
Submitted:
24 July 2024
Posted:
25 July 2024
You are already at the latest version
Abstract
Over the past decade, the production of biofuels from lignocellulosic biomass has steadily increased to offset the use of fuels from petroleum. To make biofuels cost-competitive, however, it is necessary to add value to the “ligno-” components (up to 30% by mass) of the biomass. The properties of lignin, in terms of molecular weight (MW), chemical functionality, and mineral impurities often vary from biomass source and biorefinery process, resulting in a challenging precursor for product development. Activated carbon (AC) is a feasible target for the lignin-rich byproduct streams because it can be made from nearly any biomass and it has a market capacity large enough to use much of the lignin generated from the biorefineries. However, it is not known how the variability in the lignin affects the key properties of AC, because until now, they could not be well controlled. In this work, various fractions of ultraclean (< 0.6 % ash) lignin are created with refined MW distributions using Aqueous Lignin Purification using Hot Agents (ALPHA) and used as precursors for AC. AC is synthesized via zinc chloride activation and characterized for pore structure and adsorption capacity. We show that AC surface area and the adsorption capacity increase when using lignin with increasing MW, and furthermore, that reducing the mineral content of lignin can significantly enhance the AC properties. The surface area of the AC from the highest MW lignin can reach ~1830 m2/g (absorption capacity). Furthermore, the single step activation carbonization using zinc chloride allows for minimal carbon burnoff (< 30%) capturing most of the lignin carbon compared to traditional burn off methods in biorefineries for heat generation.
Keywords:
Activated Carbon
; Lignin
; Bioproducts
; Lignin Molecular Weight
; Lignocellulosic
; Biorefinery
1. Introduction
Further development and utilization of energy crops for the purpose of producing lignocellulosic biofuels will result in a dramatic reduction in atmosphere CO2 levels by replacing or substituting fuels derived from petroleum. The effect of CO2 reduction can be compounded when the “ligno-” component of the biomass can be converted into a solid product rather than burning it for its heating value, thereby results in a net-negative CO2 output. By 2030, the global biofuels market is anticipated to expand significantly, reaching a value of 176.5 billion US dollars [1]. Instead of relying on expensive and edible biomass as feedstock, using lignocellulosic biomass is a better option for producing biofuels. This type of biomass is readily available in nature and is much more cost-effective [2,3,4,5]. During the biofuels production process, biorefineries extract cellulose and hemicellulose to produce biofuels, while separating lignin as a byproduct [4,5,6]. Currently, most of the separated lignin from biorefineries is burned as a low-value fuel for heat and electricity generation, thereby returning the carbon back to the environment. A similar situation occurs in paper mills, where cellulose is used to produce paper and lignin is burned. In all, nearly 100 million tons of lignin are separated annually from biorefineries and paper mills, while 98% of them are combusted for energy recovery [7].
Lignin, which comprises up to 30% of the mass of the lignocellulosic biomass [8,9], is the most abundant natural biopolymer that is rich in aromatic functionality. Instead of combusting lignin for heat or electricity, it can be used to produce chemicals and incorporated into various materials, such as hydrogels [10], polyurethane foams [11], epoxy resin [12], and phenolic powder resin [12]. Moreover, its high carbon content makes it suitable for carbon-rich products, including carbon fiber [13] and activated carbon [14]. Notably, lignin is a natural polymer, and its use as an alternative to replace petrochemical precursors for these products can significantly reduce the need for fossil fuel. However, the application of lignin is sometimes less than perfect. The main issue is that the variability in the properties of lignin, such as impurity content and heterogeneous molecular weight (MW), is inevitable as they arise from different natural environments where the lignin originates. These inconsistencies pose challenges to the industrial application of lignin. For instance, carbon fiber production requires very clean lignin precursors with high molecular weight; otherwise, its mechanical performance will be poor [15].
Activated carbon (AC), also called activated charcoal, is a kind of processed carbon characterized by high specific surface area and controllable porosity [14]. It can be produced from carbonaceous source materials such as wood [16], coal [17], biomass [18,19,20], or petroleum pitch [21] through physical or chemical activation. Physical activation, which is a two-step process, involves carbonizing the material and then activating it at high temperatures using steam, carbon dioxide, or air as the activating agent [14], resulting in a wider pore size distribution but higher burn-off and lower yield. On the other hand, chemical activation is a one-step process that simultaneously activates and carbonizes the carbonaceous material at low temperature with a chemical activating agent like zinc chloride, potassium hydroxide, or phosphoric acid. This method leads to a narrower pore size distribution, lower burn-off, and generally higher yields compared to physical activation
The porous structure of AC gives rise to high specific surface areas and particularly high adsorption capacities, making it widely used in water/gas purification [22,23,24], food/beverage processing [25], heterogeneous catalysis [26], and electronics [27,28,29]. The global AC market size was estimated at 4.92 billion US dollars in 2023 and is projected to grow at a compound annual growth rate of 6.0% from 2024 to 2030[30]. This large market volume is important because of the mass quantities of available lignin byproduct. In addition to the inherent demand for AC for the applications noted above, converting lignin from energy crops into solid carbon serves a dual purpose of carbon capture and storage. Theoretically, the amount of AC that can be produced from lignin indicates the amount of carbon that can be captured and stored. But that is not all; an increase in supply of AC, specifically for the purpose of water purification, will result in lower cost clean water. Since AC does not have strict limitations on its precursor quality, theoretically, any lignin can be carbonized and activated. Lignin with high impurity content and varying molecular weight, which cannot be utilized in other applications, can be converted into AC. Such flexibility allows lignin with inconsistent properties to be utilized in AC production, expanding its potential uses across various industries.
The general goal of synthesizing AC is to achieve the highest surface area at the lowest cost (higher AC yield). To achieve this, particularly with a biopolymer like lignin which inherently exhibits inconsistent properties, it is necessary to understand the relationship between lignin properties and the key performance metrics of AC. Previous studies on using lignin as a precursor for AC has shown a wide range in properties, with surface area calculations from <10 to 2753 m2/g, substantial variations in overall product yields, and differences in pore size and distribution [31]. The discrepancies in results arises because of variations in activation processes (either physical or chemical) and lignin molecular properties. Until recently, previous studies typically focused on evaluating the effects of activation agents and/or carbonization conditions on AC properties, and rarely consider how the properties of lignin affect the resulting activate carbon.
