Submitted:
26 July 2024
Posted:
26 July 2024
You are already at the latest version
Abstract
Cleomella serrulata is a native flowering plant found in North America. Even though this plant is of ecological and native medicinal importance, very little is known about the genomic makeup of Cleomella and the Cleomaceae family at large. Here we report the complete chloroplast genome of Cleomella serrulata and provide an evolutionary comparison to other chloroplast genomes from Cleomaceae and closely related families. This study confirms the taxonomic placement of Cleomella as a distinct genus, but also provides phylogenetic insights that imply potential adaptive strategies and evolutionary mechanisms driving genomic diversity of the Cleomella genus. Whole genome-based and ANI comparisons indicate that the Cleomella species form a distinct clade that is about equidistant from the other Cleomaceae genera as it is from the genera from the nearby Capparaceae and Brassicaceae. This is the first complete chloroplast-based phylogenetic comparison of Cleomella species to other related genera and helps refine the complex taxonomic distinctions of the Cleomaceae.
Keywords:
Cleomella
; native plant
; medicinal plant
; chloroplast evolution
; Nebraska
; Rocky Mountain bee plant
; stinking clover
; phylogeny
1. Introduction
In the arid landscapes of North America, among the vast stretches of desert flora, one can encounter the delicate yet resilient Cleomella serrulata. Commonly known as the Rocky Mountain bee plant or stinking clover, this unassuming member of the Cleomaceae family captivates with its intricate biology and ecological significance [1].
The Cleomaceae are a small family of flowering plants in the order Brassicales, comprising about 270 species in 17 accepted genera (USDA-ARS GRIN Taxonomy, 2024; [2]), that are found widespread on various continents. Cleomella serrulata is native to the American prairies and thrives in various arid habitats, including rocky slopes, desert washes, and sandy plains, primarily in the western United States and northern Mexico. Its range spans from Arizona and New Mexico to Utah, Colorado, and Wyoming, where it adds a splash of color to the rugged terrain, especially during its flowering season in late spring and early summer [3]. This plant’s ability to adapt to harsh environmental conditions underscores its resilience and ecological importance in maintaining biodiversity within arid ecosystems.
Cleomella serrulata has several intriguing characteristics that contribute to its ecological niche. One notable feature is its unique relationship with pollinators. The plant produces nectar-rich flowers with a distinct scent, attracting a diverse array of pollinators, including bees, butterflies, and hummingbirds [4,5]. This symbiotic relationship highlights the plant’s role as a crucial food source for local pollinator populations, emphasizing its importance in maintaining ecosystem stability.
Cleomella serrulata stands as a testament to the resilience and intricacies of desert flora. Its modest appearance belies a rich tapestry of ecological interactions, cultural significance, and taxonomic nuances. As our knowledge of this enigmatic species continues to evolve, so does our appreciation for the wonders of the natural world and the imperative to safeguard its diversity for future generations. Moreover, Cleomella serrulata possesses medicinal properties that indigenous communities have recognized for centuries. Historically, Native American tribes utilized various parts of the plant for medicinal purposes, from treating skin ailments to alleviating respiratory issues [6]. These traditional uses underscore the cultural significance and ethnobotanical value of Cleomella serrulata within local communities.
The taxonomic classification of Cleomella serrulata has undergone several revisions over time, reflecting advancements in botanical research and molecular techniques. Initially classified within the Cleome genus, recent phylogenetic studies have prompted taxonomic reevaluations, leading to the establishment of the Cleomella genus as a distinct lineage within the Cleomaceae family [7]. Unfortunately, this restructuring of the Cleomaceae was done purely based on morphological and geographical characteristics and did not include genomic comparisons. However, previous gene-based phylogenetic studies (using chloroplast genes and ITS), that were performed before the restructuring [5,8] appear to be in agreement with the reformation of the Cleomaceae. At the time there was insufficient chloroplast or nuclear genomic information available to further validate the taxonomic restructuring using whole genome data. Our current study forms the genomic base for a more detailed genomic analysis of the genera within Cleomaceae and their relationship with other closely related plant families, with particular emphasis on the North American Clemoids. This evolving understanding of its taxonomic position underscores the dynamic nature of botanical classification and the importance of interdisciplinary approaches in discovering more about plant diversity and evolutionary relationships.
Furthermore, ongoing research into Cleomella serrulata’s genetic makeup and ecological interactions continues to unravel its complexities, shedding light on its evolutionary history and adaptive strategies [8]. Collaborative efforts between botanists, ecologists, and geneticists have enabled a more comprehensive understanding of this species’ ecological role and conservation needs, emphasizing the importance of interdisciplinary approaches in addressing contemporary challenges in biodiversity conservation. The Cleomaceae have been central to several important ecological and evolutionary studies on floral morphology and development [9,10], the evolution of C4 photosynthesis [11,12,13,14,15], pollination biology [4], and comparative genomics [16,17] and transcriptomics [18,19]. The scientific interest in Cleomaceae is highly augmented by the close sister relationship to the Brassicaceae because the latter family includes the model organism Arabidopsis thaliana. However, Cleomaceae studies have been hindered by the lack of genomic information needed for taxonomic, evolutionary and molecular biological studies. Although the Cleomaceae is a family comprising of some 270 species [2], before the current study, there was only one other Cleomella chloroplast genome in the NCBI Genbank database, NC_049613 from Cleomella lutea, which was deposited without publication or genomic analysis, and only two other Cleomaceae complete chloroplast genomes, of African origin, were recently described: MT948188 Thulinella chrysantha (= Cleome chrysantha), and NC_054213 Dipterygium glaucum (= Cleome pallida) [20]. In addition, two more distant Cleomaceae of African origin were also found in the NCBI database: NC_054276 Gynandropis gynandra and NC_066812 Coalisina paradoxa. That leaves the North American Cleomaceae very underrepresented as far as chloroplast genomes sequences and genome-based phylogenetic studies.
