
Submitted:
21 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Systemic Lupus Erythematosus (lupus) and B-cell lymphoma (lymphoma) are serious diseases. Lupus and lymphoma co-occur at higher-than-expected rates and primarily depend on B-cells for their pathology, implicating shared inflammation-related B-cell molecular mechanisms as a potential cause of co-occurrence. We consequently implemented a novel Immune Imbalance Transcriptomics (IIT) algorithm and applied IIT to lupus, lymphoma, and healthy B-cell RNA-seq data to find shared- and contrasting mechanisms potentially targetable for treatment. We observed 8,562 genes significantly dysregulated in both lupus and lymphoma. Of those genes, we found 5,335 to have a significant immune imbalance, defined as significant dysregulation by both diseases, as determined by IIT. Gene Ontology (GO) term and pathway enrichment of IIT genes yielded immune-related “Neutrophil Degranulation” and “Adaptive Immune System”, validating that the IIT algorithm isolates biologically relevant genes in immunity and inflammation. We found that 389 IIT gene products are drug-targetable with established or repurposed drugs. Among our results, we found 40 known and 349 novel lupus targets, along with 151 known and 238 novel lymphoma targets. Known disease drug targets in our IIT results further validate that IIT isolates genes with disease-relevant mechanisms. We anticipate the IIT algorithm and shared and contrasting gene mechanisms uncovered here will contribute to the development of immune-related therapeutic options for lupus and lymphoma patients.
Keywords:
immune imbalance
; Immune Imbalance Transcriptomics
; IIT
; B-cell lymphoma
; Systemic Lupus Erythematosus (SLE)
; RNA-seq
; algorithm
; drug discovery
; cancer
; autoimmune disease
1. Introduction
The Systemic Lupus Erythematosus (lupus) is a debilitating autoimmune disease with widely varying clinical manifestations, affecting an estimated 3.41 million people [1,2]. The pathology of lupus is perpetrated in part by autoreactive B-cells, which produce auto-antibodies against self-antigens such as DNA, nuclear proteins, and other damage-associated molecular patterns (DAMPs) that remain when cells are damaged or undergo apoptosis [3]. Lupus has a complex diagnostic process, and many patients wait for years for a firm diagnosis. Lupus also has a substantial impact on the daily life of patients and their families due to severe symptoms (kidney failure, severe rash, depression, chronic fatigue, chronic pain, etc.) and limited treatment options, sometimes with undesirable side effects. Hydroxychloroquine, glucocorticosteroids, immunosuppressive drugs are effective at attenuating lupus symptoms and have been the standard of treatment for decades, with the recent addition of biologics such as belimumab. Hydroxychloroquine is still considered the cornerstone of most lupus treatment, despite the long-term risk of retinal toxicity, which can contribute to patient blindness [4]. Glucocorticoids are impressively effective at treating lupus symptoms in the short-term, but can cause permanent damage to multiple organ systems, cataracts, osteoporosis, and coronary artery disease after long-term use or at higher than standard dosing [5,6,7].
Lymphomas are estimated to make up 5% of malignancies worldwide [8], with ~450,000 new cases of Non-Hodgkin’s Lymphoma annually, leading to 240,000 deaths per year [9]. In addition, up to 90% of Non-Hodgkin’s lymphomas originate from B-cells [10]. Many B-cell Non-Hodgkin’s lymphomas (lymphomas), including diffuse large B-cell lymphoma, are aggressive and difficult to treat without severe side effects [8]. The slower-moving indolent lymphomas are typically considered incurable, and while patients are subjected to milder treatments such as rituximab-bendamustine (R- Benda), they run the long-term risk of cumulative toxicity from repeated chemotherapies and tumor progression into an aggressive lymphoma subtype [8]. The first-line therapy for fast-moving lymphomas is aggressive combination chemotherapy, such as R-CHOP (rituximab, cyclophosphamide, vincristine, and prednisone). In the case of relapse, the first-line treatments are followed by anti-CD19 chimeric antigen receptor (CAR) T-cell therapy or more intense chemotherapy regimes, which all require in-patient hospital stays, such as R-ICE (rituximab, ifosfamide, carboplatin, etoposide) or high dose chemotherapy with autologous stem cell rescue (HD-SCT) regimens such as the Nordic protocol (administration of maxi R–CHOP alternating with rituximab and high-dose cytarabine) [8]. Though some sources estimate lymphoma patient survival to be as much as 72%, the price paid to reach survival takes a heavy toll on the body and life quality of the patient as they deal with rampant toxicity from chemotherapies or B-cell aplasia and immune compromise from currently-approved anti-CD19 CAR T-cell therapies [8,11].
Patients with lupus are seven times more likely to develop B-cell lymphomas than healthy patients, suggesting a possible mechanistic relationship between the two diseases [12]. An important aspect of both lupus and lymphoma progression and of their relationship is immune involvement. The immune system is generally responsible for tolerating self-tissue, for removing non-self threats, including cancers, bacteria, and viruses, and for healing damaged self-tissue upon clearance of non-self threats. Healthy immune function begins and ends with a non-inflamed homeostasis which could be termed ‘immune balance’ [13,14]. Immune activation against threats is primarily accomplished via inflammatory signaling (including but not limited to eicosanoids including B- and E-prostaglandins and leukotrienes, vasoactive amines, complement cascade, kinins, and depending on the circumstances, cytokines such as TNFA, IFNG, IL1, IL2, IL6, IL12, IL17, IL18, and IL23) [15], while tolerance of self-tissue and wound healing/cleanup are accomplished via anti-inflammatory/pro-resolving signaling (including but not limited to nitric oxide, adenosine, cortisol, steroids, eicosanoids including lipoxins, resolvins, and D-prostaglandins, protectins, IL10, and TGFB) [16]. Conditions such as cancer or autoimmune diseases develop when this immune balance is disrupted by a failure of immune activation (cancer) or a failure of immune tolerance and resolution (autoimmune disease). Intense inflammation associated with immune activation from several causes (infection, allergy, trauma, etc.) can set the stage for autoimmune development [17,18]. Failure to remove inflammation-inducing, damage-associated molecular patterns (DAMPs) and/or pathogen-associated molecular patterns (PAMPs) can lead to the loss of self-tolerance [19]. In particular, pathogen molecular mimicry and the creation of cross-reactive and/or DAMP-targeted antibodies (e.g., anti-DNA) are known mechanisms of autoimmune disease development [17,18]. The failure of immune tolerance is integral to autoimmune development. In contrast, the inability of the immune system to effectively identify tumor cells can result in tolerance that enables the growth and progression of cancer [20]. Cancer develops as tumor cells create subclones and strive to avoid immune surveillance via immune-silencing strategies such as PD1/PDL1 blockade [21,22]. Cancer development is dependent upon the high immune tolerance of the tumor, which develops via natural selection as non-immune-avoidant cancer cells are killed and successful immune-evading cancer subclones proliferate [20,22]. The failure of sustained immune activation is integral to cancer development. Though there is some contrast suggested between lupus and lymphoma, lupus and lymphoma depend on immune function going awry pathologically. This similarity may contribute to the co-occurence of lupus and lymphoma.
Despite the higher-than-expected rate of lupus and lymphoma co-occurrence, to our knowledge, minimal studies have compared and contrasted lupus and lymphoma gene expression using RNA-sequencing. Additionally, there are no datasets in the NCBI gene expression omnibus (GEO) database that include bulk or single-cell RNA sequencing (RNA-seq) samples from patients with both conditions in the same study [23].
Given the lack of an available dataset containing lupus, lymphoma, and healthy B-cell RNA-seq data, the current study aimed to conduct a survey of existing gene expression data from publicly available datasets in an attempt to understand better the mechanistic transcriptional similarities and differences between these two conditions. We then used these RNA-seq results as a use-case for our novel Immune Imbalance Transcriptomics (IIT) algorithm. This algorithm aims to provide a reproducible workflow to determine the top gene candidates that best represent the immune imbalance for a consistent tissue type (e.g. B-cells). We anticipate that identifying the shared and contrasting underlying mechanisms between these two contrasting B-cell related diseases can be used to determine the relevant mechanisms that contribute to immune imbalance and can therefore be used in the identification of candidate therapeutics that can be developed or repurposed to target these diseases.
2. Materials and Methods
The Public RNA-sequencing datasets from the GEO database [23], originally generated from clinical material, were manually screened to find primary B-cell lymphoma, lupus B-cell, and healthy B-cell control samples. As performed previously [24], B-cell lymphoma RNA-sequencing samples were acquired using the search term “B-cell lymphoma” with the goal of finding B-cell non-Hodgkin’s lymphoma (lymphoma) samples and healthy B-cell controls. All 208 studies matching the phrase “B-cell lymphoma” were manually reviewed. All 34 studies matching the phrase “(B-cell SLE) OR (B-cell lupus)” were also manually reviewed. In addition, the top 238 of 842 studies retrieved using the “(B-cell) NOT ‘cell line’” query were manually reviewed to increase the number of B-cell healthy control samples. Specifically, before manual review, the query results were automatically filtered using the built-in GEO filters “Homo sapiens” to include only samples collected from humans and “Expression profiling by high-throughput sequencing” to include only RNA-seq datasets. To minimize the downstream impact of irrelevant data, RNA-seq samples from cell lines, formalin-fixed paraffin-embedded tissues, gene expression microarray experiments, single-cell RNA-sequencing experiments, xenografts, metastases, adjacent normal tissues, sequencing performed on platforms other than Illumina, samples known to be infected (EBV, KSHV, HIV) and samples which contained more diverse tissue types (i.e. whole blood, PBMCs, brain, etc.) were manually identified and excluded. While a subset of the healthy control samples was obtained from the same studies as the cancer samples, other healthy control samples were obtained from other studies to create roughly equivalent-sized cancer and healthy groups. The final dataset that was assembled for secondary analysis consisted of 361 samples (136 lymphoma samples, 138 lupus samples, and 88 healthy B-cell samples) from 16 studies (Table 1) [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39].
2.1. Preprocessing of RNA-Sequencing Data
Following the manual curation of the RNA-seq samples, the FASTQ sequencing files were pre-processed as previously described [24,40,41,42]. In brief, FASTQ files containing RNA-sequencing data were downloaded from the Sequence Read Archive (SRA) using the sra-tools software (version 2.10.8) [43]. The FASTQ files, the associated metadata file (Supplementary File S1), and a configuration file were used as inputs to the Automated Reproducible MOdular workflow for preprocessing and differential analysis of RNA-seq data (ARMOR) workflow (version 1.3.0) [44,45]. More specifically, the ARMOR Snakemake-based workflow includes the following steps: trimming sequencing adapters and poor-quality regions of the reads with TrimGalore! [46], generating quality control metrics with FastQC [47], mapping and quantifying reads to the human GRCh38 transcriptome with Salmon (version 1.3.0) [48], calculating significant differential gene expression using edgeR (version 4.0.16) [49], and performing Gene Ontology (GO) enrichment against terms from the Molecular Signatures Database (MSigDB) [50] using the Camera algorithm (version 3.50.3) [51]. Differentially expressed genes calculated by ARMOR were evaluated with log2 fold-change (log2FC) and false discovery rate (FDR)-adjusted p-values.
