
Submitted:
19 July 2024
Posted:
22 July 2024
You are already at the latest version
Abstract
Head and neck squamous cell carcinoma (HNSCC) is a highly heterogeneous and the most common form of head and neck cancer, posing significant challenges for disease management. Single-cell RNA sequencing (sc-RNAseq) has emerged as a valuable tool for addressing these challenges by enabling detailed characterization of the tumor microenvironment (TME) at the cellular level. This review assesses the utility of sc-RNAseq in HNSCC research, compiling current strategies employing single-cell technologies. For HNSCC etiology, sc-RNAseq allows for the construction of cellular atlases, characterization of different cell types, and investigation of key genes and processes involved in cancer initiation, development, and progression within the TME. In terms of HNSCC diagnosis and prognosis, the high resolution offered by sc-RNAseq enables the identification of cell type-specific signatures, enhancing prognostic models and disease stratifiers for more accurate patient outcome assessments. Regarding HNSCC treatment, sc-RNAseq provides insights into cellular responses to various treatments, including radiotherapy, chemotherapy, and immunotherapy, contributing to improved patient stratification and personalized treatment strategies. This review highlights the unique contributions of sc-RNAseq to HNSCC research, addressing its cellular and biological complexity, and emphasizes its potential for advancing research and clinical practice in other cancer types.
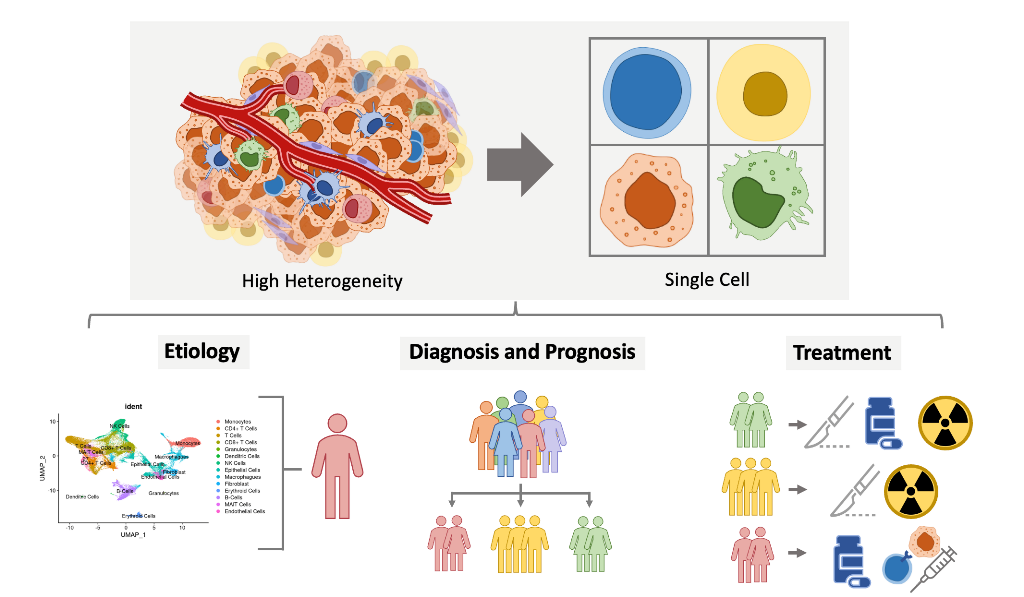
Keywords:
HNSCC
; single-cell RNA sequencing
; tumor microenvironment
1. Introduction
Head and neck squamous cell carcinoma (HNSCC) represent a significant burden in the global landscape of cancer, accounting for more than 90% of the head and neck cancers (HNCs). While advancements have been made in the treatment and prognosis of HNSCC in recent decades, the chances of a relapse within two years are above 50% and the five-year overall survival (OS) rate remains below 50% indicating substantial room for further improvement [1]. A major challenge in dealing with HNSCC lies in the high degree of intra-tumor and inter-tumor heterogeneity, which heightens the difficulty of administrating proper standard treatment [2,3].
In the last decade, transcriptomics has been based in bulk RNA sequencing, where all cells in a sample are sequenced together to produce one gene expression profile of the whole tumor. This is evidenced by collaborative initiatives like The Cancer Genome Atlas (TCGA) and The National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC), encompassing an extensive collection of tumor samples analyzed at various levels, including the transcriptomic one [4,5]. However, this approach fails to uncover the cellular sources of variation, as it combines multiple cell types in different developmental states, thereby obscuring critical molecular events and signals occurring within distinct cell subpopulations, which could be key for therapeutical targeting [6,7]. Single-cell transcriptomics has emerged as a solution, providing a gene expression profile for each individual cell within the sample. This enables a granular characterization of the tumor cells and their tumor microenvironment (TME) at the cellular level, shedding light on the intrinsic properties of distinct cell populations within one tumor, thereby facilitating the identification of therapeutic targets specific to different subpopulations [8].
The current literature illustrates the substantial impact of single-cell transcriptomics through all the three HNSCC research stages [2,9]. Cancer research stages include cancer etiology, cancer diagnosis and prognosis, and cancer treatment. The first stage, cancer etiology, involves fundamental research done to understand the variables and mechanisms underlaying cancer initiation, development, and progression. HNSCC etiology is a remarkably complex disease etiology, it comprises a wide range of factors, from the composition of the TME to genetic mutations and environmental exposures. The second stage, cancer diagnosis and prognosis, utilizes the insights obtained from fundamental research to identify the presence, location, and severity of the disease. This information is vital in the search of key nodes that enable to stratify patients according to their risk. The stratification is important because it allows for a more specific treatment plan according to the characteristics of the selected group. Lastly, cancer treatment, uses a combination of basic knowledge about the cancer and the targets found to stratify the groups to develop tailored treatment strategies and study the responses.
This comprehensive review evaluates the use of single-cell transcriptomics in HNSCC, emphasizing its unique contributions and the challenges it poses in advancing our understanding and management of this complex disease.
2. HNSCC Etiology
Single-cell transcriptomic data can be used to unravel and provide a deeper understanding on the biological mechanisms and factors driving cancer initiation, development, and progression. Fundamental research is essential to lay the foundation in which cancer diagnosis, prognosis and treatment will rest. The use of single-cell analysis introduces a new layer of complexity to our understanding of the TME, taking the information to the cellular level and allowing to examine the changing patterns in tumor cells and their surrounding microenvironment [10].
Head and neck squamous cell carcinoma (HNSCC) represents a biologically diverse disease that exhibits a substantial level of histological and molecular heterogeneity. This includes intra-tumoral heterogeneity, which refers to the presence of diverse subpopulations within a single tumor sample, and inter-tumoral heterogeneity, which describes the differences observed between tumor samples from distinct anatomic sites within the same patient, as well as between different patients or tumor types [9,11]. In current literature, several strategies have been proposed to give further insight into cancer heterogeneity through single-cell transcriptomics. These approaches include atlas construction, cell type characterization and expression of process related genes (Figure 1).
3.1. Atlas Construction
One of the approaches to exploring the complexity of HNSCC is the generation of HNSCC cell atlases, creating cell-type maps or collections of cell-specific gene expression profiles. The application of these atlases allows for an in-depth exploration of HNSCC cellular diversity across sites, states, and patients, trying to reach the highest degree of resolution that is financially and technically possible.
Puram et al. published the first HNSCC atlas, in which ~6,000 cells from 18 HNSCC patient, including primary tumors and lymph node metastasis samples, were analyzed to allow for comparisons in tumor progression [12]. This study aimed to investigate intra-tumoral heterogeneity, defining different cell types composing the TME across patients and differentiating between malignant and non-malignant cell types. The study also encompassed inter-tumoral heterogeneity, with results showing similarities in the expression profiles of defined cell types throughout different patients. In a more recent study, the focus shifted to profiling oropharyngeal squamous cell carcinoma (OPSCC), including both HPV-positive and HPV-negative tumors, expanding the cell count to 70,970. This comprehensive analysis revealed extensive cellular diversity within and between tumors and uncovered a subset of malignant cells in HPV-positive tumors lacking detectable HPV expression, which also exhibited reduced HPV-associated phenotypes [13]. Another example is the HNSCC atlas by Kürten et al. which provides a broad overview of immune and non-immune cells in HNSCC [14]. The samples include matched CD45+ peripheral blood leukocytes (PBL) as control sample and tumor-infiltrating leukocyte (TIL) samples, as well as CD45- non-immune cells obtained from primary tumors of 15 HNSCC patients. An in-depth characterization of tumor populations and differences in inflammation levels in HPV+ versus HPV- HNSCC samples was performed. HPV viral gene expression was shown to be traceable in single cells. The breadth of the dataset allowed the characterization of multiple cell types, with a particular focus on epithelial cells and CD8+ T cells and their dysfunctional state, which has been targeted as one of the main effectors of anti-cancer immunity. In another HNSCC atlas presented by Choi et al., the aim was to profile the progression of HNSCC cell types and cell-cell interactions with samples ranging from normal tissue to metastatic tumors in the lymph node, including precancerous leukoplasia and primary tumor [10]. Immune cells, specially Tregs and CD154-CD4+ T cells infiltration were positively correlated with tumor progression. The proportional changes in cell types were proven to reflect cancer progression according to microenvironment alteration. HNSCC atlases can have a fewer number of samples but a much higher cell number per sample. In the atlas proposed by Song et al. two samples from the larynx were analyzed and ~14,000 cells were retrieved, allowing for the characterization of the immune compartment and the assessment of tumor cell heterogeneity of the samples [15]. The largest patient cohort for scRNAseq in HNSCC is presented in the atlas by Bill et al., profiling 52 fresh tumor tissues from 51 patients, encompassing 187,399 cells and a wide range of clinical characteristics. By addressing the heterogeneity of HNSCC, the study revealed significant insights into tumor-associated macrophage (TAM) polarization and its impact on the tumor microenvironment (TME), highlighting the potential for new therapeutic interventions across multiple cancer types [16].
