
Submitted:
26 July 2024
Posted:
26 July 2024
You are already at the latest version
Abstract
Oxidative stress constitutes a main molecular mechanism underlying cardiovascular diseases (CVDs). This pathological mechanism can be triggered by NADPH oxidases (NOXs), which produce reactive oxygen species (ROS). In fact, the different NOXs have been associated with myocardial infarction, atherothrombosis, and stroke. More specifically, we will focus on the implications of NOXs in atherothrombotic stroke. Each NOX member participates in a different way in the several stages of this disease: endothelial dysfunction, immune cell infiltration, foam cell genesis, vascular smooth muscle cells (VSMC) proliferation, and atherosclerotic plaque formation. Additionally, some NOXs are involved in plaque instability, thrombosis, ischemic stroke, and ischemia-reperfusion injury (IRI). Interestingly, the effects of NOXs in this pathology depend on the specific homologue, the cell type in which they are activated, and the stage of the disease. In this review we summarize the most up-to-date information about the implications of vascular NOXs in each of these processes. Finally, we highlight some limitations and future perspectives of the study of NOXs in CVDs.
Keywords:
Oxidative stress
; NADPH oxidases
; NOX
; atherosclerosis
; thrombosis
; stroke
; vessel wall
; atherothrombosis
1. Introduction
Atherothrombotic stroke is a cardiovascular disease (CVD) with a high global incidence. It occurs when a thrombus forms after the rupture of an atherosclerotic plaque and it travels across the bloodstream, finally obstructing blood flow in the brain parenchyma [1]. This disease progresses through several phases, beginning with the development of an atherosclerotic plaque. Initially, endothelial cells are damaged by humoral stimuli, leading to inflammation and the transfer of low-density lipoproteins (LDL) into the vessel intima [2]. Reactive oxygen species (ROS) produced from different cell types oxidize these LDLs (oxLDLs). Immune cells attracted by the damaged endothelium infiltrate and accumulate oxLDLs, becoming foam cells [3]. These cells accumulate and secrete growth factors, causing the invasion of the subendothelial space by vascular smooth muscle cells (VSMC). VSMC proliferate and switch their phenotype (sometimes becoming foam cells), contributing to plaque development. This plaque can either occlude the artery, or rupture, leading to thrombus formation, which could potentially cause an ischemic stroke [1].
One of the most relevant molecular mechanism underlying atherothrombotic stroke is oxidative stress, characterized by an imbalance between ROS levels and antioxidant defenses in the cells. In this context, ROS overreact, modifying lipids, proteins, and DNA, and cause cell damage, which may trigger different pathologies [4]. ROS are mainly produced by the mitochondrial electron chain, the NADPH oxidase family (NOX), nitric oxide synthases, xanthine oxidase, and cytochrome P450 [5]. Interestingly, the NOX family is the primary ROS source in the vasculature and its overactivation is a key factor in several CVDs, such as atherothrombotic stroke [6].
2. NADPH Oxidases in the Vessel Wall
NOXs constitute a family of enzymes that produce ROS as their main catalytic product. This family is composed of seven different members: five homologues (NOX1-NOX5) and two dual oxidases (DUOX1, DUOX2). These members differ in their regulatory subunits, their expression across different cell types, and their intracellular localization [7]. Specifically, NOX1, NOX2, NOX4 and NOX5 are expressed in the vascular wall and monocytes/macrophages, while DUOXs present lower levels [8], and NOX3 is restricted to the inner ear [9]. In this section we summarize the distribution of the main NOX members in endothelial cells, VSMC and monocytes/macrophages (Figure 1).
NOX1 was described in 2000 in mammalian cell lines [10]. Its activation requires of regulatory subunits (p47phox, NOXA1), and its main product is O2•-. NOX1 is expressed by endothelial cells [11] and VSMC [12] in the vascular wall, and by monocytes [13]. Intracellularly, NOX1 has been detected in the nucleus, the plasma membrane, endosomes, and peroxisomes [14]. In VSMC, NOX1 is expressed in caveolin-coated areas of the plasma membrane [15]. Little is known about its localization in endothelial cells and monocytes. NOX1 upregulation has been associated with CVDs such as atherosclerosis, hypertension, and diabetes [16,17,18].
NOX2 (gp91phox or phagocytic NOX) was the first described member and it produces O2•-. It is a highly glycosylated protein, and its activation depends on phosphorylation and the recruitment of p22phox, p47phox and p67phox subunits. NOX2 is expressed in endothelial cells, VSMC and monocytes in different subcellular localizations [14]. For instance, in macrophages, NOX2 is located at intracellular membranes at baseline, translocating to the plasma membrane upon activation [19]. In endothelial cells, NOX2 associates with the actin cytoskeleton and cellular protrusions [20]. By contrast, NOX2 localization in VSMC remains unknown. NOX2 has been associated with diabetes, myocardial infarction, and thrombosis [21,22].
NOX4 (initially named “Renox”) was firstly described in the kidney [23]. NOX4 only requires p22phox subunit to produce ROS and appears to be active under physiological conditions [24]. The primary effect of this oxidase is mediated by H2O2. NOX4 is expressed in endothelial cells [25], VSMC [16], and monocytes [26]. In endothelial cells and macrophages, it locates in the endoplasmic reticulum [20,26]; and in VSMCs, in focal adhesions, mitochondria, and the nucleus [15,27,28]. There is some debate about the role of NOX4 in CVDs, as it has been described to display both protective and damaging roles [29].
NOX5 was the last discovered member of the family. It is highly expressed in the testis, spleen, and lymph nodes [30], but it is absent in the rodent genome, which complicates its study in the experimental rodent models. Interestingly, NOX5 is the only member regulated by Ca++ levels. In the human vasculature, NOX5 is present at all vascular segments [31]: endothelial cells [32], VSMC [33] and monocytes [34]. It locates in the nucleus, endoplasmic reticulum, and plasma membrane [35]. In VSMC it is abundant in cholesterol-rich areas of the plasma membrane and translocates to rafts upon activation [36]. In dendritic cell-derived monocytes, NOX5 is expressed on the outer membrane of the mitochondria [37]. Recently, NOX5 has been found to interact with the actin cytoskeleton [38]. NOX5 is implicated in several CVDs such as diabetes, atherosclerosis and myocardial infarction [39].
3. NADPH Oxidases in Atherosclerosis
3.1. NADPH Oxidases and Endothelial Dysfunction
NOX members are activated by humoral factors associated with CVDs, leading to endothelial dysfunction. For example, Ang II stimulates NOX2 [40,41], NOX4 [42], and NOX5 [43,44] activity/expression in human endothelial cells. ET-1 activates NOX2 and NOX5-derived ROS in porcine and human endothelial cells respectively [43,45], contributing to endothelial dysfunction. Moreover, oxLDLs induce NOX2 activity and endothelial dysfunction in human primary coronary artery endothelial cells [46]. The role of NOX4 in endothelial dysfunction in response to LDL/oxLDL stimulation is debated; some studies describe a pathological role in human umbilical vein endothelial cells (HUVEC) [47,48], while others suggest a protective role in LDL-receptor knock-out mice [49]. Finally, lysophosphatidylcholine activates NOX5 increasing Ca++ levels in human aortic endothelial cells (HAEC) [50].
In addition, NOXs are involved in endothelial cell apoptosis. For instance, NOX1 regulates apoptosis in human sinusoidal endothelial cells [11]. The localization of NOX2 and NOX4 appears closely linked to apoptosis in HUVEC. Specifically, caspase 3 activity associates with NOX2- and NOX4-derived ROS production in response to homocysteine [51]. Finally, our group previously showed that NOX5 induces apoptosis in HAECs and immortalized human brain microvascular endothelial cells (hBMECs) [52,53].
These proapoptotic effects of NOXs create a proinflammatory environment by releasing cytokines from endothelial cells. For instance, NOX2 in endothelial cells mediates the in vivo production of granulocyte-monocyte colony stimulating factor (GM-CSF), likely through toll-like receptor (TLR) stimulation [54]. Likewise, NOX4 potentiates the secretion of IL-8 and monocyte chemoattractant protein 1 (MCP-1) upon TLR-4 activation in human endothelial cells, as well as induces IL-6 production in response to palmitate [55,56,57]. Moreover, endothelial NOX5 in diabetic Akita mice increased inflammation via MCP-1 and TLR4 [58]. Tobacco smoke extract induced CX(3)CL1 production via NOX5 activation in HUVEC, increasing immune cell adhesion [59]. Prostaglandins (PGs) are also implicated in this process. In HUVEC, NOX4 inhibits the protective effects of PGI2, related with different vasculopathies [60]. Finally, in immortalized HAEC (teloHAEC), NOX5 increases PGE2 production by COX-2 activation [44]. All the mentioned studies are summarized in Figure 2.
