
Submitted:
19 July 2024
Posted:
19 July 2024
You are already at the latest version
Abstract
In this work, we report the synthesis of a new thiosemicarbazone-based drug of N'-(di(pyridin-2-yl)methylene)-4-(thiazol-2-yl)piperazine-1-carbothiohydrazide (HL) featuring a thiazole spectator for the efficient coordination with Cu(II) to give [CuCl(L)]2 (1) and [Cu(NO3)(L)]2 (2). Both 1 and 2 exhibit dimeric structures ascribed to the presence of di-2-pyridylketone moieties that demonstrate dual functions of chelation and intermolecular bridging. HL, 1 and 2 are highly toxic against hepatocellular carcinoma cell lines Hep-G2, PLC/PRF/5, and HuH-7, with half maximal inhibitory concentration (IC50) values as low as 3.26 nmol/mL (HL), 2.18 nmol/mL (1), and 2.54 × 10−5 nmol/mL (2) for PLC/PRF/5. While the free ligand HL may elicit its anticancer effect via sequestration of bio-relevant metal ions (i.e. Fe3+ and Cu2+), 1 and 2 are also capable of generating cytotoxic reactive oxygen species (ROS) to inhibit cancer cell proliferation. Our preliminary pharmacokinetic studies revealed that oral administration (per os, PO) of HL has a significantly longer half-life t1/2 of 21.61 ± 9.4 h, nearly doubled as compared to that of the intravenous (i.v.) administration of 11.88 ± 1.66 h, certifying HL as an effective chemotherapeutic drug via PO administration.
Keywords:
Thiosemicarbazone
; anticancer drug
; copper complex
; reactive oxygen species
; pharmacokinetics
1. Introduction
Thiosemicarbazones (TSCs) are a class of Schiff-base derivatives that exhibit diverse biologically beneficial activities, such as antibacterial, antiviral, and enzyme inhibitory activities [1,2]. Most notably, TSCs have long been reported as a highly promising class of drugs for a broad-spectrum of cancers through a wide range of anticancer mechanisms, such as ribonuclease reductase and topoisomerase II inhibition [3,4,5,6]. Other mechanisms include the quick sequestration of cell-proliferative-dependent ions (i.e., Fe3+ and Cu2+) to elicit the anticancer effect [4]. The anticancer effect of TSCs is primarily linked to, and profoundly affected by, their structures that feature an S, N chelator [4,5]. In this regard, additional introduction of an N coordination site, such as α-pyridyl serves as an advantage as it forms a more stable N, N, S pincer-like chelation that results in more effective ion removal [7]. In this sense, TSCs are particularly effective for treating iron overload diseases such as leukemia and neuroblastoma [8]. Recently findings also suggested that, upon capturing of metal ions by TSCs, a different anticancer mechanism based on reactive oxygen species (ROS) chemistry can be commenced by taking advantage of the unique characteristics of the tumor microenvironment (TME), such as the overexpression of H2O2 and GSH [9,10,11,12]. The ROS generation can subsequently strengthen the chemotherapeutic outcome of TSCs.
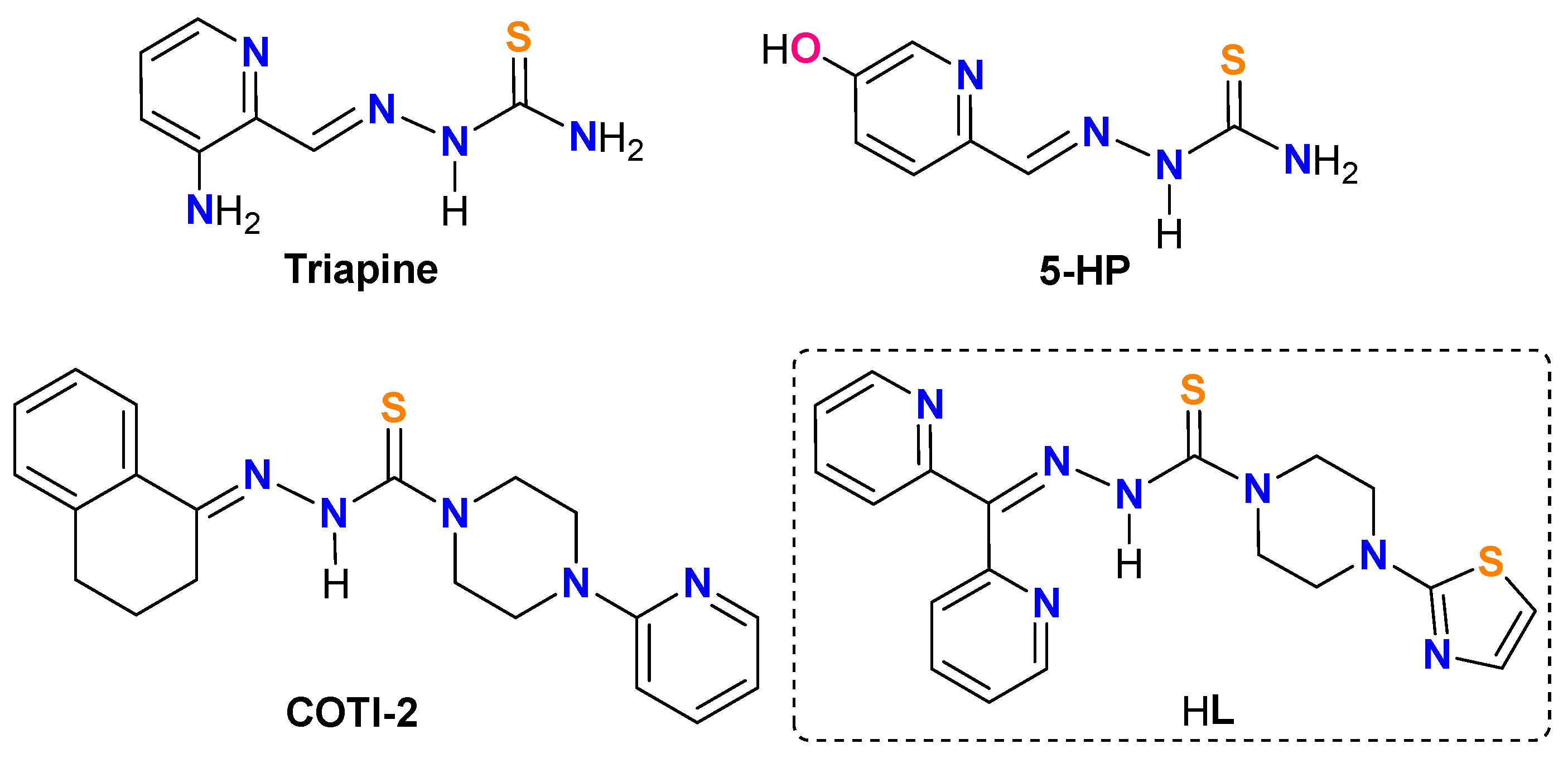
The promising potential of TSCs as anticancer drugs has been manifested by several drugs that successfully entered clinical trials, such as 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine), 5-hydroxypicolinaldehyde thiosemicarbazone (5-HP), and 4-(pyridine-2-yl)-N-([(8E)-5,6,7,8-tetrahydroquinolin-8-ylidene]amino)piperazine-1-carbothioamide (COTI-2) (Chart 1). Nevertheless, numerous side effects were also associated with these drugs, i.e., the formation of methemoglobin hypoxia by Triapine[13], severe hematological and gastrointestinal side effects for 5-HP[14], and drug resistance and other adverse effects (i.e., nausea, vomiting, and fatigue) for COTI-2 [15,16].
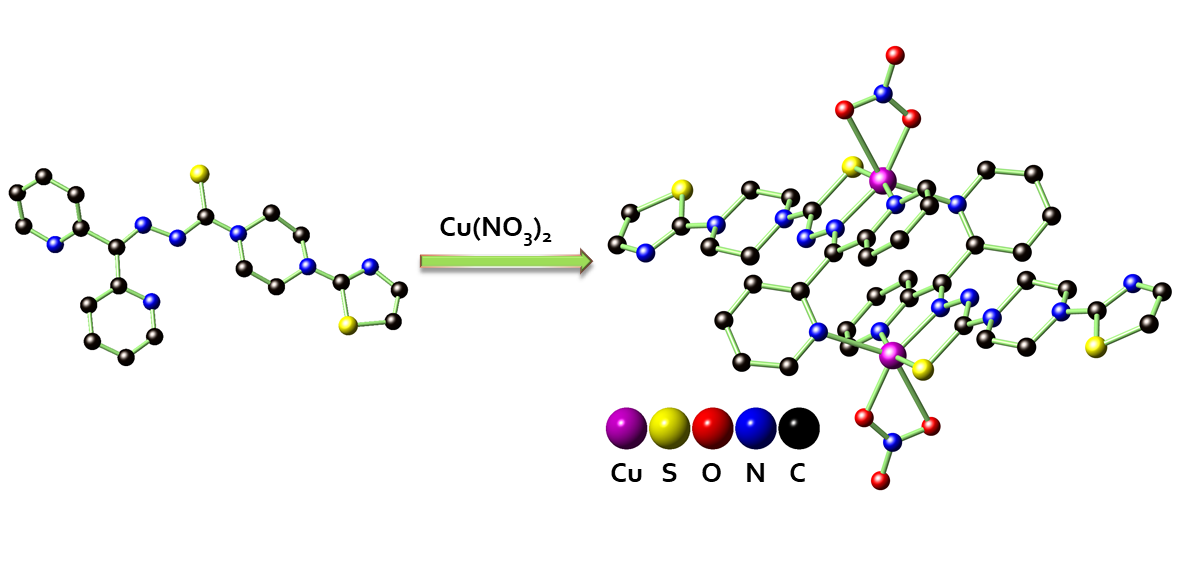
In this work, we report the synthesis of a novel TSC-based anticancer drug of N′-(di(pyridin-2-yl)methylene)-4-(thiazol-2-yl)piperazine-1-carbothiohydrazide (HL) featuring di-2-pyridylketone for chelating-bridging and a thiazole spectator (Chart 1). Ligand HL can readily associate Cu2+ to give the corresponding coordination complexes of [CuCl(L)]2 (1), and [Cu(NO3)(L)]2 (2). The structures of HL, 1, and 2 were confirmed by various spectroscopic techniques and ultimately authenticated by single-crystal X-ray diffraction studies. HL, 1, and 2 are highly toxic against hepatocellular carcinoma cell lines Hep-G2, PLC/PRF/5, and HuH-7. Notably, these species are extremely toxic for PLC/PRF/5, with the corresponding half maximal inhibitory concentration (IC50) values as low as 3.26 nmol/mL (HL), 2.18 nmol/mL (1), and 2.54 × 10−5 nmol/mL (2). Our preliminary pharmacokinetic studies of HL further revealed that oral administration (per os, PO) exhibits a significantly longer half-life t1/2 of 21.61 ± 9.4 h as compared to that of the intravenous (i.v.) administration of 11.88 ± 1.66 h. These results highlight that HL and its relevant functional coordination complexes can be promising chemotherapeutic drugs for oral administration.
