
Submitted:
25 July 2024
Posted:
26 July 2024
You are already at the latest version
Abstract
Seven patients in our clinical program who were diagnosed with high burden (>10%) premature ventricular contractions (PVCs) and concomitant significant upper GI disease with no other significant cardiac history demonstrated a significant reduction in the burden of PVCs following surgical or procedural interventions of the upper GI tract [68.34% reduction, p=0.024). Furthermore, in all cases, the origin of the PVCs was from the base of the right ventricular outflow tract (RVOT). This is the first report in the literature that we are aware of to report this unique association for which we propose a dual mechanism of action of vagally mediated upper GI driven PVCs and a direct anatomical effect of these upper GI anatomies on the right ventricular outflow tract (RVOT) of the heart. These are very prelimary findings that warrant larger clinical and mechanistic studies that may ultimately define a new subset of PVCs for which we propose a term, “E-PVCs”.
Keywords:
Premature ventricular contractions
; vagus nerve
; esophageal disease
1. Introduction
Premature ventricular contractions (PVCs) are common in the general population. In fact, if monitored for more than a few hours, PVCs are observed broadly, as demonstrated in a population-based cohort of adults aged 25-41 years old who had 24-hour Holter monitoring. Of all participants, 69% had at least one PVC (Premature ventricular contraction) with a median count of 2 PVCs, and the 95th percentile had 193 PVCs [1]. In the Cardiovascular Health Study that followed patients for 14 years, patients aged 65 years and older in the general population underwent 24-hour Holter monitoring and were found to have PVCs comprising a median of 0.011% of all heart beats [2]. This trend increases markedly in the elderly population, where 99.5% of those aged 75 years and older were found to have at least one PVC in the Multi-Ethnic Study of Atherosclerosis. Though PVCs are often viewed as benign by clinicians, a multiracial community-based cohort of more than 15,000 individuals revealed a higher risk of coronary artery disease events and death in those with the presence of a PVC [3]. The Cardiovascular Health Study also found that a higher frequency of PVCs was linked with a 5-year reduction in left ventricular systolic function (measured by left ventricular ejection fraction), increased risk for heart failure, and increased risk for death [2].
2. Physiologic Mechanisms
The mechanism of PVCs are multifactorial, related to triggered activity, automaticity, and reentry [4]. Triggered mechanisms include behavioral (sympathetic stress) or from underlying diseases, induced by increased intracellular calcium in the plateau phase of the action potential during prolonged repolarization. PVCs generated in this setting may cause torsade's de pointes or acquired long QT syndrome [5]. The increased intracellular calcium is typically a result of activation of cAMP-dependent protein Kinase A, so this mechanism is often a target of induction or termination of PVCs. For example, caffeine can result in delayed after-depolarization due to calcium release from the sarcoplasmic reticulum [6]. On the other hand, adenosine inhibits the production of adenylyl cyclase, which in turn reduces cAMP release, and therefore, is useful in the termination of PVCs [7]. Catecholamines also play a role in the induction of calcium release, so b-blockers are a useful targeted therapy for triggered activity [8]. Likewise, non-dihydropyridine calcium channel blockers prevent intracellular calcium accumulation via blockage of L-type calcium channels [8], and are sometimes utilized for therapy. PVCs related to automaticity are present with parasystole, occurring at the same cycle length [9]. The automaticity mechanism is multifactorial, ranging from an exaggerating automaticity that normal cardiomyocytes have to electric isolation like that seen in fibrosis. PVCs occurring through reentry require the presence of 2 electrical pathways and a temporary or permanent unidirectional block in one pathway. This may involve pathology in the fascicles, producing a fascicular PVC, or an area of fibrosis, where a series of cardiomyocytes are electrically connected creating slower conduction pathways, resulting in a PVC.
Idiopathic PVCs have often been linked to increased sympathetic nervous system activity [10]. Gillis et al. demonstrated that increased ventricular irritability in cats was associated with increased firing of the sympathetic nervous impulses in the cardiac system [11]. Estes and Izlar showed that ventricular tachycardia episodes in a patient were terminated by bilateral cardiac sympathectomy [12]. Although the impact of the sympathetic nervous system has been extensively studied, the role of the parasympathetic system in PVCs is still largely unexplored. Data so far indicates that some patients have vagally suppressible PVCs, while others have vagally inducible PVCs. Weiss et al demonstrated that 5 out of 10 patients had a statistically significant decrease in PVCs with phenylephrine-induced increase in reflex vagal tone [13]. However, in the same study, atropine, a muscarinic antagonist that blocks cardiac vagal nerve activity, also reduced the burden of PVCs. He et al. suggested that PVCs were evoked by sudden fluctuations in autonomic balance, and when separated by type, fast rate-dependent PVC (F-PVC) might be facilitated by sympathetic activation, while slow rate-dependent PVC (S-PVC) might be induced by vagal activation [14]. In this case series, patients presented with pathologies (GERD, achalasia, and gastric lap-band placement) in the distal esophagus that may have triggered a neural reflex mediated by the vagus nerve. The reflex allows changes in gastric and esophageal mechanoreceptors to send powerful vagal-mediated inhibition to the lower esophagus and lower esophageal sphincter, which results in lower parasympathetic vagal input to the alimentary and cardiovascular systems [15]. We postulate that this mechanism results in a high burden of S-PVCs via suppression of the parasympathetic nervous system.
3. Cardiovascular Physiologic Consequences of PVCs
The electromechanical coupling of cardiac function hinges upon the orchestrated stimulation initiated by the sinoatrial (SA) node, which causes the sequential contraction of atrial myocardium. Following this, a transient delay at the atrioventricular (AV) node facilitates ventricular filling, crucial for optimizing cardiac output. Subsequently, the propagated electrical impulse traverses the bundle of His and Purkinje fibers, stimulating ventricular contraction. This process is known as excitation–contraction coupling (ECC). [16]
The synchronization of ECC is paramount, as it ensures adequate ventricular filling, requisite for optimal stroke volume and cardiac output. [17] Premature ventricular contractions disrupt this synchrony, precipitating ventricular depolarization prior to complete diastolic filling, thereby compromising stroke volume [18], and when PVC burden is significant, decrease cardiac output. [19] This hemodynamic disturbance can result in reduced arterial pressure, impeding tissue perfusion, and precipitating clinical manifestations such as palpitations, syncope, and fatigue (Figure 1), [20,21].
4. Clinical Features and Therapies
PVCs are typically diagnosed as an incidental finding or during evaluation of clinical symptoms, including palpitations, chest discomfort, presyncope, dyspnea, and fatigue [22]. This is usually due to the strong heartbeat that follows the increased filling time after the PVC. Patients may also present with abrupt syncope, and very rarely, sudden arrhythmic death from PVC-induced ventricular fibrillation. A family history screening for sudden death may be useful. On physical exams, one may find apparent bradycardia ascertained by a palpable pulse alone, occurring from a poorly perfused PVC. PVCs are diagnosed via a 12-lead electrocardiogram (ECG). The gold standard for assessing PVC frequency used to be a 24-hour Holter monitor, however recent evidence has demonstrated that due to substantial daily variation, the duration of monitoring should be extended (>5 days) [23].
Current guidelines suggest that management of PVCs may be pursued if the patient is symptomatic, presents with a high burden of PVC, or has PVCs accompanied by structural heart disease. First-line medical therapy involves either b-blockers or and calcium channel blockers. However, patients with F-PVC are more likely to benefit from this treatment, likely because it is mediated by the sympathetic nervous system [24]. In general, catheter ablation procedures have a success rate of 80-95%, while b-blockers only have a success rate between 12-24% [25]. If pharmacologic treatment is ineffective for higher burden (>15%), catheter ablation is considered, providing superior effectiveness but also requiring acquiescence to procedural risks [26,27,28]. Patients who are opposed to cardiac ablation, have failed the procedure, or are unfavorable candidates may be trialed on additional antiarrhythmics [26,27,28].