3.2. NADPH Oxidases, Immune Cell Infiltration and Foam Cells
In this proinflammatory context, endothelial cells upregulate adhesion molecules to facilitate immune cell adhesion and infiltration. NOX activity and expression closely correlates with adhesion molecule expression. NOX2 overexpression in murine endothelial cells increases leukocyte adhesion and VCAM-1 expression. In mice with cardiac hypertrophy produced by Ang II infusion, NOX2 enhances immune cell infiltration into the heart [61]. Similarly, salusin-β (a prohypertensive peptide) increases monocytic adhesion in VSMC through NOX2 [62]. By contrast, the overexpression of NOX2 in monocytes does not promote adhesion to endothelial cells [63]. Again, NOX4 overexpression in endothelial cells exhibits a protective role, reducing the recruitment of immune cells caused by Ang II in vivo [64]. However, NOX4 also promotes the expression of ICAM-1 and VCAM-1 in response to TNF-α in HUVEC [65]. Finally, NOX5 increases VCAM-1 and ICAM-1 expression, and mononuclear cell infiltration in HUVEC [66]. This pathway could be induced by oxLDLs [50]. In a myocardial infarction model, mice overexpressing NOX5 in endothelial cells upregulate cardiac VCAM-1 levels [67].
Changes in the extracellular matrix (ECM) can facilitate immune cell infiltration into the subendothelial space of the vessel wall. Interestingly, p22phox regulates vascular wall elastic fiber composition in vivo, correlating with the MMP-12/TIMP-1 ratio [68]. More interactions between NOX enzymes and matrix metalloproteinases (MMPs) are known. For instance, Ang II-induced NOX1 activation promotes VSMC migration and proliferation via MMP-9 [69]. NOX2 activation by Ang II induces vascular remodeling in VSMC and adventitial fibroblasts [70,71]. Besides, NOX2 is implicated in the development of the atherosclerotic plaque in vivo, which seems to be related with a reduction in the MMP-9 activity [72]. Finally, the Ang II-NOX4 axis influences the effects of adventitial fibroblasts in the vascular ECM [73]. The relationship between NOX5 and MMP expression/activity in the vascular context remains unclear.
NOXs also play a crucial role in foam cell formation by mediating lipid oxidation. NOX1 may play a dual role in this process. On the one hand, NOX1 activation by TLRs increases ROS production and foam cell generation [74,75]. On the other hand, NOX1 participates in atherosclerotic plaque propagation by mediating LDL pinocytosis [76]. NOX2 has been implicated in macrophage apoptosis during efferocytosis, contributing to increased inflammation and immune cell infiltration into the plaque [77]. Furthermore, NOX2 promotes lipid accumulation in VSMC, another source of foam cells [62]. There is no literature suggesting a direct relationship between NOX4 or NOX5 and foam cells, although NOX5 has been associated with VSMC migration in hypertensive patients [78]. All these studies are summarized in Figure 3.
3.3. NADPH Oxidases, Plaque Development, Plaque Rupture, and Thrombosis
Foam cells stimulate neighboring VSMC, promoting their migration and proliferation within the atherosclerotic plaque, finally increasing its size. NOX1A mediates VSMC migration, proliferation, and differentiation into macrophage-like cells in ApoE-/- mice [79]. These effects of NOX1 on VSMC have been widely described in vitro and in vivo [80,81,82]. Interestingly, NOX2 activity follows NOX1 in VSMC, promoting cell migration through a second peak of ROS [70]. Although the effect of NOX2 on VSMC migration was already described [62], this study highlights a temporal relationship between two NOXs in a specific cell type.
NOX2 expressed in endothelial cells contributes to vascular remodeling by increasing VSMC proliferation, indicating a paracrine effect derived from NOX activity [83]. NOX2 in VSMC induces IL-17A production, potentially promoting a phenotypic switch [84]. The precise role of NOX4 in VSMC migration and differentiation remains unclear, some studies suggest an anti-synthetic and anti-proliferative role [85], while others propose a pro-migratory one [86]. Although limited information exists on NOX5 and VSMC, great progress has been made recently. VSMC extracted from hypertensive patients present greater migratory capacity mediated by NOX5 activation [87]. Regarding vascular calcification, NOX5 promotes the switch of VSMC to a synthetic phenotype that secretes Ca++-loaded vesicles [88]. This effect of NOX5 in VSMC-mediated vascular calcification may appear in humans in response to smoking [33].
Several in vivo models of atherosclerosis served to evaluate the effects of NOXs on plaque development. NOX1 produces pro-atherosclerotic effects in diabetic mice, as its deletion decreases chemokines secretion, immune infiltration, and profibrotic markers expression [89]. Interestingly, ET-1 production from endothelial cells is linked to NOX1 pro-atherosclerotic effects in vivo [90]. Dietary supplementation with blackberry prevented NOX1 pathological effects in vivo [91]. NOX2 plays a pro-atherosclerotic role in early stages of the disease by reducing NO availability [92]. NOX2 inhibition by gp91ds-tat causes the regression of the atherosclerotic plaques in ApoE-/- mice [72]. NOX4 deletion in ApoE-/- mice accelerates atherosclerosis in response to partial carotid artery ligation under a high-fat diet, suggesting a protective role [93]. Conversely, in LDLr-/- mice, NOX4 exhibits a pathological profile in response to a high-fat diet, increasing plaque burden [49]. NOX4 deletion in VSMC prevents plaque development in response to Ang II [94]. In coronary artery disease patients, NOX5 appears overexpressed in atherosclerotic plaques, localizing in endothelial cells in early lesions and in VSMC in advanced lesions [31]. Furthermore, NOX5 colocalizes with immune cells in plaques from patients with atherosclerosis [95]. Regarding experimental atherosclerosis, endothelial overexpression of human NOX5 in knock-in mice does not affect plaque development [96]. However, NOX5 deletion in rabbits aggravates atherosclerosis in response to high-fat diet [97]. Therefore, it is unclear if NOX5 promotes pathology or acts in response to vascular damage.
NOXs can also modulate the stability of atherosclerotic plaques. NOX1 activation in VSMC promotes the development of unstable plaques [98]. In a model combining carotid branch ligation, renal artery constriction and high fat diet in ApoE-/- mice, NOX2 inhibition favors the production of more stable plaques [99]. NOX4 expression in patients with carotid artery stenosis directly correlates with plaque stability and inversely correlates with caspase-3 activity, suggesting a protective role [100]. The role of NOX5 in plaque stability remains unknown.
The instability of atherosclerotic plaques leads to rupture and thrombosis. In this stage of the disease, NOXs play a crucial role. Pharmacological inhibition or genetic depletion of NOX1 reduces collagen-dependent thrombosis in a FeCl3-induced carotid occlusion model [101]. However, studies where NOX1 is specifically depleted from platelets suggest that NOX1 is involved in thrombin-induced thrombosis. By contrast, NOX2 seems to be involved in collagen-induced thrombosis [102]. On the contrary, another group describes that NOX2 is dispensable for arterial thrombosis in large vessels [103]. Finally, in a carotid occlusion model comparing a triple knock-out mouse (NOX1-/-, NOX2-/- and NOX4-/-) to NOX-specific knock-out mice, it was concluded that NOX4 does not play a relevant role in pulmonary thromboembolism and ex vivo platelet aggregation, while NOX1 and NOX2 act as prothrombotic enzymes [104]. The role that NOX5 plays in thrombosis remains unclear. As it can be observed, there is still some controversy regarding the role of NOXs in thrombosis. All the described studies are summarized in Figure 4.
4. NADPH Oxidases in Thrombosis and Stroke
4.1. NADPH Oxidases, BBB Disruption and Stroke
Once the thrombus occludes a cerebral artery, an ischemic stroke occurs. In this context, NOX1 seems to alter BBB permeability. Oxygen/glucose deprivation increases the ROS production by NOX1 in hBMEC, reducing the expression of adherent proteins and promoting permeability [105]. GKKT136901, a NOX1/4 inhibitor, prevents the increased permeability of hBMEC caused by methamphetamine, restoring ZO-1 and VE-cadherin expression [106]. In a model of traumatic brain injury, NOX1 is upregulated in endothelial cells from the neurovascular unit, overproducing O2•– and increasing permeability by the dysregulation of the TJs [107]. However, the relevance of NOX1 in middle cerebral artery occlusion (MCAO) models seems to be limited [108,109].
