
Submitted:
19 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
New strategies for enhancing drug delivery to the blood–brain barrier (BBB) represents a major challenge in treating cerebral diseases. Nano-emulsion based nanocarriers represent an ideal candidate to improve drug delivery thanks to their versatility in functionalization and cargo protection. In this work, a paclitaxel loaded nano-emulsion has been firstly functionalized and stabilized with two layers constituted of chitosan and hyaluronic acid, and, secondly, the latter has been conjugated to the CRT peptide. CRT is a bioactive peptide that selectively recognizes bEnd.3 cells, model of the BBB, thanks to its interactions with transferrin (Tf) and its receptor (TfR).
Cytotoxic results showed a higher uptake of CRT functionalized nano-emulsion 41.5 % more than the negative control, demonstrating the ability of this novel tool to be accumulated on the brain endothelium tissue. Thanks to these results, our approach can be fully generalizable to the design of multifunctional nanocarriers for delivery of therapeutic agents to the central nervous systems.
Keywords:
active targeting
; CRT peptide
; drug delivery
; cancer
; nanoemulsion
1. Introduction
Effective cancer therapy for the treatment of brain tumours and central nervous system (CNS) diseases remains one of the most challenging areas in drug delivery research. One of the major issues is represented by their inability to cross the physical obstacle of the blood–brain barrier (BBB) [1,2]. Identifying routes for non-invasive drug delivery to brain and developing targeting strategies to transport biologics into the brain represent a research area of growing importance. It is known from literature that enhancing lipophilicity and positive charge is a possible strategy to increase passive diffusion in the same way as glucose, water, amino acids and small lipophilic molecules do that is crucial to neural function [3]. This is for instance the penetration mechanism of cell penetrating peptides possessing multiple positive charges. However, these modifications generally lead to higher unspecific uptake in many tissues often resulting in off-target effects since they are not selective. A promising strategy for overcoming the BBB to deliver biologics is the targeting of endogenous receptor-mediated transport (RMT) systems that engage vesicular trafficking to transport ligands across the BBB endothelium. DDS modified with appropriate targeting ligands, could improve the access to the brain via RMT and release its cargo.
The transferrin receptor (TfR) is one of the first RMT systems studied for BBB drug delivery applications [4]. TfR is ubiquitously overexpressed on the brain capillary endothelial cells because it mediates iron delivery to the brain, via binding and intracellular trafficking of the iron-binding protein transferrin (Tf). Most importantly, compared to healthy brain cells, TfR has much higher expression levels in human glioblastoma because it is required for cancer cell proliferation [5,6]. The use of Tf as targeting ligand has been demonstrated [7]. Unfortunately, in vivo there is a competitive binding to TfR between the endogenous Tf and the Tf-modified DDS, thus inducing insufficient delivery to the tumour site [8]. One approach to overcome this issue is the use of targeting moieties whose TfR recognition is mediated by a different molecular pathway. A recently published disulfide-bridged cyclic peptide, CRTIGPSVC (CRT), was discovered by selection of a phage display peptide library in vivo [9]. CRT functionally mimics iron by binding to apo-Tf and causes the adoption of the iron-bound holo-Tf conformation, and thereby gain access to the brain through the Tf-TfR interaction. This peptide exhibited promising results for the treatment of brain tumours by delivering the herpes simplex virus thymidine kinase gene to a mouse model of human glioma [9,10]. The delivery was accomplished via intravenous administration of a CRT-targeted adeno-associated virus and phage hybrid vector and resulted in significant tumour shrinkage. Another example of the therapeutic prospective of CRT for glioblastoma involved the treatment with paclitaxel loaded CRT-NPs of diseased mice, for which a remarkably prolonged median survival was observed [11]. However, although biodegradable, solid nanoparticles are classified as nanomaterials and there are some concerns on their use with heavy regulatory paths. Here we propose, an alternative liquid-based nanocarrier, which is an oil in water nanoemulsion (O/W NE). O/W NE is a versatile tool in drug delivery field, which can be easily functionalized with several targeting moieties and protect at the same time drugs solubilized in their lipophilic phase, as in case of solid NPs [12]. In this work, we prepare O/W NEs that are stabilized by two polymeric layers of chitosan and biotinylated hyaluronic acid, the latter being functionalized with the CRT bioactive peptide to promote the NEs accumulation on BBB. An easy additive decoration strategy that exploits biotin−streptavidin physical interaction [10] is adopted to conjugate the peptide outside the system. CRT is linked to a biotinylated poly(ethylene glycol) (PEG) chain in order to inhibit the NEs clearance by reticuloendothelial system (RES) and expose the peptide at the external side. In order to verify its specificity toward cells over-expressing TfR receptor, biological tests of peptide functionalized O/W NEs have been carried out. We chose a mouse brain cell line (bEnd.3) as model of blood-brain-barrier, and paclitaxel (PTX), a well-known cytotoxic drug, to appreciate peptide-mediated accumulation on bEnd.3 cells by the induced cytotoxicity. Even if PTX is an anticancer drug, its purpose in this context is mainly to assess NEs ability accumulate a harmful substance toward healthy BBB cells thanks to the CRT peptide. Our results suggest that the designed vector is ready for targeted pharmacophore delivery and that further integration on the surface of a cell-penetrating peptide [13] will lead to BBB safe crossing of the carrier to reach the tumour site.
2. Materials and Methods
2.1. Materials
Soybean oil and surfactant Lipoid E80 (egg lecithin powder 80-85% enriched with phosphatidylcholine and 7–9.5% content in phosphatidylethanolamine) were purchased from Lipoid GmbH and used without further purification. For preparation of all nanoemulsions and solutions, Millipore Milli-Q water was used. Chitosan (CT, LMW 90-150 kDa, DDA 84% determined via 1H-NMR), 1-hydroxybenzotriazole hydrate (HOBt), N,N′-Diisopropylcarbodiimide (DIC), N,N-Diisopropylethylamine (DIEA), trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), dichloromethane (DCM), anhydrous N,N-dimethyl-formamide (DMF), 1,2-Ethanedithiol (EDT), Triisopropylsilane (TIS), piperidine, acetone, diethyl ether, dimethyl sulfoxide (DMSO), were purchased from Sigma Aldrich. Hyaluronic acid 250 kDa and biotin-PEG2k-COOH were purchased from Creative PEGWorks. N-α-Fmoc amino acids were provided by NovaBiochem. Paclitaxel was purchased from Discovery Fine Chemicals Ltd.
2.2. Peptide Synthesis and Purification
CRT (βA-CRTIGPSVC-βA-K) peptide were synthesized using the standard solid-phase-9-fluorenyl methoxy carbonyl (Fmoc) procedure and were obtained with good overall yields (50-60%). The syntheses were performed by using the Biotage®Syro WaveTM peptide synthesizer (Biotage, Uppsala, The Netherlands). The peptide scale synthesis was 0.1 mmol. It was assembled on Rink Amide resin with a substitution level of 0.71 mmol/g. The following protected amino acids were used to synthesize the peptide:
Fmoc-Lys(Boc)-OH; Fmoc-Ile-OH; Fmoc-Gly-OH; Fmoc- Ser(tBu)-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Pro-OH; Fmoc-Cys(Trt)-OH; Fmoc-Ala-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH
The synthetic procedure can be summarized as follow:
- Deprotection: Fmoc group was removed at the beginning of cycle with a 20% piperidine solution in DMF. After deprotection, the resin was washed with DMF to remove the residual piperidine. The peptide resin was then ready for coupling.
- Activation: the carboxyl group of each Fmoc-amino acid was activated by addition of HBTU (2 eq.)/Oxima Pure (2 eq.)/ DIEA (4 eq.).
- Coupling: the pre-activated Fmoc-amino acid reacted with the free amino-terminal group of the growing peptide chain on the resin using DMF as the reaction solvent
- Capping: this reaction was performed after each coupling step, using a solution of Ac2O 20% and DIEA 5% in DMF. Capping cycle was introduced to prevent deletion byproducts.
Deprotection, coupling and capping steps were repeated for each subsequent amino acid, until the chain assembly was completed. When the coupling was complete, the resin was washed with DMF. At completion of the synthesis, the resin was washed several times with DMF and finally dried. The peptide was cleaved from the resin by treating it with 94% TFA/2.5% EDT/2.5% water/1% TIS for 2 hours at room temperature. The mixture was then concentrated and transferred to glass centrifugal tubes for compound precipitation using ice-cold diethyl ether, which was performed repeatedly. Purified CRT peptide was obtained by preparative RP-HPLC with a Vydac C18 column (22 mm x 250 cm; 10 μm), eluted with a linear gradient (solvent A, H2O 0.1% TFA; solvent B, ACN 0.1% TFA) from 20 to 70% B over 58 minutes at flow rate of 23 mL·min-1. All analyses were performed at detection wavelength of 220 nm and reported after blank chromatogram subtraction.
2.3. Peptide Cyclization
CRT peptide was dissolved in an aqueous solution at a concentration of 0.1 mM. Then, DMSO was added dropwise until its final concentration was 5%. The reaction mixture was kept open to atmosphere under vigorous magnetic stirring over-night. The product was monitored by LC-MS analysis. When the reaction was completed, the water was evaporated; the peptide was precipitated in cold ethyl acetate and lyophilized.
