
Submitted:
24 July 2024
Posted:
26 July 2024
You are already at the latest version
Abstract
Smith-Magenis syndrome is a complex neurobehavioral genetic disorder. The spectrum of SMS phenotype is broad but commonly includes craniofacial, neurobehavioral, and otolaryngologic features. While the etiology of SMS is commonly attributed to one copy interstitial deletion in the 17p11.2 region (90-95% of cases), variants identified by sequence analysis in RAI1 have also been reported in 5-10% of cases. The phenotypic spectrum of patients with RAI1 sequence alterations is not clear. In this study, we report a 9-year-old male with global cognitive and psychomotor developmental delay, musculoskeletal and cardiovascular abnormalities, and dysmorphic craniofacial features. Whole genome quad analysis of the proband, unaffected parent, and unaffected brother identified a novel de novo RAI1:c.2736delC variant. This is the first report of this variant in the literature. This report also highlights the details of the genome analysis and this patient's phenotypic spectrum.
Keywords:
Smith-Magenis syndrome
; retinoic acid-induced 1 protein
; neurodevelopmental disorders
; joint whole genome sequencing analysis
1. Introduction
Smith-Magenis syndrome (SMS; OMIM #182290) is a complex neurobehavioral disorder with an estimated prevalence of 1:15,000. This rare syndrome is characterized by neurodevelopmental delay, coarse facial features evolving with age, and maladaptive behaviors that include prolonged temper tantrums and self-injurious behaviors, including hand biting, trichotillomania, and polyembolokoilamania [1]. During infancy, hypotonia, hyporeflexia, and feeding difficulties are commonly seen neurological findings [2,3,4,5]. The maladaptive behavior phenotype is typically recognized after 18 months of age. The classic neurological features include intellectual disability, delayed speech, sensory integration problems, decreased pain sensitivity, and abnormalities in sleep patterns [1]. Additional system involvements, such as musculoskeletal and cardiovascular, are also reported as part of SMS [2,3,4,5,6].
The underlying genomic etiology of SMS is interstitial deletion at 17p11.2 in 90-95% of patients. This genomic location encompasses the retinoic acid-induced 1 (RAI1) gene. RAI1 is the key gene in SMS and plays a regulatory role in healthy skeletal and nervous system development, behavioral maturation, and circadian rhythm. Pathogenic de novo single nucleotide variants (SNVs) in RAI1 are reported in 5-10% of patients with SMS [2,5,6,7]. In this category, missense, nonsense, and splice site variants in RAI1 have been reported [4]. The diagnosis of the SMS syndrome is based on clinical molecular diagnostics tests. The first-tier molecular test is chromosomal microarray analysis (CMA), and if negative, a sequencing-based test is typically performed. Genome studies have reported SMS-like features, including ID, sleep disturbances, and self-injuries behaviors in patients without an SMS molecular diagnosis who harbored deleterious variants in KMTD2, MECP2, KDM5C, IQSEC2, and DEAF1 genes [8,9].
Here, we report a 9-year-old male with global cognitive and psychomotor developmental delay and facial, musculoskeletal, and cardiovascular abnormalities. The patient’s initial chromosomal microarray and karyotype tests were normal. The quad joint WGS analysis of the proband, parents, and brother detected a pathogenic novel de novo variant, which has not been reported previously. This report highlights the importance of sequencing analysis in patients clinically suspected of SMS and with negative conventional diagnostic tests. Additionally, sharing the genotype and detailed phenotype of these relatively rare SMS cases helps understand the genetic and phenotypic spectrum of the disease.
2. Material and Methods
2.1. Participants
The index patient, a 9-year-old male, his parents, and his younger brother were enrolled in the CROseq genome program. The CROseq Genome Program is a collaborative research program between Brigham and Women’s Hospital (BWH) (Boston, USA), and the Department of Pediatrics, University Hospital Centre Zagreb (Zagreb, Croatia), supported by the Mila Za Sve Foundation (Rijeka, Croatia). This program aims to investigate the genomic etiology of disease in patients with complex phenotypes and negative conventional genetic tests. Patient consent, enrollment, and clinical assessment were performed at the University Hospital Center Zagreb.