2.2. Principal Component Analysis
Following the mapping and quantification of gene read counts with Salmon (version 1.3.0) [48], transcript read counts were aggregated to each relevant gene, and gene read counts were normalized per gene to control for outlier genes with high read counts before performing principal component analysis (PCA) to determine the relatedness of included samples. PCA results were graphed using ggplot2 (version 3.5.0) [52] to visualize disease groups and potential batch effects.
2.3. Immune Imbalance Determination
The IIT algorithm was then used to identify the shared and contrasting mechanism(s) that may include novel therapeutic targets across the disease states (lupus and lymphoma). In brief, the IIT algorithm (Supplementary File S2) reads the sample metadata and transcript-level read counts that are each produced by the ARMOR workflow (Supplementary File S3-S4), and runs differential gene expression analysis internally using edgeR (version 4.0.16) [49]. IIT uses the log2 fold-change (log2FC) and false discovery rate (FDR) adjusted p-value data from one cancer vs. healthy (lymphoma vs. healthy) differentially expressed gene (DEG) comparison and one autoimmune vs. healthy (lupus vs. healthy) DEG comparison, all from the same tissue type (B-cells). This enables the algorithm to effectively determine shared and contrasting mechanisms between both comparisons that are relevant to the biology of the tissue type studied. To efficiently weight the fold-change values by the FDR p-values, the algorithm multiplies the log2FC values by -log10FDR p-value for each gene, which increases the numerical value for highly significant results. To allow for easier comparison, the (log2FC)*(-log10FDR p-value) products from both comparisons (lupus vs. healthy; lymphoma vs. healthy) were z-score normalized. All products were squared to remove negative values and then square-rooted to minimize amplification of outliers, generating the equivalent of an absolute value. The square-rooted results from the two comparisons (lupus vs. healthy; lymphoma vs. healthy) were then summed together to yield an intermediate score metric for each gene. Ratios of the square-rooted results for each of the two preceding comparisons were computed to determine which comparison contributed more to the intermediate score metric for each gene. To appropriately weight genes differentially expressed with similar intensity in both disease conditions (compared to healthy controls), the intermediate score metric was multiplied by the smallest of the ratios for each gene. This analysis' resulting final immune imbalance scores were then stored in tabular form.
To calculate a p-value for these final immune imbalance scores, the metadata labels (e.g. lymphoma, lupus, or healthy) were randomly permuted for all samples in the original datasets. This iterative permutation and re-calculation of immune imbalance scores was performed for 1,000 replicates to create a null distribution of the immune imbalance scores. The entire null distribution of 18,486,468 IIT scores from 1,000 permutations, as well as the original analysis, was normalized via z-score, and p-values were calculated for the upper tail of the distribution. Following the p-value calculation, a Bonferroni adjustment was applied to correct for multiple hypothesis testing and reduce the likelihood of false positives [53]. Gene frequency counts were taken to control for batch effects in the significant IIT results to see how often each gene showed up in the IIT-significant portion of the null distribution. The frequency counts were then z-score normalized and p-values were calculated for each gene frequency count. Based on this p-value, genes with significantly high gene frequency counts (p < 0.05) were removed from the significant IIT results to minimize the noise of batch effects.
2.4. Immune Imbalance Validation
Enrichment analysis was then performed, using enrichR (version 3.2) [54,55,56] to identify the significant Gene Ontology (GO terms) and intracellular signaling pathways for these IIT results [57,58,59,60,61]. Enrichment results were visualized by quadrant using the ggVennDiagram (version 1.5.2) and UpSetR (version 1.4.0) packages [62,63,64]. GO terms were also compiled as supplementary material for each significant IIT gene result using the biomaRt (version 2.58.2) package [65,66].
Significant genes were then compared against the Open Targets Platform database (version 24.06) target-disease association profiles [67] for “systemic lupus erythematosus” (EFO: MONDO_0007915) and “B-cell non-Hodgkins lymphoma” (EFO: EFO_1001938) [67]. Known targets for each disease were obtained by manually downloading the “Known Drugs” table from the Open Targets Platform (version 24.06) disease profile pages and extracting the gene identifiers associated with targets hit by the drugs. This list was then compared to the IIT significant gene results to evaluate the presence of IIT results among the known drug targets. Results that overlapped between the significant IIT results and the known drug targets were sorted according to their IIT scores, with the highest IIT-scoring genes considered first.
and Methods should be described with sufficient details to allow others to replicate and build on the published results. Please note that the publication of your manuscript implicates that you must make all materials, data, computer code, and protocols associated with the publication available to readers. Please disclose at the submission stage any restrictions on the availability of materials or information. New methods and protocols should be described in detail while well-established methods can be briefly described and appropriately cited.
Research manuscripts reporting large datasets that are deposited in a publicly available database should specify where the data have been deposited and provide the relevant accession numbers. If the accession numbers have not yet been obtained at the time of submission, please state that they will be provided during review. They must be provided prior to publication.
Interventionary studies involving animals or humans, and other studies that require ethical approval, must list the authority that provided approval and the corresponding ethical approval code.
3. Results
We began by identifying the appropriate FASTQ files from relevant studies (e.g., cancer and autoimmune disease samples each compared to healthy controls) that we would process with our IIT algorithm to predict targetable gene products in the relevant tissue (Figure 1, Supplementary File S3). To do so, we searched the GEO database for all eligible lupus and lymphoma samples, healthy B-cell control samples originating from the lupus and lymphoma studies, and additionally gleaned healthy B-cell controls from other human RNA-sequencing studies (Table 1, Supplementary File S1). During the screening, we excluded non-human samples, non-primary samples, non-B-cell samples, and other criteria that would negatively bias or affect our results. Our final dataset included 361 human patient samples, with 136 lymphoma samples, 138 lupus samples, and 88 healthy B-cell samples, from 16 studies [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39].
After identifying the FASTQ files for the relevant human samples, the sequencing reads were trimmed, mapped, and quantified against the human reference transcriptome. Transcript-level read counts were aggregated to calculate gene read counts. We applied a z-score normalization to the gene read count table (by gene) to avoid amplifying the impact of genes with high read counts and then ran a principal component analysis (PCA) to observe the relationship between samples and evaluate their similarity (Figure 2). We found that the samples from different disease statuses (healthy, lupus, or lymphoma patients) generally clustered together (except one lymphoma sample), and with some overlap between the phenotypic groups (Figure 2A). We next colored samples by study of origin and observed that the samples from each disease/control group were interspersed from multiple sequencing projects, suggesting a degree of similarity among samples from the same phenotypic groups, with relatively few batch effects among studies or phenotypes (Figure 2B-D, Supplementary Figure S1). These files were then assessed for differential gene expression using the Automated Reproducible Modular workflow for preprocessing and differential analysis of RNA-seq data (ARMOR) [44,45], making two comparisons: lupus vs. healthy and lymphoma vs. healthy. We found ~12,000 genes that displayed significant differential expression in the lupus vs. healthy analysis (Supplementary File S5), and ~13,000 significant genes in the lymphoma vs. healthy analysis (Supplementary File S6).
After computing the DEGs for the two comparisons (lupus vs. healthy and lymphoma vs. healthy), we then applied the IIT algorithm to find genes that have statistically significant differential expression in both comparisons. Briefly, the IIT algorithm includes the following steps: 1) for each of the input DEG sets, the log2 fold-change values (log2FCs) are multiplied by their corresponding -log10FDR-adjusted p-values to weight the expression of each gene by its statistical significance. Next, the weighted scores for each comparison are z-score normalized prior to taking the square root of the squared normalized value (to eliminate negative values and reduce outliers). These intermediate scores, which still included genes expressing opposite directions, are then summed and normalized using the smaller ratio of either the intermediate score of the lupus vs. healthy comparison or the intermediate score of the lymphoma vs. healthy comparison to generate a final immune imbalance score. To determine the significance of our results and minimize potential batch effects, we calculated a null distribution for these final immune imbalance scores by performing 1,000 permutations of the original DEG analysis (Supplementary Files S7-S10), by randomly assigning samples to disease groups in each permutation and calculating IIT values for all genes, resulting in 18,468,000 permuted IIT final scores (Supplementary File S11). This null distribution aids with controlling for batch effects by assigning statistical significance only to IIT scores which have a strong enough signal to rise above the statistical noise. We then z-score normalized the IIT null distribution to assign significant p-values (p < 0.05) to the highest IIT scores on the upper tail of the null distribution, where the most intensely immune-imbalanced results should be located. We then performed multiple hypothesis testing corrections using the stringent Bonferroni method. Following the assignment of IIT score p-values based on the null distribution, we conducted one additional test to remove noise. Using the ~18 million data points in the null distribution, we tallied up how many times a gene was considered IIT significant in one of the 1,000 data permutations to remove genes with abundant frequency counts from the IIT-significant portion of the null distribution. When visualizing frequency counts for all genes in the IIT-significant portion of the null distribution, we found that the average gene frequency count was 205, meaning that the average gene garnered a significant IIT score in 205 permutations (Figure 3A). We performed z-score normalization on the gene frequency counts, then calculated the p-value indicating which genes occurred at significantly abundant frequency counts and therefore had significantly high noise. We removed the genes above this frequency gate from further analysis to prevent noise from batch effects.
We then assessed the DEGs from the lupus vs. healthy and lymphoma vs. healthy differential expression analyses using the Immune Imbalance Transcriptomics (IIT) algorithm. Out of the initial overlapping significantly differentially expressed pool of 8,562 genes, we observed that 5,507 had statistically significant immune imbalance (Bonferroni p-value < 0.05; Supplementary File S12). Following application of the frequency gate as described above, 5,335 significant genes were left for consideration (Supplementary File S13). When we visualized overlapping gene results from the lupus and lymphoma differential gene comparisons using log2FCs as the axis scales and one scatter-point per gene, we observed that the overlapping genes fall into four quadrants, with the significantly immune-imbalanced genes found by IIT localized to the outer edges (representing higher log2FCs) and non-significant IIT genes near the center (representing lower log2FCs), with genes filtered out by the frequency gate scattered throughout (Figure 3B).