The primary objective of this study was to acquire the chloroplast genome of Cleomella serrulata for comparative analysis within the Cleomaceae family. By obtaining and analyzing the chloroplast genome, we aimed to identify evolutionary relationships, genetic variation, and potential adaptive traits within this taxonomic group.
2. Materials and Methods
2.1. Isolation and DNA Extraction
The Cleomella serrulata used in this study was collected in July 2023 from plants growing in our native garden at Bellevue University (41.15128 N, 95.91927 W). The native garden was established in 2020 and has been maturing for 4-5 years now. Cleomella species were not part of the initial intentional planting and have appeared naturally during the establishment of the native garden. Cleomella serrulata was identified using the USDA Plants Database (https://plants.usda.gov/home/plantProfile?symbol=CLSE) and the Minnesota Wildflower database (https://www.minnesotawildflowers.info/flower/rocky-mountain-beeplant). Plants were about 1 meter tall and blooming at the time of sample collection. Leaf and stem samples were collected and frozen at -80 C in sterile tubes.
Pigment extraction was performed by taking about 4 flower petals and cutting them up in small fragments in a 2 ml sterile tube. For water extractions, 1 ml of sterile water was added to the fragmented petals and incubated at room temperature for 15 min. The suspension was vortexed for 2 min and centrifuged for 5 min at 4,000xg. Spectra were collected using an Evolution 300 UV-vis spectrophotometer (Thermo Scientific).
For DNA isolation, we used several plant leaves that were first cut up using sterile scissors and then ground to a fine powder using a sterile mortar and pestle. A total of 300 mg of ground up Cleomella powder was used for total DNA extraction using the DNeasy Plant Mini kit (Qiagen). The following adaptations were made to the manufacturer protocol. The sample was subjected to bead beating for 2 min using 1.5 mm high impact Zironium beads (BenchmarkScientific) in a BeadBug Microtube Homogenizer (model D1030, LABRepCo) at a speed of 3000 rpm, after adding AP1 solution, to optimize tissue disruption. The incubation period at 65 °C was increased from 10 minutes to one hour. Subsequently, the sample was refrigerated at 4°C overnight before adding Buffer P3, to increase cell lysis. DNA analysis using Qubit and NanoDrop showed a DNA concentration of 46 ng/L, with a 260/280 nm absorbance ratio of 1.53. A total of 460 ng of DNA was used for whole genome sequencing.
2.2. DNA Sequencing, Mapping and Annotation
The sequencing library was prepared using the Illumina DNA Library Prep kit. The genome was sequenced by an Illumina MiniSeq, using 500 µL of a 1.8 pM library. Paired-end (2 × 150 bp) sequencing generated 910,668 reads and 137.5 Mbps of sequencing data. The sequence read length distribution was 35–151 with >90% of the read lengths above 149 bp. Quality control of the reads was performed using FastQC (version 1.0.0) [21] within Basespace (Illumina), using a k-mer size of 5 and contamination filtering for overrepresented sequences against the default contamination list. We assembled the genome de novo using SPAdes (version 3.9.0) [22] within BaseSpace.
To isolate the specific reads that belong to chloroplast DNA, the Illumina reads and assembled contigs were reassembled using Minimap2 (v2.24) [23] within Geneious Prime (v2024.0.3), with the Cleomella lutea chloroplast genome (NC_049613.1) as a reference genome. This aligned 25,476 reads to produce a consensus sequence of 154,482 bp.
The consensus sequence derived from the alignment and mapping process was annotated using into AGORA, a bioinformatics platform specialized in chloroplast annotation [24]. Some of the coding regions were manually refined using BLAST comparison with reference chloroplast genomes from NCBI Genbank.
2.3. Phylogenetic Trees and ANI Calculations
To initiate the comparative analysis of chloroplast genomes within the Cleomaceae family, an NCBI BLAST (Basic Local Alignment Search Tool) search was conducted using, a segment of the Cleomella chloroplast genome to identify homologous sequences within the Cleomaceae family. Subsequently, the multiple sequence alignment and phylogenetic tree construction were performed using MEGA X software [25]. The alignment process utilized the ClustalW algorithm to align sequences, accommodating for potential variations in sequence divergence. The resulting alignment was then used to construct a phylogenetic tree employing Maximum Likelihood, providing insights into the evolutionary relationships among Cleomella species. The evolutionary history was inferred by using the Maximum Likelihood method and General Time Reversible model [26]. The tree with the highest log likelihood (-451022.84) is shown in Figure 3. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 11 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. There were a total of 168261 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 [25]. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format [27].
The structural features of the chloroplast genomes of C. serrulata (NC_088033), C. lutea (NC_049613), Dipterygium glaucum (NC_054213), Thulinella chrysantha (MT948188) and Pachycladon cheesemanii (NC_021102) were compared using the mVISTA program [28] and the annotation of C. serrulata was used as reference in the Shuffle-LAGAN mode [29].