2. Results and Discussion
2.1. Synthesis and X-ray Structure Characterization of HL, [CuCl(L)]2 (1), and [Cu(NO3)(L)]2 (2)
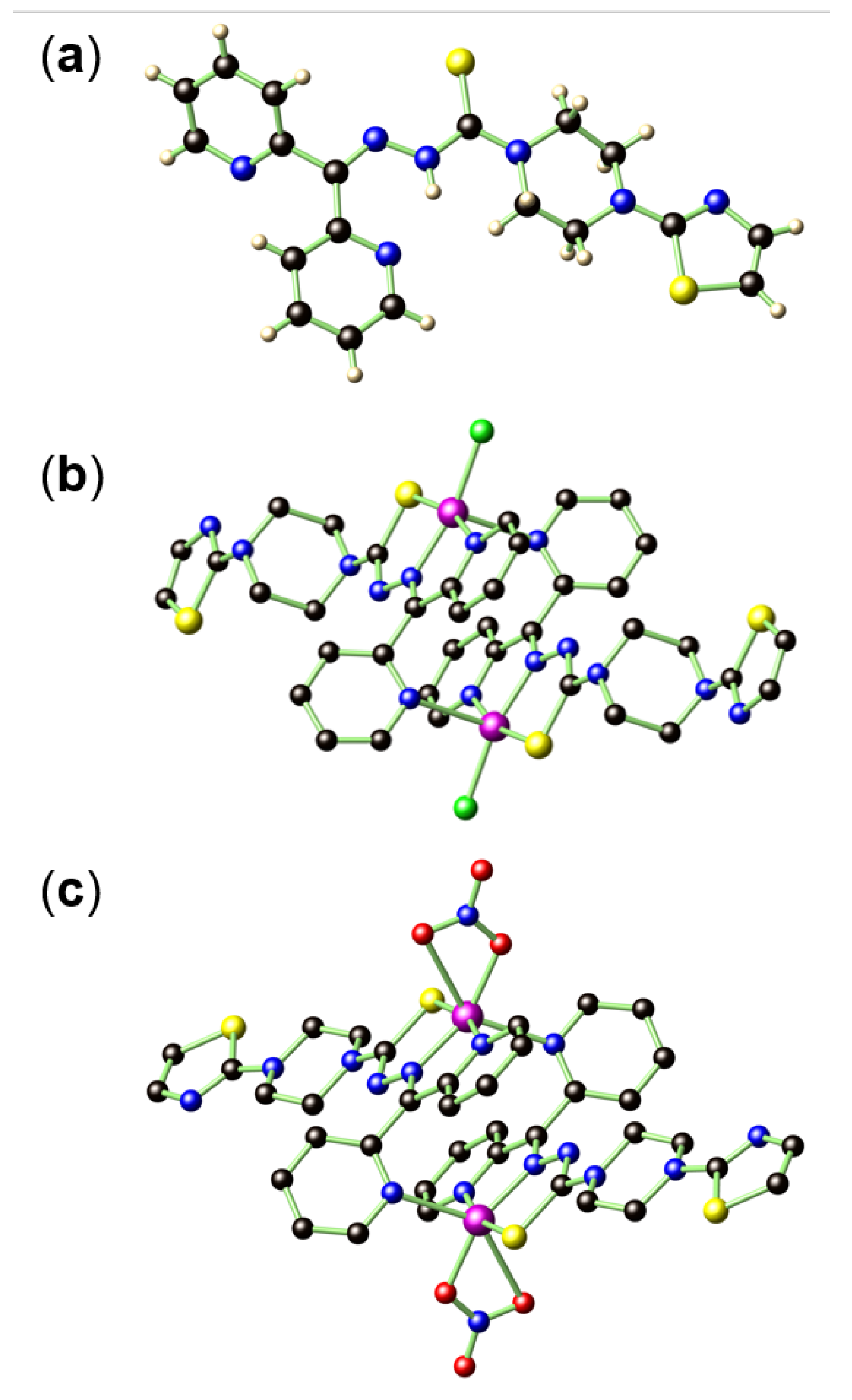
The synthesis of ligand HL was achieved by a five-step protocol to give an overall yield of 9.3% (Scheme 1). The purity of HL was confirmed by microanalysis, 1H, and 13C nuclear magnetic resonance (NMR) spectroscopy (Figure S1). The assumed connectivity of HL was also unambiguously determined by single-crystal X-ray diffraction studies (Figure 1a).
Coordination complexes of 1 and 2 were prepared accordingly by reacting HL and the respective Cu(II) sources under appropriate conditions (Scheme 2). Single crystal X-ray structure data for 1 (Figure 1b) and 2 (Figure 1c) indicated that both compounds form dimeric structures in the solid state, enabled by the head-to-tail complementary Cu−N coordination. In 1, each Cu2+ shows a square pyramidal coordination geometry, fulfilled by an S, one azomethinic N, one α-pyridyl N atom from an L ligand, and one Cl− that defines the square plane, in addition to one pyridyl N atom from adjacent L ligand that occupies the apical position. A similar connectivity is also found in 2 except that one Cl− is replaced by a chelating NO3−, yielding a distorted octahedral coordination geometry for Cu2+.
Notably, the ligands in both 1 and 2 undergo a change of conjugation with the loss of hydrazinic proton and the conversion of C=S into C−S bond. Such structure variation is seen in a variety of transition metal complexes of α-pyridyl thiosemicarbazone chelators, such as Triapine complexes of Fe3+/Ga3+[17,18], di-2-pyridyl ketone 4,4-dimethyl-3-thiosemicarbazone (HDp44mT) complexes of Fe3+[19], as well as several Cu2+ complexes [20]. Bormio Nunes et al. also reported that, for Fe3+/Cu2+ complexes of COTI-2, the N−N single bond of the ligand is inherited in the final chelating complexes [16]. As a result of the C=S to C−S bond variation, the C=S bond distance in HL of 1.681(5) Å increased to 1.739(4) Å in 1 and 1.744(6) Å in 2 (Table S1). Nevertheless, the N−N distance in HL of 1.354(5) Å remained nearly unaltered as compared to the N=N distances in 1 (1.354(4) Å) and 2 (1.351(6) Å) due to the additional coordination of one N to Cu2+.
2.2. Spectroscopic Characterizations of HL, 1, and 2
Energy dispersive X-ray spectroscopy (EDS) revealed an atomic ratio of Cu : Cl : S of 2.0 : 2.2 : 4.2 in 1 and Cu : S of 2.0 : 4.6 in 2 (Figure S2), consistent with the derived ratio of Cu : Cl : S of 1.0 : 1.0 : 2.0 (1) and Cu : S of 1.0 : 2.0 (2) from single-crystal data, and the elements Cu, S, N and C are evenly distributed in their elemental mapping diagrams (Figure S2). The Fourier-transform infrared (FT-IR) spectra show that upon coordination with Cu2+, the C=N bond vibration peaked at 1571 cm−1 in HL shifts to 1593 cm−1 for both 1 and 2 (Figure S3). Meanwhile, bands at 850 cm−1 (1) and 856 cm−1 (2) which were assignable as C=S bond vibrations, also shifted as compared to that of HL (869 cm−1) upon bonding with Cu2+ [21,22]. In the ultraviolet-visible (UV-Vis) spectroscopy (Figure 2), ligand-to-metal charge transfer (LMCT) bands in 1 and 2 are found at 426 nm as compared to HL [23]. Meanwhile, the ligand-centered UV-Vis absorption band of the HL undergoes a significant blue shift due to the conjugation change and charge transfer reactions. The LMCT leads to the partial reduction of Cu2+ which is favorable for Cu+-induced reactions, such as the ROS generation via the Fenton-like process [24]. The Cu 2p X-ray photoelectron spectroscopy (XPS) of both 1 and 2 revealed two sets of binding energies (Figure 3). Peaks at 933.8/953.5 eV (1) and 933.8/953.8 eV (2) are assignable as the spin-orbit splitting of Cu 2p3/2 and 2p1/2 of Cu2+ [25,26,27]. Two satellite peaks at 943.3/963.3 eV (1) and 943.4/963.0 eV (2) are also attributed to Cu2+ ions due to its d9 electronic configuration that is susceptible to photoreduction to give a more stable d10 configuration.
2.3. Hydroxyl Radical (•OH) Production
The generation of •OH from H2O2 by HL, 1, and 2 was assayed by 3,3′,5,5′-tetramethylbenzidine (TMB) [12,28,29]. The colorless TMB can be quickly oxidized by •OH to give a characteristic blue charge-transfer complex oxTMB with intense absorption at 652 nm [30,31]. Different concentrations of HL (5, 10, 20, 30 μg/mL) or 1 and 2 containing equivalent HL were treated with (TMB) solution containing 100 μM H2O2, which is assumed as approximately the concentration reported to be present in tumor cells, or five times that in normal cells [32,33]. After 20 min of incubation with HL, there is no oxTMB found as suggested by both the UV-Vis and the direct observation of solution colors (Figure S4). In sharp contrast, both 1 and 2 can induce the generation of •OH under otherwise identical conditions in a concentration-dependent manner with 2 further outperforms 1 (Figure 4, and Figure S4). In addition, the MeOH solutions of HL, 1, and 2 at HL / L concentrations of 30 μg/mL remained stable upon keeping for 72 h (Figure S5), as evidenced by their unaltered UV-Vis absorption patterns throughout the experiments.
