
Submitted:
19 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Idiopathic pulmonary fibrosis remains a relevant problem in the field of healthcare with an unfavorable prognosis for patients due to progressive fibrous remodeling of the pulmonary parenchyma. Starting with the reactions damaging the epithelial lining of alveoli, pulmonary fibrosis is implemented through a cascade of complex mechanisms, the crucial of which is the TGF-β/SMAD mediated pathway, involving various cell populations. Considering that a number of the available drugs (pirfenidone and Nintedanib) have only limited effectiveness in slowing the progression of fibrosis, the search and justification of application of other approaches aimed at regulating the immune response, cellular aging processes, programmed cell death and transdifferentiation of cell populations remains relevant. This literature review presents the key modern concepts concerning molecular genetics and cellular mechanisms of lung fibrosis development, based mainly on in vitro and in vivo studies in experimental models of bleomycin-induced pulmonary fibrosis, as well as the latest data on metabolic features, potential application points and effects of vitamin D and its metabolites.
Keywords:
lungs
; fibrosis
; inflammation
; vitamin D
; vitamin D receptors
1. Introduction
Pulmonary fibrosis (idiopathic pulmonary fibrosis) is a stage-by-stage, progressive and irreversible lesion of the lung tissue with increasing respiratory failure and an unfavorable prognosis [1]. At the same time, the etiopathogenesis of this disease remains insufficiently studied. Consequently, the available therapeutic approaches are also significantly limited, with the presence of only one approach that has proven effective in relation to patient survival – lung transplantation [2]. The median of the expected life expectancy of patients with idiopathic pulmonary fibrosis is only 2–3 years [3], which indicates an aggressive and extremely progressive lesion of the respiratory parts of the lungs, with a constant and increasing load on the system from year to year for public health. In this regard, the search for new highly effective and safe approaches to pharmacotherapy for this pathological condition, designed to improve the quality of life of patients with the restoration of the structure of lung tissue, or at least, slowing down the rate of progression of fibrotic changes, does not stop. Some of these approaches to therapy were the use of immunosuppressants (glucocorticoid therapy, azathioprine, cyclophosphamide, etc.) [4], anticoagulants [5], endothelin receptor antagonists (bosentan, ambrisentan, macitentan) [6], various antifibrotic and immunomodulatory drugs (interferon gamma, etanercept, imatinib, etc.) [7,8], which, in general, have proven ineffective in randomized phase II or III clinical trials. At the same time, currently, inhibition of transforming growth factor beta (TGF-β) is considered as one of the promising approaches, whose crucial role in the development of pulmonary fibrosis has been repeatedly confirmed, both in experimental and clinical studies [9], as well as the suppression of some other growth factors, including vascular endothelial growth factors (VEGFR-1, VEGFR-2 and VEGFR-3), fibroblast growth factors (FGFR-1, FGFR-2 and FGFR-3) and platelet growth factors (PDGRF-α and PDGRF-β) [10]. In particular, pirfenidone (an inhibitor TGF-β) [11] and nintedanib (inhibitor of vascular endothelial growth factor receptors, platelet growth factor and fibroblast growth factor) [12] are the only medicines that are officially approved for treatment of idiopathic pulmonary fibrosis. At the same time, despite a slight slowdown in fibrotic changes in the lungs and an improvement in the quality of life of patients, these drugs are still unable to change the overall progression of the disease and high mortality rates during the first 3-5 years from the moment of diagnosis [13]. In this regard, a detailed study of the pathogenesis of the disease and the search for possible treatment methods with pathogenetically justified application points continues, with the continued interest of researchers in nature-based therapeutic approaches, among which one of the key places is occupied by vitamin D and its analogues with effects realized through ligand-associated activation of vitamin D receptors.
This review attempts to systematize the data available in the literature on key factors and features of pathogenesis of pulmonary fibrosis in terms of potential application points of effects of ligand-associated activation of vitamin D receptors, as well as to present existing experimental and clinical data on the effects of vitamin D and its analogues in conditions of progressive pulmonary fibrosis.
2. Key Pathogenetic Mechanisms and Cell Populations in the Development of Pulmonary Fibrosis
Pulmonary fibrosis is a complex and multi-component pathological process of unexplained etiology, probably associated with the chronic exposure of damaging factors of various nature and origin to the respiratory tract of the lungs [14]. Starting with the damage to the alveolar epithelium, followed by a cascade of coagulation reactions, inflammatory response and regeneration with remodeling of lung tissue, pulmonary fibrosis appears to be a pathological process with dysregulation of reparative regeneration and excessive progressive growth of fibrous connective tissue. As with regard to etiology, although assumptions are made about the role of key risk factors such as smoking, exposure to ionizing radiation, etc. [15,16], potential molecular mechanisms that underlie in the development of these disorders remain not fully clarified. As an illustration of our lack of understanding of the role of certain factors in the development of pulmonary fibrosis, population-based studies on tobacco smoking can be taken as an example, the results of which are often multidirectional [17,18]. At the same time, the role of inflammation and its proliferative phase with a shift in the system of intercellular regulation towards proliferation, activation and differentiation of collagen- synthesizing fibroblastic cells is obvious, including due to the mechanism of the epithelial-mesenchymal transition, with excessive deposition of extracellular matrix components [19].
According to classical concepts, pulmonary fibrosis is a stage-by-stage process with a sequential transition from the phase of damage to the phase of inflammatory response, followed by reparative regeneration and remodeling of lung tissue. Genetic factors, including various single-nucleotide polymorphisms of genes (in particular, in the gene of a protein interacting with Toll- like receptors, which is a suppressor of the TGF-β signaling pathway) are currently considered as initial predisposing factors [20,21], the effect of environmental factors (such as smoking and various bacterial, viral or fungal infections) [17,18,22], age-related changes with increased secretion of proinflammatory cytokines and antiapoptotic effects on fibroblastic cells [23]. In this regard, both the effect of external damaging factors and age-related changes are accompanied by a number of epigenetic modifications, including DNA methylation, various histone modifications, as well as changes in the expression of various non-coding RNAs (microRNAs) [24]. In particular, an increase in expression of miRNA-21 and miRNA-199 with a decrease in the expression level of miRNA-31 and miRNA-200 was demonstrated against the background of idiopathic pulmonary fibrosis [25,26]. These epigenetic factors may play a decisive role in starting a cascade of pathological reactions. In particular, miRNA-21 has been shown to be able to induce epithelial- mesenchymal transition and promote the development of TGF-β- mediated fibrosis [27], whereas miRNA-200, on the contrary, stimulates the process of transdifferentiation into type I alveolar epithelium [28]. Among other epigenetic factors that could potentially be involved in the regulation of lung tissue remodeling, other small non-coding RNAs have also been identified, including miRNA-145, miRNA-424, miRNA- 301a, miRNA-29 and others [29]. The data obtained in the framework of these studies, among other things, open up new prospects for therapeutic approaches based on blocking specific miRNAs [30].
Starting with damage to the alveolar epithelium and vascular endothelium of the microcirculatory bed with a proinflammatory response and activation of coagulation cascade, the pathological process enters a phase of progression with predominantly TGF-β-mediated cell activation of fibroblastic series and formation of typical foci of fibrosis. At the same time, all major cell populations are involved in the formation of pulmonary fibrosis , including alveolocytes of type I and II, cells of stromal microenvironment (fibroblasts, myofibroblasts and vascular endothelium) and inflammatory cells (including neutrophils, cells of monocyte- macrophage series and cells of lymphoid series).
2.1. Alveolar Epithelium
According to most researchers, damage to the alveolar epithelium is an invariable trigger point in the development of idiopathic pulmonary fibrosis. Type I alveolar epithelium lines more than 90% of the alveolar surface, and its damage triggers the activation program of type II alveolocytes, which are surfactant-producing cells, as well as progenitor cells under conditions of reparative regeneration [31]. Under normal conditions, damage to type I alveolocytes followed by type II alveolocyte hyperplasia is completed by the restoration of the epithelial lining of the alveoli due to transdifferentiation of type II alveolocytes into type I alveolar epithelium [32]. At the same time, this transdifferentiation from type II alveolocytes to type I alveolocytes is carried out through an intermediate cellular form – a population of so-called basaloid cells. In vivo and in vitro studies have demonstrated that effects on this intermediate cell population can both stimulate and inhibit their further differentiation into type I alveolocytes. It is also shown that in the foci of fibrosis in patients with idiopathic pulmonary fibrosis, the number of these cells is significantly increased [33], and that this cell population is able to transdifferentiate into keratin-5 positive basal cells (Krt5+) with subsequent differentiation into bronchial epithelium, which may explain the phenomenon of bronchialization of alveoli in idiopathic pulmonary fibrosis [34]. In addition, another type of transdifferentiation of type II alveolocytes is likely due to activation of epithelial-mesenchymal transition mechanisms [35]. In general, in the case of damage to the basement membrane, as well as against the background of a pronounced increase in vascular permeability due to damage to the endothelium, reepithelization does not occur and type II alveolocytes continue to secrete numerous growth factors, proinflammatory and profibrotic cytokines. At the same time, the alveolar epithelium is the main cell population responsible for the production of platelet growth factor (PDGF), transforming growth factor beta (TGF-β) and tumor necrosis factor alpha (TNF-α), which are key orchestrators in the system of intercellular interaction and the subsequent development of pulmonary fibrosis [31]. A combination of external damaging factors and internal factors, such as cell aging with telomere shortening, mitochondrial dysfunction, endoplasmic reticulum stress, disorders of apoptosis and autophagy processes lead to ineffective restoration of damaged epithelium due to type II alveolocytes. Ultimately, this leads to the activation and proliferation of fibroblasts and myofibroblasts and excessive deposition of extracellular matrix components.