NOX2 presents similar effects on BBB permeability and stroke. At oxygen and glucose deprivation conditions, NOX2-derived ROS promote hBMEC permeability [110]. Likewise, methamphetamine induces NOX2 activation, dysregulating ZO-1, occludin and claudin 5, and increasing BBB permeability in rat microvascular endothelial cells in vivo [111]. In an ischemic stroke model, NOX2 expression increased in the brain twelve hours, one day and two days after the reperfusion. Interestingly, NOX2 knock-out mice subjected to ischemic stroke present lower infarct volume and edema, and improved neurological outcomes compared to their control littermates [112]. Nevertheless, other studies suggest a limited role for NOX2 in these models [108].
Little information is available about the role of endothelial NOX4 in BBB permeability. NOX4 expression increases after stroke in human patients and experimental models. NOX4 knock-out mice subjected to MCAO are prevented from BBB leakage and neuronal apoptosis. Similar effects are observed when using VAS2870, a specific NOX4 inhibitor [108]. Finally, endothelial NOX4 knock-out mice exhibit smaller infarct sizes after MCAO [113].
Finally, the role of NOX5 in BBB disruption and stroke remains poorly understood. Our group showed that in aged endothelial NOX5 knock-in mice, ZO-1 and occludin expression decreased, which was accompanied by memory loss [114].
4.2. NADPH Oxidases and Immune Infiltration in the Brain
After the onset of ischemia, immune cells infiltrate the brain in coordinate temporal waves. Initially, infiltrative immune cells present a proinflammatory phenotype, turning into anti-inflammatory resolving cells at later stages of the disease [115]. Scarce information is available about NOXs effects at this point. Immortalized hBMEC exposed to the organic pollutant PCB153 increased ICAM-1 and VCAM-1 expression through a NOX-dependent mechanism. Interestingly, apocynin and diphenyleneiodonium chloride, two flavoprotein inhibitors, prevented these effects [116]. NOX2 is related with the developmental process of microglia in the cerebral cortex of mice. More specifically, NOX2 promotes the infiltration of macrophages in the developing tissue, indicating a role in peripheral immune cells infiltration into the brain [117]. In a model of chronic restrain stress, several NOX subunits and proinflammatory markers appear upregulated in the cerebrovascular endothelium, indicating a relationship between NOXs and cerebral inflammation during adult life [118].
In other CVDs the findings are similar. In a subarachnoid hemorrhage model, splenectomy reduces the infiltration of inflammatory cells and NOX2 expression in cardiac and cerebral tissues [119]. NOX activity seems to mediate ATP-derived NETosis after MCAO, an activity produced by infiltrating neutrophils [120]. Besides, NOX2 negatively regulates TIPE-2, a protein that inhibits the infiltration of peripheral immune cells [121]. Although scarce, the works carried out to date suggest that NOXs might play a role in the infiltration of immune cells during ischemic stroke.
4.3. NADPH Oxidases and Ischemia-Reperfusion Injury
NOXs have been deeply studied in the ischemia-reperfusion injury (IRI), a damage associated with an excessive production/accumulation of ROS after the recanalization of the infarcted area. Some authors suggest that NOX1, NOX2 and NOX4 play a relevant role in this process, since their expression increase after reperfusion in a nylon-induced MCAO model. In this study, PI3Kγ-/- mice inhibit the upregulation of NOXs, which resulted in reduced neutrophil infiltration, MMP-9 expression and brain damage [122]. Nonetheless, in a similar model of IRI in rats, NOX2 and NOX4 expression, but not NOX1, were upregulated at protein levels in the ischemic tissue [123].
Surprisingly, ROS produced during IRI may develop a dual role. In a mouse model of MCAO and reperfusion, NOX-inhibition by apocynin after damage displayed different effects depending on the timing. After the resolution of stroke, apocynin reduces inflammation and promotes angiogenesis. However, one week and two weeks after the event, it increases inflammation and reduces angiogenesis [124].
NOX1 mediates ROS production after oxygen/glucose deprivation and reoxygenation in murine brain endothelial cells [105]. In male rats subjected to MCAO followed by reperfusion, NOX1 expression increases in neurons of the peri-infarcted region. Interestingly, NOX1 inhibition increases newborn cells survival in this region, improving functional recovery [125].
In a similar model, NOX2 knock-out mice present increased revascularization of the infarcted area after three days [126]. Then, miR-652 reduced NOX2 expression, ROS production and tissue injury one day after reperfusion in the brain of rats subjected to MCAO [127]. NOD2 seems to activate NOX2 during IRI in vivo, increasing the secretion of proinflammatory mediators in the initial days after reperfusion [128]. Moreover, NOX4 targeting by miR-454 reduced ROS production in neuron-like SH-SY5Y cells, exhibiting a protective effect. Likewise, this miRNA reduced infarct size, edema, and cell death in the brain of rats one day after reperfusion [129]. Besides, NOX4 has been associated with TLR4 activation one day after IRI. TLR4 inhibition reduced NOX4 expression, oxidative stress, and neuronal apoptosis in vivo [130]. Interestingly, several drugs that exert neuroprotective effects against IRI in rodents seem to act by downregulating NOX4 [131,132,133]. Additionally, apocynin and NADPH treatment protected the brain tissue from inflammation and injury in an IRI model, a protection mediated by NOX2 and NOX4 downregulation [134]. NOX2 and NOX4 overexpression seem to be acute responders to IRI, as their levels increase three hours after reperfusion, returning to baseline after one day [135].
There is little information available about NOX5 implications in IRI. Nonetheless, Casas et al., recently described a key role for this oxidase. NOX5 produces ROS in the first hour after reoxygenation, while NOX4 acts later with a peak of activity after four hours. Interestingly, hypoxia increased NOX5 expression in hBMEC. Besides, in an immune and endothelial cell knock-in model for NOX5, BBB leakage and infarct volume were increased, and the neurological outcomes worsened after reperfusion [136].
5. Conclusions
The atherothrombotic stroke is strongly influenced by NOX activity in each stage. NOX members promote endothelial dysfunction, immune cell infiltration, foam cell genesis, atherosclerotic plaque development, thrombosis, BBB disruption, cerebral inflammation and IRI. Although NOXs have been widely studied, there is still a great lack of information from different approaches:
- (i)
- More cell type-specific knock-out/knock-in in vivo models would help to improve the current knowledge.
- (ii)
- More integrative studies that deep in the interconnection between different NOXs or their paracrine effects should be performed.
- (iii)
- here is an urgent need to develop isoform-specific NOX inhibitors and study these enzymes as potential therapeutical targets in CVDs.
To sum up, although great efforts have been made to study NOXs in atherothrombotic stroke, there is still a lot to discover.
Funding
This research was funded by the Government of Navarra through “Ayudas a centros tecnológicos y organismos de investigación para la realización de proyectos I+D colaborativos” (PC159-160-161 NOXICTUS), and from the Ministry of Economy and Competitiveness, Spain (SAF2016-79151-R, SAF2013-49088-R). Javier Marqués was funded by “Asociación de Amigos de la Universidad de Navarra” and by FPU PhD grants (FPU19/01807) from the Spanish Ministry of Education.