5. Genetic Associations
Premature Ventricular Contractions (PVCs) emerge as a result of intricate interactions between genetic and acquired factors, delineating the multifaceted nature of their origin. The genetic underpinnings of PVCs encompass variations within genes pivotal for cardiac ion channels and the intricate electrical signaling within cells. Notably, genetic mutations in genes such as SCN5A, integral to sodium channels, and KCNQ1/KCNE1, which form potassium channels, are key contributors to PVC development. These mutations have the potential to disrupt the finely orchestrated cardiac rhythm, thereby precipitating the premature contractions characteristic of PVCs. However, PVCs can also be influenced by non-genetic factors, such as structural heart disease, electrolyte imbalances, and stimulants, further underscoring the intricate blend of factors at play [29]. Within this genetic landscape, SCN5A serves as a critical determinant, encoding the sodium channel Nav1.5 that plays a pivotal role in orchestrating the rapid depolarization phase during the cardiac action potential. Genetic variations within SCN5A have the potential to perturb the flow of sodium ions, culminating in irregular electrical activity and the emergence of arrhythmias like PVCs. KCNQ1 and its interacting partner KCNE1 form the potassium channel Kv7.1, indispensable for efficient repolarization of cardiac cells. Genetic mutations affecting these genes can potentially prolong the repolarization phase, creating an environment conducive to arrhythmias [29,30,31].
To probe these genetic contributions, a study of 100 post-MI patients included clinical genetic assessments alongside a range of more common cardiac diagnostic evaluation. Notably, the examination of genetic polymorphisms revealed significant findings within SCN5A, KCNE1, and KCNQ1 genes. Specifically, polymorphisms like H558R in SCN5A, S38G in KCNE1, and intronic variations in KCNQ1 were identified. Remarkably, these polymorphisms were closely linked to anomalies in the QT interval, characterized by both QT prolongation and an increase in QT dispersion. Despite these associations, the study's intriguing outcome illuminated that these genetic variations, while influencing repolarization patterns, did not manifest a direct correlation with complex ventricular arrhythmias, sudden cardiac arrest, or sudden cardiac death in post-MI patients [31]. These collective findings underscore the intricate relationship between genetic predisposition, the intricate cardiac electrophysiological landscape, and the development of PVCs and related conditions. While genetic variations significantly contribute to the subtle nuances of repolarization patterns and gastrointestinal dysfunctions, the genesis of PVCs emerges from a complex interplay involving genetic factors, acquired conditions, and external triggers. This intricate interplay highlights the layered complexity in the origination and progression of cardiac arrhythmias and related conditions, epitomizing the multifaceted nature of these events.
6. Autonomic Nervous System and PVCs
The autonomic nervous system has been hypothesized to play a significant role in pathogenesis, maintenance, and interference in ventricular arrhythmias. The vagus nerve contributes to the formation of the esophageal plexus, innervating the smooth muscle of the esophagus, and notably gives off cardiac branches, regulating heart rate through the parasympathetic nervous system. However, the role of the autonomic nervous system is unclear as in some reports increased sympathetic activity and, therefore, decreased vagal tone are observed prior to PVC onset and, in others, increased vagal tone is associated with the pathogenesis of PVCs. One case study discusses the association between cardiac arrhythmias and vagus nerve stimulation in patients with GERD symptoms [32]. They observed a patient with GERD-LIKE symptoms that developed PVC and postulated that the potential pathophysiology of PVC is due to either reflux stimulating the vagus nerve that caused bradycardia and arrhythmia or that the inflammation causes the arrhythmia.
The vagus nerve is the tenth cranial nerve and is a major component of the parasympathetic nervous system, which is responsible for slowing the heart rate and reducing cardiac output [33]. The right and left vagi originate in the medulla oblangata, passing through the jugular foramen, traveling alongside carotid through the superior thoracic aperture the into the thoracic cavity [34]. There, they join into the anterior and posterior vagal trunks of the esophageal plexus. The vagal trunks course around the esophagus and innervate the heart at the level of the atria, where they directly communicate with the SA and AV nodes [34] (Figure 2A). The AV node subsequently communicates with the bundle of His thereby controlling ventricular heart rate. It's important to note that the precise anatomical relationships can vary somewhat among individuals, and there may be some variability in the proximity of the vagus nerve branches and the right ventricle. When stimulated, the vagus nerve releases the neurotransmitter acetylcholine, and subsequently binds to muscarinic receptors largely in the SA and AV node, leading to both a negative chronotropic and inotropic effect. The primary effects of vagal stimulation on the heart, including changes in heart rate and conduction, are generally associated with its interactions with the AV node [33]. While a principle use of vagal nerve stimulation is for cessation of epileptic disorders, vagal nerve stimulation has also been shown to help with various cardiac abnormalities including heart failure, dysrhythmia, and cardiac arrest. The use of vagal nerve stimulators showcases the vital role that the vagal nerve plays in cardiac function [33].
A portion of the vagus nerve continues past the heart, following the path of the esophagus to innervate some of the contents of the abdominal cavity. The vagal nerve esophageal plexus courses around the esophagus, directly innervating the esophagus and governing its peristalsis [35]. The anterior and posterior vagal trunks pass into the abdomen through the esophageal hiatus of the diaphragm at the level of T10, sharing this opening with esophagus [36] (Figure 2A). Hiatal hernias are a protrusion of a portion of the stomach through the esophageal hiatus, into the thoracic cavity. This can cause increased pressure on the vagal nerve, particularly as the size of the hernia increases. This can propel the vagal nerve cranially, leading to increased pressure on and irritation of the vagal nerve by the hiatal hernia [37] (Figure 2B).
Figure 2.
(A) Normal anatomic relationship of diaphragm, esophagus, vagus nerve, and heart. Note the very close anatomical proximity of the esophageal component of the vagus nerve to the right heart and particularly the posterior aspect of the right ventricular outflow tract to the lower esophagus. (B) Hiatal hernia compressing the vagal nerve along with direct proximity to the right ventricle and posterior aspect of the RVOT [38].
Figure 2.
(A) Normal anatomic relationship of diaphragm, esophagus, vagus nerve, and heart. Note the very close anatomical proximity of the esophageal component of the vagus nerve to the right heart and particularly the posterior aspect of the right ventricular outflow tract to the lower esophagus. (B) Hiatal hernia compressing the vagal nerve along with direct proximity to the right ventricle and posterior aspect of the RVOT [38].