For the 18S rRNA and ITS analyses, the evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [30]. The trees with the highest log likelihood are shown (-2828.01 for 18SrRNA and -1857.22 for ITS). The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.0500 for 18SrRNA and parameter = 0.9221 for ITS)). The trees are drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 10 nucleotide sequences for 18S rRNA and 23 for ITS. There were a total of 1821 positions (18S rRNA) and 285 positions (ITS) in the final datasets. Evolutionary analyses were conducted in MEGA11 [25]. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format [27].
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank with the following accession number NC_088033. The small ribosomal and ITS1 sequences have been submitted to Genbank and the corresponding accession number is PP512521.
3. Results and Discussion
3.1. Sampling and Identification
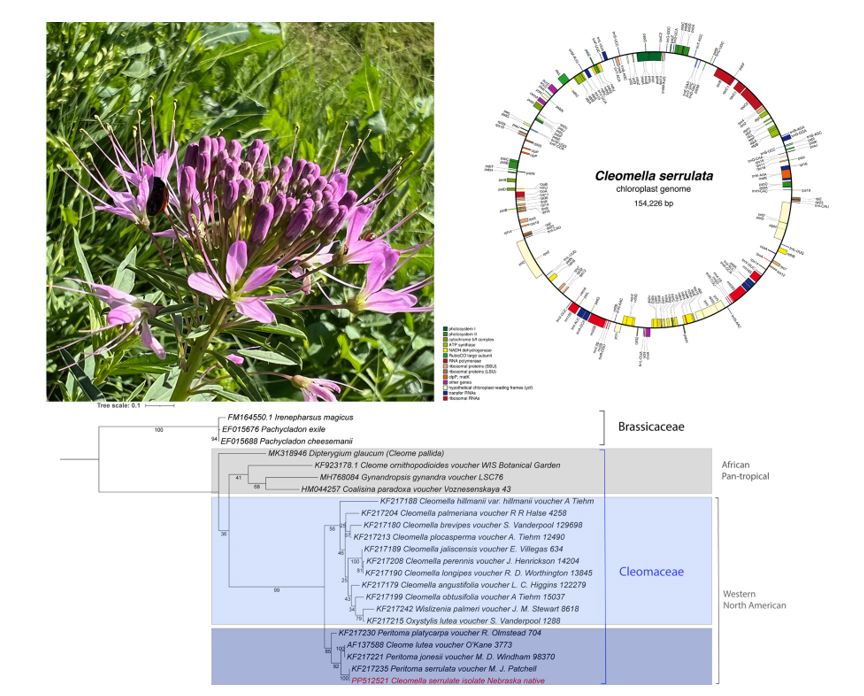
Cleomella serrulata samples were collected from a strain that grows in the Bellevue University Native Garden area (Figure 1) and designated as isolate ‘Nebraska_native’. For the initial species identification, based on morphological features, we used the descriptions provided by the the USDA Plants Database (https://plants.usda.gov/home/plantProfile?symbol=CLSE) and the Minnesota Wildflower database (https://www.minnesotawildflowers.info/flower/rocky-mountain-beeplant). Distinguising features were the elongated clusters of stalked purple flowers, with many flowers blooming in a rounded cluster. The flowers are composed of 4 sepals forming a bowl with 4 triangular leaves (Figure 1). The leaves are mostly compounded in sets of three and leaflets are 3-5 cm long and about 1 cm wide. Plants were about 1 meter tall and blooming at the time of sample collection. Leaf and stem samples were collected and frozen at -80 C in sterile tubes and used for total DNA extractions. Flower leafs were also collected and used for spectral analysis. Water extracted pigments had a broad maximum at 546 nm, indicating that they likely belong to the anthocyanine family (supplemental Figure 1 and [9]). In late June the plants appear to form slender, dangling fruit pods that are about 5 cm long (Figure 1, top right). The pods contained 12-22 seeds per pod with an elongated egg shape. Leaves and stem material was used for DNA extraction.
3.2. Genome Sequencing and Chloroplast Structural Analysis
After Illumina paired-end sequencing, a total of 910,668 reads and 137.5 Mbps of genomic data was obtained. These reads were assembled using SPAdes which yielded 307 contigs (>1000 bp) with an assembly length of 1,461,102 bp and a G+C molar percent of 39.23. The largest contig was 110,028 bp in length. Both the assembled contigs and the raw sequencing reads were used in MiniMap2 to assemble into a consensus chloroplast complete genome, using the Cleomella lutea chloroplast genome (NC_049613) as a reference. The total size of the assembled C. serrulata chloroplast genome was 154,226 bp.