2.2. Cells of the Stromal Microenvironment – Fibroblasts, Myofibroblasts and Vascular Endothelium
After damage to the alveolar epithelium with a violation of reparative regeneration processes and the formation of proinflammatory and profibrotic cytokine background, fibroblasts are activated and their transdifferentiation into myofibroblasts that actively secrete collagen [36]. Under normal conditions, a resolution phase occurs in which myofibroblasts are deactivated, fibroblast recruitment is suppressed, and cellular detritus along with excess amounts extracellular matrix are resorbed by macrophages. In the case of idiopathic pulmonary fibrosis, this process is disrupted by a central mechanism of TGF-β-mediated signaling pathway. Meanwhile, the expression of TGF-β is observed in almost all cell populations, including alveolar epithelium (including in aberrant basaloid cells), alveolar macrophages, fibroblastic cells and cells of the immune system [37]. Under these conditions, the transdifferentiation of fibroblasts into myofibroblasts with increased expression of alpha - smooth muscle actin (α-SMA) creates conditions for active collagen synthesis and excessive accumulation of extracellular matrix [38]. Differentiation of myofibroblasts and inhibition of apoptosis in this cell population is supported by many factors, including TGF-β, integrin aVβ6, PDGF, VEGF, various coagulation factors, Wnt- mediated signaling pathway, etc. [39,40]. Similar factors lead to an increase in the expression of mesenchymal and corresponding suppression of the expression of epithelial markers, which act as fundamentals of the implementation of the epithelial-mesenchymal transition. Usually, the epithelial-mesenchymal transition is characterized by the loss of epithelial markers (for example, E-cadherin) and the appearance of expression of mesenchymal markers (N-cadherin, vimentin or α-SMA). In conditions of idiopathic pulmonary fibrosis, the population of fibroblastic cells is not the result of epithelial transdifferentiation. In this case, the epithelial-mesenchymal transition leads to the expression of mesenchymal markers in the alveolar epithelium, which contributes to the progression of fibrosis [41].
Endothelial cells are also involved in the pathogenesis of idiopathic pulmonary fibrosis, being an integral part of stromal cell microenvironments. Firstly, damage to the endothelium in the respiratory parts of the lungs leads to the activation of the coagulation cascade that, along with other cytokines, released by the alveolar epithelium, contributes to the development of an inflammatory response [42,43]. In the areas of connective tissue proliferation in pulmonary fibrosis, there is an increase in the expression of VEGF and IL-8, both in endothelial cells and in type II alveolocytes [44]. Meanwhile, VEGF is one of the key mediators of angiogenesis, suppressing endothelial apoptosis and stimulating its proliferation and differentiation. Also, similar to the epithelial-mesenchymal transition, there is experimental data indicating the ability of endothelial cells to transdifferentiate into fibroblasts and myofibroblasts, with activation of this mechanism due to TGF-β and Ras/MAPK signaling pathway [45].
2.3. Immunocompetent Cells
Neutrophils, being a cell population that is one of the first to respond to damage to the alveolar epithelium, according to most researchers, are not key effectors of a dysregulatory inflammatory response and progressive fibrosis. However, neutrophils are able to secrete a number of profibrotic factors (in particular, IL-8/CXCL8), metalloproteinases and take an active part in the remodeling of the extracellular matrix [46,47]. In addition, neutrophils are capable of forming so-called extracellular neutrophils traps, the appearance of which is associated with the stimulation of fibroblasts and the induction of fibrosis [48].
Unlike neutrophils, macrophages represent one of the key cell populations involved in the implementation of the resolving phase of inflammation and subsequent reparative regeneration. Various subpopulations of macrophages in the foci of damage are able to secrete a number of cytokines (including IL-1, IL-6, TNF-α, TGF-β, various metalloproteinases and growth factors), thereby regulating activation cells of the stromal microenvironment, the degree of deposition of extracellular matrix and angiogenesis processes [49]. Meanwhile, the emphasis in studying the population of monocyte-macrophage cells, whether alveolar or interstitial macrophages, is to identify two key subpopulations of cells: proinflammatory macrophages type 1 (M1) with the classical activation pathway and anti-inflammatory macrophages type 2 (M2) with an alternative activation pathway [50], although these two subpopulations themselves are not similar [51]. The induction of M1 macrophages is associated with the action of bacterial lipopolysaccharides and TNF-α, whereas the M2 subpopulation responds to IL-4 and IL-10 stimuli and is actively involved in the resolving phase of the inflammatory process and the occurrence of fibrosis [52]. A number of studies have shown that in conditions of pulmonary fibrosis, M2 macrophages predominate, actively secreting such profibrotic growth factors as TGF-β, fibroblast growth factor, PDGFa, IGF-1, and VEGF, stimulating excessive deposition of extracellular matrix [53,54]. This is also confirmed in experimental studies with bleomycin-induced pulmonary fibrosis, in which an increase in the number of subpopulation of macrophages M2 was observed on the background of increased expression of IL-1β with subsequent transdifferentiation of type II alveolocytes into type I alveolocytes [55]. Thus, M2 macrophages are the key orchestrators of the dysregulatory regeneration of the alveolar epithelium and the launch of a cascade of profibrotic tissue remodeling in conditions of progressive pulmonary fibrosis.
Like macrophages, lymphocytes also appear to be one of the key cellular subpopulations involved in the implementation of the inflammatory response and subsequent fibrosis, primarily due to modulation of the immune response. The role of various subpopulations of T-helpers has been studied quite well, in particular, their distribution into type 1 T-helpers (Th1) and type 2 T-helpers (Th2). Th1 cells are inducers of a pro-inflammatory response, secreting a number of cytokines (for example, IFN-γ and IL-12) and contributing to the realization of the exudative phase of inflammation [56], whereas Th2 cells contribute to the development of proliferative reactions in the foci of inflammation by secreting IL-4, IL-5, IL-9, IL-13 and a number of other cytokines [57]. It has been established that such profibrotic cytokines as IL-13 and IL-4, contribute to the differentiation and activation of myofibroblasts [58]. Thus, it can be considered that the shift towards Th2 cells is one of the key pathogenetic links in progressive pulmonary fibrosis. The start of this cellular mechanism of adaptive immune response is apparently triggered by damaged epithelial cells and monocyte-macrophage cells by active secretion of TGF-β, IL-1β and other cytokines, followed by recruitment of T- lymphocytes. At the same time, the role of other T-lymphocyte subpopulations is also being studied in the development of pulmonary fibrosis, in particular T-regulatory cells, Th17, Th9 and γδT cells, for which multidirectional modulating effects have been demonstrated [59,60].
In particular, the cascade of IL-17 mediated immune reactions is currently considered as one of the central mechanisms in the pathogenesis of pulmonary fibrosis from the point of view of intercellular interactions [61,62]. Although the IL-17 family (IL-17A – IL-17F) plays an important role in the formation of an anti-infectious immune response, dysregulation of the expression of individual representatives of this family of cytokines (mainly IL-17A) is one of the main factors, triggering profibrotic changes and contributing to their progression [63]. Although it is currently established that the secretion cytokines of the IL-17 family can be carried out by epithelial cells, dendritic cells, macrophages, various subpopulations of lymphocytes (including γδT lymphocytes, cytotoxic CD8+ T lymphocytes, etc.), the key cellular subpopulation remains specialized T-helpers of the 17th type (Th17), differentiation which is carried out from naive CD4+ lymphocytes in conditions of cytokine co-stimulation due to IL-1β, TGF-β, IL-6 and IL-23 [64]. In experimental studies on a model of bleomycin-induced pulmonary fibrosis, it was shown that IL-17A is secreted by Th17 and acts as one of the inducers and factors of progression of fibrous remodeling of the pulmonary parenchyma [65]. Similar data on the role of the Th17-dependent immune response were obtained for IL-1β-induced pulmonary fibrosis, as well as pulmonary fibrosis induced by the graft-versus-host reaction [66,67].
Some significant interactions between vitamin D / VDR and different subpopulations of T-cells are shown in Figure 1.
Likewise, the role of the B-lymphocytic link in implementation of the profibrotic tissue response is actively discussed. In particular, under conditions of pulmonary fibrosis development, activation of B lymphocytes with an increase in the release of a number of cytokines and metalloproteinases has been demonstrated, which contributes to dysregulation of the resolving phase of inflammation and excessive deposition of extracellular matrix [68,69,70].
Vit D – vitamin D and its metabolites; IL – interleukins; Th1 – T-helper type 1; Th2 –T-helpers type 2; Th17 –T-helpers type17; Treg – regulatory T-cells; IFNγ – interferon gamma; TNFα – tumor necrosis factor alpha. Solid arrows – stimulation / enhancing effects; dashed arrows – inhibitory effects.
2.4. Cell Aging and Apoptosis
Despite advances in understanding the key pathogenetic mechanisms that lead to the development of a dysregulatory immune response in the lung parenchyma, followed by progressive fibrosis, the trigger mechanisms are still poorly understood. Currently, as such, a combination of factors includes cell aging (primarily, the epithelial lining of the alveoli), as well as a violation of the regulation of the processes of programmed cell death (apoptosis).
In this aspect, cellular aging is a complex regulated process, including the so-called replicative aging and stress-induced premature aging of cells, caused in two ways by both genetic and epigenetic factors, and damaging environmental factors [71], accompanied by typical changes in the form of telomere shortening, DNA damage and epigenetic modifications, mitochondrial dysfunction and oxidative stress [72,73]. Both the age-related physiological and pathologically conditioned process of cellular aging are accompanied by the formation of the so-called senescence-associated secretory phenotype (SASP), characterized, among other things, by an increase in the secretion of cytokines such as IL-1, IL-6, TGF-β, TNF- α, MMP-2 and MMP-9 [74]. In recent studies, IL-11, which is part of the IL-6 family and is actively secreted by fibroblasts in response to profibrotic stimulation, including TGF-β, IL-13 and fibroblast growth factors, has also been assigned a key role [75,76].