2.4. CCK-8 Assay for HL, 1, and 2
The cytotoxicity of HL, 1, and 2 against three hepatocellular carcinoma cell lines Hep-G2, PLC/PRF/5, and HuH-7 were evaluated by cell counting kit-8 (CCK-8) assay. As shown in Figure 5, all three complexes demonstrate superior cytotoxicity originating from the ligand, particularly for the PLC/PRF/5 cell line. Notably, 1 and 2 demonstrated higher cytotoxicity as compared to the HL, presumably due to the additional ROS generation induced by the overexpressed H2O2 in the cancer cells. The IC50 values for HL, 1, and 2 against PLC/PRF/5 were as low as 3.26 nmol/mL, 2.18 nmol/mL, and 2.54 × 10−5 nmol/mL, which is favorably lower than those against Hep-G2 (78.83 nmol/mL for HL, 38.11 nmol/mL for 1, and 16.86 nmol/mL for 2), and HuH-7 (192.20 nmol/mL for HL, 176.60 nmol/mL for 1, and 87.53 nmol/mL for 2) cell lines (Table S2).
2.5. Intracellular ROS Generation by HL, 1, and 2
Given the efficient •OH production in water induced by 1 and 2, we next investigated in vitro ROS generation by 1 and 2 in the representative HuH-7 cell line using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as a fluorescence probe, and compared to that of HL [34,35]. The non-emissive DCFH-DA can readily be uptaken by the cell, where it is hydrolyzed into 2,7-dichlorodihydrofluorescein (DCFH) by cellular enzymes. Upon oxidation by ROS, DCFH can convert to 2,7-dichlorofluorescein (DCF) which gives bright green fluorescence [29,36]. As shown in Figure 6, HL alone produced negligible ROS. In sharp contrast, 1 and 2 can induce the formation of strong fluorescence characterizing the effective generation of ROS in the HuH-7 cells.
2.6. In Vivo Pharmacokinetic Study of HL.
For pharmacokinetic studies, CD®(SD) IGS Rats (5−6 weeks old) were randomly divided into two groups (3 for each group) for oral administration (per os, PO) or intravenous administration (i.v.) of HL with dosages of 30 mg kg−1 and 0.5 mg kg−1, respectively (Tables S4 and S5). The blood was then collected from the orbital sinus with a heparinized syringe at different time intervals (0.5, 1.0, 2.0, 4.0, 8.0, and 24 h for PO, and 0.03, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h for i.v.). High-performance liquid chromatography (HPLC) gives the mean area under the curve (AUC0-inf) values of 10409.01 ± 5313.4 ng h mL−1 for PO, and 5701.36 ± 2647.15 ng h mL−1 for i.v. HL achieved the largest Cmax value of 425.93 ± 176.1 ng mL−1 at a Tmax of 1.0 h for PO, while for i.v., the Cmax value of 806.43 ± 46.66 ng mL−1 was immediately observed at Tmax of 0.03 h, which is expected for i.v. administration. Notably, the PO administration exhibits a significantly longer half-life t1/2 of 21.61 ± 9.4 h, nearly doubled as compared to that of the i.v. administration of 11.88 ± 1.66 h (Figure 7), certifying that HL has a favorable pharmacokinetic via PO administration.
3. Materials and Methods
3.1. General
tert-Butyl piperazine-1-carboxylate (BOC-PIP; ≥99%, Aladdin), 2-bromothiazole (98%, Crgent Biotech), di-2-pyridyl ketone (98%, Crgent Biotech), NH2NH2 (≥95%, adamas), Cu(NO3)2 (≥99%, Aladdin), CuO (≥97%, Aladdin), DMF (≥99%, Aladdin), CH2Cl2 (≥99%, Aladdin), trifluoroacetic acid (TFA; ≥99%, Aladdin), i-PrOH (≥99%, Aladdin), methyl tert-butyl ether (MTBE, ≥99%, Aladdin), NaHCO3 (>99%, Crgent Biotech), EtOH (≥95%), and Cs2CO3 (≥99%, Crgent Biotech) were commercially available and used without further purifications.
Hep-G2, and PLC/PRF/5, and HuH-7 cell lines were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Phosphate buffer solution (PBS) was purchased from Shanghai Basal Media Technologies Co., Ltd. Cell culturing medium MEM (with NEAA + 10% FBS + 1% P/S), MEM (with NEAA + 1% P/S), DMEM (10% FBS + 1% P/S), McCoy’s 5A (10% FBS + 1% P/S), DMEM (1% P/S), and 0.25% trypsin solution (containing EDTA, dissolved in PBS) were purchased from Procell Life Science & Technology Co., Ltd. The cell counting kit-8 (CCK-8) and the reactive oxygen species detection kit were from APEXBIO and Shanghai Beyotime Biotechnology Co., Ltd., respectively.
1H nuclear magnetic resonance (NMR) spectra were obtained on a Varian UNITY plus-400/plus-600 NMR spectrometer (Varian, Inc., Palo Alto, CA, USA). Fourier transform infrared (FT-IR) spectra were measured on a Bruker VERTEX 70+HYPERION 2000 FT-IR spectrometer (Bruker AXS GmbH, Germany) using the attenuated total refraction (ATR) technique. Elemental analyses for C, H, and N were measured on a Carlo-Erba CHNO-S microanalyzer (Carlo Erba, Waltham, MA, USA). Ultraviolet-visible (UV-Vis) spectroscopy was obtained on a Varian Cary-50 UV-visible spectrophotometer (Varian, Inc., Palo Alto, CA, USA). X-ray photoelectron spectroscopy (XPS) was performed on an EXCALAB 250 XI X-ray photoelectron spectrometer (Thermo Scientific, Waltham, MA, USA). The high performance liquid chromatography (HPLC) were carried out on an Angilient 1260 Infinity II Bio-SEC system (Agilent Technologies, Inc., CA, USA). The CCK-8 cytotoxicity assay was conducted on a multifunction microplate detector by recording the absorption at 450 nm using a TECAN M1000PRO microplate reader (Tecan, Zürich, Switzerland).
3.2. Synthetic Steps for HL
Ligand HL can be synthesized via a five-step process as indicated below.
Step 1. Synthesis of S1. t-Butyloxy carbonyl (BOC) protected piperazine (BOC-PIP; 400.0 mg, 2.148 mmol) and 2-bromothiazole (529.0 mg, 3.221 mmol) were dissolved in DMF (4 mL) and stirred for 5 min. Cs2CO3 (1400 mg, 4.295 mmol) was added as a solid and the mixture heated overnight at 120°C. The formed product was extracted from H2O using ethyl acetate to obtain S1 as a white solid. Yield (290 mg, 50% based on BOC-PIP). 1H NMR (400 MHz, CDCl3) δ 7.209 (s, 1H), 6.605 (s, 1H), 3.566 (s, 4H), 3.466 (s,4H), 1.483 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 172.096, 154.601, 139.567, 107.814, 80.273, 48.456, 28.396.
Step 2. Synthesis of S2. Compound S1 (290.0 mg, 1.078 mmol) was dissolved in CH2Cl2 (4 mL) to obtain a light yellow-brown solution and trifluoroacetic acid (TFA, 12 mL) was subsequently added, and the solution quickly turned yellow upon stirring. The mixture was stirred for 2 h and the solvent evaporated, DCM / MeOH = 2 : 1 and NaHCO3 were consequently introduced to obtain S2 as a light yellow powder. Yield (180 mg, 99% based on S1). 1H NMR (400 MHz, DMSO-d6) δ 9.845 (s, 1H), 7.360 (s, 1H), 7.088 (s, 1H), 5.039 (s, 7H), 3.843 (s, 4H), 3.257 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.534, 133.871, 110.024, 46.268, 41.908.
Step 3. Synthesis of S3. Complex S2 (180.0 mg, 1.065 mmol) was added to CH2Cl2 (5 mL) to form a suspension, and Cs2CO3 (342.0 mg, 1.917 mmol) was introduced to give a yellow solution. The mixture was stirred at r.t. for 15 h, and the product was washed with H2O and extracted with CH2Cl2. The organic layers were combined and dried. The pure yellow compound of S3 can be isolated by chromatography using CH2Cl2/i-PrOH (25 : 1, v/v) as the eluent. Yield (270 mg, 91% based on S2). 1H NMR (400 MHz, CDCl3) δ 7.904 (s, 1H), 7.234 (s, 2H), 7.111 (s, 1H), 6.683 (s, 1H), 4.040 (s, 4H), 3.656 (s, 4H). 13C NMR (101 MHz, CDCl3) δ 179.348, 170.996, 139.720, 137.427, 130.303, 119.252, 108.941, 50.694, 48.032.
Step 4. Synthesis of S4. Compound S3 (270.0 mg, 0.968 mmol) was added to EtOH (4 mL) to form a suspension, and NH2NH2 (0.0063 mL, excess) was subsequently added. The mixture was heated to reflux at 80°C to yield a white suspension of S4 which was purified by chromatography using CH2Cl2/MeOH (25 : 1, v/v) as the eluent. Yield (175 mg, 74% based on S3). 1H NMR (400 MHz, DMSO-d6) δ 9.225 (s, 1H), 7.189 (s, 1H), 6.881 (s, 1H), 4.802 (s, 2H), 3.882 (s, 4H), 3.417 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 183.080, 171.525, 139.917, 108.901, 48.052, 46.913.
Step 5. Synthesis of HL. Compound S4 (40.0 mg, 0.165 mmol) was dissolved in EtOH (10 mL) and stirred for 10 min. Di-2-pyridyl ketone (34.0 mg, 0.181 mmol) was subsequently introduced and the mixture was heated to reflux at 80°C overnight. Upon cooling to r.t., the crude powder of HL was recrystallized with MeOH / MTBE (5 : 1, v/v) to give a pure product of yellow-green solid of HL. Yield (18.8 mg, 28% based on S4). Single crystals of HL were grown by slow evaporation of a MeOH/DMSO (v/v = 1 : 1) solution of HL. Anal. Calcd for C19H19N7S2: C 55.72, H 4.68, N 23.94; found: C 55.26; H 4.93; N, 23.49. IR (ATR, cm−1): 3053(w), 2842(w), 1572(m), 1519(vs), 1487(s), 1464(s), 1382(s), 1339(m), 1305(vs), 1279(vs), 1220(vs), 1204(vs), 1130(vs), 1104(s), 1048(s), 1016(vs), 995(vs), 962(s), 916(s), 898(w), 870(m), 858(w), 802(vs), 785(s), 762(m), 743(vs), 723(vs), 693(vs), 669(m), 656(m), 642(s), 618(s). 1H NMR (400 MHz, DMSO-d6) δ 14.64 (s, 1H), 8.89 (s, 1H), 8.61 (s, 1H), 7.98 (s, 3H), 7.60 (s, 3H), 7.20 (s, 1H), 6.89 (s, 1H), 4.16 (s, 4H), 3.32 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.77, 148.40, 147.95, 139.47, 137.79, 137.30, 126.84, 124.64, 123.78, 108.38, 48.33, 47.45.
