
Submitted:
20 July 2024
Posted:
24 July 2024
You are already at the latest version
Abstract
The generation of forensic DNA profiles consisting of single nucleotide polymorphisms (SNPs) is now being facilitated by wider adoption of next-generation sequencing (NGS) methods in casework laboratories. At the same time, and in part because of this advance, there is an intense focus on the generation of SNP profiles from evidentiary specimens for so-called forensic or investigative genetic genealogy (FGG or IGG) applications. However, FGG methods are constrained by the algorithms for genealogical database searches, which were designed for use with single-source profiles, and the fact that many forensic samples are mixtures. To enable the use of two-person mixtures for FGG, we developed a workflow, MixDeR, for the deconvolution of mixed SNP profiles. MixDeR, a flexible and easy to use R package and Shiny app, processes ForenSeq Kintelligence® (QIAGEN, Inc.) SNP genotyping results and directs deconvolution of the profiles in EuroForMix (EFM). MixDeR then filters the EFM outputs to produce inferred single-source genotypes in reports formatted for use with GEDmatch® PRO. An optional MixDeR output includes metrics that assist with testing and validation of the workflow. As the Shiny app provides a graphical user interface and the software is designed to be run offline, MixDeR should be suitable for use by any laboratory developing FGG capabilities, no matter their bioinformatic resources or expertise.
Keywords:
mixtures
; SNPs
; forensic genetic genealogy (FGG)
; investigative genetic genealogy (IGG)
; Kintelligence
; software
1. Introduction
It has long been recognized that there are some general advantages to the use of panels of single nucleotide polymorphisms (SNPs) for human forensic genetic applications, including their relative abundance in the genome, their lower susceptibility to genotyping failures resulting from DNA degradation, and the absence of common PCR artifacts encountered in short tandem repeat (STR) typing such as stutter [1,2,3,4]. Since the mid-2000s, SNP genotyping has been used in the forensic context to generate identity-informative data, inform biogeographic ancestry estimations, and predict externally visible characteristics [5,6,7,8,9,10,11,12,13,14,15,16], and is now widely accepted as a viable DNA-based approach [17,18]. Yet, prior to the last decade, the methodologies used for forensic SNP genotyping generally limited the number of markers that could be simultaneously typed to a few dozen or less [19].
In recent years, some forensic laboratories have implemented or have begun validating next-generation sequencing (NGS) technologies due to their technological and/or power of discrimination advantages for multiple marker types, including STRs, mitochondrial DNA, and microhaplotypes [20,21,22,23,24,25,26,27,28]. Compared to older forensic assays that often relied upon capillary electrophoresis, the use of NGS platforms has also significantly expanded the number of informative SNP loci that can be genotyped concurrently, whether by targeted methods such PCR-based assays or by whole genome sequencing. Though microarrays have been used for many years to genotype SNPs, and at present remain the most cost-effective and high-throughput approach for genotyping dense SNP panels when high quantities of pristine DNA are available, array-based systems are not well-suited for many evidentiary samples [29,30,31]. NGS, however, can be used to simultaneously genotype hundreds to millions of SNPs from even very low DNA input quantities and poor-quality templates, using the same instrumentation that forensic laboratories are already implementing for other marker types.
A new approach to generating investigative leads in criminal and missing persons casework, termed forensic genetic genealogy or investigative genetic genealogy (FGG or IGG), utilizes DNA profiles comprised of many thousands of SNPs [32,33,34]. The process entails developing a large-scale SNP profile from a forensic specimen, a search of the profile against one or more public DNA databases, and traditional genealogical research and investigative work to evaluate the database search results. The databases contain information derived from direct-to-consumer (DTC) SNP testing kits and were built to facilitate DNA-based identification of biological relatives in personal genealogy research. However, as some of the databases can be used to perform the same type of kinship analysis using a SNP profile generated from an evidentiary sample, the approach has now been used to assist in several hundred forensic cases [35,36].
The ForenSeq® Kintelligence Kit (QIAGEN, Inc.), a PCR-based assay that targets 10,230 SNPs, was the first commercial forensic kit designed specifically for FGG applications. In practice, SNP profiles generated using the Kintelligence kit and the associated ForenSeq® Universal Analysis Software (UAS; QIAGEN, Inc.) are searched against a portion of the publicly accessible GEDmatch® database, using the law enforcement-specific GEDmatch® PRO portal [37,38].
Though DNA mixtures are common in forensic casework, the database search algorithms utilized for FGG, including those implemented in GEDmatch® PRO, are meant for single-source SNP profiles. To enable the use of mixtures for FGG applications, we developed and performed initial testing of an approach for deconvolution of Kintelligence SNP profiles generated from mixed DNA samples [39]. Here we describe an expanded and comprehensive workflow for SNP mixture deconvolution implemented in a new open-source R package, MixDeR (Mixture Deconvolution in R). The MixDeR workflow starts from Kintelligence SNP profiles, which are pre-processed before deconvolution using the open-source probabilistic genotyping software EuroForMix (EFM; [40]). Following MixDeR-directed mixture deconvolution from the EFM command line using automatically formatted files, EFM allele probability outputs are filtered to maximize accuracy of the inferred genotype(s), which are in turn formatted in the manner of GEDmatch® PRO reports. Finally, MixDeR implements a user interface through a Shiny app, allowing even those with no coding experience to easily and efficiently run the SNP mixture deconvolution workflow.
2. Materials and Methods
MixDeR is an R package that exists as both a Shiny app and a command line tool. Several R packages must be installed prior to installing MixDeR, including dplyr, euroformix, glue, prompter, readxl, rlang, shiny, shinyFiles, shinyjs, tibble, and tidyr. EuroForMix version 4.0 or later is required. It is highly recommended to use RStudio for MixDeR. MixDeR is open source and the source code is available on GitHub (https://www.github.com/bioforensics/mixder).
The MixDeR workflow is summarized in Figure 1. The process begins from Kintelligence Sample Reports exported directly from the UAS. Once input files containing the requisite information and/or data are properly formatted, mixture deconvolution is performed using EFM. Following deconvolution, either validation metrics are calculated or GEDmatch® PRO-formatted reports are created. The following sections describe in greater detail the important components, features, and modules of MixDeR.
2.1. Input Files
MixDeR requires the use of a sample manifest, in which each row contains either a single sample identifier (ID), or a single sample ID plus a replicate sample ID. The manifest enables the workflow to be run on multiple mixture samples, one after another. This batch processing significantly reduces hands-on time and guarantees consistency of analysis across samples in the same batch. Currently, MixDeR’s functionality is limited to processing two-person mixtures.
EFM requires specific formats for mixture (evidence) profiles and reference profiles [40]. MixDeR provides the flexibility to input Kintelligence mixture data in either 1) CSV files pre-formatted for EFM, or 2) Sample Reports exported directly from the UAS (version 2.5 or earlier). If UAS Sample Reports are imported, MixDeR converts the data to the correct EFM format. For proper EFM deconvolution, MixDeR removes the X and Y chromosome loci from the Kintelligence SNP set. Thus, X and Y chromosome loci should also be removed from CSV files manually created for use with the MixDeR workflow.
Given the large number of markers in the Kintelligence panel, there is significant inter-locus variation in SNP read counts (see Figure S1 and [38]), presumably due to differences in amplification efficiency. During initial development of the MixDeR workflow, this variation proved challenging at the EFM deconvolution step. To account for the variation and improve deconvolution outcomes, a method was developed to group SNPs of similar read counts into separate datasets (bins), deconvolute the divided datasets through EFM separately, then combine the resulting genotypes after deconvolution [39]. MixDeR will perform these steps (division of SNPs into bins, independent deconvolution of each SNP set in EFM, and compilation of the results into a single profile) automatically using a user-defined value for the number of bins.
If performing a conditioned deconvolution or calculating validation metrics, reference genotypes are required. The reference genotypes may be in the form of either individual Sample Reports exported from the UAS or compiled into a single CSV file. In the Shiny app, the user specifies a folder containing the reference genotypes. If a folder has been specified and “Conditioned Analysis” is selected in the app, a dropdown menu will populate with the sample IDs of the provided references; the user may then select one or more references on which to condition the deconvolution. If more than one reference is selected, separate conditioned deconvolutions will be performed using each reference.