2.4. Biotin-PEG2k-COOH Peptide Conjugation
The peptide was conjugated at the N-term with Biotin-PEG2k-COOH directly on the resin. Firstly, Fmoc protecting group was removed with a 20% piperidine solution in DMF, followed by several washing steps. Then, the coupling reaction with Biotin-PEG2k-COOH was conducted directly on the resin with DIC/HOBt/DIEA (1:1:2) 0.1 M; using DMF as solvent, overnight under nitrogen flow. At completion of the synthesis, the resin was washed several times with DMF, NMP, DCM, isopropanol and methanol, and finally dried. Biotin-PEG2k-peptide was cleaved from the resin by treating with 94% TFA/2.5% EDT/2.5% water/1% TIS for 2 hours, precipitated in ice-cold diethyl ether and lyophilized.
2.5. Peptides Analysis and Purification
The identity of crude peptides was analysed by analytical RP-HPLC–ESI-MS. The LC-MS was performed with a Shimadzu LC-10ADvp equipped with an SPDM10Avp diode-array detector. ESI-MS spectra were recorded on a Shimadzu LC-MS-2010EV system with ESI interface and Shimadzu LC-MS solution Workstation software for the data processing. A Q-array-octapole-quadrupole mass analyzer was used as the detector. Argon was used as ion gas in the CID cell and data were analyzed by Shimadzu LC-MS solution Workstation software. The optimized MS parameters were selected as followed: curved desolvation line (CDL) temperature 200°C; block temperature 200°C; probe temperature 200°C; detector gain 1.6 kV; probe voltage +4.5 kV; CDL voltage -15 V. Nitrogen served as nebulizer gas (flow rate: 1.5 L·min-1). All analyses were performed with a Vydac C18 column (4.6 mm x 150 mm; 5μm), eluted with a linear elution gradient from 1% to 70% B over 35 minutes at a flow rate 1 mL·min-1). The running eluents were: solvent A, H2O 0.1% TFA and solvent B, ACN 0.1% TFA.
The crude non-cyclic peptide was further purified by preparative RP-HPLC with a Vydac C18 column (22 mm x 250 cm; 10 μm), eluted with a linear gradient (solvent A, H2O 0.1% TFA; solvent B, ACN 0.1% TFA) from 20 to 80% B over 58 minutes at flow rate of 23 mL·min-1. All analyses were performed at detection wavelength of 220 nm. The pooled fractions, containing the desired products, were lyophilized. The peptides homogeneity was assessed by analytical HPLC and by ESI mass spectrometry. The crude Biotin-PEG2k-peptide was purified by preparative flash chromatography, using a Biotage ISOLERA flash purification system, ISO-1SW model, equipped with a diode-array detector. The product was eluted with a linear gradient (solvent A, H2O 0.1% TFA; solvent B, ACN 0.1% TFA) from 0% to 95% B over 20 column volumes, using SNAP C18 12g as column. The pooled fractions, containing the desired products were analysed by analytical RP-HPLC–ESI-MS.
2.6. MALDI-TOF Analysis of PEGylated Peptides
PEGylated peptide was characterized by matrix-assisted laser desorption/ionization mass spectrometry coupled to two times of flight analysers (MALDI-TOF-TOF). The sample was prepared with a final concentration of ~ 2 pmol/µL in the matrix by mixing the peptide with a solution 60% of α-cyano-4-hydroxycinnamic acid (CHCA) and 40% of 5-Dihydroxybenzoic acid (DHB).
The two matrix solutions were prepared as follows:
- 20 mg/mL of CHCA in a solution of H2O 5% formic acid in ACN (30/70 v/v)
- 20 mg/mL of DHB in a solution of H2O 0.1% TFA in ACN (30/70 v/v)
Approximately, 0.25 µL of the sample was deposited on the MALDI plate, after a layer deposition of a saturated solution of CHCA in acetone and allowed to dry prior to analysis. The mass spectra were recorded on an AB SCIEX TOF/TOF 5800 instrument operated in the reflector positive mode. MALDI-TOF MS analyses were conducted at a laser intensity of 4287 units and laser pulse rate of 400 Hz with a set mass range of 1000 to 6000 Da. A continuous stage motion set in a random pattern at 600 μm/s was used for sampling. Calibration was performed using Cal mix 5 from AB SCIEX as calibrants, which contained des-Arg1-Bradykinin, Angiotensin I, Glu1-Fibrinopeptide B, adrenocorticotropic hormone ACTH (1–17 clip), ACTH (18–39 clip) and ACTH (7–38 clip) resulting in a mass accuracy of 50 ppm. Each spectrum represents the sum of 2040 laser pulses from randomly chosen spots per sample position. Raw data were analysed using TOF/TOF Series Explorer software provided by the manufacturer and are reported as monoisotopic masses.
2.7. Paclitaxel Loaded Oil in Water Nanoemulsion
Firstly, a 20-wt % oil in water pre-emulsion was prepared. 5.8 g of lecithin Lipoid E 80 (egg lecithin powder 80-85% enriched with phosphatidyl choline and 7-9.5% content in phosphatidyl ethanolamine) were dissolved in 24 mL of soybean oil (density at 20 °C of 0.922 g·mL-1) at 60 °C using the immersion sonicator (Ultrasonic Processor VCX500 Sonic and Materials), performing runs of 10 seconds for 1 minute at 10% of sonication amplitude (microtip screwed). Then, 1 mL of ethanol solution of PTX (5 mg/mL) was added to the oil phase and kept for 1 hours at 70 °C to evaporate the ethanol. Subsequently, the oil phase was added to the aqueous phase (Milli-Q water), and mixed using the immersion sonicator with runs of 10 seconds for 8 minutes at 70% of amplitude (a pulse-on and a pulse-off respectively of 10 seconds). The pre-emulsion was finally homogenized for 3 single cycles and 200 steps at a pressure of 2000 bar by a high-pressure homogenizer (110P series microfluidizer) to obtain the final nanoemulsion.
2.8. Polymers Multilayer Deposition above Paclitaxel Loaded O/W NEs
Firstly, a layer of chitosan was deposited around the oil template with a final concentration of oil and chitosan of 10 wt% and 0.1 wt%, respectively. 0.1 M acetic acid solution of chitosan (0.125 wt%) was prepared with a final pH=4. Nanoemulsion 20 wt% oil was added quickly to the chitosan solution under vigorous stirring and kept under stirring for 15 minutes to allow uniform chitosan deposition. The nanoemulsion with the first positive layer of chitosan was passed through a high-pressure valve homogenizer at 700 bars for 100 continuous steps. The next hyaluronic acid layer was prepared by aid of two syringe pumps (HARVARD APPARATUS 11 PLUS) and an ultrasonic bath (FALC INSTRUMENTS). Starting from the secondary nanoemulsion 10 wt% oil - 0.1 wt% CT, a negative charged polymer layer was deposited by mixing 1:1 (v:v) of a 0.24 wt% aqueous solution of biotinylated hyaluronic acid, with the secondary nanoemulsion suspension. The two liquid phases were injected at the same flow rate (0.4 mL min-1) through two polymicro flexible fused silica micrometric capillaries (inner diameter of 200 µm) interfaced at their extremities (Molex). Each drop was then collected inside a glass tube immersed in the ultrasonic bath at room temperature, 59 kHz and 100% power for 15 minutes. The NCs were characterized at each step of preparation by DLS analysis.
2.9. Nanocarrier Assembly
The streptavidin solution was prepared by dissolving 1 mg in 1 mL of Milli-Q water (16,6 μM). It was added to the HA-Biotin 0.12 wt%-CT 0.05 wt%-O/W NEs 5 wt% oil, under sonication for 15 minutes and T= 20 °C, at a final concentration of 5.69 μM. In the same way the compound CRT-PEG2k-biotin was added under sonication for 15 minutes and T= 20 °C to the streptavidin-HA-Biotin-CT-O/W NEs at a molar ratio 2:1 between CRT-PEG2k-biotin and the streptavidin. The final concentrations were 3.2 μM and 6.4 μM for streptavidin and CRT respectively, while the final oil weight percentage was 2.78 wt%. The NCs were characterized at each step of preparation measuring the size and Z-potential by dynamic light scattering as described previously.
2.10. Particle Size and Z-Potential Measurements
All nanoemulsions and their successive functionalization were characterized at each step of preparation by measuring size and polydispersity index (PdI), using Zetasizer Nano ZS device (Malvern Instruments) with a 4 mW He-Ne ion laser at the wavelength of 633 nm and a photodiode detector at an angle of 173°. All the samples were diluted to a droplet concentration of 0.025 wt% by using acetic acid 20 mM at pH 4 for monolayer, and Milli-Q water for emulsions and bilayer suspensions. The calculation of the particle size distribution was performed using a default refractive index ratio (1.59) and 5 runs for each measurement (1 run lasting 100 s), at least 3 times for each sample. A particle electrophoresis instrument (Zetasizer zs nano series ZEN 3600, Malvern Instruments Ltd., Malvern, U.K.) was used for the Z-potential determinations. Samples were diluted as for the particle size analysis. Setting 50 runs for each measurement carried out the Z-potential analysis. Samples were collected into polystyrene cuvettes and measured three times and the results presented are the averages of these measurements. Experiments were carried out at 25 °C. Zetasizer software (Malvern Instruments) was used to obtain the data. Cumulate analysis was used to give the Z-average value, hydrodynamic diameter, polydispersity index and the intensity size distribution graphs.
