
Submitted:
23 July 2024
Posted:
23 July 2024
You are already at the latest version
Abstract
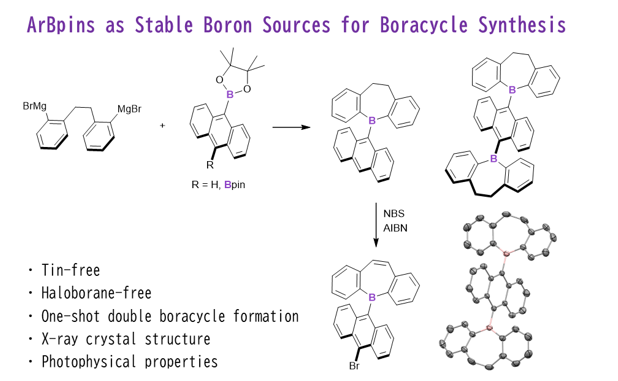
The general synthesis of boron-containing cyclic compounds (boracycles) necessitates toxic organotin precursors or highly reactive boron halides. Here we report the synthesis of seven- and five-membered boracycles utilizing arylboronic acid pinacol esters (ArBpins) as stable boron sources. Grignard reagents generated from 2,2′-dibromodibenzyl or 2,2′-dibromobiphenyl reacted with ArBpins, where Ar = 9-anthryl (Anth), 2,4,6-trimethylphenyl (Mes), 2,4,6-triisopropylphenyl (Tip), to give 10,11-dihydro-5H-dibenzo[b,f]borepins or dibenzoborole derivatives. This Bpin-based method was successfully applied to a one-shot double boracycle formation, providing a dihydrodibenzoborepin–anthracene–dihydrodibenzoborepin triad molecule in a good yield. The dihydrodibenzoborepin bearing the Anth group was directly converted to the unsaturated borepin by NBS/AIBN. All products were characterized by NMR, HRMS, and in some cases, single-crystal X-ray diffraction analysis. Additionally, the photophysical properties of the products are also reported.
Keywords:
10
; 11-dihydrodibenzoborepin
; dibenzoborole
; dibenzoborepin
; synthetic method
; boronic acid pinacol ester
1. Introduction
Boron-containing unsaturated cyclic compounds (hereafter referred to as boracycles) have been studied intensively in many fields, including materials science[1,2,3,4] and aromatic/antiaromatic chemistry.[5,6] This is due to their unique conjugation system involving the vacant p orbital of the boron atom. Of particular interest are fused boroles and borepins, five- and seven-membered boracycles, respectively, owing to their 4π-antiaromaticity and 6π-aromaticity as well as fluorescent properties.[7,8,9,10]
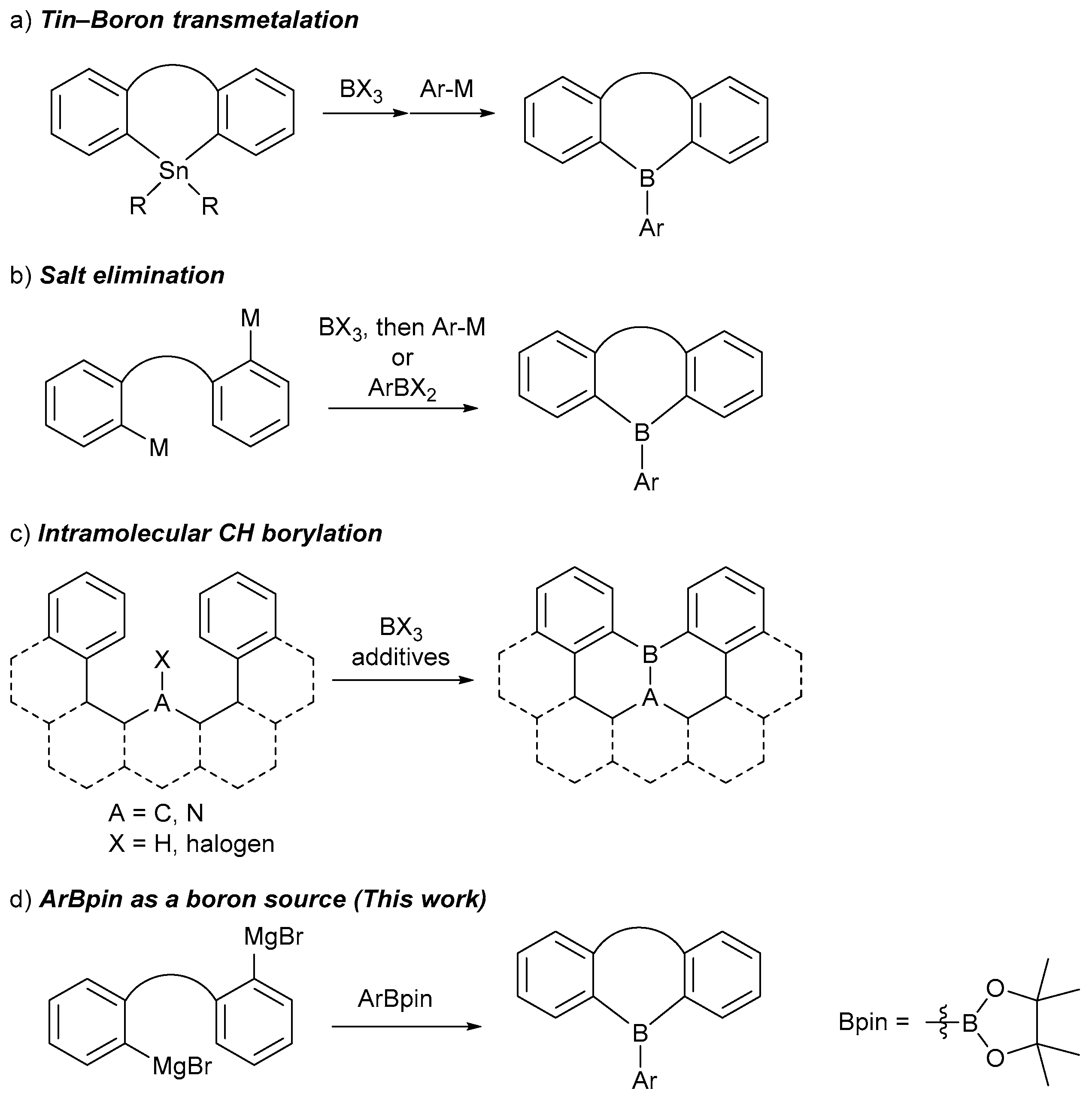
Synthetic methods for boracycles can be roughly divided into three types: 1) tin–boron transmetallation, 2) salt-elimination reaction, and 3) intramolecular CH borylation (Scheme 1).[11,12,13,14] The tin–boron transmetallation method has been a reliable route to many types of boracycles including dibenzoboroles and dibenzoborepins.[8,15,16,17,18] Similarly to the synthesis of other heterocycles,[19] the salt-elimination reaction between metalated carbons, e.g. organolithiums and Grignard reagents, and haloboranes (BX3 and RBX2, where X = Cl, Br) is also widely employed.[15] Additionally, intramolecular cyclization via CH borylation has been developed as a convenient approach, particularly for the synthesis of boron-containing PAHs (polycyclic aromatic hydrocarbons) in the last decade.[20,21,22,23] However, the aforementioned methods rely on toxic organotin compounds and highly reactive haloboranes. Therefore, alternative boracycle synthesis without these unpleasant chemicals is highly demanded.