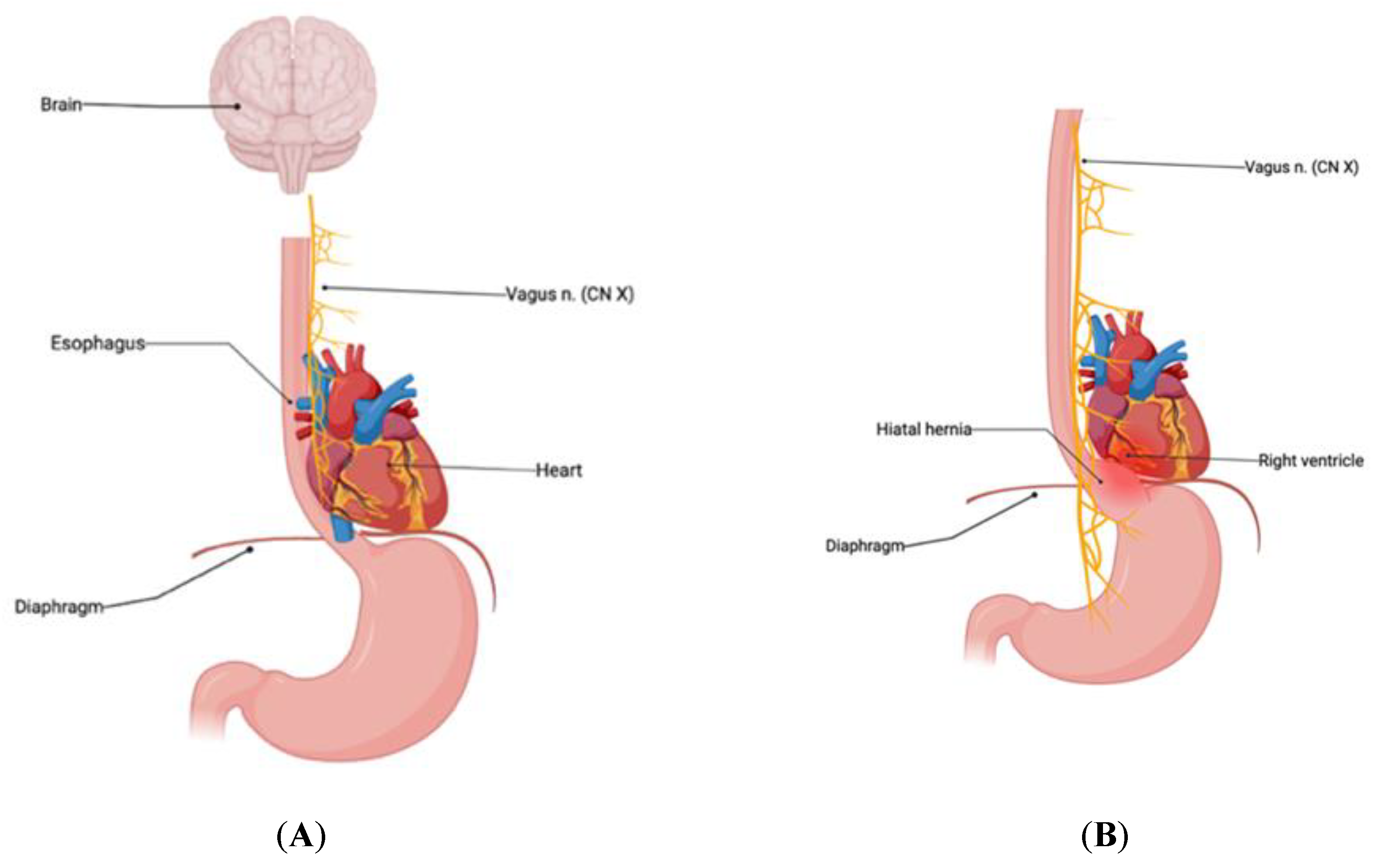
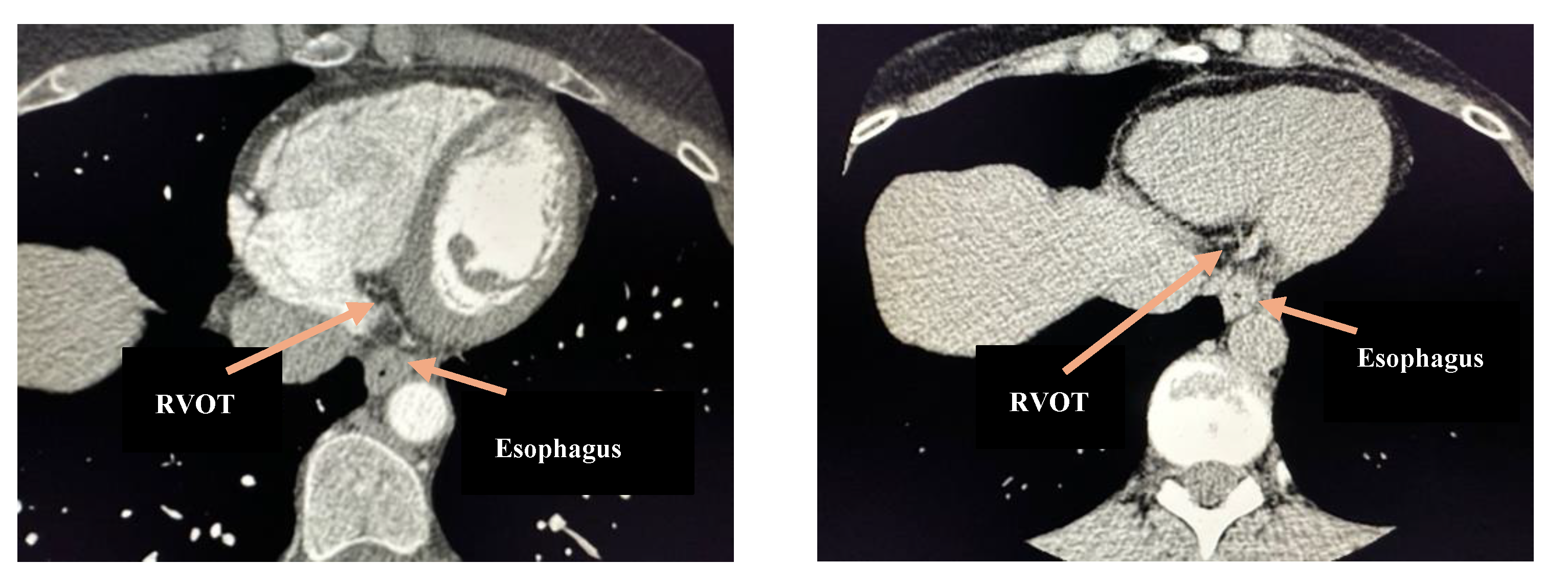
Figure 3.
Standard thoracic CT, axial images with and without contrast. Note the anatomical proximity between the lower esophagus and the right ventricular outflow tract (RVOT).
Figure 3.
Standard thoracic CT, axial images with and without contrast. Note the anatomical proximity between the lower esophagus and the right ventricular outflow tract (RVOT).
In this report, we present seven cases of a significant reduction in PVCs following esophageal/upper GI procedures and/or surgery, presenting evidence for the first time to our knowledge of this novel association between cardiac electrophysiology and gastrointestinal anatomical diseases.
7. Methods
7.1. Clinical Subject Selection
After obtaining IRB approval from the TCU IRB, we reviewed the charts of subjects who had high PVC burden (defined in this study as >10%) who also had concomitant upper GI disease requiring procedural or surgical intervention. We reviewed their clinical symptoms, progress, and cardiac data pre and post procedure, with a focus on continuous rhythm monitoring (Holter) data.
7.2. Statistics
Descriptive statistics were utilized to summarize patient characteristics and PVC burden distributions. The primary analysis focused on assessing the mean reduction in PVC burden post-intervention. A paired t-test or Wilcoxon signed-rank test was employed to compare baseline and post-intervention PVC burden. Statistical significance was determined at a threshold of p < 0.05 (see Table 1 and Supplementary Material).