2.11. Cryo-TEM Characterization
For the preparation of the frozen-hydrated sample the plunge freezing method was performed. Briefly a drop of 3 μL of the samples were put on a previously glow-discharged 200 mesh holey carbon grids (Ted Pella, USA) after that the grid was inserted in the chamber of a FEI Vitrobot Mark IV (FEI company, the Netherlands) at 4°C and 90% of humidity. The droplet of sample was blotted with filter paper for 1 s, (blot force 1, drain time 0,5 s) and then the grid was plunged into the liquid propane. The grid was then stored in liquid nitrogen in a grid box until it was finally transferred to a cryo-specimen 626 holder (Gatan, Inc., USA) and loaded into the cryo-transmission electron microscope for imaging. To obtain the image of the nanoparticles we used a Tecnai G2 20, a cryo-tomo transmission electron microscope (FEI company, the Netherland) equipped with LaB6 emitter (acceleration voltage of 200 kV) and recorded at with a 2 k × 2 k CCD-Eagle 2HS camera. The Frozen-hydrated sample is radiation-sensitive material, so to avoid damaging; the observation was carried out in Low Dose Mode.
2.12. Cell Culture
bEnd.3 cells were grown in DMEM (10% FBS, 1% L-Glu, 1% Streptomycin pennicyllin). Cell culture were always performed at 37 °C in 5% CO2 and 100% relative humidity (RH). Cells were used from passages 23 to 30.
2.13. Cytotoxicity Analysis
Cell viability was quantified by the PrestoBlue Assay (Invitrogen) and compared to non- treated cells, which were used as a control. Briefly, 1 × 104 bEnd.3 cells were seeded in a 96-well and incubated for several times (30min, 2h, and 4h) with PTX loaded CRT-PEG2k O/W NEs, PEG2k -O/W NEs and free PTX, diluted 1:5 in cells, at a final PTX concentration of 1.4 µM. PrestoBlue Assay was performed according to the manufacturer’s procedure, after 24 hours. Fluorescence of PrestoBlue reagent solution (excitation 535 nm) was read at 615 nm by using a spectrofluorometer (Wallac 1420 Victor2, Perkin–Elmer, USA). All experiments were performed in triplicate.
2.14. Uptake of PTX Loaded NCs
bEnd.3 cells were grown in DMEM (10% FBS, 1% L-Glu, 1% Streptomycin pennicyllin). After seeding, cells (1 × 104) were left overnight to allow attachment. Then were incubated and treated with PTX loaded CRT-PEG2k O/W NEs and PEG2k-O/W NEs (using rhodaminated streptavidin during NCs assembly) for 4 hours in cell specific medium at 37°C. Cells were then washed twice with PBS. Cells were fixed for 20 minutes in 4% of paraformaldehyde PFA. Nuclei and cell shape were labeled by DRAQ5 (excitation 633 nm) and WGA 555 (labels cellular membrane), respectively. The fluorescence intensity was analyzed by Zeiss LSM 700 confocal microscope. Images were reconstructed by ImageJ.
3. Results and Discussion
3.1. Synthesis, Purification, and Characterization of CRT
One of the most promising peptides that are able to recognize the TfR receptor is the cyclic 9-mer peptide CRT, whose sequence is CRTIGPSVC. The amino acid sequence is here slightly modified by introducing a non-α-amino acid (β-alanine) at the N-tem and C-term, and a lysine at the C-term as the last amino acid (βA-CRTIGPSVC-βA-K). β-alanine acts as a spacer to reduce the influence of the nanocapsule surface on the peptide conformation, while the Lys was inserted to exploit its side chain (-NH2) for further functionalization (i.e. fluorophore labelling). The peptide synthesis was performed exploiting solid-phase protocols (SPPS), using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry and a super acid labile resin. The resulting peptide was deprotected and cleaved from the resin. The crude peptide purity was assessed by analytical RP-LC-MS, whose chromatogram and mass spectra are reported in Figure 1a. Purified CRT peptide was cyclized by dissolving it in aqueous solution containing 5% DMSO to the recommended dilute concentration of the thiol moieties (0.01–0.1 mM), and thus to favoring the intra-chain disulphide bond formation between the two cysteine residues over dimer formation, which is a frequent side reaction during cyclization [14,15]. Unambiguous indication of peptide cyclization was accomplished by LC-MS analysis (Figure 1b). A slight shift in the retention time of non-cyclic and cyclic peptides can be observed, followed by a two units difference in the [M+H]+ values corresponding to the expected loss of two protons upon disulphide bridge formation.
Peptide Biotin/PEGylation was performed to anchor the cyclic peptide to our nanocapsules by exploiting biotin-streptavidin affinity (Figure 2).
A biotin functionalized PEG2k linker (biotin-PEG2k -COOH) was conjugated at the N-term of the peptide sequence, directly on the resin, via amide bond formation with carboxyl group.
Solid-phase synthesis was preferred to simplify the removal of unreacted biotin-PEG2k-COOH from PEGylated peptide by several wash steps with a set of solvents (NMP-DCM-iPrOH-Et2O). Once the peptide coupling with the biotin-PEG2k -COOH linker was done, the biotin-PEG2k–CRT peptide (CRT-PEG2k) has been deprotected and cleaved from the resin. The crude CRT-PEG2k was purified by preparative flash chromatography, obtaining the pure product with a final yield of approximately 5%. The pooled fractions, containing the desired products were analysed by analytical RP-HPLC, whose chromatogram was reported in Figure 3.
Identification of the product at Rt = 23.78 min was performed by matrix-assisted laser desorption/ionization mass spectrometry coupled to two time of flight analyzers (MALDI-TOF-TOF). Comparison of the centroid mass peaks of biotin-PEG2k-COOH linker acquired before and after peptide conjugation confirms the reaction outcome (Figure 4).
The intense polydisperse Gaussian distribution and the typical expected ethylene oxide repeat units of 44 Da were observed, corroborating the presence of PEG within the samples. The increment of mass for CRT-PEG2k perfectly corresponds to the CRT peptide, a clear evidence that the reaction took place. The observed mass increment (2589.11 ± n × 44 Da) with respect to the expected theoretical isotopic mass of CRT-PEG2k (2566.22 ± n × 44 Da) is due to the +23 m/z sodiated species (MNa)+. Peptide cyclization was then performed as described before directly on the obtained pegylated peptide.
3.2. Nanocarrier Assembly
The CRT-PEG2k was then linked to the multi-layered O/W NEs (CRT-PEG-NEs), with the aim to exploit its recognition to overexpressed TfR on the brain endothelium. We used a previously optimized decoration strategy [12], featuring an outer shell of biotinylated hyaluronic acid (Biot-HA-NEs), with the only difference that the oil core of the nanoemulsion is loaded with paclitaxel. Briefly, the streptavidin (SAV) was added to Biot-HA-NEs under sonication, followed by addition of CRT-PEG2k. The system has been characterized at all stages by DLS (Table 1 and Figure 5). At all stages narrowly monodispersed functionalized nanocapsules were obtained with a net negative charge according to the Z-potential measurements.
The morphological characterization of the paclitaxel-loaded nanocapsule was performed by cryo-TEM analysis (Figure 5b). The microscopy clearly shows a homogeneous sample fixed in its frozen hydrated state confirming the size distribution observed by DLS. The image displays a well-defined electron-dense core, corresponding to the PTX-loaded in the oil core that makes it difficult to observe the polymer layers around the O/W NEs.
DLS periodical measurements were performed to evaluate the size evolution over time. Figure 6 shows that the hydrodynamic diameter of CRT-PEG2k-NEs is quite stable within the one-month detection window, with only minimal coalescence in the last 5 days (~20% radius increase).
3.3. Preliminary Biological Tests
bEnd.3 cells, an immortalized mouse brain endothelial cell line represent an attractive candidate as model of the BBB due to their rapid growth, maintenance of BBB characteristics and formation of functional barriers [16]. It was demonstrated in our group, that the number of TfRs per bEnd3 cells was 100-fold higher than in HUVEC cells, confirming a high value expression of TfRs [17]. In order to understand the best conditions to appreciate the selective activity of CRT-PEG2k-NEs toward bEnd.3 cells, thanks to CRT-TfRs interaction, we investigated the PTX effect on this cell line using the same procedure of Falanga et al. [18].
A confluent monolayer of bEnd.3 cells were incubated with PTX loaded CRT-PEG2k-NEs, diluted 1:5 in cells, at a final PTX concentration of 1.4 µM for several times (30 min, 2h and 4h). Moreover, cells were treated with cell medium alone as positive control, free PTX as negative control and un-functionalized PEG2k- NEs that act as blank. After incubation, the cells were washed and a quantitative evaluation of cell viability (normalized to positive control, which is set to 100%) was obtained by PrestoBlue® assay after 48 hours (Figure 7).