One possible solution is using boronate esters [B(OR)3 and R′B(OR)2] as boron sources, and indeed, such boracycle formation has been reported. For example, the Yamaguchi’s group has reported the synthesis of dibenzoboroles and dibenzoborepins using ArB(OMe)2 as a boron source.[10,24] Additionally, stepwise boracycle formations involving arylboronic acid esters have also been developed.[7,25,26,27] Given the widespread application of arylboronic acid pinacol esters (ArBpins) in cross-coupling chemistry owing to their stability and moderate reactivity,[28] we envisage that ArBpins can be useful and stable boron sources for boracycle synthesis. However, to the best of our knowledge, ArBpins have never been used as boron sources for boracycles, although the usage of B2pin2 for the synthesis of Mes2BBpin has been reported.[29] An additional advantage of using ArBpin is that its synthetic method has been well-established: Miyaura borylation,[30,31] CH borylation,[32,33] and nucleophilic borylation. Therefore, the synthesis of boracycles using ArBpin would enable facile access to various boracycles which could serve as acceptor units in donor–acceptor type molecules. In this report, we present the synthesis of seven- and five-membered boracycles utilizing ArBpin as boron sources. This method enables the straightforward synthesis of Donor–Acceptor and Acceptor–π–Acceptor type molecules with borepin acceptors. The photophysical properties of the newly synthesized borepin derivatives are also documented.
2. Results and Discussion
2.1. Synthetic Studies
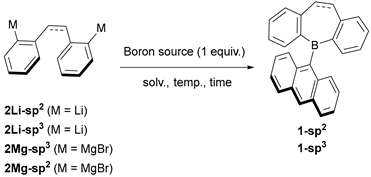
The synthesis of 5-(9-anthryl)-5H-dibenzo[b,f]borepin 1-sp2 and its saturated analog 1-sp3 using AnthBpin (Anth = 9-anthryl) as a boron source was investigated (Table 1). The treatment of AnthBpin and dilithium reagents [(Z)-2,2′-dilithiostilbene (2Li-sp2) and 2,2′-dilithiodibenzyl (2Li-sp3)] did not provide 1-sp2 nor 1-sp3 (entries 1 and 2). Switching the nucleophile to the Grignard reagent (2Mg-sp3) resulted in the isolation of 1-sp3 in 62% yield (entry 3). Therefore, the type of nucleophile is important in this ArBpin-based boracycle synthesis. It is noteworthy that 1-sp3 was successfully purified by column chromatography although they have a 9-antrhyl group that is sterically less demanding compared to widely used protecting groups such as Mes (2,4,6-trimethylphenyl) and Tip (2,4,6-triisopropylphenyl). When AnthB(OMe)2 was employed as a boron source, the yield of 1-sp3 decreased to 31% (entry 4), indicating that the bidentate character of the pinacol group stabilizes a reaction intermediate and/or suppresses undesired side reactions. Our attempts to synthesize 1-sp2 by this method are unsuccessful because the preparation of Grignard reagent 2Mg-sp2 from (Z)-2,2′-dibromostilbene and magnesium causes the concomitant formation of phenanthrene and the E-isomer (entry 5 and Table S1). This type of Z/E isomerization of stilbene by alkaline metals and a FeI complex has been reported.[34,35,36]
To explore the substrate scope of this ArBpin-based boracycle synthesis, other reagents were next subjected to the reaction (Scheme 2). When MesBpin[37] and TipBpin[38] were allowed to react with 2Mg-sp3 in THF under reflux, the yields of the corresponding dihydrodibenzoborepins 3 and 4 were only 16 and 4%, respectively. However, using cyclopentyl methyl ether (CPME) as a solvent improved the yields (59% for 3 and 46% for 4).[39] The higher boiling point of CPME (106 °C) compared to that of THF (66 °C) would be a key factor in the efficient reaction when bulky Ar groups are employed. The reaction using 9-Br-10-Bpin-anthracene[40] provided 10-bromoanthryl derivative 5-sp3 (64% yield), the bromo substituent of which can be used for further functionalization. Moreover, the one-shot-double boracycle formation was achieved when 9,10-(Bpin)2-anthracene was used as a substrate to generate a dihydrodibenzoborepin–anthracene–dihydrodibenzoborepin triad molecule 6-sp3 in a 66% yield. On the contrary, our attempts to synthesize 6-sp3 using the tin-boron transmetallation were unsuccessful; the reactions of 5,5-dimethyl-5H-dibenzo[b,f]stannepin and BCl3 and subsequent 9,10-dilithioanthracene provided a complex mixture, highlighting the usefulness of the Bpin-based method.
We next investigated the conversion of 1-sp3 to the corresponding unsaturated one (1-sp2) because the direct synthesis of 1-sp2 from the reaction in Table 1 failed (Scheme 3). According to the literature, bromination of a dihydroborepin with N-bromosuccinimide (NBS) and subsequent elimination reaction using a base would be a promising route.[41] However, bromination of 1-sp3 using 1.1 equiv. NBS and 0.25 equiv. azobis(isobutyronitrile) (AIBN) did not yield benzyl bromide 1Br-sp3 but 5-sp3 in 68% yield. Thus, bromination of 5-sp3 under the same conditions was next carried out, which provided a 3:2 mixture of 5-sp3 and 5-sp2, and again, no benzyl bromide was obtained. Increasing the amount of AIBN to 0.5 equiv. allowed the isolation of 5-sp2 in a 55% yield. Notably, one-pot synthesis of 5-sp2 was also achieved by a treatment of 1-sp3 with 2 equiv. of NBS and 0.5 equiv. of AIBN (73% yield). The bromo substituent in 5-sp2 was replaced by a hydrogen atom via a lithiation/protonation process to afford 1-sp2 in 89% yield.
This ArBpin-based method is also applied for the synthesis of a dibenzoborole. We selected a Tip borole 7 as the target molecule because this molecule can be purified by column chromatography owing to the bulky Tip group.[24] Grignard reagent 8Mg[42] generated from 2,2′-dibromobiphenyl reacted with TipBpin in THF under reflux to afford 7 in 19% (Scheme 4). Although this yield is inferior to the reported method using 8Mg and TipB(OMe)2 (45%),[24] it is worth noting that TipBpin can also be used as a stable boron source for the dibenzoborole synthesis. In this case, the reaction in CPME instead of THF under reflux did not give 7 at all, probably due to the less thermal stability of antiaromatic 7 at higher temperatures.
2.2. Single Crystals X-ray Diffraction Analysis
Figure 1 illustrates the crystal structures of 3, 5-sp3 and 6-sp3 and that of 4 is shown in Figure S1. Each asymmetric unit of 3, 4 and 6-sp3 contains two independent molecules which are structurally similar. The dihydrodibenzoborepin skeleton of 5-sp3 is partially disordered over two positions with a ratio of 58:42.
Table 2 shows the selected bond lengths and angles for these dihydrodibenzoborepins. Each boron atom has a planar three-coordinated structure with the sum of the C–B–C angles of about 360°. These compounds have highly twisted structures with C1−B1−C15−C16 torsion angles (80.2(2)–89.80(14)°) being larger than those in related 9-diarylborylanthracenes and 9,10-bis(diarylboryl)anthracenes (ca. 45–63°), where Ar = Mes and 2,6-dimethyl-4-trimethylammoniumphenyl.[43,44] Therefore, the bridging ethylene units in the dihydrodibenzoborepin skeletons induce a large dihedral angle between the dihydrodibenzoborepins and π- or donor units, which hinders effective conjugation between the vacant p orbital of the boron atom and the π electrons of the Mes and 9-anthryl groups. Accordingly, the B1−C15 bonds are slightly longer than the B1−C1/C6 bonds (ca. 1.59 vs. 1.57 Å). The dihydrodibenzoborepin skeletons are distorted with the C1−C2−C5−C6 torsion angles around 20°, in contrast to the only slightly bent structures of aromatic dibenzoborepins.[8,45]
2.3. Photophysical Properties and Theoretical Calculations
Although the photophysical properties of aromatic dibenzoborepins are well-investigated,[8,46] little is known about those of dihydrodibenzoborepins. Therefore, we explored the absorption and emission properties of 1-sp3 and 6-sp3 as well as 1-sp2. Figure 2 shows absorption and emission spectra of 1-sp3 and 1-sp2 recorded in different solvents (hexane, toluene, THF, CHCl3 and CH2Cl2). Spectra for 6-sp3 were recorded only in CH2Cl2 due to the poor solubility in non-halogenated solvents (Figure S2). These photophysical data are summarized in Table 3.