Acknowledgments
Authors thank Dr. Íñigo Izal for providing the artwork of the present review.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative Stress and Vascular Disease. Arterioscler Thromb Vasc Biol 2005, 25, 29–38. [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 2016, 118, 620–636. [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid Med Cell Longev 2019, 2019. [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat Rev Mol Cell Biol 2020, 21, 363–383. [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat Rev Mol Cell Biol 2014, 15, 411–421. [CrossRef]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev 2007, 87, 245–313. [CrossRef]
- Ogboo, B.C.; Grabovyy, U. V.; Maini, A.; Scouten, S.; van der Vliet, A.; Mattevi, A.; Heppner, D.E. Architecture of the NADPH Oxidase Family of Enzymes. Redox Biol 2022, 52, 102298. [CrossRef]
- Touyz, R.M.; Briones, A.M. Reactive Oxygen Species and Vascular Biology: Implications in Human Hypertension. Hypertens Res 2011, 34, 5–14. [CrossRef]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a Superoxide-Generating NADPH Oxidase of the Inner Ear. J Biol Chem 2004, 279, 46065–46072. [CrossRef]
- Bánfi, B.; Maturana, A.; Jaconi, S.; Arnaudeau, S.; Laforge, T.; Sinha, B.; Ligeti, E.; Demaurex, N.; Krause, K.H. A Mammalian H+ Channel Generated through Alternative Splicing of the NADPH Oxidase Homolog NOH-1. Science 2000, 287, 138–142. [CrossRef]
- Kobayashi, S.; Nojima, Y.; Shibuya, M.; Maru, Y. Nox1 Regulates Apoptosis and Potentially Stimulates Branching Morphogenesis in Sinusoidal Endothelial Cells. Exp Cell Res 2004, 300, 455–462. [CrossRef]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel Gp91(Phox) Homologues in Vascular Smooth Muscle Cells : Nox1 Mediates Angiotensin II-Induced Superoxide Formation and Redox-Sensitive Signaling Pathways. Circ Res 2001, 88, 888–894. [CrossRef]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A Crucial Role for Reactive Oxygen Species in RANKL-Induced Osteoclast Differentiation. Blood 2005, 106, 852–859. [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox Proteins in Signal Transduction. Free Radic Biol Med 2009, 47, 1239–1253. [CrossRef]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 2004, 24, 677–683. [CrossRef]
- Niu, X.L.; Madamanchi, N.R.; Vendrov, A.E.; Tchivilev, I.; Rojas, M.; Madamanchi, C.; Brandes, R.P.; Krause, K.H.; Humphries, J.; Smith, A.; et al. Nox Activator 1: A Potential Target for Modulation of Vascular Reactive Oxygen Species in Atherosclerotic Arteries. Circulation 2010, 121, 549–559. [CrossRef]
- Tabet, F.; Schiffrin, E.L.; Callera, G.E.; He, Y.; Yao, G.; Östman, A.; Kappert, K.; Tonks, N.K.; Touyz, R.M. Redox-Sensitive Signaling by Angiotensin II Involves Oxidative Inactivation and Blunted Phosphorylation of Protein Tyrosine Phosphatase SHP-2 in Vascular Smooth Muscle Cells from SHR. Circ Res 2008, 103, 149–158. [CrossRef]
- Wendt, M.C.; Daiber, A.; Kleschyov, A.L.; Mülsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J.F.; Lassègue, B.; Walter, U.; et al. Differential Effects of Diabetes on the Expression of the Gp91phox Homologues Nox1 and Nox4. Free Radic Biol Med 2005, 39, 381–391. [CrossRef]
- Borregaard, N.; Heiple, J.M.; Simons, E.R.; Clark, R.A. Subcellular Localization of the B-Cytochrome Component of the Human Neutrophil Microbicidal Oxidase: Translocation during Activation. J Cell Biol 1983, 97, 52–61. [CrossRef]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid Redox Signal 2005, 7, 308–317. [CrossRef]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes Aggravates Myocardial Ischaemia Reperfusion Injury via Activating Nox2-Related Programmed Cell Death in an AMPK-Dependent Manner. J Cell Mol Med 2020, 24, 6670–6679. [CrossRef]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL Oxidation by Platelets Propagates Platelet Activation via an Oxidative Stress-Mediated Mechanism. Atherosclerosis 2014, 237, 108–116. [CrossRef]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of Renox, an NAD(P)H Oxidase in Kidney. Proc Natl Acad Sci U S A 2000, 97, 8010–8014. [CrossRef]
- Ellmark, S.H.M.; Dusting, G.J.; Fui, M.N.; Guzzo-Pernell, N.; Drummond, G.R. The Contribution of Nox4 to NADPH Oxidase Activity in Mouse Vascular Smooth Muscle. Cardiovasc Res 2005, 65, 495–504. [CrossRef]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circulation 2004, 109, 227–233. [CrossRef]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 Is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidized Low Density Lipoprotein-Induced Macrophage Death. Circ Res 2010, 106, 1489–1497. [CrossRef]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased Mitochondrial NADPH Oxidase 4 (NOX4) Expression in Aging Is a Causative Factor in Aortic Stiffening. Redox Biol 2019, 26. [CrossRef]
- Perrotta, I.; Sciangula, A.; Perrotta, E.; Donato, G.; Cassese, M. Ultrastructural Analysis and Electron Microscopic Localization of Nox4 in Healthy and Atherosclerotic Human Aorta. Ultrastruct Pathol 2011, 35, 1–6. [CrossRef]
- Rajaram, R.D.; Dissard, R.; Jaquet, V.; De Seigneux, S. Potential Benefits and Harms of NADPH Oxidase Type 4 in the Kidneys and Cardiovascular System. Nephrol Dial Transplant 2019, 34, 567–576. [CrossRef]
- Bánfi, B.; Molnár, G.; Maturana, A.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.H. A Ca(2+)-Activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J Biol Chem 2001, 276, 37594–37601. [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium Dependent Nox5 NADPH Oxidase Contributes to Vascular Oxidative Stress in Human Coronary Artery Disease. J Am Coll Cardiol 2008, 52, 1803. [CrossRef]
- Pai, W.Y.; Lo, W.Y.; Hsu, T.; Peng, C.T.; Wang, H.J. Angiotensin-(1-7) Inhibits Thrombin-Induced Endothelial Phenotypic Changes and Reactive Oxygen Species Production via NADPH Oxidase 5 Downregulation. Front Physiol 2017, 8. [CrossRef]
- Petsophonsakul, P.; Burgmaier, M.; Willems, B.; Heeneman, S.; Stadler, N.; Gremse, F.; Reith, S.; Burgmaier, K.; Kahles, F.; Marx, N.; et al. Nicotine Promotes Vascular Calcification via Intracellular Ca2+-Mediated, Nox5-Induced Oxidative Stress, and Extracellular Vesicle Release in Vascular Smooth Muscle Cells. Cardiovasc Res 2022, 118, 2196–2210. [CrossRef]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human Monocytes and Macrophages Express NADPH Oxidase 5; a Potential Source of Reactive Oxygen Species in Atherosclerosis. Biochem Biophys Res Commun 2015, 461, 172–179. [CrossRef]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.L. NOX5: Molecular Biology and Pathophysiology. Exp Physiol 2019, 104, 605. [CrossRef]