Data have shown an increase of cell mortality both for free PTX and for CRT-PEG2k O/W NEs over time, with no significant difference between two and four hours. Very interestingly, it was possible to observe a significant cytotoxicity effect of CRT-PEG2k O/W NEs compared to blank (PEG2k O/W NEs). This is an evident consequence of peptide capability to accumulate the nanocarrier on the cells surface, and allow its internalization, thanks to ligand-receptor recognition. For all the incubation times, CRT-PEG2k O/W NEs exhibit an increase of cell death respect to PEG2k O/W NEs and respect to free PTX. For instance, at 30 min of incubation, CRT-PEG2k O/W NEs exhibit an increase of 38% of cell death respect to PEG2k O/W NEs and almost 17% respect to free PTX. Making an average over all the times, respect to un-functionalized PEG2k- O/W NEs, CRT-PEG2k-O/W NEs induced an increased cells death of 33.05 ± 4.42 %.
To confirm the effect due to the targeted accumulation, cellular uptake assays by confocal microscopy was performed. A confluent monolayer of bEnd.3 cells were incubated with CRT-PEG2k O/W NEs and PEG2k O/W NEs, for 4 h at the same experimental condition of cytotoxicity test described before. Rhodaminated streptavidin was used during the nanocapsules assembling to detect their fluorescence. In addition, the control (CTRL) consisted in cells treated with cell medium alone. Figure 8 shows confocal microscopic images of bEnd.3 cell monolayer after NCs uptakes, which shed light on the previous cytotoxicity results. The presence of functionalized nanocapsules was shown in red, while the cells cytoplasm and the nuclei were in green and blue respectively. In Figure 9, it is reported the plot of rhodaminated-NCs ‘mean’ fluorescence intensity normalized for the bEnd.3 cell number.
The uptake data are in accordance with the previous cytotoxicity results by showing a clear fluorescence difference between CRT-PEG2k O/W NEs and PEG2k O/W NEs. However, no fluorescence can be detected from the images of the control cells. Indeed, the fluorescence intensity of peptide-functionalized NCs was approximately 40% higher compared to the negative control. Furthermore, a slight amount of PEG2k O/W NEs was detected in the cells.
4. Conclusions
In this study we demonstrate the versatility of engineered layer by layer nanocapsules in active targeting. To develop a novel nanocapsule able to target the BBB our O/W NEs ahve been functionalized with the CRT peptide, which is able to recognize the BBB cells thanks to Tf/TfR mechanism. O/W NEs are stabilized by a double layer of chitosan and biotinylated hyaluronic acid, and, thanks to their long shelf-life, represent an ideal candidate for further decoration of a bioactive peptide on the outer layer by an additive strategy, without purification steps. CRT-functionalized NEs preserved a narrow distributed hydrodynamic diameter below 150 nm, stable over time. Preliminary cytotoxic tests were carried out using mouse brain cell line (bEnd.3) as model of endothelial brain tissue. Results show an increased cellular death of 33.05 ± 4.42 % for CRT-PEG2k-NEs compared to undecorated PEG2k-NEs that act as a control, while the uptake of CRT-PEG2k-NEs increased 41.5 ± 24 % with respect to the negative control. We proved in vitro the ability of CRT peptide to target the brain endothelium tissue while conjugated to the proposed O/W NE-based carrier. The proposed platform paves the way to the design of novel multifunctional nanocarriers for delivery of therapeutic agents to the CNS which are made of a vegetable oil core easily degraded and absorbed with no concerns in terms of accumulation in the body in contrast with many solid based nanomaterials. Further development will concern the integration of a cell penetrating peptide, like gH625, able to cross the BBB. Moreover, for anticancer therapeutic purposes, potentially new therapeutics19 or the well-established curcumin instead of PTX can be used, the latter being used in this case only as a cytotoxic molecule to test NCs accumulation ability. Indeed, curcumin not only has been demonstrated to be a good anticancer substance, but also it was specific for tumor cells.12 Therefore, it may be an ideal candidate for blood-brain-barrier treatments, because, although a fraction of NCs will break and release curcumin in the endothelial cells instead of crossing them, such a molecule has no activity toward these cells, avoiding side effect to healthy sites.
Acknowledgements
This research was supported by EU funding within the MUR PNRR “National Center for Gene Therapy and Drugs based on RNA Technology” (Project no. CN00000041 CN3 Spoke #8 “Platforms for RNA/DNA delivery”).
References
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chemical Society Reviews 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.R.; et al. Surface charge, glycocalyx, and blood-brain barrier function. Tissue Barriers 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Friden, P.M.; et al. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 4771–4775. [Google Scholar] [CrossRef] [PubMed]
- Recht, L.; Torres, C.O.; Smith, T.W.; Raso, V.; Griffin, T.W. Transferrin receptor in normal and neoplastic brain tissue: Implications for brain-tumor immunotherapy. J. Neurosurg. 1990, 72, 941–945. [Google Scholar] [CrossRef]
- Gomme, P.T.; McCann, K.B. Transferrin: Structure, function and potential therapeutic actions. Drug Discovery Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Loureiro, J.A.; Coelho MA, N.; Pereira, M.C. Transferrin Receptor-Targeted Nanocarriers: Overcoming Barriers to Treat Glioblastoma. Pharmaceutics 2022. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Staquicini, F.I.; et al. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J. Clin. Invest. 2011, 121, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; et al. A Hybrid Vector for Ligand-Directed Tumor Targeting and Molecular Imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; et al. Enhancing Glioblastoma-Specific Penetration by Functionalization of Nanoparticles with an Iron-Mimic Peptide Targeting Transferrin/Transferrin Receptor Complex. Mol. Pharm. 2015, 12, 2947–2961. [Google Scholar] [CrossRef] [PubMed]
- De Capua, A.; et al. Active targeting of cancer cells by CD44 binding peptide-functionalized oil core-based nanocapsules. RSC Adv. 2021, 11, 24487–24499. [Google Scholar] [CrossRef] [PubMed]
- Fotticchia, T.; et al. Enhanced Drug Delivery into Cell Cytosol via Glycoprotein H-Derived Peptide Conjugated Nanoemulsions. ACS Nano 2017, 11, 9802–9813. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford Univ. Press 2000, 1–341. [Google Scholar]
- Chen, L.; Annis, I.; Barany, G. Disulfide Bond Formation in Peptides. Curr. Protoc. Protein Sci. 2001, 23. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Morris, A.P.; O’Neil, R.G. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007, 1130, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Fotticchia, I.; et al. Energetics of ligand-receptor binding affinity on endothelial cells: An in vitro model. Colloids Surfaces B Biointerfaces 2016, 144, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; et al. Design, Synthesis and Characterization of Novel Co-Polymers Decorated with Peptides for the Selective Nanoparticle Transport across the Cerebral Endothelium. Molecules 2018, 23, 1655. [Google Scholar] [CrossRef]
- De Fenza, M.; Esposito, A.; D'alonzo, D.; Guaragna, A. Synthesis of piperidine nucleosides as comformationally restricted immucillin mimics. Molecules 2021, 26, 1652. [Google Scholar] [CrossRef]
Figure 1.
(a) RP-HPLC chromatogram of crude CRT. ESI-MS spectrum relative to the peak at Rt = 27.86 min that corresponds to [M+H]+ of CRT (theoretical mass: 1246.50 Da; observed mass: 1246.65 Da). (b) RP-HPLC chromatogram of pure cyclic CRT. ESI-MS spectrum relative to the peak at Rt = 28.14 min that corresponds to [M+H]+ of cyclic-CRT (theoretical mass: 1244.49 Da; observed mass: 1244.60 Da).
Figure 1.
(a) RP-HPLC chromatogram of crude CRT. ESI-MS spectrum relative to the peak at Rt = 27.86 min that corresponds to [M+H]+ of CRT (theoretical mass: 1246.50 Da; observed mass: 1246.65 Da). (b) RP-HPLC chromatogram of pure cyclic CRT. ESI-MS spectrum relative to the peak at Rt = 28.14 min that corresponds to [M+H]+ of cyclic-CRT (theoretical mass: 1244.49 Da; observed mass: 1244.60 Da).