No significant solvent dependency was observed in absorption spectra of 1-sp3 and 1-sp2. Dihydrodibenzoborepin derivatives 1-sp3 and 6-sp3 show a broad and weak absorption ranging from ca. 450 to 400 nm resulting from charge transfer (CT) from the anthracene unit to the dihydrodibenzoborepin acceptor, whereas such CT absorption is weak in 1-sp2 probably due to the aromatic character of borepin which reduces the Lewis acidity of the boron atom.[47] Three absorption peaks around 350–400 nm originating from the anthracene unit are slightly redshifted compared to those of anthracene by ca. 10 nm,[48] the reason of which would be the inductive effect by the electropositive boron atoms (vide infra).
As mentioned above, the dihydrodibenzoborepin units are almost perpendicular to the central anthracene core, which causes a less effective conjugation between them. Therefore, it is important to compare the photophysical properties of 1-sp3, 1-sp2, 6-sp3 and related borylanthracenes. The absorption maxima of 9-[B(Mes)2]anthracene and 9,10-bis[B(Mes)2]anthracene whose anthracene unit tilts ca. 53° with respect to the boron-centered plane are 420 and 455 nm,[49,50] redshifted compared to those of 1-sp3 and 6-sp3 (390 and 398 nm). This difference is rationalized by the different degree of π(anthracene)–p*(boron) conjugation.
In contrast to the absorption spectra, emission spectra of 1-sp3 and 1-sp2 are highly dependent on the solvents, as commonly found in Donor–Acceptor type molecules.[51,52,53] Stokes shifts of 1-sp3 varied from ca. 3500 to 6300 cm−1, being larger than those of 1-sp2, as the solvent polarity increases. These larger Stokes shifts in 1-sp3 suggests that the dihydrodibenzoborepin skeleton is more flexible than the dibenzoborepins, allowing a greater structural relaxation in the excited state. Compound 1-sp3 shows two types of emission peaks; 1) anthracene-based emission with three peak tops around 360 to 430 nm and 2) a broadened CT emission peak ranging from 450 to 530 nm. Interestingly, strength of these two emissions depends on the solvent; the anthracene-based emission is dominant in THF, whereas the CT emission is a major contributor in the other solvents. The lower contribution of the CT emission in THF can be rationalized by the coordination of THF to the vacant p orbital of the boron atoms. Although a similar trend is found in the spectra of 1-sp2, the anthracene-based emission peaks are broadened, the reason for which is not clear at this point.
To better understand the electronic structures, frontier molecular orbitals (MOs) for 1-sp3, 1-sp2, 6-sp3 as well as anthracene were calculated with CAM-B3LYP/6-31G(d,p)//B3LYP/6-31G(d) (Figure 3).[54,55] As expected from the highly twisted structures, π(anthracene)–p*(boron) conjugation is not found in the MOs. Importantly, the energy levels for the anthracene-based MOs increase as the number of the borepin unit increases, for example, −6.45 eV (anthracene), −6.2 eV (1-sp3 and 1-sp2), −6.06 eV (6-sp3) in the HOMOs, due to the σ-donating character of the boron atoms.[56] The effective cyclic conjugation in the dibenzoborepin markedly raises the energy levels of the MOs derived from the dibenzoborepin moiety in 1-sp2 compared to those of the corresponding MOs in 1-sp3 [−6.99 eV (HOMO−1 in 1-sp2) vs. −7.82 eV (HOMO−2 in 1-sp3) and −0.58 eV (LUMO in 1-sp2) vs. −0.72 eV (LUMO in 1-sp3)]. These differences account for the strong absorption around 315 and 330 nm found only in 1-sp2.
3. Materials [M1] and Method
3.1. General Considerations
All manipulations were performed under an argon atmosphere by using standard Schlenk techniques. Et2O, THF, hexane, benzene, toluene, CPME were dehydrated by 4A molecular sieves. All reagents were purchased from Sigma-Aldrich Chemical Co., FUJIFILM Wako Pure Chemical Corporation, Tokyo Chemical Industry Co., LTD., Kanto Chemical Co., Inc. or Nacalai Tesque and used as received unless otherwise stated. Column chromatography was carried out using Wakogel silica 60N (particle size: 40–100 μm). (Z)-2,2′-Dibromostilbene,[8] 2,2′-dibromodibenzyl,[57] 2-(9-anthryl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (AnthBpin),[58] 9,10-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)anthracene [Anth(Bpin)2],[59] AnthB(OMe)2,[60] 2-(2,4,6-trimethylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (MesBpin),[37] 2-(2,4,6-triisopropylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (TipBpin)[61] were synthesized according to the literature. 1H, 13C{1H}, 11B{1H} NMR spectra were recorded on a JEOL ECZL-500R spectrometer at 20 °C unless otherwise stated. Chemical shifts are reported in δ and referenced to residual 1H and 13C{1H} NMR signals of the deuterated solvents as internal standards or to the 11B NMR signal of BF3·Et2O in CDCl3 (δ 0) as an external standard. Multiplicities are abbreviated as singlet (s), doublet (d), triplet (t), quartet (q), septet (sept), multiplet (m) and broad (br). The HRMS data were obtained by a Bruker ultrafleXtreme using 9-nitroanthracene as a matrix. Diffraction data were collected on a Bruker APEX II (for 3 and 6-sp3) or Bruker D8 QUEST (for 4 and 5-sp3) with Mo Kα radiation (λ = 0.71075 Å) at –110 to −80 °C. The structures were solved by direct methods using SHELXS. The refinements were performed using SHELXL-2019/3.[62] The positions of the non-hydrogen atoms were determined by SHELXT 2018/2.[63] All non-hydrogen atoms were refined on Fo2 anisotropically by full-matrix least-square techniques. All hydrogen atoms were placed at the calculated positions with fixed isotropic parameters. UV-vis absorption and emission spectra were recorded using a JASCO V-650 and JASCO FP-6600 spectrometers. Theoretical calculations were performed using the Gaussian 16 program.[64] The optimized structures of 1-sp3 and 6-sp3 are in good agreement with the corresponding X-ray structures. All local minima were confirmed by the vibrational frequency calculations with zero imaginary frequency.
3.2. Preparation of 2Mg-sp3
THF (5.0 mL) was added to a 25-mL Schlenk tube containing Mg turning (109.4 mg, 4.500 mmol, 3.0 equiv.) and 2,2′-dibromodibenzyl (510.0 mg, 1.500 mmol) at room temperature. The reaction mixture was refluxed for 2 h. After cooling to room temperature, insoluble materials were filtered off under argon to give a gray solution of 2Mg-sp3 in THF (about 0.3 M).
3.3. Synthesis of 5-(9-anthryl)-5H-9,10-dihydrodibenzo[b,f]borepin (1-sp3) Using AnthBpin
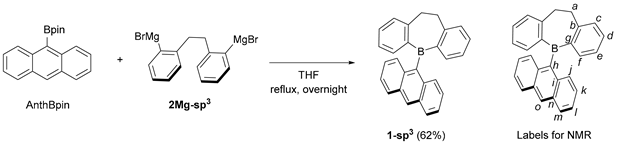
AnthBpin (71.0 mg, 0.193 mmol, 1.0 equiv.) in THF (3.0 mL) was added to a solution of 2Mg-sp3 (0.3 M in THF, 0.64 mL, 0.19 mmol). The reaction mixture was refluxed overnight, after which the solvent was removed in vacuo. The product was purified by column chromatography (hexane:CH2Cl2 = 9:1) to yield 1-sp3 (44.1 mg, 0.120 mmol, 62 %) as a yellow solid. 1H NMR (500 MHz, CDCl3): δ = 8.47 (s, 1H, o), 8.05 (d, 3JHH = 8.5 Hz, 2H, m), 7.46 (dd, 3JHH = 8.5 Hz, 4JHH = 1.0 Hz, 2H, j), 7.40 (ddd, 3JHH = 8.5 Hz, 3JHH = 6.5 Hz, 4JHH = 1.0 Hz, 2H, l), 7.36 (td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 2H, d), 7.33 (dd, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 2H, c), 7.29 (dd, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 2H, f), 7.21 (ddd, 3JHH = 8.5 Hz, 3JHH = 6.5 Hz, 4JHH = 1.0 Hz, 2H, k), 6.91 (td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 2H, e), 3.45 (s, 4H, a); 13C{1H} NMR (126 MHz, CDCl3): δ = 152.5 (4°, b), 146.3 (4°, h), 143.2 (3°, f), 140.1 (4°, g), 133.7 (3°, d), 133.3 (4°, i), 131.3 (4°, n), 130.3 (3°, j), 128.81 (4°, c), 128.78 (3°, m), 126.00 (3°, e), 125.95 (3°, o), 125.1 (3°, l), 124.6 (3°, k), 37.9 (2°, a); 11B{1H} NMR (160 MHz, CDCl3): δ = 71.8; HRMS m/z calcd for C28H21B+ [M]+: 368.1736, found: 368.1736; Mp: 142 °C (decomp.).
