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia and B-Cell Non–Hodgkin Lymphoma. Cancer Discovery 2024, 14, 66–75. [Google Scholar] [CrossRef]
- Tadmor, T.; Eyre, T.A.; Benjamini, O.; Chaudhry, A.; Shen, J.; Leng, S.; Farooqui, M.Z.H.; Lavie, D. BELLWAVE-011: Phase 3 Randomized Trial of Nemtabrutinib versus Ibrutinib or Acalabrutinib in Untreated Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. JCO 2024, 42, TPS7088–TPS7088. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N Engl J Med 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed]
- On-Target BTK Mutations Promote Resistance to Noncovalent BTK Inhibitors. Cancer Discovery 2022, 12, 1179. [CrossRef]
- Dobrovolsky, D.; Wang, E.S.; Morrow, S.; Leahy, C.; Faust, T.; Nowak, R.P.; Donovan, K.A.; Yang, G.; Li, Z.; Fischer, E.S.; et al. Bruton Tyrosine Kinase Degradation as a Therapeutic Strategy for Cancer. Blood 2019, 133, 952–961. [Google Scholar] [CrossRef]
- Robbins, D.W.; Noviski, M.A.; Tan, Y.S.; Konst, Z.A.; Kelly, A.; Auger, P.; Brathaban, N.; Cass, R.; Chan, M.L.; Cherala, G.; et al. Discovery and Preclinical Pharmacology of NX-2127, an Orally Bioavailable Degrader of Bruton’s Tyrosine Kinase with Immunomodulatory Activity for the Treatment of Patients with B Cell Malignancies. J Med Chem 2024, 67, 2321–2336. [Google Scholar] [CrossRef]
- Danilov, A.; Tees, M.T.; Patel, K.; Wierda, W.G.; Patel, M.; Flinn, I.W.; Latif, T.; Ai, W.; Thompson, M.C.; Wang, M.L.; et al. A First-in-Human Phase 1 Trial of NX-2127, a First-in-Class Bruton’s Tyrosine Kinase (BTK) Dual-Targeted Protein Degrader with Immunomodulatory Activity, in Patients with Relapsed/Refractory B Cell Malignancies. Blood 2023, 142, 4463. [Google Scholar] [CrossRef]
- Searle, E. Initial Findings from a First-in-Human Phase 1a/b Trial of NX-5948, a Selective Bruton’s Tyrosine Kinase (BTK) Degrader, in Patients with Relapsed/Refractory B Cell Malignancies; ASH, 2023 December 11. [Google Scholar]
- Seymour, J.F.; Cheah, C.Y.; Parrondo, R.; Thompson, M.C.; Stevens, D.A.; Lasica, M.; Wang, M.L.; Kumar, A.; Trotman, J.; Alwan, M.; et al. First Results from a Phase 1, First-in-Human Study of the Bruton’s Tyrosine Kinase (BTK) Degrader Bgb-16673 in Patients (Pts) with Relapsed or Refractory (R/R) B-Cell Malignancies (BGB-16673-101). Blood 2023, 142, 4401. [Google Scholar] [CrossRef]
Figure 1.
BCR signaling in a B-cell lymphocyte.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