Functionally, these gene profiles can be categorized into four quadrants. For clarity, we label these quadrants as “cancer” (lymphoma in this use-case) and “autoimmune disease” (lupus in this use-case), knowing that this algorithm can be applied to different cancer-autoimmune pairings. The four quadrants are as follows: upregulated in both lymphoma (Cancer) and lupus (Autoimmune disease; Quadrant I, C+A+), downregulated in both lymphoma and lupus (Quadrant III, C-A-), upregulated in lymphoma but downregulated in lupus (Quadrant II, C+A-), and downregulated in lymphoma but upregulated in lupus as compared to healthy (Quadrant IV, C-A+). The top four immune-imbalanced genes from each quadrant are represented in Table 2. Among the top four IIT genes per quadrant are several gene products that have previously been shown in other studies to be involved in lupus and/or lymphoma, including C1QB (C+A+ IIT quadrant) [24,68,69,70], IFI27 (C+A+ IIT quadrant) [71,72,73,74], and TSC22D3 (C-A+ IIT quadrant) [75].
To identify the relevant functional terms and signaling cascades in the significant IIT results, we ran separate Gene Ontology (GO) and intracellular pathway hypergeometric enrichment analyses on the significant IIT genes using the EnrichR package [54]. We retrieved all GO terms individually for significant IIT genes, compiled them for easy reference (Supplementary File S14), and utilized a text search to determine how many GO term descriptions mentioned “immune” or “immunity” per gene. We ran GO term enrichments on the genes in each quadrant (Figure 3A-C, Supplementary File S15), and an additional hypergeometric enrichment on the entire significant IIT gene set (Table 3, Supplementary File S15). We were somewhat surprised to observe relatively few overlapping terms between quadrants in these GO enrichment results (Figure 3A-C), suggesting that genes in different quadrants potentially play unique functional roles. Interestingly, we also found many quadrant-specific results overlapping with the GO enrichment terms obtained from enriching all significant IIT genes together (Figure 3D). Most quadrant-specific GO enrichments contained more results than the total IIT gene set analysis, potentially suggesting stronger signal and lower noise in quadrant-specific genes (Figure 3D).
We found 659 significantly enriched signaling pathways from the four databases used in the analysis (Table 4, Supplementary File S16, Supplementary Figure S2). We noted four pathways with immune-related function among the top 10 significantly enriched, including “Neutrophil Degranulation,” “Immune System,” “Adaptive Immune System,” and “Innate Immune System.” The presence of these significant immune-related terms among our top results of significantly enriched pathways involved in immune function supports our hypothesis that our IIT algorithm can isolate genes relevant to immune function. It also introduces the possibility that other significant IIT genes with no currently known involvement in immune effector function and immune signaling may play uncharacterized roles in the immune function of B-cells.
We also observed multiple pathways relating to cell cycle regulation and cell division in our top ten enriched pathway results, including control of cell cycle by Rho GTPases such as “Signaling By Rho GTPases” and “Signaling By Rho GTPases, Miro GTPases And RHOBTB3”; as well as pathways involved in increasing transcription and translation such as “Eukaryotic Translation Elongation” and “Peptide Chain Elongation”, suggesting heightened activity that could be attributed to cell division. These findings augment our confidence in these results due to the well-known high rate of cell division in lupus auto-reactive B-cells and the high rate of aggressive division in B-cell lymphoma. This serves as an internal control that the IIT algorithm finds biologically relevant genes to B-cell function in the context of lupus and lymphoma pathology.
Following the pathway enrichment analysis, we analyzed the list of significant immune-imbalanced genes to identify therapeutic targets for potential drug repurposing. Using the OpenTargets database [67] and an adaptation of the Pathway2Targets algorithm [76], we predicted targets that could be utilized against lupus and lymphoma. A promising outcome of our target prediction analysis is that we found 389 (out of 5,335 significant immune-imbalanced genes) with known drugs that are either approved or in development for at least one indication (Table 5, Supplementary File S17-S18). Lupus currently has 172 targets with known drugs, 40 of which are significantly immune-imbalanced in this study. Lymphoma currently has 437 targets with drugs in testing, and 151 of these targets appear on our significantly immune-imbalanced list. These known lupus and lymphoma drug targets in our significant immune-imbalanced results grant further veracity to the biological relevance of the IIT gene results to lupus and lymphoma treatment. Additionally, we found 349 novel targets for lupus and 238 novel targets for lymphoma using IIT, which may be useful in the future.
4. Discussion
In this work, we identified ~13,000 genes differentially expressed between lymphoma and healthy B-cells and ~12,000 genes between lupus B-cells and healthy B-cells, and the top results are biologically relevant to the pathology of these B-cell diseases. We used the DEG lists from these two comparisons as a use case to demonstrate the ability to identify robust and biologically relevant mechanisms in lupus and lymphoma using the significant 5,335 gene products identified by the null distribution within our novel Immune Imbalance Transcriptomics (IIT) algorithm, as well as biologically relevant enrichment results and potential therapeutic targets Previous bioinformatic approaches have investigated the shared transcriptomic mechanisms of disease between an autoimmune disease and a cancer from the same tissue type [77], but have not accounted for potential contrasting mechanisms, a novel attribute of IIT.
More and more evidence shows that cancer and autoimmune diseases can have a causal or correlative relationship, with the development of one creating a greater risk for the development of the other in the same patient [19,77,78,79,80,81,82,83,84,85]. The rise in co-occurrence risk suggests that the two diseases likely share some underlying causes and common mechanisms, such as chronic inflammation [85]. Though inflammation is classically activated by infection or injury, other causes exist, including underlying tissue malfunction, which induces para-inflammation as the body attempts to return to homeostasis [86]. Para-inflammation has previously been implicated as the cause of chronic inflammatory conditions, including cancer and autoimmune disease [86]. The rising occurrence of chronic inflammatory diseases and their co-incidence justifies a new research approach: finding, characterizing, and modifying the shared underlying disease pathologic mechanisms.
Among novel disease target candidates for antagonists in both lupus and lymphoma patient treatments are Complement C1qB Chain (C1QB, C+A+ quadrant), Interferon Alpha Inducible Protein 27 (IFI27, C+A+ quadrant), Insulin Like Growth Factor Binding Protein 2 (IGFBP2, C+A+ quadrant), and Transcobalamin 2 (TCN2, C+A+ quadrant). Each is a good candidate for knock-down treatment in pathogenic lupus and lymphoma cells due to their known involvement in lupus and lymphoma progression via overexpression, although care should be taken to minimize adverse events with these potential targets. Increased C1QB expression decreases the effectiveness of combined Ibrutinib and Venetoclax in patients with Mantle-Cell lymphoma (a B-cell lymphoma) and is associated with a worse prognosis [68]. High IFI27 expression has been shown to drive cell proliferation and to spark B-cell lymphomagenesis [72] and is a biomarker of B-cell cancer progression [73]. IFI27 is part of the Interferon-I signature assessment used to determine SLE subtype during the process of lupus diagnosis [87] and is a known SLE biomarker [74]. Additionally, targeting and neutralizing IGFBP2 (C+A+ quadrant) as a lupus nephritis biomarker shows potential as a new option in treating patients with lupus nephritis, an advanced complication of SLE targeting the kidneys with few workable treatments [88,89]. TCN2 may be a predictive biomarker in primary large B-cell lymphoma of immune-privileged sites, making it a good early target to prevent metastasis [90].
A subset of potential agonistic targets could benefit patients due to their significant downregulation in both lupus and lymphoma B-cells. However, very little is known about many of these significant IIT genes (located in Quadrant III) in the context of lupus and lymphoma B-cells. OTUD1 has been shown to repress type-I interferon-mediated disease, which includes lupus, in vivo [91] and to have loss-of-function in SLE [92], suggesting the promise of treatments that increase the activity of OTUD1 in lupus B-cells. Transmembrane Anterior Posterior Transformation 1 (TAPT1, C-A- quadrant), and Proline Rich Nuclear Receptor Coactivator 1 (PNRC1, C-A- quadrant) are novel results which, as underlying mechanistic genes shared by lupus and lymphoma, could be leveraged as possible therapeutic targets.
Perhaps more interesting is the potential application of contrasting immune imbalance mechanisms. It is well-known that tumors can be treated by inducing cancer-specific autoimmunity [93,94], suggesting that, to some degree, cancers and autoimmune diseases can be regulated by contrasting mechanisms. This contrast is seen by the apparent opposite utilization of the two complementary aspects of the immune system: immune activation (autoimmunity) and immune tolerance (cancer) [13,14]. Appropriate immune homeostasis, which is characterized by proper immune activation and subsequent resolution, is not present in either autoimmune disease (failure to tolerate self and resolve inflammation) or cancer (failure to activate sufficiently to detect and clear non-self tumor) [15,16]. Some of the most apparent examples of the “opposite mechanisms” at play in cancer and autoimmune disease involve immunotherapies (steroids, T-cell therapies, checkpoint inhibitors, other monoclonal antibody therapies, oncolytic viruses, etc.) [77,95]. The growing utilization of immunotherapies in cancer has revealed a fascinating conundrum: the rare occurrence of immune-related adverse events, specifically, loss of self-tolerance leading to autoimmunity [77,95]. The occurrence of cancer immunotherapy-related adverse immune events, such as cytokine storms (and other immune-related inflammatory phenomena) or induction of tissue-specific or systemic autoimmunity, point to a new and largely untouched area of research: the relationship between cancer and autoimmunity and what factors determine whether adverse immune events develop in a patient treated with immunotherapy [96]. Conversely, some patients do not respond strongly enough to cancer immunotherapies to clear or inhibit tumor growth [97]. In autoimmunity, anti-inflammatory immunotherapies can potently improve patient quality of life, but increases the risk of infection. The immunotherapeutic silencing of immune activation can also contribute to the development of cancer in autoimmune patients who are treated with anti-inflammatory drugs for long periods [7]. Though successful immunotherapies and drugs affecting immune function are seminal achievements of substantial research efforts in the fields of cancer and autoimmunity, the failure of some patients to return to healthy immune homeostasis demonstrates that there is still room for improvement.
A potential alternative to the current immunotherapeutic treatments is harnessing the power of contrasting immune imbalance mechanisms, such as genes described in this study, to pull patients toward a healthy center. A gene upregulated in lupus B-cells may provide clues about how to modulate the immune microenvironment of B-cell lymphomas; likewise, an immuno-modulatory gene upregulated by B-cell lymphoma could possess the power to dampen the rampant inflammation of lupus. Many experimental methods for in vivo activation or inhibition of gene expression are currently under study and consideration, including gene delivery by engineered viruses [98] and CRISPR methods (e.g., CRISPRa, CRISPRI, etc.) in pathogenic cells to increase or decrease gene expression [99]. A current limitation in the field of in vivo gene editing is that it’s difficult to target the right cells without off-target effects. However, we anticipate that with further development, such approaches could enable in vivo gene therapy to treat immune-related diseases including cancer and autoimmunity using immune-imbalanced targets. Good examples of a significantly immune-imbalanced gene already in use as a drug target for both lupus and lymphoma are HRH1 (Quad I), JAK1 (Quad III), and PTGS1 (Quad IV).