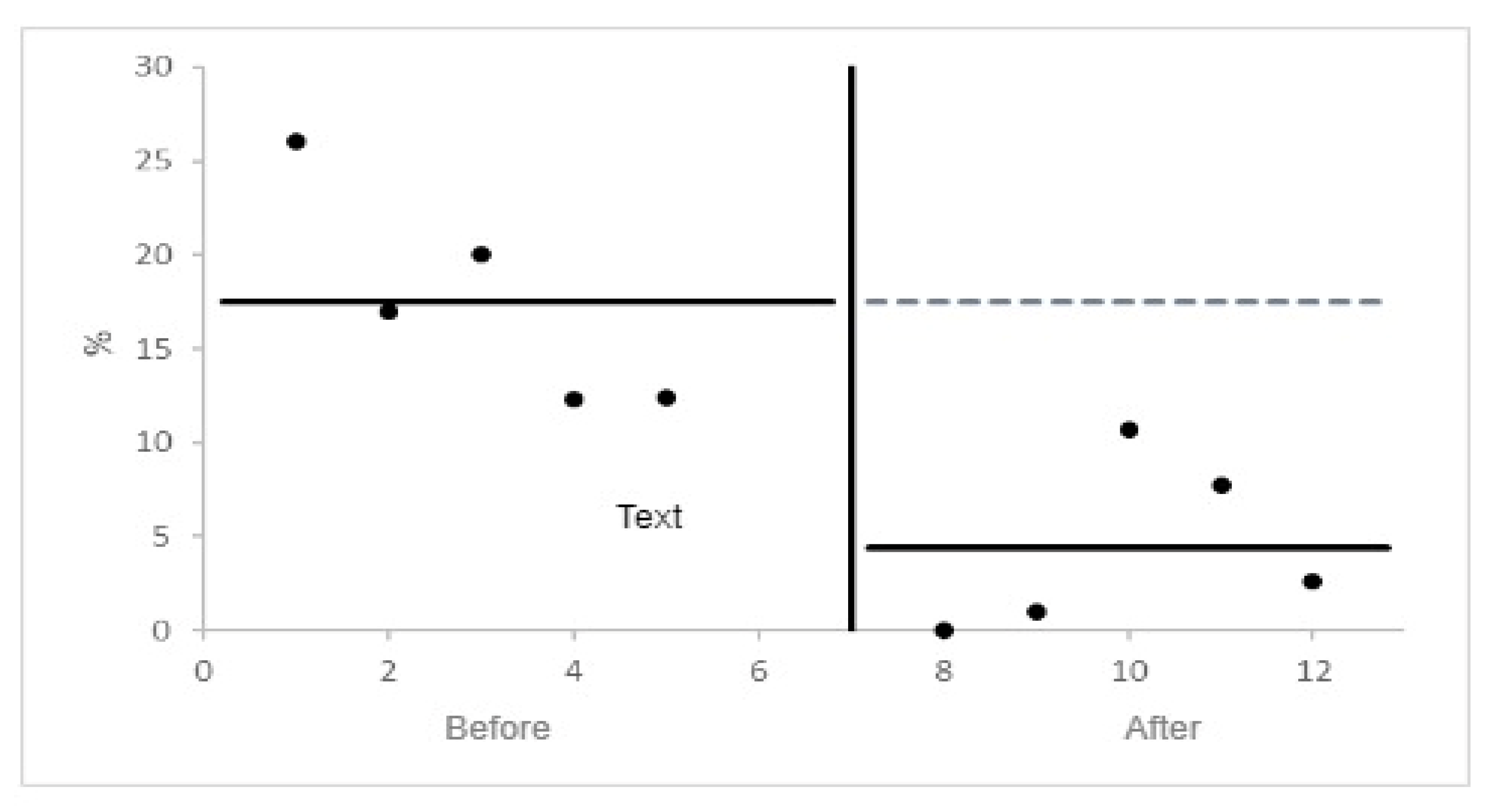
Ordinary least squares interrupted time series analysis was also employed to examine the temporal trends in PVC burden before and after esophageal interventions. It considers potential confounders and provides insight into the longitudinal effects of interventions on PVCs (see Figure 5). This was made possible by the data in Table 1 and draw.io software.
8. Case Descriptions
8.1. Subject 1 – Index Case
A 60-year-old female with no previous cardiac history presented with an abnormal heart rate reading of 40 bpm, but devoid of any cardiac symptoms. Her relevant history included gastroesophageal reflux disease and gastritis with no family history of arrhythmias or sudden cardiac death. Her initial cardiac evaluation in another program showed a 12-lead EKG with sinus rhythm without proarrhythmic features and normal conduction intervals but with frequent, unifocal RVOT basal PVCs. An initial echocardiogram and nuclear perfusion stress test were reported as normal, the index Holter demonstrated a unifocal PVC burden of 26%, and she did not tolerate an initial trial of b-blockers. She had severe gastroesophageal reflux disease (GERD) secondary to a large hiatal hernia that was refractory to conservative therapies, and subsequently underwent a LINX surgical procedure, which is a magnetic sphincter augmentation system (MSA) is used in the treatment of refractory GERD [39]. She established care in our program, and in a follow-up visit after the LINX she reported a significant decline in her palpitations. Curious, we repeated a Holter study, showing a <1% burden of PVCs, followed by a Holter one year later demonstrating a <0.01% burden of PVCs. There were no interval changes in medications, laboratory data, or clinical conditions that accounted for this difference. We believed that her PVCs may have been associated with her esophageal pathology and began observing other patients for similar patterns in an IRB approved study.
8.2. Subject 2
A 46-year-old female with history of achalasia and hypothyroidism presented with palpitations. Her resting 12-lead EKG demonstrated EKG demonstrated sinus rhythm with no proarrhythmic features and normal conduction intervals but frequent PVCs that appeared to originate from the base of the right ventricular outflow tract. Her echocardiogram demonstrated a structurally and functionally normal heart, and her stress echocardiogram was without ischemia and notable for exercise phase suppression of PVCs. Her index Holter monitor demonstrated a PVC burden of 17%. During this time, the patient elected to undergo an esophageal dilation to treat her achalasia. At a subsequent follow-up visit with us, our patient reported a significant improvement in her symptomatic palpitations. A repeat 7-day patch Holter revealed a near resolution of PVCs with a burden of <1%. There were no interval changes in medications, laboratory data, or clinical conditions.
8.3. Subject 3
A 62-year-old female with a history of hypothyroidism and remote history of a lap-band presented with intermittent palpitations and moderate to severe reflux. Her resting 12-lead EKG demonstrated sinus with no proarrhythmic features and normal conduction intervals with frequent PVCs originating from the base of the RVOT. A Holter demonstrated a 18% unifocal PVC burden, and her echocardiogram demonstrated a structurally and functionally normal heart. A high resolution coronary computed tomography angiogram (CCTA) demonstrated no obstructive epicardial coronary artery stenosis, myocardial bridging, or coronary anomalies. The study did identify a dilated fluid-filled esophagus concerning obstruction at the level of gastric lap-band. We trialed a b-blocker, without any significant symptom relief, and eventually referred her for surgical removal of her lap-band which resulted in a significant reduction in her symptomatic palpitations. A repeat Holter showed a decline in PVCs to 10.7% and we anticipate a further reduction in PVCs in her next study.
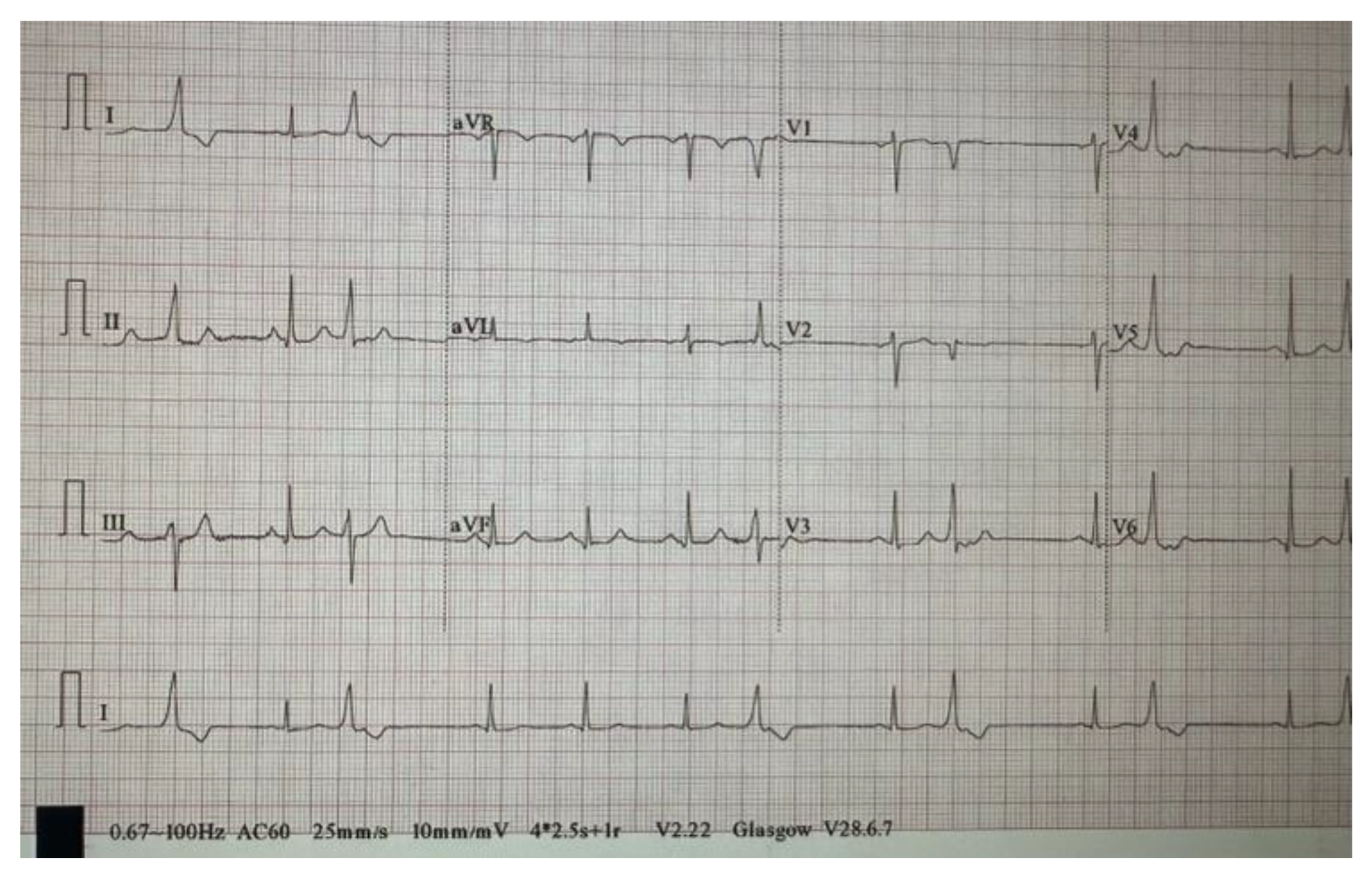
Figure 4.