- Anagnostopoulou, A.; Camargo, L.L.; Rodrigues, D.; Montezano, A.C.; Touyz, R.M. Importance of Cholesterol-Rich Microdomains in the Regulation of Nox Isoforms and Redox Signaling in Human Vascular Smooth Muscle Cells. Sci Rep 2020, 10. [CrossRef]
- Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Tlili, A.; Marie, J.C.; Monteiro, R.C.; Gougerot-Pocidalo, M.A.; Dang, P.M.C.; El-Benna, J. NOX5 and P22phox Are 2 Novel Regulators of Human Monocytic Differentiation into Dendritic Cells. Blood 2017, 130, 1734–1745. [CrossRef]
- Richter, S.M.; Massman, L.C.; Stuehr, D.J.; Sweeny, E.A. Functional Interactions between NADPH Oxidase 5 and Actin. Front Cell Dev Biol 2023, 11. [CrossRef]
- Marqués, J.; Cortés, A.; Pejenaute, Á.; Zalba, G. Implications of NADPH Oxidase 5 in Vascular Diseases. Int J Biochem Cell Biol 2020, 128. [CrossRef]
- Li, M.; Liu, X.; He, Y.; Zheng, Q.; Wang, M.; Wu, Y.; Zhang, Y.; Wang, C. Celastrol Attenuates Angiotensin II Mediated Human Umbilical Vein Endothelial Cells Damage through Activation of Nrf2/ERK1/2/Nox2 Signal Pathway. Eur J Pharmacol 2017, 797, 124–133. [CrossRef]
- Liang, G.Z.; Cheng, L.M.; Chen, X.F.; Li, Y.J.; Li, X.L.; Guan, Y.Y.; Du, Y.H. ClC-3 Promotes Angiotensin II-Induced Reactive Oxygen Species Production in Endothelial Cells by Facilitating Nox2 NADPH Oxidase Complex Formation. Acta Pharmacol Sin 2018, 39, 1725–1734. [CrossRef]
- Hong, O.K.; Lee, S.S.; Yoo, S.J.; Choi, S.H.; Lee, M.K.; Cha, B.Y.; Kim, M.K.; Baek, K.H.; Song, K.H.; Kwon, H.S. Effects of DA-9801 on the Inflammation and Apoptosis Induced by Angiotensin II in Human Dermal Microvascular Endothelial Cells. J Pharmacol Sci 2021, 145, 52–59. [CrossRef]
- Montezano, A.C.; Burger, D.; Paravicini, T.M.; Chignalia, A.Z.; Yusuf, H.; Almasri, M.; He, Y.; Callera, G.E.; He, G.; Krause, K.H.; et al. Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 5 (Nox5) Regulation by Angiotensin II and Endothelin-1 Is Mediated via Calcium/Calmodulin-Dependent, Rac-1-Independent Pathways in Human Endothelial Cells. Circ Res 2010, 106, 1363. [CrossRef]
- Marqués, J.; Cortés, A.; Pejenaute, Á.; Ansorena, E.; Abizanda, G.; Prósper, F.; Martínez-Irujo, J.J.; Miguel, C. de; Zalba, G. Induction of Cyclooxygenase-2 by Overexpression of the Human NADPH Oxidase 5 (NOX5) Gene in Aortic Endothelial Cells. Cells 2020, 9. [CrossRef]
- Su, E.; Zhao, L.; Yang, X.; Zhu, B.; Liu, Y.; Zhao, W.; Wang, X.; Qi, D.; Zhu, L.; Gao, C. Aggravated Endothelial Endocrine Dysfunction and Intimal Thickening of Renal Artery in High-Fat Diet-Induced Obese Pigs Following Renal Denervation. BMC Cardiovasc Disord 2020, 20. [CrossRef]
- Valente, A.J.; Irimpen, A.M.; Siebenlist, U.; Chandrasekar, B. OxLDL Induces Endothelial Dysfunction and Death via TRAF3IP2: Inhibition by HDL3 and AMPK Activators. Free Radic Biol Med 2014, 70, 117–128. [CrossRef]
- Chen, B.; Zhao, J.; Zhang, S.; Wu, W.; Qi, R. Aspirin Inhibits the Production of Reactive Oxygen Species by Downregulating Nox4 and Inducible Nitric Oxide Synthase in Human Endothelial Cells Exposed to Oxidized Low-Density Lipoprotein. J Cardiovasc Pharmacol 2012, 59, 405–412. [CrossRef]
- Zhao, W.; Li, C.; Gao, H.; Wu, Q.; Shi, J.; Chen, X. Dihydrotanshinone I Attenuates Atherosclerosis in ApoE-Deficient Mice: Role of NOX4/NF-ΚB Mediated Lectin-Like Oxidized LDL Receptor-1 (LOX-1) of the Endothelium. Front Pharmacol 2016, 7. [CrossRef]
- Langbein, H.; Brunssen, C.; Hofmann, A.; Cimalla, P.; Brux, M.; Bornstein, S.R.; Deussen, A.; Koch, E.; Morawietz, H. NADPH Oxidase 4 Protects against Development of Endothelial Dysfunction and Atherosclerosis in LDL Receptor Deficient Mice. Eur Heart J 2016, 37, 1753–1761. [CrossRef]
- da Silva, J.F.; Alves, J. V.; Silva-Neto, J.A.; Costa, R.M.; Neves, K.B.; Alves-Lopes, R.; Carmargo, L.L.; Rios, F.J.; Montezano, A.C.; Touyz, R.M.; et al. Lysophosphatidylcholine Induces Oxidative Stress in Human Endothelial Cells via NOX5 Activation - Implications in Atherosclerosis. Clin Sci (Lond) 2021, 135, 1845–1858. [CrossRef]
- Sipkens, J.A.; Hahn, N.; van den Brand, C.S.; Meischl, C.; Cillessen, S.A.G.M.; Smith, D.E.C.; Juffermans, L.J.M.; Musters, R.J.P.; Roos, D.; Jakobs, C.; et al. Homocysteine-Induced Apoptosis in Endothelial Cells Coincides with Nuclear NOX2 and Peri-Nuclear NOX4 Activity. Cell Biochem Biophys 2013, 67, 341–352. [CrossRef]
- Cortés, A.; Pejenaute, Á.; Marqués, J.; Izal, Í.; Cenoz, S.; Ansorena, E.; Martínez-Irujo, J.J.; Miguel, C.; Zalba, G. Nadph Oxidase 5 Induces Changes in the Unfolded Protein Response in Human Aortic Endothelial Cells and in Endothelial-Specific Knock-in Mice. Antioxidants 2021, 10. [CrossRef]
- Marqués, J.; Fernández-Irigoyen, J.; Ainzúa, E.; Martínez-Azcona, M.; Cortés, A.; Roncal, C.; Orbe, J.; Santamaría, E.; Zalba, G. NADPH Oxidase 5 (NOX5) Overexpression Promotes Endothelial Dysfunction via Cell Apoptosis, Migration, and Metabolic Alterations in Human Brain Microvascular Endothelial Cells (HCMEC/D3). Antioxidants (Basel) 2022, 11. [CrossRef]
- Schuett, J.; Schuett, H.; Oberoi, R.; Koch, A.K.; Pretzer, S.; Luchtefeld, M.; Schieffer, B.; Grote, K. NADPH Oxidase NOX2 Mediates TLR2/6-Dependent Release of GM-CSF from Endothelial Cells. FASEB J 2017, 31, 2612–2624. [CrossRef]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH Oxidase 4 in Lipopolysaccharide-Induced Proinflammatory Responses by Human Aortic Endothelial Cells. Cardiovasc Res 2006, 72, 447–455. [CrossRef]
- Maloney, E.; Sweet, I.R.; Hockenbery, D.M.; Pham, M.; Rizzo, N.O.; Tateya, S.; Handa, P.; Schwartz, M.W.; Kim, F. Activation of NF-KappaB by Palmitate in Endothelial Cells: A Key Role for NADPH Oxidase-Derived Superoxide in Response to TLR4 Activation. Arterioscler Thromb Vasc Biol 2009, 29, 1370–1375. [CrossRef]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl Sulfate Stimulates Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells by Inducing Oxidative Stress through Activation of the NADPH Oxidase-Nuclear Factor-ΚB Pathway. Circ J 2010, 74, 2216–2224. [CrossRef]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or Vascular Smooth Muscle Cell-Specific Expression of Human NOX5 Exacerbates Renal Inflammation, Fibrosis and Albuminuria in the Akita Mouse. Diabetologia 2019, 62, 1712–1726. [CrossRef]
- Rius, C.; Company, C.; Piqueras, L.; Cerdá-Nicolás, J.M.; González, C.; Servera, E.; Ludwig, A.; Morcillo, E.J.; Sanz, M.J. Critical Role of Fractalkine (CX3CL1) in Cigarette Smoke-Induced Mononuclear Cell Adhesion to the Arterial Endothelium. Thorax 2013, 68, 177–186. [CrossRef]
- Muzaffar, S.; Jeremy, J.Y.; Angelini, G.D.; Shukla, N. NADPH Oxidase 4 Mediates Upregulation of Type 4 Phosphodiesterases in Human Endothelial Cells. J Cell Physiol 2012, 227, 1941–1950. [CrossRef]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J Am Coll Cardiol 2014, 63, 2734–2741. [CrossRef]