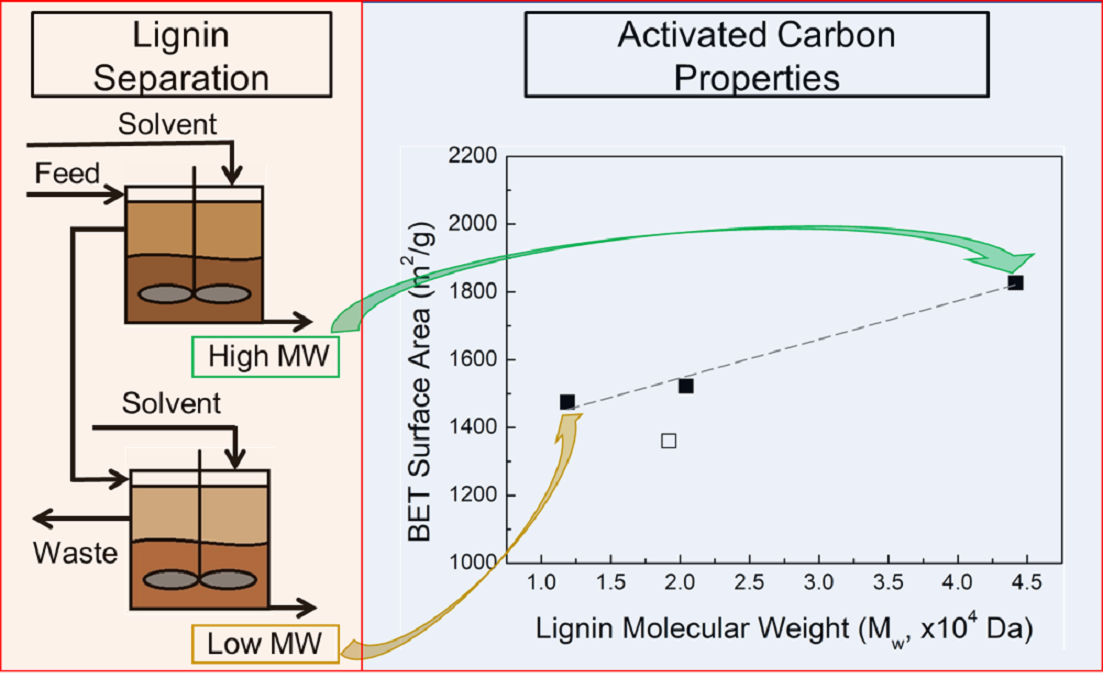
In our previous work, we presented clear correlations between the composition of lignocellulosic biomass – lignin, cellulose/hemicellulose, and ash content – and the AC surface area, pore width, pore distribution, and carbon fractional conversion [32]. Despite these findings, it is less clear how the lignin and its properties affect the resulting activate carbon, because controlling lignin properties at the molecular level is challenging. In response to the difficulty in using lignin as a precursor for carbon-based products, the Aqueous Lignin Purification using Hot Agents (ALPHA) process [13,33,34] (Figure 1) has been developed to purify and fractionate lignin based on molecular properties. The ALPHA process involves mixing the lignin sample with an organic solvent at elevated temperature to create a liquid-liquid equilibrium, resulting in the formation of a lignin-rich phase and a solvent-rich phase. In this process, higher MW lignin is generally directed to the lignin-rich phase, while lower MW goes to the solvent-rich phase. By adjusting the operation conditions, different ultraclean lignin fractions with refined MWs and distributions can be tailored during the ALPHA process. This method enables some degree of control over the lignin MW.
Here, we investigate the relationship between lignin MW and mineral content on the key performance metrics of AC, including surface area, pore volume, pore size, and carbon fractional conversion. Pure lignin samples with different MWs and narrow MW distributions were isolated from a commercial Kraft lignin feedstock using the ALPHA process [13,33,34]. AC was synthesized from these lignin samples using a ZnCl2 activation process with relatively low-temperature carbonization. We found that the surface area of AC is correlated with lignin, and pore enlargement is promoted in lignin sample with lower MW. Because lignin from biorefineries and paper mills typically contains considerable amount of ash content, and the effect of lignin MW on the AC product is often coupled with the effect of ash content when directly using feedstock lignin. Therefore, we also demonstrate how the lignin ash content affects AC properties by controlling for lignin MW. The surface area and total pore volume of AC are lower when synthesized from lignin samples with higher ash content, and the higher ash content also promotes pore enlargement.
2. Experimental
2.1. Lignin sample preparation
The lignin feedstock (LF) was obtained from a commercial source, named Biochoice® lignin, which is supplied by Domtar Corp. The LF was fractionated through the ALPHA (Aqueous Lignin Purification using Hot Agents) process [13,33,34]. In all cases, a 50 mL reactor (model 4593, Parr Instrument) was used for ALPHA processing, and ~10 g of the LF was charged into the reactor along with ~30 g of solvent. The mixture was stirred and heated to a specific temperature, allowing it to mix for at least 15 mins. After the mixing time, the reactor was opened, revealing a liquid lignin-rich (LR) and solvent-rich (SR) phase. The LR phase adhered to the impeller of the reactor and was then collected into a sample pan, while the SR phase was decanted into a separate pan. Both phases were dried to remove the solvent and ground into a powder with a mortar and pestle. For all fractions, acetone/water mixtures were used as a solvent. All further references to compositions will be denoted on a weight basis.
Three lignin fractions of different molecular weights (MW) were obtained independently from the ALPHA process. They are a high MW lignin fraction (LH), a medium MW lignin fraction (LM), and a low MW lignin fraction (LL).
LH was obtained from the LR phase using a solvent composition of 5:5 (acetone to water), a temperature of 45 °C, and a solvent-to-lignin ratio of 3:1. LM was also obtained from the LR phase, utlizing a solvent composition of 3:7, a temperature of 45 °C, and a solvent-to-lignin ratio of 3:1. The low MW lignin fraction (LL) was acquired via a two-stage ALPHA process. Initially, a SR phase was isolated using 50% acetone at 45 °C. This SR phase was then dried to a powder to recover a low MW fraction. This fraction was charged to stage 2 where it was contacted with a 40/60 w/w acetone/water solution with the LR phase then recovered as the final product. This second stage was operated at 25 °C. Lignin yields from the LF of the high MW fraction, the medium MW fraction, and the low MW fraction were controlled at 14%, 84%, and 50% respectively at their own individual runs.
2.2. Lignin Characterization
The molecular weight (MW) of lignin was determined using SEC-MALS (size exclusion chromatography, followed by multi-angle light scattering). Lignin was first dissolved at a nominal concentration of 3 mg/mL in a solution of 0.05 M LiBr in dimethyl formamide (DMF). The mixture was sonicated for 30 min and filtered through 0.20 μm PTFE syringe filters. The filtered mixture was then injected into an Agilent 1200 series HPLC system, with a 0.05M LiBr in DMF mobile phase, flowing at 0.6 mL/min. A stationary phase of one HT5 Styragel (WAT045945, Waters) followed by one Polargel-L (PL1117-6830, Agilent) was used for separation, in conjunction with an Optilab-WREX-08 differential refractometer and a Wyatt Technology DAWN MALS instrument (with filtered detectors) used for detection.
Ash content in lignin was determined via Thermogravimetric Analysis (TGA). ~10 mg of lignin was placed in a platinum holder of a thermogravimetric analyzer (Q5000, TA Instruments), heated to 100 °C, held for 15 mins, and then heated to 800 °C. The purge gas was air and the heating ramp was 10 °C/min. The lignin ash content was defined as W/W0, where W0 is the initial lignin mass and W is the remaining mass of the sample at the end of the run.
Metal contents of the lignin and lignin fractions were measured via Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES, model ACROS, Spectro Analytical Instruments). Before measurement, a weight of 100 mg dried lignin was digested in 5 mL concentrated nitric acid at 25 °C for 30 mins and then further digested by heating to 125 °C for 90 mins, followed by adding 3 mL hydrogen peroxide (H2O2) and heating at 125 °C for 60 mins. Afterward, 3 mL additional H2O2 was added, and the sample was kept heated at 125 °C for 60 mins. Finally, the sample was air dry at 200 °C for 1 h and the dried sample was diluted in 10 mL 1.6 M nitric acid and another 50 mL deionized water after cooling. The resulting liquid was transferred to the ICP tube for detection.
2.3. Activated Carbon Synthesis
1.8 g of lignin was dried in a vacuum oven at 60 °C for 12 h, and then sieved through a 60-mesh sieve. The lignin was mixed with ZnCl2 solution at a ratio of 2.5:1 by weight (ZnCl2 anhydrous:lignin) under stirring at 350 RPM for 24 h. The ZnCl2 solution was made by anhydrous ZnCl2 (98+%, Alfa Aesar) and enough DI water to obtain a ratio of 2.0 mL water per gram of total solids (ZnCl2 and lignin). The lignin-ZnCl2 mixture was then dried by rotary evaporation and by vacuum oven at 110 °C for 24 h. The dried mixture was packed in graphite foil and placed in the center of a horizontal quartz tube in an electric furnace (Lindberg/Blue M, Thermo Scientific). The tube was purged with high purity (+99.99%) Nitrogen (N2) at a flow rate of 1000 cm3/min for 30 min and next the N2 flow rate was adjusted to 300 cm3/min for carbonization. The lignin was heated at 10 °C/min to 500 °C and held for 1 h. After cooling to ambient temperature, the AC was washed with 150 mL 3M hydrochloric acid (HCl) for 1 h under stirring at 350 RPM. The HCl acid was filtered (Nylon filter, 0.45μm, Sigma Aldrich) and the AC was then rinsed by 60℃ DI water and filtered repeatedly until the conductivity of the washing water was near the conductivity of the DI water (~0.6 μS/cm). The AC was finally dried in vacuum at 110 °C for 24 h.