2.2. Performing Mixture Deconvolution
EFM requires additional information to perform mixture deconvolution, including analytical thresholds (ATs) and allele frequency data [40]. For Kintelligence data analysis in the UAS, the minimum read depth is 10 reads, and a user-specified AT is subsequently applied as a percentage of the total read count for a locus [41]. As a result, the effective AT applied during UAS analysis may differ for each SNP. However, in EFM an AT must be specified as a single value (read count) per locus. While working with smaller SNP panels would not be as cumbersome, manually generating the effective AT used for each Kintelligence locus during UAS analysis and specifying these in a config file for EFM is both error-prone and impractical. For this reason, MixDeR was programmed to automatically calculate the effective AT per locus when static (single value read count) and dynamic (percentage-based) ATs are specified by the user in MixDeR. For each locus, MixDeR will utilize the higher of the two read count values produced by the static and dynamic ATs to create the effective AT values table for EFM analysis.
Allele frequency data are also required for mixture deconvolution in EFM. MixDeR provides the user the option to select from two pre-loaded allele frequency datasets: (1) allele frequencies of all populations from the 1000 Genomes phase 3 dataset [42] and (2) allele frequencies of all populations from the gnomAD version 4 dataset [43]. In addition, MixDeR can also utilize custom allele frequency data. However, EFM requires allele frequencies to be in a specific format: each column contains a single SNP locus, while the rows contain the corresponding frequencies for each allele. Given the large number of SNPs in the Kintelligence panel, creating this file manually is cumbersome. While MixDeR will accept an EFM-formatted allele frequency file, it can also accept a CSV file with each row containing frequency information for one locus (i.e., reference and alternate alleles with their corresponding frequencies) and will properly format the frequencies for use in EFM.
EFM permits combined analysis of replicate samples to improve mixture deconvolution results [40]. Accordingly, MixDeR was programmed to permit analysis of up to two samples at once. When a replicate sample is specified in the sample manifest, MixDeR creates the SNP sets for the first sample, then ensures the same SNPs are contained in each set for the corresponding replicate. Further, the effective AT values table accounts for the variation in ATs by sample by using the highest AT applied for each SNP (as required for proper analysis in EFM). While using replicates for mixture deconvolution can be extremely useful, creating the appropriate data for the EFM analysis can be unmanageable for large SNP datasets. MixDeR is able to easily accomplish the required efforts in a fast, automated, and consistent manner.
2.3. Inferring SNP Genotypes from EFM Results
EFM outputs potential genotypes for each unknown contributor, along with associated posterior probabilities [40]. While there are multiple options included in the EFM deconvolution output, MixDeR utilizes the allele probabilities (from the EFM “All Marginal (A)” table) to infer the SNP genotypes for each unknown contributor.
To generate the inferred genotypes, MixDeR applies probability thresholds separately to each allele, such that 1) any SNP with an allele 1 probability below the allele 1 probability threshold is removed from the final dataset, 2) any SNP with an allele 2 probability below the allele 2 probability threshold is reported as homozygous for allele 1 (analogous to dropping the allele 2 call), and 3) any SNP with allele 1 and allele 2 probabilities at or above the thresholds is reported as heterozygous. Examples are displayed in Figure 2. In the Shiny app, the user specifies either a single probability threshold each for allele 1 and allele 2 (for GEDmatch® PRO-formatted report creation, section 2.4), or a range of probability thresholds for each allele (for calculation of validation metrics, section 2.5). The thresholds can range between 0 and 1 in increments of 0.01.
Additionally, MixDeR includes an option to specify a minimum number of SNPs in the inferred genotypes. When the application of the user-specific probability thresholds does not result in the minimum number of SNPs, a genotype is instead inferred using the minimum number of SNPs with the highest allele 1 probabilities, and the specified allele 2 probability threshold(s) are used to determine whether each locus is homozygous or heterozygous.
2.4. Creating GEDmatch® PRO-Formatted Reports
MixDeR uses the three settings specified in section 2.3 (allele 1 probability threshold, allele 2 probability threshold, and minimum number of SNPs) to infer the single-source SNP genotypes from the EFM output. Using GRCh37 coordinates, the inferred genotypes are then formatted to match the GEDmatch® PRO Reports typically exported directly from the UAS when the “GEDmatch PRO Report” option is selected in MixDeR.
2.5. Calculating Validation Metrics
To assist with the testing and validation of the workflow implemented in MixDeR, the software can use the user-specified probability thresholds and minimum number of SNPs described in section 2.3 to calculate metrics helpful for evaluating inferred genotypes (“Calculate Metrics” option in MixDeR). These metrics include the total number of SNPs in an inferred genotype, the accuracy of the inferred genotype (i.e., the percentage of SNPs matching a provided known genotype), and the percent heterozygosity of the inferred genotype. When the “Calculate Metrics” option is selected, MixDeR creates two final output files: one file contains metrics for each combination of allele 1 and allele 2 probability thresholds in the ranges specified by the user, and a second file contains metrics for each allele 2 probability threshold in combination with the minimum number of SNPs. These validation metrics are instrumental during workflow testing to determine the best threshold combinations for maximizing genotype accuracy while at the same time including enough SNPs for downstream genealogical database searches.
2.6. Verification of Software Functions and Generation of Example Results
To verify that the MixDeR workflow performs as expected, we used Kintelligence data from a 1:5 mixture previously developed and described in [44], along with reference profiles for each contributor. In the 1:5 mixture, sample NA24143 (HG004; from source NIST RM 8392 (National Institute of Standards and Technology, Gaithersburg, MD) as described in [44]) was the major contributor and sample NA24631 (HG005; obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research (Camden, NJ) as described in [44]) was the minor contributor. The mixture was amplified in duplicate using the ForenSeq® Kintelligence Kit with 1 ng total DNA as the starting input, and a Kintelligence profile was generated using a 1.5% AT for UAS analysis. The files used for the testing are provided with this paper as Supplementary Material.
Using MixDeR, data from the mixture Kintelligence Sample Reports were divided into either one or 10 SNP sets, and EFM mixture deconvolution (using version 4.0.7) was performed using the 1000 Genomes general population allele frequency data. The mixture profiles were analyzed both individually and as replicates using three different methods: 1) unconditioned, 2) conditioned on HG004, and 3) conditioned on HG005. Validation metrics were calculated using allele 1 probability threshold ranges of 0 to 1, allele 2 probability threshold ranges of 0 to 1, and 6,000 as the minimum number of SNPs. Finally, the GEDmatch® PRO reports were generated using the MixDeR default settings, allowing for verification of the inferred genotypes and report format.
3. Results and Discussion
3.1. MixDeR Workflow
MixDeR was built around the use of EFM for the deconvolution of Kintelligence SNP mixtures. A few prior studies have successfully utilized EFM to calculate likelihood ratios for known contributors to SNP mixtures [45,46], and even to deconvolute mixtures profiles using either a combination of both STRs and SNPs [47] or a small set of SNPs only [48]. To date, though, there have been no papers describing the use of EFM for deconvolution of mixture profiles consisting of a large number of SNPs into individual contributor genotypes, either for FGG applications or other purposes. As a result, the use of EFM for the deconvolution of Kintelligence mixture profiles comprised of more than 10,000 SNPs required extensive up-front work and testing to produce a viable workflow. Key components of the workflow include 1) the use of EFM output allele probabilities rather than genotype probabilities, 2) the separation of the large panel of Kintelligence SNPs into sets, 3) the use of independent allele 1 and allele 2 probability thresholds, and 4) the application of a minimum number of SNPs.