3.4. Synthesis of 9,10-bis(5H-9,10-dihydrodibenzo[b,f]borepin-5-yl)anthracene (6-sp3)
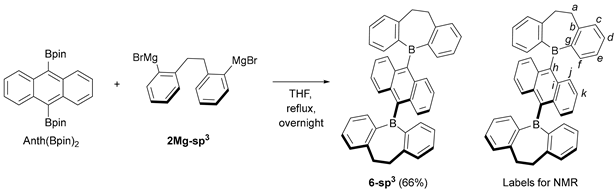
A solution of 2Mg-sp3 (0.3 M in THF, 9.27 mL, 2.78 mmol, 2.0 equiv.) was added to a solution of Anth(Bpin)2 (598.6 mg, 1.392 mmol, 1.0 equiv.) in THF (24.0 mL). The reaction mixture was refluxed overnight, during which a yellow solid precipitated out. The yellow solid insoluble in THF was filtered off and washed with CH2Cl2 to yield 6-sp3 (414.7 mg, 0.9187 mmol, 66 %) as a yellow solid. 1H NMR (500 MHz, CDCl3): δ= 7.50 (dd, 3JHH = 8.0 Hz, 4H, j), 7.48 (dd, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 4H, f), 7.41 (td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 4H, d), 7.35 (td, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 4H, c), 7.12 (dd, 3JHH = 7.0 Hz, 4H, k), 7.02 (td, 4H, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, e), 3.48 (s, 8H, a); 13C{1H} NMR (126 MHz, CDCl3): δ= 152.4 (4°, b), 143.3 (3°, f), 133.7 (3°, d), 132.8 (4°, i), 130.8 (3°, j), 128.8 (3°, c), 126.1 (3°, e), 124.1 (3°, k), 37.9 (2°, a). Two signals (g and h) were not observed due to the low solubility to CDCl3 as well as the quadrupolar relaxation caused by 11B. 11B{1H} NMR signals could not be observed also due to the low solubility. HRMS m/z calcd for C42H32B2+ [M]+: 558.2690, found: 558.2721. Mp: 151 °C (decomp.).
4. Conclusions
We have developed a novel synthetic method for dihydrodibenzoborepin and dibenzoborole skeletons utilizing ArBpins as stable boron sources. This method enabled one-shot-double formation of boracycles to give 6-sp3 that could not be obtained by a well-established transmetallation route using an organotin compound. The dihydrodibenzoborepins 1-sp3 and 5-sp3 were directly converted into dibenzoborepin 5-sp2 under refluxing with NBS/AIBN. In the crystalline state, compounds 3, 4, 5-sp3 and 6-sp3 adopt a highly twisted structure because of the steric hindrance between the bulky Ar substituents and the dihydrodibenzoborepins. Spectroscopic studies reveal that 1-sp3 and 1-sp2 exhibit the solvatofluorochromic properties.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Synthetic procedures and analytical data for 3, 4, 5-sp3, 5-sp2 and 1-sp2; Table S1: Preparation and quenching reactions of 2Mg-sp2; Figure S1: Molecular structures of 4; Figure S2: Absorption (solid line) and emission spectra (broken line) of 6-sp3; Table S2. Crystallographic data for 3, 4, 5-sp3 and 6-sp3; Cartesian coordinates for 1-sp3, 1-sp2 and 6-sp3; 1H and 13{1H} NMR spectra of the new compounds.
Author Contributions
Conceptualization, T.K.; methodology, H.K.; validation, H.K., F.K. and N.M.; investigation, H.K., F.K. and N.M.; resources, T.K.; data curation, T.K.; writing—original draft preparation, T.K.; writing—review and editing, H.K., K.F., N.M.; visualization, T.K.; supervision, T.K.; project administration, T.K.; funding acquisition, T.K. and H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI, grant number 22K05078 and The Sasakawa Scientific Research Grant from The Japan Science Society.
Data Availability Statement
Crystallographic data for the reported compounds in this article have been deposited at the Cambridge Crystallographic Data Centre: CCDC 2371388 (3), 2371389 (4), 2371390 (5-sp3), 2371391 (6-sp3). These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
Acknowledgements
The authors thank Ms. Kana Kobayashi in Ochanomizu University for her help on measuring HRMS for some products.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ji, L.; Griesbeck, S.; Marder, T.B. Recent developments in and perspectives on three-coordinate boron materials: a bright future. Chem. Sci. 2017, 8, 846–863. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Wakamiya, A. Boron as a key component for new π-electron materials. Pure Appl. Chem. 2006, 78, 1413–1424. [Google Scholar] [CrossRef]
- Hirai, M.; Tanaka, N.; Sakai, M.; Yamaguchi, S. Structurally Constrained Boron-, Nitrogen-, Silicon-, and Phosphorus-Centered Polycyclic π-Conjugated Systems. Chem. Rev. 2019, 119, 8291–8331. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.; Dewhurst, R.D.; Ignat'ev, N.V.; Finze, M.; Braunschweig, H. Boron: Its Role in Energy-Related Processes and Applications. Angew. Chem., Int. Ed. 2020, 59, 8800–8816. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, H.; Kupfer, T. Recent developments in the chemistry of antiaromatic boroles. Chem. Commun. 2011, 47, 10903–10914. [Google Scholar] [CrossRef]
- Messersmith, R.E.; Tovar, J.D. Assessment of the aromaticity of borepin rings by spectroscopic, crystallographic and computational methods: a historical overview. J. Phys. Org. Chem. 2015, 28, 378–387. [Google Scholar] [CrossRef]
- Iida, A.; Yamaguchi, S. Thiophene-Fused Ladder Boroles with High Antiaromaticity. J. Am. Chem. Soc. 2011, 133, 6952–6955. [Google Scholar] [CrossRef] [PubMed]
- Mercier, L.G.; Piers, W.E.; Parvez, M. Benzo- and Naphthoborepins: Blue-Emitting Boron Analogues of Higher Acenes. Angew. Chem., Int. Ed. 2009, 48, 6108–6111. [Google Scholar] [CrossRef]
- Messersmith, R.E.; Siegler, M.A.; Tovar, J.D. Aromaticity Competition in Differentially Fused Borepin-Containing Polycyclic Aromatics. J. Org. Chem. 2016, 81, 5595–5605. [Google Scholar] [CrossRef]
- Ando, N.; Kushida, T.; Yamaguchi, S. A planarized B-phenyldibenzoborepin: impact of structural constraint on its electronic properties and Lewis acidity. Chem. Commun. 2018, 54, 5213–5216. [Google Scholar] [CrossRef]