2.4. Activated Carbon Characterization
N2 adsorption and desorption isotherms of AC were measured at 77 K by using an automated gas sorption analyzer (Autosorb iQ, Quantachrome Instruments), after samples of approximately 150 mg were degassed at 250 °C for 7.5h under vacuum. Based on the Brunauer-Emmett-Teller (BET) theory, the gas sorption behavior was connected to the porosity of the AC. Total pore volume was determined by the amount of N2 adsorption expressed in liquid form at 77 K and a relative pressure (P/P0) of approximately 0.95. Average pore width was calculated using the specific surface area and the total pore volume by the Gurwitch rule. Pore distribution data were obtained by analyzing N2 desorption data using density functional theory (DFT).
Aqueous adsorption capacity of AC was determined by iodine (I2) number and methylene blue (MEB) value. 60 mg of AC and 20 mg of AC were respectively mixed with 20 mL 0.1 M standard I2 solution and 20 mL 600 mg/L MEB solution under stirring at 125 RPM for 24 h. The mixtures were centrifuged, and clear I2 and MEB solutions were decanted. The clear I2 solution was titrated by 0.025 M sodium thiosulfate (Na2S2O3) and the concentration of the clear MEB solution was measured by an ultraviolet-visible (UV-Vis) spectrophotometer (Agilent BioTek Microplate Readers, Agilent) compared against calibration curve.
2.5. Carbon Fractional Conversion
The carbon fractions in lignin and AC were determined by elemental analysis of combustion products using automatic analyzers in Atlantic Microlab, Inc (Atlanta, Georgia, USA). Before the combustion, lignin was vacuum dried (0.015 mmHg) at 120℃ for 2 h and AC was vacuum dried (0.015 mmHg) at 250℃ for 4 h before combustion. The carbon fractional conversion is defined as the mass of carbon in AC divided by the mass of carbon in lignin. The overall yield is defined as the AC mass divided by the lignin mass.
3. Results and Discussion
3.1. Lignin Samples for Activated Carbon
A commercial lignin, named Biochoice® lignin, was used as the feed lignin (LF) in this work to study the relationship between molecular weight (MW) and ash content of lignin and the properties of AC. This lignin is classified as a sugar-free, low-ash lignin, produced from Southern Pine trees via the Kraft process. Aqueous Lignin Purification using Hot Agents (ALPHA) process, described in greater detail elsewhere [13,33,34]. was used to fractionate the LF to obtain clean lignin with different MWs by manipulating the organic solvent/water ratio and temperature. Typically, during the ALPHA process, the higher MW lignin goes to the lignin-rich phase, while the lower MW lignin and ash remain in the solvent-rich phase. By separating the lignin-rich phase from the solvent-rich phase, a clean, high MW lignin can be obtained. The solvent-rich phase can then be subject to a second stage of ALPHA processing to extract a clean, low MW lignin.
Figure 2a shows the MW of the lignin samples, as determined from size exclusion chromatography followed by multi-angle light scattering (SEC-MALS), as well as the polydispersity index (PDI) in the inset. Through the ALPHA process, we were successful in producing a range of lignin samples with different MWs. Compared to the feed lignin (LF), which had a weight-average MW (Mw) of 19,200 Da, the resulting lignin samples spanned a range of Mw from 11,900 Da to 44,200 Da. Specifically, three distinct lignin samples were obtained: the low MW lignin (LL) with an Mw of 11,900 Da, the medium MW lignin (LM) with an Mw of 20,400 Da, and the high MW lignin (LH) with an Mw of 44,200 Da. Furthermore, The PDI of the three ALPHA-processed lignin samples are similar to the PDI of the LF, indicating that the ALPHA process does not apparently alter the distribution of lignin MW. These three samples all have narrow MW distributions.
Metal content (wt %) of the lignin samples was determined by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) and is shown in Figure 2b. The primary metal in the LF is sodium, which came from the delignification process (Kraft process for Kraft lignin). After the ALPHA process, nearly 90% of the metals are reduced, resulting in LL, LM, and LH all exhibiting very low metal contents lower than 1200 ppm. Figure 2c displays thermogravimetric analysis (TGA) profiles of the LF and the three ALPHA-processed lignin samples. The ash content of the lignin samples, determined as the residue remaining after TGA at 700°C, is presented in Figure 2d. Similar to the metal content, ash in the lignin is much reduced after the ALPHA process. Compared to the ash content of 2.8% in LF, all the ash contents in the three ALPHA-processed lignin samples are lower than 0.6%.
3.2. Activated Carbon Yield and Fractional Conversion
Taking into account the goal of saving energy and achieving the highest possible overall AC yield, AC was synthesized from the LF and the three ALPHA-processed lignin samples using constant ZnCl2 activation with low-temperature carbonization condition. Figure 3a shows overall yield of AC and carbon fractional conversion plotted as a function of MW of lignin. The overall yield of AC is the amount of AC obtained from the lignin, defined as the mass of AC divided by the mass of the starting lignin sample. The carbon fractional conversion is the proportion of carbon in lignin that ends up in AC, defined as the mass of carbon in AC divided by the mass of carbon in lignin. In Figure 3a, the overall AC yields and the carbon fractional conversions of the three ALPHA-processed lignin samples are fairly high, all higher than 50% and 70%. This is due to ZnCl2 as an activating agent inhibits the gasification and tar formation in the carbonaceous material of the lignin [35,36,37]. At the same time, the overall AC yields and the carbon fractional conversions of the three ALPHA-processed lignin samples are relatively consistent, ranging between 55-58% and 71-74%, respectively. This suggests that variations in lignin MW have little effect on the overall AC yield and the carbon fractional conversion, which corresponds to the result of our previous work that the clean lignin samples have the same carbon fractional conversions [32]. Compared to the ALPHA-processed samples, AC derived from the LF exhibits a lower overall AC yield and carbon fractional conversion. Considering that the LF has a high ash content (2.7%), the lower overall AC yield and carbon fractional conversion indicate that ash negatively impacts the conversion from lignin to AC. One possible explanation for this phenomenon could be that the ash, during the preparation of AC, functions as a catalyst, accelerating the burn off of the lignin carbon [38,39].
3.3. Pore Structure of Activated Carbon
3.3.1. Effect of MW of Lignin on Pore Properties
Figure 3 display surface area, pore volume, and average pore width of AC as a function of MW of lignin in panel b, c, and d, respectively. The surface area of AC is interpreted from the nitrogen (N2) adsorption isotherm obtained at 77K, based on the Brunauer-Emmett-Teller (BET) theory. The total pore volume is determined based on the N2 adsorption amount, expressed in liquid form at 77 K and a relative pressure (P/P0) of approximately 0.95. The average pore width is calculated from the total pore volume and the surface area of AC, using the Gurwitch rule. As the lignin MW increases, the surface area of AC has a linear growth, ranging from 1430 to 1830 m2/g for lignin sample with MW value varying from 12,000 to 44,000 Da. The total pore volume of AC also increases with the increasing MW, with the lignin of the highest MW having the highest total pore volume of 1.27 cm3/g. Notably, the LL sample and the LM sample have comparable total pore volumes. As shown in Figure 3d, the average pore width of AC is much greater in lignin samples with low MW, while the average pore width of AC from the medium and high MW lignin samples remains relatively consistent.