EKG demonstrates sinus rhythm with frequent PVCs originating from the base of the RVOT.
8.4. Subject 4
A 74-year-old African American male with a history of gastritis, hypercholesterolemia, hypertension presented with occasional palpitations, dysphagia, and chest discomfort. His EKG demonstrated sinus with no proarrhythmic features and normal conduction intervals without PVCs. An initial 7-day Holter monitor revealed a PVC burden of 12.3%. An echocardiogram demonstrated mild aortic valve insufficiency, but normal LV (Left Ventricle) dimensions and intracardiac pressures. He underwent a coronary CTA (Computed Tomography Angiography) demonstrating an elevated calcium score (>500) and moderate to severe angiographic disease; subsequent cardiac catheterization revealed no significant intraluminal coronary artery disease with calcium being largely extraluminal. We advised conservative management of his PVCs. Given his ongoing dysphagia, we referred him to our GI colleagues, who identified esophageal disease requiring esophageal dilation resulting in amelioration of all symptoms. A repeat 7-day Holter monitor post esophageal dilation revealed a decline in PVC burden to 7.7%.
8.5. Subject 5
A 67-year-old female patient with a history of reflux was referred to our practice for evaluation of palpitations and angina. Her initial EKG demonstrated sinus with frequent PVCs originating from the base of the RVOT. Her echo demonstrated a structurally and functional normal study. Given her symptom profile and overall risk assessment, we elected non-invasive coronary anatomical and flow risk assessment with a high-resolution coronary CTA, which demonstrated angiographically normal coronaries with normal flow. An index 7-day patch Holter demonstrated a high burden of unifocal PVCs matching EKG morphology at 12.4% and no other arrhythmias. She did not respond to initial b-blocker therapies. Her CT demonstrated a large hiatal hernia, and after additional GI evaluation, she was referred for surgical intervention. After surgery she noted a clinical improvement in her palpitations, and a repeat Holter examination demonstrated a decline in PVC burden to 2.6%.
8.6. Subject 6
A 69-year-old African American patient established patient in our practice with a history of HTN, hiatal hernia s/p repair noted chest pressures with anginal type features. His EKG demonstrated sinus with frequent PVCs originating from the base of the RVOT, his repeat echo demonstrated no changes from a prior structurally and functional normal study. A Holter demonstrated a 24% unifocal PVC burden without other arrhythmias. We proceeded with a high-resolution coronary CTA, which demonstrated angiographically normal coronaries with normal flow and no significant extraluminal coronary artery calcium. We suspected an esophageal etiology for his symptoms and after GI evaluation, underwent an esophageal dilation. He noted an immediate improvement in palpitations, and resolution of chest pains. A repeat Holter demonstrated a decline in PVC burden to 11%.
Figure 5.
PVC burden before and after Upper GI/esophageal interventions by Ordinary Least Squares Interrupted Time Series Analysis [43].
Figure 5.
PVC burden before and after Upper GI/esophageal interventions by Ordinary Least Squares Interrupted Time Series Analysis [43].
9. Discussion
Our study reviews the case of seven patients with esophageal disease who presented with idiopathic PVC burden. These PVCs notably arose from the base of the RVOT in all patients. All were thoroughly evaluated for cardiac etiology of PVC, but no cardiac disease was found. However, when these patients’ esophageal diseases were treated with surgical intervention, they had a spontaneous reduction in PVC burden. After procedural intervention of the esophageal/Upper GI disorder, we observed a mean 68.34 % decline in PVC burden in this cohort, as well as a decline in symptoms as reported by the patients. Due to this noteworthy PVC burden reduction after gastrointestinal intervention, it is important to explore possible mechanisms by which treating esophageal disease subsequently improves cardiac conduction.
The esophagus, vagus nerve, and the right ventricle are all adjacent structures and so it stands to reason that pathology in one could have detrimental effects on the other structures. There are reports of esophageal pathology stimulating vagal nerve parasympathetic action on the heart. For example, deglutition syndrome or swallow syncope, is a condition in which patients with gastrointestinal disease such as hiatal hernia, achalasia, and esophageal cancer had syncopal episodes following the ingestion of foods or liquids [40]. The pathophysiologic mechanism of swallow syncope is thought to be vagal stimulation from increased esophageal pressure causing reflex bradycardia and hypotension [41]. However, some reports describe deglutition syndrome resulting in complete heart block or supraventricular tachycardia [42,43]. With larger paraoesophageal hernias there can be increased pressure on the left atrium and therefore decreased cardiac output [37].
Other studies have shown a correlation between hiatal hernia with GERD and atrial fibrillation (AF) [44]. The treatment of hiatal hernia and GERD by either surgical intervention or proton pump inhibitor was found to relieve AF in several cases [44,45,46]. One proposed mechanism for how GERD contributes to AF is that esophagus inflammation releases cytokines that stimulate the vagus nerve, affecting cardiac rhythm [47]. Another hypothesis is that a hiatal hernia can physically irritate the vagus nerve or heart by direct contact thereby causing dysrhythmia [32]. While there have been discussions of atrial arrhythmias due to gastrointestinal disorders, there is a paucity in the literature discussing ventricular arrythmias and upper GI disorders.
Our patients all demonstrated PVCs in relation to upper GI disorders, as evidenced by relief of cardiac symptoms after treatment of GI disease. One commonality is that all of these patients’ GI dysfunction can lead to an increase in esophageal size, especially post prandially. The increased esophageal size places direct anatomical pressure on the vagal nerve, leading to vagal nerve stimulation resulting in parasympathetic cardiac effects (Figure 2 and Figure 3). Two of the patients in our study underwent esophageal dilation, with complete relief of PVCs symptoms after, suggesting that direct effects on the vagal nerve played a role in their symptomatic PVCs. While we cannot ascertain if this anatomical pressure caused stimulation or inhibition, we believe that pressure on the vagus nerve leads to stimulation of mechanoreceptors resulting in bradycardia-induced PVCs [48]. All of our patients experienced a significant decline and/or symptomatic relief of PVCs after a surgical intervention that decreased almost surely decreased mechanical pressure on the vagus nerve [49].