Several immune imbalance genes in this analysis are good candidates for therapeutic agonists. Decreased expression of TPM2 (C+A- quadrant) in childhood-onset SLE contributes to musculoskeletal system symptoms [100], suggesting potential benefits of an agonist. TSC22 Domain Family Member 3 (TSC22D3, C-A+ quadrant) is a proapoptotic gene in some cell types and is positively regulated by Estrogen-Related Receptor-Beta (ESRRB) [75]. Previous studies have shown that ESRRB repression in acute lymphoblastic leukemias, which are closely related to B-cell lymphomas, contributes to treatment resistance. This suggests that activating TSC22D3 would improve the success of malignant B-cell treatment [75]. KLF6 (C-A+ quadrant) is a key part of recruiting macrophages to the inflammation site and the immune response subsequent success, which is lacking in cancers, including B-cell lymphomas [101]. These promising targets for gene-upregulation treatment present new possibilities for patients.
Additionally, many of the top immune-imbalanced genes could be potential targets for antagonists. Clinical analyses showed that tumors expressing increased levels of NFE2-Like bZIP Transcription Factor 3 (NFE2L3, C+A- quadrant) indicated a worse prognosis [102], making it an ideal candidate for future knockdown studies. It is thought that upregulation of TSC22 Domain Family Member 3 (TSC22D3, C-A+ quadrant) and glucocorticoid-induced leucine zipper (GILZ; encoded in the same locus) work with interferon-1 to induce glucocorticoid resistance in SLE patients, dampening their treatment response [103]. Protein Phosphatase 1 Regulatory Subunit 15A (PPP1R15A, C-A+ quadrant) is also upregulated by SLE and assists with the production of pro-inflammatory cytokines such as interferons, which contribute particularly to the pathogenesis of interferon-high cases of lupus [104]. Treatment-induced antagonism or knockdown of the gene products in these cases could also improve patient outcomes. Parathymosin (PTMS, C+A- quadrant), Plexin A1 (PLXNA1, C+A- quadrant), ADP Ribosylation Factor Like GTPase 4A (ARL4A, C-A+ quadrant), are novel immune imbalance results with no known mechanisms and with promising potential to change pathogenic properties of lupus and lymphoma B-cells, which justifies future experimentation.
While our IIT results can be useful, it is important to explicitly recognize at least some of the inherent limitations of this study. First and foremost, we recognize that not all types of B-cell lymphoma were studied since our sample pool was limited to publicly available datasets on the Gene Expression Omnibus (GEO). Due to it being one of the most common types of B-cell Non-Hodgkin’s lymphoma, and one of the most aggressive, the majority of lymphoma samples included were from the diffuse large B-cell lymphoma subtype. As such, we expect that these results will be especially applicable to diffuse large B-cell lymphoma, with limited relevance being likely for the other lymphoma subtypes, due to fewer contributed samples.
Another potential area of limitation arises from including multiple sequencing projects in the same differential gene expression analysis. As no studies contained the representative samples we needed, we decided to combine multiple studies and indirectly account for batch effects within the algorithm itself. The gene-specific batch effects are particularly important, which would boost or lower read counts for a specific gene. We have adjusted for this Using two strategies: 1) generating a null distribution and calculating a rigorous p-value from the 18,486,468 data points included in the null distribution and 2) analyzing the frequency of each gene appearing in the significant portion of the null distribution and removing those that appeared the most. We generated the null distribution by making 1,000 permutations of the initial dataset, randomly shuffling sample phenotypes before the analysis, then deriving the final IIT scores for all genes across all permutations and including these with the original gene results. This null distribution can drown out noise by providing ~18.5 million additional data points, making a p-value cutoff much more rigorous. Results with higher statistical noise and lower IIT scores (and presumably less biological relevance) become insignificant when using the null distribution p-value as a cutoff. Additionally, we applied a frequency gate to control for genes with high statistical noise that could be attributed to batch effects. The frequency gate removed genes that appeared abundantly, at high frequency, within the null distribution’s significant results. The rigor of this null-distribution and frequency-gate approach increases our confidence in the significant IIT results.
Our novel algorithm, Immune Imbalance Transcriptomics (IIT) identifies the shared and unique transcriptional mechanisms between cancer and autoimmunity in the same tissue (or cell) type. The biological use case for this algorithm comparing lupus and lymphoma B-cells is an important step forward and suggests that an algorithmic approach may enable the ability to glean additional mechanistic insights and continue to advance the state of diagnostics and treatment for cancer and autoimmune patients alike. The IIT approach allows for a more robust consideration of the effects of immune-related gene products on identifying therapeutic targets that can help to return the lupus and lymphoma phenotypes to a more immune-balanced, non-inflamed homeostasis. We anticipate that the identified mechanisms and targets will aid with future efforts to develop improved therapeutics for both lupus and lymphoma. As one of the first reproducible methods for comparing cancer and autoimmune transcriptional effects, our IIT algorithm potentially provides a novel approach to a better understanding of the mechanisms of lupus and lymphoma disease and how to target these mechanisms without upsetting the immune balance. We expect that this approach can improve the quality of life for patients by providing effective treatment options with fewer and less severe side effects.
5. Patents
N.R.-S. and B.E.P. are filing a patent on the Immune Imbalance Transcriptomics algorithm.
Supplementary Materials
The following supporting information can be downloaded at DOI: 10.5281/zenodo.12789186, File S1: Metadata for Samples Used in RNA-Seq Analysis; Figure S1: Principal Component Analysis (PCA) of All Analyzed Samples by Study; File S2: Immune Imbalance Transcriptomics Algorithm; File S3: Lupus txiMeta Object for Algorithm; File S4: Lymphoma txiMeta Object for Algorithm; File S5: Lupus edgeR Results; File S6: Lymphoma edgeR Results; File S7: Lupus permutation deck; File S8: Lymphoma permutation deck; File S9: EdgeR results for all lupus permutations; File S10: EdgeR results for all lymphoma permutations; File S11: Null distribution with ~18.5 million IIT scores from original analysis and 1,000 permutations; File S12: genes that were IIT-significant after null distribution p-value determination and removal of noisy genes using frequency gate; File S13: All significant IIT genes, including those excluded by the frequency gate; File S14: All GO terms by gene for easy reference; File S15: GO enrichment results; File S16: pathway enrichment results; Figure S2: Pathway Enrichment Venn Diagrams; File S17: Drugs Known to interact with significant immune-imbalanced genes; File S18: Immune Imbalance Genes which are also known drug targets.
Author Contributions
Conceptualization, Naomi Rapier-Sharman and Brett E. Pickett; Data curation, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow and Michael T. Told; Formal analysis, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow and Michael T. Told; Investigation, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow and Michael T. Told; Methodology, Naomi Rapier-Sharman, Lane Fischer, Liesl Fawson, Stephen R. Piccolo and Brett E. Pickett; Project administration, Brett E. Pickett; Resources, Kim L. O’Neill; Software, Naomi Rapier-Sharman, Stephen R. Piccolo and Brett E. Pickett; Supervision, Brett E. Pickett; Validation, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow, Stephen R. Piccolo and Brett E. Pickett; Visualization, Naomi Rapier-Sharman; Writing – original draft, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow, Joseph Parry and Brett E. Pickett; Writing – review & editing, Naomi Rapier-Sharman, Sehi Kim, Maddie Mudrow, Joseph Parry, Brian D. Poole, Kim L. O’Neill, Stephen R. Piccolo and Brett E. Pickett. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The human RNA-sequencing samples used in this study were acquired from the Gene Expression Omnibus (GEO) from publicly-available, previously-released, de-identified sequencing projects curated by other lab groups.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets analyzed during the current study are available in the Gene Expression Omnibus (GEO) repository. All GEO accession numbers for included sequencing projects can be found in Table 1, while individual SRA identifiers for included samples can be viewed in Supplementary File S1. All supplementary materials are cited within the text and are accessible at DOI: 10.5281/zenodo.12789186.
Acknowledgments
We thank the Mary Lou Fulton Office of Research Computing at Brigham Young University for their gracious provision of a supercomputing environment for initial sample processing.
Conflicts of Interest
N.R.-S. and B.E.P. have filed a provisional patent on the Immune Imbalance Transcriptomics algorithm.