Generally, during low temperature carbonization with ZnCl2 activation of the lignocellulosic biomass, the evolution of the porous structure of AC is driven by the release of functional groups from the carbon skeleton as volatiles, leaving vacancies behind [38,40,41,42]. This process is governed by two primary factors: generation of new pores or enlargement of existing pores. Once a new pore is formed, whether the chemical activation agent (ZnCl2) continues to act on this pore to enlarge its size, or acts elsewhere to create new pores, largely depends on the sample properties of the lignocellulosic biomass. Typically, these two processes occur simultaneously, only to different degrees. The pore properties of AC are essentially a reflection of these two processes. Our previous study has discussed that an evolution of porous structure favoring pore creation typically results in a higher surface area [32]. According to the pore properties of AC in Figure 3, it can be observed that when the lignin is in high and medium MW range (approximately greater than 2 × 104 Da), a higher MW of lignin is more conducive to new pore creation, thus obtaining a higher specific surface area. However, when the lignin is in low MW range (approximately less than 2 × 104 Da), a decrease in lignin MW progressively enhances pore enlargement in the existing pores. This leads to the AC derived from LL exhibiting a larger average pore width.
Despite the LF and LM samples having similar lignin MWs, the AC derived from the LF exhibits a lower surface area and pore volume, but a higher average pore width compared to the AC from the LM. This disparity could potentially be attributed to the high ash content of the LF. Specifically, the ash might be reducing the surface area and total pore volume of the AC, while simultaneously enlarging the pores, as indicated by the higher average pore width in AC derived from LF. As reported in the literature [43], high ash content can obstruct the entry of chemical activating agents into lignin particles, thereby reducing the contact area between the activation reagent and the lignin. Moreover, ash content can also block the channel for the off-gases releasing, which is unfavorable to the development of pores. Therefore, the surface area and total pore volume of the higher ash content sample are lower. For the higher average pore width, it is because the ash can also impede the penetration of ZnCl2 into the lignin particle, causing pore enlargement of exterior pores. Furthermore, ash weakens the intermolecular forces between lignin molecules, making it easier for pores to enlarge. Metals, such as Na, can act as catalysts during the carbonization process, accelerating the thermal decomposition of lignin and contributing to the enlargement of existing pores [38,39].
3.3.2. Effect of MW of Lignin on Pore-Size Distribution
To investigate how lignin MW affects pore size distribution of AC, the N2 desorption data were analyzed using density functional theory (DFT). Figure 5a shows the pore size distribution of AC of the ALPHA-processed lignin with varying MW, represented as differential pore volumes at each pore width. Three distinct peaks in pore size distribution are observed at approximately 1.8 nm, 2.7 nm, and 3.6 nm, representing the primary pore widths within the AC. In other words, a peak value in differential pore volume around a specific pore width indicates that the AC contains more pores at this size. As the MW of the lignin increases, the pore volume of micropores (<2.0 nm) and small mesopores (~2.7 nm) increases. However, the pore volume of the larger mesopores exhibits a different trend. The peak shapes observed for the larger mesopores around 3.6 nm of the LM and LH samples are similar, but the LL sample displays a much higher and broader peak compared to the higher MW samples.
Figure 4.
(a) Pore size distributions in AC from lignin samples (represented as differential pore volumes at each pore width) with varying MW of lignin. (b) Peak values of differential pore volume as a function of MW of lignin at pore widths of ~1.8 nm, ~2.6 nm, and ~3.6 nm.
Figure 4.
(a) Pore size distributions in AC from lignin samples (represented as differential pore volumes at each pore width) with varying MW of lignin. (b) Peak values of differential pore volume as a function of MW of lignin at pore widths of ~1.8 nm, ~2.6 nm, and ~3.6 nm.
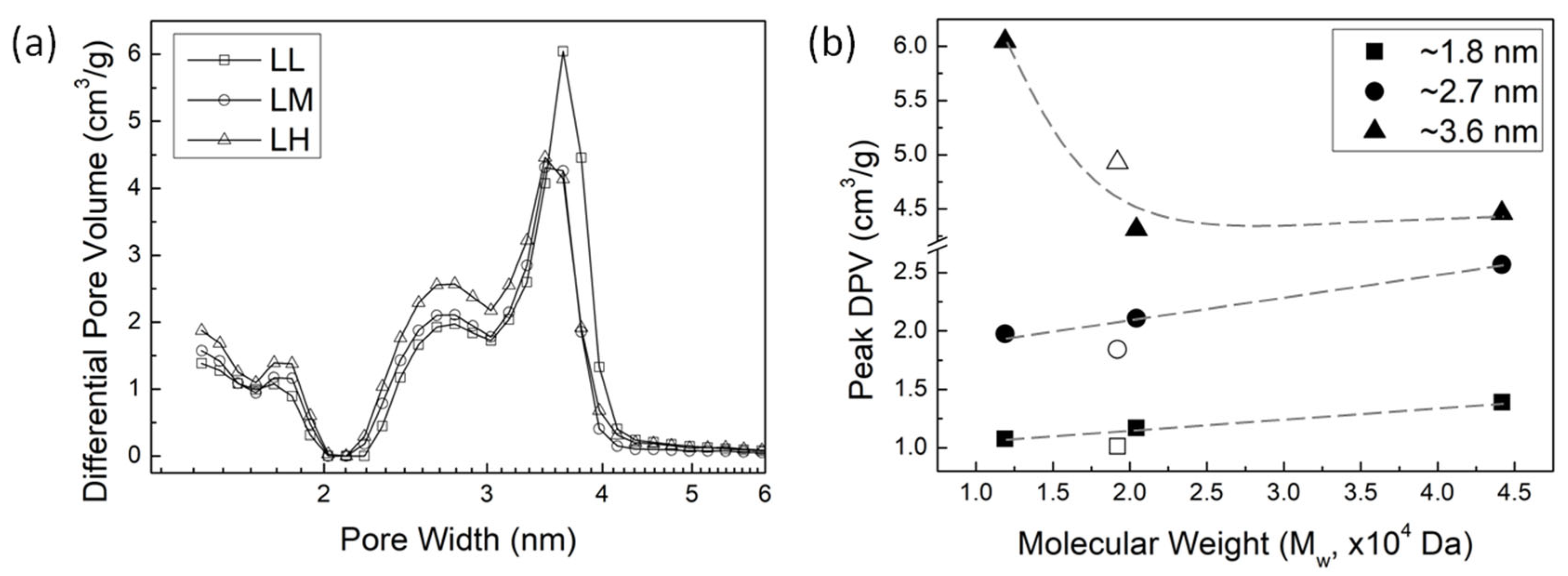
Figure 5.
Pore size distributions in AC from lignin feedstock (LF) and ALPHA-processed medium MW lignin sample (LM).
Figure 5.
Pore size distributions in AC from lignin feedstock (LF) and ALPHA-processed medium MW lignin sample (LM).