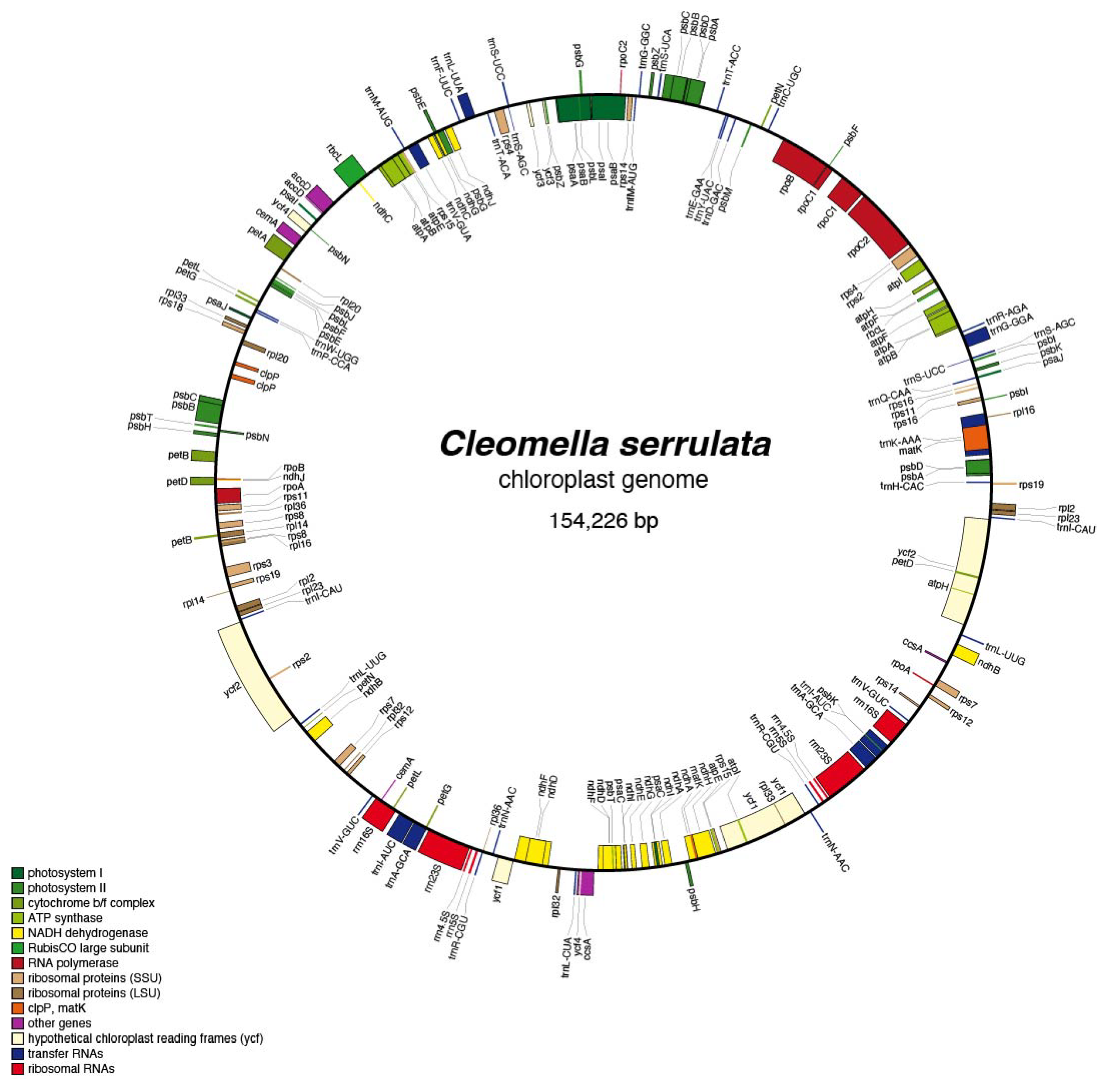
The assembled chloroplast genome of Cleomella serrulata revealed a typical chloroplast genome organization consistent with other angiosperms. The chloroplast genome is circular, double-stranded DNA with characteristic gene arrangements for plant chloroplasts (Figure 2 and supplemental Table 1). The chloroplast contained photosynthesis-related genes, ribosomal RNA genes (5s, 16S, and 23S), 14 ribosomal rps genes, RuBisCo (rbc), maturaseK gene (MatK), 10 NADH dehydrogenase subunits (ndhA-J), and 37 transfer RNA genes, amongst other conserved chloroplast genes (Figure 2). There are 107 CDS on the negative strand, while the remaining 123 CDS are encoded on the positive strand. There are introns in 6 of the tRNA sequences (trnK, trnG, trnL, trnV, trnI, and trnA), 4 of the ribosomal proteins (rps12, rps16, rpl2, and rpl16), and 8 other protein coding genes: atpF, rpoC1, pafI, clpP, ndhB, ndhA, petB, and petD. The overall chloroplast genome sequence has an average percent G+C of 36.5%. The low average %GC is identical to the closely related Cleomella lutea chloroplast genome (the only other Cleomella genome currently in the database).
Figure 2.
Complete chloroplast genome and gene organization of the annotated Cleomella serrulata chloroplast. The genome was annotated using AGORA.
Figure 2.
Complete chloroplast genome and gene organization of the annotated Cleomella serrulata chloroplast. The genome was annotated using AGORA.
3.3. Comparative Genomics
Our study employed a whole genome-based comparative genomics approach to gain insights into evolutionary relationships and genetic divergence within the Cleomella genus. The phylogenetic analysis revealed a high degree of similarity between the chloroplast genomes of Cleomella serrulata and other members of the Cleomaceae family (Figure 3). There is currently only one other Cleomella chloroplast genome available in the database, from Cleomella lutea (NC_049613), and the whole-genome comparison showed this to be the closest relative, consistent with the taxonomic identification of our species based on morphological features. A more detailed comparative analyses demonstrated conserved gene content, gene order, and overall genome structure, indicative of a close evolutionary relationship among these two species.
Figure 3.
Phylogenetic tree of the complete chloroplast genomes for all available Cleomaceae and related families. The new genome is marked in red. Accession numbers from Genbank are included with the names. The tree was generated by using the Maximum Likelihood method and General Time Reversible model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to visualize the phylogenetic tree format.