3.3. Synthesis and Characterization of [CuCl(L)]2 (1)
Method 1: HL (100 mg, 0.244 mmol) was introduced in 20 mL MeOH to give a yellow suspension, and CuCl2•2H2O (42 mg, 0.246 mmol) in 10 mL MeOH was subsequently introduced dropwise. The formed mixture was stirred at 40°C for 8 h. The solvent was removed, and the formed brown powder was recrystallized in MeOH and Et2O to obtain the crystal of 1.
Method 2: CuO (21.4 mg, 0.269 mmol) and HCl (19.6 mg, 0.537 mmol) were mixed in 3 mL of MeOH and the mixture stirred at 40°C. With the gradual dissolution of CuO, the solution turned pale green. After 10 min, HL (100 mg, 0.244 mmol) was introduced and the solution became dark brown. The mixture was stirred for an additional 2 h and then filtered, washed with a mixture of MeOH/MTBE (1 : 1, v/v; MTBE = methyl tert-butyl ether) to give a brown powder of [CuCl(L)]2 which was dried under vacuo. Yield (220 mg, 89% based on HL). Anal. Calcd for C38H40Cl2Cu2N14S4: C 44.79, H 3.96, N 19.24; found: C 44.18, H 3.37, N 18.96. IR (ATR, cm−1): 3077(w), 2993(w), 2645(w), 1592(s), 1563(m), 1476(w), 1458(w), 1440(vs), 1418(s), 1383(s), 1364(vs), 1318(w), 1289(m), 1275(s), 1247(vs), 1193(m), 1169(w), 1130(s), 1089(w), 1058(w), 1012(s), 982(w), 966(w), 956(w), 923(s), 850(m), 812(m), 795(s), 772(w), 738(m), 709(m), 686(w), 664(s), 644(s), 612(m).
3.4. Synthesis and Characterization of [Cu(NO3)(L)]2 (2)
Cu(NO3)2 (50.5 mg, 0.269 mmol) and HL (100 mg, 0.244 mmol) were mixed in 3 mL MeOH and the mixture was stirred at 40°C to obtain a dark brown solution, and upon further stirring for 2 h, the solution turned dark green. The mixture was filtered and washed with a mixture of MeOH/MTBE (1 : 1, v/v; MTBE = methyl tert-butyl ether) to give a brown powder of 2 which was dried under vacuo. Single crystals were obtained by slow diffusion of Et2O into a MeOH solution of 2. Yield (235 mg, 90% based on HL). Anal. Calcd for C38H36Cu2N16O6S4: C 42.73, H 3.40, N 20.98; found: C 42.38, H 3.33, N 20.81. IR (ATR, cm−1): 3080(w), 2846(w), 1592(m), 1520(s), 1483(s), 1461(vs), 1424(vs), 1380(s), 1338(w), 1278(vs), 1239(vs), 1203(vs), 1140(vs), 1102(m), 1052(w), 1012(vs), 920(s), 897(w), 855(m), 819(m), 792(s), 744(m), 724(w), 700(m), 665(m), 615(m).
3.5. Single Crystal X-ray Crystallography
Diffraction data for HL, 1, and 2 were acquired either on a Bruker APEX II CCD X-ray diffractometer (Bruker AXS GmbH, Germany) using Mo-Kα (λ = 0.71073 Å) (HL) or Ga-Kα (λ = 1.34138 Å) irradiation (1 and 2). Refinement and reduction of the collected data were achieved using the program Bruker SAINT and absorption corrections were performed using a multi-scan method [37]. All the crystal structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques with SHELXTL-2016 [38]. Crystallographic data for HL, 1, and 2 have been deposited in the Cambridge Crystallographic Data Center (CCDC) as supplementary publication numbers 2341904 (HL), 2341905 (1), and 2341906 (2). These data can be obtained free of charge either from the CCDC via www.ccdc.cam.ac.uk/data_request/cif or from the Supporting Information. A summary of the key crystallographic data for HL, 1, and 2 is listed in Table 1. Selected bond distances and angles were listed in Table S1.
3.6. In Vitro Cytotoxicity Evaluation by CCK-8 Assay
The HuH-7 cell line was cultured in DMEM + 10% FBS + 1% P/S and DMEM + 1% P/S. Cells grew as a monolayer and were detached upon confluence using trypsin (0.5% w/v in PBS). The cells were harvested from the cell culture medium by incubating in trypsin solution for 3 min, and then centrifuged with the supernatant subsequently discarded. 3 mL portion of serum-supplemented cell culture medium was added to neutralize any residual trypsin. The cells were re-suspended in serum-supplemented DMEM at a concentration of 5 × 104 cells/mL. Cells were cultured at 37°C and 5% CO2 for the CCK-8 studies. HuH-7 cells were seeded at a density of 2 × 104 cells per well in 90 µL of culture medium (DMEM + 10% FBS + 1% P/S), and cultured for 24 h at 37°C and 5% CO2 for attachment. The culture medium was then replaced by a serum-free medium (DMEM + 1% P/S) containing various concentrations of HL, 1, and 2 (Pharmaceuticals solubilized with 2 parts per thousand DMSO). All experiments were carried out with three replicates (n = 3), and the untreated cells served as the 100% cell viability control, while the cell-free medium (DMEM + 10% FBS + 1% P/S + CCK-8) served as the blank. HuH-7 cells were directly incubated for a period of 72 h. After incubation, 100 μL of culture medium (DMEM + 10% FBS + 1% P/S) and 10 μL CCK-8 was introduced and cultured for an additional 2 h before spectrophotometric measurement at 450 nm on a microplate reader. The relative cell viability (%) related to control cells was calculated using the equation:
in which V% is the percentage of cell viability, [A]experimental is the absorbance of the wells culturing the treated cells, [A]blank is the absorbance of the blank, and [A]control is the absorbance of the wells culturing untreated cells. The cytotoxicity assessment for Hep-G2 and PLC/PRF/5 resembled that of HuH-7 except that the culturing media was different, viz MEM (NEAA) + 10% FBS + 1% P/S and MEM (NEAA) + 1% P/S for both Hep-G2 and PLC/PRF/5.
3.7. Detection of Intracellular Reactive Oxygen Species
HuH-7 cells were introduced into a 12-well plate (4 × 105 per well) in 1 mL of growing media (DMEM + 10% FBS + 1% P/S) and the cells were incubated at 37°C under 5% CO2 for 24 h for attachment. The growing media was removed and the wells were replenished with HL, 1, and 2 in 1 mL growing media (with 2 ppm of DMSO) with equivalent HL concentrations of 2 μg/mL. The cells were incubated for an additional 2 h and then the culturing media was replaced with 10 μmol/mL DCFH-DA in DMEM + 1% P/S (1 mL), prepared by diluting (1 : 1000) a concentrated DCFH-DA following the supplied protocols. The cells were incubated for a further 20 min and then the culturing media was removed. The cells were further rinsed with culturing media (DMEM + 1% P/S) three times to remove the residual DCFH-DA, and observed under a BD5000 inverted microscope to estimate and compare the ROS generation abilities of these materials.
3.8. Pharmacokinetics of HL
CD®(SD) IGS Rats (5−6 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. All animal experiments were used in accordance with the requirements of the Experimental Animal Welfare Ethics Committee of Transcenta Diagnostic Technology (Suzhou) Co., LTD (PZ-20211210001). For pharmacokinetic experiments, SD rats were randomly divided into two groups, with 3 for PO administration (30 mg kg−1) and i.v. administration (0.5 mg kg−1), respectively. Plasma was collected from the jugular vein with the presence of a heparinized syringe at different time intervals, viz. 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h for PO, and 0.03, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h for i.v. Plasma was first centrifuged at 3500 rpm for 10 min, then 0.05mL of the plasma was extracted, then trifluoroacetic acid was used to acid precipitate protein, and NaOH to neutralize the solution. The mixture was diluted using MeCN: H2O (5 : 5 v/v). After 10 min of precipitating, supernatant fluids were collected by centrifugation at 10000 rpm for 5 min and filtered with a syringe through a 0.22 μm hydrophilic membrane filter and measured by HPLC method. For the HPLC assay, the analytical column was an Agilent ZORBAX SB C18 column (4.6 mm × 150 mm, 5 μm. The mobile phase was MeCN: H2O (5:5 v/v), the flow rate of the mobile phase was 0.8 mL min−1 and the UV detector was at 245 nm.
4. Conclusions
In this work, we report that the thiosemicarbazone derivative of HL and its Cu(II) complexes 1 and 2 demonstrated superior anticancer performances against hepatocellular carcinoma cell lines Hep-G2, PLC/PRF/5, and HuH-7. For 1 and 2, the involvement of ROS as catalyzed by the Fenton-like process is also obvious. Intriguingly, the pharmacokinetic studies revealed that HL can be successfully absorbed via PO administration, with a favorable half-life of 21.61 ± 9.4 h, almost double that of via i.v. administration, rendering HL promising clinical potential. We believe that the association of such type of ligand with diverse metal ions may further equip the drug with additional beneficial features, such as the modulation of acute toxicity, toxicity via ROS, and on-demand metal supplements during the curation process.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, additional figures and tables.