In summary, the broadness of the data obtained from SC atlases enables an unprecedent amount of information that can be extracted from a single experiment. Moreover, the importance of a proper clinical annotation of the patients is demonstrated in the wider range of questions well annotated data can address. Such annotations can include factors like sex, HPV-status, or exposure to environmental factors like alcohol or tobacco. The imperative for research transparency has propelled these studies to render their data openly accessible, thereby facilitating utilization by fellow researchers who might be constrained in their capacity to generate SC data independently or whose inquiries could be addressed through preexisting datasets. The development of integration methods that assess biological variation across samples has created opportunities to unify data from different studies. This allows for the creation of a ‘universal atlas’ as a comprehensive reference map for HNSCC, overcoming current limitations such as the small number of cases processed per study. A list of available HNSCC sc-RNA-seq datasets is provided in Table 1.
3.2. Cell Type Specific Characterization
A different approach to unraveling the HNSCC characteristics is to focus on specific subsets to gain a deeper understanding of their dynamics, location, and interactions. Such an approach has proven highly relevant for characterizing the TME and identifying key targets for personalized treatment. Several studies have pursued this task as one of the analyses performed after generating an atlas [15,17,18]. Other publications use publicly available atlas data to explore unstudied subpopulations, such as subtypes of cancer-associated fibroblasts (CAFs) or tumor cells [19]. These publications may supplement the study with data of their own [20]. A third option is the generation of new data that is specifically oriented toward a particular subpopulation, for instance B cells or CD8 T cells selected through cell sorting [21,22,23].
In current literature, the main methods employed to analyze scRNA-seq data comprise sub-clustering of a designated cell type to distinguish cell states based on expression profiles, followed by a detailed characterization using differential expression (DE) or pathway analysis, cell-cell communication (CCC) analysis to uncover putative intercellular interactions within the selected group and communication pathways with its microenvironment and differentiation trajectory reconstruction (DTR) analysis to reconstruct the developmental course of a related group of states. Two main cell compartments have been explored using this methodology in literature, the tumor cell compartment and the immune compartment.
3.2.1. Tumor Cell Characterization and Crosstalk with Stromal Compartments
Tumor cells, which possess a proliferative advantage over neighboring cells, along with their surrounding stromal cells are a key player in cancer development. The significant heterogeneity observed among tumor cells, as well as of the components of the stromal compartment, may lead to differences in morphological and physiological features of HNSCC [15]. A comprehensive characterization of these subpopulations is needed to understand their behavior and devise effective therapeutic strategies that specifically target them. One of the main limitations is proper cancer cell annotation. Although some methods use manual selection of clusters based on expression of marker genes or in their copy number variation status (CNV), novel machine learning-based methods are emerging that automatize the process of tumor cell selection [24,25].
In order to gain a deeper understanding of a cell type an initial step is the employment of sub-clustering methods to find subtypes or substates that may arise as a result of different cancer stages and interactions with the environment. One study focused on sub-clustering tumor cells, selected by cluster marker genes, and identified five sub-clusters: keratinocyte-like, proliferative, immortal, metastasis, and immune cell clusters [15]. The clusters were labelled according to the gene ontology (GO) pathways that were enriched for each sub-cluster. In another study, tumor cells were detected as a sub-cluster of epithelial cells and further divided into three cluster based on cluster marker genes with previously characterized functions: partial epithelial-mesenchymal transition, cycling G2/M, cycling S [19]. A cancer stem cell (CSC) sub-cluster was selected among 12 cancer cell sub-clusters due to an enrichment in stem cell marker expression, such as CD44, CD98, CD47, and CD276, the presence of the markers in some of the other sub-clusters demonstrated that CSCs could be found in a variety of places [20].
DTR analysis can be useful to understand the chronological evolution of a cell population. In the five sub-cluster study by Song et al. the trajectory analysis showed the proliferative sub-cluster at the beginning of the trajectory, branching into immortal tumor cells or keratinocytes-like tumor cells, placing a timeline in the development of cancer cell types. Keratinocytes-like and proliferative tumor cells were observed to have distinct spatial location, with the latter being associated with worse patient prognosis [15].
CCC can be employed to understand how tumor cells interact with their surroundings. For instance, results of such analysis suggested that the expression of TNFSF12 (TWEAK – an apoptosis-inducing ligand –) in cancer-associated fibroblasts (CAFs) has an opposite expression pattern in primary tumors that have latter metastasized or not, a high expression is related with promotion of cancer progression through proliferation, invasion and EMT in the tumor cells of their surroundings and is correlated with a low HNSCC survival probability [19]. Sub-clustering of CAFs in HNSCC has shown differences in gene expression profiles, with the presence of specific sub-clusters being associated with metastasis and bad prognosis. These findings highlight the relevance of CAFs subtypes in HNSCC [19,26]. CSCs and myeloid cells were found to interact through cell-cell communication analysis, this suggested that CSCs contribute to immune-escape signaling through promoting maturation and differentiation of myeloid cells [20]. HPV status assessment was performed at single cell level in tumor cells, CCC analysis correlated HPV+ tumor cells with infiltrated T-cells, which may account for the difference in prognosis according to HPV status [27].
3.2.2. Immune Landscape
The immune component of the TME is the responsible for tumor-related humoral and cellular immune response, including immune surveillance and tumor growth control [28]. HNSCC cancer cells have proven to be highly evasive to the immune system as well as having immune suppressive features [29]. However, the HNSCC immune landscape has a high degree of plasticity, HPV+ tumors have exhibited a more immune-active or "hot" phenotype while HPV- tumors are usually characterized by a more immune-suppressed or "cold" phenotype [30]. Patients with a colder tumor type exhibit worse prognosis and less response to immunotherapy [31].Therefore, the classification of a tumor into hot or cold phenotype is crucial for developing effective treatment strategies. Proper dissection at single cell level of the immune component is essential in determining the appropriate approach for each patient.
In Chen et al. sub-clustering on T cell population 14 sub-clusters were identified and further characterized according to their cluster marker gene expression. The comparison between states, normal tissue versus tumor tissue, allowed for the identification of a higher proportion of exhausted T cell substates, with less cytotoxic potential, in tumor sample making the blockage of their immunosuppressive role a target for immunotherapy [21]. A more specific study on HPV+ CD8+ T cells allowed for their classification in three sub-clusters each one enriched in cluster markers for a different developmental stage. These clusters were labelled as stem-like, transitory and differentiated stages [23]. In a different study of Huang et al. four sub-clusters of macrophages were detected. DE analysis between the groups also allowed for their classification according to developmental stage in early, transforming and mature groups [32]. Sub-clustering of B cells from healthy and HNSCC donors allowed the differentiation of 6 B cell sub-clusters. Assessment of the expression of multiple transmembrane cytotoxic tumor necrosis factor (TNF) super family (TNFSF) ligands through the different sub-clusters showed differential expression by all these sub-clusters. These findings suggest that specific sub-clusters of tissue-resident B cells, especially germinal center B cells, might possess elevated intrinsic cytotoxic capabilities compared to their counterparts in the circulatory system [18].