- Sun, H.J.; Zhao, M.X.; Liu, T.Y.; Ren, X.S.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. Salusin-β Induces Foam Cell Formation and Monocyte Adhesion in Human Vascular Smooth Muscle Cells via MiR155/NOX2/NFκB Pathway. Sci Rep 2016, 6. [CrossRef]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular Cell Adhesion Molecule-1 Expression and Signaling During Disease: Regulation by Reactive Oxygen Species and Antioxidants. Antioxid Redox Signal 2011, 15, 1607. [CrossRef]
- Wang, M.; Murdoch, C.E.; Brewer, A.C.; Ivetic, A.; Evans, P.; Shah, A.M.; Zhang, M. Endothelial NADPH Oxidase 4 Protects against Angiotensin II-Induced Cardiac Fibrosis and Inflammation. ESC Heart Fail 2021, 8, 1427–1437. [CrossRef]
- Xia, F.; Wang, C.; Jin, Y.; Liu, Q.; Meng, Q.; Liu, K.; Sun, H. Luteolin Protects HUVECs from TNF-α-Induced Oxidative Stress and Inflammation via Its Effects on the Nox4/ROS-NF-ΚB and MAPK Pathways. J Atheroscler Thromb 2014, 21, 768–783. [CrossRef]
- Escudero, P.; De Marañón, A.M.; Collado, A.; Gonzalez-Navarro, H.; Hermenegildo, C.; Peiró, C.; Piqueras, L.; Sanz, M.J. Combined Sub-Optimal Doses of Rosuvastatin and Bexarotene Impair Angiotensin II-Induced Arterial Mononuclear Cell Adhesion through Inhibition of Nox5 Signaling Pathways and Increased RXR/PPARα and RXR/PPARγ Interactions. Antioxid Redox Signal 2015, 22, 901–920. [CrossRef]
- Cortés, A.; Marqués, J.; Pejenaute, Á.; Ainzúa, E.; Ansorena, E.; Abizanda, G.; Prósper, F.; de Miguel, C.; Zalba, G. Endothelial NOX5 Overexpression Induces Changes in the Cardiac Gene Profile: Potential Impact in Myocardial Infarction? J Physiol Biochem 2023, 79, 787–797. [CrossRef]
- Wang, H.; Albadawi, H.; Siddiquee, Z.; Stone, J.M.; Panchenko, M.P.; Watkins, M.T.; Stone, J.R. Altered Vascular Activation Due to Deficiency of the NADPH Oxidase Component P22phox. Cardiovascular Pathology 2014, 23, 35–42. [CrossRef]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.V.P.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II Enhances AT1-Nox1 Binding and Stimulates Arterial Smooth Muscle Cell Migration and Proliferation through AT1, Nox1, and Interleukin-18. Am J Physiol Heart Circ Physiol 2012, 303, 282–296. [CrossRef]
- Moraes, J.A.; Frony, A.C.; Dias, A.M.; Renovato-Martins, M.; Rodrigues, G.; Marcinkiewicz, C.; Assreuy, J.; Barja-Fidalgo, C. Alpha1beta1 and Integrin-Linked Kinase Interact and Modulate Angiotensin II Effects in Vascular Smooth Muscle Cells. Atherosclerosis 2015, 243, 477–485. [CrossRef]
- Li, X.; Wang, H.F.; Li, X.X.; Xu, M. Contribution of Acid Sphingomyelinase to Angiotensin II-Induced Vascular Adventitial Remodeling via Membrane Rafts/Nox2 Signal Pathway. Life Sci 2019, 219, 303–310. [CrossRef]
- Quesada, I.M.; Lucero, A.; Amaya, C.; Meijles, D.N.; Cifuentes, M.E.; Pagano, P.J.; Castro, C. Selective Inactivation of NADPH Oxidase 2 Causes Regression of Vascularization and the Size and Stability of Atherosclerotic Plaques. Atherosclerosis 2015, 242, 469–475. [CrossRef]
- Haurani, M.J.; Cifuentes, M.E.; Shepard, A.D.; Pagano, P.J. Nox4 Oxidase Overexpression Specifically Decreases Endogenous Nox4 MRNA and Inhibits Angiotensin II-Induced Adventitial Myofibroblast Migration. Hypertension 2008, 52, 143–149. [CrossRef]
- Lee, S.H.; Park, D.W.; Park, S.C.; Park, Y.K.; Hong, S.Y.; Kim, J.R.; Lee, C.H.; Baek, S.H. Calcium-Independent Phospholipase A2beta-Akt Signaling Is Involved in Lipopolysaccharide-Induced NADPH Oxidase 1 Expression and Foam Cell Formation. J Immunol 2009, 183, 7497–7504. [CrossRef]
- Lee, J.G.; Lim, E.J.; Park, D.W.; Lee, S.H.; Kim, J.R.; Baek, S.H. A Combination of Lox-1 and Nox1 Regulates TLR9-Mediated Foam Cell Formation. Cell Signal 2008, 20, 2266–2275. [CrossRef]
- Csányi, G.; Feck, D.M.; Ghoshal, P.; Singla, B.; Lin, H.; Nagarajan, S.; Meijles, D.N.; Al Ghouleh, I.; Cantu-Medellin, N.; Kelley, E.E.; et al. CD47 and Nox1 Mediate Dynamic Fluid-Phase Macropinocytosis of Native LDL. Antioxid Redox Signal 2017, 26, 886–901. [CrossRef]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 Protect against Oxidative Stress-Induced Macrophage Apoptosis during Efferocytosis. Circ Res 2010, 106, 1861–1869. [CrossRef]
- Yu, P.; Han, W.; Villar, V.A.M.; Yang, Y.; Lu, Q.; Lee, H.; Li, F.; Quinn, M.T.; Gildea, J.J.; Felder, R.A.; et al. Unique Role of NADPH Oxidase 5 in Oxidative Stress in Human Renal Proximal Tubule Cells. Redox Biol 2014, 2, 570–579. [CrossRef]
- Vendrov, A.E.; Sumida, A.; Canugovi, C.; Lozhkin, A.; Hayami, T.; Madamanchi, N.R.; Runge, M.S. NOXA1-Dependent NADPH Oxidase Regulates Redox Signaling and Phenotype of Vascular Smooth Muscle Cell during Atherogenesis. Redox Biol 2019, 21. [CrossRef]
- Al Ghouleh, I.; Rodríguez, A.; Pagano, P.J.; Csányi, G. Proteomic Analysis Identifies an NADPH Oxidase 1 (Nox1)-Mediated Role for Actin-Related Protein 2/3 Complex Subunit 2 (ARPC2) in Promoting Smooth Muscle Cell Migration. Int J Mol Sci 2013, 14, 20220–20235. [CrossRef]
- Abhijit, S.; Bhaskaran, R.; Narayanasamy, A.; Chakroborty, A.; Manickam, N.; Dixit, M.; Mohan, V.; Balasubramanyam, M. Hyperinsulinemia-Induced Vascular Smooth Muscle Cell (VSMC) Migration and Proliferation Is Mediated by Converging Mechanisms of Mitochondrial Dysfunction and Oxidative Stress. Mol Cell Biochem 2013, 373, 95–105. [CrossRef]
- Zhao, Q.; Zhang, J.; Wang, H. PGC-1α Limits Angiotensin II-Induced Rat Vascular Smooth Muscle Cells Proliferation via Attenuating NOX1-Mediated Generation of Reactive Oxygen Species. Biosci Rep 2015, 35. [CrossRef]
- Zhang, J.; Chen, C.; Li, L.; Zhou, H.J.; Li, F.; Zhang, H.; Yu, L.; Chen, Y.; Min, W. Endothelial AIP1 Regulates Vascular Remodeling by Suppressing NADPH Oxidase-2. Front Physiol 2018, 9. [CrossRef]
- Pietrowski, E.; Bender, B.; Huppert, J.; White, R.; Luhmann, H.J.; Kuhlmann, C.R.W. Pro-Inflammatory Effects of Interleukin-17A on Vascular Smooth Muscle Cells Involve NAD(P)H- Oxidase Derived Reactive Oxygen Species. J Vasc Res 2011, 48, 52–58. [CrossRef]
- Fernandes, D.C.; Wosniak, J.; Gonçalves, R.C.; Tanaka, L.Y.; Fernandes, C.G.; Zanatta, D.B.; de Mattos, A.B.M.; Strauss, B.E.; Laurindo, F.R.M. PDIA1 Acts as Master Organizer of NOX1/NOX4 Balance and Phenotype Response in Vascular Smooth Muscle. Free Radic Biol Med 2021, 162, 603–614. [CrossRef]
- Meng, D.; Lv, D.D.; Fang, J. Insulin-like Growth Factor-I Induces Reactive Oxygen Species Production and Cell Migration through Nox4 and Rac1 in Vascular Smooth Muscle Cells. Cardiovasc Res 2008, 80, 299–308. [CrossRef]
- Camargo, L.L.; Montezano, A.C.; Hussain, M.; Wang, Y.; Zou, Z.; Rios, F.J.; Neves, K.B.; Alves-Lopes, R.; Awan, F.R.; Guzik, T.J.; et al. Central Role of C-Src in NOX5- Mediated Redox Signalling in Vascular Smooth Muscle Cells in Human Hypertension. Cardiovasc Res 2022, 118, 1359–1373. [CrossRef]