3.1.1. Use of EFM Allele Probabilities Rather Than Genotype Probabilities
The EFM mixture deconvolution output includes four different tables of posterior probabilities; three provide per-locus genotype probabilities, while the fourth includes per-locus probabilities for each allele [40]. During initial testing with smaller SNP panels, use of the top genotype probabilities was sufficient for inference of accurate genotypes in some circumstances. However, once testing was extended to the significantly larger Kintelligence SNP set, accuracy of the inferred genotypes decreased, and a genotype probability threshold could not be identified that maximized inferred genotype accuracy across different mixtures. However, by switching to use of the allele probabilities (the “All Marginal (A)” table output by EFM), accuracy of the inferred Kintelligence genotypes improved, and the outcomes for different mixtures were more consistent [unpublished data]. Use of the allele probabilities enables application of separate thresholds for allele 1 and allele 2, and consequently more control over the inferred SNP genotypes. The use of the EFM-output allele probabilities is a fixed feature in MixDeR.
3.1.2. Separation of SNPs into Sets
As described above (see Section 2.1), the significant inter-locus variation in SNP read counts with the Kintelligence assay led us to develop a method within MixDeR to group SNPs of similar read counts into separate datasets for EFM deconvolution. Figure 3 demonstrates the significant improvement in allele 1 probabilities when SNPs are ordered by read depth and then divided into 10 smaller sets (or ~1,006 SNPs per set). An analysis of a 1:5 mixture (described in section 2.6) conditioned on the major contributor resulted in allele 1 probabilities for the minor contributor ranging from 0.7561 to 1.0 with a median of 0.9976 when the SNPs were separated into 10 sets. Though the range for the allele 1 probabilities was nearly identical when the SNPs were deconvoluted in a single set (0.7567-1.0), the median was notably lower at 0.9143. The difference was more pronounced when comparing the probabilities from an unconditioned analysis of the same mixture. While the allele 1 probabilities had a wider range for 10 sets of SNPs than a single set of SNPs (0.5238-1.0 and 0.7932-1.0, respectively), the median allele 1 probability for the 10 sets (0.9979) was substantially higher than the median probability resulting from deconvolution of the SNPs in a single set (0.8685). Results obtained when using the default minimum number of SNPs are presented in Figure S2.
More importantly, binning the SNPs into 10 sets for deconvolution resulted in substantial improvements in the accuracy of the inferred genotypes for unknown contributors, regardless of the type of deconvolution performed (conditioned or unconditioned) and whether the unknown was a major or minor contributor (Table 1). For example, when the default MixDeR settings (see section 3.1.3) were applied to the analysis described above, the accuracy of the inferred minor contributor genotype was 62% for the unconditioned deconvolution and 73% for the conditioned deconvolution when SNPs were grouped into a single set. However, when the SNPs were binned into 10 smaller sets, the accuracy of the inferred minor contributor genotypes improved to 83% (unconditioned) and 84% (conditioned). An even more dramatic improvement in accuracy was observed for the major contributor when the deconvolution was unconditioned. When the SNPs were combined in a single bin, accuracy of the inferred genotype was just 62%; however binning the SNPs into 10 sets resulted in an inferred genotype accuracy of nearly 99%.
While MixDeR allows the user to specify the number of SNP sets and will divide the SNPs equally among the sets, it will also detect files created by the user within the specified input folder, assuming the files are named and formatted properly. This feature allows the user to manually create the files for EFM deconvolution using any binning strategy.
3.1.3. Use of Independent Allele 1 and Allele 2 Probability Thresholds
In the EFM “All Marginal (A)” table, the allele 1 probability is always the higher of the two probabilities for a given SNP locus (unless the allele probabilities are equal or only one allele is inferred, i.e. called homozygous). The allele 1 probability threshold used to filter the EFM deconvolution output in MixDeR is positively correlated with genotype accuracy: the higher the allele 1 probability threshold is set, the more likely it is that the inferred genotype will be correct. This can be observed in Figure 4, which displays results obtained for the same 1:5 mixture analysis described previously and shows genotype accuracy improving as the allele 1 probability threshold increases from 0.00 to 0.99. For this reason, the allele 1 probability threshold is used by MixDeR to determine whether to include a locus in the final inferred genotype: any locus with an allele 1 probability below the user-specified threshold is excluded.
Subsequently, MixDeR uses the allele 2 probability threshold to determine whether to keep the EFM-output allele 2 for a locus (if above the threshold) or to drop it and infer a homozygous result for the locus. For example, consider a scenario in which the EFM output includes a SNP locus with potential alleles A (allele 1) and C (allele 2), with an allele 2 posterior probability of 0.6000. If an allele 2 probability threshold of 0.50 was applied in MixDeR, the inferred genotype for the locus would be A,C; if instead a threshold of 0.80 was applied, the inferred genotype would be A,A. Further examples are shown in Figure 2.
Given how MixDeR applies the thresholds for each allele, the relationship between allele 2 probability thresholds and inferred genotype accuracy is not the same as is observed for the allele 1 probability thresholds. As Figure 4 shows, accuracy of the inferred genotype is lowest when using low or high allele 2 probability thresholds, and highest in the middle of the allele 2 probability threshold range.
Based on testing performed during the development of MixDeR, the default allele 1 and allele 2 probability thresholds in the software are 0.99 and 0.60, respectively. However, as previously described, a user may set different allele 1 and allele 2 probability thresholds, or a range of thresholds for each allele for testing and validation of the workflow.
3.1.4. Use of a Minimum Number of SNPs
As described above, the higher the allele 1 probability threshold used, the greater the accuracy of the inferred genotype. However, as the allele 1 probability threshold increases, more loci are eliminated, and thus fewer SNPs are present in the final inferred genotype. This is, in fact, the reason that a default value of 0.99 was selected for the allele 1 probability threshold in MixDeR: using a value of 1.00 results in the highest accuracy but eliminates too many (and depending on the mixture profile, potentially all) SNPs, rendering the threshold essentially useless. Though a threshold of 0.99 proved robust across a broad range of mixture types during the development of MixDeR, it is also the case that for some mixtures and/or contributors, the number of SNPs retained will be low when the default threshold is used.
As downstream analyses such as genealogical database searching may require a minimum number of SNPs in the profile, or a specified number of SNPs may be deemed optimal (for maximizing true positive and minimizing false positive matches to relatives, for instance), MixDeR was programmed to permit users to set a minimum number of SNPs. When a minimum number of SNPs, N, is set, MixDeR first uses the allele 1 and allele 2 probability thresholds in the normal manner to produce the inferred genotype, and then considers the number of SNPs in the inferred genotype. If the number of SNPs in the inferred genotype does not meet or exceed the value of N, MixDeR then generates a new inferred genotype using the N SNPs with the highest allele 1 probabilities (using the allele 2 probability threshold to determine whether each locus is homozygous or heterozygous as normal). In the latter instance, the GEDmatch® PRO-formatted report will be generated using the specified minimum number of SNPs. However, the two validation metrics files output by the software will always consist of (1) all combinations of allele 1 and allele 2 probability thresholds within the ranges specified by the user, and (2) the specified minimum number of SNPs combined with all allele 2 probability thresholds specified by the user. Based on the upload criteria for GEDmatch® PRO [38], the software defaults to a minimum of 6,000 SNPs.
3.2. MixDeR GUI
A central feature of MixDeR is the graphical user interface (GUI) provided via the Shiny app. Although EFM is commonly run via a GUI, using the EFM GUI to perform the multiple deconvolutions that may be required for analysis of large SNP panels and/or many samples is tedious and requires significant hands-on time. Conversely, while the command line version of EFM allows for less hands-on time and provides the option for batch processing, use of the command line necessitates a nontrivial amount of experience with R coding. For these reasons, the MixDeR package includes a user interface to perform batched mixture deconvolution using custom parameters for each run (e.g., different loci and different AT values per locus) without requiring any R experience on the part of the user.
The files required for EFM mixture deconvolution - including evidence and reference profiles, configuration files that include the specific SNPs and locus-specific ATs utilized for analysis, and allele frequency files - are challenging to manually curate for large panels of SNPs. Within the MixDeR GUI, a user can direct automatic formatting of many of the EFM-required files. By incorporating these formatting functions into MixDeR, the mixture deconvolution step of the workflow is performed more efficiently and consistently, significantly reducing the possibility of user error.