2.2. Clinical Assessment of Brain Structure and Activity
The brain abnormalities were assessed by MRI scanning (MAGNETOM Trio, A Tim System 3T eco, Zagreb, Croatia). The patient was under general anesthesia during the scan. Age-adjusted repetition time and time-to-echo were applied to create multiplanar T1- and T2-weighted images. The electroencephalogram (Nihon Kohden, Neurofax EEG-1200K; Zagreb, Croatia) examination was performed to evaluate the brain activity. During the record, the sampling rate was 200-10000 Hz. The patient’s forehead is cleaned with alcohol to lower the impedance less than 5 Ω for each electrode. The electrodes were set up in FPz-F9 and FPz-AF7 of the international 10-20 system, which focuses on the brain in the same hemisphere.
2.3. Traditional Genetic Testing
Karyotyping was performed to analyze chromosomal abnormalities according to standard procedures at University Hospital Centre Zagreb. Briefly, the lymphocytes were purified from the peripheral blood sample. Cells were arrested at metaphase and fixed before being harvested for slide preparation. The number and morphology of chromosomes were assessed in at least 20 metaphases. The karyotype result was reported per The International System for Human Cytogenomic Nomenclature (ISCN) 2020.
Chromosome microarray (CMA) was performed according to standard procedures at University Hospital Centre Zagreb. Briefly, genomic DNA was isolated with the FlexiGene DNA Kit (Qiagen) and CMA was performed using Agilent 4x180k aCGH+SNP array (Agilent Technologies, Santa Clara, CA, USA). The genomic DNA of the test and control samples were labeled with fluorescent dyes and hybridized to the array. The array was scanned with a DNA Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA). Data were obtained using the “CytoGenomics 5.1.1.15” software (Agilent Technologies, Santa Clara, CA, USA) to generate the final plots.
2.4. Sample Preparation and Whole Genome Sequencing (WGS)
DNA extraction and WGS were performed at the Medical College of Wisconsin (Milwaukee, USA). Genomic DNA was isolated from peripheral blood samples (2ml) with a purity ratio of 1.75 – 2.0. After robotic DNA library construction, sequencing was conducted on the NovaSeq 6000 platform, with an average depth of 40X.
2.5. Quad Joint Whole Genome Analysis
Genome analyses were performed at the BWH, USA. The quad whole genome analysis was performed on proband, unaffected parents, and the unaffected proband’s brother. Following the quad analysis, a phenotype-based analysis was performed using human phenotype ontology (HPO) terms of the patient’s indications. Variants were prioritized by the gene’s association with the phenotype and variant type. The HPO terms were: Neurodevelopmental delay (HP:0012758), moderate intellectual disability (HP:0002342), delayed speech and language development (HP:0000750), attention deficit hyperactivity disorder (HP:0007018), bicuspid aortic valve (HP:0001647) and stenosis (HP:0001650), failure to thrive (HP:0001508), abnormal facial shape (HP:0001999), unilateral ptosis (HP:0007687), frontal bossing (HP:0002007), relative macrocephaly (HP:0004482), abnormality of mitochondrial metabolism (HP:0003287).
All variants across the genome were included in the investigation. Following the technical assessment, medium, high, and very high-quality variants were further evaluated, and low-quality variants were excluded. The variant frequencies were annotated by gnomAD exome and genome population allele frequencies. The in silico prediction algorithms for missense variants included CADD, REVEL, Polyphen, SIFT, MutationTaster, Mutation Assessor, FATHMM, FITCONS, GENOCANYON, dbscSNV ADA, and dbscSNV RF. The spliceAI prediction score was used in the evaluation of splice variants.
Zygosity analysis was performed by identifying and evaluating compound heterozygous and homozygous variants in the proband. The recessive variants in the proband inherited from unaffected parents and related to the patient’s HPOs were assessed. Separately, de novo analysis was performed to assess variants present in the proband and related to the patient’s HPOs that are absent in the unaffected parents and the brother.
The variant classification was performed according to ACMG-AMP guidelines (25741868). In the attribution of PM2 and BS1 criteria, the gene-specific threshold was applied based on gnomAD (v2.1.1) aggregated allele frequency. The aggregated in silico prediction score was indicative of a deleterious effect if it was higher than 0.7; scores less than 0.15 were assigned for a benign effect. The genes’ tolerance to the loss of function variants was evaluated by using gene constraint metrics of pLI=1 and/or o/e<0.35.
3. Results
3.1. Clinical Description
The proband is a 9-year-old male presented with indications of global cognitive and psychomotor developmental delay, musculoskeletal and cardiovascular abnormalities, and dysmorphic craniofacial features. During his infancy, the patient had generalized hypotonia and feeding difficulties. His brain ultrasound, which was performed at four months of age, was normal. Following intensive physical therapy, he started walking at two years old. The psychomotor assessment revealed that the patient has severe psychomotor retardation. The brain MRI performed at 3.5 years of age showed perinatal hypoxic-ischemic lesions. The electroencephalogram was unremarkable (data not shown).