When comparing between carcino-mediated or virus-driven immune landscape in HNSCC, notable differences in transcriptional profiles, relative composition, and developmental trajectories have been identified primarily within CD4+Tconv cells, B cells, and myeloid cells. These differences are hypothesized to arise from the activation of the innate immune system mediated by the presence of the virus [17]. B cell-related pathways seemed to be important in HPV+ HNSCC in contrast to HPV- tumors, as well as genes revealed by DE and survival analysis: upregulation of AREG and TGFBI (markers of plamacytoid dendritic cells (pDC)) was associated to worse survival and downregulation of CD27, CXCR3 (expressed in T cell-related cells) MS4A1, and CD19 (expressed in B näive cells) was related to improved survival [33]. In another recent study sub-clustering of macrophages in 8 sub-clusters allowed for a more intricate classification of this cell type than the classical M1 (anti-tumor) or M2 (pro-tumor) like macrophage classification described in literature. This study validated that the infiltration ratio of M1 like sub-clusters was increased in HPV+, a TCR+ tumor-promoting subpopulation described in previous literature was also detected and SC resolution allowed for a deeper characterization [34].
DTR analysis in CD8+ T cells proved a similar behavior between HPV+ and HPV- tumors, suggesting a potential similarity in the response of CD8+ T cell therapy across these distinct tumor types [17]. The same analysis in a broader T cell population coming from healthy and primary tumor samples was able to demonstrate different developmental branches according to the “T cell activation signal” the initially non-functional T cells were receiving [21]. Macrophage DTR analysis in HPV+ versus HPV- conditions demonstrated similarities in the developmental trajectories between the two [34]. DTR analysis validated developmental trajectories from DE-derived stage-related genes. In a study on HNSCC-associated TAMM populations, three sub-clusters were categorized by developmental stages using cluster marker genes. Remarkably, the DTR analysis confirmed the identical trajectory, reinforcing its validity [23,32]
Cell-cell communication analysis showed an increased cross-talk in the immune component of the TME versus normal control [17]. CCC analysis applied to TCR+ versus TCR- macrophages showed differences in immune cell interactions, which suggest a reason for their difference in aggressiveness [34].
In summary, in-depth investigations into distinct cell types offer a wealth of insights into their behavior under specific conditions, developmental trajectories, and intercellular interactions. This could serve as a resource for identifying biomarkers and targets directed towards distinct cell types implicated in specific conditions, surpassing bulk methods in resolution and advancing personalized treatment. However, it is vital to note that scRNAseq provides only a transcriptional depth layer. To achieve a comprehensive understanding, validation through wet lab experiments, and correlation with complementary data sources such as functional and spatial annotations, patient survival data, bulk RNAseq, histology, or spatial transcriptomics is imperative.
3.3. Expression of Process Related Genes
The study of critical genes relevant to the HNSCC process can greatly benefit from evaluating their expression patterns in distinct cell types using scRNA-seq. Such data have been key in characterizing processes like ferroptosis and cuproptosis, where cancer stem cells were identified as the core cluster related to ferroptosis, and epithelial cells showed high expression of CDKN2A, a gene associated with copper-induced cell death [35,36]. Utilizing scRNA-seq data from atlases, which mitigates the high economic burden of generating new data, allows researchers to answer pivotal questions. Tools like CancerSEA and TISH2 facilitate these analyses by linking gene expression to functional states across various cancer types [37,38]. For example, CancerSEA was used to investigate perineural invasion (PNI) in HNSCC, revealing that fibroblasts, which exhibit high expression of PNI-related genes, play a significant role in processes like EMT, metastasis, and invasion [39]. Furthermore, the Cornichon Family AMPA Receptor Auxiliary Protein 4 (CNIH4) has been identified as a key cancer marker associated with stemness, cell cycle, DNA repair, invasion, and proliferation, with high expression in tumor cells [40]. Additionally, scRNA-seq data have shown that genes involved in mitochondrial dynamics and store-operated calcium channels are heterogeneously expressed across cell types, highlighting fibroblasts and cancer cells as potential cancer drivers [41]. Genes like APOBEC3B, APOBEC3C, and YTHDC1, highly expressed in tumor cells, were linked to tumor stemness and correlated with markers such as SOX2 and BM1, and pathways promoting stemness [42,43]. SEC11A, characterized as an oncogene and valuable prognostic biomarker, showed expression across multiple cell types, particularly correlating with immunosuppressive immature myeloid cells (MDSCs) [44]. Chloride intracellular channel 4 (CLIC4), a tumor suppressor, was detected as predominantly expressed in tumor-associated fibroblasts and endothelial cells, supporting its role in growth arrest, differentiation, and apoptosis [45]. Moreover, scRNA-seq has been used to characterize the expression of genes involved in tumor growth, angiogenesis, invasion, and metastasis through pathways like the CXCL12/CXCR4 axis [46]. By integrating these insights, scRNA-seq data provides a comprehensive understanding of the cellular and molecular landscape of HNSCC, uncovering new therapeutic targets and biomarkers for personalized treatment strategies.
4. HNSCC Diagnosis and Prognosis
Despite advances in current treatments for HNSCC, patients with advance stages of the disease only have a 50% five-year survival rate and, in the past few decades, there has been little improvement in this regard [47,48]. Novel therapies, targeting immune checkpoint inhibitors programmed cell death-1 (PD-1)/PD-1 ligand (PD-L1), have proven to be effective in only a small sub-group of patients. In order to guide more personalized treatments, strategies that help in the prediction of HNSCC progression and selection of molecular biomarkers related to patient prognosis are highly important [49,50]. The deep resolution offered by SC technologies allows for a more refined selection of cell type-related signatures, which could prove of great relevance for the construction of prognostic models and disease stratifiers. The information can be transferred from SC data to bulk data or vice versa in diverse ways, paving the way for multi-level data integration and allowing for maximal usage of all available data (Figure 2).
Prognostic gene signatures established from bulk-RNAseq analysis can be further characterized using sc-RNAseq analysis. 8 methylation/autophagy-related genes (MARGs) and 2 immune cells infiltrated in the tumor were selected to build a prognostic risk scoring system (pRS). Clustering analysis of the 8 pRS genes divided single cells into high or low risk cells, with fibroblasts being mainly present in the high-risk cluster. Because of the 8 pRS expression heterogeneity across cell types, a neural network-based deep learning model was built based on scRNA-seq data which was able to infer cell types [49]. Due to the prognostic value of FGFR-signal, hypoxia, and glycolysis a 6 gene fibrosis-hypoxia-glycolysis-related prognostic classifier was constructed based on bulk RNA-seq. Through SC analysis, the expression of this gene signature was found to be higher in monocytes, this cell type also showed a higher infiltration in HNSCC patient samples than in healthy donors which points to its association to poor prognosis [50]. To verify the relation between genes and model in a 6 gene matrisome-associated prognostic model, single cell data was used. A risk score for each cell was calculated and higher risk was associated with tumor cells compared to the non-tumor cells [51].
A different approach to integrating SC and bulk-RNA seq data relays in the importance of particular cell types to cancer development and, therefore, to its outcome. In a study about mast cells (MC), a cell type playing a pro-tumorigenic role by promoting several cancer inducing functions, MC cluster marker genes extracted from SC analysis were selected to construct a MC-related prognostic model using bulk-RNAseq data [3]. HPV+ CD8+ T cluster marker genes were selected using DE analysis comparing HPV+ and HPV- CD8+ T cell populations established through SC analysis. These genes were subsequently utilized to establish a prognostic gene signature able to divide HPV+ patients in high and low risk groups [47]. In a further study A CAF-related gene signature was developed based on cluster marker genes associated with poor prognosis from a CAF subpopulation identified via SC analysis. The high-risk group was associated with low levels of anti-tumor immune cells and less sensitivity to conventional chemotherapy and immunotherapy, highlighting immune cell type infiltration as a main driver of the risk differences [26]. To assess the prognostic value of CD4+ T follicular helper (TFH) related genes, the signature was analyzed in bulk-RNAseq data paired with survival analysis finding its enrichment associated with better progression free survival (PFS) [17]. Deconvolution algorithms can be utilized to computationally estimate cell-type proportions in bulk expression data using information extracted from SC data [52]. For example, TCGA bulk-RNA seq was deconvoluted using single cell data, giving a cell fraction score for each of the 10 cell types detected in the SC dataset. An 8 gene prognostic model was constructed using genes correlating with a higher CD8+ T cell content. Risk divided populations were then characterized finding low-risk group as a more immune-active or hot type than the high-risk group, being the low-risk group better responder to immunotherapies [47].
The prognostic value of cell differentiation-related genes (HDRGs) from scRNA-seq trajectory analysis has proven useful for HNSCC molecular stratification and therapy selection [53]. For example, a study identified 159 prognostic HDRGs, resulting in three subtypes—active stroma, active metabolic, and active immune—each with distinct therapeutic implications [7]. DTR in SC data also revealed LN-related genes, leading to a five-gene prognostic model that identifies molecular subtypes and informs therapeutic decisions [54]. Finally, CCC analysis in scRNA-seq identified key interactions across HNSCC samples, leading to a five-gene model by multifactorial stepwise regression, highlighting the importance of these interactions in cancer progression [55].