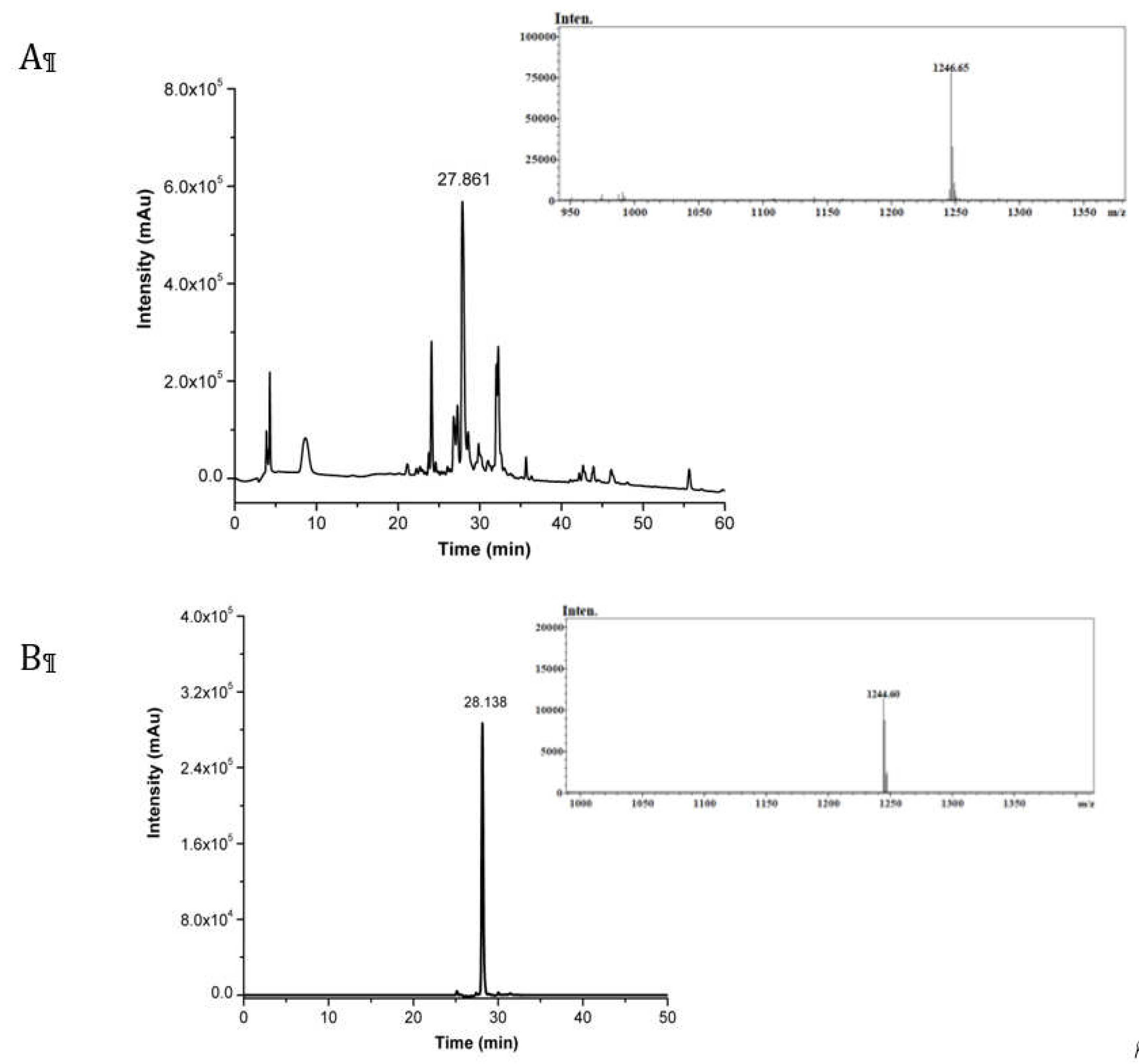
Figure 2.
Schematic representation of the solid-phase synthetic strategy of CRT peptide PEGylation.
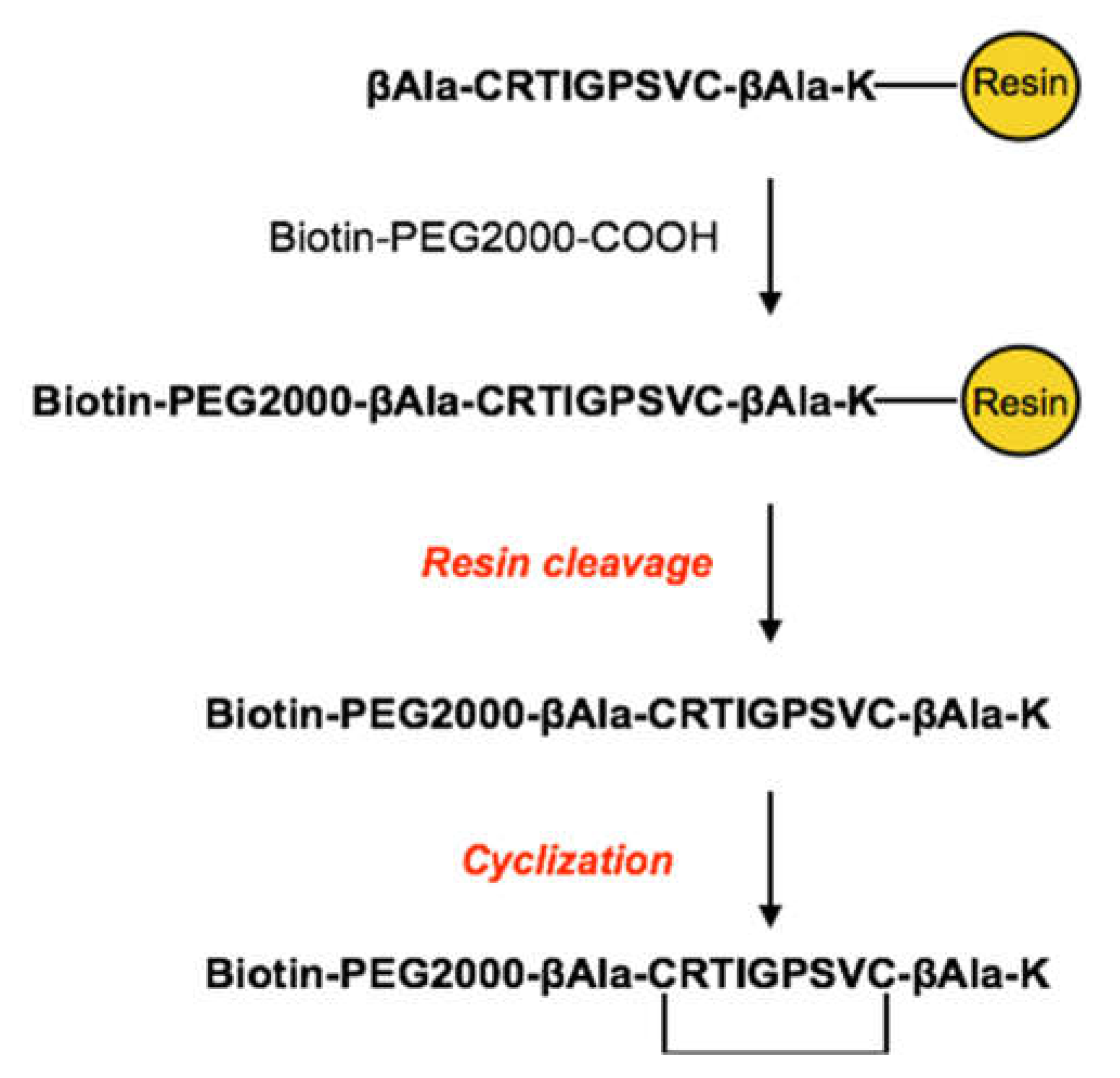
Figure 3.
Analytical RP-HPLC chromatogram of purified CRT-PEG2k, detected at λ = 220 nm.
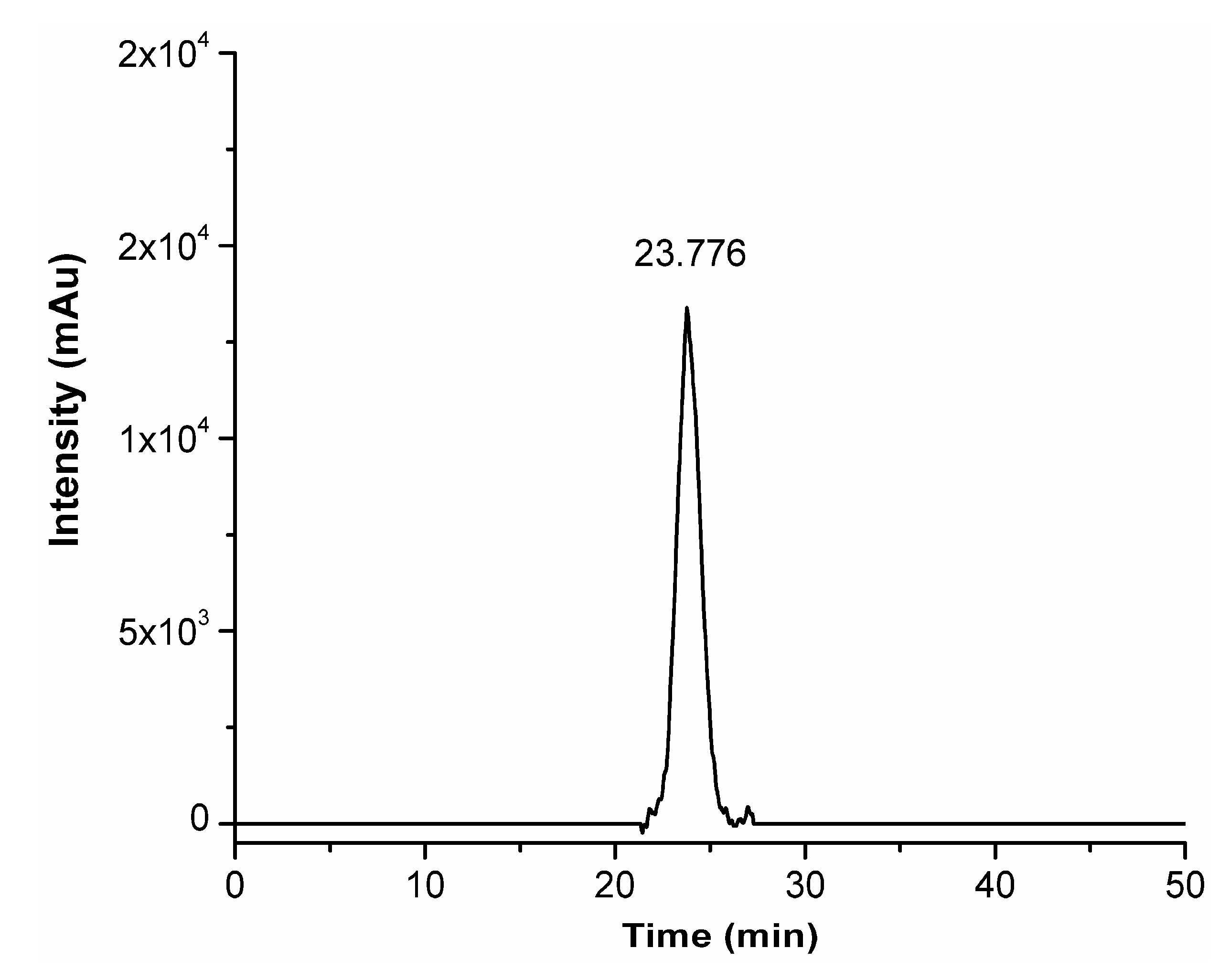
Figure 4.