The detailed clinical evaluation at 9 years of age indicated brachycephaly, broad face, hypertelorism, wide nasal bridge, short and full-tipped nose, midface retrusion, deeply set ears, tented and down-turned upper lip, everted upper lip vermilion, micrognathia, and low hairline (Figure 1). He exhibited global cognitive and psychomotor developmental delay, severe ID, and developmental language disorder. The pervasive developmental disorder was evident with the lack of speech development and poor contact. Behavioral phenotypes included aggression toward others self-injurious episodes, and polyembolokoilamania. He did not acquire sphincter control and exhibited incontinence. He had sleep disturbances, including night awakening, with difficulty falling back asleep. There were skeletal abnormalities including mild scoliosis, thoracic kyphosis, short palm and foot, brachydactyly, and partial syndactyly of both hands’ second and third fingers (Figure 1). He reportedly had a bicuspid aortic valve with mild stenosis and congenital left eyelid ptosis.
3.2. Genomic Findings
CMA and Karyotype Analysis
Karyotype testing was 46, XY. CMA analysis of the proband detected a maternally inherited 2.1 Mb duplication in 22q11.21 (Supplementary Figure S1 and S2). This region contains OMIM genes SLC25A1, CDC45, GP1BB, TBX1, TXNRD2, COMT, TANGO2, RTN4R, SCARF2, PI4KA, SERPIND1, SNAP29, and LZTR1. The mother is unaffected, therefore, this 22q11.21 gain was considered unrelated to the patient’s clinical findings of SMS. The patient was enrolled in the CROseq genome program, and a quad WGS joint analysis was performed.
Quad WGS Joint Analysis
After WGS, a quad-WGS analysis of the proband, unaffected brother, and parents was performed. HPO-based analysis revealed 10,290 variants in 2,567 genes. Compound heterozygous analysis interrogated possible causative variants inherited from the unaffected parents. Quad analysis did not identify deleterious homozygous or compound heterozygous candidate variants in the proband.
De novo analysis interrogated variants related to the HPO that are present in the proband, but absent in the parents and the unaffected sibling. This analysis identified the variant NM_030665.4 (RAI1):c.2736delC (p.Gly913Alafs*37), located in exon 3 of RAI1 (Figure 2). This variant is a 1bp deletion that results in a frameshift and a premature stop codon. Due to its location, this variant likely results in a premature translational stop signal and nonsense-mediated mRNA decay. The gene constraint metrics for RAI1 are pLI = 1, o/e = 0.04. The variant has not been reported in gnomAD, ClinVar, or any published literature. The phasing analysis by quad joint WGS showed that the parents and younger brother did not harbor this variant. No other deleterious variants in genes related to the patient’s HPO list were identified.
Discussion
Smith-Magenis Syndrome, also known as 17p11.2 microdeletion syndrome, was first discovered in 1986 [10]. Since then, the genomic and clinical spectrum of more than 500 SMS patients has been reported [11]. Most cases of SMS are caused by microdeletion in 17p11.2, while 5-10% of cases are due to pathogenic RAI1 sequence variants [2]. When the phenotypic findings suggest SMS, the first molecular testing approach is generally CMA, followed by a single gene or multiplanel sequence analysis that includes RAI1.
The RAI1 has a regulatory role in embryogenesis through the transcriptional regulation of many genes [12,13,14]. These include the skeletal development genes PSTPIP2 and ANGH [15]; lipid metabolism genes LIPE, HMGCS1, and INSIG1 [15]; neurological development genes ZIC1, PSEN2, RXRB, CLN8, SMA4, NF1, and KMT2A; behavioral function gene SCN12A 19236431 [15]; circadian activity genes NR1D2, PER2, PER3, CRY1, and ARNTL 22578325; cellular growth and cell cycle regulation genes SPTBN1, POLDIP3, PPP1R14D, GLI3, KMT2A, and ADD3 [15]; and insulin regulation genes INSIG1, PIK3R1, ZNF236, and LIPE [15]. As a result, patients with SMS commonly have moderate to severe neurodevelopmental and behavioral abnormalities, metabolic problems, sleep problems, and skeletal abnormalities [3].