- Iqbal, S.A.; Pahl, J.; Yuan, K.; Ingleson, M.J. Intramolecular (directed) electrophilic C–H borylation. Chem. Soc. Rev. 2020, 49, 4564–4591. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Bartholome, T.A.; Tidwell, J.R.; Pujol, A.; Yruegas, S.; Martinez, J.J.; Martin, C.D. 9-Borafluorenes: Synthesis, Properties, and Reactivity. Chem. Rev. 2021, 121, 4147–4192. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Hatakeyama, T. Development of One-Shot/One-Pot Borylation Reactions toward Organoboron-Based Materials. Bull. Chem. Soc. Jpn. 2021, 94, 950–960. [Google Scholar] [CrossRef]
- He, J.; Rauch, F.; Finze, M.; Marder, T.B. (Hetero)arene-fused boroles: a broad spectrum of applications. Chem. Sci. 2021, 12, 128–147. [Google Scholar] [CrossRef] [PubMed]
- Eisch, J.J.; Hota, N.K.; Kozima, S. Synthesis of pentaphenylborole, a potentially antiaromatic system. J. Am. Chem. Soc. 1969, 91, 4575–4577. [Google Scholar] [CrossRef]
- Eisch, J.J.; Galle, J.E.; Kozima, S. Bora-aromatic systems. Part 8. The physical and chemical consequences of cyclic conjugation in boracyclopolyenes. The antiaromatic character of pentaarylboroles. J. Am. Chem. Soc. 1986, 108, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chase, P.A.; Piers, W.E.; Patrick, B.O. New Fluorinated 9-Borafluorene Lewis Acids. J. Am. Chem. Soc. 2000, 122, 12911–12912. [Google Scholar] [CrossRef]
- Wakamiya, A.; Mishima, K.; Ekawa, K.; Yamaguchi, S. Kinetically stabilized dibenzoborole as an electron-accepting building unit. Chem. Commun. 2008, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Saito, M. 3.17 - Siloles, Germoles, Stannoles, and Plumboles. In Comprehensive Heterocyclic Chemistry IV, Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Oxford, 2022; pp. 798–832. [Google Scholar]
- Hatakeyama, T.; Hashimoto, S.; Seki, S.; Nakamura, M. Synthesis of BN-Fused Polycyclic Aromatics via Tandem Intramolecular Electrophilic Arene Borylation. J. Am. Chem. Soc. 2011, 133, 18614–18617. [Google Scholar] [CrossRef]
- Hirai, H.; Nakajima, K.; Nakatsuka, S.; Shiren, K.; Ni, J.; Nomura, S.; Ikuta, T.; Hatakeyama, T. One-Step Borylation of 1,3-Diaryloxybenzenes Towards Efficient Materials for Organic Light-Emitting Diodes. Angew. Chem., Int. Ed. 2015, 54, 13581–13585. [Google Scholar] [CrossRef]
- Nakatsuka, S.; Yasuda, N.; Hatakeyama, T. Four-Step Synthesis of B2N2-Embedded Corannulene. J. Am. Chem. Soc. 2018, 140, 13562–13565. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-Shot Multiple Borylation toward BN-Doped Nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Shirasaka, T.; Akiyama, S.; Tamao, K. Dibenzoborole-Containing π-Electron Systems: Remarkable Fluorescence Change Based on the “On/Off” Control of the pπ−π* Conjugation. J. Am. Chem. Soc. 2002, 124, 8816–8817. [Google Scholar] [CrossRef]
- Iida, A.; Sekioka, A.; Yamaguchi, S. Heteroarene-fused boroles: what governs the antiaromaticity and Lewis acidity of the borole skeleton? Chem. Sci. 2012, 3, 1461–1466. [Google Scholar] [CrossRef]
- Brend'amour, S.; Gilmer, J.; Bolte, M.; Lerner, H.-W.; Wagner, M. C-Halogenated 9,10-Diboraanthracenes: How the Halogen Load and Distribution Influences Key Optoelectronic Properties. Chem. Eur. J. 2018, 24, 16910–16918. [Google Scholar] [CrossRef]
- Urban, M.; Durka, K.; Górka, P.; Wiosna-Sałyga, G.; Nawara, K.; Jankowski, P.; Luliński, S. The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes. Dalton Trans. 2019, 48, 8642–8663. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. [Google Scholar] [CrossRef] [PubMed]
- The converstion of a B2pin2 to pinBBAr2 has been reported; Asakawa, H. ; Lee, K.-H.; Lin, Z.; Yamashita, M. Facile scission of isonitrile carbon–nitrogen triple bond using a diborane(4) reagent. Nat. Commun. 2014, 5, 4245. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Metal-catalyzed reactions of diborons for synthesis of organoboron compounds. Chem. Rec. 2004, 3, 271–280. [Google Scholar] [CrossRef]
- Hartwig, J.F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C−H Activation for the Construction of C−B Bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef]
- Levin, G.; Ward, T.A.; Szwarc, M. Electron-transfer catalyzed cis-trans isomerization of stilbene. Stability of sodium cis-stilbenide and the existence of sodium salts of cis- and of trans-stilbene dianions. J. Am. Chem. Soc. 1974, 96, 270–272. [Google Scholar] [CrossRef]
- Ito, S.; Kuwabara, T.; Ishii, Y. A Tin Analogue of the Cycloheptatrienyl Anion: Synthesis, Structure, and Further Reduction to Form a Dianionic Species. Organometallics 2020, 39, 640–644. [Google Scholar] [CrossRef]
- Sieg, G.; Müller, I.; Weißer, K.; Werncke, C.G. Taming the stilbene radical anion. Chem. Sci. 2022, 13, 13872–13878. [Google Scholar] [CrossRef] [PubMed]
- Grenz, D.C.; Schmidt, M.; Kratzert, D.; Esser, B. Dibenzo[a,e]pentalenes with Low-Lying LUMO Energy Levels as Potential n-Type Materials. J. Org. Chem. 2018, 83, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Suzuki, N.; Mellerup, S.K.; Wang, X.; Yamaguchi, S.; Wang, S. Pyridyl Directed Catalyst-Free trans-Hydroboration of Internal Alkynes. Org. Lett. 2016, 18, 720–723. [Google Scholar] [CrossRef] [PubMed]
- , After obtaining this results, we investigated the synthesis of 1-sp3 in CPME under reflux condition. However, the yield was similar to that in THF (60%). Thus, CPME is specifically effective for bulky ArBpins such as TipBpin.