MALDI mass spectra (centroid) of Biotin-PEG2k-COOH linker on the top and CRT-PEG2k peptide at the bottom. Insets report the zoomed centroid section of the spectra.
Figure 4.
MALDI mass spectra (centroid) of Biotin-PEG2k-COOH linker on the top and CRT-PEG2k peptide at the bottom. Insets report the zoomed centroid section of the spectra.
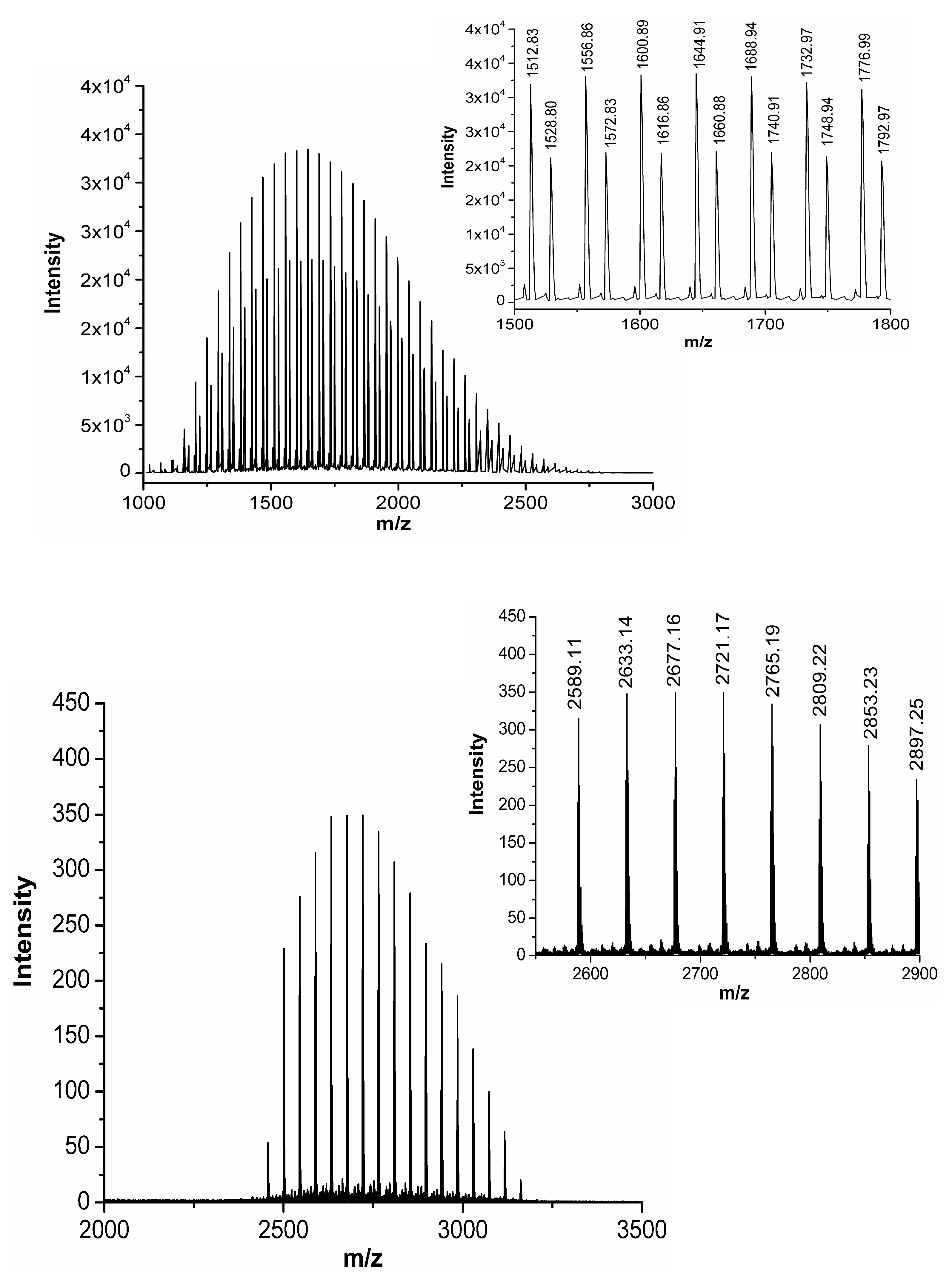
Figure 5.
(a) Overlapping of mean hydrodynamic size of each component deposited around NEs. (b) Cryo-TEM projection image of paclitaxel loaded HA-CT-O/W NEs. Scale bar corresponds to 500 nm.
Figure 5.
(a) Overlapping of mean hydrodynamic size of each component deposited around NEs. (b) Cryo-TEM projection image of paclitaxel loaded HA-CT-O/W NEs. Scale bar corresponds to 500 nm.
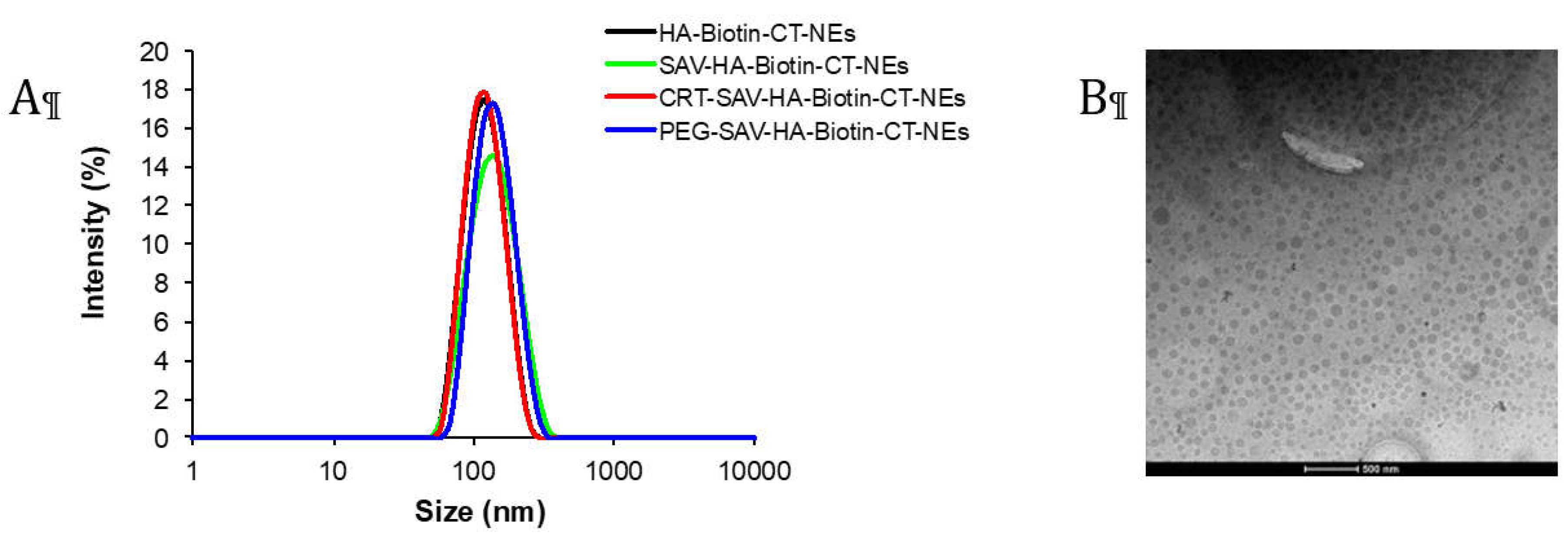
Figure 6.
Dimensional behaviour over time for PTX loaded CRT-NEs measured by DLS analysis.