At the same time, the role of cellular aging as a trigger mechanism extends to various cell populations, including alveolar epithelial cells, fibroblasts and endothelium, although the key importance, as before, is assigned to the aging of alveolocytes type II and basaloid cells [77] with corresponding changes in their genetic apparatus (including, among other things, an increase in the expression of plasminogen activator inhibitor 1 (PAI-1) and the p53 signaling pathway) [78]. The role of cellular aging in microenvironment cells with increased expression of alpha smooth muscle actin (α-SMA) and increased secretion of extracellular matrix in fibroblasts has also been established [79]. According to Yanai et al. fibroblasts in conditions of pulmonary fibrosis show pronounced signs of replicative cellular aging, but unlike epithelial cells, they are resistant to oxidative stress and apoptosis [80]. Clinical observations confirm the role of cellular aging. In particular, it has been shown that age is one of the most significant risk factors, with a twofold increase in the frequency of development of pulmonary fibrosis for every decade over the age of 50 [81], consistent with the theory of cellular aging. Besides, both in experimental models [82,83] and in pilot clinical studies [84] it has been demonstrated that the elimination of cellular subpopulations with signs of aging or therapy aimed at leveling the effects of cellular aging (in particular, with usage of a combination of dasatinib and quartzetine) [85], leads to a significant slowdown in the progression of pulmonary fibrosis and restoration of lost function.
Invariably related to the issue of cellular aging and renewal of cellular subpopulation, the role of programmed cell death (apoptosis) in the conditions of development of pulmonary fibrosis is also important. At the same time, according to in vitro and in vivo studies, changes in the regulation of apoptosis in various cell populations are often multidirectional [86,87]. In particular, alveolocytes in conditions of pulmonary fibrosis development are characterized by a significantly higher level of readiness for apoptotic death, manifested, among other things, by significant increase in the expression of the proapoptotic protein p53 [88]. Hypoxia-induced endoplasmic stress, TGF-β/p38 MAPK-mediated signaling pathway, angiotensin receptor signaling pathway, and classical Fas-mediated mechanism are considered as possible mechanisms and signaling pathways of apoptosis activation in this cell population [89]. Unlike type II alveolocytes, fibroblasts and myofibroblasts in conditions of pulmonary fibrosis development, being less sensitive to oxidative stress; on the contrary, exhibit increased resistance to apoptosis [90,91]. Meanwhile, these multidirectional changes of alveolocytes and cells of mesenchymal origin in conditions of pulmonary fibrosis in both cases, are associated with cellular aging and changes in gene expression, including PAI-1 and p53 [92], although detailed mechanisms of acquisition by various cellular subpopulations of different sensitivity to the action of proapoptotic signals still remain unexplored. Among others, there is evidence indicating the role of various epigenetic mechanisms, in particular, miRNA-34a [93,94].
3. Effects of Vitamin D and Its Analogues Implemented Through Ligand-Associated Activation of Vitamin D Receptors
3 Vitamin D is currently considered as a biologically active substance, which has a wide range of effects that are not limited to the regulation of calcium and phosphorus homeostasis [95]. The so-called "non-classical" effects of vitamin D which include regulation of cell proliferation and differentiation, apoptosis, intercellular adhesion, oxidative stress and inflammatory response are of particular interest to researchers [96]. Entering the body from the outside with food or synthesized endogenously in the skin under the action of UV irradiation, vitamin D is metabolized into its active form (1,25-dihydroxy vitamin D) due to specific enzymes - hydroxylases (25–hydroxylase and 1α-hydroxylase), which exhibits the full range of its biological effects through interaction with the vitamin D receptor (VDR), which is a ligand-dependent transcription factor [97]. At the same time, the possibility of metabolic formation of active form of vitamin D is not limited to the liver and kidneys, and currently both specific hydroxylases and VDR expression have been detected in almost all tissues and cell populations, including epithelial alveolar cells, alveolar macrophages [98,99] and immune cells [100], which emphasizes the important physiological role vitamin D in helping to ensure tissue homeostasis and the functioning of the immune system due to auto- and paracrine effects. Relatively recently, an alternative pathway of vitamin D metabolism has also been identified with the participation of an enzyme of the cytochrome P450 – CYP11A1 family with the formation of numerous sets of hydroxyl derivatives, among which 20(OH)D3, 20,23(OH)2D3, 1,20(OH)2D3, 1,20,23(OH)3D3 and 17,20,23(OH)3D3 are of higher importance, all of which are not inferior, but often surpass the classical active form of vitamin D, 1,25(OH)2D3, in terms of physiological activity, while having unexpressed or no calcemic effects at all [101,102]. As well as the enzymes of the classical metabolic pathway, the expression of CYP11A1 was detected in many cells, including immune cells [103]. Similar to 1.25(OH)2D3, CYP11A1-mediated derivatives are able to bind to a specific VDR receptor [104], as well as, equally important, to other nuclear receptors, including retinoic acid receptors (RXR), liver X-receptors (LXR) and other receptors representing transcription factors, which expands the potential range of effects of these ligands [105].
Currently, available data from in vitro and in-vitro studies on various cell populations, including immunocompetent cells, allow us to consider vitamin D, its endogenous metabolites (including CYP11A1-mediated ones) and a number of its synthetic analogues, that are the agonists of vitamin D receptors (paricalcitol), as promising agents for the prevention and treatment of pulmonary fibrosis.
The potential antifibrotic mechanisms of ligand-associated activation of vitamin D receptors are based on various effects on key signaling pathways, including SMAD [106], p38 MAPK [107], NF-kB [108], JAK/STAT [109], PPAR-α/γ [110,111] and calcineurin/NFAT [112]. In addition to this, ligand-dependent transcriptional effects of VDR on components of other signaling pathways, including Nrf2 [113] are explained. As one of the mechanisms mediated by some of the above signaling pathways and implemented under conditions of fibrous remodeling of the pulmonary parenchyma, the mechanism of VDR-mediated inhibition of the renin-angiotensin system (RAS) by suppressing renin expression, on one hand, and inhibition of TGF-β/SMAD and p38 MAPK signaling pathways, on the other hand is described [114]. It is shown that the RAS components (mainly, angiotensin II) stimulate the formation of extracellular matrix, increase the expression of TGF-β, contributing to the development of pulmonary fibrosis [115]. In general, the vitamin D/VDR complex, acting as a transcription factor, through various application points, controls the processes of cellular differentiation and proliferation, and is also involved in the regulation of the immune response (Table 1).
In terms of interaction with the above-mentioned key signaling pathways, in relation to the pathogenetic links in the development of pulmonary fibrosis, a number of immunotropic effects of ligand-associated activation of VDR have been demonstrated in studies. In general, vitamin D ((ligand-associated activation of VDR) is assigned a suppressor function with the redistribution of T helper subpopulations from Th1 cells towards Th2 and Treg. It is assumed that vitamin D can play a dual role: along with the late effects of stimulating the resolving phase of inflammation, the effects of the initial activation of the inflammatory response are also manifested [116,117]. Meanwhile, the value of vitamin D as an antifibrotic agent in conditions of pulmonary fibrosis from this point of view may be ambiguous, given one of the key roles of Th2 cells in the implementation of the profibrotic response [118]. On the other hand, a study by Joshi S et al. demonstrated a transcription-mediated effect of 1,25(OH)2D3 with a decrease in IL-17A expression by blocking (NFAT), suppression of histone deacetylase and transcription factor Runx1 with an increase in Foxp3 expression and a shift in the Th17/Treg ratio towards immunosuppressive T-regulatory cells [119].
Given the key role of the alveolar epithelium in triggering the cascade of pulmonary parenchyma remodeling, the data obtained using alveolocyte cell cultures is of particular interest. In a study by Heejae Han et al. obesity in laboratory animals with the development of vitamin D deficiency, accompanied by increased expression of TGF-β, components of the renin-angiotensin system and proinflammatory cytokines (IL-1β, IL-6, TNFa). At the same time, the addition of vitamin D to the culture of human bronchial epithelial cells and alveolocytes of laboratory mice significantly reduced the expression of TGF-β1 [120], which can be regarded as one of the key effects from a pathogenetic point of view in terms of preventing progressive fibrosis. In an in vitro study by Allan M. Ramirez et al. a dose-dependent suppression of the key profibrotic cytokine expression (TGF-β1) has also been demonstrated in the culture as fibroblasts and epithelial cells of human lung tissue under conditions of application of 1,25(OH)2D3 [121]. In vitro experiments also found that vitamin D significantly suppressed expression of the PSAT1 gene and activation of the MAPK-mediated signaling pathway in human pulmonary fibroblast culture [107]. In addition, it was found that an increase in the expression of vitamin D receptors in fibroblasts in conditions of pulmonary fibrosis is, apparently, one of the protective and adaptive mechanisms that limit the proliferation and activation of fibroblastic cells by suppressing the JAK1/STAT3 signaling pathway [122].