Curiously, all of our patients’ PVCs originated from the base of RVOT, which is close in proximity to the esophagus. Compression from the esophagus/upper GI abnormality directed against the right ventricle resulting in RVOT irritation is theoretically feasible. This affected tissue in the RVOT may potentially create a reentry circuit, which is a known cause of PVCs [3]. All of our patients’ RVOT basal PVCs resolved with procedural GI management, lending to our hypothesis that esophageal/Upper GI anatomical abutment of the RV induced a potential reentry circuit leading to these stereotypic PVCs. Remediation of the esophageal/upper GI disorder directly relieved this mechanical pressure on the RVOT, resulting in the consistently observed reduction in PVCs.
10. Limitations
Limitations of our study includes a very sample size of cases that would require a significantly larger series of clinical studies and more definitive mechanistic investigation before generalization to a larger population. Furthermore, there may be a natural fluctuation in PVC burden; patients are often evaluated at high symptom and PVC burden with a reduction over time which may simply be due to regression to the mean. However, the novelty of the data reported in this paper should generate collaborative interest to pursue these larger studies to confirm this novel and preliminary finding. In several of our subjects, there was a residual burden of PVCs remaining after GI intervention, suggesting more than one pathway of PVC pathogenesis in these subjects.
11. Conclusions
In our series of patients, we believe that procedural/surgical intervention for esophageal diseases resulted in a significant reduction or near resolution of PVCs. Our mechanistic hypothesis is two-fold; the first through vagal mediation, specifically within the lower esophagus, and the second being esophageal/upper GI mechanical induction of PVCs through direct effects on the RVOT of the right ventricle. These are highly preliminary findings that need further investigation, but if confirmed through mechanistic investigation and larger clinical studies as we have suggested, the assessment of esophageal and upper GI pathologies may become a part of the standard paradigm for the evaluation and treatment of high burden PVCs, leading to a new subcategory of PVCs for which propose a new term esophageal PVC (E-PVCs).
Author Contributions
Conceptualization, M.S.; methodology, M.S..; formal analysis, M.S., F.K., E.H, C.K., P.M., P.M., A.R., A.W; investigation, F.K., E.H, C.K., M.S..; data curation, F.K., E.H, C.K.,P.M., M.S.; writing—original draft preparation F.K., E.H, C.K., P.M., P.M; writing—review and editing, M.S., F.K., E.H, C.K., P.M., P.M., A.R., A.W; visualization, P.M.; supervision, M.S..; project administration, M.S..; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Approval/ Human Subject Informed Consent
TCU IRB#2022-204.
Informed Consent Statement
Informed consent was obtained from the subject involved in the study and is stored at CCMS-FW.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to patient privacy considerations.
Acknowledgments
The authors wish to thank the staff of Consultants in Cardiovascular Medicine and Science – Fort Worth for clinical subject and records coordination. .
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Source of Funding
None.
Supplementary Material
Statistical calculations
Difference Scores Calculations
Treatment 1
N1: 5
df1 = N - 1 = 5 - 1 = 4
M1: 17.54
SD1: 5.74
SEM1: 2.5
Treatment 2
N2: 5
df2 = N - 1 = 5 - 1 = 4
M2: 4.4
SD2: 4.6
SEM2: 2.06
P-value Calculation
dof = n-1 = 4
XD = 13.14
SXD =√1/(n-1) Σ(XDi – XD)2 = 7.38
t = XD/(SD/√n) = 13.14/(7.38/√5) = 3.98
The calculated t exceeds the critical value (3.98 > 2.78) so the means are statistically significant (p < 0.05). [50]
References
- von Rotz, M., et al., Risk factors for premature ventricular contractions in young and healthy adults. Heart, 2017. 103(9): p. 702-707.
- Dukes, J.W., et al., Ventricular Ectopy as a Predictor of Heart Failure and Death. J Am Coll Cardiol, 2015. 66(2): p. 101-9. [CrossRef]
- Massing, M.W., et al., Usefulness of ventricular premature complexes to predict coronary heart disease events and mortality (from the Atherosclerosis Risk In Communities cohort). Am J Cardiol, 2006. 98(12): p. 1609-12. [CrossRef]
- Marcus, G.M., Evaluation and Management of Premature Ventricular Complexes. Circulation, 2020. 141(17): p. 1404-1418. [CrossRef]
- Brachmann, J., et al., Bradycardia-dependent triggered activity: relevance to drug-induced multiform ventricular tachycardia. Circulation, 1983. 68(4): p. 846-56. [CrossRef]
- Schlotthauer, K. and D.M. Bers, Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res, 2000. 87(9): p. 774-80. [CrossRef]
- Lerman, B.B., et al., Adenosine-sensitive ventricular tachycardia: evidence suggesting cyclic AMP-mediated triggered activity. Circulation, 1986. 74(2): p. 270-80. [CrossRef]
- Lerman, B.B., Mechanism, diagnosis, and treatment of outflow tract tachycardia. Nat Rev Cardiol, 2015. 12(10): p. 597-608. [CrossRef]
- Murakawa, Y., et al., Reappraisal of the coupling interval of ventricular extrasystoles as an index of ectopic mechanisms. Br Heart J, 1992. 68(6): p. 589-95. [CrossRef]
- Zimmermann, M., Sympathovagal balance prior to onset of repetitive monomorphic idiopathic ventricular tachycardia. Pacing Clin Electrophysiol, 2005. 28 Suppl 1: p. S163-7. [CrossRef]
- Gillis, R.A., Cardiac sympathetic nerve activity: changes induced by ouabain and propranolol. Science, 1969. 166(3904): p. 508-10. [CrossRef]
- Estes, E.H., Jr. and H.L. Izlar, Jr., Recurrent ventricular tachycardia. A case successfully treated by bilateral cardiac sympathectomy. Am J Med, 1961. 31: p. 493-7. [CrossRef]
- Weiss, T., G.M. Lattin, and K. Engelman, Vagally mediated suppression of premature ventricular contractions in man. Am Heart J, 1975. 89(6): p. 700-7. [CrossRef]
- He, W., et al., Autonomic involvement in idiopathic premature ventricular contractions. Clin Res Cardiol, 2013. 102(5): p. 361-70. [CrossRef]
- Chen, J.-H., Ineffective esophageal motility and the vagus: current challenges and future prospects. Clinical and experimental gastroenterology, 2016. 9: p. 291-299. [CrossRef]
- Pfeiffer ER, Tangney JR, Omens JH, McCulloch AD. Biomechanics of Cardiac Electromechanical Coupling and Mechanoelectric Feedback. J Biomech Eng. 2014;136(2):0210071-02100711. [CrossRef]
- Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102(5):369-392. [CrossRef]
- Ho Tse ZT, Dumoulin CL, Clifford G, et al. Real-ECG extraction and stroke volume from MR-Compatible 12-lead ECGs; testing during stress, in PVC and in AF patients. Journal of Cardiovascular Magnetic Resonance. 2011;13(1):P6. [CrossRef]
- Cohn K, Kryda W. The influence of ectopic beats and tachyarrhythmias on stroke volume and cardiac output. J Electrocardiol. 1981;14(3):207-218. [CrossRef]
- Farzam K, Richards JR. Premature Ventricular Contraction. In: StatPearls. StatPearls Publishing; 2023. Accessed August 26, 2023. http://www.ncbi.nlm.nih.gov/books/NBK532991/.