References
- Tian, J.; Zhang, D.; Yao, X.; Huang, Y.; Lu, Q. Global Epidemiology of Systemic Lupus Erythematosus: A Comprehensive Systematic Analysis and Modelling Study. Ann. Rheum. Dis. 2023, 82, 351–356. [Google Scholar] [CrossRef]
- Dörner, T.; Furie, R. Novel Paradigms in Systemic Lupus Erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Gershwin, M.E. The Consequences of Apoptosis in Autoimmunity. J. Autoimmun. 2008, 31, 257–262. [Google Scholar] [CrossRef]
- Take a Look at the Eyes in Systemic Lupus Erythematosus: A Novel Point of View. Autoimmun. Rev. 2019, 18, 247–254. [CrossRef]
- Zonana-Nacach, A.; Barr, S.G.; Magder, L.S.; Petri, M. Damage in Systemic Lupus Erythematosus and Its Association with Corticosteroids. Arthritis Rheum. 2000, 43, 1801–1808. [Google Scholar] [CrossRef]
- Gladman, D.D.; Urowitz, M.B.; Rahman, P.; Ibañez, D.; Tam, L.-S. Accrual of Organ Damage over Time in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2003, 30, 1955–1959. [Google Scholar]
- Basta, F.; Fasola, F.; Triantafyllias, K.; Schwarting, A. Systemic Lupus Erythematosus (SLE) Therapy: The Old and the New. Rheumatol Ther 2020, 7, 433–446. [Google Scholar] [CrossRef]
- Jamil, A.; Mukkamalla, S.K.R. Lymphoma. In StatPearls [Internet]; StatPearls Publishing, 2023. [Google Scholar]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C. ; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol 2018, 4, 1553–1568. [Google Scholar]
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin Lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile Strategy for Controlling the Specificity and Activity of Engineered T Cells. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E450–E458. [Google Scholar] [CrossRef]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The Risk of Lymphoma Development in Autoimmune Diseases: A Meta-Analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef]
- The Resolution of Inflammation: Principles and Challenges. Semin. Immunol. 2015, 27, 149–160. [CrossRef]
- Rogovskii, V. Immune Tolerance as the Physiologic Counterpart of Chronic Inflammation. Front. Immunol. 2020, 11, 2061. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The Crucial Roles of Inflammatory Mediators in Inflammation: A Review. Vet World 2018, 11, 627–635. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N.; Stockinger, B.; Tak, P.P. The Resolution of Inflammation. Nat. Rev. Immunol. 2012, 13, 59–66. [Google Scholar] [CrossRef]
- Delogu, L.G.; Deidda, S.; Delitala, G.; Manetti, R. Infectious Diseases and Autoimmunity. J. Infect. Dev. Ctries. 2011, 5, 679–687. [Google Scholar] [CrossRef]
- Kim-Hellmuth, S.; Bechheim, M.; Pütz, B.; Mohammadi, P.; Nédélec, Y.; Giangreco, N.; Becker, J.; Kaiser, V.; Fricker, N.; Beier, E.; et al. Genetic Regulatory Effects Modified by Immune Activation Contribute to Autoimmune Disease Associations. Nat. Commun. 2017, 8, 266. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer Immunosuppression and Autoimmune Disease: Beyond Immunosuppressive Networks for Tumour Immunity. Immunology 2006, 119, 254–264. [Google Scholar] [CrossRef]
- Ghorani, E.; Swanton, C.; Quezada, S.A. Cancer Cell-Intrinsic Mechanisms Driving Acquired Immune Tolerance. Immunity 2023, 56, 2270–2295. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Menter, T.; Tzankov, A. Mechanisms of Immune Evasion and Immune Modulation by Lymphoma Cells. Front. Oncol. 2018, 8, 344644. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Rapier-Sharman, N.; Clancy, J.; Pickett, B.E. Joint Secondary Transcriptomic Analysis of Non-Hodgkin’s B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers. J Bioinform Syst Biol 2022, 5, 119–135. [Google Scholar] [CrossRef]
- Faramand, R.; Jain, M.; Staedtke, V.; Kotani, H.; Bai, R.; Reid, K.; Lee, S.B.; Spitler, K.; Wang, X.; Cao, B.; et al. Tumor Microenvironment Composition and Severe Cytokine Release Syndrome (CRS) Influence Toxicity in Patients with Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Clin. Cancer Res. 2020, 26, 4823–4831. [Google Scholar] [CrossRef]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.-W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer Sequence Variants and Transcription-Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B-Cell Lymphoma. Immunity 2015, 42, 186–198. [Google Scholar] [CrossRef]
- Raju, S.; Kretzmer, L.Z.; Koues, O.I.; Payton, J.E.; Oltz, E.M.; Cashen, A.; Polic, B.; Schreiber, R.D.; Shaw, A.S.; Markiewicz, M.A. NKG2D-NKG2D Ligand Interaction Inhibits the Outgrowth of Naturally Arising Low-Grade B Cell Lymphoma In Vivo. J. Immunol. 2016, 196, 4805–4813. [Google Scholar] [CrossRef]
- Teater, M.; Dominguez, P.M.; Redmond, D.; Chen, Z.; Ennishi, D.; Scott, D.W.; Cimmino, L.; Ghione, P.; Chaudhuri, J.; Gascoyne, R.D.; et al. AICDA Drives Epigenetic Heterogeneity and Accelerates Germinal Center-Derived Lymphomagenesis. Nat. Commun. 2018, 9, 222. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-Cell Lymphomas in Patients with Myelofibrosis Receiving JAK1/2 Inhibitor Therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-Oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef] [PubMed]
- Rouhigharabaei, L.; Ferreiro, J.F.; Tousseyn, T.; van der Krogt, J.-A.; Put, N.; Haralambieva, E.; Graux, C.; Maes, B.; Vicente, C.; Vandenberghe, P.; et al. Non-IG Aberrations of FOXP1 in B-Cell Malignancies Lead to an Aberrant Expression of N-Truncated Isoforms of FOXP1; 2015. [Google Scholar]
- Panwar, B.; Schmiedel, B.J.; Liang, S.; White, B.; Rodriguez, E.; Kalunian, K.; McKnight, A.J.; Soloff, R.; Seumois, G.; Vijayanand, P.; et al. Multi-Cell Type Gene Coexpression Network Analysis Reveals Coordinated Interferon Response and Cross-Cell Type Correlations in Systemic Lupus Erythematosus. Genome Res. 2021, 31, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, G.; Lanata, C.M.; Trupin, L.; Paranjpe, I.; Jain, T.S.; Nititham, J.; Taylor, K.E.; Combes, A.J.; Maliskova, L.; Ye, C.J.; et al. Transcriptomic Analysis of Immune Cells in a Multi-Ethnic Cohort of Systemic Lupus Erythematosus Patients Identifies Ethnicity- and Disease-Specific Expression Signatures. Commun Biol 2021, 4, 488. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, S.; Ehrenberg, P.K.; Geretz, A.; Yum, L.; Kundu, G.; May, K.; Fourati, S.; Nganou-Makamdop, K.; Williams, L.D.; Sawant, S.; et al. Monocyte-Derived Transcriptome Signature Indicates Antibody-Dependent Cellular Phagocytosis as a Potential Mechanism of Vaccine-Induced Protection against HIV-1. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, C.; Zhao, K.; Yang, Y.; Rassadkina, Y.; Fajnzylber, J.; Regan, J.; Li, J.Z.; Lichterfeld, M.; Yu, X.G. Immune-Profiling of SARS-CoV-2 Viremic Patients Reveals Dysregulated Innate Immune Responses. Front. Immunol. 2022, 13, 984553. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Wang, X.; Campbell, M.R.; Cho, H.-Y.; Pittman, G.S.; Martos, S.N.; Bell, D.A. Epigenomic Profiling of Isolated Blood Cell Types Reveals Highly Specific B Cell Smoking Signatures and Links to Disease Risk. Clin. Epigenetics 2023, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M.; Pyfrom, S.C.; Schmidt, J.A.; Koues, O.I.; Kowalewski, R.A.; Grams, N.R.; Sun, J.J.; Berman, L.R.; Duncavage, E.J.; Lee, Y.-S.; et al. Loss of Synergistic Transcriptional Feedback Loops Drives Diverse B-Cell Cancers. EBioMedicine 2021, 71, 103559. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Ren, Z.; Zhang, B.; Mao, L.; Zhu, G.; Gao, L.; Su, J.; Ye, J.; Long, Z.; Zhu, Y.; et al. Insufficient Epitope-Specific T Cell Clones Are Responsible for Impaired Cellular Immunity to Inactivated SARS-CoV-2 Vaccine in Older Adults. Nat Aging 2023, 3, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Rapier-Sharman, N.; Krapohl, J.; Beausoleil, E.J.; Gifford, K.T.L.; Hinatsu, B.R.; Hoffmann, C.S.; Komer, M.; Scott, T.M.; Pickett, B.E. Preprocessing of Public RNA-Sequencing Datasets to Facilitate Downstream Analyses of Human Diseases. Data 2021, 6, 75. [Google Scholar] [CrossRef]
- Transcriptomics Secondary Analysis of Severe Human Infection with SARS-CoV-2 Identifies Gene Expression Changes and Predicts Three Transcriptional Biomarkers in Leukocytes. Comput. Struct. Biotechnol. J. 2023, 21, 1403–1413. [CrossRef]
- Moreno, C.; Bybee, E.; Tellez Freitas, C.M.; Pickett, B.E.; Weber, K.S. Meta-Analysis of Two Human RNA-Seq Datasets to Determine Periodontitis Diagnostic Biomarkers and Drug Target Candidates. Int. J. Mol. Sci. 2022, 23, 5580. [Google Scholar] [CrossRef]
- GitHub - Ncbi/sra-Tools: SRA Tools. Available online: https://github.com/ncbi/sra-tools (accessed on 19 July 2024).
- Orjuela, S.; Huang, R.; Hembach, K.M.; Robinson, M.D.; Soneson, C. ARMOR: An Automated Reproducible MOdular Workflow for Preprocessing and Differential Analysis of RNA-Seq Data. G3 Genes|Genomes|Genetics 2019, 9, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake--a Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics - Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 8 March 2023).