However, the data available in the literature regarding the effects of vitamin D is contradictory. In particular, in a study by Trinidad Guijarro et al. (2018) using representative cell models of type II alveolocytes and myofibroblasts in vitro, as well as a model of bleomycin-induced pulmonary fibrosis in vivo, it was shown that the use of 1,25(OH)2D3 induced cellular aging and aggravated histopathological changes in the lungs [123]. The authors associated the obtained results with a potential violation of DNA repair in the presence of vitamin D in conditions of bleomycin-induced damage. At the same time, a later study by the same research group confirmed the absence of similar negative effects in in vitro and in vivo models for hypocalcemic vitamin D analogues (paricalcitol and calcipotriol), making them preferred candidates for leveling bleomycin- induced damage [124]. At the same time, it is worth noting that there is practically no data on CYP11A1-mediated vitamin D derivatives in relation to pulmonary fibrosis.
With reference to the epithelial-mesenchymal transition in conditions of pulmonary fibrosis, in the study by Fei Jiang et al., it was shown that incubation of human alveolar epithelial cells (A549) with TGF- β (5 ng/ml) led to an increase in the expression of mesenchymal markers (N-cadherin and vimentin) in them with a corresponding suppression of the expression of epithelial markers (E-cadherin). Meanwhile, the addition of 1α.25-dihydroxy vitamin D3 (50 nmol) to the cell culture prevented these changes with suppression of the expression of transcription factors, associated with epithelial-mesenchymal transition (Snail and β-catenin) and normalization of mRNA levels of fibronectin and type I collagen genes [125]. Similar effects of vitamin D related to suppression of epithelial-mesenchymal transition have also been demonstrated in another study of alveolar cell culture [126].
Experimental models of pulmonary fibrosis in rodents, mainly using intratracheal instillation [127,128] or aerosol inhalation of bleomycin [129,130], have introduced significant understanding of the cellular and molecular genetic mechanisms of pulmonary fibrosis and potential application points of vitamin D and its analogues. In particular, in an experimental study on rodents, it was found that the use of vitamin D contributed to a significant decrease in the manifestations of bleomycin-induced fibrosis with a decrease in hydroxyproline levels, expression of smooth muscle actin and TGF-β, and the severity of histopathological and ultrastructural signs of lung tissue damage [131]. Application of vitamin D in conditions of bleomycin-induced pulmonary fibrosis contributed to a decrease in the expression levels of mRNA collagen type I, type III and smooth muscle actin with the restoration of bleomycin-induced decrease in VDR mRNA expression. Suppression of phosphorylation of TGF-β1-induced SMAD was one of the mechanisms of antifibrotic effects of vitamin D [132].
In another study using an experimental model of bleomycin-induced pulmonary fibrosis in C57BL/6 mice, it was shown that the use of a vitamin D receptor agonist (paricalcitol) significantly prevented body weight loss and contributed to less pronounced fibrotic changes in lung tissue, whereas vitamin D deficiency, on the contrary, was accompanied by significantly more pronounced damage to the lung parenchyma. The use of paricalcitol led to suppression of the expression of TGF β, α SMA, type I collagen and fibronectin, as well as components of the renin- angiotensin system (angiotensinogen, angiotensin II and type I angiotensin receptors) [114].
Key mechanisms of pulmonary fibrosis development and potential application points of vitamin D and its metabolites, which implement their effects through VDR-dependent modulation of signaling pathways, are schematically presented in Figure 2 based on in vitro and in vivo research data.
In contrast to experimental studies in vitro and in vivo, data from clinical trials, including multicenter randomized trials, are extremely heterogeneous and, for the most part, indicate a lack of evidence regarding the role of vitamin D and its potential effects on the risk of developing and progression of pulmonary fibrosis.
Effects of vitamin D and its metabolites / VDR: a – inhibition of the secretion of proinflammatory cytokines (IL-1β, TGFß), including by suppressing the renin-angiotensin system; b – suppression of the epithelial- mesenchymal transition; c – modulation of the polarization of macrophages on M1 and M2 subpopulations; d, e – an increase in the subpopulation of Th2 cells and a shift from Th17 towards T- regulatory lymphocytes.
In particular, a recent large-scale study using Mendelian randomization revealed the absence of any significant links between genetically determined levels of circulating vitamin D (25(OH)D) and the risk of developing idiopathic pulmonary fibrosis [133]. Meanwhile, a number of other studies have established a relationship between vitamin D deficiency and the development of a number of pulmonary pathologies, including respiratory infectious diseases and chronic obstructive pulmonary disease [134,135]. In another clinical study involving a few subjects, it was demonstrated that the use of a combination of vitamins D, C and E in patients with idiopathic pulmonary fibrosis, leads to an improvement in respiratory function and a decrease in inflammatory response and oxidative stress [136].
4. Conclusions
The data obtained so far regarding the nature and key mechanisms of the development of pulmonary fibrosis suggest significant progress in understanding the pathogenesis of fibrous remodeling of the pulmonary parenchyma. Starting with damage reactions, the pathological process is realized through a cascade of complex molecular genetic mechanisms, the key of which is TGF-β/SMAD an indirect pathway involving various cell populations, including alveolocytes of type I and II, cells of the mesenchymal microenvironment such as fibroblasts and myofibroblasts, antigen-presenting cells and a wide range of T-lymphocyte subpopulations. In this regard, it is the dysregulatory immune response with a profibrotic inflammatory response vector that is the key target of both existing and potential therapeutic approaches aimed at leveling the fibrous transformation of the pulmonary parenchyma, restoration of respiratory function and improvement of survival rates and quality of life in patients with pulmonary fibrosis.
From the point of view of regulating the immune response, based on the data of in vitro and in vivo studies, the use of vitamin D and its metabolites seems promising (including CYP11A1-mediated derivatives), which realize their effects through VDR-dependent modulation of the signaling pathways TGF-β/SMAD, p38 MAPK, NF-kB, JAK/STAT, PPAR- α/γ, NFAT, Nrf2 and others, manifested, among other things, in a change in the distribution key cellular subpopulations, cytokine profile, the processes of cellular aging and programmed cell death. Meanwhile, the absence of significant evidence to date of the effectiveness of vitamin D and its derivatives in idiopathic pulmonary fibrosis in clinical studies may be associated with both genetic polymorphism, epigenetic modifications, dietary and lifestyle features on the one hand, and possible imperfection of existing generally accepted models of bleomycin-induced pulmonary fibrosis on the other hand. However, accumulated preclinical data and the understanding of key application points of VDR-mediated effects in remodeling conditions of pulmonary parenchyma emphasizes the need for further study of this issue and resolution of the problem of inconsistency between preclinical and clinical data.
Author Contributions
Conceptualization: M.K. writing—original draft preparation: M.K. and D.E.; writing—review and editing: T.S. and V.M.; drawing: M.K. and T.S. All authors are equally responsible for plagiarism, self-plagiarism or other ethical transgressions. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the Russian Science Foundation (project No. 23-15-20015).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic Pulmonary Fibrosis: Current and Future Treatment. Clinical Respiratory J. 2022, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Cox, I.A.; Campbell, J.A.; Xia, Q.; Otahal, P.; De Graaff, B.; Corte, T.J.; Teoh, A.K.Y.; Walters, E.H.; Palmer, A.J. Mortality and Survival in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. ERJ Open Res. 2022, 8, 00591–02021. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Luppi, F.; Ferrara, G.; Mura, M. Immunomodulatory Treatment of Interstitial Lung Disease. Ther. Adv. Respir. Dis. 2022, 16, 175346662211170. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Nakayama, K.; Yanai, M.; Suzuki, T.; Yamaya, M.; Watanabe, M.; Sasaki, H. Anticoagulant Therapy for Idiopathic Pulmonary Fibrosis. Chest 2005, 128, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pan, Y.; Xin, W.; Yan, C. The Potential Benefit of Endothelin Receptor Antagonists’ Therapy in Idiopathic Pulmonary Fibrosis: A Meta-Analysis of Results from Randomized Controlled Trials. Medicine 2022, 101, e29981. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Costabel, U.; Cottin, V.; Du Bois, R.M.; Lasky, J.A.; Thomeer, M.; Utz, J.P.; Khandker, R.K.; McDermott, L.; et al. Treatment of Idiopathic Pulmonary Fibrosis with Etanercept: An Exploratory, Placebo-Controlled Trial. Am. J. Respir. Crit. Care Med. 2008, 178, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G. Idiopathic Pulmonary Fibrosis: Lessons from Clinical Trials over the Past 25 Years. Eur. Respir. J. 2017, 50, 1701209. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Wang, Z.; Zhu, H.; Liu, W.; Zhao, M.; Jiang, X.; Zhao, J.; Ren, C.; Zhang, Y.; Luo, L. Research Progress on Drugs Targeting the TGF-β Signaling Pathway in Fibrotic Diseases. Immunol. Res. 2022, 70, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and Adjunctive Treatment for Idiopathic Pulmonary Fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in Patients with Progressive Fibrotic Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis (RELIEF): A Double-Blind, Randomised, Placebo-Controlled, Phase 2b Trial. The Lancet Respiratory Medicine 2021, 9, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Alonzi, V.; d’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini, P.; Bargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.; Fabiano, A.; Mota, P.C.; Rodrigues, I.; Carvalho, D.; Melo, N.; Novais-Bastos, H.; Alexandre, A.T.; Moura, C.S.; Guimarães, S.; et al. The Impact of Nintedanib and Pirfenidone on Lung Function and Survival in Patients with Idiopathic Pulmonary Fibrosis in Real-Life Setting. Pulmonary Pharmacology & Therapeutics 2023, 83, 102261. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Jarzebska, N.; Karetnikova, E.S.; Markov, A.G.; Kasper, M.; Rodionov, R.N.; Spieth, P.M. Scarred Lung. An Update on Radiation-Induced Pulmonary Fibrosis. Front. Med. 2021, 7, 585756. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Evangelou, E. Tobacco Smoking and Risk for Pulmonary Fibrosis. Chest 2021, 160, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.; Lee, C.-H.; Lee, J.; Kim, Y.W.; Han, K.; Choi, S.M. Impact of Smoking on the Development of Idiopathic Pulmonary Fibrosis: Results from a Nationwide Population-Based Cohort Study. Thorax 2022, 77, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.-Y.; Kim, H.; Bae, Y.; Song, J.W. Smoking Status and Clinical Outcome in Idiopathic Pulmonary Fibrosis: A Nationwide Study. Respir. Res. 2024, 25, 191. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.E.; Schwartz, D.A. Genetic Risk Factors for Idiopathic Pulmonary Fibrosis: Insights into Immunopathogenesis. JIR 2021, 13, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.N.; Invernizzi, R.; Laskowska, J.W.; O’Neill, A.; Doroudian, M.; Moghoofei, M.; Mostafaei, S.; Li, F.; Przybylski, A.A.; O’Dwyer, D.N.; et al. Candidate Role for Toll-like Receptor 3 L412F Polymorphism and Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 205, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Maher, T.M. The Role of Infection in the Pathogenesis of Idiopathic Pulmonary Fibrosis. European Respiratory Review 2013, 22, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. The American Journal of the Medical Sciences 2019, 357, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Schwartz, D.A. Epigenetics of Idiopathic Pulmonary Fibrosis. Translational Research 2015, 165, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Perera, U.E.; Derseh, H.B.; Dewage, S.N.V.; Stent, A.; Wijayarathna, R.; Snibson, K.J. Evaluation of microRNA Expression in a Sheep Model for Lung Fibrosis. BMC Genomics 2021, 22, 827. [Google Scholar] [CrossRef] [PubMed]
- Cadena-Suárez, A.R.; Hernández-Hernández, H.A.; Alvarado-Vásquez, N.; Rangel-Escareño, C.; Sommer, B.; Negrete-García, M.C. Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition. IJMS 2022, 23, 6613. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 Mediates Fibrogenic Activation of Pulmonary Fibroblasts and Lung Fibrosis. Journal of Experimental Medicine 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Moimas, S.; Salton, F.; Kosmider, B.; Ring, N.; Volpe, M.C.; Bahmed, K.; Braga, L.; Rehman, M.; Vodret, S.; Graziani, M.L.; et al. miR-200 Family Members Reduce Senescence and Restore Idiopathic Pulmonary Fibrosis Type II Alveolar Epithelial Cell Transdifferentiation. ERJ Open Res. 2019, 5, 00138–02019. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Henket, M.; Remacle, C.; Cambier, M.; Struman, I.; Winandy, M.; Moermans, C.; Louis, E.; Malaise, M.; Ribbens, C.; et al. Systematic Review of Overlapping microRNA Patterns in COVID-19 and Idiopathic Pulmonary Fibrosis. Respir. Res. 2023, 24, 112. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Su, Y.; Hsia, I.; Xu, Y.; Vincent-Chong, V.K.; Mojica, W.; Seshadri, M.; Zhao, R.; Wu, Y. Delivery of Anti-microRNA-21 by Lung-Targeted Liposomes for Pulmonary Fibrosis Treatment. Molecular Therapy - Nucleic Acids 2023, 32, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. IJMS 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed]
- Selman, M. Role of Epithelial Cells in Idiopathic Pulmonary Fibrosis: From Innocent Targets to Serial Killers. Proceedings of the American Thoracic Society 2006, 3, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-Cell RNA-Seq Reveals Ectopic and Aberrant Lung-Resident Cell Populations in Idiopathic Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human Alveolar Type 2 Epithelium Transdifferentiates into Metaplastic KRT5+ Basal Cells. Nat. Cell. Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human Alveolar Epithelial Cells Type II Are Capable of TGFβ-Dependent Epithelial-Mesenchymal-Transition and Collagen-Synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Homps-Legrand, M.; Crestani, B.; Mailleux, A.A. Origins of Pathological Myofibroblasts in Lung Fibrosis: Insights from Lineage Tracing Mouse Models in the Single-Cell RNA Sequencing Era. American Journal of Physiology-Lung Cellular and Molecular Physiology 2023, 324, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Hu, Y. TGF β1: Gentlemanly Orchestrator in Idiopathic Pulmonary Fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Decaris, M.L.; Schaub, J.R.; Chen, C.; Cha, J.; Lee, G.G.; Rexhepaj, M.; Ho, S.S.; Rao, V.; Marlow, M.M.; Kotak, P.; et al. Dual Inhibition of Avβ6 and Avβ1 Reduces Fibrogenesis in Lung Tissue Explants from Patients with IPF. Respir. Res. 2021, 22, 265. [Google Scholar] [CrossRef] [PubMed]
- Bahram Yazdroudi, F.; Malek, A. Optimal Controlling of Anti-TGF-β and Anti-PDGF Medicines for Preventing Pulmonary Fibrosis. Sci. Rep. 2023, 13, 15073. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C. Procoagulant Signalling Mechanisms in Lung Inflammation and Fibrosis: Novel Opportunities for Pharmacological Intervention? Pharmacology 2008, 153. [Google Scholar] [CrossRef] [PubMed]
- May, J.; Mitchell, J.A.; Jenkins, R.G. Beyond Epithelial Damage: Vascular and Endothelial Contributions to Idiopathic Pulmonary Fibrosis. Journal of Clinical Investigation 2023, 133, e172058. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Habiel, D.M.; Hohmann, M.; Camelo, A.; Shang, H.; Zhou, Y.; Coelho, A.L.; Peng, X.; Gulati, M.; Crestani, B.; et al. Antifibrotic Role of Vascular Endothelial Growth Factor in Pulmonary Fibrosis. JCI Insight 2017, 2, e92192. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial–Mesenchymal Transition in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Xaubet, A.; Agustí, C.; Luburich, P.; Barberá, J.A.; Carrión, M.; Ayuso, M.C.; Roca, J.; Rodriguez-Roisin, R. Interleukin-8 Expression in Bronchoalveolar Lavage Cells in the Evaluation of Alveolitis in Idiopathic Pulmonary Fibrosis. Respiratory Medicine 1998, 92, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, J.; Zhang, C.; Zhang, X.; Gao, P. Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front. Med. 2021, 8, 616200. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil Extracellular Traps Promote Differentiation and Function of Fibroblasts. The Journal of Pathology 2014, 233, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Pokhreal, D.; Crestani, B.; Helou, D.G. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways. Cells 2023, 12, 2193. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, Functional, and Plasticity Features of Classical and Alternatively Activated Human Macrophages. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or Foe in Idiopathic Pulmonary Fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tian, H.; Liu, H.; Xie, R. The Role of Macrophage-Derived TGF-Β1 on SiO2-Induced Pulmonary Fibrosis: A Review. Toxicol. Ind. Health 2021, 37, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Valencia, S.; Zhou, B. Roles of Macrophages and Endothelial Cells and Their Crosstalk in Acute Lung Injury. Biomedicines 2024, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Jian, Z.; Xu, T.; Li, F.; Deng, H.; Zhou, Y.; Lai, S.; Xu, Z.; Zhu, L. Macrophage Polarization: An Important Candidate Regulator for Lung Diseases. Molecules 2023, 28, 2379. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.J.; Yu, G.; Loh, K.S.; Savir, A.; Borczuk, A.; Kahloon, R.; Juan-Guardela, B.; Deiuliis, G.; Tedrow, J.; Choi, J.; et al. Cytokine-Like Factor 1 Gene Expression Is Enriched in Idiopathic Pulmonary Fibrosis and Drives the Accumulation of CD4+ T Cells in Murine Lungs. The American Journal of Pathology 2012, 180, 1963–1978. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; McKenzie, A.N.J. TH2 Cell Development and Function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Huang, T.; Zhang, L. Correction to: T Cells in Idiopathic Pulmonary Fibrosis: Crucial but Controversial. Cell Death Discov. 2023, 9, 74. [Google Scholar] [CrossRef]