Author Contributions
Conceptualization, W.H.Z. and G.Y.; methodology, S.Y.L. and C.M.Z.; software, W.H.Z. and C.M.Z.; validation, C.M.Z., P.X., W.H.Z. and G.Y.; formal analysis, S.Y.L. C.M.Z., and P.X.; data curation, Y.N. and M.L.D.; writing—original draft preparation, S.Y.L. and C.M.Z.; writing—review and editing, W.H.Z., and G.Y.; supervision, W.H.Z.; project administration, W.H.Z. and G.Y.; funding acquisition, W.H.Z. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to SuZhou LongMab Biosciences., Ltd. for the final support of this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Klayman, D.L.; Scovill, J.P.; Bartosevich, J.F.; Mason, C.J. 2-Acetylpyridine thiosemicarbazones. 2. N4,N4-Disubstituted derivatives as potential antimalarial agents. J. Med. Chem. 1979, 22, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, H.; Gambinob, D. The wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini. Rev. Med. Chem. 2004, 4, 31–39. [Google Scholar] [PubMed]
- Kalinowski, D.S.; Quach, P.; Richardson, D.R. Thiosemicarbazones: The new wave in cancer treatment. Future Med. Chem. 2009, 1, 1143–1151. [Google Scholar] [CrossRef]
- Shakya, B.; Yadav, N.P. Thiosemicarbazones as potent anticancer agents and their modes of action. Mini. Rev. Med. Chem. 2020, 20, 638–661. [Google Scholar] [CrossRef]
- Serda, M.; Kalinowski, D.S.; Rasko, N.; Potůčková, E.; Mrozek-Wilczkiewicz, A.; Musiol, R.; Małecki, J.G.; Sajewicz, M.; Ratuszna, A.; Muchowicz, A. , et al. Exploring the anti-cancer activity of novel thiosemicarbazones generated through the combination of retro-fragments: Dissection of critical structure-activity relationships. Plos One 2014, 9, e110291. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, J.R.; Hueting, R. Metal complexes of thiosemicarbazones for imaging and therapy. Inorg. Chim. Acta 2012, 389, 3–15. [Google Scholar] [CrossRef]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel di-2-pyridyl–derived iron chelators with marked and selective antitumor activity: In vitro and in vivo assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef]
- Richardson, D.R. Iron chelators as therapeutic agents for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Hu, P.; Shi, J. Nanomedicine remodels tumor microenvironment for solid tumor immunotherapy. J. Am. Chem. Soc. 2024, 146, 10217–10233. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Zou, Y.-M.; Li, R.-T.; Yu, L.; Huang, T.; Peng, J.; Meng, W.; Sun, B.; Zhang, W.-H.; Jiang, Z.-H.; Chen, J. , et al. Reprogramming of the tumor microenvironment using a PCN-224@IrNCs/d-Arg nanoplatform for the synergistic PDT, NO, and radiosensitization therapy of breast cancer and improving anti-tumor immunity. Nanoscale 2023, 15, 10715–10729. [Google Scholar] [CrossRef]
- Hou, Y.-K.; Zhang, Z.-J.; Li, R.-T.; Peng, J.; Chen, S.-Y.; Yue, Y.-R.; Zhang, W.-H.; Sun, B.; Chen, J.-X.; Zhou, Q. Remodeling the tumor microenvironment with core–shell nanosensitizer featuring dual-modal imaging and multimodal therapy for breast cancer. ACS Appl. Mater. Interfaces 2023, 15, 2602–2616. [Google Scholar] [CrossRef]
- Ma, B.; Goh, B.C.; Tan, E.H.; Lam, K.C.; Soo, R.; Leong, S.S.; Wang, L.Z.; Mo, F.; Chan, A.T.C.; Zee, B. , et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine®) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest. New Drugs 2008, 26, 169–173. [Google Scholar] [CrossRef]
- DeConti, R.C.; Toftness, B.R.; Agrawal, K.C.; Tomchick, R.; Mead, J.A.R.; Bertino, J.R.; Sartorelli, A.C.; Creasey, W.A. Clinical and pharmacological studies with 5-hydroxy-2-formylpyridine thiosemicarbazone. Cancer Res. 1972, 32, 1455–1462. [Google Scholar] [PubMed]
- Westin, S.N.; Nieves-Neira, W.; Lynam, C.; Salim, K.Y.; Silva, A.D.; Ho, R.T.; Mills, G.B.; Coleman, R.L.; Janku, F.; Matei, D. Abstract CT033: Safety and early efficacy signals for COTI-2, an orally available small molecule targeting p53, in a phase I trial of recurrent gynecologic cancer. Cancer Res. 2018, 78, CT033–CT033. [Google Scholar] [CrossRef]
- Bormio Nunes, J.H.; Hager, S.; Mathuber, M.; Pósa, V.; Roller, A.; Enyedy, É.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P. , et al. Cancer cell resistance against the clinically investigated thiosemicarbazone COTI-2 is based on formation of intracellular copper complex glutathione adducts and ABCC1-mediated efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef]
- Kowol, C.R.; Trondl, R.; Heffeter, P.; Arion, V.B.; Jakupec, M.A.; Roller, A.; Galanski, M.S.; Berger, W.; Keppler, B.K. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J. Med. Chem. 2009, 52, 5032–5043. [Google Scholar] [CrossRef] [PubMed]
- Kowol, C.R.; Berger, R.; Eichinger, R.; Roller, A.; Jakupec, M.A.; Schmidt, P.P.; Arion, V.B.; Keppler, B.K. Gallium(III) and iron(III) complexes of α-N-heterocyclic thiosemicarbazones: Synthesis, characterization, cytotoxicity, and interaction with ribonucleotide reductase. J. Med. Chem. 2007, 50, 1254–1265. [Google Scholar] [CrossRef]
- Richardson, D.R.; Sharpe, P.C.; Lovejoy, D.B.; Senaratne, D.; Kalinowski, D.S.; Islam, M.; Bernhardt, P.V. Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J. Med. Chem. 2006, 49, 6510–6521. [Google Scholar] [CrossRef]
- Bai, X.-G.; Zheng, Y.; Qi, J. Advances in thiosemicarbazone metal complexes as anti-lung cancer agents. Front. Pharmacol. 2022, 13, 1018951. [Google Scholar] [CrossRef]
- Quiroga, A.G.; M. Pérez, J.; López-Solera, I.; Montero, E.I.; Masaguer, J.R.; Alonso, C.; Navarro-Ranninger, C. Binuclear chloro-bridged palladated and platinated complexes derived from p-isopropylbenzaldehyde thiosemicarbazone with cytotoxicity against cisplatin resistant tumor cell lines. J. Inorg. Biochem. 1998, 69, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.A.C.; Costa, W.R.P.; de F. Faria, E.; Bessa, M.A.d.S.; Menezes, R.d.; Martins, C.H.G.; Maia, P.I.S.; Deflon, V.M.; Oliveira, C.G. Copper(II) complexes based on thiosemicarbazone ligand: Preparation, crystal structure, Hirshfeld surface, energy framework, antiMycobacterium activity, in silico and molecular docking studies. J. Inorg. Biochem. 2021, 223, 111543. [Google Scholar] [CrossRef] [PubMed]
- Carcelli, M.; Tegoni, M.; Bartoli, J.; Marzano, C.; Pelosi, G.; Salvalaio, M.; Rogolino, D.; Gandin, V. In vitro and in vivo anticancer activity of tridentate thiosemicarbazone copper complexes: Unravelling an unexplored pharmacological target. Eur. J. Med. Chem. 2020, 194, 112266. [Google Scholar] [CrossRef] [PubMed]
- Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: An emerging tool for sustainable organic synthesis. ChemCatChem 2022, 14, e202200916. [Google Scholar] [CrossRef]
- Cui, Y.; Ding, Y.; Molina, S.E.V.; Li, Y. A mixed-valence CuII/CuI coordination polymer based on bridged thiocyanate and in situ formed di(N-heterocyclic) sulfide: Synthesis, structure, and magnetic properties. J. Mol. Struct. 2021, 1241, 130623. [Google Scholar] [CrossRef]
- Meda, L.; Ranghino, G.; Moretti, G.; Cerofolini, G.F. XPS detection of some redox phenomena in Cu-zeolites. Surf. Interface Anal. 2002, 33, 516–521. [Google Scholar] [CrossRef]
- Zeng, C.-M.; Luo, S.-Y.; Wang, X.; Cao, F.-L.; Zhang, Z.-S.; Zhang, W.-H.; Dai, C.-L.; Young, D.J. A porphyrin-based 3D metal-organic framework featuring [Cu8Cl6]10+ cluster secondary building units: Synthesis, structure elucidation, anion exchange, and peroxidase-like activity. Chem. Asian J. 2024, 19, e202400237. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-J.; Yu, X.-Z.; Zhang, S.-Q.; Zhang, Y.-X.; Chen, X.-L.; Long, Z.-J.; Hu, H.-Z.; Xie, D.-H.; Zhang, W.-H.; Chen, J.-X. , et al. Hydrogel with ROS scavenging effect encapsulates BR@Zn-BTB nanoparticles for accelerating diabetic mice wound healing via multimodal therapy. iScience 2023, 26, 106775. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, B.-W.; Zou, Y.-M.; Niu, R.-J.; Chen, J.-X.; Zhang, W.-H.; Young, D.J. Nanoscale two-dimensional FeII- and CoII-based metal−organic frameworks of porphyrin ligand for the photodynamic therapy of breast cancer. Molecules 2023, 28, 2125. [Google Scholar] [CrossRef] [PubMed]
- Josephy, P.D.; Eling, T.; Mason, R.P. The horseradish peroxidase-catalyzed oxidation of 3,5,3′,5′-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J. Biol. Chem. 1982, 257, 3669–3675. [Google Scholar] [CrossRef]
- Yuan, B.; Chou, H.-L.; Peng, Y.-K. Disclosing the origin of transition metal oxides as peroxidase (and catalase) mimetics. ACS Appl. Mater. Interfaces 2022, 14, 22728–22736. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Yang, J.; Zheng, W.; Sun, J.; Wang, W.; Qian, H. Recent advances on modulation of H2O2 in tumor microenvironment for enhanced cancer therapeutic efficacy. Coord. Chem. Rev. 2023, 481, 215049. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, F.; Li, X.; Niu, G.; Yang, Y.; Li, H.; Jiang, Y. Tumor microenvironment-responsive Fenton nanocatalysts for intensified anticancer treatment. J. Nanobiotechnology 2022, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.-H.; Zeng, C.-M.; Zhang, Z.-S.; Cao, F.-L.; Zhang, W.-H.; Chen, J.-X.; Young, D.J. [FeIIICl(TMPPH2)][FeIIICl4]2: A stand-alone molecular nanomedicine that induces high cytotoxicity by ferroptosis. Molecules 2024, 29, 2495. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Tang, X.-Y.; Meng, W.; Lai, Y.-H.; Zhou, X.; Yu, X.-Z.; Zhang, W.-H.; Chen, J.-X. Immunogenic radiation therapy for enhanced antitumor immunity via a core–shell nanosensitizer-mediated immunosuppressive tumor microenvironment modulation. ACS Nano 2023, 17, 19853–19864. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, R.; Ye, Q.; Zou, Y.; Lu, X.; Zhang, W.; Chen, J.; Zhao, Y. Mn3O4 nanoshell coated metal–organic frameworks with microenvironment-driven O2 production and GSH exhaustion ability for enhanced chemodynamic and photodynamic cancer therapies. Adv. Healthc. Mater. 2023, 12, 2202280. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS (Version 2.03): Program for empirical absorption correction of area detector data; University of Göttingen: Germany, 1996. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
Chart 1.