5. HNSCC Treatment
The final step in HNSCC cancer research is to actively attack the cancer via a selected treatment. Treatment for HNSCC with a curative intend includes tumor resection, radiotherapy, chemotherapy or their combination [56]. However, tumor relapse has still an incidence of about 50% within 2 years and the 5-year survival remains quite unfavorable at rates of less than 50% [1]. In the last decades, due to the appearance of immunotherapy and better selection of target groups, treatment of HNSCC has steadily improved [57]. Intertumoral heterogeneity in HNSCC is among the reasons for the unfavorable outcomes, accounting for the variability in therapeutical responses among patients [58]. The use of sc-RNAseq contributes to understand specific cellular responses to treatments, including radiotherapy, chemotherapy, and immunotherapy, which could, in turn, contribute to an improvement in patient stratification and a more personalized selection of treatment strategy.
Single-cell analysis has the potential to greatly improve the monitoring of treatment responses in longitudinal human studies. It allows tracking of various cell populations at the clonal level, opening a new way to investigate treatment effects. Although this approach is still in its early stages, a few studies have started this exploration, with more expected in the future. In the areas covered by this review, the use of single-cell analysis for monitoring patient treatment response remains relatively untouched. In the two human studies described in this section, the proposed strategy involved comparing primary tumor samples before and after treatment to observe changes in cell populations in response to treatment. Using SC analysis on samples taken before and after treatment with nivolumab, CAFs were identified as the cell population exhibiting the greatest changes in abundance. This study led to the identification of a CAF subpopulation related with immuno-activation, T cell–stimulating CAF (tsCAF). This knowledge allows for the development of strategies to force CAF differentiation toward the proinflammatory tsCAF phenotype rather than the immunosuppressive phenotype, leading to an improvement in tumor response to immunotherapy [59]. Additionally, SC analysis of samples treated with induction chemotherapy (ICT), chemotherapy administered before surgical removal of the tumor, further highlighted the importance of CAFs in tumor treatment. In the study, CAFs showed upregulation of EMT related pathways induced by chemotherapy, recognizing them as drivers of response and potential targets during therapy. A limitation presented in this study is that samples coming from only one patient were used [60].
Other examples of strategies followed to study HNSCC response to treatment using sc-RNAseq can be found in preclinical studies. In some of these studies, analysis is conducted comparing before and after treatment states, while in others, silencing or knock-out models of target proteins are performed as way to mimic the treatment effect. In all cases a pre-treatment reference is utilized to track changes in cell population behavior. B cells from HNSCC tumor samples induced by subcutaneous injection of B16-OVA and AT-84-E7-OVA cells, which reflects HPV+ HNSCC in mice, were analyzed before and after anti-PD-L1 checkpoint blockade therapy (CBI), radiotherapy and CBI combined with radiotherapy. A higher percentage of germinal center (GC) B cells in CBI combined with radiotherapy were detected. GCs are the hallmark of B-cell–mediated adaptive immunity so their presence is a sign of effectiveness of treatment [61]. Evaluation of IFNAR-1 blocking as a potential therapy was assessed by SC analysis of samples from HNSCC tumors generated by subcutaneous implantation of empty vector or shIfnar-1 MOC2-E6/E7 cells in mice. The study of cell population composition led to the conclusion that IFNAR-1 blockade in tumor cells expands stem-like effector T-cells, restricts MDSCs and reduces tumor burden, proving itself a valid therapeutical option [62]. Effect of ICB therapies anti-PD1 and anti-CTLA4 on TILs of different murine oral carcinoma (MOC) models that recapitulate HNSCC was evaluated using sc-RNAseq analysis. The study focused on the T cell compartment, specifically the CD8+ T cell sub-cluster. In samples showing the best response to the treatment, T cells had differentiated from näive/memory like to PD1+ activated states, unraveling tumor characteristics necessary for successful anti-PD1/anti-CTLA4 therapy [63]. To study the therapeutical effect of tRNA m7G methyltransferase METTL1 blockade, sc-RNAseq analysis of samples from Mettl1-knockout (KO) mice with 4NQO induced HNSCC was performed. Alterations in the immune landscape were shown to happen in Mettl1-KO mice, with a decrease in CD4+ T exhaustion and Tregs, making METTL1 blockade a suitable treatment strategy [64].
6. Outlook
The advancement of sc-RNAseq technology still faces significant challenges such as comparability of results. One of the main obstacles is the accurate characterization of cell types and states, given that cells exist in a dynamic state of continuous change, while sc-RNAseq captures only a snapshot of this process. Cell labelling is a critical step in sc-RNAseq analysis, as it directly impacts the obtained results. However, various studies employ their own definitions of cell types and states, making it difficult to integrate and consolidate the findings [11]. Even more, labelling can be limited by the current biological knowledge, finding cells in states that have not yet been defined [34]. The development of automated tools such as AI-driven labelling tools and a correct pruning and curation of the data represent a promising way to achieve standardization of results derived from sc-RNAseq analyses [65]. For instance, the scGate labelling tool effectively filters and annotates single-cell RNA sequencing data, enhancing the accuracy and reproducibility of cell type identification [66]. Nevertheless, the proliferation of tool development can also pose some challenges. With many tools emerging daily, each claiming to offer improved results, researchers face difficulty in selecting an appropriate analysis method. As a result, each study tends to have its own unique approach to analysis. Community efforts like "Single-Cell Best Practices" and "scRNA-tools" aim to combine and consolidate knowledge and provide general guidelines which could also contribute to standardization in the analysis process and improve result consistency [67,68]. Additional issues associated with sc-RNAseq include the limited depth of data, confined to the transcriptome dimension. Although in some cases the transcriptomic information is paired with cell surface markers, due to cells being sorted by fluorescence-activated cell sorting (FACS), the overall information is still quite limited. To overcome these limitations, the integration of complementary SC technologies, such as spatial technologies which trace transcripts to their localization within tissue or SC genomics or epigenetics, can provide a multi-layered insight alongside sc-RNAseq data [69].
Costs and manpower limitations are still relevant in the SC field. SC techniques are complex, needing qualified professionals, thereby adding to the already substantial expenses associated with the technology, including material costs [6]. A significant advantage of these technologies is that available public data can be used to address a wide range of research questions, potentially reducing project costs. Furthermore, computational methods can facilitate the reusability of the previously existing data. For example, the practice of pseudobulking, which involves converting sc-RNAseq data into bulk-RNAseq data, allows the use of existing tools established for bulk analysis in sc-RNAseq. Pseudobulking enables the extraction of multiple smaller and more specific datasets from a larger dataset and it offers the possibility of increasing the versatility of the data by generating subsets that are tailored to address distinct research questions or objectives [70]. SC data integration tools are also valuable for effectively utilizing existing data by combining information from different sources, patients, and studies. The reason for this is that these integration tools can assess for technical biases between samples while preserving the relevant biological bias necessary for accurate analysis [71,72].
One of the emerging hot topics in cancer research is the prevention stage, which remains relatively unexplored for HNSCC. sc-RNAseq) has the potential to significantly contribute to this field by providing a detailed understanding of the earliest cellular changes that occur during cancer initiation. For instance, sc-RNAseq could be used to identify and characterize premalignant lesions at the single-cell level, revealing key molecular pathways involved in the transition from normal to malignant states. Additionally, sc-RNAseq can further study and validate preventive strategies such as HPV vaccination [73] by analyzing the cellular and molecular impact of these strategies on the organism. By elucidating the mechanisms through which prevention strategies impede cancer development, sc-RNAseq can help refine and optimize prevention protocols, ultimately contributing to more effective cancer prevention strategies.
7. Conclusions
The use of sc-RNAseq technologies provides a high level of granularity, which can be crucial in deciphering cell-dependent processes. Due to the high heterogeneity of HNSCC, the application of SC technologies is quite promising, as was shown in this review. Further questions that could be address using this kind of methods include processes that can be traced back to each individual cell. An example of this is the effect of the HPV virus on cellular behavior and the relevance of cell type and state in this context. Similarly, the study of sex chromosome behavior, including their presence, expression, and function across different cell types and states, can uncover critical factors that contribute to the composition and interaction of the tumor microenvironment (TME), ultimately driving cancer initiation, establishment, and progression [74,75]. Therefore, to fully exploit the potential of sc-RNAseq technology, it is essential to assess the specific requirements of each research question and use complementary tools that provide deeper understanding of cellular processes. By facilitating a comprehensive characterization of distinct cellular behaviors, sc-RNAseq enables a deeper comprehension of HNSCC, thereby allowing an enhanced patient stratification and personalized treatment selection.