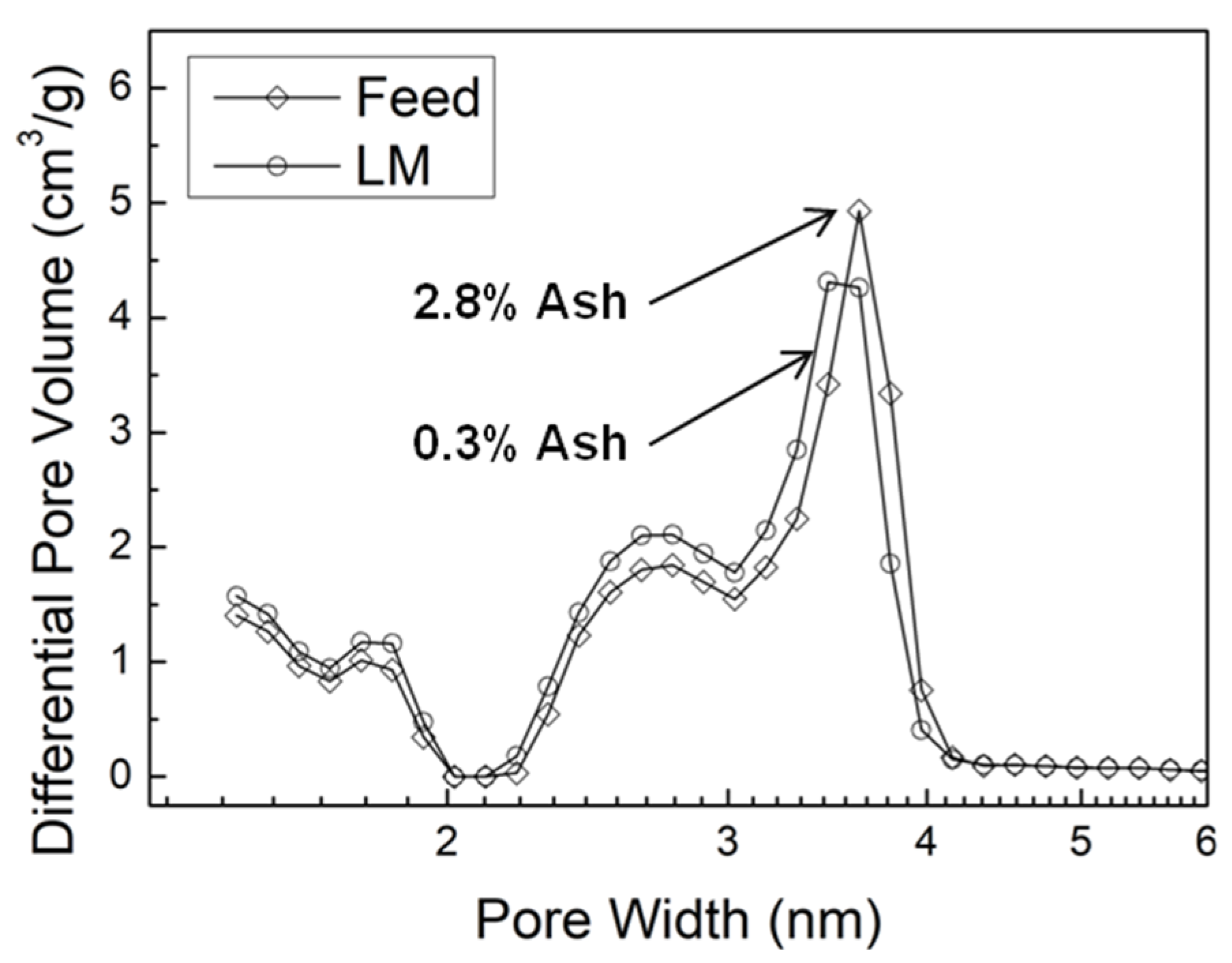
Figure 4b illustrates peak values in differential pore volume vs. MW of lignin. It shows a linear increase in the differential pore volume of micropores (<2.0 nm) and smaller mesopores (~2.7 nm) as the MW of lignin increases. Considering that the micropores and smaller mesopores contribute significantly to the surface area and total pore volume of the AC [32], the increase in the surface area and total pore volume of AC observed in Figure 3b,c can be attributed to the development of these micropores and smaller mesopores. Moreover, the trend in the differential pore volume peak of larger mesopores (~3.6 nm) mirrors the trend of the average pore width with increasing lignin MW, as observed in Figure 3d. The notably larger average pore width of the LL sample in Figure 3d is due to the higher pore volume of larger mesopores, as indicated by the broader and higher peak around 3.6 nm.
From the pore size distribution in AC from lignin samples with varying MW, we can deduce how lignin MW affects the pore properties of AC. When the MW of lignin is in medium and high MW range (greater than 2 × 104 Da), the development of micropores and smaller mesopores remains in a certain balance with pore enlargement, leading to a consistent average pore width in AC (~2.78 nm as shown in Figure 3d). When the MW of lignin is in low MW range (less than 2 × 104 Da), the pore enlargement significantly enhances with the decrease in lignin MW. This enhanced pore enlargement does not affect the linear growth of the surface area of AC with lignin MW, but it results in a relatively higher pore volume in the LL sample, causing the pore volume of AC not to linearly increase with the MW of lignin like the surface area does. (shown in Figure 3c)
In fact, the underlying mechanism of the development of micropores and smaller mesopores is the generation of new pores. Our previous study found that once a new pore forms, whether the chemical activation agent (ZnCl2) continues to act on this pore to enlarge it or act elsewhere to create new pores depends on the reactivity of the lignocellulosic biomass sample [32]. If the sample is more reactive, then the process of generating new pores is more pronounced. While if the sample is less reactive, then the enlargement of existing pores becomes more significant. This is because once a pore forms and disrupts the aromatic structure, it becomes more favorable for reactions to occur within this disrupted carbon system than to initiate the formation of new pores. As the MW of lignin increases, the more pronounced impact of the generation of new pores suggests that higher MW lignin is more reactive. However, when dealing with lignin of lower MW, the significant role of pore enlargement implies that lower MW lignin is less reactive.
Considering that the oxygen-containing groups and aliphatic structures in lignocellulosic biomass exhibits more reactivity than non-oxygen-containing groups and aromatic structures [32], the pore enlargement observed in lower molecular weight lignin could be attributed to: (1) decreased oxygen content, (2) reduced presence of aliphatic carbons, or perhaps a combination of both factors. During synthesis of AC, oxygen-containing groups, aliphatic carbons, or aliphatic oxygen-containing groups in higher MW lignin are easier to be converted to volatiles and released in the off-gas, facilitating the creation of new (small) pores. While lower MW lignin is relatively less reactive, once a pore is created and the aromatic structure is disrupted, reactions involving the disrupted carbon system become more favorable compared to generating new pores. This results in the enlargement of existing pores rather than the formation of new pores. Furthermore, literature also reported that polymer with lower MW tend to have closer contact with each other, thus yielding higher density [44]. Lower MW lignin may possess a stacked aromatic structure, which impedes the penetration of chemical-activating agents into the lignin particles. Therefore, the pores created on the outer surface of the lignin particles experience enlargement in the presence of the excess ZnCl2.
Because LF, the original lignin feed, and LM have similar MW values, and the ash content in the ALPHA-processed LM sample is much lower, it’s possible to determine the impact of ash content on AC pore properties. The pore size distributions of AC from the LF sample and the LM sample are shown in Figure 5. AC derived from the LF (higher ash content) exhibits a smaller volume of micropores and small mesopores (2.7 nm), as indicated by the lower peak values around 1.8 nm and 2.7 nm, respectively. However, they exhibit a larger volume of large mesopores (3.6 nm), signified by the higher peak value around 3.6 nm, compared to the LL sample that has extremely low ash content. This observation is consistent with the previously discussed conclusion, explaining why AC derived from LF exhibits a higher average pore width, as shown in Figure 3d. In other words, the presence of ash facilitates the enlargement of existing pores more readily than the formation of new pores, as discussed above.
3.4. Aqueous Adsorption Capacity of Activated Carbon
The aqueous adsorption capacity of AC is often evaluated using either Iodine number or methylene blue (MEB) value, which represent the amount of each chemical that can be absorbed on a given carbon surface. Figure 6 shows the results for the iodine and MEB adsorption measurements in panels a and b, respectively. Adsorption values are reported as mg of species absorbed per g of AC. Regardless of the size of the molecular adsorbate (iodine is small and MEB is fairly large), the aqueous adsorption capacity of the AC increases as the lignin MW increases in a manner consistent with the change in surface area with lignin MW. This trend indicates that the aqueous adsorption capacity of AC is determined by surface area of the AC, rather than by the total pore volume.