- Furmanik, M.; Chatrou, M.; Van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; Van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ Res 2020, 127, 911–927. [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; De Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus-Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [CrossRef]
- Ouerd, S.; Idris-Khodja, N.; Trindade, M.; Ferreira, N.S.; Berillo, O.; Coelho, S.C.; Neves, M.F.; Jandeleit-Dahm, K.A.; Paradis, P.; Schiffrin, E.L. Endothelium-Restricted Endothelin-1 Overexpression in Type 1 Diabetes Worsens Atherosclerosis and Immune Cell Infiltration via NOX1. Cardiovasc Res 2021, 117, 1144–1153. [CrossRef]
- Serino, A.; Zhao, Y.; Hwang, J.; Cullen, A.; Deeb, C.; Akhavan, N.; Arjmandi, B.; Salazar, G. Gender Differences in the Effect of Blackberry Supplementation in Vascular Senescence and Atherosclerosis in ApoE-/- Mice. J Nutr Biochem 2020, 80. [CrossRef]
- Judkins, C.P.; Diep, H.; Broughton, B.R.S.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct Evidence of a Role for Nox2 in Superoxide Production, Reduced Nitric Oxide Bioavailability, and Early Atherosclerotic Plaque Formation in ApoE-/- Mice. Am J Physiol Heart Circ Physiol 2010, 298. [CrossRef]
- Schürmann, C.; Rezende, F.; Kruse, C.; Yasar, Y.; Löwe, O.; Fork, C.; Van De Sluis, B.; Bremer, R.; Weissmann, N.; Shah, A.M.; et al. The NADPH Oxidase Nox4 Has Anti-Atherosclerotic Functions. Eur Heart J 2015, 36, 3447–3456. [CrossRef]
- Yu, W.; Xiao, L.; Que, Y.; Li, S.; Chen, L.; Hu, P.; Xiong, R.; Seta, F.; Chen, H.; Tong, X. Smooth Muscle NADPH Oxidase 4 Promotes Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis by Regulating Osteopontin. Biochim Biophys Acta Mol Basis Dis 2020, 1866. [CrossRef]
- Vlad, M.L.; Manea, S.A.; Lazar, A.G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Histone Acetyltransferase-Dependent Pathways Mediate Upregulation of NADPH Oxidase 5 in Human Macrophages under Inflammatory Conditions: A Potential Mechanism of Reactive Oxygen Species Overproduction in Atherosclerosis. Oxid Med Cell Longev 2019, 2019. [CrossRef]
- Ho, F.; Watson, A.M.D.; Elbatreek, M.H.; Kleikers, P.W.M.; Khan, W.; Sourris, K.C.; Dai, A.; Jha, J.; Schmidt, H.H.H.W.; Jandeleit-Dahm, K.A.M. Endothelial Reactive Oxygen-Forming NADPH Oxidase 5 Is a Possible Player in Diabetic Aortic Aneurysm but Not Atherosclerosis. Sci Rep 2022, 12. [CrossRef]
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ Res 2021, 128, 1320–1322. [CrossRef]
- Cho, D.I.; Ahn, M.J.; Cho, H.H.; Cho, M.; Jun, J.H.; Kang, B.G.; Lim, S.Y.; Yoo, S.J.; Kim, M.R.; Kim, H.S.; et al. ANGPTL4 Stabilizes Atherosclerotic Plaques and Modulates the Phenotypic Transition of Vascular Smooth Muscle Cells through KLF4 Downregulation. Exp Mol Med 2023, 55, 426–442. [CrossRef]
- Wang, Y.; Liu, X.Y.; Zhao, W.X.; Li, F.D.; Guo, P.R.; Fan, Q.; Wu, X.F. NOX2 Inhibition Stabilizes Vulnerable Plaques by Enhancing Macrophage Efferocytosis via MertK/PI3K/AKT Pathway. Redox Biol 2023, 64. [CrossRef]
- Hofmann, A.; Frank, F.; Wolk, S.; Busch, A.; Klimova, A.; Sabarstinski, P.; Gerlach, M.; Egorov, D.; Kopaliani, I.; Weinert, S.; et al. NOX4 MRNA Correlates with Plaque Stability in Patients with Carotid Artery Stenosis. Redox Biol 2022, 57. [CrossRef]
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH Oxidase 1 Is a Novel Pharmacological Target for the Development of an Antiplatelet Drug without Bleeding Side Effects. FASEB J 2020, 34, 13959–13977. [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler Thromb Vasc Biol 2016, 36, 846–854. [CrossRef]
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J.; Fasano, M.B.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH Oxidase Is Dispensable for Platelet Activation or Arterial Thrombosis in Mice. Blood Adv 2019, 3, 1272–1284. [CrossRef]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arterioscler Thromb Vasc Biol 2021, 41, 683. [CrossRef]
- Zhang, B.; Li, J. Phoenixin-14 Protects Human Brain Vascular Endothelial Cells against Oxygen-Glucose Deprivation/Reoxygenation (OGD/R)-Induced Inflammation and Permeability. Arch Biochem Biophys 2020, 682. [CrossRef]
- Hwang, J.S.; Cha, E.H.; Ha, E.; Park, B.; Seo, J.H. GKT136901 Protects Primary Human Brain Microvascular Endothelial Cells against Methamphetamine-Induced Blood-Brain Barrier Dysfunction. Life Sci 2020, 256. [CrossRef]
- Kuriakose, M.; Younger, D.; Ravula, A.R.; Alay, E.; Rama Rao, K. V.; Chandra, N. Synergistic Role of Oxidative Stress and Blood-Brain Barrier Permeability as Injury Mechanisms in the Acute Pathophysiology of Blast-Induced Neurotrauma. Sci Rep 2019, 9. [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-Stroke Inhibition of Induced NADPH Oxidase Type 4 Prevents Oxidative Stress and Neurodegeneration. PLoS Biol 2010, 8. [CrossRef]
- Jackman, K.A.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Importance of NOX1 for Angiotensin II-Induced Cerebrovascular Superoxide Production and Cortical Infarct Volume Following Ischemic Stroke. Brain Res 2009, 1286, 215–220. [CrossRef]
- Alwjwaj, M.; Kadir, R.R.A.; Bayraktutan, U. Outgrowth Endothelial Progenitor Cells Restore Cerebral Barrier Function Following Ischaemic Damage: The Impact of NOX2 Inhibition. Eur J Neurosci 2022, 55, 1658–1670. [CrossRef]
- Namyen, J.; Permpoonputtana, K.; Nopparat, C.; Tocharus, J.; Tocharus, C.; Govitrapong, P. Protective Effects of Melatonin on Methamphetamine-Induced Blood-Brain Barrier Dysfunction in Rat Model. Neurotox Res 2020, 37, 640–660. [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J Cereb Blood Flow Metab 2014, 34, 660–667. [CrossRef]
- Casas, A.I.; Geuss, E.; Kleikers, P.W.M.; Mencl, S.; Herrmann, A.M.; Buendia, I.; Egea, J.; Meuth, S.G.; Lopez, M.G.; Kleinschnitz, C.; et al. NOX4-Dependent Neuronal Autotoxicity and BBB Breakdown Explain the Superior Sensitivity of the Brain to Ischemic Damage. Proc Natl Acad Sci U S A 2017, 114, 12315–12320. [CrossRef]
- Cortés, A.; Solas, M.; Pejenaute, Á.; Abellanas, M.A.; Garcia-Lacarte, M.; Aymerich, M.S.; Marqués, J.; Ramírez, M.J.; Zalba, G. Expression of Endothelial Nox5 Alters the Integrity of the Blood-Brain Barrier and Causes Loss of Memory in Aging Mice. Antioxidants 2021, 10. [CrossRef]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front Immunol 2021, 12. [CrossRef]
- Eum, S.Y.; Andras, I.; Hennig, B.; Toborek, M. NADPH Oxidase and Lipid Raft-Associated Redox Signaling Are Required for PCB153-Induced Upregulation of Cell Adhesion Molecules in Human Brain Endothelial Cells. Toxicol Appl Pharmacol 2009, 240, 299–305. [CrossRef]