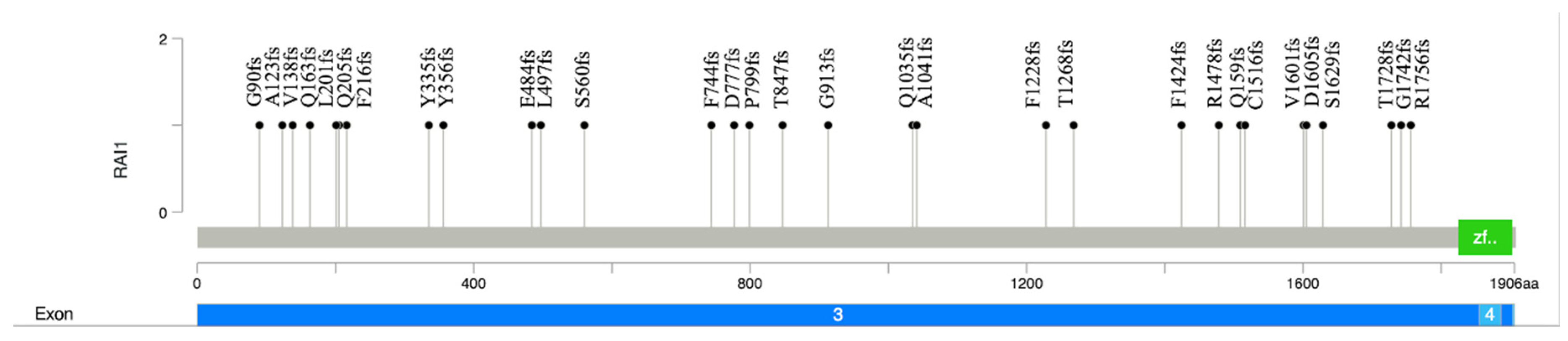
Here, we report a patient with a spectrum of Smith-Magenis syndrome and the pathogenic de novo novel NM_030665.4 (RAI1):c.2736delC variant. This is variant is located in exon 3 (of 6 exons). Most reported pathogenic RAI1 variants are located in exon 3 [4] which encodes nearly 98% of the protein. There are reports of mutational hot spots in this exon [16]. The RAI1-encoded nuclear protein has a zinc finger homology structural domain, which is indicative of the transcription regulatory activity of RAI1 [17]. The truncating variants lead to aberrant cytoplasmic subcellular localization and, therefore, the inability of transcription activation [18]. To date, there are 78 frameshift, 29 nonsense, and 4 missense variants reported as pathogenic or likely pathogenic in ClinVar. Most of these variants, including the (RAI1):c.2736delC variant in our case, are located in exon 3 (Figure 2).
The broad phenotypical variation of SMS stems from the genetic pathophysiology of the genomic region. Patients with the 17p11.2 microdeletion are more likely to have severe cognitive impairment, hearing loss, cardiac abnormalities, and hypotonia. These features are associated with the deletion of genes residing within the region [19]. Patients with normal copy17p number who carry a deleterious sequence variant in RAI1 reportedly exhibit polyembolokoilamania (inserting foreign objects into orifices), skin picking, and self-hugging behavior [19]. Consistent with these reports, our patient exhibited these behaviors. Our patient also exhibited severe intellectual disability and cardiac abnormalities, as well as hypotonia during infancy. The joint analysis did not reveal any additional alterations in the genome consistent with these phenotypes.
In conclusion, we report a detailed clinical description of a patient with SMS and the novel pathogenic RAI1 sequence variant. Studies reporting clinical findings of SMS and pathogenic RAI1 variants help expand the understanding of the genetic pathophysiology of this complex syndrome.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Chromosomal microarray of the proband. Interstitial duplication of the long arm of chromosome 22 in the region 22q11.21 (18996947_21086225), 2.1 Mb in size (arr[GRCh38] 22q11.21(18996947-21086225)x3 mat). Figure S2: Chromosomal microarray of the mother. Interstitial duplication of the long arm of chromosome 22 in the q11.21 region (18996947-21454704), 2.46 Mb in size (arr[GRCh38]22q11.21 (18996947_21454704)x3.
Funding
This investigation was done in collaboration with Brigham and Women’s Hospital, Inc/University Hospital Centre Zagreb, with funding support provided by Mila za Sve Foundation.
Institutional Review Board Statement
Institutional Review Board at BWH and University Hospital Centre Zagreb approved this study (Class: 8.1-21/6-2; Reg. No.: 02/21 AG). Written informed consent was obtained from all the participants at the University Hospital Centre Zagreb, Croatia. This study is compliant with the General Data Protection Regulation (GDPR), approved by BWH, Mila Za Sve Foundation, and the University Hospital Centre Zagreb, Croatia.