Additionally, the MixDeR GUI enables a user to set and automatically apply the desired allele probability thresholds and minimum number of SNPs to the EFM mixture deconvolution output, and to choose the MixDeR process type. For deconvolution of a Kintelligence evidence profile, a user can select the option to generate a GEDmatch® PRO-formatted report. Importantly, though, for testing and/or validation of the workflow, a user can select the option to generate metrics that facilitate evaluation of deconvolution outcomes for Kintelligence mixture profiles developed from known genotypes. The compiled tables produced via this MixDeR option enable the user to easily compare results obtained:
- using different allele probability threshold combinations;
- using different minimum SNP numbers;
- from mixture profiles developed using differing DNA inputs;
- from mixtures with differing contributor ratios;
- from conditioned versus unconditioned deconvolutions; and
- from single versus replicate profiles.
As the EFM deconvolution step can be time consuming, MixDeR was programmed to allow loading of deconvoluted data from previous EFM runs. This feature enables a user to skip the file preparation and mixture deconvolution steps of the workflow, and instead specify only the allele filtering options and MixDeR output file type (GEDmatch® PRO-formatted report or validation metrics files). Of note, all files used by or generated by MixDeR are maintained in the folders specified by the user, enabling traceability of the workflow steps and outcomes, and manual review of all files if necessary.
3.3. Software Verification and Example Results
The software testing described in section 2.6 produced all expected outputs, including 1) evidence and reference input files for EFM with SNPs grouped into one or 10 sets, 2) EFM allele probability tables, 3) validation metrics files, and 4) files formatted in the manner of GEDmatch® PRO reports. All output files are included with this paper as Supplementary Material.
Additionally, to verify the functionality of the MixDeR option to use deconvoluted data from a previous EFM run, the option was used to generate a second set of validation metrics files and GEDmatch® PRO reports from the EFM output files produced during the first round of software testing. The two sets of MixDeR output files were identical when compared.
The software verification testing included using the default settings in MixDeR to process single or replicate Kintelligence profiles developed from a 2-contributor mixed sample with a 1:5 mixture ratio using 1 ng of total DNA for PCR. The deconvolutions were first conditioned on the major contributor to infer the genotype of the minor contributor, then also conditioned on the minor contributor to infer the genotype of the major contributor. Lastly, unconditioned deconvolutions were performed, inferring the genotypes of both the major and minor contributors. Key metrics from each deconvolution are summarized in Table 2.
Overall, the inferred genotypes for the major contributor had the highest accuracy and heterozygosity while also including the most SNPs, regardless of the deconvolution type. Using replicate profiles in the deconvolution was most beneficial for inference of the minor contributor genotype, in both the conditioned (genotype accuracy improvement from 84% to 90%) and unconditioned (improvement from 83% to 90%) scenarios. Given the already high accuracy of the genotypes inferred for the major contributor using single profiles (>98%), the use of replicates resulted in only minor improvements in genotype accuracy.
3.4. Challenges
A few issues were encountered while performing SNP mixture deconvolution in EFM that can affect the inferred genotypes:
- If a mixture ratio of exactly 1:1 is predicted by EFM, the alleles and allele probabilities for both contributors in the EFM output will be the same. However, even when the EFM-predicted mixture ratio was not exactly 1:1, we encountered instances in which the alleles and allele probabilities for both contributors in the EFM output were identical.
- In the EFM “All Marginal (A)” output, the contributor 1 allele probabilities should always be higher than contributor 2 allele probabilities. However, we encountered instances in which the opposite occurred (the contributor 2 allele probabilities were higher than the contributor 1 allele probabilities).
MixDeR was programmed to detect these problematic scenarios when they appear in the EFM results. When MixDeR finds that one of the issues has occurred, the software automatically re-runs the EFM deconvolution step on the offending SNP set until either 1) the output from the deconvolution appears as expected (i.e., the output for the two contributors is not identical, and the contributor 1 allele probabilities are higher than the contributor 2 allele probabilities), or 2) the SNP set has been run ten separate times. If the SNP set has been run ten times but still does not appear as expected, MixDeR excludes the offending SNP set from the final inferred genotype.
3.5. Limitations
The MixDeR workflow for SNP mixture deconvolution has a few limitations. At present, MixDeR can only analyze two-person mixtures, and only when the mixture ratio is not exactly 1:1. While the effect of assuming a two-contributor mixture when the actual number of contributors is higher has not been evaluated for SNP mixture deconvolution, underestimating the number of contributors to SNP mixtures has been shown to result in significantly underestimated LRs for true contributors [46]. We hypothesize that underestimating the number of contributors would also affect deconvolution results, likely leading to lower posterior probabilities and reduced accuracy of the inferred genotypes. The user is strongly encouraged to be highly confident in their assessment of the number of contributors for the analyzed mixtures. Moving forward, new tools developed specifically for use with SNPs, such as the recent Demixtify [49], may assist in the estimation of the number of contributors to SNP mixtures.
As MixDeR was designed to process SNPs from the ForenSeq® Kintelligence Kit, at present most files (e.g., allele frequency files, GEDmatch® PRO-formatted reports) pertain specifically to the Kintelligence SNP set. Additionally, MixDeR utilizes a set method for dividing the SNPs into individual datasets prior to mixture deconvolution; while the user specifies how many datasets to create, the method behind the partitioning is fixed. Since MixDeR is open source, it could theoretically be adapted by users to address some of these limitations (e.g., to process three-person mixtures or perform mixture deconvolution on a different set of SNPs). Future development of MixDeR may address one or more of these limitations as well.
The last step in the MixDeR workflow generates an inferred single-source SNP profile formatted for upload to GEDmatch® PRO. However, GEDmatch® PRO requires a hash for direct upload to the portal, ordinarily added to the report header during generation in the UAS (and presumably intended to prevent users from uploading mocked Kintelligence profiles that were not generated using the ForenSeq® Kintelligence Kit and the UAS). Given that MixDeR is not able to replicate this hash, the MixDeR-generated report cannot be uploaded directly to GEDmatch® PRO. Thus, at present, coordination with QIAGEN is required to upload a MixDeR-generated report to GEDmatch® PRO.
3.6. Current and Future Work
A comprehensive evaluation of several aspects of the mixture deconvolution workflow, performed using MixDeR, is currently underway. These include but are not limited to the SNP binning strategy, use of different allele frequency datasets, and performance expectations for different mixture types with regards to contributor ratios and DNA quantities. A significant additional component of the study is the ability to detect genetic relatives of the inferred single source SNP profiles in the GEDmatch® database via GEDmatch® PRO queries. The authors invite users of the Kintelligence assay to contact the corresponding author if they are interested in making contributions to this study in the form of Kintelligence mixture data.
In the future, MixDeR will continue to be developed to meet new software and analysis needs, including as updated versions of R, the required R packages, the UAS, or EFM are deployed that affect the functional or performance aspects of MixDeR. Updated versions of MixDeR with comprehensive documentation of changes will be made available at https://www.github.com/bioforensics/mixder.
4. Conclusions
MixDeR provides forensic laboratories with a workflow for performing deconvolution of Kintelligence SNP profiles developed from mixed DNA samples, along with a user-friendly GUI that simplifies and automates the workflow steps. In addition to providing an inferred single-source evidence file output designed for use with the GEDmatch® PRO portal, the software includes features to assist users in testing and validating the workflow on their own laboratory-developed Kintelligence mixture data. As MixDeR is an open-source, publicly available R package and Shiny app that requires little coding expertise, it should have utility for any laboratory that is interested in using the ForenSeq® Kintelligence Kit for FGG applications. Additionally, MixDeR is designed to be used offline, making it suitable for use by laboratories that may prefer to avoid employing a web-based application. Current and future versions of MixDeR can be downloaded from https://www.github.com/bioforensics/mixder.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Inter-locus balance; Figure S2: Distribution of allele 1 probabilities using a minimum number of SNPs; Supplementary Data.