Data Availability Statement
No new data were created in this study. All the data reported in this review were found in original articles cited in the text.
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2018, 68, 394–424, . [CrossRef]
- Jawa, Y.; Yadav, P.; Gupta, S.; Mathan, S.V.; Pandey, J.; Saxena, A.K.; Kateriya, S.; Tiku, A.B.; Mondal, N.; Bhattacharya, J.; et al. Current Insights and Advancements in Head and Neck Cancer: Emerging Biomarkers and Therapeutics with Cues from Single Cell and 3D Model Omics Profiling. Front. Oncol. 2021, 11, . [CrossRef]
- Cai, Z.; Tang, B.; Chen, L.; Lei, W. Mast cell marker gene signature in head and neck squamous cell carcinoma. BMC Cancer 2022, 22, 1–16, . [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120, . [CrossRef]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713, . [CrossRef]
- Stampe, H.; Jakobsen, K.K.; Bendtsen, S.K.; Grønhøj, C.; von Buchwald, C. Systematic review on the current knowledge and use of single-cell RNA sequencing in head and neck cancer. APMIS 2021, 129, 619–625, . [CrossRef]
- Huang, Z.-D.; Liu, Z.-Z.; Liu, Y.-Y.; Fu, Y.-C.; Lin, L.-L.; Hu, C.; Gu, H.-Y.; Wei, R.-X. Molecular Subtypes Based on Cell Differentiation Trajectories in Head and Neck Squamous Cell Carcinoma: Differential Prognosis and Immunotherapeutic Responses. Front. Immunol. 2021, 12, 791621, . [CrossRef]
- Qi, Z.; Barrett, T.; Parikh, A.S.; Tirosh, I.; Puram, S.V. Single-cell sequencing and its applications in head and neck cancer. Oral Oncol. 2019, 99, 104441–104441, . [CrossRef]
- Cao, S.; Wang, J.R.; Ji, S.; Yang, P.; Dai, Y.; Guo, S.; Montierth, M.D.; Shen, J.P.; Zhao, X.; Chen, J.; et al. Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nat. Biotechnol. 2022, 40, 1624–1633, . [CrossRef]
- Choi, J.-H.; Lee, B.-S.; Jang, J.Y.; Lee, Y.S.; Kim, H.J.; Roh, J.; Shin, Y.S.; Woo, H.G.; Kim, C.-H. Single-cell transcriptome profiling of the stepwise progression of head and neck cancer. Nat. Commun. 2023, 14, 1–13, . [CrossRef]
- Yu, X.; Chen, Y.A.; Conejo-Garcia, J.R.; Chung, C.H.; Wang, X. Estimation of immune cell content in tumor using single-cell RNA-seq reference data. BMC Cancer 2019, 19, 1–11, . [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24, doi:10.1016/j.cell.2017.10.044.
- Puram, S.V.; Mints, M.; Pal, A.; Qi, Z.; Reeb, A.; Gelev, K.; Barrett, T.F.; Gerndt, S.; Liu, P.; Parikh, A.S.; et al. Cellular states are coupled to genomic and viral heterogeneity in HPV-related oropharyngeal carcinoma. Nat. Genet. 2023, 55, 640–650, . [CrossRef]
- Kürten, C.H.L.; Kulkarni, A.; Cillo, A.R.; Santos, P.M.; Roble, A.K.; Onkar, S.; Reeder, C.; Lang, S.; Chen, X.; Duvvuri, U.; et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat. Commun. 2021, 12, 1–16, . [CrossRef]
- Song, L.; Zhang, S.; Yu, S.; Ma, F.; Wang, B.; Zhang, C.; Sun, J.; Mao, X.; Wei, L. Cellular heterogeneity landscape in laryngeal squamous cell carcinoma. Int. J. Cancer 2020, 147, 2879–2890, . [CrossRef]
- Bill, R.; Wirapati, P.; Messemaker, M.; Roh, W.; Zitti, B.; Duval, F.; Kiss, M.; Park, J.C.; Saal, T.M.; Hoelzl, J.; et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 2023, 381, 515–524, . [CrossRef]
- Cillo, A.R.; Kürten, C.H.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9, . [CrossRef]
- Janjic, B.M.; Kulkarni, A.; Ferris, R.L.; Vujanovic, L.; Vujanovic, N.L. Human B Cells Mediate Innate Anti-Cancer Cytotoxicity Through Concurrent Engagement of Multiple TNF Superfamily Ligands. Front. Immunol. 2022, 13, 837842, . [CrossRef]
- Huynh, N.C.-N.; Huang, T.-T.; Nguyen, C.T.-K.; Lin, F.-K. Comprehensive Integrated Single-Cell Whole Transcriptome Analysis Revealed the p-EMT Tumor Cells—CAFs Communication in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 6470, . [CrossRef]
- Xiao, M.; Zhang, X.; Zhang, D.; Deng, S.; Zheng, A.; Du, F.; Shen, J.; Yue, L.; Yi, T.; Xiao, Z.; et al. Complex interaction and heterogeneity among cancer stem cells in head and neck squamous cell carcinoma revealed by single-cell sequencing. Front. Immunol. 2022, 13, 1050951, . [CrossRef]
- Chen, J.; Yang, J.; Li, H.; Yang, Z.; Zhang, X.; Li, X.; Wang, J.; Zhang, Y.; Chen, S.; Song, M. Single-Cell Transcriptomics Reveal the Intratumoral Landscape of Infiltrated T-Cell Subpopulations in Oral Squamous Cell Carcinoma. Mol. Oncol. 2021, 15, 866–886.
- Wieland, A.; Patel, M.R.; Cardenas, M.A.; Eberhardt, C.S.; Hudson, W.H.; Obeng, R.C.; Griffith, C.C.; Wang, X.; Chen, Z.G.; Kissick, H.T.; et al. Defining HPV-specific B cell responses in patients with head and neck cancer. Nature 2021, 597, 274–278, . [CrossRef]
- Eberhardt, C.S.; Kissick, H.T.; Patel, M.R.; Cardenas, M.A.; Prokhnevska, N.; Obeng, R.C.; Nasti, T.H.; Griffith, C.C.; Im, S.J.; Wang, X.; et al. Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature 2021, 597, 279–284, . [CrossRef]
- Dohmen, J.; Baranovskii, A.; Ronen, J.; Uyar, B.; Franke, V.; Akalin, A. Identifying tumor cells at the single-cell level using machine learning. Genome Biol. 2022, 23, 1–23, . [CrossRef]
- Yu, X.; Wang, Z.; Zeng, T. Essential gene expression pattern of head and neck squamous cell carcinoma revealed by tumor-specific expression rule based on single-cell RNA sequencing. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2020, 1866, 165791, . [CrossRef]
- Zhang, Q.; Wang, Y.; Xia, C.; Ding, L.; Pu, Y.; Hu, X.; Cai, H.; Hu, Q. Integrated analysis of single-cell RNA-seq and bulk RNA-seq reveals distinct cancer-associated fibroblasts in head and neck squamous cell carcinoma. Ann. Transl. Med. 2021, 9, 1017–1017, . [CrossRef]
- Meng, L.; Lu, H.; Li, Y.; Zhao, J.; He, S.; Wang, Z.; Shen, J.; Huang, H.; Xiao, J.; Sooranna, S.R.; et al. Human papillomavirus infection can alter the level of tumour stemness and T cell infiltration in patients with head and neck squamous cell carcinoma. Front. Immunol. 2022, 13, 1013542, . [CrossRef]
- Chen, S.M.Y.; Krinsky, A.L.; Woolaver, R.A.; Wang, X.; Chen, Z.; Wang, J.H. Tumor immune microenvironment in head and neck cancers. Mol. Carcinog. 2020, 59, 766–774, . [CrossRef]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52, . [CrossRef]
- Gameiro, S.F.; Evans, A.M.; Mymryk, J.S. The Tumor Immune Microenvironments of HPV+ and HPV- Head and Neck Cancers. WIREs Mech Dis 2022, 14, e1539.