Figure 3.
Phylogenetic tree of the complete chloroplast genomes for all available Cleomaceae and related families. The new genome is marked in red. Accession numbers from Genbank are included with the names. The tree was generated by using the Maximum Likelihood method and General Time Reversible model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to visualize the phylogenetic tree format.

No other Cleomella genomes are currently available in Genbank, which limits furthertaxonomic mitogenome-based analysis at the species level. The closest genera that have chloroplast data available were Thulinella (Cleomaceae), Dipterygium (Cleomaceae), Cadaba (Capparaceae), Crateva (Capparaceae), Pachycladon (Brassicaceae), Irenepharsus (Brassicaceae), and Arabidella (Brassicaceae) (Figure 3). A pairwise comparison of the bidirectional average nucleotide identity (ANIb) between Cleomella serrulata and the closest relatives, showed it to have a close relationship with the other Cleomella species (lutea) with an ANI of 99.6%. The ANI of C. serrulata with the other Cleomaceae: Thulinella chrysantha, Cleome chrysantha and Dipterygium glaucum, was lower: 94.5%, 94.5%, and 94.1%. Similaraly, the ANIb with the more divergent Cleomaceae, Gynandropis gynandra and Coalisina paradoxa, was 94.4% and 94.3%, while these showed ANI values of 95-96% to each other and to Thulinella chrysantha. However, similar ANIb values were obtained when comparing the Cleomella serrulata chloroplast to the species from the other families: Cadaba glandulosa 93.9 %, Crateva religiosa 94.3%, Pachycladon cheesemanii 93.4%, Irenepharsus magicus 93.2%, and Arabidella filifolia 93.3%. This indicates that Cleomella serrulata is evolutionary nearly equidistant from its fellow Cleomaceae as it is to the species from the nearby families, which is consistent with the clade distribution in Figure 3.
The structural characteristics of the DNA divergence between the different chloroplast genomes was analyzed by performing an mVISTA alignment of the closest Cleomaceae genomes (Figure 4). The Cleomella serrulata annotation was added as a reference and one of the Brassicaceae genomes (Pachycladon cheesemanii) was added for comparison. The alignment shows the close relationship between the two Cleomella species, but also indicates highly conserved genomes between the genera with few variations. As expected, the noncoding regions were less conserved than the coding regions (Figure 4), although four genes, atpF, ndhF, ndhA, ycf1, and to a lesser extent rpoC1, show a higher variability in their gene content. This indicates more evolutionary variation in these proteins and may imply more functional diversity in these proteins in C. lutea and C. serrulata.
Both the ANI and whole chloroplast-based phylogenetic tree analyses show that the two Cleomella species clearly belong to a separate clade and validate their distinction as a separate genus. However, they are about equidistant from the other Cleomaceae genera (Thulinella, Cleome and Dipterygium) as they are from the Capparaceae and Brassicaceae representatives. This indicates an earlier evolutionary separation of the Cleomella genus then the other genera in the Cleomaceae family. As more chloroplast genomes become available in this family in the future, a deeper evolutionary comparison should be performed, and this may potentially warrant a further refinement in the Cleomaceae taxonomy based on genomic comparisons.
2.3. Chloroplast versus Nuclear DNA Evolution
In addition to the larger chloroplast isolation, we also identified an 18S rRNA sequence as part of an 8672 bp contig. This contig contained the native small subunit ribosomal RNA gene and internal transcribed spacer 1 (ITS1). When performing an NCBI BLAST we found the 18S rRNA to be 99.94% identical to a previously isolated Cleomella serrulata (voucher Ahrendsen, KT459185) (1806/1807 bp) isolated from Nebraska grasslands, which is additional confirmation of our species identification. There are currently no other Cleomella 18S rRNA sequences in the Genbank database and the closest relative in the NCBI BLAST analysis was Arabidella chrysodema (voucher PERTH 05393264; OL339508) with 98.57% identity (1788/1814 bp) for the 18S rRNA. A phylogenetic tree using a wider divergence of available 18S rRNA sequences from other families (Figure 5), confirms that our isolate belongs to the Cleomella genus and is clearly genetically separated from the other genera. The closest available relatives based on this 18S rRNA comparison were Leiospora (Brassicaceae), Cakile (Brassicaceae), and Camelina (Brassicaceae) (Figure 5).
A more commonly used genetic marker than 18S rRNA in plants and fungi is the internal transcribed spacer 1 (ITS1). There are more Cleomaceae ITS fragments in NCBI Genbank than 18S rRNA sequences, which allows for a more detailed placement of our isolate amongst the Cleomaceae. Figure 6 is a phylogenetic tree constructed with the closest ITS fragments available in the database. Note that many of the ITS fragments still contain the ‘Peritoma’ genus name in the database, which was updated to Cleomella in the last taxonomic revision [7]. Cleome ornithopioides is the type species of the genus Cleome and was also included in this tree [2]. This analysis clearly places our isolate as a strain of Cleomella serrulata amongst the Cleomaceae, with C. lutea as the nearest relative. The species distribution in Figure 6 is consistent with the geographic distribution and with earlier single gene-based analyses of some of these species [8]. The Western North American Cleomaceae genera form an obvious separate clade from the African/Pan-tropical genera (blue and gray boxes in Figure 6). In addition, a clear separation is formed by the Cleomella lutea and Cleomella serrulata clade within the Western North America group (light and darker blue boxes in Figure 6). This clade also contains Cleomella (Peritoma) platycarpa, found from northeastern California to Idaho) and Cleomella (Peritoma) jonesii, found in California, Arizona and Mexico (https://powo.science.kew.org/).