- Pospiech, S.; Bolte, M.; Lerner, H.-W.; Wagner, M. Diborylated Magnesium Anthracene as Precursor for B2H5−-Bridged 9,10-Dihydroanthracene. Chem. Eur. J. 2015, 21, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- van Tamelen, E.E.; Brieger, G.; Untch, K.G. Synthesis of a borepin. Tetrahedron Lett. 1960, 1, 14–15. [Google Scholar] [CrossRef]
- Matsuoka, W.; Ito, H.; Sarlah, D.; Itami, K. Diversity-oriented synthesis of nanographenes enabled by dearomative annulative π-extension. Nat. Commun. 2021, 12, 3940. [Google Scholar] [CrossRef]
- Förster, C.; Seichter, W.; Schwarzer, A.; Weber, E. Supramolecular behaviour of bulky arylboranes in the crystalline state. Supramol. Chem. 2010, 22, 571–581. [Google Scholar] [CrossRef]
- Griesbeck, S.; Ferger, M.; Czernetzi, C.; Wang, C.; Bertermann, R.; Friedrich, A.; Haehnel, M.; Sieh, D.; Taki, M.; Yamaguchi, S.; et al. Optimization of Aqueous Stability versus π-Conjugation in Tetracationic Bis(triarylborane) Chromophores: Applications in Live-Cell Fluorescence Imaging. Chem. Eur. J. 2019, 25, 7679–7688. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Saito, S.; Sasamori, T.; Yamaguchi, S. Borylated Dibenzoborepin: Synthesis by Skeletal Rearrangement and Photochromism Based on Bora-Nazarov Cyclization. Angew. Chem., Int. Ed. 2013, 52, 3760–3764. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Harhausen, M.; Liedtke, R.; Bussmann, K.; Fukazawa, A.; Yamaguchi, S.; Petersen, J.L.; Daniliuc, C.G.; Fröhlich, R.; Kehr, G.; et al. Dibenzopentalenes from B(C6F5)3-Induced Cyclization Reactions of 1,2-Bis(phenylethynyl)benzenes. Angew. Chem., Int. Ed. 2013, 52, 5992–5996. [Google Scholar] [CrossRef] [PubMed]
- Ashe, A.J.; Klein, W.; Rousseau, R. Evaluation of the aromaticity of borepin: synthesis and properties of 1-substituted borepins. Organometallics 1993, 12, 3225–3231. [Google Scholar] [CrossRef]
- Jones, R.N. The Ultraviolet Absorption Spectra of Anthracene Derivatives. Chem. Rev. 1947, 41, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Akiyama, S.; Tamao, K. Tri-9-anthrylborane and Its Derivatives: New Boron-Containing π-Electron Systems with Divergently Extended π-Conjugation through Boron. J. Am. Chem. Soc. 2000, 122, 6335–6336. [Google Scholar] [CrossRef]
- Charlot, M.; Porrès, L.; Entwistle, C.D.; Beeby, A.; Marder, T.B.; Blanchard-Desce, M. Investigation of two-photon absorption behavior in symmetrical acceptor–π–acceptor derivatives with dimesitylboryl end-groups. Evidence of new engineering routes for TPA/transparency trade-off optimization. Phys. Chem. Chem. Phys. 2005, 7, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Kosower, E.M. Intramolecular donor-acceptor systems. 9. Photophysics of (phenylamino)naphthalenesulfonates: a paradigm for excited-state intramolecular charge transfer. Acc. Chem. Res. 1982, 15, 259–266. [Google Scholar] [CrossRef]
- Kuwabara, T.; Orii, J.; Segawa, Y.; Itami, K. Curved Oligophenylenes as Donors in Shape-Persistent Donor–Acceptor Macrocycles with Solvatofluorochromic Properties. Angew. Chem., Int. Ed. 2015, 54, 9646–9649. [Google Scholar] [CrossRef]
- Hermann, M.; Wassy, D.; Esser, B. Conjugated Nanohoops Incorporating Donor, Acceptor, Hetero- or Polycyclic Aromatics. Angew. Chem., Int. Ed. 2021, 60, 15743–15766. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Zhu, J.; Lin, Z.; Marder, T.B. Trans Influence of Boryl Ligands and Comparison with C, Si, and Sn Ligands. Inorg. Chem. 2005, 44, 9384–9390. [Google Scholar] [CrossRef] [PubMed]
- Kupracz, L.; Kirschning, A. Multiple Organolithium Generation in the Continuous Flow Synthesis of Amitriptyline. Adv. Synth. Catal. 2013, 355, 3375–3380. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, T.; Zhao, Y.; Fan, D.; Xia, Y.; Zhang, P. Synthesis, crystal structure, photo- and electro-luminescence of 3-(4-(anthracen-10-yl)phenyl)-7-(N,N′-diethylamino)coumarin. Synth. Met. 2010, 160, 1642–1647. [Google Scholar] [CrossRef]
- Zhu, Y.; Rabindranath, A.R.; Beyerlein, T.; Tieke, B. Highly Luminescent 1,4-Diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole- (DPP-) Based Conjugated Polymers Prepared Upon Suzuki Coupling. Macromolecules 2007, 40, 6981–6989. [Google Scholar] [CrossRef]
- Filthaus, M.; Oppel, I.M.; Bettinger, H.F. Supramolecular structures and spontaneous resolution: the case of ortho-substituted phenylboronic acids. Org. Biomol. Chem. 2008, 6, 1201–1207. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, X.; Wang, S. Cascade Dehydrogenative Hydroboration for the Synthesis of Azaborabenzofulvenes. Org. Lett. 2018, 20, 1617–1620. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev.A.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
Scheme 1.
(a–c) General synthesis of boracycles and (d) ArBpin based synthesis (this work). M = Li, MgBr; X = halogen.
Scheme 1.
(a–c) General synthesis of boracycles and (d) ArBpin based synthesis (this work). M = Li, MgBr; X = halogen.
Scheme 2.
Reactions of 2Mg-sp3 and ArBpins. a: yields in THF. b: yields in CPME.
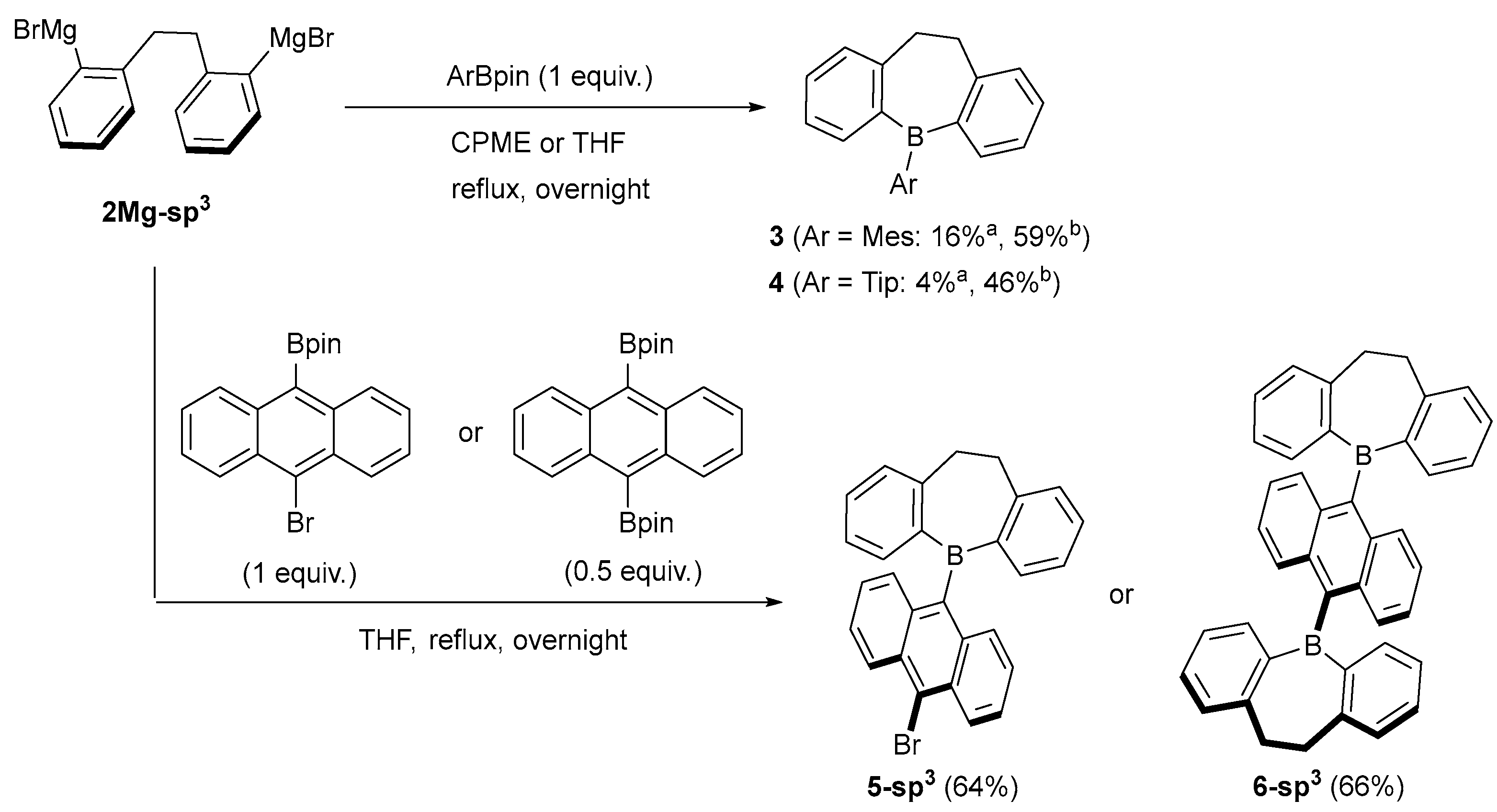
Scheme 3.
Reactions of saturated borepins and NBS/AIBN, and synthesis of 1-sp2.