Data Availability Statement
Data for this manuscript are subject to BWH institutional and GDPR privacy policies and restricted from inclusion in repositories. They may be available upon request.
Acknowledgments
We thank our patient and their family for their participation in the CROseq Genome Program and for permitting us to anonymously share the clinical details.
References
- Elsea, S.H.; Girirajan, S. Smith-Magenis syndrome. Eur J Hum Genet. 2008, 16, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Duncan, W.C.; Smith, A.C. Neurologic and developmental features of the Smith-Magenis syndrome (del 17p11.2). Pediatr Neurol. 2006, 34, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Feldman, J.; Mirzaa, G.M.; et al. GeneReviews. 1993.
- Rinaldi, B.; Villa, R.; Sironi, A.; Garavelli, L.; Finelli, P.; Bedeschi, M.F. Smith-Magenis Syndrome-Clinical Review, Biological Background and Related Disorders. Genes (Basel). 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Yeetong, P.; Vilboux, T.; Ciccone, C.; et al. Delayed diagnosis in a house of correction: Smith-Magenis syndrome due to a de novo nonsense RAI1 variant. Am J Med Genet A. 2016, 170, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Mora, P.; Encina, C.A.; Canales, C.P.; et al. Functional and cellular characterization of human Retinoic Acid Induced 1 (RAI1) mutations associated with Smith-Magenis Syndrome. BMC Mol Biol 2010, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Slager, R.E.; Newton, T.L.; Vlangos, C.N.; Finucane, B.; Elsea, S.H. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003, 33, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Loviglio, M.N.; Beck, C.R.; White, J.J.; et al. Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med. 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.I.; Ciccone, C.; Simon, K.L.; et al. Exome analysis of Smith-Magenis-like syndrome cohort identifies de novo likely pathogenic variants. Hum Genet. 2017, 136, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; McGavran, L.; Robinson, J.; et al. Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet. 1986, 24, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.L.; Gropman, A.L.; Martin, S.C.; et al. Neurodevelopment of children under 3 years of age with Smith-Magenis syndrome. Pediatr Neurol. 2009, 41, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Tahir, R.; Kennedy, A.; Elsea, S.H.; Dickinson, A.J. Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mech Dev 2014, 133, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Yan, J.; Shi, X.; et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum Mol Genet. 2007, 16, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Falco, M.; Amabile, S.; Acquaviva, F. gene mutations: mechanisms of Smith-Magenis syndrome. Appl Clin Genet. 2017, 10, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Truong, H.T.; Blanchard, C.L.; Elsea, S.H. A functional network module for Smith-Magenis syndrome. Clin Genet. 2009, 75, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, C.; Bonnet-Brilhault, F.; Toutain, A.; et al. Identification of Nine New RAI1-Truncating Mutations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions. Mol Syndromol. 2014, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Saifi, G.M.; Shaw, C.J.; et al. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004, 115, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Mora, P.; Canales, C.P.; Cao, L.; et al. RAI1 transcription factor activity is impaired in mutants associated with Smith-Magenis Syndrome. PLoS One. 2012, 7, e45155. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Vlangos, C.N.; Szomju, B.B.; et al. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006, 8, 417–427. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Dysmorphic features of the proband at the age of 9 years. A) Brachycephaly, broad face, hypertelorism, wide nasal bridge, midface retrusion, deeply set ears, tented and down-turned upper lip, everted upper lip vermilion, light-colored hair; B) Brachycephaly, prominent forehead, short and full-tipped nose, deeply set ears, midface retrusion, micrognathia, tented and down-turned upper lip, everted upper lip vermilion, light-colored hair; C) Short foot, brachydactyly; D) Short palm, brachydactyly.
Figure 1.
Dysmorphic features of the proband at the age of 9 years. A) Brachycephaly, broad face, hypertelorism, wide nasal bridge, midface retrusion, deeply set ears, tented and down-turned upper lip, everted upper lip vermilion, light-colored hair; B) Brachycephaly, prominent forehead, short and full-tipped nose, deeply set ears, midface retrusion, micrognathia, tented and down-turned upper lip, everted upper lip vermilion, light-colored hair; C) Short foot, brachydactyly; D) Short palm, brachydactyly.