A notable observation from our study was the comparison between the chloroplast genome and nuclear DNA (18S gene and ITS) sequences (Fig 3 and 5). While the chloroplast genome exhibited consistency and similarity across Cleomella species, the nuclear 18S rRNA sequences displayed more significant divergence. However, this likely results from a lack of 18S rRNA and overall genetic information about other species closely related to Cleomella. The ITS phylogenetic comparison on the other hand, allows for a broader comparison, and clearly shows C. serrulata and C. lutea as a distinct clade of the Cleomaceae family. This is in agreement with the whole chloroplast genome comparison (Figure 3), however the latter analysis shows that the C. serrulata/lutea clade may actually be equidistant from the other Cleomaceae than the other nearby families Brassicaceae and Capparaceae. Given the fact that the chloroplast genome comparison encompasses a much larger genetic fraction (as opposed to a single marker), this is likely to provide a deeper evolutionary comparison. Alternatively, this difference might also be due to the differences in using chloroplast versus nuclear DNA genetic markers, which may have different evolutionary rates. Having more chloroplast genomes and eventually nuclear genomes available in the future will certainly help clarify this issue.
Nevertheless, the Cleomaceae family is clearly closest related to the Brassicaceae and possibly even closer to the Capparaceae family (Figure 3), however there is a need for more extensive genetic and genomic information for these family in order to finetune this taxonomy. The present study is a first step in that direction and is already providing a deeper insight into the Cleomaceae and its genera in this complex and ancient plant evolution. This initial analysis shows that the Cleomaceae may not be as monophyletic as has been expected.
Our findings show that the isolated chloroplast genome sequence belongs to the Cleomella genus, and additionally, it corroborates previous studies suggesting that Cleomella is its own valid genus. Comparative analyses using chloroplast genomic data with earlier research, support the taxonomic classification of Cleomella serrulata. Moreover, our results align with previous assertions regarding the evolutionary relationships and taxonomic placement of Cleomella species within the Cleomaceae family [5]. While our study focused on a limited set of Cleomella species, future research endeavors could benefit from expanding the scope of comparative genomics analyses. Including a broader range of genomes from Cleomella taxa and incorporating additional molecular markers beyond chloroplast genomes and nuclear marker sequences will undoubtedly enhance our understanding of evolutionary patterns, species relationships, and genetic diversity within the genus, and may warrant further rearrangement of the Cleomaceae and specifically the phylogenetic positioning of the Cleomella genus.
4. Conclusions
In conclusion, our study presents a comprehensive analysis of the chloroplast genomes within the Cleomella genus, shedding light on their structural features, genetic content, and evolutionary relationships. By utilizing advanced sequencing technologies, bioinformatic tools, and comparative genomics approaches, we have elucidated key insights into the evolutionary dynamics shaping this diverse plant group. Our findings highlight a strong conservation of chloroplast genome organization and gene content across Cleomella species, indicative of their close evolutionary relationship and shared ancestry. Comparative analyses with other angiosperm taxa further underscore the uniqueness of chloroplast evolution within the Cleomaceae family and suggest an earlier evolutionary split of the Cleomella genus from the other genera in the Cleomaceae. This implies potential adaptive strategies and evolutionary mechanisms driving genomic diversity within the Cleomella genus that could be revealed by further genomic sequencing and biochemical characterization studies.
Furthermore, comparing chloroplast and nuclear DNA sequences underscores the importance of integrating multiple molecular markers to comprehensively understand evolutionary patterns and species relationships. While nuclear DNA sequences provide valuable insights into species divergence and phylogenetic relationships, chloroplast genomes offer additional layers of genetic information, enriching our understanding of evolutionary processes. Looking ahead, our study sets the stage for future research endeavors to explore the evolutionary history, genetic diversity, and adaptive traits within the Cleomella genus. Expanding comparative genomic studies to include a broader range of taxa and incorporating additional molecular markers can further unravel the intricate evolutionary dynamics shaping plant biodiversity.
Compared to crop and agricultural plant species, the genomic information of native plant species is still very limited, which hampers more detailed plant evolutionary studies. Overall, our findings contribute to the growing body of knowledge surrounding plant evolution and biodiversity, emphasizing the importance of interdisciplinary approaches and collaborative efforts in advancing our understanding of the natural world.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Absorption spectra of Cleomella serrulata extracts; Table S1: Summary of genetic features located on the complete Cleomella serrulata chloroplast genome.
Author Contributions
Conceptualization, JAK and JR; methodology, KJT, EKS, and JAK; software, EKS, KJT, and JAK; validation, KJT, EKS, and JAK; formal analysis, MKV, KJT, EKS, and JAK ; investigation, MKV, KJT, EKS, and JAK; resources, JAK and JR.; data curation, MKV, KJT, and JAK; writing—original draft preparation, MKV and JAK.; writing—review and editing, MKV, EKS, KJT, JR, and JAK; visualization, MKV and JAK; supervision, JAK; project administration, JAK.; funding acquisition, JAK. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank with the following accession number NC_088033. The small ribosomal and ITS1 sequences have been submitted to Genbank and the corresponding accession number is PP512521.