- da Silva RC, Gondim MC, Melo GM, et al. Decreased cardiac output: an integrative review. Rev Bras Enferm. 76(2):e20220265. [CrossRef]
- Lee, A., R. Denman, and H.M. Haqqani, Ventricular Ectopy in the Context of Left Ventricular Systolic Dysfunction: Risk Factors and Outcomes Following Catheter Ablation. Heart Lung Circ, 2019. 28(3): p. 379-388. [CrossRef]
- Loring, Z., P. Hanna, and C.N. Pellegrini, Longer Ambulatory ECG Monitoring Increases Identification of Clinically Significant Ectopy. Pacing Clin Electrophysiol, 2016. 39(6): p. 592-7. [CrossRef]
- Hamon, D., et al., Premature ventricular contraction diurnal profiles predict distinct clinical characteristics and beta-blocker responses. J Cardiovasc Electrophysiol, 2019. 30(6): p. 836-843. [CrossRef]
- Zhong, L., et al., Relative efficacy of catheter ablation vs antiarrhythmic drugs in treating premature ventricular contractions: a single-center retrospective study. Heart Rhythm, 2014. 11(2): p. 187-93. [CrossRef]
- Ling, Z., et al., Radiofrequency ablation versus antiarrhythmic medication for treatment of ventricular premature beats from the right ventricular outflow tract: prospective randomized study. Circ Arrhythm Electrophysiol, 2014. 7(2): p. 237-43.
- Stec, S., et al., Benign symptomatic premature ventricular complexes: short- and long-term efficacy of antiarrhythmic drugs and radiofrequency ablation. Kardiol Pol, 2012. 70(4): p. 351-8.
- Cronin, E.M., et al., 2019 HRS/EHRA/APHRS/LAHRS expert consensus statement on catheter ablation of ventricular arrhythmias. Heart Rhythm, 2020. 17(1): p. E2-e154.
- Olszak-Waśkiewicz, M., et al., The association between SCN5A, KCNQ1 and KCNE1 gene polymorphisms and complex ventricular arrhythmias in survivors of myocardial infarction. Kardiologia Polska, 2008. 66(8): p. 845-853; discussion 854-855.
- Chen, G.-X., et al., Clinical characteristics and electrophysiologic properties of SCN5A variants in fever-induced Brugada syndrome. EBioMedicine, 2023. 87: p. 104388. [CrossRef]
- Leung, J., et al., Clinical Characteristics, Genetic Findings and Arrhythmic Outcomes of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia from China: A Systematic Review. Life (Basel, Switzerland), 2022. 12(8): p. 1104. [CrossRef]
- Noom, M.J., A. Dunham, and C.G. DuCoin, Resolution of Roemheld Syndrome After Hiatal Hernia Repair and LINX Placement: Case Review. Cureus, 2023. 15(4): p. E37429. [CrossRef]
- Capilupi, M.J., S.M. Kerath, and L.B. Becker, Vagus Nerve Stimulation and the Cardiovascular System. Cold Spring Harbor Perspectives in Medicine, 2020. 10(2): p. A034173. [CrossRef]
- Kenny, B.J. and B. Bordoni, Neuroanatomy, Cranial Nerve 10 (Vagus Nerve), in StatPearls. 2023, StatPearls Publishing: Treasure Island (FL).
- Browning, K.N., S. Verheijden, and G.E. Boeckxstaens, The vagus nerve in appetite regulation, mood and intestinal inflammation. Gastroenterology, 2017. 152(4): p. 730-744. [CrossRef]
- Shahid, Z. and B. Burns, Anatomy, Abdomen and Pelvis: Diaphragm, in StatPearls. 2023, StatPearls Publishing: Treasure Island (FL).
- Akçay, M. and İ. Çamlıdağ, Huge Hiatal Hernia Mimicking a Mass with Compressive Effects on the Left Atrium Causing Paroxysmal Atrial Fibrillation. The Journal of Tehran University Heart Center, 2019. 14(2): p. 90-91. [CrossRef]
- Image Created with biorender.com. Available online: http://Biorender.com (accessed on 12 March 2024).
- Skubleny, D., et al., LINX(®) magnetic esophageal sphincter augmentation versus Nissen fundoplication for gastroesophageal reflux disease: a systematic review and meta-analysis. Surg Endosc, 2017. 31(8): p. 3078-3084. [CrossRef]
- Patel, N., et al., Deglutition syncope. Proceedings (Baylor University. Medical Center), 2017. 30(3): p. 293-294.
- Kahn, A., L.M. Koepke, and S.B. Umar, Deglutition Syncope: A Case Report and Review of the Literature. ACG Case Reports Journal, 2015. 3(1): p. 20-22. [CrossRef]
- Abbood, A., et al., A Large Intrathoracic Hiatal Hernia as a Cause of Complete Heart Block. Case Reports in Cardiology, 2021. 2021: p. 6697016. [CrossRef]
- Erdoğan, H.İ., H. Gök, and M. Karanfil, Swallowing-induced atrioventricular block and syncope in a patient with achalasia. The Turkish Journal of Gastroenterology: The Official Journal of Turkish Society of Gastroenterology, 2015. 26(1): p. 75-76.
- Weigl, M., et al., Reflux esophagitis in the pathogenesis of paroxysmal atrial fibrillation: results of a pilot study. Southern Medical Journal, 2003. 96(11): p. 1128-1132. [CrossRef]
- Schilling, R.J. and G.C. Kaye, Paroxysmal atrial flutter suppressed by repair of a large paraesophageal hernia. Pacing and clinical electrophysiology: PACE, 1998. 21(6): p. 1303-1305. [CrossRef]
- Mohamed, A., et al., Gastroesophageal Reflux and Its Association With Atrial Fibrillation: A Traditional Review. Cureus. 12(9): p. E10387. [CrossRef]
- Roman, C., et al., Atrial fibrillation in patients with gastroesophageal reflux disease: A comprehensive review. World Journal of Gastroenterology, 2014. 20(28): p. 9592-9599. [CrossRef]
- Prescott, S.L. and S.D. Liberles, Internal senses of the vagus nerve. Neuron, 2022. 110(4): p. 579-599. [CrossRef]
- Zheng, L., et al., Symptomatic Premature Ventricular Contractions in Vasovagal Syncope Patients: Autonomic Modulation and Catheter Ablation. Frontiers in Physiology, 2021. 12. [CrossRef]
- Hawley, S., et al., <p>Sample size and power considerations for ordinary least squares interrupted time series analysis: a simulation study</p>. Clinical Epidemiology, 2019. Volume 11: p. 197-205.
Figure 1.
In this particular hemodynamic tracing from a catheter placed in the ascending aorta, note the typical anterograde conducted sinus beat (labeled A), with a corresponding arterial systolic pressure is 88 mmHg (labeled A). In a Post PVC beat (labeled B) the arterial pressure declines to 70 mmHg, representing a 21% decrease in perfusion pressure.
Figure 1.
In this particular hemodynamic tracing from a catheter placed in the ascending aorta, note the typical anterograde conducted sinus beat (labeled A), with a corresponding arterial systolic pressure is 88 mmHg (labeled A). In a Post PVC beat (labeled B) the arterial pressure declines to 70 mmHg, representing a 21% decrease in perfusion pressure.