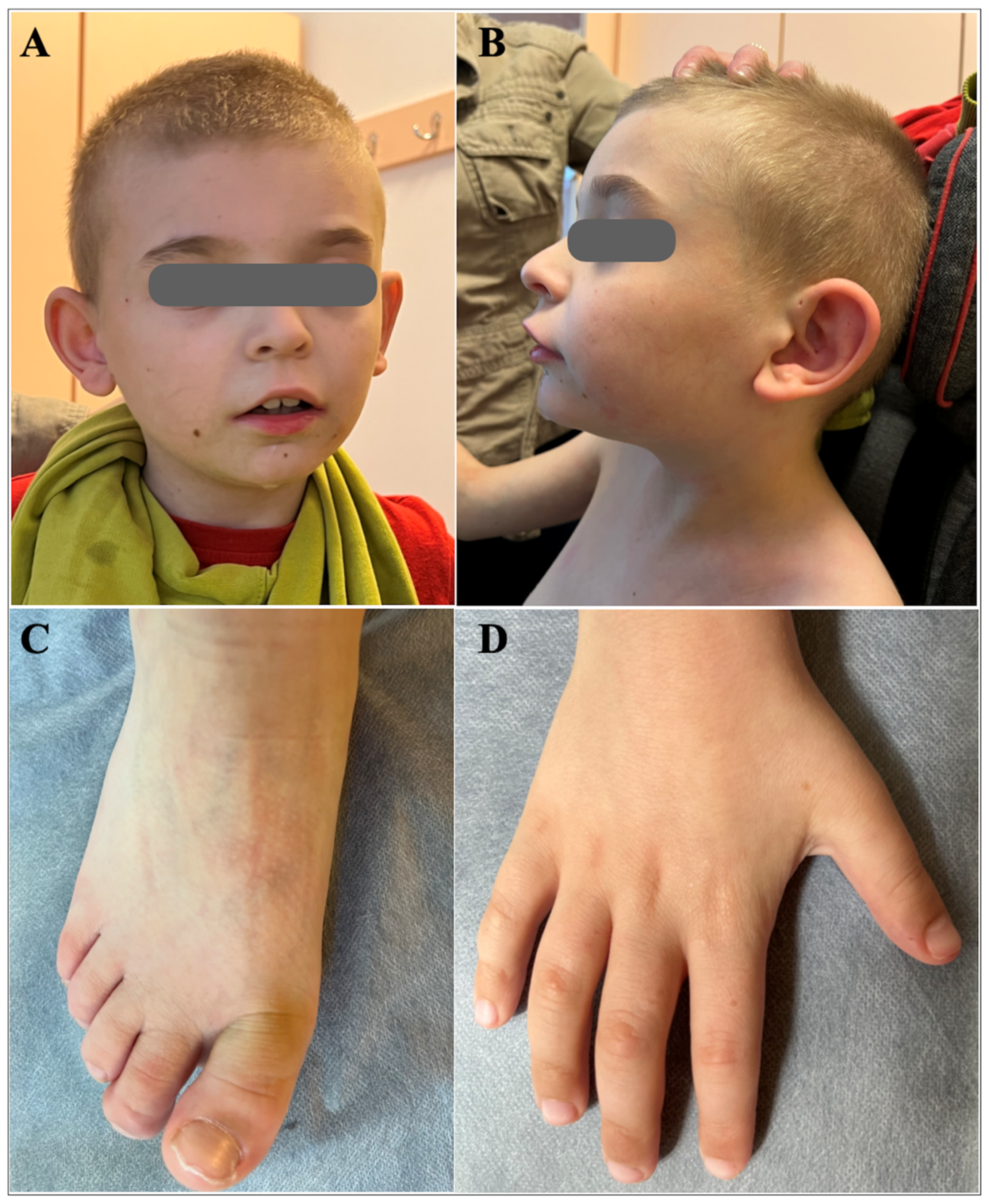
Figure 2.
Lollipop plot of the pathogenic and likely pathogenic RAI1 frameshift variants. The plot shows the p.G913fs variant identified in the proband in this study and the distribution of frameshift RAI1 variants reported in the ClinVar database to date (n=30). The Y axis shows the variant number, and the X axis illustrates the position of amino acid residues along the RAI1 protein. It includes exon 3 and exon 4, the green box denotes the PHD-like zinc-binding domain.
Figure 2.
Lollipop plot of the pathogenic and likely pathogenic RAI1 frameshift variants. The plot shows the p.G913fs variant identified in the proband in this study and the distribution of frameshift RAI1 variants reported in the ClinVar database to date (n=30). The Y axis shows the variant number, and the X axis illustrates the position of amino acid residues along the RAI1 protein. It includes exon 3 and exon 4, the green box denotes the PHD-like zinc-binding domain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