Molecular diagrams of Triapine, 5-HP, COTI-2, and HL.
Scheme 1.
The five-step synthetic procedure for HL.
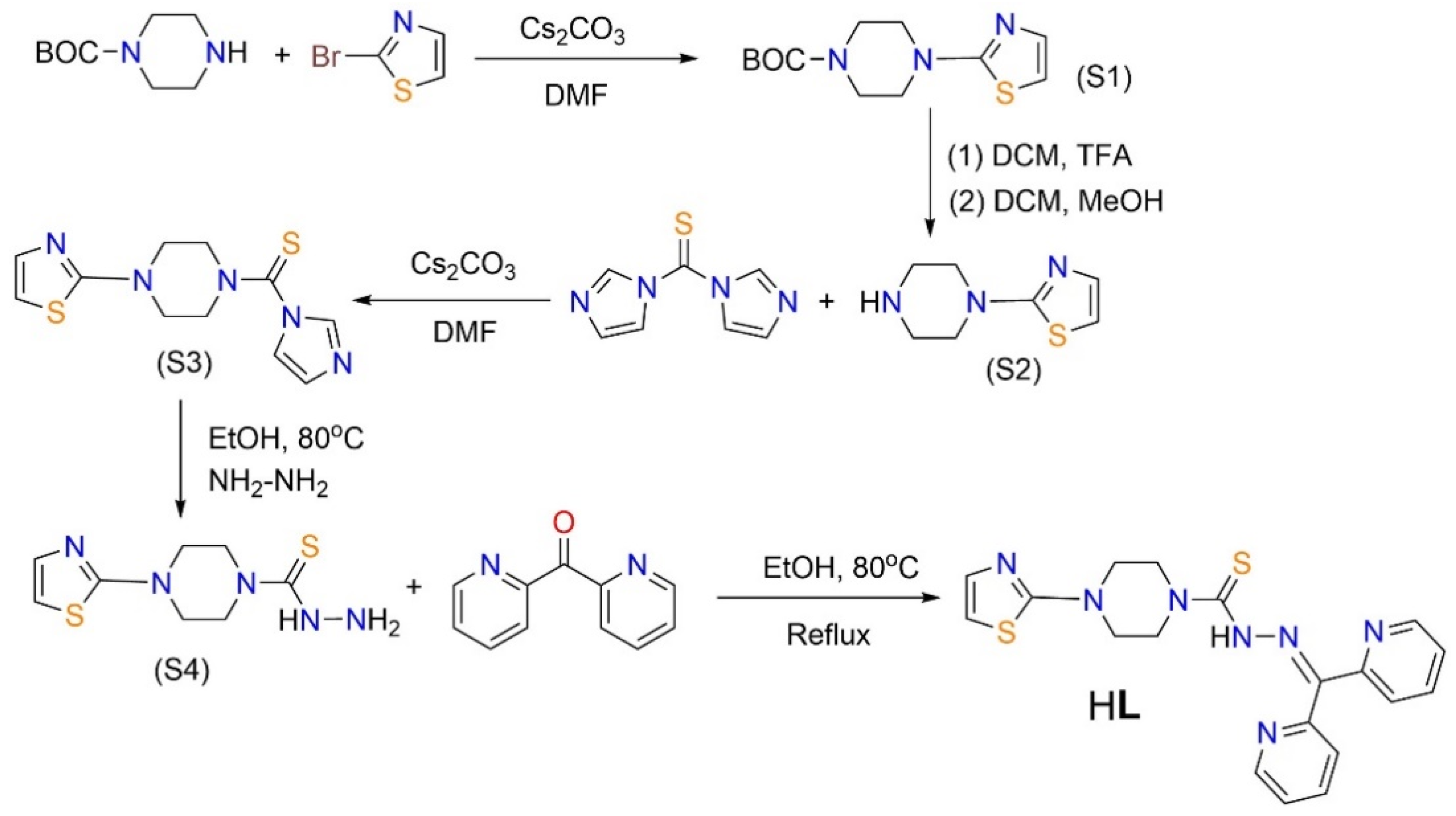
Figure 1.
The single crystal structure of HL (a), 1 (b), and 2 (c). In (b) and (c), the H atoms were also omitted for clarity. Color legend: Cu dark magenta, Cl green, S yellow, O red, N blue, C black, H light pink.
Figure 1.
The single crystal structure of HL (a), 1 (b), and 2 (c). In (b) and (c), the H atoms were also omitted for clarity. Color legend: Cu dark magenta, Cl green, S yellow, O red, N blue, C black, H light pink.
Scheme 2.
The synthetic procedure for 1 and 2 from HL.
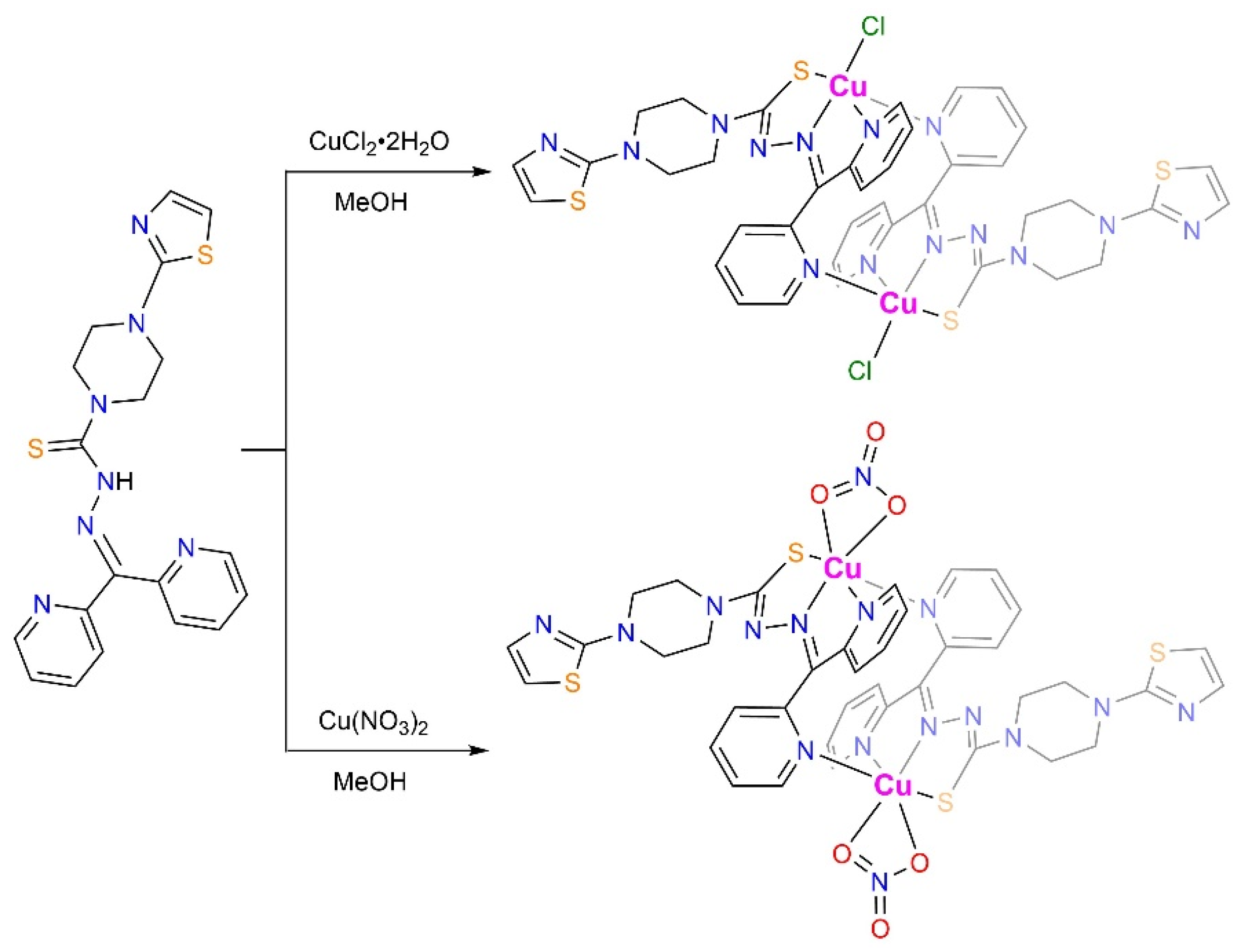
Figure 2.
The UV-Vis spectra of HL (light red), 1 (light orange), and 2 (light blue) in MeOH solutions.
Figure 2.
The UV-Vis spectra of HL (light red), 1 (light orange), and 2 (light blue) in MeOH solutions.