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168, . [CrossRef]
- Huang, Y.; Liu, H.; Liu, X.; Li, N.; Bai, H.; Guo, C.; Xu, T.; Zhu, L.; Liu, C.; Xiao, J. The Chemokines Initiating and Maintaining Immune Hot Phenotype Are Prognostic in ICB of HNSCC. Front. Genet. 2022, 13, 820065, . [CrossRef]
- Li, S.; Wang, Y.; Sun, R.; Franceschi, D.; Pan, H.; Wei, C.; Ogbuehi, A.C.; Lethaus, B.; Savkovic, V.; Gaus, S.; et al. Single-Cell Transcriptome Analysis Reveals Different Immune Signatures in HPV- and HPV + Driven Human Head and Neck Squamous Cell Carcinoma. J. Immunol. Res. 2022, 2022, 1–19, . [CrossRef]
- Jiang, Y.; Zhang, S.; Tang, L.; Li, R.; Zhai, J.; Luo, S.; Peng, Y.; Chen, X.; Wei, L. Single-cell RNA sequencing reveals TCR+ macrophages in HPV-related head and neck squamous cell carcinoma. Front. Immunol. 2022, 13, 1030222, . [CrossRef]
- Liu, F.; Tang, L.; Li, Q.; Chen, L.; Pan, Y.; Yin, Z.; He, J.; Tian, J. Single-cell transcriptomics uncover the key ferroptosis regulators contribute to cancer progression in head and neck squamous cell carcinoma. Front. Mol. Biosci. 2022, 9, 962742, . [CrossRef]
- Jiang, X.; Ke, J.; Jia, L.; An, X.; Ma, H.; Li, Z.; Yuan, W. A novel cuproptosis-related gene signature of prognosis and immune microenvironment in head and neck squamous cell carcinoma cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 203–218, . [CrossRef]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2019, 47, D900–D908, . [CrossRef]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430, . [CrossRef]
- Zhang, Z.; Liu, R.; Jin, R.; Fan, Y.; Li, T.; Shuai, Y.; Li, X.; Wang, X.; Luo, J. Integrating Clinical and Genetic Analysis of Perineural Invasion in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 434, . [CrossRef]
- Liu, H.; Li, Y. Potential roles of Cornichon Family AMPA Receptor Auxiliary Protein 4 (CNIH4) in head and neck squamous cell carcinoma. Cancer Biomarkers 2022, 35, 439–450, . [CrossRef]
- Hegde, M.; Daimary, U.D.; Jose, S.; Sajeev, A.; Chinnathambi, A.; Alharbi, S.A.; Shakibaei, M.; Kunnumakkara, A.B. Differential Expression of Genes Regulating Store-operated Calcium Entry in Conjunction With Mitochondrial Dynamics as Potential Biomarkers for Cancer: A Single-Cell RNA Analysis. Front. Genet. 2022, 13, 866473, . [CrossRef]
- Messerschmidt, C.; Obermayer, B.; Klinghammer, K.; Ochsenreither, S.; Treue, D.; Stenzinger, A.; Glimm, H.; Fröhling, S.; Kindler, T.; Brandts, C.H.; et al. Distinct Immune Evasion in APOBEC-enriched, HPV-negative HNSCC. Int. J. Cancer 2020, 147, 2293–2302.
- Weng, J.; Fan, H.; Liu, H.; Tang, S.; Zheng, Y. YTHDC1 Promotes Stemness Maintenance and Malignant Progression in Head and Neck Squamous Cell Carcinoma. Stem Cells Int. 2022, 2022, 1–13, . [CrossRef]
- Hu, C.; Fan, J.; He, G.; Dong, C.; Zhou, S.; Zheng, Y. Signal peptidase complex catalytic subunit SEC11A upregulation is a biomarker of poor prognosis in patients with head and neck squamous cell carcinoma. PLOS ONE 2022, 17, e0269166, . [CrossRef]
- Carofino, B.L.; Dinshaw, K.M.; Ho, P.Y.; Cataisson, C.; Michalowski, A.M.; Ryscavage, A.; Alkhas, A.; Wong, N.W.; Koparde, V.; Yuspa, S.H. Head and neck squamous cancer progression is marked by CLIC4 attenuation in tumor epithelium and reciprocal stromal upregulation of miR-142-3p, a novel post-transcriptional regulator of CLIC4. Oncotarget 2019, 10, 7251–7275, . [CrossRef]
- Yorozu, A.; Sekiguchi, S.; Takasawa, A.; Okazaki, F.; Niinuma, T.; Kitajima, H.; Yamamoto, E.; Kai, M.; Toyota, M.; Hatanaka, Y.; et al. CXCL12 is expressed by skeletal muscle cells in tongue oral squamous cell carcinoma. Cancer Med. 2022, 12, 5953–5963, . [CrossRef]
- Zhang, S.; Zhang, W.; Zhang, J. 8-Gene signature related to CD8+ T cell infiltration by integrating single-cell and bulk RNA-sequencing in head and neck squamous cell carcinoma. Front. Genet. 2022, 13, 938611, . [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386, doi:10.1002/ijc.29210.
- Kang, X.; Chen, Y.; Yi, B.; Yan, X.; Jiang, C.; Chen, B.; Lu, L.; Sun, Y.; Shi, R. An integrative microenvironment approach for laryngeal carcinoma: the role of immune/methylation/autophagy signatures on disease clinical prognosis and single-cell genotypes. J. Cancer 2021, 12, 4148–4171, . [CrossRef]
- Chen, Q.; Chu, L.; Li, X.; Li, H.; Zhang, Y.; Cao, Q.; Zhuang, Q. Investigation of an FGFR-Signaling-Related Prognostic Model and Immune Landscape in Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 801715, . [CrossRef]
- Huang, C.; Liang, Y.; Dong, Y.; Huang, L.; Li, A.; Du, R.; Huang, H. Novel prognostic matrisome-related gene signature of head and neck squamous cell carcinoma. Front. Cell Dev. Biol. 2022, 10, 884590, . [CrossRef]
- Qi, Z.; Liu, Y.; Mints, M.; Mullins, R.; Sample, R.; Law, T.; Barrett, T.; Mazul, A.L.; Jackson, R.S.; Kang, S.Y.; et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1230, . [CrossRef]
- Yin, J.; Zheng, S.; He, X.; Huang, Y.; Hu, L.; Qin, F.; Zhong, L.; Li, S.; Hu, W.; Zhu, J. Identification of molecular classification and gene signature for predicting prognosis and immunotherapy response in HNSCC using cell differentiation trajectories. Sci. Rep. 2022, 12, 1–17, . [CrossRef]
- Han, P.; Tan, L.; Ouyang, Q.; Yu, P.; Shi, X.; Hu, J.; Wei, W.; Lu, Z.; Ji, Q.; Qu, N.; et al. Development and validation of a gene model predicting lymph node metastasis and prognosis of oral squamous cell carcinoma based on single-cell and bulk RNA-seq analysis. J. Oral Pathol. Med. 2022, 52, 389–401, . [CrossRef]
- Wang, J.; Sun, H.-C.; Cao, C.; Hu, J.-D.; Qian, J.; Jiang, T.; Jiang, W.-B.; Zhou, S.; Qiu, X.-W.; Wang, H.-L. Identification and validation of a novel signature based on cell-cell communication in head and neck squamous cell carcinoma by integrated analysis of single-cell transcriptome and bulk RNA-sequencing. Front. Oncol. 2023, 13, 1136729, . [CrossRef]
- Caudell, J.J.; Gillison, M.L.; Maghami, E.; Spencer, S.; Pfister, D.G.; Adkins, D.; Birkeland, A.C.; Brizel, D.M.; Busse, P.M.; Cmelak, A.J.; et al. NCCN Guidelines® Insights: Head and Neck Cancers, Version 1.2022. J. Natl. Compr. Canc. Netw. 2022, 20, 224–234.
- Pulte, D.; Brenner, H. Changes in Survival in Head and Neck Cancers in the Late 20th and Early 21st Century: A Period Analysis. Oncologist 2010, 15, 994–1001, doi:10.1634/theoncologist.2009-0289.