Author Contributions
Conceptualization, Rebecca Mitchell and Rebecca Just; Data curation, Rebecca Mitchell; Formal analysis, Rebecca Mitchell and Rebecca Just; Funding acquisition, Rebecca Mitchell, Michelle Peck, Erin Gorden and Rebecca Just; Investigation, Rebecca Mitchell, Michelle Peck, Erin Gorden and Rebecca Just; Methodology, Rebecca Mitchell, Michelle Peck and Rebecca Just; Project administration, Rebecca Mitchell; Resources, Rebecca Mitchell, Michelle Peck, Erin Gorden and Rebecca Just; Software, Rebecca Mitchell; Supervision, Rebecca Mitchell and Rebecca Just; Validation, Rebecca Mitchell, Michelle Peck, Erin Gorden and Rebecca Just; Visualization, Rebecca Mitchell and Rebecca Just; Writing – original draft, Rebecca Mitchell and Rebecca Just; Writing – review & editing, Rebecca Mitchell, Michelle Peck, Erin Gorden and Rebecca Just.
Funding
This work was funded under Agreement No. HSHQDC-15-C-00064 awarded to Battelle National Biodefense Institute (BNBI) by the Department of Homeland Security (DHS) Science and Technology Directorate (S&T) for the management and operation of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of DHS or the U.S. Government. DHS does not endorse any products or commercial services mentioned in this presentation. In no event shall DHS, BNBI or NBACC have any responsibility or liability for any use, misuse, inability to use, or reliance upon the information contained herein. In addition, no warranty of fitness for a particular purpose, merchantability, accuracy or adequacy is provided regarding the contents of this document. Notice: This manuscript has been authored by Battelle National Biodefense Institute, LLC under Contract No. HSHQDC-15-C-00064 with DHS. The US Government (USG) retains and the publisher, by accepting the article for publication, acknowledges that the USG retains a non-exclusive, paid up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for USG purposes.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and the study protocol was reviewed and approved by WCG IRB (protocol #20201322, approved 29 May 2020). All research involving living individuals, their data, or their biospecimens was conducted in compliance with the Federal Policy for the Protection of Human Subjects (The Common Rule, codified for DHS as 6 CFR 46), DHS Management Directive 026-04, and any other applicable statutory requirements. Research involving human subjects has only been initiated after the following has occurred: the need for IRB review has been determined, IRB approval has been obtained as applicable, and DHS Compliance Assurance Program Office certification or concurrence has been issued.
Informed Consent Statement
The study used two purchased DNA extracts: NIST RM 8392 from the National Institute of Standards and Technology, and NA24631from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research. Informed consent was obtained from the subjects as part of the original sample collection.
Data Availability Statement
The data used for all analyses are described in section 2.6 and are available in the Supplementary Material (“Input” folder in the Supplementary Data file). Output files from the analyses, including SNP sets, EFM outputs, validation metrics, and GEDmatch® PRO reports, are also available in the Supplementary Material (“Output” folder in the Supplementary Data file). MixDeR is publicly available on GitHub at https://www.github.com/bioforensics/mixder.
Acknowledgments
The authors thank Claire Levy for assistance with data generation, and Sarah Radecke, Cassidy Robinhold, Erin Romos, June Snedecor, and Kathryn Stephens for discussion. Additionally, the authors thank Lisa Gall for critical project support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gill, P. An assessment of the utility of single nucleotide polymorphisms (SNPs) for forensic purposes. Int. J. Leg. Med. 2001, 114, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B. SNP typing strategies. Forensic Science International 2004, 2, 139–142. [Google Scholar]
- Sobrino, B.; Carracedo, A. SNP Typing in Forensic Genetics: A Review. in Methods in Molecular Biology, 2005, 297, 107–126. [Google Scholar] [CrossRef]
- Kayser, M.; de Knijff, P. Improving human forensics through advances in genetics, genomics and molecular biology. Nature Reviews 2011, 12, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.J.; Phillips, C.; Børsting, C.; Balogh, K.; Bogus, M.; Fondevila, M.; Harrison, C.D.; Musgrave-Brown, E.; Salas, A.; Syndercombe-Court, D.; et al. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis 2006, 27, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Grigorenko, E.L.; Kajuna, S.L.; Karoma, N.J.; Kungulilo, S.; Kim, J.-J.; Lu, R.-B.; Odunsi, A.; et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci. Int. 2006, 164, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Fang, R.; Ballard, D.; Fondevila, M.; Harrison, C.; Hyland, F.; Musgrave-Brown, E.; Proff, C.; Ramos-Luis, E.; Sobrino, B.; et al. Evaluation of the Genplex SNP typing system and a 49plex forensic marker panel. Forensic Sci. Int. Genet. 2007, 1, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Salas, A.; Sánchez, J.; Fondevila, M.; Gómez-Tato, A.; Álvarez-Dios, J.; Calaza, M.; de Cal, M.C.; Ballard, D.; Lareu, M.; et al. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Sci. Int. Genet. 2007, 1, 273–280. [Google Scholar] [CrossRef]
- Tomas, C.; Stangegaard, M.; Børsting, C.; Hansen, A.J.; Morling, N.; The SNPforID Consortium. Typing of 48 autosomal SNPs and amelogenin with GenPlex SNP genotyping system in forensic genetics. Forensic Sci. Int. Genet. 2008, 3, 1–6. [Google Scholar] [CrossRef]
- Sanchez, J.; Børsting, C.; Balogh, K.; Berger, B.; Bogus, M.; Butler, J.; Carracedo, A.; Court, D.S.; Dixon, L.; Filipović, B.; et al. Forensic typing of autosomal SNPs with a 29 SNP-multiplex—Results of a collaborative EDNAP exercise. Forensic Sci. Int. Genet. 2008, 2, 176–183. [Google Scholar] [CrossRef]
- Fang, R.; Pakstis, A.J.; Hyland, F.; Wang, D.; Shewale, J.; Kidd, J.R.; Kidd, K.K.; Furtado, M.R. Multiplexed SNP detection panels for human identification. Forensic Science International: Genetics Supplement Series 2009, 2, 538–539. [Google Scholar] [CrossRef]
- Kosoy, R.; Nassir, R.; Tian, C.; White, P.A.; Butler, L.M.; Silva, G.; Kittles, R.; Alarcon-Riquelme, M.E.; Gregersen, P.K.; Belmont, J.W.; et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum. Mutat. 2009, 30, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; van Duijn, K.; Vingerling, J.R.; Hofman, A.; Uitterlinden, A.G.; Janssens, A.C.J.; Kayser, M. Eye color and the prediction of complex phenotypes from genotypes. Curr. Biol. 2009, 19, R192–R193. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, A.J.; Speed, W.C.; Fang, R.; Hyland, F.C.L.; Furtado, M.R.; Kidd, J.R.; Kidd, K.K. SNPs for a universal individual identification panel. Hum. Genet. 2009, 127, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Liu, F.; Ballantyne, K.N.; van Oven, M.; Lao, O.; Kayser, M. IrisPlex: A sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci. Int. Genet. 2010, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Branicki, W.; Liu, F.; van Duijn, K.; Draus-Barini, J.; Pośpiech, E.; Walsh, S.; Kupiec, T.; Wojas-Pelc, A.; Kayser, M. Model-based prediction of human hair color using DNA variants. Hum. Genet. 2011, 129, 443–454. [Google Scholar] [CrossRef]