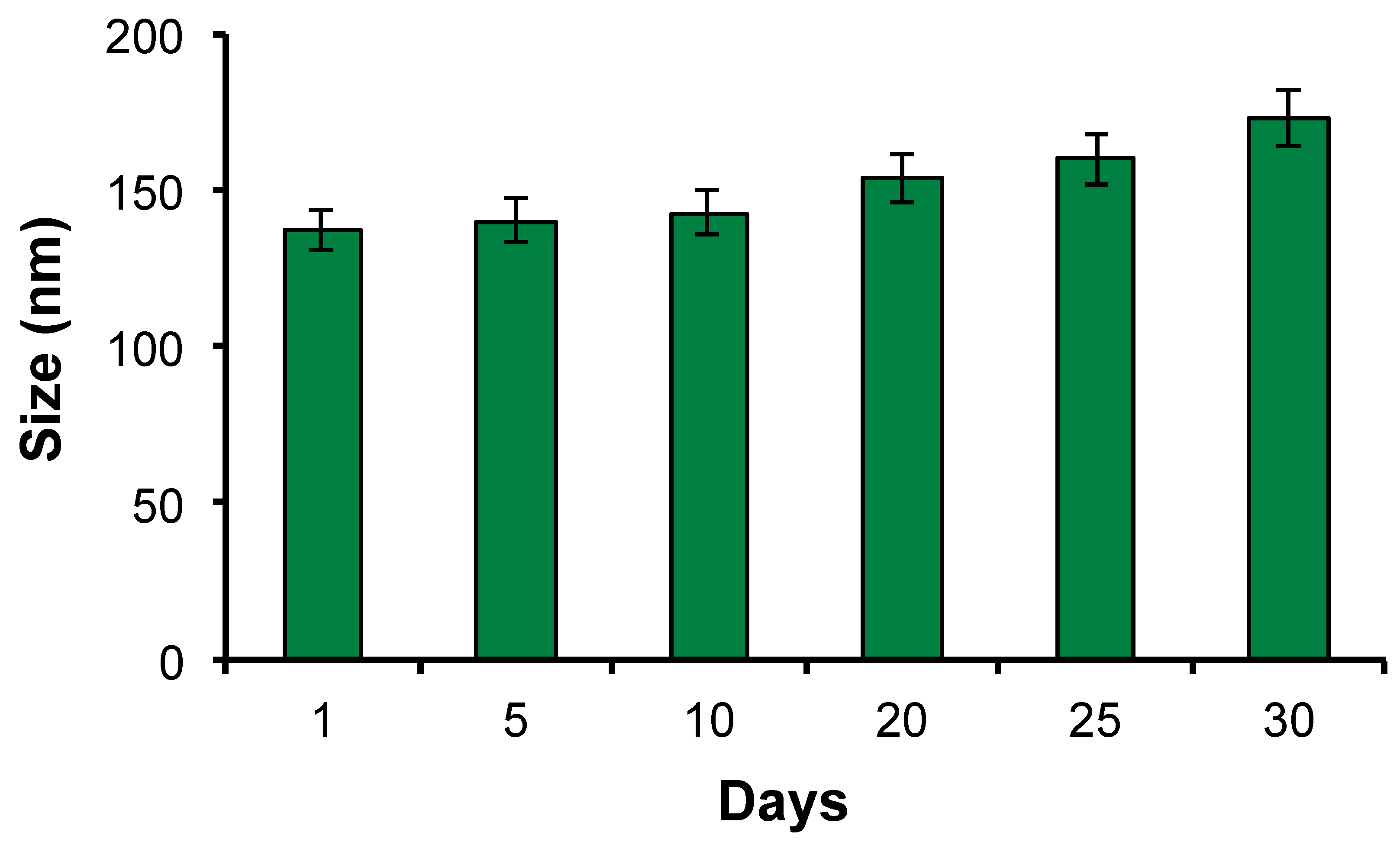
Figure 7.
Cytotoxicity assay of PTX loaded CRT-PEG2k- NEs and PEG2k-NEs, and free PTX. bEnd.3 cells were treated for several incubation times (30 min, 2h and 4h) and cell viability was evaluated after 48 h. Data are reported as mean of three independent experiment (n=3 ±SD) and expressed as percentage compared to control cells. The asterisk (*) indicates the statistical significance vs CTRL using Student’s t test; (***) p ≤ 0.001.
Figure 7.
Cytotoxicity assay of PTX loaded CRT-PEG2k- NEs and PEG2k-NEs, and free PTX. bEnd.3 cells were treated for several incubation times (30 min, 2h and 4h) and cell viability was evaluated after 48 h. Data are reported as mean of three independent experiment (n=3 ±SD) and expressed as percentage compared to control cells. The asterisk (*) indicates the statistical significance vs CTRL using Student’s t test; (***) p ≤ 0.001.
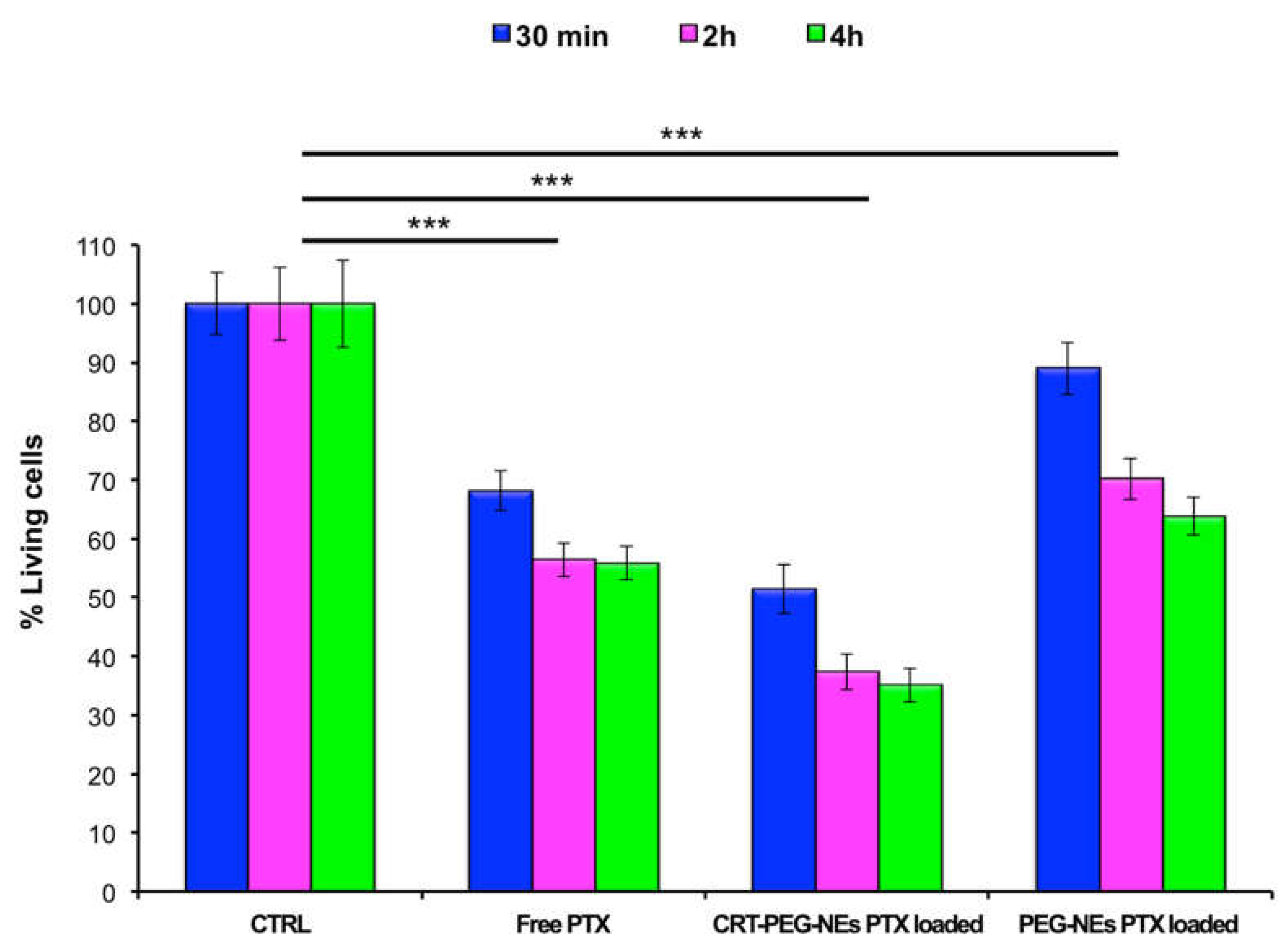
Figure 8.
Confocal images of bEnd.3 cell. Untreated, rhodaminated CRT-PEG2k O/W NEs and PEG2k O/W NEs interactions with a confluent monolayer of bEnd.3 cells. Nuclei (blue) and cellular membrane (green) of the cells were stained with DAPI and WGA 555 respectively, while red color represents rhodamine uptake. Scale bar is 100 µm.
Figure 8.
Confocal images of bEnd.3 cell. Untreated, rhodaminated CRT-PEG2k O/W NEs and PEG2k O/W NEs interactions with a confluent monolayer of bEnd.3 cells. Nuclei (blue) and cellular membrane (green) of the cells were stained with DAPI and WGA 555 respectively, while red color represents rhodamine uptake. Scale bar is 100 µm.