Acknowledgments
This work was sponsored by the Wilson Enhancement Fund for Applied Research in Science at Bellevue University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Riser, J.P., Cardinal-McTeague, W.M., Hall, J.C., Hahn, W.J., Sytsma, K.J., & Roalson, E.H. Phylogenetic relationships among the North American Cleomoids (Cleomaceae): A test of Iltis’s reduction series. American Journal of Botany 2013, 100(10), 2102–2111. [CrossRef]
- Roalson, E. H. A revised synonymy, typification and key to species of Cleome sensu stricto (Cleomaceae). Phytotaxa 2021, 496, 54–68.
- Barker, M.S., Vogel, H., & Schranz, M.E. Paleopolyploidy in the Brassicales: Analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biology and Evolution 2009, 1, 391–399. [CrossRef]
- Cane, J.H. Breeding biologies, seed production and species-rich bee guilds of Cleome lutea and Cleome serrulata (Cleomaceae). Pl. Spec. Biol. 2008, 23, 152–158.
- Patchell, M.J., Roalson, E.H., & Hall, J.C. Resolved phylogeny of Cleomaceae based on all three genomes. TAXON 2014, 63(2), 315–328. [CrossRef]
- Adams, K.R., Stewart, J.D., & Baldwin, S.J. Pottery paint and other uses of Rocky Mountain Beeweed (Cleome serrulata pursh) in the Southwestern United States: Ethnographic Data, Archæological Record, and elemental composition. KIVA 2002, 67(4), 339–362. [CrossRef]
- Roalson, E.H., Hall, J.C., Riser II, J.P., Cardinal-McTeague, W.M., Cochrane, T.S., & Sytsma, K.J.. A revision of generic boundaries and nomenclature in the North American Cleomoid clade (Cleomaceae). Phytotaxa 2015, 205(3), 129. [CrossRef]
- Hall, J.C. Systematics of Capparaceae and Cleomaceae: An evaluation of the generic delimitations of capparis and cleome using plastid DNA sequence datathis paper is one of a selection of papers published in the special issue on systematics research. Botany 2008, 86(7), 682–696. [CrossRef]
- Nozzolillo, C., Amiguet, V.T., Bily, A.C., Harris, C.S., Saleem, A., Andersen, O.M. & Jordheim, M. Novel aspects of the flowers and floral pigmentation of two Cleome species (Cleomaceae), C. hassleriana and C. serrulata. Biochem. Syst. Ecol. 2010, 38, 361–369. [CrossRef]
- Patchell, M.J., Bolton, M.C., Mankowski, P. & Hall, J.C. Comparative floral development in Cleomaceae reveals two distinct pathways leading to monosymmetry. Int. J. Pl. Sci. 2011, 172, 352–365. [CrossRef]
- Brown, N.J., Parsley, K. & Hibberd, J.M. The future of C-4 research – Maize, Flaveria or Cleome? Trends Pl. Sci. 2005, 10, 215–221. [CrossRef]
- Marshall, D.M., Muhaidat, R., Brown, N.J., Liu, Z., Stanley, S., Griffiths, H., Sage, R.F. & Hibberd, J.M. Cleome, a genus closely related to Arabidopsis, contains species spanning a developmental progression from C-3 to C-4 photosynthesism. Plant J. 2007, 51, 886–896. [CrossRef]
- Voznesenskaya, E.V., Koteyeva, N.K., Chuong, S.D.X., Ivanova, A.N., Barroca, J., Craven, L.A. & Edwards, G.E. Physiological, anatomical and biochemical characterisation of photosynthetic types in genus Cleome (Cleomaceae). Funct. Pl. Biol. 2007, 34, 247–267. [CrossRef]
- Feodorova, T.A., Voznesenskaya, E.V., Edwards, G.E. & Roalson, E.H. Biogeographic patterns of diversification and the origins of C-4 in Cleome (Cleomaceae). Syst. Bot. 2010, 35, 811–826. [CrossRef]
- Koteyeva, N.K., Voznesenskaya, E.V., Roalson, E.H. & Edwards, G.E. Diversity in forms of C-4 in the genus Cleome (Cleomaceae). Ann. Bot. (Oxford) 2011, 107, 269–283. [CrossRef]
- Schranz, M.E. & Mitchell-Olds, T. Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. Pl. Cell 2006, 18, 1152–1165. [CrossRef]
- Barker, M.S., Vogel, H. & Schranz, M.E. Paleopolyploidy in the Brassicales: Analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biol. Evol. 2009, 1, 391–399. [CrossRef]
- Brautigam, A., Kajala, K., Wullenweber, J., Sommer, M., Gagneul, D., Weber, K.L., Carr, K.M., Gowik, U., Mass, J., Lercher, M.J., Westhoff, P., Hibberd, J.M. & Weber, A.P.M. An mRNA Blueprint for C-4 photosynthesis derived from comparative transcriptomics of closely related C-3 and C-4 species. Pl. Physiol. 2011a, 155, 142–156. [CrossRef]
- Brautigam, A., Mullick, T., Schliesky, S. & Weber, A.P.M. Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C-3 and C-4 species. J. Exp. Bot. 2011b, 62, 3093–3102. [CrossRef]
- Alzahrani D, Albokhari E, Yaradua S, Abba A. Complete chloroplast genome sequences of Dipterygium glaucum and Cleome chrysantha and other Cleomaceae Species, comparative analysis and phylogenetic relationships. Saudi J Biol Sci. 2021, 28(4), 2476-2490. [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 31 May 2024).