- Qu, X.; Yi, X.; Zhong, H.; Ruan, W.; Huang, D. Effect and Mechanism of Imbalance via Th9 Cells and Th17/Treg Cells in Inflammatory and Fibrotic Phases of Pulmonary Fibrosis in Mice. Biotechnology and Genetic Engineering Reviews 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Segawa, S.; Goto, D.; Iizuka, A.; Kaneko, S.; Yokosawa, M.; Kondo, Y.; Matsumoto, I.; Sumida, T. The Regulatory Role of Interferon-γ Producing Gamma Delta T Cells via the Suppression of T Helper 17 Cell Activity in Bleomycin-Induced Pulmonary Fibrosis. Clinical and Experimental Immunology 2016, 185, 348–360. [Google Scholar] [CrossRef]
- Nie, Y.-J.; Wu, S.-H.; Xuan, Y.-H.; Yan, G. Role of IL-17 Family Cytokines in the Progression of IPF from Inflammation to Fibrosis. Military Med. Res. 2022, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Biswas, P.S. Interleukin-17: Friend or Foe in Organ Fibrosis. Cytokine 2019, 120, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Li, Z.; Yang, H.-Z.; Liu, H.; Wang, J.-P.; Ma, Y.-G.; Wang, X.-X.; Liu, H.-Z.; Sun, W.; Hu, Z.-W. Blocking IL-17A Promotes the Resolution of Pulmonary Inflammation and Fibrosis Via TGF-Β1–Dependent and –Independent Mechanisms. The Journal of Immunology 2011, 187, 3003–3014. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y. Biology of Interleukin-17 and Its Pathophysiological Significance in Sepsis. Front. Immunol. 2020, 11, 1558. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, E.; Fisher, A.J.; Gu, H.; Mickler, E.A.; Agarwal, M.; Wilke, C.A.; Kim, K.K.; Moore, B.B.; Vittal, R. IL-17A Deficiency Mitigates Bleomycin-induced Complement Activation during Lung Fibrosis. The FASEB Journal 2017, 31, 5543–5556. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1β–Mediated Pulmonary Fibrosis Is IL-17A Dependent. Journal of Experimental Medicine 2010, 207, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Martinu, T.; McManigle, W.C.; Kelly, F.L.; Nelson, M.E.; Sun, J.; Zhang, H.L.; Kolls, J.K.; Gowdy, K.M.; Palmer, S.M. IL-17A Contributes to Lung Fibrosis in a Model of Chronic Pulmonary Graft-versus-Host Disease. Transplantation 2019, 103, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, R.J.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic Partitioning of Interleukin-6 Signalling in Mice Dissociates Stat3 from Smad3-mediated Lung Fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.; Chiba, H. B Cells in Idiopathic Pulmonary Fibrosis: Targeting Immune Cells with Antifibrotic Agents. Am. J. Respir. Cell. Mol. Biol. 2021, 64, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Prêle, C.M.; Miles, T.; Pearce, D.R.; O’Donoghue, R.J.; Grainge, C.; Barrett, L.; Birnie, K.; Lucas, A.D.; Baltic, S.; Ernst, M.; et al. Plasma Cell but Not CD20-Mediated B-Cell Depletion Protects from Bleomycin-Induced Lung Fibrosis. Eur. Respir. J. 2022, 60, 2101469. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gonzalez, F.; Faner, R.; Rojas, M.; Agustí, A.; Serrano, M.; Sellarés, J. Cellular Senescence in Lung Fibrosis. IJMS 2021, 22, 7012. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ding, Q.; Lin, Z.; Chen, X.; Chen, S.; Zhu, Y. New Insights of Epigenetics in Vascular and Cellular Senescence. Journal of Translational Internal Medicine 2021, 9, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Shreeya, T.; Ansari, M.S.; Kumar, P.; Saifi, M.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Senescence: A DNA Damage Response and Its Role in Aging and Neurodegenerative Diseases. Front. Aging 2024, 4, 1292053. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lu, Q.; Liu, X. Advances in Cellular Senescence in Idiopathic Pulmonary Fibrosis (Review). Exp. Ther. Med. 2023, 25, 145. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Misso, N.L.A.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse Effects of Interleukin-6 on Apoptosis of Fibroblasts from Pulmonary Fibrosis and Normal Lungs. Am. J. Respir. Cell. Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 Signaling Underlies Fibrosis, Parenchymal Dysfunction, and Chronic Inflammation of the Airway. Exp. Mol. Med. 2020, 52, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Jiang, C.; Banerjee, S.; Yi, N.; Zmijewski, J.W.; Liu, G.; Liu, R.-M. PAI-1 Regulation of P53 Expression and Senescence in Type II Alveolar Epithelial Cells. Cells 2023, 12, 2008. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Zeische, R.; Fraifeld, V.E. Cellular Senescence-like Features of Lung Fibroblasts Derived from Idiopathic Pulmonary Fibrosis Patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Asai, A.; Kawagishi, H.; Mikawa, R.; Iwashita, Y.; Kanayama, K.; Sugimoto, K.; Sato, T.; Maruyama, M.; Sugimoto, M. Elimination of p19ARF-Expressing Cells Enhances Pulmonary Function in Mice. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic Drugs Target Alveolar Epithelial Cell Function and Attenuate Experimental Lung Fibrosis Ex Vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, A.; Kellogg, D.; Justice, J.; Goros, M.; Gelfond, J.; Pascual, R.; Hashmi, S.; Masternak, M.; Prata, L.; LeBrasseur, N.; et al. Senolytics Dasatinib and Quercetin in Idiopathic Pulmonary Fibrosis: Results of a Phase I, Single-Blind, Single-Center, Randomized, Placebo-Controlled Pilot Trial on Feasibility and Tolerability. eBioMedicine 2023, 90, 104481. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J. Evolving Concepts of Apoptosis in Idiopathic Pulmonary Fibrosis. Proceedings of the American Thoracic Society 2006, 3, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.M.; Hernady, E.B.; Reed, C.K.; Johnston, C.J.; Groves, A.M.; Finkelstein, J.N. Apoptosis Resistance in Fibroblasts Precedes Progressive Scarring in Pulmonary Fibrosis and Is Partially Mediated by Toll-Like Receptor 4 Activation. Toxicological Sciences 2019, 170, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, K.; Jiang, S.; Fu, L.; Shi, Y.; Tan, R.; Cui, J.; Zhou, Y. P53: A Key Protein That Regulates Pulmonary Fibrosis. Oxidative Medicine and Cellular Longevity 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- The Role of Apoptosis in Pulmonary Fibrosis. Histology and Histopathology 2004, 867–881. [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative Expression Profiling in Pulmonary Fibrosis Suggests a Role of Hypoxia-Inducible Factor-1α in Disease Pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A.; Hogaboam, C.M. Quercetin Enhances Ligand-Induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Liu, G.; Cai, L.; Deshane, J.; Antony, V.; Thannickal, V.J.; Liu, R.-M. Divergent Regulation of Alveolar Type 2 Cell and Fibroblast Apoptosis by Plasminogen Activator Inhibitor 1 in Lung Fibrosis. The American Journal of Pathology 2021, 191, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Disayabutr, S.; Kim, E.K.; Cha, S.-I.; Green, G.; Naikawadi, R.P.; Jones, K.D.; Golden, J.A.; Schroeder, A.; Matthay, M.A.; Kukreja, J.; et al. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0158367. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Ge, J.; Xie, N.; Banerjee, S.; Zhou, Y.; Liu, R.-M.; Thannickal, V.J.; Liu, G. miR-34a Promotes Fibrosis in Aged Lungs by Inducing Alveolarepithelial Dysfunctions. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017, 312, L415–L424. [Google Scholar] [CrossRef] [PubMed]
- Pludowski, P.; Grant, W.B.; Konstantynowicz, J.; Holick, M.F. Editorial: Classic and Pleiotropic Actions of Vitamin D. Front. Endocrinol. 2019, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.-G.; Kerlan, V.; Desailloud, R. Non-Classical Effects of Vitamin D: Non-Bone Effects of Vitamin D. Annales d’Endocrinologie 2021, 82, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Vitamin D and Its Target Genes. Nutrients 2022, 14, 1354. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.S.; Singer, F.R.; Gacad, M.A.; Sharma, O.P.; Hayes, M.J.; Vouros, P.; Holick, M.F. Isolation and Structural Identification of 1,25- Dihydroxyvitamin D3 Produced by Cultured Alveolar Macrophages in Sarcoidosis. The Journal of Clinical Endocrinology & Metabolism 1985, 60, 960–966. [Google Scholar] [CrossRef]