- Babraham Bioinformatics - FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 March 2023).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Smyth, G.K. Camera: A Competitive Gene Set Test Accounting for Inter-Gene Correlation. Nucleic Acids Research 2012, 40, e133–e133. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Science & Business Media, 2009; ISBN 9780387981413. [Google Scholar]
- Shaffer, J.P. Multiple Hypothesis Testing. Annu. Rev. Psychol. 1995, 46, 561–584. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinformatics 2013, 14, 128. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc 2021, 1, e90. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-Scale Gene Function Analysis with the PANTHER Classification System. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Website Available online. [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.-H.; Chen, C.; Akyol, T.; Dusa, A.; Yu, G.; Cao, B.; Cai, P. ggVennDiagram: Intuitive Venn Diagram Software Extended. Imeta 2024, 3, e177. [Google Scholar] [CrossRef]
- Gao, C.-H.; Yu, G.; Cai, P. ggVennDiagram: An Intuitive, Easy-to-Use, and Highly Customizable R Package to Generate Venn Diagram. Front. Genet. 2021, 12, 706907. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Baker, J.; Malangone, C.; Lopez, I.; Miranda, A.; Cruz-Castillo, C.; Fumis, L.; et al. The next-Generation Open Targets Platform: Reimagined, Redesigned, Rebuilt. Nucleic Acids Res. 2023, 51, D1353–D1359. [Google Scholar] [CrossRef]
- Davis, J.; Handunnetti, S.M.; Sharpe, C.; Turner, G.; Anderson, M.A.; Roberts, A.W.; Seymour, J.F.; Tam, C.S.; Ritchie, D.; Koldej, R. Long Term Responses to Venetoclax and Ibrutinib in Mantle Cell Lymphoma Are Associated with Immunological Recovery and Prognostic Changes in Inflammatory Biomarkers. Blood 2019, 134, 2791–2791. [Google Scholar] [CrossRef]
- Guo, J.; Gao, Y.; Wang, Y.; Zou, Y.; Du, Y.; Luo, C.; Shi, Y.; Yang, Y.; Wu, X.; Su, Y.; et al. Investigation of C1-Complex Regions Reveals New C1Q Variants Associated with Protection from Systemic Lupus Erythematosus, and Affect Its Transcript Abundance. Sci. Rep. 2018, 8, 8048. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, R.D.; Racila, E.; Racila, D.M. C1q: Its Functions within the Innate and Adaptive Immune Responses and Its Role in Lupus Autoimmunity. J. Invest. Dermatol. 2005, 125, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Suomela, S.; Cao, L.; Bowcock, A.; Saarialho-Kere, U. Interferon Alpha-Inducible Protein 27 (IFI27) Is Upregulated in Psoriatic Skin and Certain Epithelial Cancers. J. Invest. Dermatol. 2004, 122, 717–721. [Google Scholar] [CrossRef]
- Sagou, K.; Sato, Y.; Okuno, Y.; Watanabe, T.; Inagaki, T.; Motooka, Y.; Toyokuni, S.; Murata, T.; Kiyoi, H.; Kimura, H. Epstein-Barr Virus Lytic Gene BNRF1 Promotes B-Cell Lymphomagenesis via IFI27 Upregulation. PLoS Pathog. 2024, 20, e1011954. [Google Scholar] [CrossRef] [PubMed]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole-Blood Transcriptional Profiling of Interferon-Inducible Genes Identifies Highly Upregulated IFI27 in Primary Myelofibrosis. Eur. J. Haematol. 2011, 87, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, L.; Wang, J.; Zhang, M.; Song, Z.; Ni, B.; You, Y. Identification of Key Biomarkers and Immune Infiltration in Systemic Lupus Erythematosus by Integrated Bioinformatics Analysis. J. Transl. Med. 2021, 19, 1–17. [Google Scholar]
- Gallagher, K.M.; Roderick, J.E.; Tan, S.H.; Tan, T.K.; Murphy, L.; Yu, J.; Li, R.; O’Connor, K.W.; Zhu, J.; Green, M.R.; et al. ESRRB Regulates Glucocorticoid Gene Expression in Mice and Patients with Acute Lymphoblastic Leukemia. Blood Adv 2020, 4, 3154–3168. [Google Scholar]
- Dobbs Spendlove, M.; M Gibson, T.; McCain, S.; Stone, B.C.; Gill, T.; Pickett, B.E. Pathway2Targets: An Open-Source Pathway-Based Approach to Repurpose Therapeutic Drugs and Prioritize Human Targets. PeerJ 2023, 11, e16088. [Google Scholar] [CrossRef]
- Adil, S.; Paracha, R.Z.; Tariq, S.; Nisar, M.; Ijaz, S.; Siddiqa, A.; Hussain, Z.; Amir, A. A Computational Systems Analyses to Identify Biomarkers and Mechanistic Link in Psoriasis and Cutaneous Squamous Cell Carcinoma. Front. Immunol. 2021, 12, 662528. [Google Scholar] [CrossRef]
- Liu, X.; Ji, J.; Forsti, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Autoimmune Disease and Subsequent Urological Cancer. J. Urol. 2013, 189, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Liu, X.; Ji, J.; Sundquist, K.; Sundquist, J. Subsequent COPD and Lung Cancer in Patients with Autoimmune Disease. Eur. Respir. J. 2011, 37, 463–465. [Google Scholar] [CrossRef]
- Hemminki, K.; Liu, X.; Ji, J.; Sundquist, J.; Sundquist, K. Autoimmune Disease and Subsequent Digestive Tract Cancer by Histology. Ann. Oncol. 2012, 23, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the Autoimmune Disease Scleroderma with an Immunologic Response to Cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef]
- Shiokawa, M.; Kodama, Y.; Yoshimura, K.; Kawanami, C.; Mimura, J.; Yamashita, Y.; Asada, M.; Kikuyama, M.; Okabe, Y.; Inokuma, T.; et al. Risk of Cancer in Patients with Autoimmune Pancreatitis. Am. J. Gastroenterol. 2013, 108, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Rahat, M.A.; Shakya, J. Parallel Aspects of the Microenvironment in Cancer and Autoimmune Disease. Mediators Inflamm. 2016, 2016, 4375120. [Google Scholar] [CrossRef] [PubMed]
- Rahat, M.A.; Coffelt, S.B.; Granot, Z.; Muthana, M.; Amedei, A. Macrophages and Neutrophils: Regulation of the Inflammatory Microenvironment in Autoimmunity and Cancer. Mediators Inflamm. 2016, 2016, 5894347. [Google Scholar] [CrossRef]
- Jiménez-Morales, S.; Ren, X.; Dean, M. Editorial: The Genetic Causes Underlying Immune Mediated Disease: A Focus on Autoimmunity and Cancer. Front. Genet. 2022, 13, 889160. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Demers-Mathieu, V. Optimal Selection of IFN-α-Inducible Genes to Determine Type I Interferon Signature Improves the Diagnosis of Systemic Lupus Erythematosus. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Li, H.; Liang, J.; Gao, Y.; Liu, M.; Xia, N.; Kong, W.; Zheng, L.; Zhang, Y.; Li, Z.; Chen, H.; et al. IGFBP2 Function as a Novel Biomarker for Active Lupus Nephritis. J. Mol. Med. 2022, 100, 1479–1491. [Google Scholar] [CrossRef]
- Mok, C.C.; Ding, H.H.; Kharboutli, M.; Mohan, C. Axl, Ferritin, Insulin-Like Growth Factor Binding Protein 2, and Tumor Necrosis Factor Receptor Type II as Biomarkers in Systemic Lupus Erythematosus. Arthritis Care Res. 2016, 68, 1303–1309. [Google Scholar] [CrossRef]
- Iorgulescu, J.B.; Medeiros, L.J.; Patel, K.P. Predictive and Prognostic Molecular Biomarkers in Lymphomas. Pathology 2024, 56, 239–258. [Google Scholar] [CrossRef]
- Beck, D.B.; Werner, A.; Kastner, D.L.; Aksentijevich, I. Disorders of Ubiquitylation: Unchained Inflammation. Nat. Rev. Rheumatol. 2022, 18, 435–447. [Google Scholar] [CrossRef]
- Lu, D.; Song, J.; Sun, Y.; Qi, F.; Liu, L.; Jin, Y.; McNutt, M.A.; Yin, Y. Mutations of Deubiquitinase OTUD1 Are Associated with Autoimmune Disorders. J. Autoimmun. 2018, 94, 156–165. [Google Scholar] [CrossRef]
- Pardoll, D.M. Inducing Autoimmune Disease to Treat Cancer. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5340–5342. [Google Scholar] [CrossRef]
- Pennell, C.A.; Barnum, J.L.; McDonald-Hyman, C.S.; Panoskaltsis-Mortari, A.; Riddle, M.J.; Xiong, Z.; Loschi, M.; Thangavelu, G.; Campbell, H.M.; Storlie, M.D.; et al. Human CD19-Targeted Mouse T Cells Induce B Cell Aplasia and Toxicity in Human CD19 Transgenic Mice. Mol. Ther. 2018, 26, 1423–1434. [Google Scholar] [CrossRef]
- Seligman, C.; Chang, Y.-M.; Luo, J.; Garden, O.A. Exploring the Role of Immune Checkpoint Inhibitors in the Etiology of Myasthenia Gravis and Lambert-Eaton Myasthenic Syndrome: A Systematic Review. Front. Neurol. 2022, 13, 1004810. [Google Scholar] [CrossRef]
- Young, A.; Quandt, Z.; Bluestone, J.A. The Balancing Act between Cancer Immunity and Autoimmunity in Response to Immunotherapy. Cancer Immunol Res 2018, 6, 1445–1452. [Google Scholar] [CrossRef]
- Hoos, A.; Eggermont, A.M.M.; Janetzki, S.; Hodi, F.S.; Ibrahim, R.; Anderson, A.; Humphrey, R.; Blumenstein, B.; Old, L.; Wolchok, J. Improved Endpoints for Cancer Immunotherapy Trials. J. Natl. Cancer Inst. 2010, 102, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Rivadeneira, D.B.; Lontos, K.; Dean, V.G.; Gunn, W.G.; Watson, M.J.; Yao, T.; Wilfahrt, D.; Hinck, C.; Wieteska, L.; et al. An Oncolytic Virus-Delivered TGFβ Inhibitor Overcomes the Immunosuppressive Tumor Microenvironment. J. Exp. Med. 2023, 220. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in Cancer Biology and Therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Zhang, Y.; Jiang, X.; Li, Y.; Cui, J.; Liao, Y. Single-Cell Analysis with Childhood and Adult Systemic Lupus Erythematosus. Autoimmunity 2024, 57, 2281228. [Google Scholar] [CrossRef]
- Kim, G.-D.; Das, R.; Goduni, L.; McClellan, S.; Hazlett, L.D.; Mahabeleshwar, G.H. Kruppel-like Factor 6 Promotes Macrophage-Mediated Inflammation by Suppressing B Cell Leukemia/Lymphoma 6 Expression. J. Biol. Chem. 2016, 291, 21271–21282. [Google Scholar] [CrossRef]
- Waku, T.; Katayama, H.; Hiraoka, M.; Hatanaka, A.; Nakamura, N.; Tanaka, Y.; Tamura, N.; Watanabe, A.; Kobayashi, A. NFE2L1 and NFE2L3 Complementarily Maintain Basal Proteasome Activity in Cancer Cells through CPEB3-Mediated Translational Repression. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef]
- Dankers, W.; Northcott, M.; Bennett, T.; D’Cruz, A.; Sherlock, R.; Gearing, L.J.; Hertzog, P.; Russ, B.; Miceli, I.; Scheer, S.; et al. Type 1 Interferon Suppresses Expression and Glucocorticoid Induction of Glucocorticoid-Induced Leucine Zipper (GILZ). Front. Immunol. 2022, 13, 1034880. [Google Scholar] [CrossRef] [PubMed]
- Letter to the Editor: Protein Phosphatase 1 Subunit Ppp1r15a/GADD34 Is Overexpressed in Systemic Lupus Erythematosus and Related to the Expression of Type I Interferon Response Genes. Autoimmun. Rev. 2019, 18, 211–213. [CrossRef]
Figure 1.
Rationale and Workflow Visualization. (a) The rationale of the Immune Imbalance Transcriptomics (IIT) algorithm. Using cancer, autoimmune, and healthy control data from the same tissue type, IIT seeks to identify differentially expressed genes from four quadrant profiles, which can be described in two distinct groupings: 1) two profiles that both indicate similar gene expression, including genes upregulated in both cancer and autoimmunity (C+A+) and genes downregulated in both cancer and autoimmunity (C-A-); and 2) two profiles that show opposite gene expression, that is, genes upregulated in cancer but downregulated in autoimmunity (C+A-) and genes downregulated in cancer but upregulated in autoimmunity (C-A+). The results and discussion refer to these four immune imbalance quadrant profiles. (b) A visual representation of the IIT algorithm workflow (see methods for additional detail).
Figure 1.
Rationale and Workflow Visualization. (a) The rationale of the Immune Imbalance Transcriptomics (IIT) algorithm. Using cancer, autoimmune, and healthy control data from the same tissue type, IIT seeks to identify differentially expressed genes from four quadrant profiles, which can be described in two distinct groupings: 1) two profiles that both indicate similar gene expression, including genes upregulated in both cancer and autoimmunity (C+A+) and genes downregulated in both cancer and autoimmunity (C-A-); and 2) two profiles that show opposite gene expression, that is, genes upregulated in cancer but downregulated in autoimmunity (C+A-) and genes downregulated in cancer but upregulated in autoimmunity (C-A+). The results and discussion refer to these four immune imbalance quadrant profiles. (b) A visual representation of the IIT algorithm workflow (see methods for additional detail).