- Davis-Marcisak, E.F.; Sherman, T.D.; Orugunta, P.; Stein-O'Brien, G.L.; Puram, S.V.; Torres, E.T.R.; Hopkins, A.C.; Jaffee, E.M.; Favorov, A.V.; Afsari, B.; et al. Differential Variation Analysis Enables Detection of Tumor Heterogeneity Using Single-Cell RNA-Sequencing Data. Cancer Res. 2019, 79, 5102–5112, . [CrossRef]
- Obradovic, A.; Graves, D.; Korrer, M.; Wang, Y.; Roy, S.; Naveed, A.; Xu, Y.; Luginbuhl, A.; Curry, J.; Gibson, M.; et al. Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin. Cancer Res. 2022, 28, 2094–2109, . [CrossRef]
- Song, H.; Lou, C.; Ma, J.; Gong, Q.; Tian, Z.; You, Y.; Ren, G.; Guo, W.; Wang, Y.; He, K.; et al. Single-Cell Transcriptome Analysis Reveals Changes of Tumor Immune Microenvironment in Oral Squamous Cell Carcinoma After Chemotherapy. Front. Cell Dev. Biol. 2022, 10, 914120, . [CrossRef]
- Kim, S.S.; Shen, S.; Miyauchi, S.; Sanders, P.D.; Franiak-Pietryga, I.; Mell, L.; Gutkind, J.S.; Cohen, E.E.; Califano, J.A.; Sharabi, A.B. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin. Cancer Res. 2020, 26, 3345–3359, . [CrossRef]
- Gong, W.; Donnelly, C.R.; Heath, B.R.; Bellile, E.; Donnelly, L.A.; Taner, H.F.; Broses, L.; Brenner, J.C.; Chinn, S.B.; Ji, R.-R.; et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. OncoImmunology 2021, 10, 1997385, . [CrossRef]
- Zhou, L.; Zeng, Z.; Egloff, A.M.; Zhang, F.; Guo, F.; Campbell, K.M.; Du, P.; Fu, J.; Zolkind, P.; Ma, X.; et al. Checkpoint blockade-induced CD8+ T cell differentiation in head and neck cancer responders. J. Immunother. Cancer 2022, 10, e004034, . [CrossRef]
- Chen, J.; Li, K.; Chen, J.; Wang, X.; Ling, R.; Cheng, M.; Chen, Z.; Chen, F.; He, Q.; Li, S.; et al. Aberrant Translation Regulated by METTL1/WDR4-Mediated TRNA N7-Methylguanosine Modification Drives Head and Neck Squamous Cell Carcinoma Progression. Cancer Commun. 2022, 42, 223–244.
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172, . [CrossRef]
- Andreatta, M.; Berenstein, A.J.; Carmona, S.J. scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets. Bioinformatics 2022, 38, 2642–2644, . [CrossRef]
- Zappia, L.; Theis, F.J. Over 1000 tools reveal trends in the single-cell RNA-seq analysis landscape. Genome Biol. 2021, 22, 1–18, . [CrossRef]
- Heumos, L.; Schaar, A.C.; Lance, C.; Litinetskaya, A.; Drost, F.; Zappia, L.; Lücken, M.D.; Strobl, D.C.; Henao, J.; Curion, F.; et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 2023, 24, 550–572, . [CrossRef]
- Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-omics Analysis for Drug-Target Identification. Biomolecules 2024, 14, 692, . [CrossRef]
- Murphy, A.E.; Skene, N.G. A balanced measure shows superior performance of pseudobulk methods in single-cell RNA-sequencing analysis. Nat. Commun. 2022, 13, 1–4, . [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296, . [CrossRef]
- Andreatta, M.; Hérault, L.; Gueguen, P.; Gfeller, D.; Berenstein, A.J.; Carmona, S.J. Semi-supervised integration of single-cell transcriptomics data. Nat. Commun. 2024, 15, 1–13, . [CrossRef]
- Shapiro, G.K. HPV Vaccination: An Underused Strategy for the Prevention of Cancer. Curr. Oncol. 2022, 29, 3780–3792, . [CrossRef]
- Vermeulen, M.C.; Pearse, R.; Young-Pearse, T.; Mostafavi, S. Mosaic loss of Chromosome Y in aged human microglia. Genome Res. 2022, 32, 1795–1807, . [CrossRef]
- Hollows, R.; Wei, W.; Cazier, J.; Mehanna, H.; Parry, G.; Halford, G.; Murray, P. Association between loss of Y chromosome and poor prognosis in male head and neck squamous cell carcinoma. Head Neck 2019, 41, 993–1006, . [CrossRef]
Figure 1.
Illustration of strategies utilized in single-cell transcriptomics to address cancer heterogeneity. The figure highlights methods such as atlas construction, subclustering, cell type characterization, cell-cell communication analysis, differentiation trajectory reconstruction, and expression profiling of process-related genes. These approaches collectively enhance our understanding of the diverse cellular components and interactions within the tumor microenvironment.
Figure 1.
Illustration of strategies utilized in single-cell transcriptomics to address cancer heterogeneity. The figure highlights methods such as atlas construction, subclustering, cell type characterization, cell-cell communication analysis, differentiation trajectory reconstruction, and expression profiling of process-related genes. These approaches collectively enhance our understanding of the diverse cellular components and interactions within the tumor microenvironment.
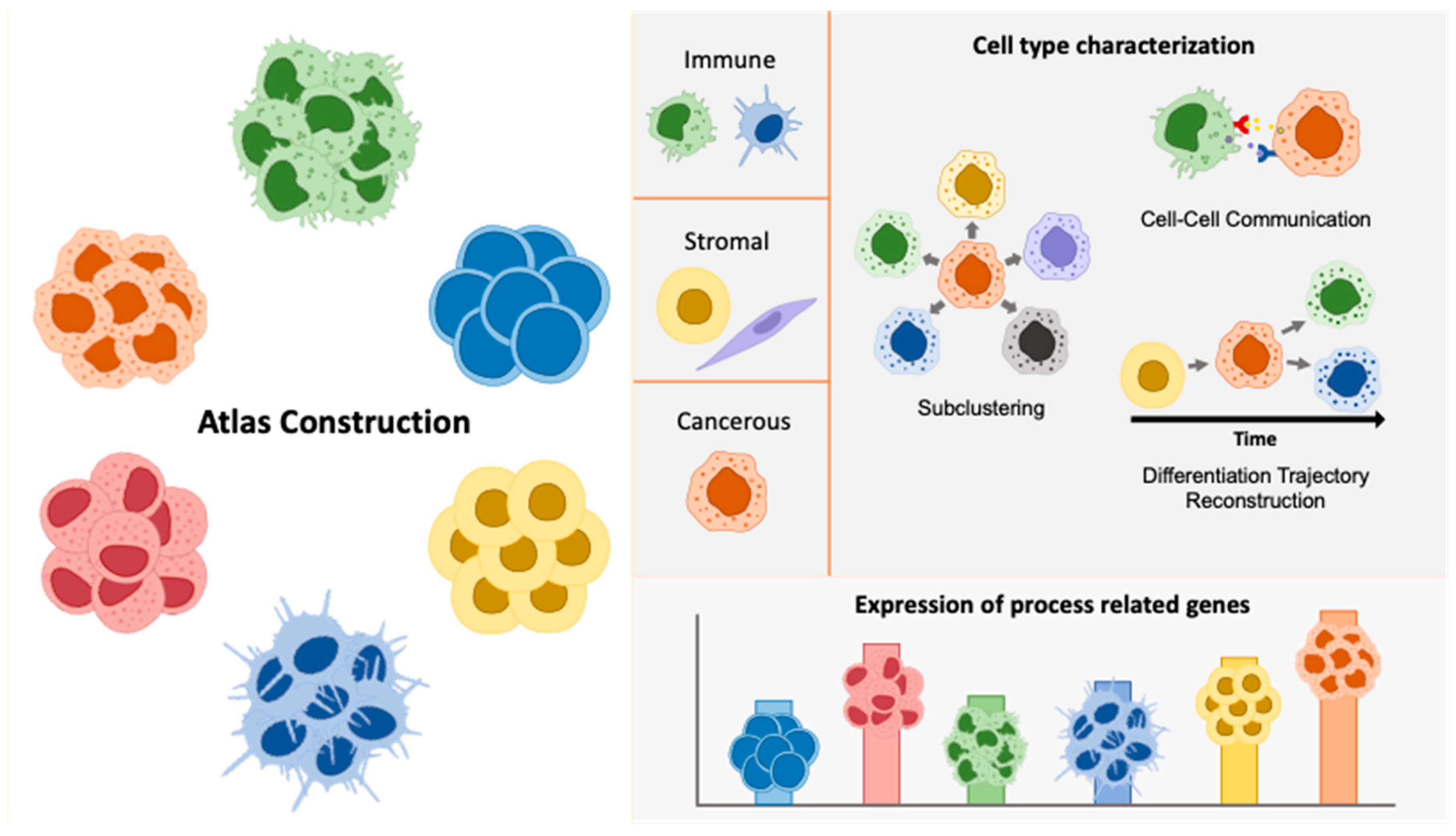
Figure 2.
Workflow illustrating the integration of single-cell RNA sequencing (sc-RNAseq) and bulk RNA sequencing (bulk-RNAseq) data for the development of prognostic gene signatures. This process can include gene selection, differential transcriptome (DTR) analysis, and validation using survival data. The integration allows for comprehensive multi-level data analysis, enhancing the utility and accuracy of prognostic models by leveraging detailed cellular insights from sc-RNAseq alongside broader bulk-RNAseq data.
Figure 2.
Workflow illustrating the integration of single-cell RNA sequencing (sc-RNAseq) and bulk RNA sequencing (bulk-RNAseq) data for the development of prognostic gene signatures. This process can include gene selection, differential transcriptome (DTR) analysis, and validation using survival data. The integration allows for comprehensive multi-level data analysis, enhancing the utility and accuracy of prognostic models by leveraging detailed cellular insights from sc-RNAseq alongside broader bulk-RNAseq data.