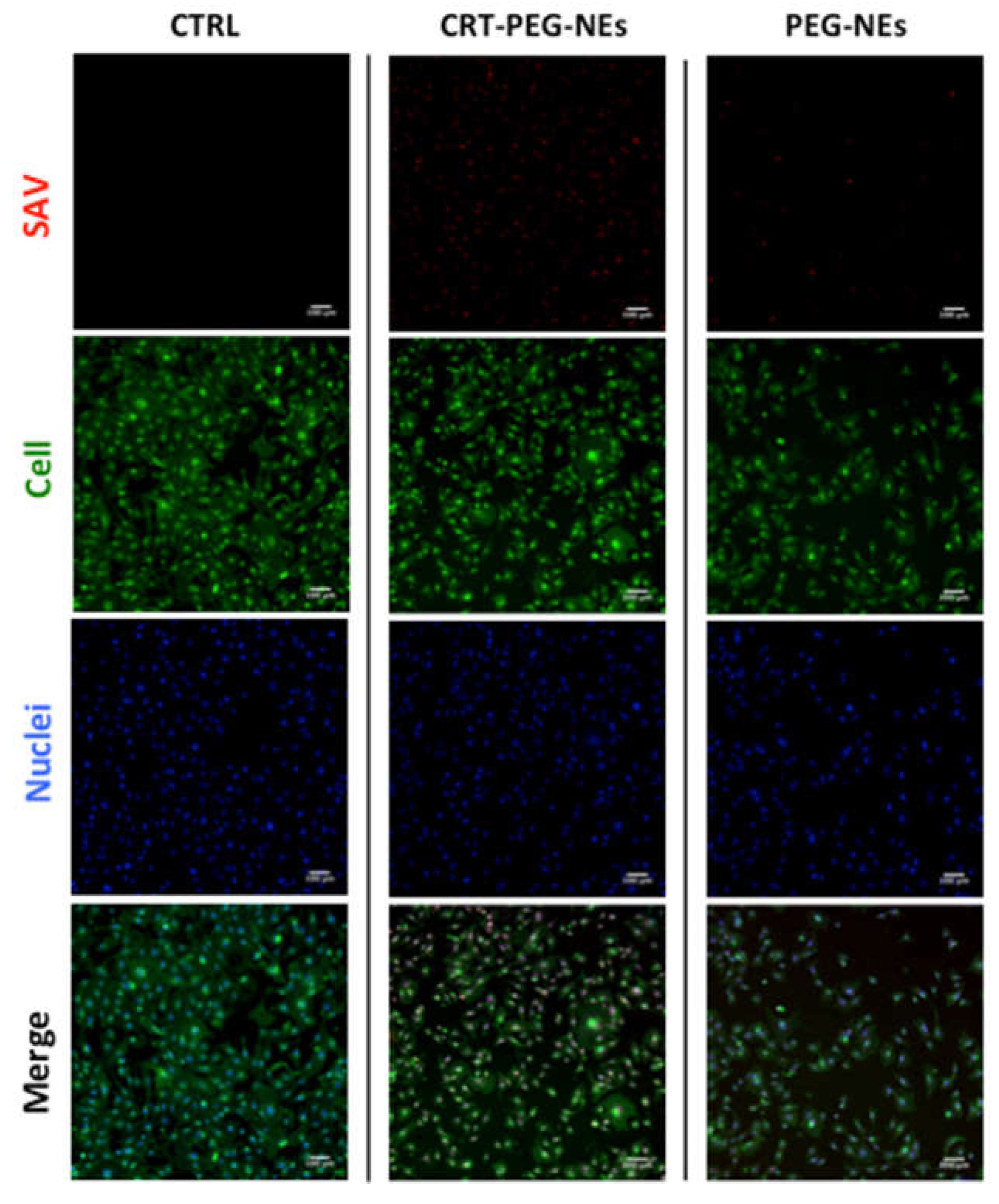
Figure 9.
Plot of mean fluorescence intensity of rhodaminated nanocapsules normalized to cells number. bEnd.3 cells were treated with CRT-PEG2k- NEs and PEG2k-NEs . Data are reported as mean (n=3) ± SD.
Figure 9.
Plot of mean fluorescence intensity of rhodaminated nanocapsules normalized to cells number. bEnd.3 cells were treated with CRT-PEG2k- NEs and PEG2k-NEs . Data are reported as mean (n=3) ± SD.
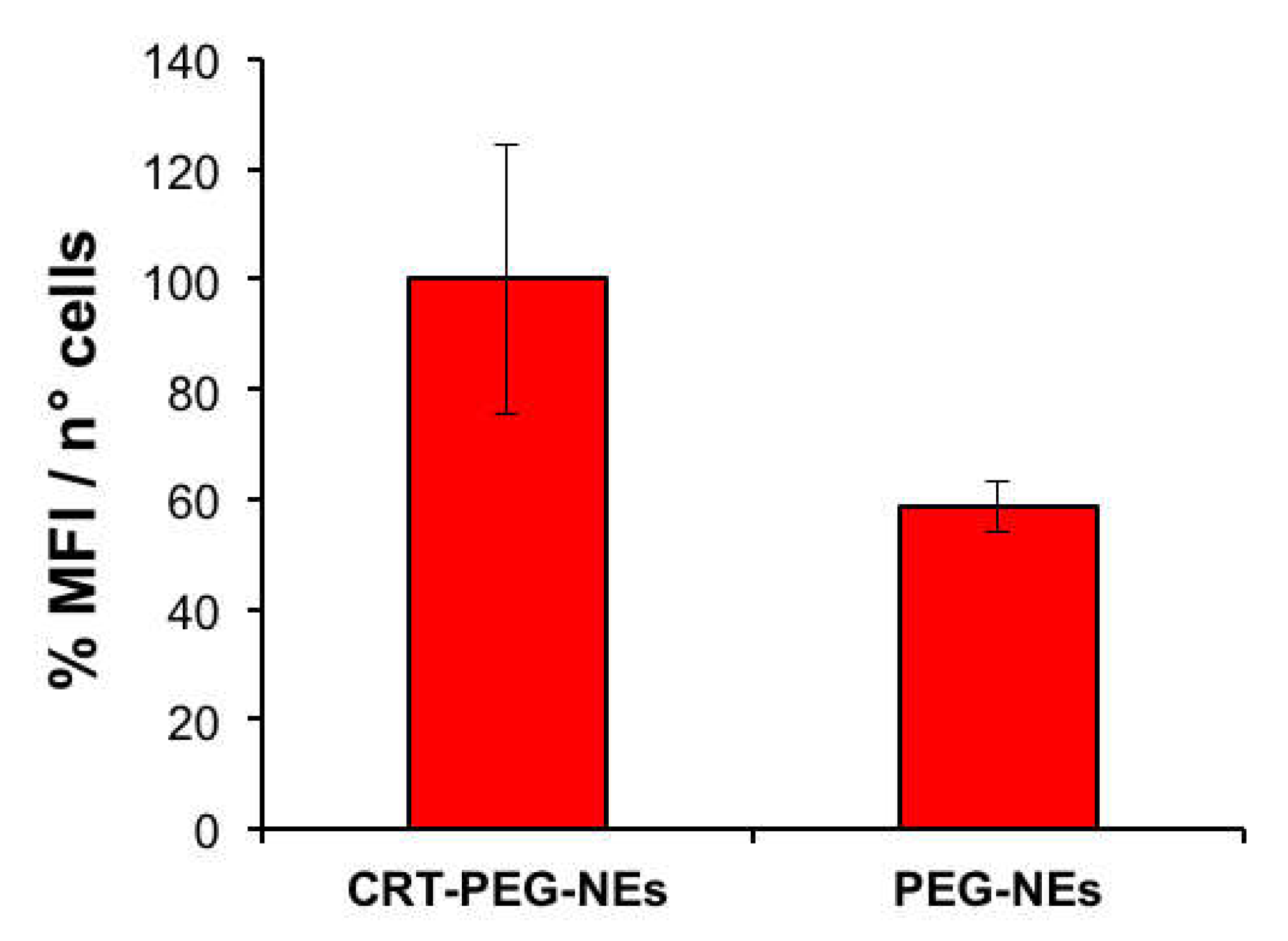

Table 1.
Size, PdI and Z-potential measurement of each NCs component, loaded with paclitaxel, during the several steps of assembly. Data are reported as mean (n=3) ± SD.
Table 1.
Size, PdI and Z-potential measurement of each NCs component, loaded with paclitaxel, during the several steps of assembly. Data are reported as mean (n=3) ± SD.
| Size (nm) | PdI | Z-potential (mV) | |
|---|---|---|---|
| Nes | 89 ± 1. | 0.080 ± 0.015 | -27 ± 4 |
| CT- Nes | 93 ± 2 | 0.086 ± 0.003 | +25 ± 1 |
| HA-biotin-CT-NEs | 113 ± 9 | 0.094 ± 0.015 | -31 ± 2 |
| SAV-HA-biotin-CT-NEs | 131 ± 7 | 0.145 ± 0.017 | -28 ± 3 |
| CRT-PEG2k-SAV-HA-biotin-CT- NEs | 137 ± 7 | 0.152 ± 0.002 | -30 ± 1 |
| PEG2k -SAV-HA-biotin CT-NEs | 138 ± 5 | 0.139 ± 0.002 | -30 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