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S., Lesin, V.M., Nikolenko, S.I., Pham, S., Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 2012, 19, 455–477.
- Li H. Minimap2: pairwise alignment for nucleotide sequences, Bioinformatics 2018, 34(18), 3094–3100. [CrossRef]
- Jung J., Kim, J.I., Jeong, Y-S., Yi, G. AGORA: organellar genome annotation from the amino acid and nucleotide references, Bioinformatics 2018, 34(15), 2661–2663. [CrossRef]
- Tamura K., Stecher G., and Kumar S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Molecular Biology and Evolution 2021, 38(7), 3022–3027. [CrossRef]
- Nei M. and Kumar S. (2000). Molecular Evolution and Phylogenetics. Oxford University Press, New York.
- Letunic I, Bork P Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 2019, 47, 256-259. [CrossRef]
- Mayor C., Brudno M., Schwartz J.R., Poliakov A., Rubin E.M., Frazer K.A., Pachter L.S., Dubchak I. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16(11), 1046-1047. [CrossRef]
- Frazer K.A., Pachter L., Poliakov A., Rubin E.M., Dubchak I., VISTA: computational tools for comparative genomics. Nucleic Acids Research 2004, 32, W273–W279. [CrossRef]
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 1980, 16, 111-120.
Figure 1.
Cleomella serrulata as found in the Natural Sciences Native Garden at Bellevue University in Nebraska, US.
Figure 1.
Cleomella serrulata as found in the Natural Sciences Native Garden at Bellevue University in Nebraska, US.

Figure 4.
Comparison of four chloroplast genomes in the Cleomaceae family and one of the Brassicaceae (Pachycladon). Cleomella serrulata was used as a reference and its annotation is shown on top. The horizontal axis indicates the coordinates in the chloroplast genome and the vertical axis indicates the percentage identity (between 50 and 100%).
Figure 4.
Comparison of four chloroplast genomes in the Cleomaceae family and one of the Brassicaceae (Pachycladon). Cleomella serrulata was used as a reference and its annotation is shown on top. The horizontal axis indicates the coordinates in the chloroplast genome and the vertical axis indicates the percentage identity (between 50 and 100%).

Figure 5.
Phylogenetic tree based on 18S rRNA sequences for all available Cleomaceae and closest families. The new isolate is marked in red. Accession numbers are included. The phylogenetic tree was generated by using the Maximum Likelihood method and Kimura 2-parameter model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format.
Figure 5.
Phylogenetic tree based on 18S rRNA sequences for all available Cleomaceae and closest families. The new isolate is marked in red. Accession numbers are included. The phylogenetic tree was generated by using the Maximum Likelihood method and Kimura 2-parameter model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format.
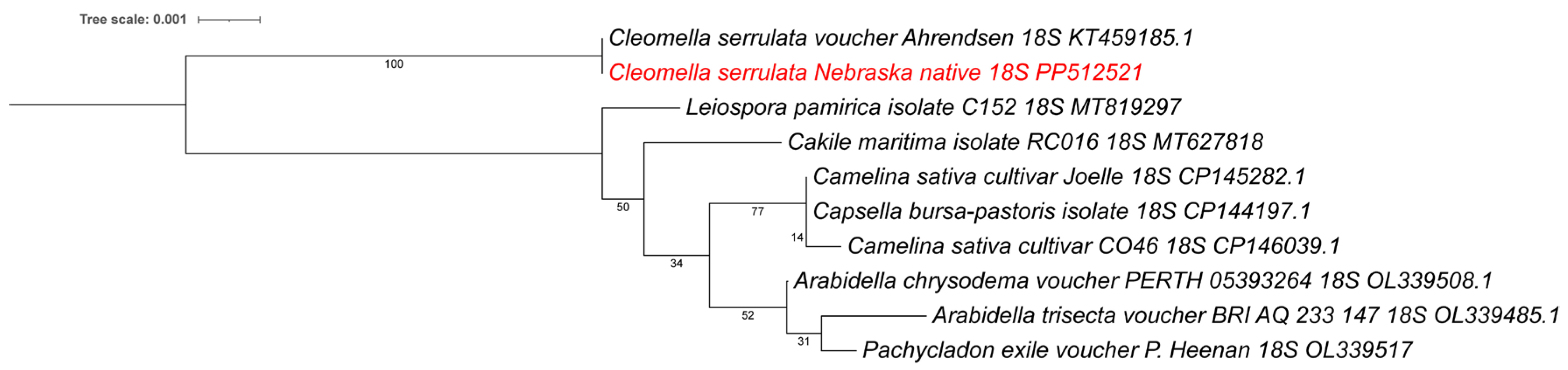
Figure 6.
Phylogenetic tree based on ITS1 sequences for Cleomaceae. The new isolate is marked in red. Accession numbers are included. The phylogenetic tree was generated by using the Maximum Likelihood method and Kimura 2-parameter model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format.
Figure 6.
Phylogenetic tree based on ITS1 sequences for Cleomaceae. The new isolate is marked in red. Accession numbers are included. The phylogenetic tree was generated by using the Maximum Likelihood method and Kimura 2-parameter model within MEGA 11. Bootstrap values were inferred from 500 replicates. iTOL was used to draw the phylogenetic trees expressed in the Newick phylogenetic tree format.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