4. Conclusions
Using ZnCl2 activation with low temperature carbonization condition, AC was synthesized from lignin with varying MW controlled by the ALPHA process. We found that the surface area and total pore volume of AC increase as the lignin MW increases. AC derived from lignin with the highest MW of 44,200 Da possessed the highest surface area of 1830 m2/g and the highest total pore volume of 1.27 cm3/g. When lignin is in low MW range, pore enlargement is more significant with the decrease in lignin MW, leading to an expansion in the average pore width of AC. Moreover, we observed that the overall yield and carbon fractional conversion from lignin to AC are not influenced by the lignin MW, but rather depend on the elemental composition of the sample. Regarding the impact of ash on AC properties, the surface area and total pore volume of AC are lower when synthesized from lignin samples with higher ash content. Higher ash content also promotes pore enlargement. The understanding of these effects of lignin properties on AC properties is instructive for the optimization of industrial-scale AC production from lignin. This knowledge can substantially contribute to achieving the highest surface area for AC, all while maintaining the lowest possible production cost.
Data Availability Statement
All raw data and processed data is available upon request from the corresponding author.
Acknowledgments
This material is based on work supported by the U.S. Department of Energy’s Office of Energy Efficiency and Renewable Energy (EERE) (under Bioenergy Technology Office Award No. DE-EE0008502). Graham Tindall would also like to acknowledge support by the Department of Education under the Graduate Assistance in Areas of National Needs Fellowship (GAANN) Fellowship (Award No. P200A180076).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Statista. Available online: https://www.statista.com/statistics/217179/global‐biofuels‐market‐size (access on 14 July 2024).
- Luque, R.; Herrero-Davila, L.; Campelo, J.M.; Clark, J.H.; Hidalgo, J.M.; Luna, D.; Marinas, J.M.; Romero, A.A. Biofuels: A Technological Perspective. Energy & Environmental Science 2008, 1, 542–564.
- Demirbas, A. Progress and Recent Trends in Biofuels. Progress in energy and combustion science 2007, 33, 1–18. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic Biomass: A Sustainable Platform for the Production of Bio-Based Chemicals and Polymers. Polymer chemistry 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic Catalysts for Upgrading of Biomass to Fuels and Chemicals. Chemical Society Reviews 2012, 41, 8075–8098. [Google Scholar] [CrossRef]
- Kumar, P.; Barrett, D.M.; Delwiche, M.J.; Stroeve, P. Methods for Pretreatment of Lignocellulosic Biomass for Efficient Hydrolysis and Biofuel Production. Industrial & engineering chemistry research 2009, 48, 3713–3729.
- Bajwa, D.; Pourhashem, G.; Ullah, A.H.; Bajwa, S. A Concise Review of Current Lignin Production, Applications, Products and Their Environmental Impact. Industrial Crops and Products 2019, 139, 111526. [Google Scholar] [CrossRef]
- Doherty, W.O.; Mousavioun, P.; Fellows, C.M. Value-Adding to Cellulosic Ethanol: Lignin Polymers. Industrial crops and products 2011, 33, 259–276. [Google Scholar] [CrossRef]
- Tardy, B.L.; Lizundia, E.; Guizani, C.; Hakkarainen, M.; Sipponen, M.H. Prospects for the Integration of Lignin Materials into the Circular Economy. Materials Today 2023, 65, 122–132. [Google Scholar] [CrossRef]
- Gregorich, N.; Ding, J.; Thies, M.C.; Davis, E.M. Novel Composite Hydrogels Containing Fractionated, Purified Lignins for Aqueous-Based Separations. Journal of Materials Chemistry A 2021, 9, 1025–1038. [Google Scholar] [CrossRef]
- Henry, C.; Nejad, M. Lignin-Based Low-Density Rigid Polyurethane/Polyisocyanurate Foams. Industrial & Engineering Chemistry Research 2023, 62, 6865–6873.
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.; Mondragon, I. Physico-Chemical Characterization of Lignins from Different Sources for Use in Phenol–Formaldehyde Resin Synthesis. Bioresource technology 2007, 98, 1655–1663. [Google Scholar] [CrossRef]
- Jin, J.; Ding, J.; Klett, A.; Thies, M.C.; Ogale, A.A. Carbon Fibers Derived from Fractionated–Solvated Lignin Precursors for Enhanced Mechanical Performance. ACS Sustainable Chemistry & Engineering 2018, 6, 14135–14142.
- Marsh, H.; Reinoso, F.R. Activated Carbon; Elsevier, 2006.
- Kanhere, S.V.; Tindall, G.W.; Ogale, A.A.; Thies, M.C. Carbon Fibers Derived from Liquefied and Fractionated Poplar Lignins: The Effect of Molecular Weight. Iscience 2022, 25. [Google Scholar] [CrossRef]
- Danish, M.; Ahmad, T. A Review on Utilization of Wood Biomass as a Sustainable Precursor for Activated Carbon Production and Application. Renewable and Sustainable Energy Reviews 2018, 87, 1–21. [Google Scholar] [CrossRef]
- Ahmadpour, A.; Do, D.D. The Preparation of Active Carbons from Coal by Chemical and Physical Activation. Carbon 1996, 34, 471–479. [Google Scholar] [CrossRef]
- Ioannidou, O.; Zabaniotou, A. Agricultural Residues as Precursors for Activated Carbon Production—A Review. Renewable and sustainable energy reviews 2007, 11, 1966–2005. [Google Scholar] [CrossRef]
- Jain, A.; Balasubramanian, R.; Srinivasan, M.P. Hydrothermal Conversion of Biomass Waste to Activated Carbon with High Porosity: A Review. Chemical Engineering Journal 2016, 283, 789–805. [Google Scholar] [CrossRef]
- Yahya, M.A.; Al-Qodah, Z.; Ngah, C.Z. Agricultural Bio-Waste Materials as Potential Sustainable Precursors Used for Activated Carbon Production: A Review. Renewable and sustainable energy reviews 2015, 46, 218–235. [Google Scholar] [CrossRef]
- Tekinalp, H.L.; Cervo, E.G.; Fathollahi, B.; Thies, M.C. The Effect of Molecular Composition and Structure on the Development of Porosity in Pitch-Based Activated Carbon Fibers. Carbon 2013, 52, 267–277. [Google Scholar] [CrossRef]
- Dias, J.M.; Alvim-Ferraz, M.C.; Almeida, M.F.; Rivera-Utrilla, J.; Sánchez-Polo, M. Waste Materials for Activated Carbon Preparation and Its Use in Aqueous-Phase Treatment: A Review. Journal of environmental management 2007, 85, 833–846. [Google Scholar] [CrossRef]
- Nor, N.M.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of Activated Carbon from Lignocellulosic Biomass and Its Applications in Air Pollution Control—a Review. Journal of Environmental Chemical Engineering 2013, 1, 658–666. [Google Scholar]
- Bhatnagar, A.; Hogland, W.; Marques, M.; Sillanpää, M. An Overview of the Modification Methods of Activated Carbon for Its Water Treatment Applications. Chemical Engineering Journal 2013, 219, 499–511. [Google Scholar] [CrossRef]
- Roy, G.M. Activated Carbon Applications in the Food and Pharmaceutical Industries; Routledge, 2023.
- Serp, P.; Figueiredo, J.L. Carbon Materials for Catalysis; John Wiley & Sons, 2008; ISBN 0-470-17885-X.
- Frackowiak, E. Carbon Materials for Supercapacitor Application. Physical chemistry chemical physics 2007, 9, 1774–1785. [Google Scholar] [CrossRef]
- Sevilla, M.; Mokaya, R. Energy Storage Applications of Activated Carbons: Supercapacitors and Hydrogen Storage. Energy & Environmental Science 2014, 7, 1250–1280.
- Abioye, A.M.; Ani, F.N. Recent Development in the Production of Activated Carbon Electrodes from Agricultural Waste Biomass for Supercapacitors: A Review. Renewable and sustainable energy reviews 2015, 52, 1282–1293. [Google Scholar] [CrossRef]
- Grand View Research. Avaulable online: https://www.grandviewresearch.com/industry-analysis/activated-carbon-market (accessed on 14 July 2024).