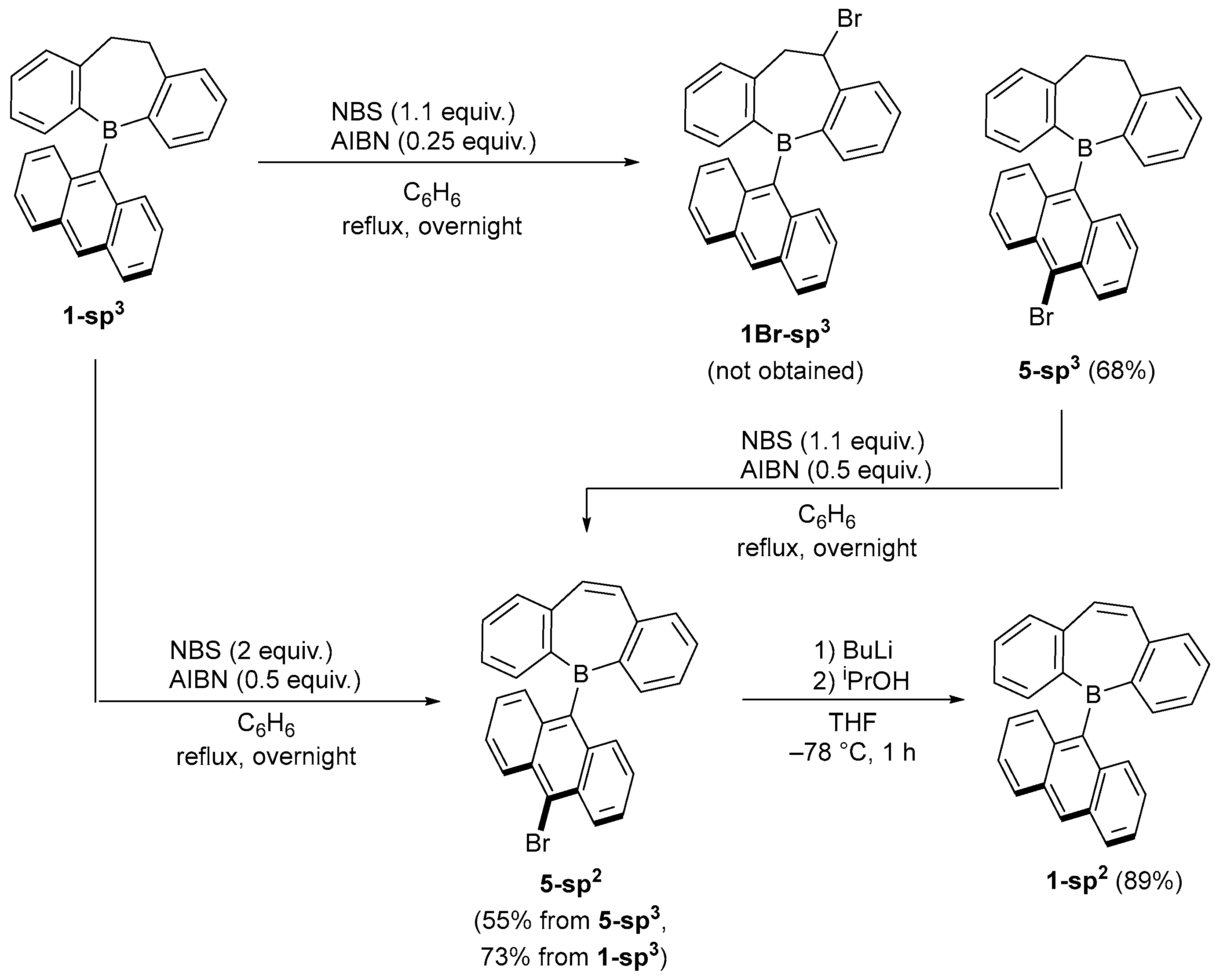
Scheme 4.
Synthesis of a dibenzoborole 7 using the ArBpin-based method.
Figure 1.
Crystal structures of 3 (left), 5-sp3 (center) and 6-sp3 (right) with thermal ellipsoid plots at 50% probability. All hydrogen atoms and a co-crystalized chlorobenzene molecule in 6-sp3 are omitted for clarity. Only one of the two independent molecules are shown for 3 and 6-sp3. Only major disordered structure of 5-sp3 is shown
Figure 1.
Crystal structures of 3 (left), 5-sp3 (center) and 6-sp3 (right) with thermal ellipsoid plots at 50% probability. All hydrogen atoms and a co-crystalized chlorobenzene molecule in 6-sp3 are omitted for clarity. Only one of the two independent molecules are shown for 3 and 6-sp3. Only major disordered structure of 5-sp3 is shown
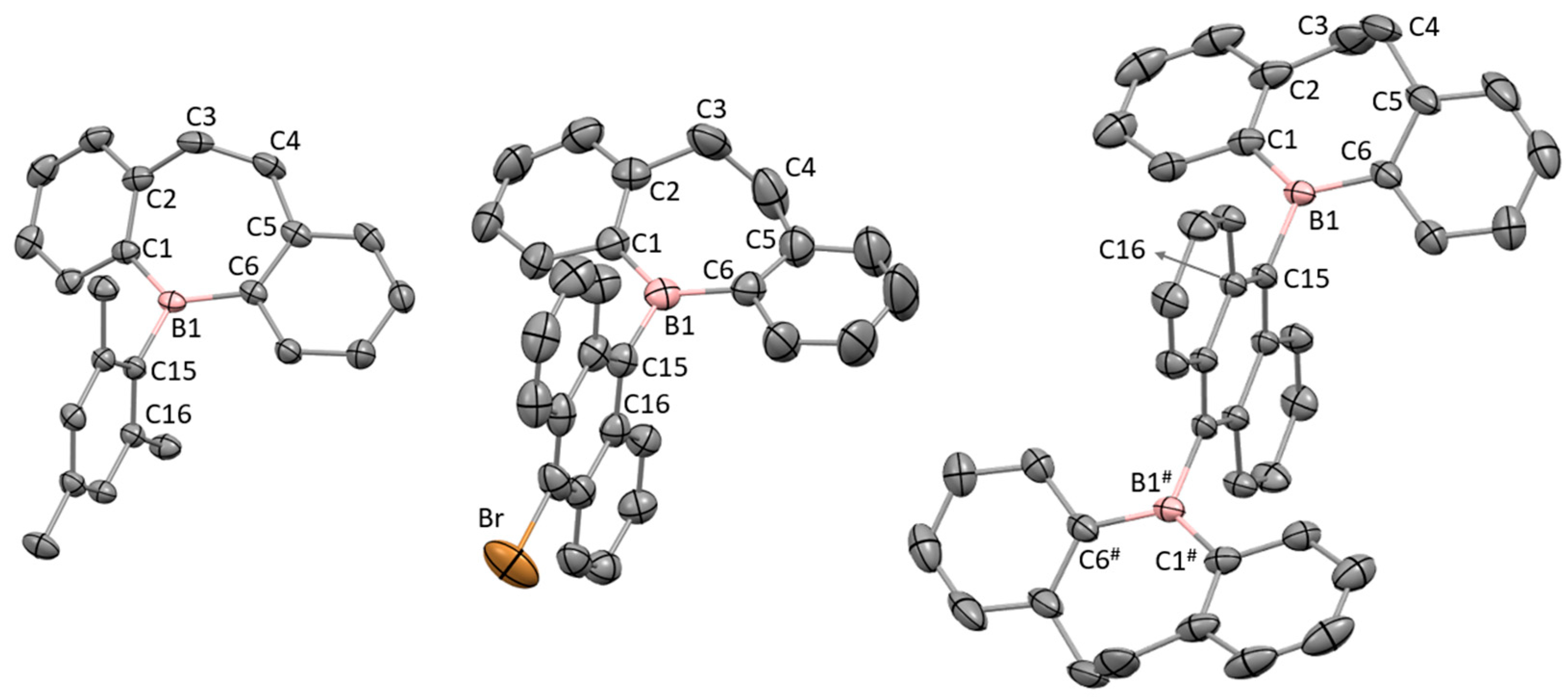
Figure 2.
UV-vis absorption and emission spectra of 1-sp3 (top) and 1-sp2 (bottom) in various solvents (excited at 350 nm. c = 2.8 × 10−5 M). The solid lines indicate absorption spectra, and the broken lines emission spectra.
Figure 2.