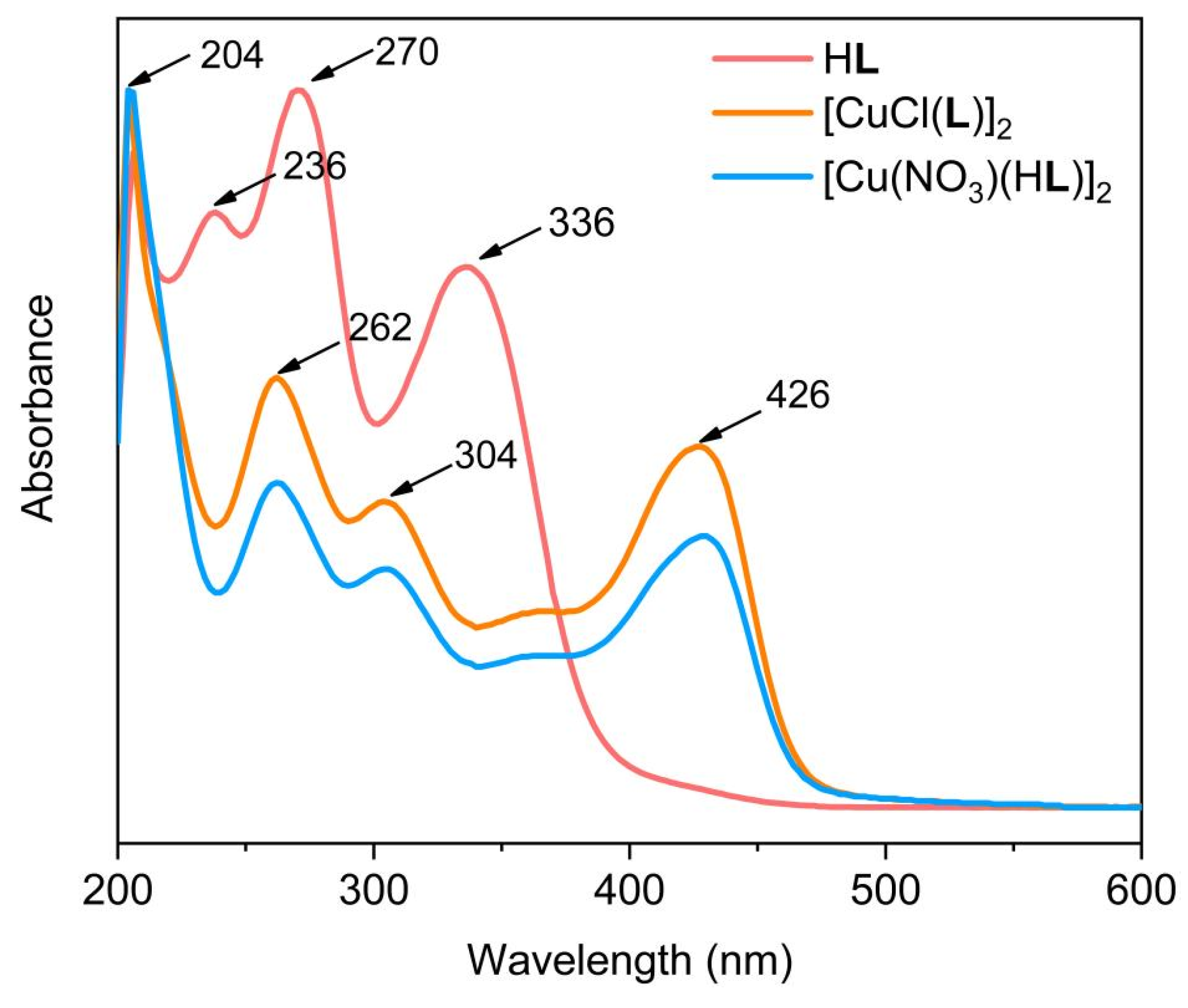
Figure 3.
The Cu 2p XPS spectra of 1 (a) and 2 (b).
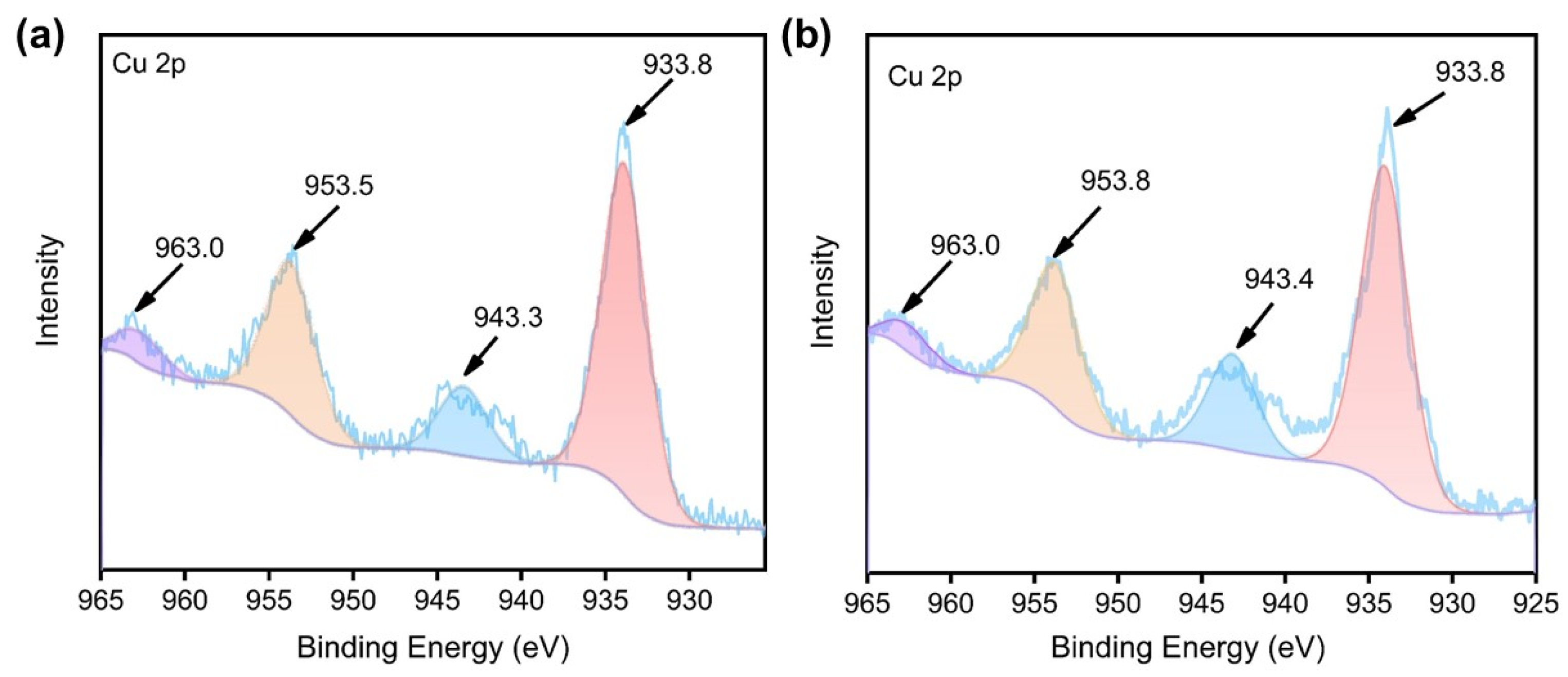
Figure 4.
Change of UV-Vis absorption intensity in the range of 500−750 nm upon treatment with different concentrations of 1 in the presence of 100 μM H2O2 (a; inset: photographs showing color change upon introducing gradient concentrations of 1). The intensity change at 652 nm (oxTMB) upon treating gradient concentrations of HL, 1, and 2 with 100 μM H2O2 (b).
Figure 4.
Change of UV-Vis absorption intensity in the range of 500−750 nm upon treatment with different concentrations of 1 in the presence of 100 μM H2O2 (a; inset: photographs showing color change upon introducing gradient concentrations of 1). The intensity change at 652 nm (oxTMB) upon treating gradient concentrations of HL, 1, and 2 with 100 μM H2O2 (b).
Figure 5.
The viability of Hep-G2 (a), PLC/PRF/5 (b), and HuH-7 (c) cells when treated with gradient concentrations of HL, 1, and 2 (n = 3).
Figure 5.
The viability of Hep-G2 (a), PLC/PRF/5 (b), and HuH-7 (c) cells when treated with gradient concentrations of HL, 1, and 2 (n = 3).
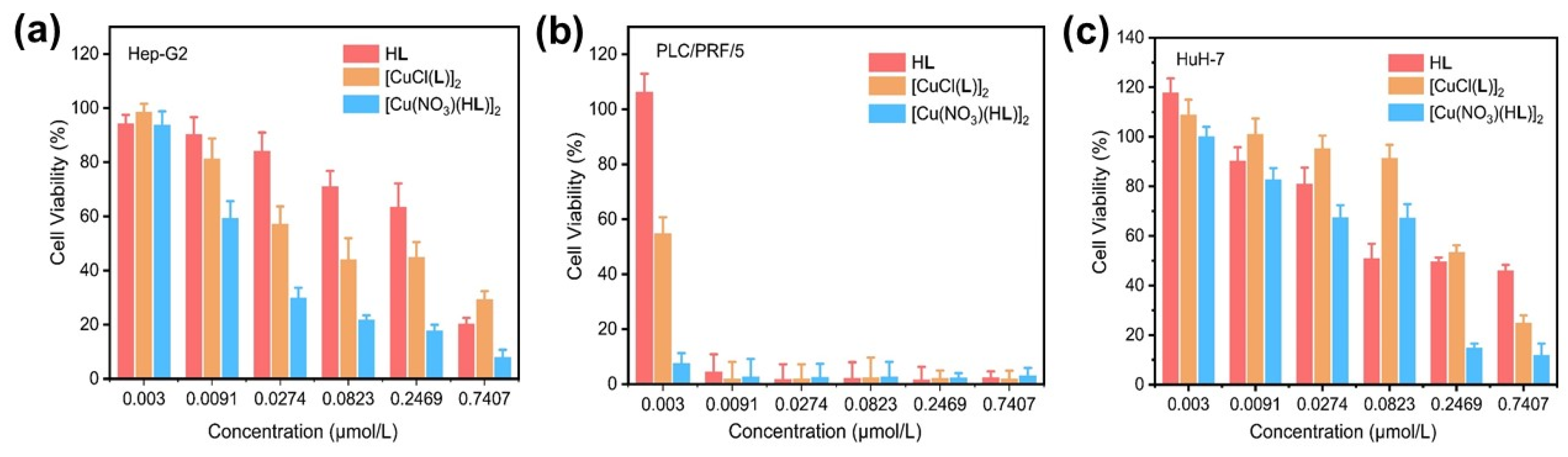
Figure 6.