- Carrott, P.J.M.; Carrott, M.R. Lignin–from Natural Adsorbent to Activated Carbon: A Review. Bioresource technology 2007, 98, 2301–2312. [Google Scholar]
- Wu, C.; Tindall, G.; Fitzgerald, C.; Thies, M.; Roberts, M. Decoupling the Role of Lignin, Cellulose/Hemi-Cellulose, and Ash on ZnCl2-Activated Carbon Pore Structure.
- Thies, M.C.; Klett, A.S.; Bruce, D.A. Solvent and Recovery Process for Lignin. US10053482B2, 2018.
- Klett, A.S.; Chappell, P.V.; Thies, M.C. Recovering Ultraclean Lignins of Controlled Molecular Weight from Kraft Black-Liquor Lignins. Chemical communications 2015, 51, 12855–12858. [Google Scholar] [CrossRef]
- Blasi, C.D.; Branca, C.; Galgano, A. Products and Global Weight Loss Rates of Wood Decomposition Catalyzed by Zinc Chloride. Energy & fuels 2008, 22, 663–670.
- Branca, C.; Di Blasi, C.; Galgano, A. Pyrolysis of Corncobs Catalyzed by Zinc Chloride for Furfural Production. Industrial & engineering chemistry research 2010, 49, 9743–9752.
- Di Blasi, C.; Branca, C.; Galgano, A.; Zenone, F. Modifications in the Thermicity of the Pyrolysis Reactions of ZnCl2-Loaded Wood. Industrial & Engineering Chemistry Research 2015, 54, 12741–12749.
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic Biomass Pyrolysis Mechanism: A State-of-the-Art Review. Progress in energy and combustion science 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Khalili, N.R.; Campbell, M.; Sandi, G.; Golaś, J. Production of Micro-and Mesoporous Activated Carbon from Paper Mill Sludge: I. Effect of Zinc Chloride Activation. Carbon 2000, 38, 1905–1915. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Zheng, C.; Lee, D.H.; Liang, D.T. In-Depth Investigation of Biomass Pyrolysis Based on Three Major Components: Hemicellulose, Cellulose and Lignin. Energy & fuels 2006, 20, 388–393.
- Siengchum, T.; Isenberg, M.; Chuang, S.S. Fast Pyrolysis of Coconut Biomass–An FTIR Study. Fuel 2013, 105, 559–565. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of Hemicellulose, Cellulose and Lignin Pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Liou, T.-H. Development of Mesoporous Structure and High Adsorption Capacity of Biomass-Based Activated Carbon by Phosphoric Acid and Zinc Chloride Activation. Chemical engineering journal 2010, 158, 129–142. [Google Scholar] [CrossRef]
- Nakano, T. Synthesis, Structure and Function of π-Stacked Polymers. Polymer journal 2010, 42, 103–123. [Google Scholar] [CrossRef]
Figure 1.
Aqueous Lignin Purification using Hot Agents (ALPHA) Process for fractionation of lignin.
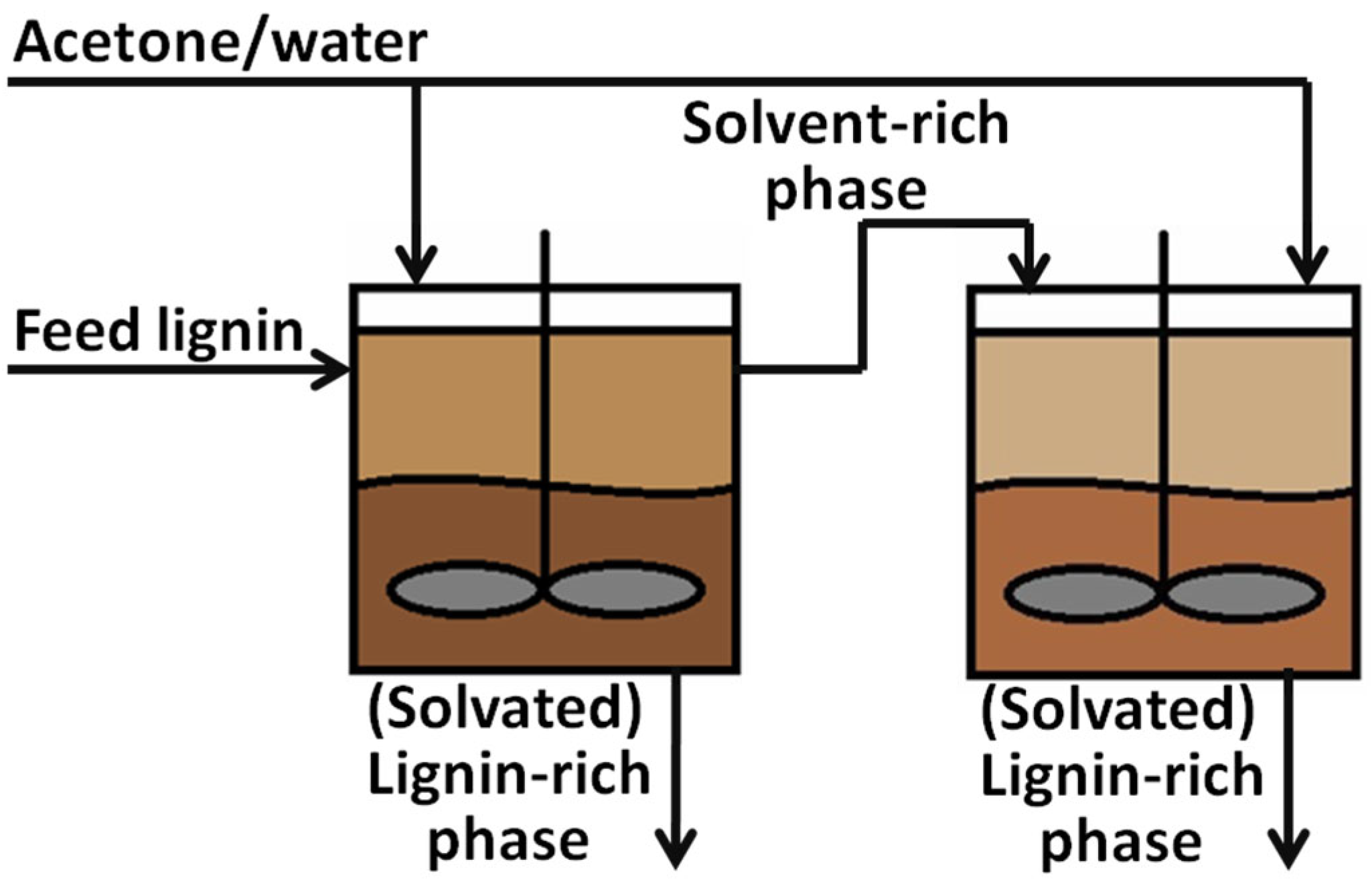
Figure 2.
(a) Weight average molecular weight (Mw), polydispersity index (PDI, inset), (b) metal content, (c) thermogravimetric (TG) curve, and (d) ash content of lignin sample.
Figure 2.
(a) Weight average molecular weight (Mw), polydispersity index (PDI, inset), (b) metal content, (c) thermogravimetric (TG) curve, and (d) ash content of lignin sample.