- Kayser, M. Forensic DNA Phenotyping: Predicting human appearance from crime scene material for investigative purposes. Forensic Sci. Int. Genet. 2015, 18, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 2015, 18, 49–65. [Google Scholar] [CrossRef]
- Daniel, R.; Santos, C.; Phillips, C.; Fondevila, M.; van Oorschot, R.; Carracedo, A.; Lareu, M.; McNevin, D. A SNaPshot of next generation sequencing for forensic SNP analysis. Forensic Sci. Int. Genet. 2014, 14, 50–60. [Google Scholar] [CrossRef]
- Köcher, S.; Müller, P.; Berger, B.; Bodner, M.; Parson, W.; Roewer, L.; Willuweit, S. Inter-laboratory validation study of the ForenSeq™ DNA Signature Prep Kit. Forensic Sci. Int. Genet. 2018, 36, 77–85. [Google Scholar] [CrossRef]
- Hollard, C.; Ausset, L.; Chantrel, Y.; Jullien, S.; Clot, M.; Faivre, M.; Suzanne, E.; Pène, L.; Laurent, F.-X. Automation and developmental validation of the ForenSeq™ DNA Signature Preparation kit for high-throughput analysis in forensic laboratories. Forensic Sci. Int. Genet. 2019, 40, 37–45. [Google Scholar] [CrossRef]
- Brandhagen, M.D.; Just, R.S.; Irwin, J.A. Validation of NGS for mitochondrial DNA casework at the FBI Laboratory. Forensic Sci. Int. Genet. 2020, 44, 102151. [Google Scholar] [CrossRef]
- Silvery, J.; Ganschow, S.; Wiegand, P.; Tiemann, C. Developmental validation of the monSTR identity panel, a forensic STR multiplex assay for massively parallel sequencing. Forensic Sci. Int. Genet. 2020, 46, 102236. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, J.C.; Amory, C.; Lagacé, R.; Roth, C.; Parson, W.; Budowle, B. Developmental Validation of a MPS Workflow with a PCR-Based Short Amplicon Whole Mitochondrial Genome Panel. Genes 2020, 11, 1345. [Google Scholar] [CrossRef]
- Cuenca, D.; Battaglia, J.; Halsing, M.; Sheehan, S. Mitochondrial Sequencing of Missing Persons DNA Casework by Implementing Thermo Fisher’s Precision ID mtDNA Whole Genome Assay. Genes 2020, 11, 1303. [Google Scholar] [CrossRef] [PubMed]
- Gandotra, N.; Speed, W.C.; Qin, W.; Tang, Y.; Pakstis, A.J.; Kidd, K.K.; Scharfe, C. Validation of novel forensic DNA markers using multiplex microhaplotype sequencing. Forensic Sci. Int. Genet. 2020, 47, 102275–102275. [Google Scholar] [CrossRef]
- Senst, A.; Caliebe, A.; Scheurer, E.; Schulz, I. Validation and beyond: Next generation sequencing of forensic casework samples including challenging tissue samples from altered human corpses using the MiSeq FGx system. J. Forensic Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, B.; Matrai, N.; Barany, G.; Tomory, G.; Heinrich, A.; Egyed, B. Internal Validation of the Precision Id Globalfiler Ngs Str Panel V2 Kit with Locus-Specific Analytical Threshold, and with Special Regard to Mixtures and Low Template DNA Detection. [CrossRef]
- de Vries, J.H.; Kling, D.; Vidaki, A.; Arp, P.; Kalamara, V.; Verbiest, M.M.; Piniewska-Róg, D.; Parsons, T.J.; Uitterlinden, A.G.; Kayser, M. Impact of SNP microarray analysis of compromised DNA on kinship classification success in the context of investigative genetic genealogy. Forensic Sci. Int. Genet. 2022, 56, 102625. [Google Scholar] [CrossRef] [PubMed]
- Davawala, A.; Stock, A.; Spiden, M.; Daniel, R.; McBain, J.; Hartman, D. Forensic genetic genealogy using microarrays for the identification of human remains: The need for good quality samples—A pilot study. Forensic Sci. Int. 2022, 334, 111242. [Google Scholar] [CrossRef]
- Russell, D.A.; Gorden, E.M.; Peck, M.A.; Neal, C.M.; Heaton, M.C.; Bouchet, J.L.; Koeppel, A.F.; Ciuzio, E.; Turner, S.D.; Reedy, C.R. Developmental validation of the illumina infinium assay using the global screening array (GSA) on the iScan system for use in forensic laboratories. Forensic Genomics 2023, 3, 15–24. [Google Scholar] [CrossRef]
- Phillis, C. The Golden State Killer investigation and the nascent field of forensic genealogy. Forensic Science International: Genetics 2018, 36, 186–188. [Google Scholar] [CrossRef]
- Wickenheiser, R.A. Forensic genealogy, bioethics and the Golden State Killer case. Forensic Sci. Int. Synerg. 2019, 1, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Kling, D.; Phillips, C.; Kennett, D.; Tillmar, A. Investigative genetic genealogy: Current methods, knowledge and practice. Forensic Sci. Int. Genet. 2021, 52, 102474. [Google Scholar] [CrossRef]
- Dowdeswell, T.L. Forensic genetic genealogy: A profile of cases solved. Forensic Sci. Int. Genet. 2022, 58, 102679. [Google Scholar] [CrossRef]
- Dowdeswell, T.L. Forensic genetic genealogy project v. 2022. Mendeley Data, vol. 1, 2023.
- Snedecor, J.; Fennell, T.; Stadick, S.; Homer, N.; Antunes, J.; Stephens, K.; Holt, C. Fast and accurate kinship estimation using sparse SNPs in relatively large database searches. Forensic Sci. Int. Genet. 2022, 61, 102769. [Google Scholar] [CrossRef]
- Antunes, J.; Walichiewicz, P.; Forouzmand, E.; Barta, R.; Didier, M.; Han, Y.; Perez, J.; Snedecor, J.; Zlatkov, C.; Padmabandu, G.; et al. Developmental validation of the ForenSeq® Kintelligence kit, MiSeq FGx® sequencing system and ForenSeq Universal Analysis Software. Forensic Science International: Genetics 2024, 71, 103055. [Google Scholar] [CrossRef]
- Mitchell, R.; Enke, S.; Eskey, K.; Ferguson, T.; Just, R. A method to enable forensic genetic genealogy investigations from DNA mixtures. Forensic Sci. Int. Genet. Suppl. Ser. 2022, 8, 159–161. [Google Scholar] [CrossRef]
- Bleka. ; Storvik, G.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2015, 21, 35–44. [Google Scholar] [CrossRef]
- Verogen. Universal Analysis Software v2.0 and above - Reference Guide. 2021.
- Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Peck, M.A.; Koeppel, A.F.; Gorden, E.M.; Bouchet, J.L.; Heaton, M.C.; Russell, D.A.; Reedy, C.R.; Neal, C.M.; Turner, S.D. Internal Validation of the ForenSeq Kintelligence Kit for Application to Forensic Genetic Genealogy. Forensic Genom. 2022, 2, 103–114. [Google Scholar] [CrossRef]
- Bleka, O.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W.; Gill, P. Open source software EuroForMix can be used to analyse complex SNP mixtures. Forensic Sci. Int. Genet. 2017, 31, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Just, R.; Parson, W.; Phillips, C.; Bleka, O. Interpretation of complex DNA profiles generated by massively parallel sequencing. In Forensic Practitioner's Guide to the Interpretation of Complex DNA Mixtures; Elsevier, 2020; pp. 419–451.
- Hwa, H.-L.; Wu, M.-Y.; Chung, W.-C.; Ko, T.-M.; Lin, C.-P.; Yin, H.-I.; Lee, T.-T.; Lee, J.C.-I. Massively parallel sequencing analysis of nondegraded and degraded DNA mixtures using the ForenSeq™ system in combination with EuroForMix software. Int. J. Leg. Med. 2018, 133, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Hwa, H.-L.; Wu, M.-Y.; Hsu, P.-M.; Yin, H.-I.; Lee, T.-T.; Su, C.-W. DNA mixture analyses of autosomal single nucleotide polymorphisms for individual identification using droplet digital polymerase-chain reaction and massively parallel sequencing in combination with EuroFormix. Aust. J. Forensic Sci. 2023, 1–14. [Google Scholar] [CrossRef]
- Woerner, A.E.; Crysup, B.; King, J.L.; Novroski, N.M.; Coble, M.D. Mixture detection with Demixtify. Forensic Science International: Genetics 2024, 69, 102980. [Google Scholar] [CrossRef]
Figure 1.
Overview of MixDeR workflow.