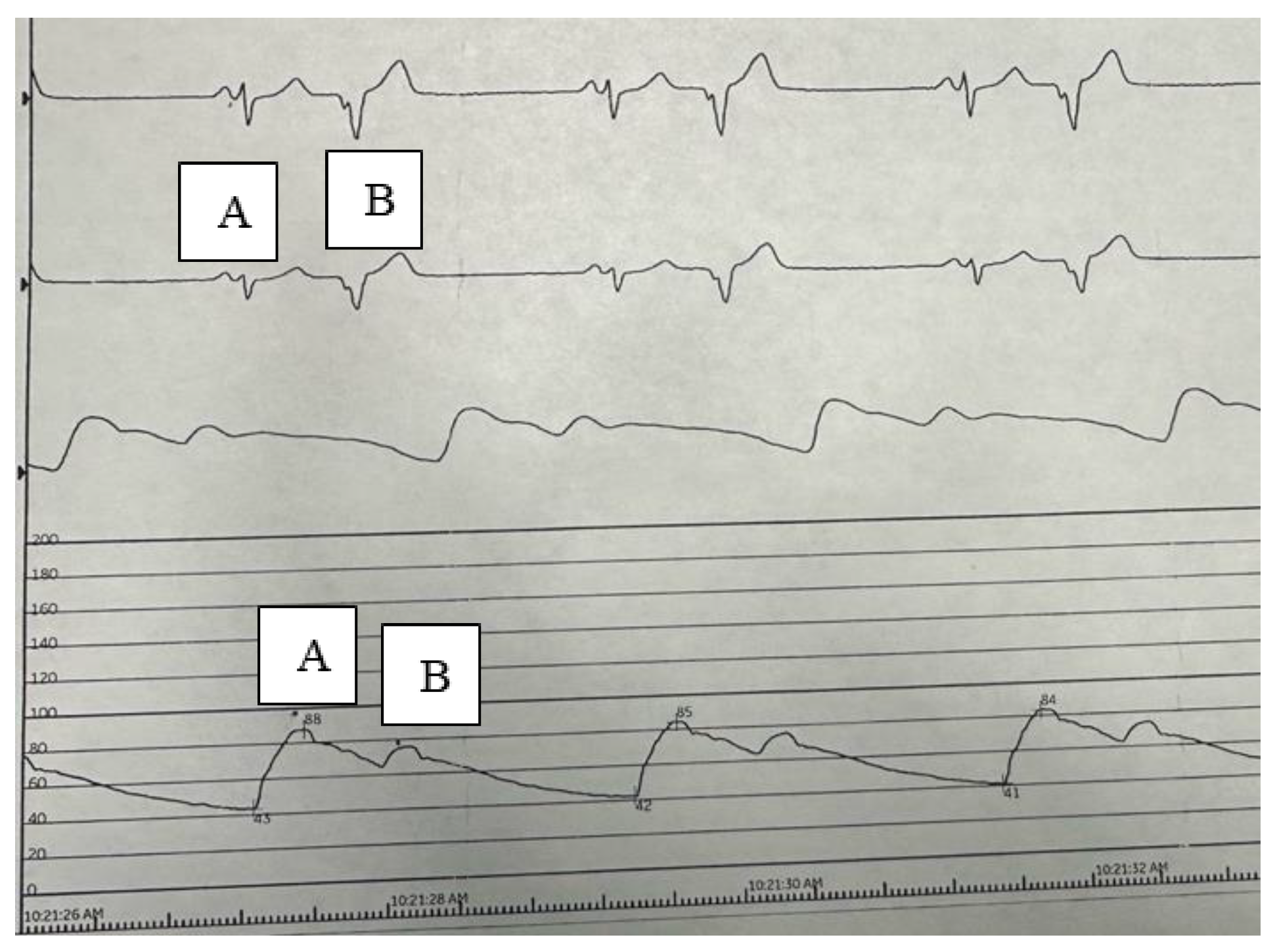
Figure 2.
Genetic factors including mutations in the SCN5A gene, encoding essential components of voltage gated sodium channels, or KCNQ1/KNCE1, which encode components of potassium channels, may be responsible for PVC formation. Other non-genetic factors that contribute to PVC formation include structural heart disease, electrolyte imbalance, and stimulant use.
Figure 2.
Genetic factors including mutations in the SCN5A gene, encoding essential components of voltage gated sodium channels, or KCNQ1/KNCE1, which encode components of potassium channels, may be responsible for PVC formation. Other non-genetic factors that contribute to PVC formation include structural heart disease, electrolyte imbalance, and stimulant use.
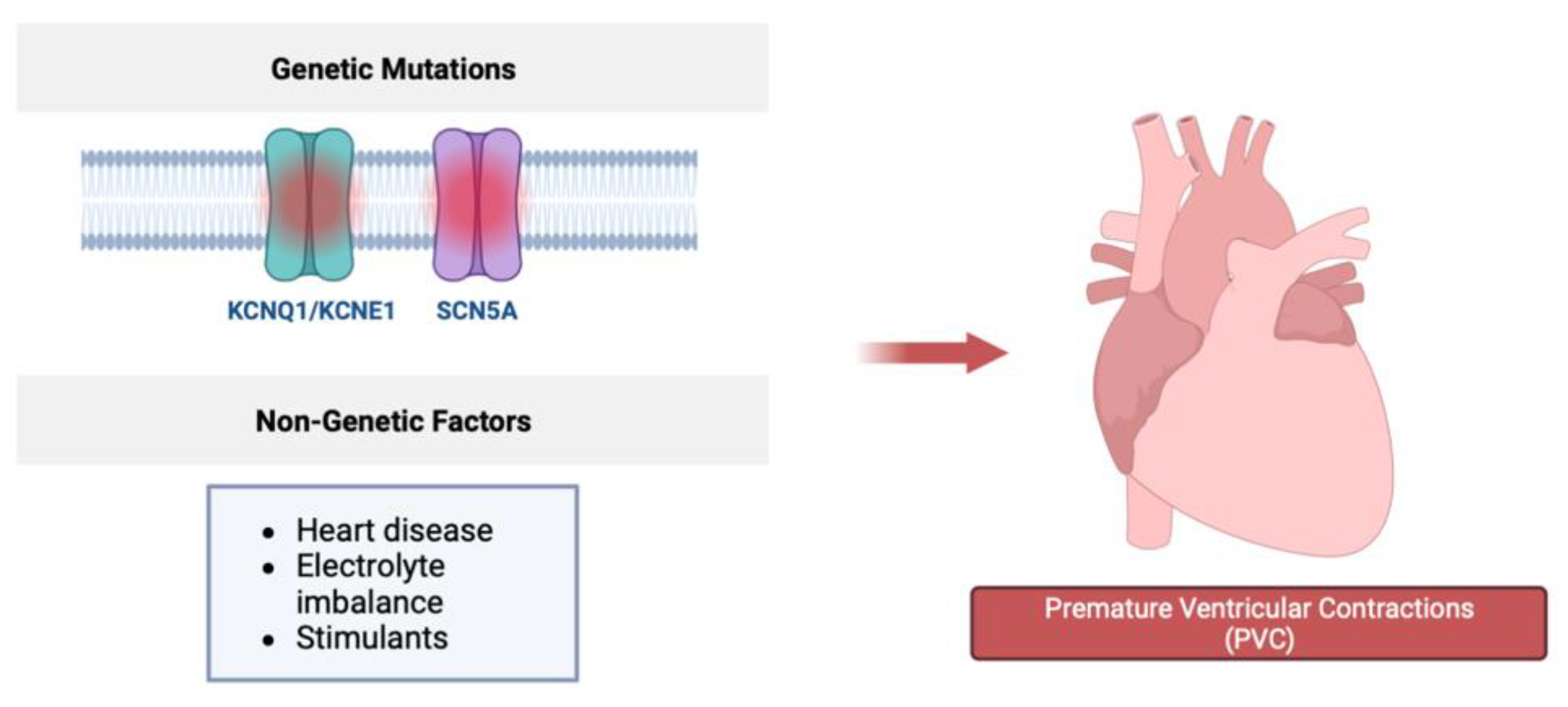

Table 1.
Reduction in PVC burden by 68.34% [p=0.024] in this patient series after Esophageal/Upper GI interventions.
Table 1.
Reduction in PVC burden by 68.34% [p=0.024] in this patient series after Esophageal/Upper GI interventions.
| Age | Sex | GI Pathology | Esophageal Intervention | Baseline PVCs (%) | Post Intervention PVCs (%) | % Decline | p-value |
|---|---|---|---|---|---|---|---|
| 60 | F | Hiatal hernia | LINX repair | 26.00 | <0.01 | 99.96 | |
| 46 | F | Achalasia | Esophageal dilation | 17.00 | <1.0 | 94.11 | |
| 62 | F | Lap-band | Lap-band removal | 20.00 | 10.70 | 46.50 | |
| 74 | M | Esophageal stenosis | Esophageal dilation | 12.29 | 7.70 | 37.34 | |
| 67 | F | Hiatal hernia | Hernia repair | 12.4 | 2.6 | 79.03 | |
| 69 | M | Esophageal stenosis | Esophageal dilation | 23.7 | 11.0 | 53.5 | |
| 68.34% | 0.024 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