UV-vis absorption and emission spectra of 1-sp3 (top) and 1-sp2 (bottom) in various solvents (excited at 350 nm. c = 2.8 × 10−5 M). The solid lines indicate absorption spectra, and the broken lines emission spectra.

Figure 3.
Frontier molecular orbitals and their energy levels (eV) for anthracene, 1-sp3, 1-sp2 and 6-sp3 at CAM-B3LYP/6-31G(d,p)//B3LYP/6-31G(d) (isovalue = 0.03). Red bars: MOs mainly from anthracene. Blue bars: MOs mainly from (dihydro)dibenzoborepins.
Figure 3.
Frontier molecular orbitals and their energy levels (eV) for anthracene, 1-sp3, 1-sp2 and 6-sp3 at CAM-B3LYP/6-31G(d,p)//B3LYP/6-31G(d) (isovalue = 0.03). Red bars: MOs mainly from anthracene. Blue bars: MOs mainly from (dihydro)dibenzoborepins.
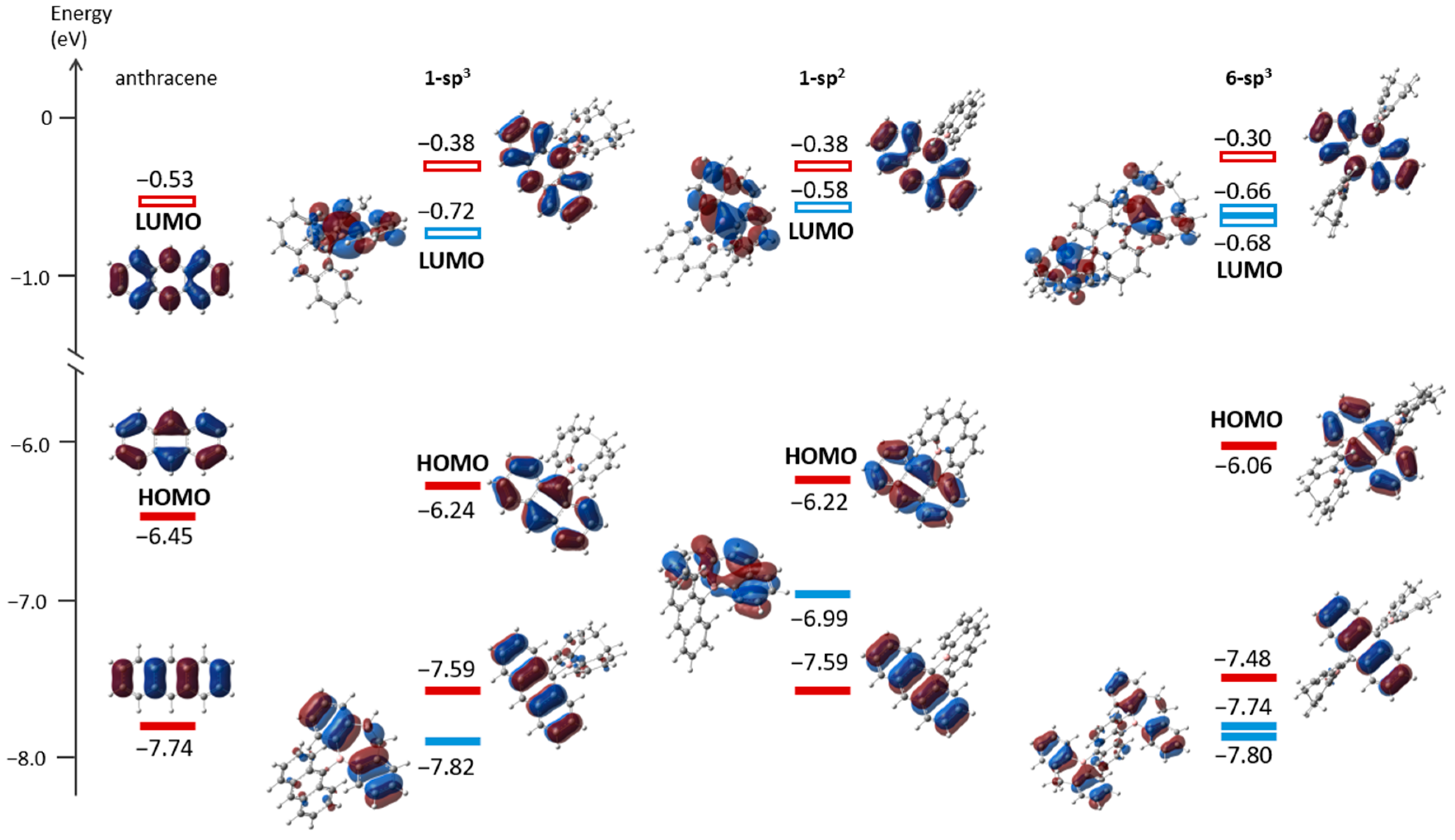

Table 1.
Synthesis of 1-sp2 and 1-sp3 using AnthBpin and AnthB(OR)2.
 | ||||
|---|---|---|---|---|
| entry | Boron source | Nucleophile | Conditions | Yield |
| 1 | AnthBpin | 2Li-sp2 | C6H6/Et2O, 5°C to r.t., overnight | 1-sp2 (–) |
| 2 | AnthBpin | 2Li-sp3 | C6H6/Et2O, 5°C to r.t., overnight | 1-sp3 (–) |
| 3 | AnthBpin | 2Mg-sp3 | THF, reflux, overnight | 1-sp3 (62%) |
| 4 | AnthB(OMe)2 | 2Mg-sp3 | THF, reflux, overnight | 1-sp3 (31%) |
| 5 | AnthBpin | 2Mg-sp2a | THF, reflux, overnight | 1-sp2 (–) |
aA mixture of 2Mg-sp2, its E-isomer and phenanthrene was used.
Table 2.
Table 2. Selected bond lengths [Å] and angles [°].
| 3 | 5-sp3 | 6-sp3 | |
|---|---|---|---|
| B1−C1/C6 | 1.5682(18)/ 1.5706(19) |
1.562(5)/ 1.567(5) |
1.572(3)/ 1.569(4) |
| B1−C15 | 1.5866(17) | 1.592(5) | 1.594(3) |
| C1−B1−C15−C16 | 89.80(14) | 89.2(4) | 80.2(2) |
| C1−C2−C5−C6 | 19.1(1) | 20.2(6) | 18.9(2) |
Table 3.
Absorption and emission properties of 1-sp3, 1-sp2 and 6-sp3.
| Solvent | λabs/nm (ε) | λemi/nm | Stokes shift/cm−1 | |
|---|---|---|---|---|
| 1-sp3 | hexane | 385 (9300), 366 (10000), 348 (8300) | 380, 400, 425, 446 | 3550 |
| toluene | 388 (9300), 369 (11000), 351 (8400) | 385, 407, 431, 487 | 5240 | |
| THF | 387 (9700), 367 (11000), 350 (8400) | 382, 404, 426, 503 | 5960 | |
| CHCl3 | 389 (7200), 369 (8300), 348 (6800) | 384, 407, 430, 508 | 6020 | |
| CH2Cl2 | 388 (9400), 369 (10000), 351 (6900) | 441, 513 | 6280 | |
| 1-sp2 | hexane | 388 (14000), 368 (10000), 350 (3900), 327 (16000), 313 (8600) |
432, 451 | 3600 |
| toluene | 391 (17000), 371 (13000), 353 (6400), 330 (21000), 316 (14000) |
471 | 4340 | |
| THF | 390 (15000), 370 (12000), 352 (5400), 329 (18000), 315 (10000) |
410, 425 | 2110 | |
| CHCl3 | 391 (15000), 372 (13000), 353 (9100), 330 (22000), 316 (16000) |
489 | 5130 | |
| CH2Cl2 | 391 (12000), 371 (9200), 353 (4000), 330 (14000), 316 (8700) |
432, 496 | 5410 | |
| 6-sp3 | CH2Cl2 | 398 (9300), 378 (10000), 360 (7900) | 532 | 6330 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