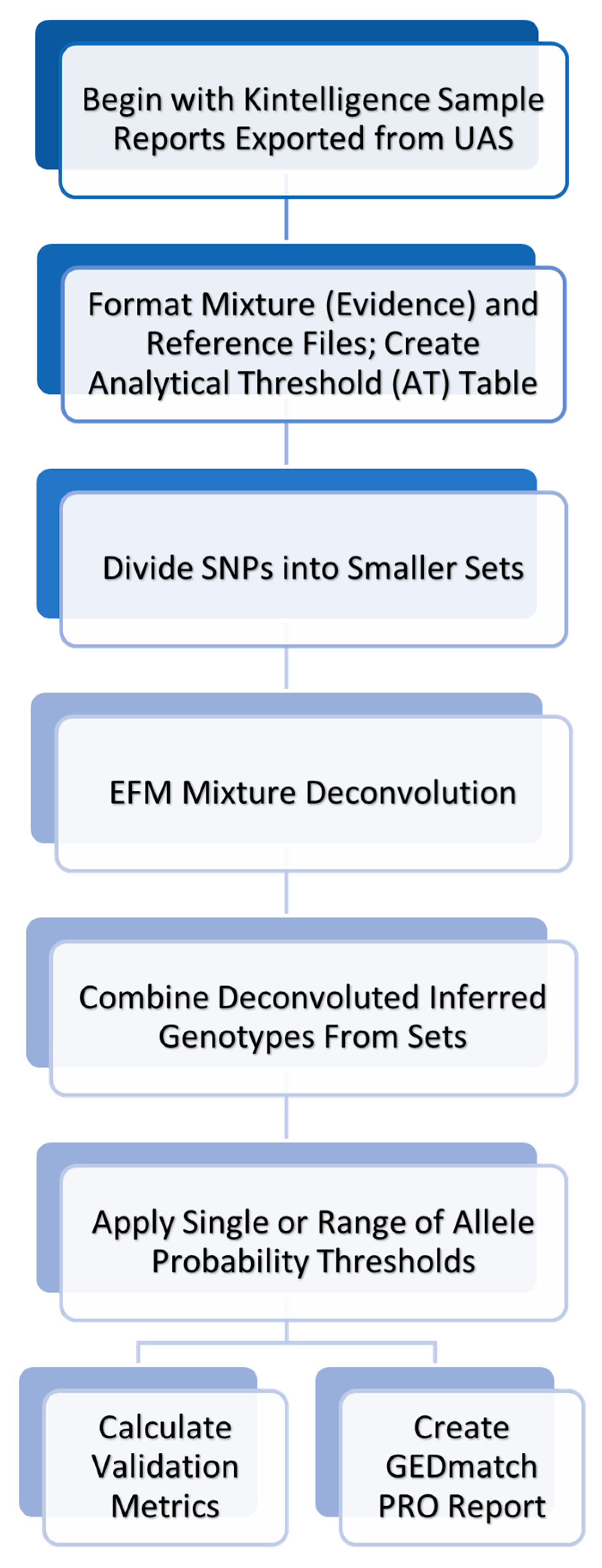
Figure 2.
MixDeR inference of genotypes using allele probability thresholds. Starting from allele probabilities in the EFM All Marginal (A) table, loci and alleles are included in or excluded from an inferred genotype based on user-specified allele 1 (A1) and allele 2 (A2) probability thresholds.
Figure 2.
MixDeR inference of genotypes using allele probability thresholds. Starting from allele probabilities in the EFM All Marginal (A) table, loci and alleles are included in or excluded from an inferred genotype based on user-specified allele 1 (A1) and allele 2 (A2) probability thresholds.
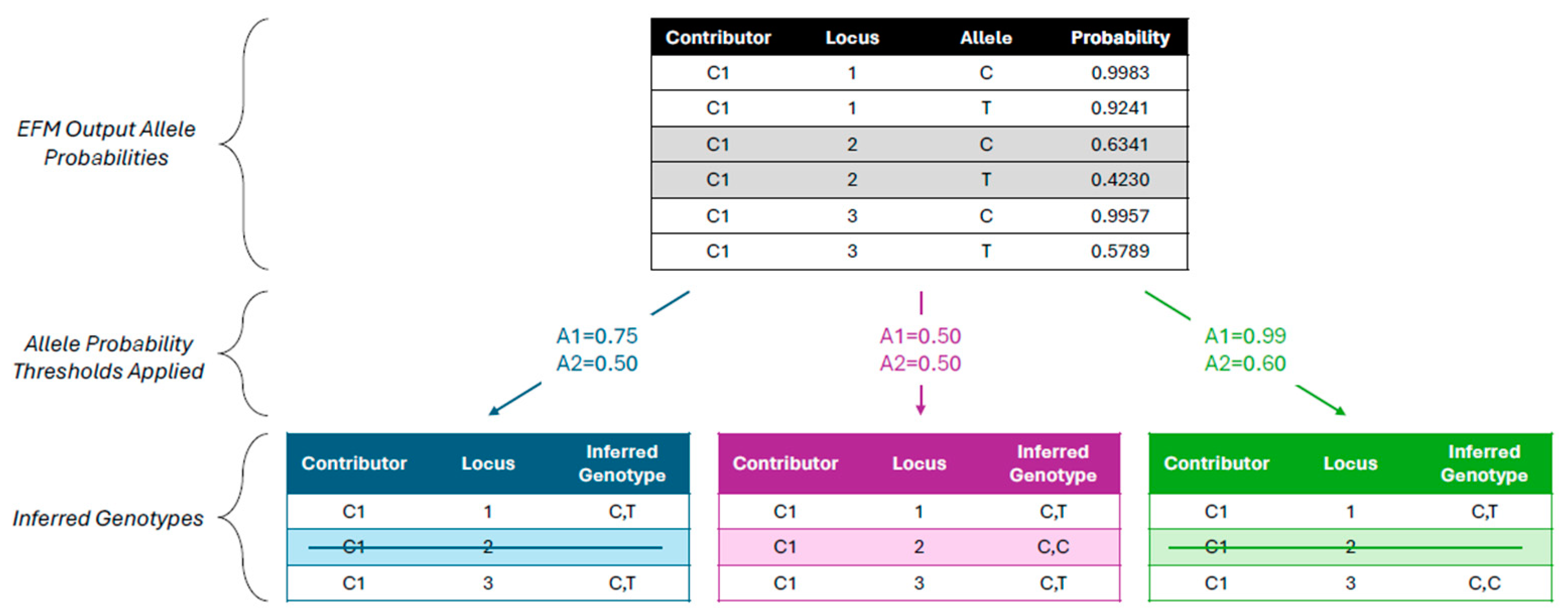
Figure 3.
Density plots showing the distribution of allele 1 probabilities using two different binning strategies. The allele 1 probabilities for the minor contributor of a 1:5 mixture were assessed after: (A) a conditioned deconvolution analysis and (B) an unconditioned deconvolution analysis, when the SNPs were divided into 10 bins (blue) and 1 bin (red). The dotted lines represent the median allele 1 probability for each dataset.
Figure 3.
Density plots showing the distribution of allele 1 probabilities using two different binning strategies. The allele 1 probabilities for the minor contributor of a 1:5 mixture were assessed after: (A) a conditioned deconvolution analysis and (B) an unconditioned deconvolution analysis, when the SNPs were divided into 10 bins (blue) and 1 bin (red). The dotted lines represent the median allele 1 probability for each dataset.
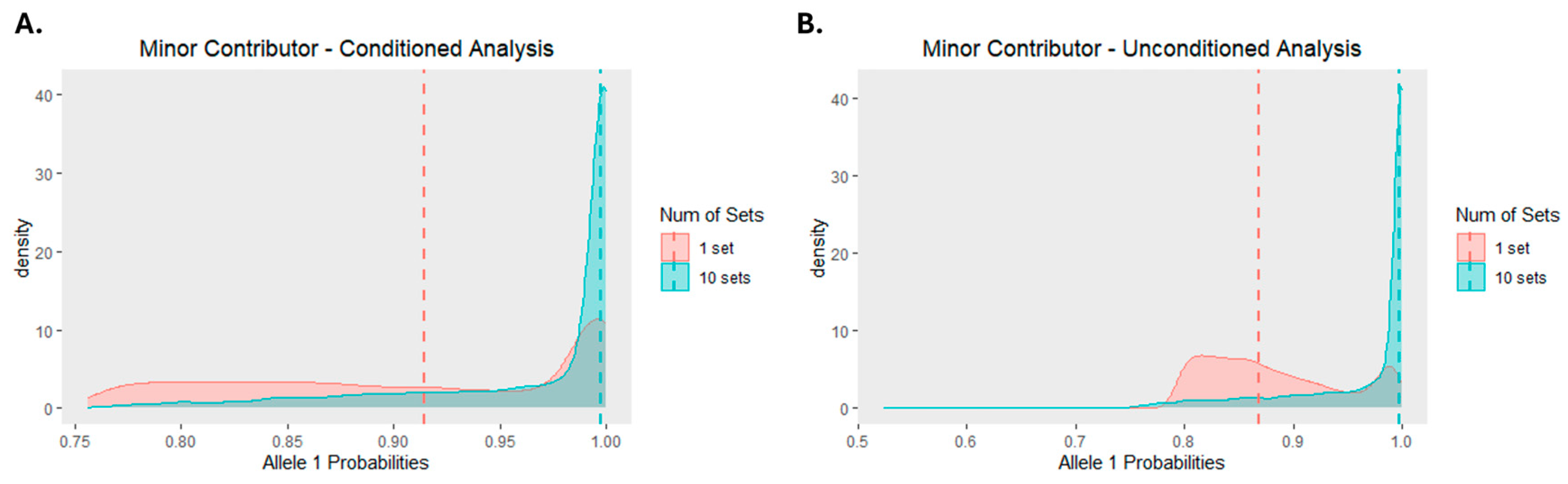
Figure 4.