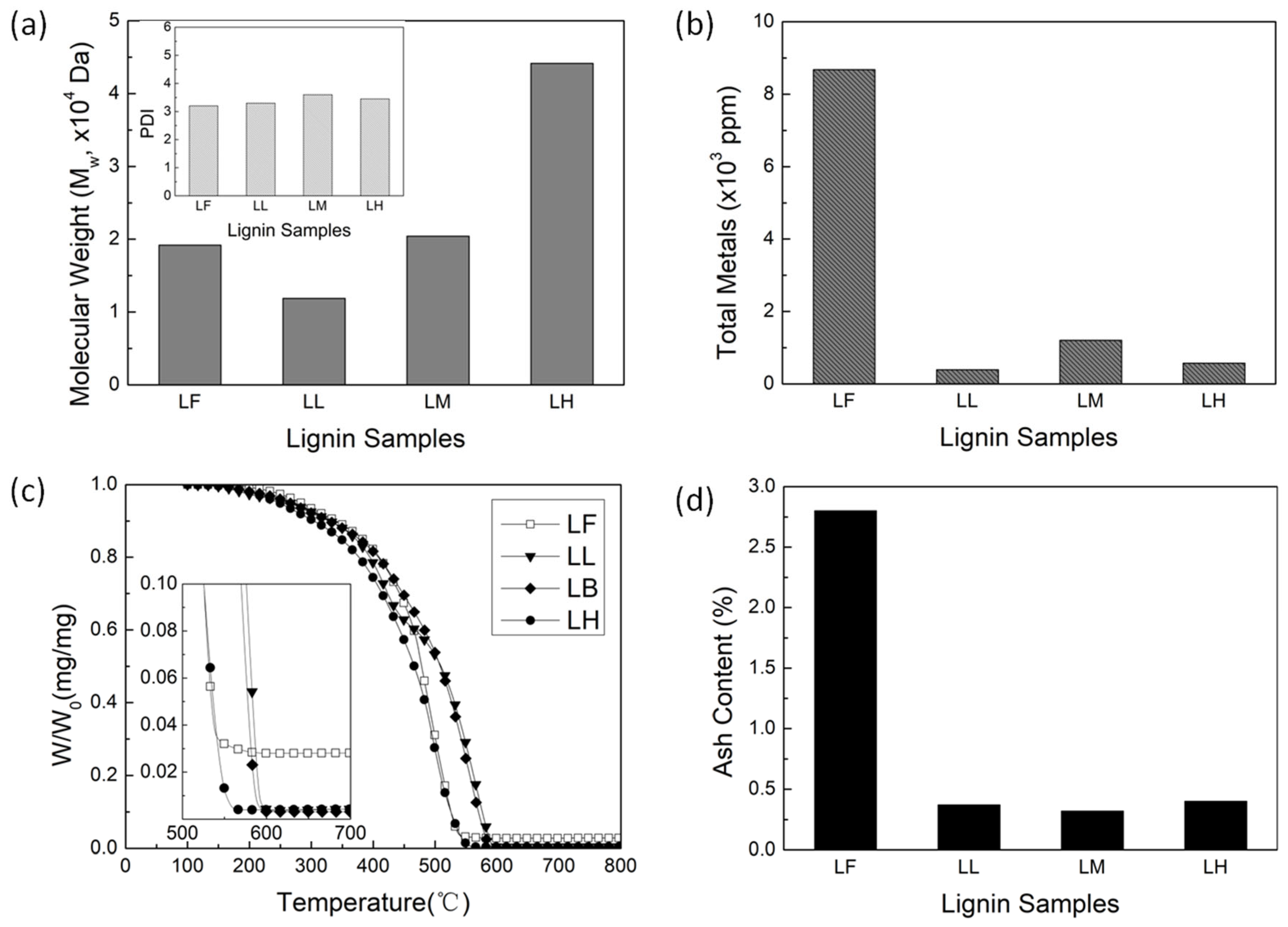
Figure 3.
(a) Overall yield (mass of activated carbon (AC)/mass of lignin) and carbon fractional conversion (mass of C in AC/mass of C in lignin) as a function of molecular weight (MW) of lignin. (b) Surface area, (c) total pore volume, and (d) average pore width of AC as a function of lignin MW.
Figure 3.
(a) Overall yield (mass of activated carbon (AC)/mass of lignin) and carbon fractional conversion (mass of C in AC/mass of C in lignin) as a function of molecular weight (MW) of lignin. (b) Surface area, (c) total pore volume, and (d) average pore width of AC as a function of lignin MW.
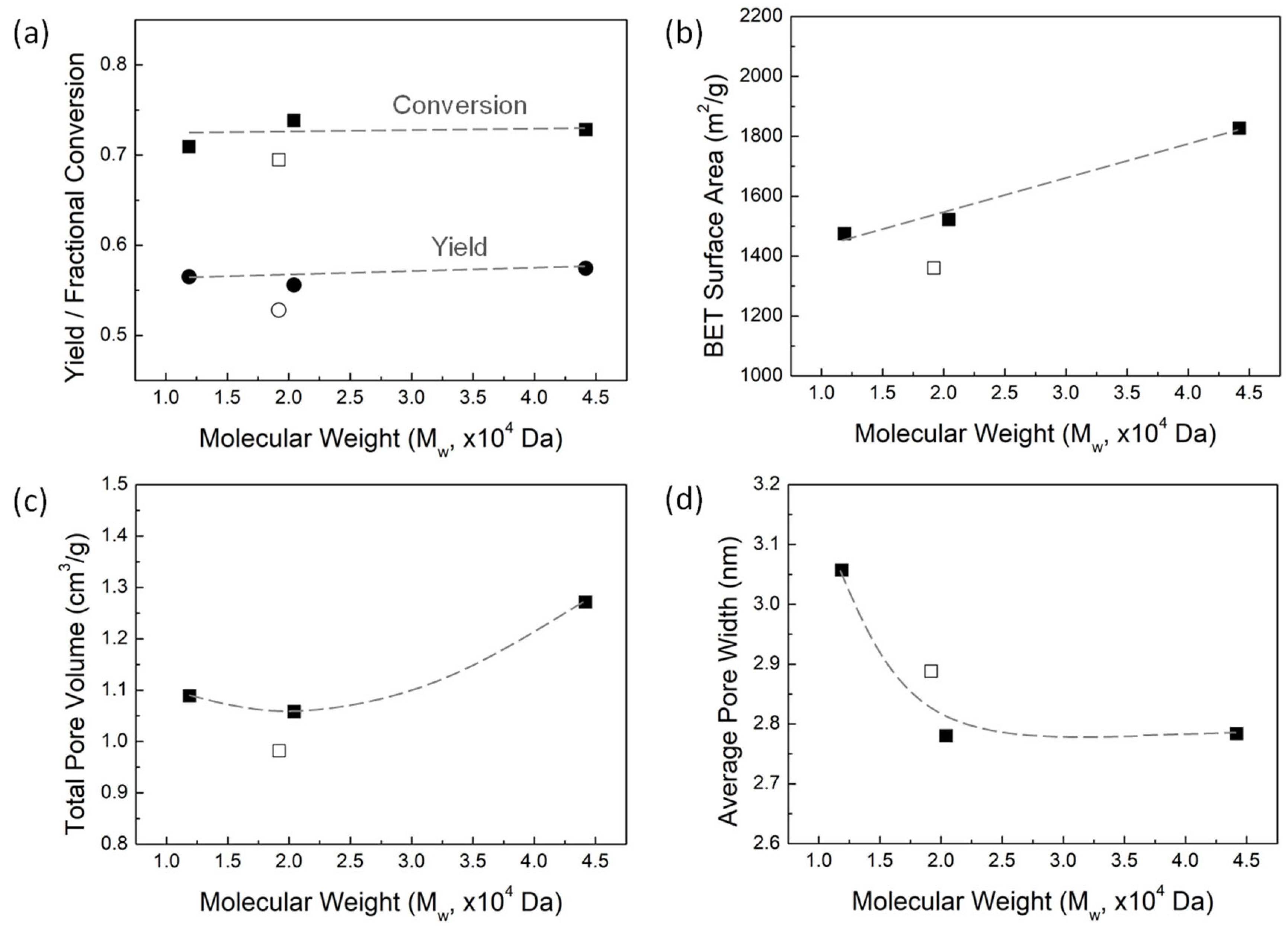
Figure 6.
(a) Iodine number and (b) methylene blue value of AC as a function of lignin MW.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