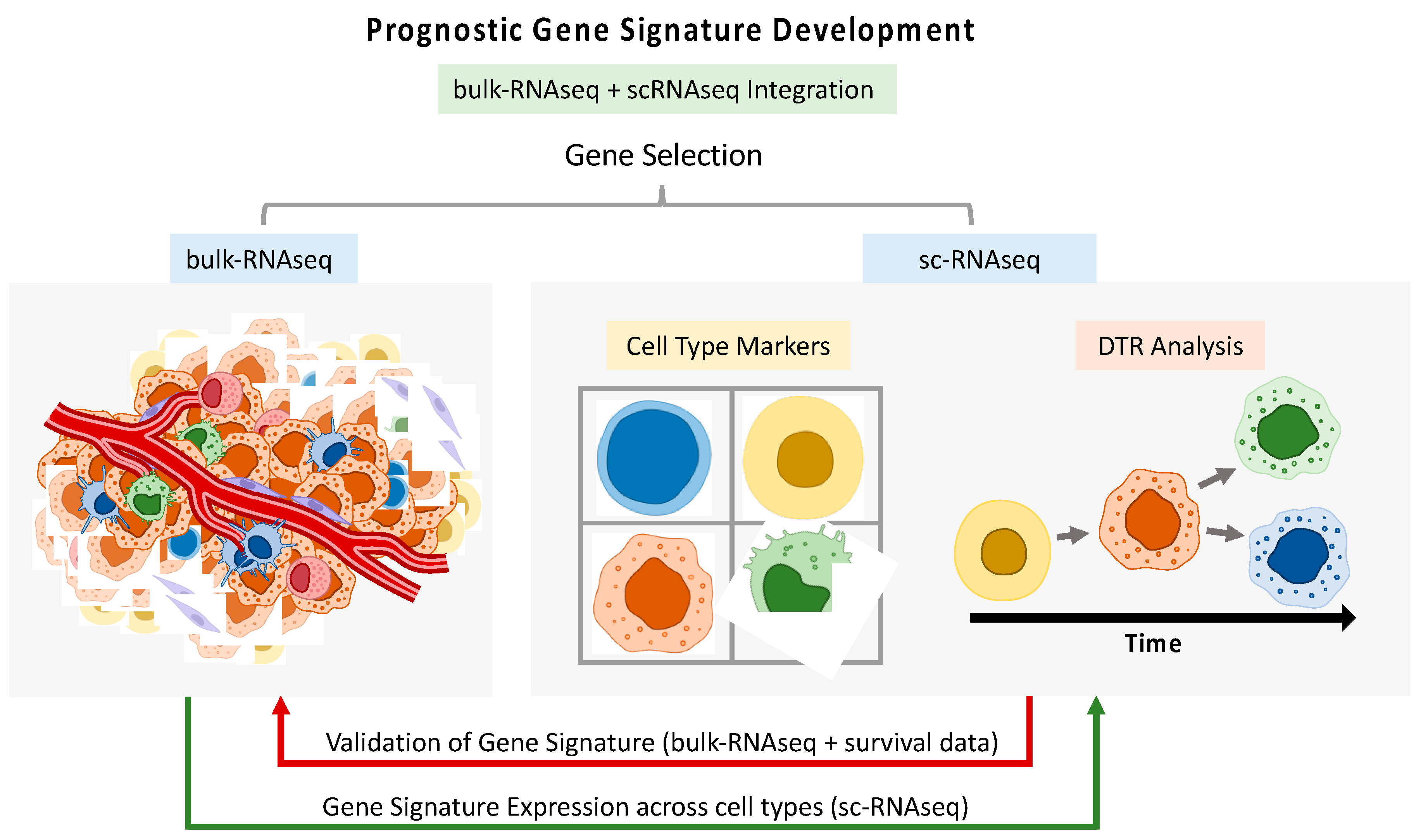

Table 1.
Summary of HNSCC scRNAseq datasets with human samples available in GEO. This table lists the datasets by accession number, publication year, authors, data type, tissue types analyzed, the number of samples, and the number of cells.
Table 1.
Summary of HNSCC scRNAseq datasets with human samples available in GEO. This table lists the datasets by accession number, publication year, authors, data type, tissue types analyzed, the number of samples, and the number of cells.
| Accession Number | Year | Publication | Data type | Tissue types | Sample Number | Cell Number |
|---|---|---|---|---|---|---|
| GSE234933 | 2023 | Bill R, Wirapati P, Messemaker M, Roh W et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 2023 Aug 4;381(6657):515-524 | scRNAseq | Primary tumor Local recurrence Distant metastasis |
52 | 87399 |
| GSE182227 | 2022 | Puram SV, Mints M, Pal A, Qi Z et al. Cellular states are coupled to genomic and viral heterogeneity in HPV-related oropharyngeal carcinoma. Nat Genet2023 Apr;55(4):640-650 | scRNAseq | Primary tumor Normal tissue |
24 | 70970 |
| GSE139324 | 2019 | Cillo AR, Kürten CHL, Tabib T, Qi Z et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020 Jan 14;52(1):183-199.e9. | scRNAseq | Peripheral/Intra-tumoral CD45+ populations | 63 | 131224 |
| GSE164690 | 2021 | Kürten CHL, Kulkarni A, Cillo AR, Santos PM et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat Commun 2021 Dec 17;12(1):7338 | scRNAseq | Primary tumor Pheripheral Blood Leucocytes |
51 | 134606 |
| GSE103322 | 2017 | Puram SV, Tirosh I, Parikh AS, Patel AP et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017 Dec 14;171(7):1611-1624.e24. | scRNAseq | Primary tumor | 18 | 5902 |
| GSE181919 | 2022 | Choi JH, Lee BS, Jang JY, Lee YS et al. Single-cell transcriptome profiling of the stepwise progression of head and neck cancer. Nat Commun 2023 Feb 24;14(1):1055. | scRNAseq | Primary tumor Normal tissue Leukoplakia Lymph-node metastasis |
37 | 54239 |
| GSE173647 | 2022 | --- | scRNAseq | Primary tumor | 2 | 13903 |
| GSE195832 | 2022 | Obradovic A, Graves D, Korrer M, Wang Y et al. Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin Cancer Res 2022 May 13;28(10):2094-2109. | scRNAseq | Primary tumor | 8 | 22906 |
| GSE140042 | 2021 | --- | scRNAseq | Primary tumor Lymph-node metastasis |
9 | --- |
| GSE200996 | 2022 | Luoma AM, Suo S, Wang Y, Gunasti L et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 2022 Aug 4;185(16):2918-2935.e29. | scRNAseq + scTCR | Peripheral/Intra-tumoral CD45+ populations | 204 | 74557 |
| GSE153559 | 2020 | Wieland A, Patel MR, Cardenas MA, Eberhardt CS et al. Defining HPV-specific B cell responses in patients with head and neck cancer. Nature 2021 Sep;597(7875):274-278. | scRNAseq | B cells Primary tumor Lymph-node metastasis Pheriphery |
7 | 8271 |
| GSE180268 | 2021 | Eberhardt CS, Kissick HT, Patel MR, Cardenas MA et al. Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature 2021 Sep;597(7875):279-284. | scRNAseq | TILs Primary tumor/Lymph-node metastasis | 39 | --- |
| GSE162025 | 2020 | Liu Y, He S, Wang XL, Peng W et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat Commun2021 Feb 2;12(1):741 | scRNAseq + scTCR | Primary tumor/Pheripheral Blood Leucocytes | 40 | 176447 |
| GSE150321 | 2020 | Song L, Zhang S, Yu S, Ma F et al. Cellular heterogeneity landscape in laryngeal squamous cell carcinoma. Int J Cancer 2020 Nov 15;147(10):2879-2890. | scRNAseq | Primary tumor | 2 | 12985 |
| GSE213047 | 2022 | Lin M, Sade-Feldman M, Wirth L, Lawrence MS et al. Single-cell transcriptomic profiling for inferring tumor origin and mechanisms of therapeutic resistance. NPJ Precis Oncol 2022 Oct 10;6(1):71. | scRNAseq | Primary tumor/Normal tissue/Lymph-node metastasis | 3 | 11470 |
| GSE172577 | 2021 | Peng Y, Xiao L, Rong H, Ou Z et al. Single-cell profiling of tumor-infiltrating TCF1/TCF7(+) T cells reveals a T lymphocyte subset associated with tertiary lymphoid structures/organs and a superior prognosis in oral cancer. Oral Oncol2021 Aug;119:105348. | scRNAseq | Primary tumor | 6 | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