Genotype accuracy at different allele 1 and allele 2 probability threshold combinations. The plot was created using the inferred genotype accuracy for a minor contributor from an unconditioned deconvolution of a 1:5 mixture. Red colored cells represent the lowest genotype accuracy values, while green colored cells indicate the highest genotype accuracy values. The general genotype accuracy pattern indicated by the heatmap in the figure was consistent across a variety of mixture conditions and deconvolution types (i.e. different mixture ratios, total DNA input, and conditioned and unconditioned deconvolutions) tested during the development of MixDeR.
Figure 4.
Genotype accuracy at different allele 1 and allele 2 probability threshold combinations. The plot was created using the inferred genotype accuracy for a minor contributor from an unconditioned deconvolution of a 1:5 mixture. Red colored cells represent the lowest genotype accuracy values, while green colored cells indicate the highest genotype accuracy values. The general genotype accuracy pattern indicated by the heatmap in the figure was consistent across a variety of mixture conditions and deconvolution types (i.e. different mixture ratios, total DNA input, and conditioned and unconditioned deconvolutions) tested during the development of MixDeR.
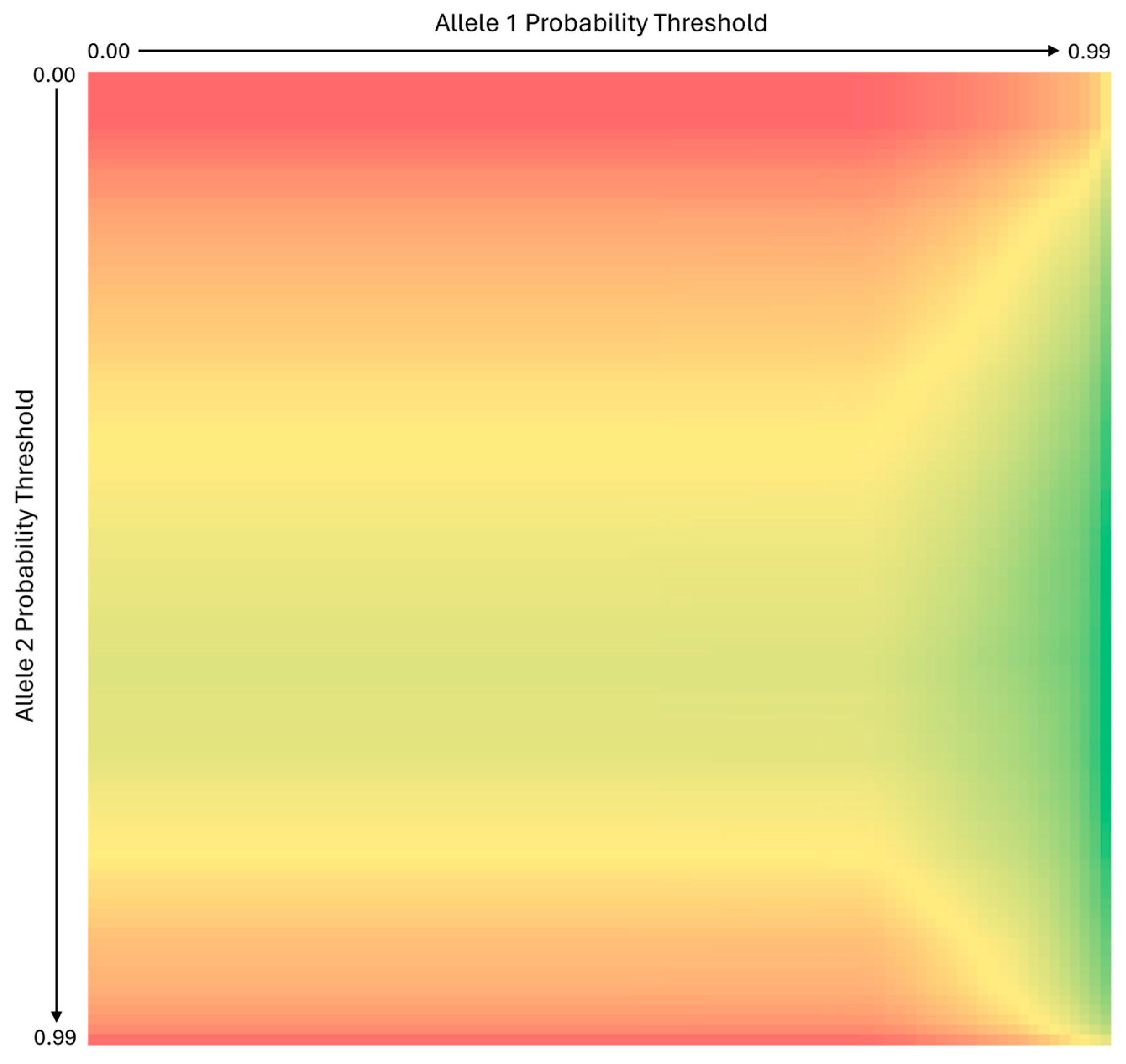

Table 1.
MixDeR results for a 1:5 mixture using two different binning strategies. Results were obtained from analysis of a single 1:5 mixture using the default settings in MixDeR.
Table 1.
MixDeR results for a 1:5 mixture using two different binning strategies. Results were obtained from analysis of a single 1:5 mixture using the default settings in MixDeR.
| Deconvolution Type | Unknown Contributor | Number of SNP Sets |
Number of SNPs in Inferred Genotype |
Inferred Genotype Accuracy |
|---|---|---|---|---|
| Conditioned | Major | 1 | 6,000 | 92.32% |
| Major | 10 | 9,898 | 99.11% | |
| Conditioned | Minor | 1 | 6,000 | 73.46% |
| Minor | 10 | 6,000 | 84.19% | |
| Unconditioned | Major | 1 | 6,000 | 62.12% |
| Major | 10 | 9,632 | 98.91% | |
| Unconditioned | Minor | 1 | 6,000 | 62.25% |
| Minor | 10 | 6,000 | 83.32% |
Table 2.
Metrics for an example 1:5 mixture processed using MixDeR.
| Deconvolution Type | Unknown Contributor | Kintelligence Profile(s) Used |
Number of SNPS in Inferred Genotypes |
Inferred Genotype Accuracy | Heterozygosity |
|---|---|---|---|---|---|
| Conditioned | Major | Single | 9,898 | 99.11% | 45.77% |
| Major | Replicates | 10,035 | 99.27% | 46.44% | |
| Conditioned | Minor | Single | 6,000 | 84.19% | 33.30% |
| Minor | Replicates | 6,000 | 90.17% | 32.90% | |
| Unconditioned | Major | Single | 9,632 | 98.91% | 44.38% |
| Major | Replicates | 10,036 | 99.05% | 46.52% | |
| Unconditioned | Minor | Single | 6,000 | 83.32% | 32.50% |
| Minor | Replicates | 6,000 | 90.45% | 32.12% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