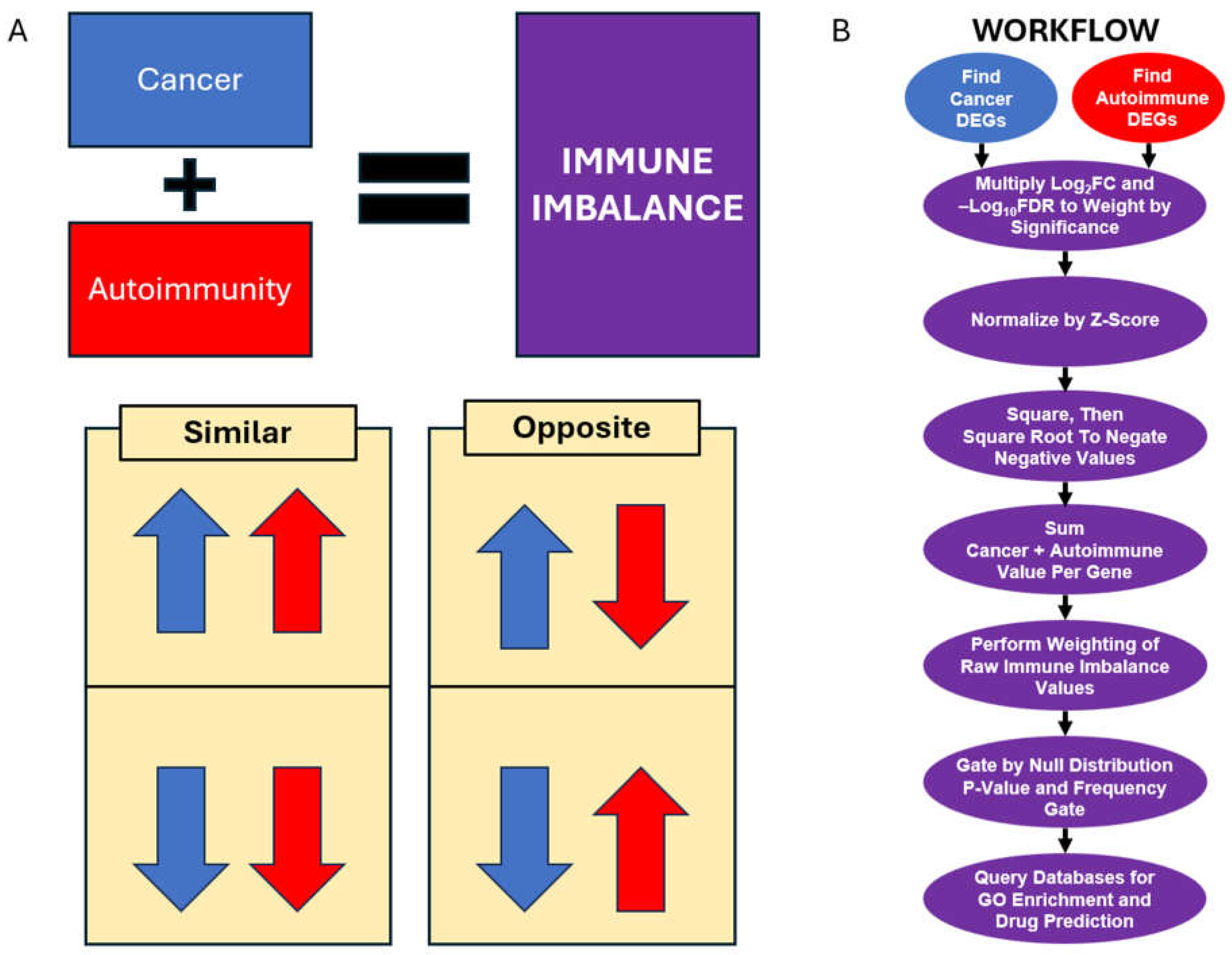
Figure 2.
Principal Component Analysis (PCA) of Analyzed Samples. Principal Component Analysis was performed on the counts tables of all samples. (a) All samples are colored by disease status. (b) Healthy samples colored by the study of origin. (c) Lymphoma samples were colored by their study of origin. (d) Lupus samples colored by the study of origin. See also Supplementary Figure S1.
Figure 2.
Principal Component Analysis (PCA) of Analyzed Samples. Principal Component Analysis was performed on the counts tables of all samples. (a) All samples are colored by disease status. (b) Healthy samples colored by the study of origin. (c) Lymphoma samples were colored by their study of origin. (d) Lupus samples colored by the study of origin. See also Supplementary Figure S1.
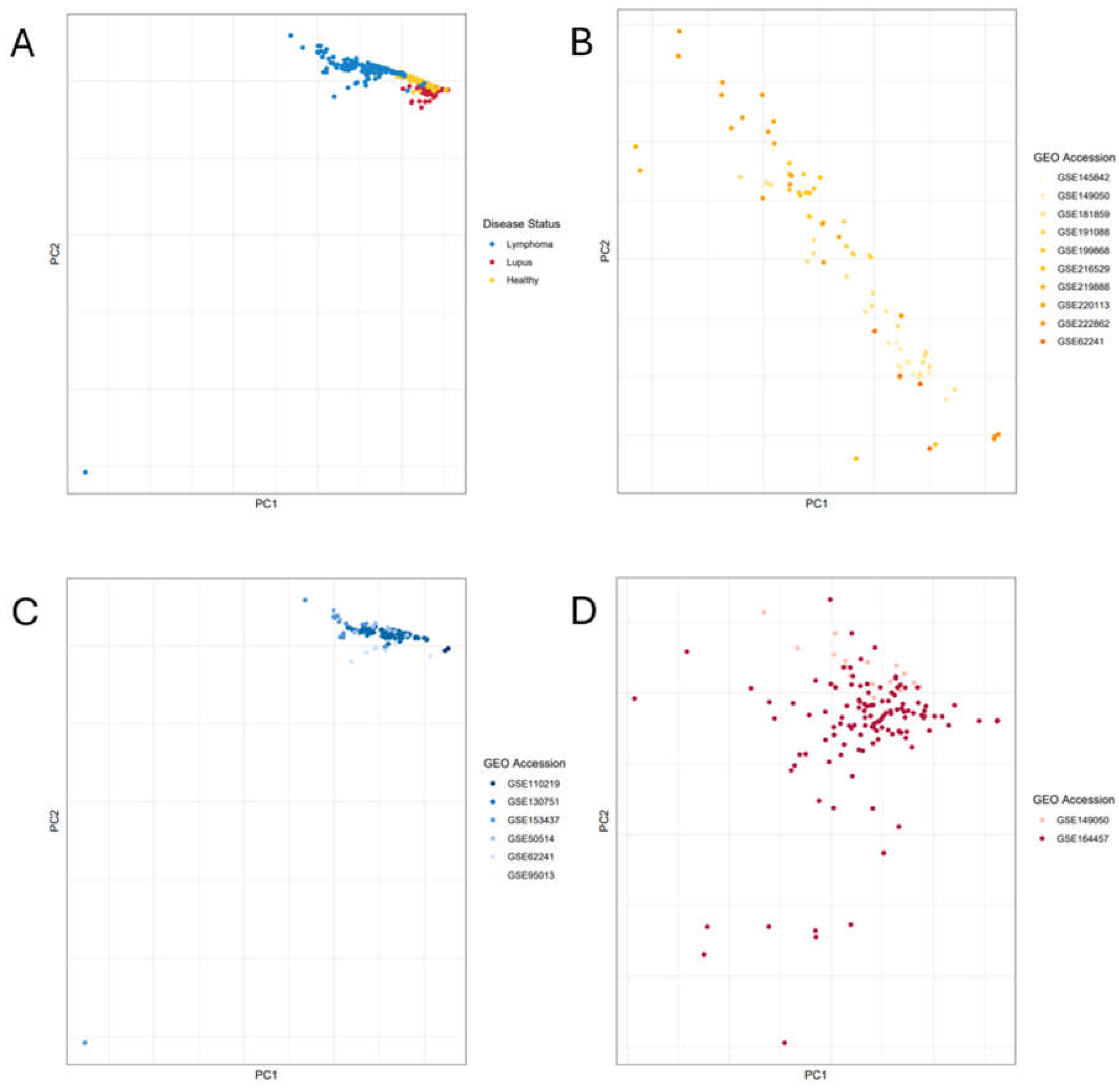
Figure 3.
Gating Lupus-Lymphoma IIT Data by Null and Frequency Gates for Significance. (a) Histogram visualization of gene frequency counts from the IIT-significant portion of the null distribution. The frequency gate, as determined by the p-value test, is marked in red on the histogram. (b) The differentially expressed genes (DEGs) that were significant in both the lupus vs. healthy and the lymphoma vs. healthy results. The lupus log2FCs are along the x-axis, while lymphoma log2FCs are on the y-axis, with each dot representing one gene. Genes with significant IIT scores are plotted in purple. Genes not significant based on the null distribution are plotted in black. Genes that were removed by the noise-detecting frequency gate are plotted in gray. Quadrants in the graph are divided by the axes.
Figure 3.
Gating Lupus-Lymphoma IIT Data by Null and Frequency Gates for Significance. (a) Histogram visualization of gene frequency counts from the IIT-significant portion of the null distribution. The frequency gate, as determined by the p-value test, is marked in red on the histogram. (b) The differentially expressed genes (DEGs) that were significant in both the lupus vs. healthy and the lymphoma vs. healthy results. The lupus log2FCs are along the x-axis, while lymphoma log2FCs are on the y-axis, with each dot representing one gene. Genes with significant IIT scores are plotted in purple. Genes not significant based on the null distribution are plotted in black. Genes that were removed by the noise-detecting frequency gate are plotted in gray. Quadrants in the graph are divided by the axes.