- Menezes, R.J.; Cheney, R.T.; Husain, A.; Tretiakova, M.; Loewen, G.; Johnson, C.S.; Jayaprakash, V.; Moysich, K.B.; Salgia, R.; Reid, M.E. Vitamin D Receptor Expression in Normal, Premalignant, and Malignant Human Lung Tissue. Cancer Epidemiology, Biomarkers & Prevention 2008, 17, 1104–1110. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Arora, J. Two Lineages of Immune Cells That Differentially Express the Vitamin D Receptor. The Journal of Steroid Biochemistry and Molecular Biology 2023, 228, 106253. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of Novel CYP11A1-Derived Secosteroids in the Human Epidermis and Serum and Pig Adrenal Gland. Sci. Rep. 2015, 5, 14875. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Marepally, S.R.; Goh, E.S.Y.; Cheng, C.Y.S.; Janjetovic, Z.; Kim, T.-K.; Miller, D.D.; Postlethwaite, A.E.; Slominski, A.T.; Tuckey, R.C.; et al. Investigation of 20S-Hydroxyvitamin D3 Analogs and Their 1α-OH Derivatives as Potent Vitamin D Receptor Agonists with Anti-Inflammatory Activities. Sci. Rep. 2018, 8, 1478. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ramirez, J.; Han, J.; Jia, Y.; Domenico, J.; Seibold, M.A.; Hagman, J.R.; Gelfand, E.W. The Steroidogenic Enzyme Cyp11a1 Is Essential for Development of Peanut-Induced Intestinal Anaphylaxis. Journal of Allergy and Clinical Immunology 2013, 132, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Hobrath, J.V.; Oak, A.S.W.; Tang, E.K.Y.; Tieu, E.W.; Li, W.; Tuckey, R.C.; Jetten, A.M. Endogenously Produced Nonclassical Vitamin D Hydroxy-Metabolites Act as “Biased” Agonists on VDR and Inverse Agonists on RORα and RORγ. The Journal of Steroid Biochemistry and Molecular Biology 2017, 173, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Qayyum, S.; Song, Y.; Janjetovic, Z.; Oak, A.S.W.; Slominski, R.M.; Raman, C.; Stefan, J.; Mier-Aguilar, C.A.; et al. Vitamin D and Lumisterol Derivatives Can Act on Liver X Receptors (LXRs). Sci. Rep. 2021, 11, 8002. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Waku, T.; Aoki, M.; Abe, R.; Nagai, Y.; Watanabe, T.; Nakajima, Y.; Ohkido, I.; Yokoyama, K.; Miyachi, H.; et al. A Nonclassical Vitamin D Receptor Pathway Suppresses Renal Fibrosis. J. Clin. Invest. 2013, 123, 4579–4594. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Ding, Q.; Wang, L.; Xu, G.; Diao, Y.; Qu, S.; Chen, S.; Shi, Y. Vitamin D3 Alleviates Pulmonary Fibrosis by Regulating the MAPK Pathway via Targeting PSAT1 Expression in Vivo and in Vitro. International Immunopharmacology 2021, 101, 108212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D Receptor Inhibits Nuclear Factor κB Activation by Interacting with IκB Kinase β Protein. Journal of Biological Chemistry 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Gouttenoire, J.; Duong, F.H.T.; Morikawa, K.; Heim, M.H.; Moradpour, D. Vitamin D Receptor and Jak–STAT Signaling Crosstalk Results in Calcitriol-Mediated Increase of Hepatocellular Response to IFN-α. The Journal of Immunology 2014, 192, 6037–6044. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kitagishi, Y. Peroxisome Proliferator-Activated Receptor and Vitamin D Receptor Signaling Pathways in Cancer Cells. Cancers 2013, 5, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hu, X.; Qi, X.; Zhu, R.; Li, C.; Zhu, Y.; Yin, S.; Cheng, L.; Zhu, R. Vitamin D Ameliorates Angiotensin II-Induced Human Endothelial Progenitor Cell Injury via the PPAR-γ/HO-1 Pathway. J. Vasc. Res. 2019, 56, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, A.J.; Kokje, V.B.; Koning, O.H.; Hamming, J.F.; Szuhai, K.; Claas, F.H.J.; Lindeman, J.H.N. Activation of the Vitamin D Receptor Selectively Interferes with Calcineurin-Mediated Inflammation: A Clinical Evaluation in the Abdominal Aortic Aneurysm. Laboratory Investigation 2016, 96, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, W.; Yang, Y.; Wei, S.; Xue, L.; Tao, S. Pharmaceutic Application of Vitamin D3 on Particle-Induced Fibrotic Effects through Induction of Nrf2 Signals. Toxicology Research 2020, 9, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nie, H.; Ge, X.; Du, J.; Liu, W.; Li, X.; Sun, Y.; Wei, X.; Xun, Z.; Li, Y.C. Vitamin D Suppresses Bleomycin-Induced Pulmonary Fibrosis by Targeting the Local Renin–Angiotensin System in the Lung. Sci. Rep. 2021, 11, 16525. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, T.; Yao, L.; Xing, Y.; Zhao, X.; Fu, J.; Xue, X. Chronic Vitamin D Deficiency Induces Lung Fibrosis through Activation of the Renin-Angiotensin System. Sci. Rep. 2017, 7, 3312. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.J.; O’Garra, A. 1α,25-Dihydroxyvitamin D3 Has a Direct Effect on Naive CD4+ T Cells to Enhance the Development of Th2 Cells. The Journal of Immunology 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A.; Rahimzadeh, M.; Brierley, C.; Gration, B.; Doree, C.; Kimber, C.E.; Plaza Cajide, A.; Lamikanra, A.A.; Roberts, D.J. The Role of Vitamin D in Increasing Circulating T Regulatory Cell Numbers and Modulating T Regulatory Cell Phenotypes in Patients with Inflammatory Disease or in Healthy Volunteers: A Systematic Review. PLoS ONE 2019, 14, e0222313. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.; Wynn, T.A. Fibrosis Is Regulated by Th2 and Th17 Responses and by Dynamic Interactions between Fibroblasts and Macrophages. American Journal of Physiology-Gastrointestinal and Liver Physiology 2011, 300, G723–G728. [Google Scholar] [CrossRef]
- Joshi, S.; Pantalena, L.-C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-Dihydroxyvitamin D 3 Ameliorates Th17 Autoimmunity via Transcriptional Modulation of Interleukin-17A. Molecular and Cellular Biology 2011, 31, 3653–3669. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chung, S.I.; Park, H.J.; Oh, E.Y.; Kim, S.-R.; Park, K.H.; Lee, J.-H.; Park, J.-W. Obesity-Induced Vitamin D Deficiency Contributes to Lung Fibrosis and Airway Hyperresponsiveness. Am. J. Respir. Cell. Mol. Biol. 2021, 64, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.M.; Wongtrakool, C.; Welch, T.; Steinmeyer, A.; Zügel, U.; Roman, J. Vitamin D Inhibition of Pro-Fibrotic Effects of Transforming Growth Factor Β1 in Lung Fibroblasts and Epithelial Cells. The Journal of Steroid Biochemistry and Molecular Biology 2010, 118, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhan, J.; Ji, H.; Xu, Y.; Xu, Q.; Zhu, X.; Liu, Y. Fibroblast Upregulation of Vitamin D Receptor Represents a Self-Protective Response to Limit Fibroblast Proliferation and Activation during Pulmonary Fibrosis. Antioxidants 2023, 12, 1634. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, T.; Magro-Lopez, E.; Manso, J.; Garcia-Martinez, R.; Fernandez-Aceñero, M.J.; Liste, I.; Zambrano, A. Detrimental Pro-Senescence Effects of Vitamin D on Lung Fibrosis. Mol. Med. 2018, 24, 64. [Google Scholar] [CrossRef] [PubMed]
- Magro-Lopez, E.; Chamorro-Herrero, I.; Zambrano, A. Effects of Hypocalcemic Vitamin D Analogs in the Expression of DNA Damage Induced in Minilungs from hESCs: Implications for Lung Fibrosis. IJMS 2022, 23, 4921. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, Y.; Xue, L.; Li, B.; Zhang, Z. 1α,25-Dihydroxyvitamin D3 Attenuates TGF-β-Induced Pro-Fibrotic Effects in Human Lung Epithelial Cells through Inhibition of Epithelial–Mesenchymal Transition. Nutrients 2017, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Sari, E.; Oztay, F.; Tasci, A.E. Vitamin D Modulates E-Cadherin Turnover by Regulating TGF-β and Wnt Signalings during EMT-Mediated Myofibroblast Differentiation in A459 Cells. The Journal of Steroid Biochemistry and Molecular Biology 2020, 202, 105723. [Google Scholar] [CrossRef] [PubMed]
- Izbicki, G.; Segel, M.J.; Christensen, T.G.; Conner, M.W.; Breuer, R. Time Course of Bleomycin-induced Lung Fibrosis. Int. J. Experimental Path. 2002, 83, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Gul, A.; Yang, F.; Xie, C.; Du, W.; Mohammadtursun, N.; Wang, B.; Le, J.; Dong, J. Pulmonary Fibrosis Model of Mice Induced by Different Administration Methods of Bleomycin. BMC Pulm. Med. 2023, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Robbe, A.; Tassin, A.; Carpentier, J.; Declèves, A.-E.; Mekinda Ngono, Z.L.; Nonclercq, D.; Legrand, A. Intratracheal Bleomycin Aerosolization: The Best Route of Administration for a Scalable and Homogeneous Pulmonary Fibrosis Rat Model? BioMed Research International 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abidi, A.; Bahri, S.; Ben Khamsa, S.; Legrand, A. A Comparative Study of Intratracheal and Aerosolization Instillations of Bleomycin Inducing Experimental Lung Fibrosis in Rat. Toxicology Mechanisms and Methods 2019, 29, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, X.; Fang, X.; Liang, A.; Yu, Z.; Gu, P.; Zeng, Y.; He, J.; Zhu, H.; Li, S.; et al. Preventive Effects of Vitamin D Treatment on Bleomycin-Induced Pulmonary Fibrosis. Sci. Rep. 2015, 5, 17638. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Bouros, E.; Barbayianni, I.; Karampitsakos, T.; Kourtidou, S.; Ntassiou, M.; Ninou, I.; Aidinis, V.; Bouros, D.; Tzouvelekis, A. Vitamin D Prevents Experimental Lung Fibrosis and Predicts Survival in Patients with Idiopathic Pulmonary Fibrosis. Pulmonary Pharmacology & Therapeutics 2019, 55, 17–24. [Google Scholar] [CrossRef]
- Lin, T.; Zhou, F.; Mao, H.; Xie, Z.; Jin, Y. Vitamin D and Idiopathic Pulmonary Fibrosis: A Two-Sample Mendelian Randomization Study. BMC Pulm. Med. 2023, 23, 309. [Google Scholar] [CrossRef] [PubMed]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D Supplementation to Prevent Acute Respiratory Tract Infections: Systematic Review and Meta-Analysis of Individual Participant Data. BMJ 2017, i6583. [Google Scholar] [CrossRef] [PubMed]
- Janssens, W.; Bouillon, R.; Claes, B.; Carremans, C.; Lehouck, A.; Buysschaert, I.; Coolen, J.; Mathieu, C.; Decramer, M.; Lambrechts, D. Vitamin D Deficiency Is Highly Prevalent in COPD and Correlates with Variants in the Vitamin D-Binding Gene. Thorax 2010, 65, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Yavari, M.; Mousavi, S.A.J.; Janani, L.; Feizy, Z.; Vafa, M. Effects of Supplementation of Vitamins D, C and E on Idiopathic Pulmonary Fibrosis (IPF): A Clinical Trial. Clinical Nutrition ESPEN 2022, 49, 295–300. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of some interactions between vitamin D and T-cells.