Comparison of the intracellular ROS generating ability of HL, 1, and 2 in HuH-7 using DCFH-DA as a fluorescence probe.
Figure 6.
Comparison of the intracellular ROS generating ability of HL, 1, and 2 in HuH-7 using DCFH-DA as a fluorescence probe.
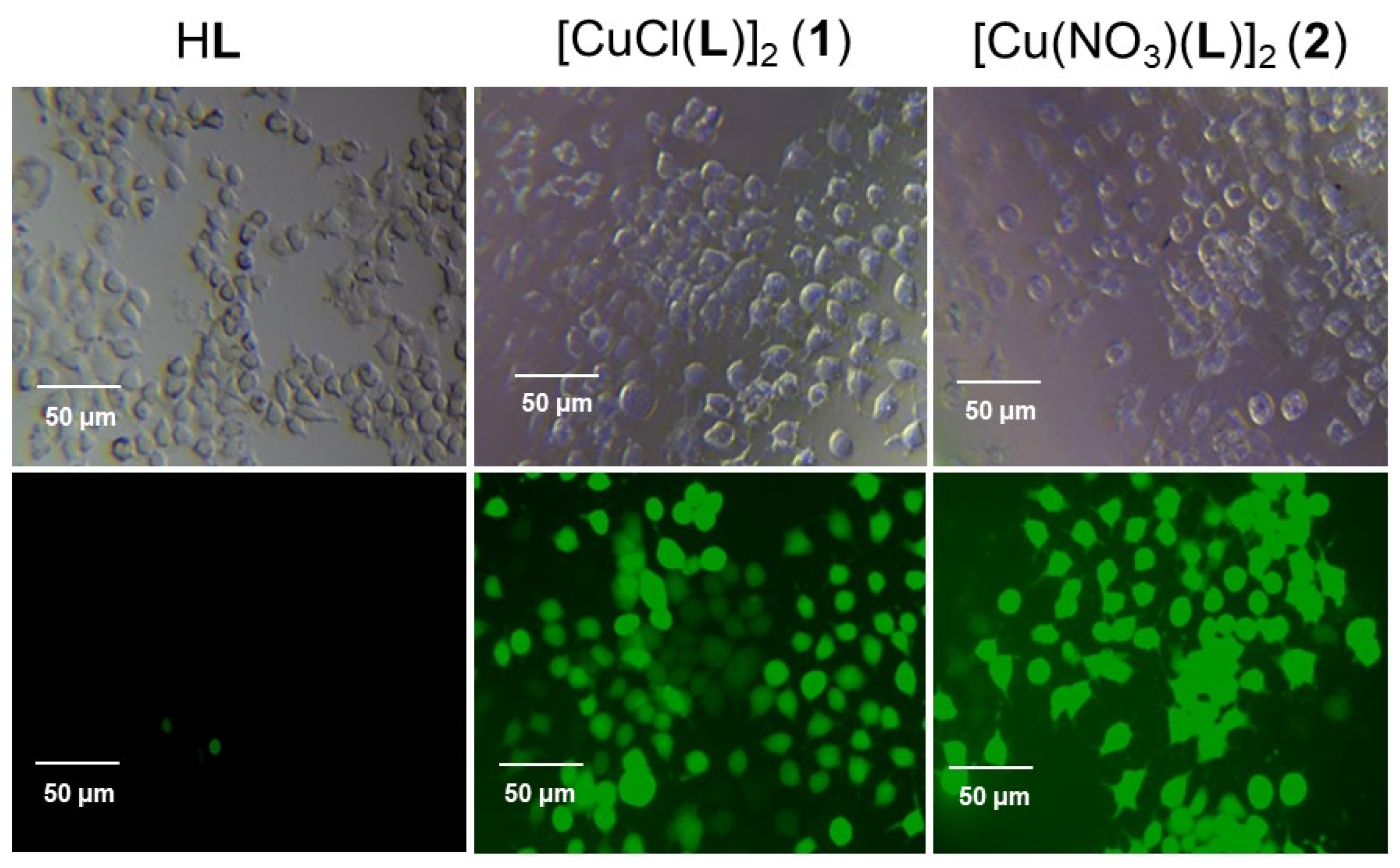
Figure 7.
The time-concentration relationships of HL via i.v. (0.5 mg kg−1) and PO (30 mg kg−1) administrations.
Figure 7.
The time-concentration relationships of HL via i.v. (0.5 mg kg−1) and PO (30 mg kg−1) administrations.
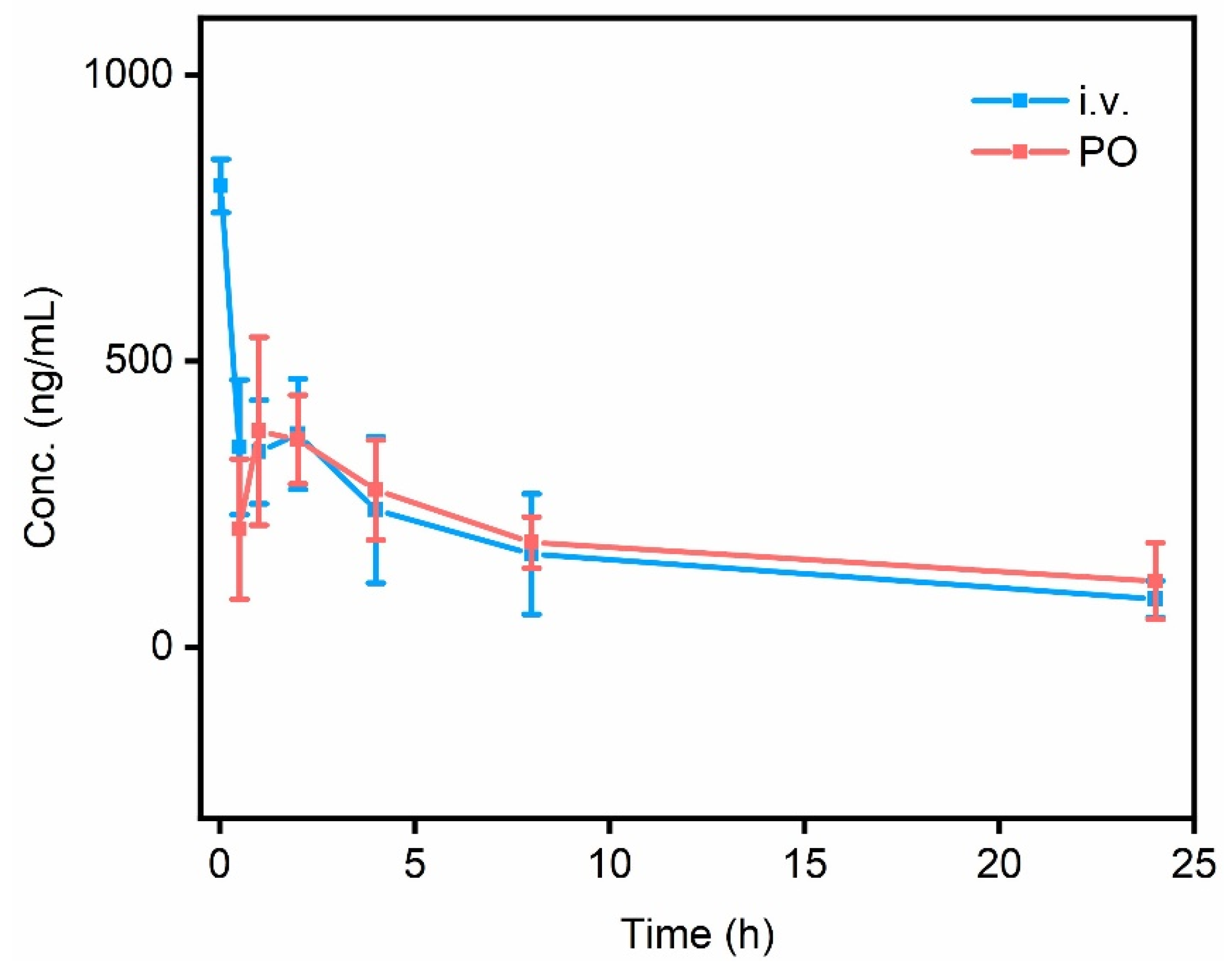

Table 1.
A summary of the cell parameters and refinement results for HL, 1, and 2.
| HL | [CuCl(L)]2 (1) | [Cu(NO3)(L)]2 (2) | |
|---|---|---|---|
| CCDC number | 2341904 | 2341905 | 2341906 |
| Formula | C19H19N7S2 | C38H40Cl2Cu2N14S4 | C38H36Cu2N16O6S4 |
| Formula weight | 409.53 | 1019.06 | 1068.15 |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group | P-1 | P-1 | P21/n |
| a/Å | 10.4251(14) | 9.342(2) | 10.7226(16) |
| b/Å | 10.7193(14) | 10.714(2) | 9.6646(15) |
| c/Å | 11.0200(15) | 11.378(3) | 21.695(3) |
| α/° | 105.347(5) | 93.542(10) | 90 |
| β/° | 103.115(5) | 109.660(10) | 104.187(2) |
| γ/° | 117.474(5) | 108.609(9) | 90 |
| V/Å3 | 962.0(2) | 998.0(4) | 2179.7(6) |
| Z | 2 | 1 | 2 |
| Dc/(g cm–3) | 1.414 | 1.696 | 1.627 |
| F(000) | 428 | 522 | 1092 |
| μ /mm–1 | 0.298 | 8.151 | 1.235 |
| Total reflns. | 25863 | 16310 | 33191 |
| Uniq. reflns. | 3928 | 4037 | 4404 |
| Observed reflns. | 1855 | 3572 | 3194 |
| Parameters | 253 | 274 | 298 |
| Rint | 0.1570 | 0.0818 | 0.0905 |
| Ra | 0.0882 | 0.0592 | 0.0677 |
| wRb | 0.1738 | 0.1658 | 0.1470 |
| GOFc | 1.029 | 1.079 | 1.048 |
| aR = Σ||Fo|–|Fc||/Σ|Fo|. b wR= {Σw(Fo2–Fc2)2/Σw(Fo2)2}1/2. c GOF = {Σw((Fo2–Fc2)2)]/(n–p)}1/2, where n = number of reflections and p = total number of parameters refined. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