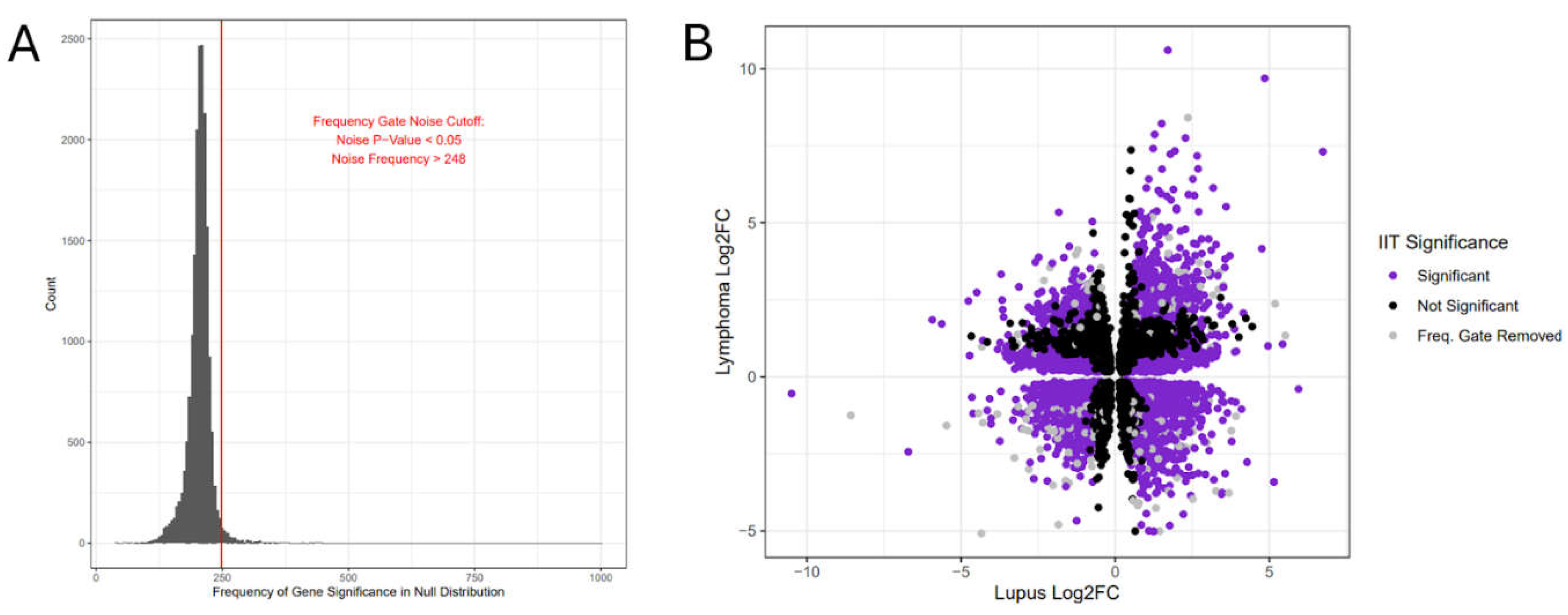
Figure 4.
Lupus-Lymphoma IIT Gene Quadrants and GO Enrichment. (a) Gene Ontology (GO) term enrichment, by quadrant, against the Biological Process GO library. (b) Gene Ontology (GO) term enrichment, by quadrant, against the Cellular Component GO library. (c) Gene Ontology (GO) term enrichment, by quadrant, against the Molecular Function GO library. (d) Upset chart (an alternative to a five-way Venn diagram) showing the overlapping GO Results from all included GO libraries between individual quadrant enrichment lists and the enrichment list of the entire significant gene dataset.
Figure 4.
Lupus-Lymphoma IIT Gene Quadrants and GO Enrichment. (a) Gene Ontology (GO) term enrichment, by quadrant, against the Biological Process GO library. (b) Gene Ontology (GO) term enrichment, by quadrant, against the Cellular Component GO library. (c) Gene Ontology (GO) term enrichment, by quadrant, against the Molecular Function GO library. (d) Upset chart (an alternative to a five-way Venn diagram) showing the overlapping GO Results from all included GO libraries between individual quadrant enrichment lists and the enrichment list of the entire significant gene dataset.

Table 1.
Sample Metadata Summary.
| Sample Phenotype | Type of Sequencing Reads | GEO Identifier | # Relevant Samples Included in Current Study |
|---|---|---|---|
| large B-cell lymphoma | paired end | GSE153437 [25] | 25 |
| follicular lymphoma | paired end | * GSE62241 [26,27] | 10 |
| diffuse large B-cell lymphoma | paired end | GSE95013 [28] | 29 |
| B-cell lymphoma | single end | GSE110219 [29] | 2 |
| diffuse large B-cell lymphoma | paired end | GSE130751 [30] | 63 |
| diffuse large B-cell lymphoma | paired end | GSE50514 [31] | 7 |
| lupus B-cells | single end | * GSE149050 [32] | 18 |
| lupus B-cells | paired end | GSE164457 [33] | 120 |
| healthy B-cells | paired end | GSE145842 [38] | 6 |
| healthy B-cells | single end | * GSE149050 [32] | 14 |
| healthy B-cells | paired end | GSE181859 [34] | 20 |
| healthy B-cells | paired end | * GSE62241 [26,27] | 4 |
| healthy B-cells | paired end | GSE191088 [39] | 6 |
| healthy B-cells | paired end | GSE199868 (currently unpublished) | 13 |
| healthy B-cells | paired end | GSE216529 [35] | 2 |
| healthy B-cells | single end | GSE219888 [36] | 2 |
| healthy B-cells | paired end | GSE220113 [37] | 17 |
| healthy B-cells | single end | GSE222862 (currently unpublished) | 3 |
* GSE62241 and GSE149050 contain both disease samples and healthy controls. For clarity, they are each included in the table twice.
Table 2.
Top Four Significantly Immune-Imbalanced Genes from Each IIT Quadrant.
| Gene Symbol | lupus* log2FC | lupus* FDR | lymphoma** log2FC | lymphoma** FDR | IIT score | IIT corrected p-value*** | |
|---|---|---|---|---|---|---|---|
|
C+A+ Quad I |
C1QB | 4.85 | 1.98E-31 | 9.69 | 7.78E-47 | 81.5 | 0 |
| IFI27 | 6.74 | 4.35E-34 | 7.31 | 2.88E-44 | 78.96 | 0 | |
| IGFBP2 | 1.99 | 3.13E-33 | 5.42 | 2.67E-31 | 38.67 | 0 | |
| TCN2 | 3.27 | 1.21E-22 | 4.29 | 1.07E-33 | 37.22 | 0 | |
|
C+A- Quad II |
PTMS | -2.59 | 1.83E-17 | 3.72 | 8.08E-27 | 25.21 | 0 |
| TPM2 | -2.48 | 2.23E-15 | 3.88 | 1.65E-25 | 21.69 | 0 | |
| NFE2L3 | -1.42 | 4.61E-21 | 1.99 | 3.27E-37 | 18.03 | 0 | |
| PLXNA1 | -1.38 | 5.76E-21 | 2.75 | 1.04E-34 | 14.91 | 0 | |
|
C-A- Quad III |
TAPT1 | -1.57 | 3.70E-30 | -2.21 | 6.12E-37 | 28.45 | 0 |
| PNRC1 | -1.72 | 4.13E-43 | -2.11 | 1.16E-43 | 27.03 | 0 | |
| OTUD1 | -1.82 | 5.75E-34 | -2.24 | 5.46E-37 | 25.54 | 0 | |
| MAP3K1 | -1.64 | 7.47E-35 | -2.15 | 1.69E-36 | 24.68 | 0 | |
|
C-A+ Quad IV |
TSC22D3 | 2.36 | 5.27E-34 | -3.22 | 2.83E-45 | 47.78 | 0 |
| KLF6 | 2.7 | 1.48E-38 | -3.13 | 2.23E-46 | 42.09 | 0 | |
| PPP1R15A | 2.93 | 1.23E-29 | -3.17 | 1.38E-40 | 38.84 | 0 | |
| ARL4A | 1.9 | 9.03E-34 | -2.69 | 1.67E-40 | 35.81 | 0 |
* Systemic Lupus Erythematosus. ** B-cell Non-Hodgkin’s Lymphoma. *** Bonferroni-corrected.
Table 3.
Top Five GO Terms from Enrichment of All Significant IIT Genes.
| Term | Overlap | Bonferroni p-value | Odds Ratio | Combined Score | GO DAG* |
|---|---|---|---|---|---|
| RNA Binding (GO:0003723) | 489/1411 | 7.58E-10 | 1.53 | 42.71 | Molecular Function |
| Protein Serine/Threonine Kinase Activity (GO:0004674) | 141/342 | 7.27E-07 | 1.98 | 40.43 | Molecular Function |
| Cytoplasmic Translation (GO:0002181) | 50/93 | 9.63E-05 | 3.26 | 58.06 | Biological Process |
| Macromolecule Biosynthetic Process (GO:0009059) | 81/183 | 3.23E-04 | 2.23 | 35.48 | Biological Process |
| Ubiquitin-Like Protein Transferase Activity (GO:0019787) | 97/240 | 4.95E-04 | 1.91 | 25.7 | Molecular Function |
* GO DAG: Gene Ontology Directed Acyclic Graph.
Table 4.
Top 10 Enriched Pathways against All Significant IIT Genes.
| Pathway | Overlap | FDR P-Value | Odds Ratio | Combined Score | Database |
|---|---|---|---|---|---|
| Neutrophil Degranulation | 190/468 | 1.66E-08 | 1.94 | 49.11 | Reactome |
| Immune System | 638/1943 | 1.66E-08 | 1.41 | 34.88 | Reactome |
| Adaptive Immune System | 271/733 | 5.95E-08 | 1.67 | 38.41 | Reactome |
| Signaling By Rho GTPases | 242/644 | 6.34E-08 | 1.71 | 38.42 | Reactome |
| Signaling By Rho GTPases, Miro GTPases And RHOBTB3 | 247/660 | 6.34E-08 | 1.7 | 38.14 | Reactome |
| Eukaryotic Translation Elongation | 52/90 | 9.11E-08 | 3.84 | 84.12 | Reactome |
| Innate Immune System | 356/1035 | 7.98E-07 | 1.5 | 29.25 | Reactome |
| Formation Of A Pool Of Free 40S Subunits | 53/98 | 1.18E-06 | 3.31 | 62.96 | Reactome |
| Peptide Chain Elongation | 48/86 | 1.23E-06 | 3.54 | 66.49 | Reactome |
| Cellular Responses To Stress | 259/722 | 1.23E-06 | 1.59 | 29.75 | Reactome |
Table 5.
Top-Scoring immune-imbalanced Targets with Known Drugs.
| Gene Symbol | Quadrant | Immune Imbalance Score | IIT corrected p-value* | Number Unique Drugs | Number Approved Drugs | Weighted Target Score |
| C3 | Quad I | 33.57 | 0 | 3 | 1 | 1357 |
| CXCR4 | Quad IV | 28.73 | 0 | 7 | 1 | 1313 |
| MAP3K1 | Quad III | 24.68 | 0 | 1 | 0 | 698 |
| IDO1 | Quad I | 22.02 | 0 | 2 | 0 | 913.5 |
| CLU | Quad I | 21.67 | 0 | 2 | 0 | 725.5 |
| CD276 | Quad I | 20.17 | 0 | 2 | 0 | 436 |
| COL1A1 | Quad I | 16.39 | 0 | 2 | 1 | 1299 |
| EPHB2 | Quad I | 16.07 | 0 | 1 | 1 | 316 |
| NR1H2 | Quad IV | 15.45 | 0 | 5 | 0 | 186 |
| TUBA1A | Quad IV | 15.17 | 0 | 24 | 1 | 295 |
* Bonferroni-adjusted p-value.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