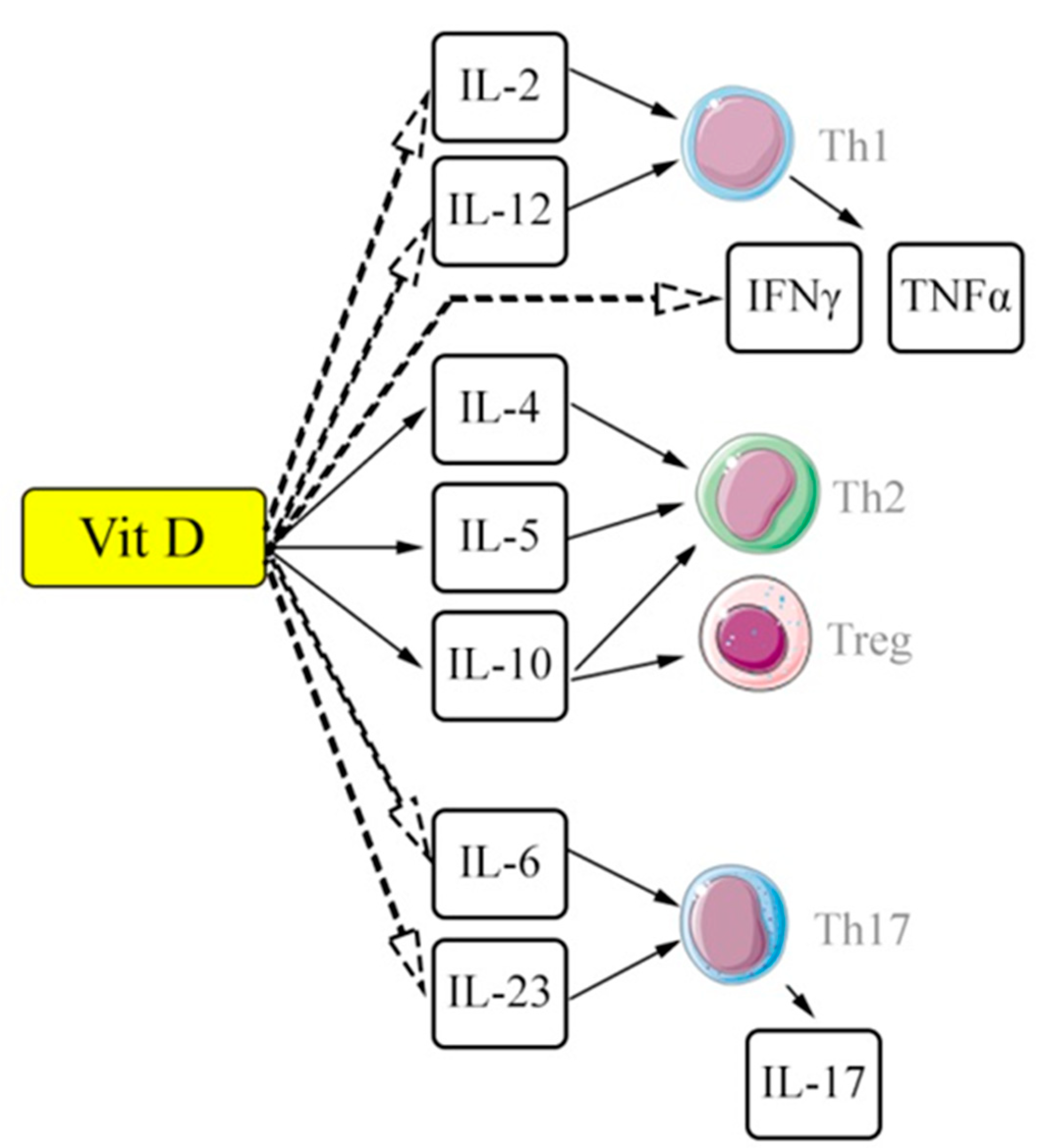
Figure 2.
Diagram of the key mechanisms of pulmonary fibrosis development and potential application points for the effects of vitamin D and its metabolites. Scheme 1. – T-helper type 1; Th2 –T-helpers type 2; Th17 –T-helpers type17; VDR – vitamin D receptor.
Figure 2.
Diagram of the key mechanisms of pulmonary fibrosis development and potential application points for the effects of vitamin D and its metabolites. Scheme 1. – T-helper type 1; Th2 –T-helpers type 2; Th17 –T-helpers type17; VDR – vitamin D receptor.
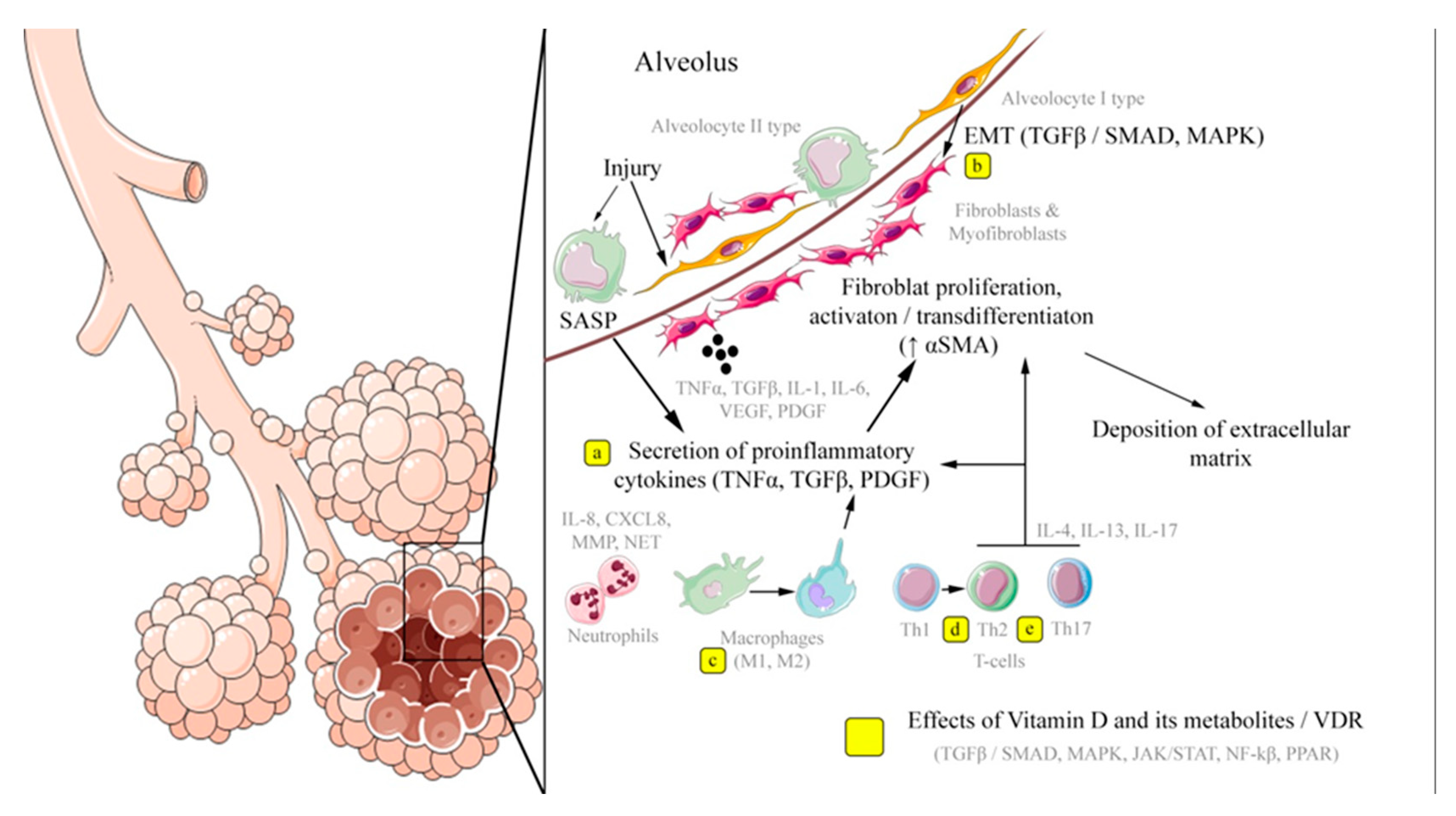

Table 1.
Some key signaling pathways and effects of ligand-associated activation of vitamin D receptors.
Table 1.
Some key signaling pathways and effects of ligand-associated activation of vitamin D receptors.
| Signaling pathway | Signaling pathway inductor | The implemented cascade of reactions | Effect of Vit D complex/VDR |
|---|---|---|---|
| SMAD | TGF-β / receptors TGF-β; angiotensin II |
Regulation of cellular cycle, differentiation (in particular of myofibroblasts), immune reactions | Suppression by reducing TGF-β expression and nuclear translocation of SMAD components |
| MARK | TGF-β; growth factors; lipopolysaccharides etc. | Regulation of cellular proliferation (↑) and differentiation, inflammatory response and elimination of cells by apoptosis (↓) | Suppression by enhancing the expression of proteinase (MAPK phosphatase-1) followed by inhibition of p38 MAPK |
| NF-κβ | Proinflammatory cytokines (IL-1. TNF α); lipopolysaccharides, growth factors | Regulation of non-specific and adaptive immunity and inflammatory response |
Suppression by inhibiting Ikkß kinase and preventing activation of NF-kß, as well as by suppressing nuclear translocation of signaling pathway components |
| JAK/STAT | Cytokines (IL-2, IL-6, IL-10, IL-12, IFN- α etc.), growth factors (EGF) | Regulation of cellular proliferation, differentiation and inflammatory response | Suppression of binding of transcription factor STAT3 to the promoter (ATF6) |
| PPAR- α/γ | Unsaturated fatty acids, eicosanoids, etc. | Regulation of metabolism of fatty acids and energy balance, regulation of immune response and cellular cycle | Competitive interaction due to binding to RXR, suppression of PPAR-α/γ promoter |
| NFAT | Ca2+ | Regulation of cellular cycle and inflammatory response | Blocking of NFAT components of Runx1 transcription factor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