- Lelli, A.; Gervais, A.; Colin, C.; Chéret, C.; de Almodovar, C.R.; Carmeliet, P.; Krause, K.H.; Boillée, S.; Mallat, M. The NADPH Oxidase Nox2 Regulates VEGFR1/CSF-1R-Mediated Microglial Chemotaxis and Promotes Early Postnatal Infiltration of Phagocytes in the Subventricular Zone of the Mouse Cerebral Cortex. Glia 2013, 61, 1542–1555. [CrossRef]
- Zhu, Y.; Haddad, Y.; Yun, H.J.; Geng, X.; Ding, Y. Induced Inflammatory and Oxidative Markers in Cerebral Microvasculature by Mentally Depressive Stress. Mediators Inflamm 2023, 2023. [CrossRef]
- Li, R.; Yuan, Q.; Su, Y.; Chopp, M.; Yan, T.; Chen, J. Immune Response Mediates the Cardiac Damage after Subarachnoid Hemorrhage. Exp Neurol 2020, 323. [CrossRef]
- Kim, S.W.; Davaanyam, D.; Seol, S.I.; Lee, H.K.; Lee, H.; Lee, J.K. Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation. Int J Mol Sci 2020, 21, 7668. [CrossRef]
- Zhang, Y.; Wei, X.; Liu, L.; Liu, S.; Wang, Z.; Zhang, B.; Fan, B.; Yang, F.; Huang, S.; Jiang, F.; et al. TIPE2, a Novel Regulator of Immunity, Protects against Experimental Stroke. J Biol Chem 2012, 287, 32546–32555. [CrossRef]
- Jin, R.; Song, Z.; Yu, S.; Piazza, A.; Nanda, A.; Penninger, J.M.; Granger, D.N.; Li, G. Phosphatidylinositol-3-Kinase Gamma Plays a Central Role in Blood-Brain Barrier Dysfunction in Acute Experimental Stroke. Stroke 2011, 42, 2033–2044. [CrossRef]
- Tuo, Y.H.; Liu, Z.; Chen, J.W.; Wang, Q.Y.; Li, S.L.; Li, M.C.; Dai, G.; Wang, J.S.; Zhang, Y.L.; Feng, L.; et al. NADPH Oxidase Inhibitor Improves Outcome of Mechanical Reperfusion by Suppressing Hemorrhagic Transformation. J Neurointerv Surg 2017, 9, 492–498. [CrossRef]
- Yingze, Y.; Zhihong, J.; Tong, J.; Yina, L.; Zhi, Z.; Xu, Z.; Xiaoxing, X.; Lijuan, G. NOX2-Mediated Reactive Oxygen Species Are Double-Edged Swords in Focal Cerebral Ischemia in Mice. J Neuroinflammation 2022, 19. [CrossRef]
- Choi, D.H.; Kim, J.H.; Lee, K.H.; Kim, H.Y.; Kim, Y.S.; Choi, W.S.; Lee, J. Role of Neuronal NADPH Oxidase 1 in the Peri-Infarct Regions after Stroke. PLoS One 2015, 10. [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Nox2 Knockout Delays Infarct Progression and Increases Vascular Recovery through Angiogenesis in Mice Following Ischaemic Stroke with Reperfusion. PLoS One 2014, 9. [CrossRef]
- Zuo, M.L.; Wang, A.P.; Song, G.L.; Yang, Z.B. MiR-652 Protects Rats from Cerebral Ischemia/Reperfusion Oxidative Stress Injury by Directly Targeting NOX2. Biomed Pharmacother 2020, 124. [CrossRef]
- Liu, H.; Wei, X.; Kong, L.; Liu, X.; Liu, X.; Cheng, L.; Yan, S.; Zhang, X.; Chen, L. NOD2 Is Involved in the Inflammatory Response after Cerebral Ischemia-Reperfusion Injury and Triggers NADPH Oxidase 2-Derived Reactive Oxygen Species. Int J Biol Sci 2015, 11, 525. [CrossRef]
- Zhang, T.; Han, H.; Zhou, Y.; Liu, Z.; Ma, T.; Cao, X. MicroRNA-454 Modulates the Oxidative Stress and Neuronal Apoptosis after Cerebral Ischemia/Reperfusion Injury via Targeting NADPH Oxidase 4 (NOX4). J Biochem Mol Toxicol 2022, 36. [CrossRef]
- Suzuki, Y.; Hattori, K.; Hamanaka, J.; Murase, T.; Egashira, Y.; Mishiro, K.; Ishiguro, M.; Tsuruma, K.; Hirose, Y.; Tanaka, H.; et al. Pharmacological Inhibition of TLR4-NOX4 Signal Protects against Neuronal Death in Transient Focal Ischemia. Sci Rep 2012, 2. [CrossRef]
- Hu, Z.Y.; Yang, Z.B.; Zhang, R.; Luo, X.J.; Peng, J. The Protective Effect of Vitexin Compound B-1 on Rat Cerebral I/R Injury through a Mechanism Involving Modulation of MiR-92b/NOX4 Pathway. CNS Neurol Disord Drug Targets 2023, 22, 137–147. [CrossRef]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin Attenuates Oxidative Stress and Neuronal Apoptosis after Ischemia/Reperfusion Injury via Nrf2-Mediated Inhibition of the NOX4/ROS/NF-ΚB Pathway. Chem Biol Interact 2018, 284, 32–40. [CrossRef]
- Lu, P.; Zhang, C.C.; Zhang, X.M.; Li, H.G.; Luo, A.L.; Tian, Y.K.; Xu, H. Down-Regulation of NOX4 by Betulinic Acid Protects against Cerebral Ischemia-Reperfusion in Mice. J Huazhong Univ Sci Technolog Med Sci 2017, 37, 744–749. [CrossRef]
- Qin, Y.Y.; Li, M.; Feng, X.; Wang, J.; Cao, L.; Shen, X.K.; Chen, J.; Sun, M.; Sheng, R.; Han, F.; et al. Combined NADPH and the NOX Inhibitor Apocynin Provides Greater Anti-Inflammatory and Neuroprotective Effects in a Mouse Model of Stroke. Free Radic Biol Med 2017, 104, 333–345. [CrossRef]
- Li, H.; Wang, Y.; Feng, D.; Liu, Y.; Xu, M.; Gao, A.; Tian, F.; Zhang, L.; Cui, Y.; Wang, Z.; et al. Alterations in the Time Course of Expression of the Nox Family in the Brain in a Rat Experimental Cerebral Ischemia and Reperfusion Model: Effects of Melatonin. J Pineal Res 2014, 57, 110–119. [CrossRef]
- Casas, A.I.; Kleikers, P.W.M.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; De Angelis, M.H.; Egea, J.; Lopez, M.G.; et al. Calcium-Dependent Blood-Brain Barrier Breakdown by NOX5 Limits Postreperfusion Benefit in Stroke. J Clin Invest 2019, 129, 1772. [CrossRef]
Figure 1.
Intracellular distribution of NOX homologues in endothelial cells, VSMC and monocytes/macrophages. Endothelial Cells. NOX1 is expressed, but its intracellular localization remains unknown. NOX2 locates in membrane protrusions and in association with the cytoskeleton. NOX4 and NOX5 locate in the endoplasmic reticulum. Vascular Smooth Muscle Cells. NOX1 locates inside clatrin-coated areas of the membrane when inactive, and translocases to the membrane upon activation. NOX2 is expressed, but its intracellular location remains unknown. NOX4 locates inside the mitochondria, at the nucleus and at focal adhesions. NOX5 locates at cholesterol-rich areas of the plasma membrane. Monocytes/Macrophages. NOX1 is expressed, but its intracellular localization remains unknown. NOX2 locates at intracellular membranes and translocases to the plasma membrane upon activation. NOX4 locates at the endoplasmic reticulum and the nucleus. NOX5 locates at mitochondria.
Figure 1.
Intracellular distribution of NOX homologues in endothelial cells, VSMC and monocytes/macrophages. Endothelial Cells. NOX1 is expressed, but its intracellular localization remains unknown. NOX2 locates in membrane protrusions and in association with the cytoskeleton. NOX4 and NOX5 locate in the endoplasmic reticulum. Vascular Smooth Muscle Cells. NOX1 locates inside clatrin-coated areas of the membrane when inactive, and translocases to the membrane upon activation. NOX2 is expressed, but its intracellular location remains unknown. NOX4 locates inside the mitochondria, at the nucleus and at focal adhesions. NOX5 locates at cholesterol-rich areas of the plasma membrane. Monocytes/Macrophages. NOX1 is expressed, but its intracellular localization remains unknown. NOX2 locates at intracellular membranes and translocases to the plasma membrane upon activation. NOX4 locates at the endoplasmic reticulum and the nucleus. NOX5 locates at mitochondria.
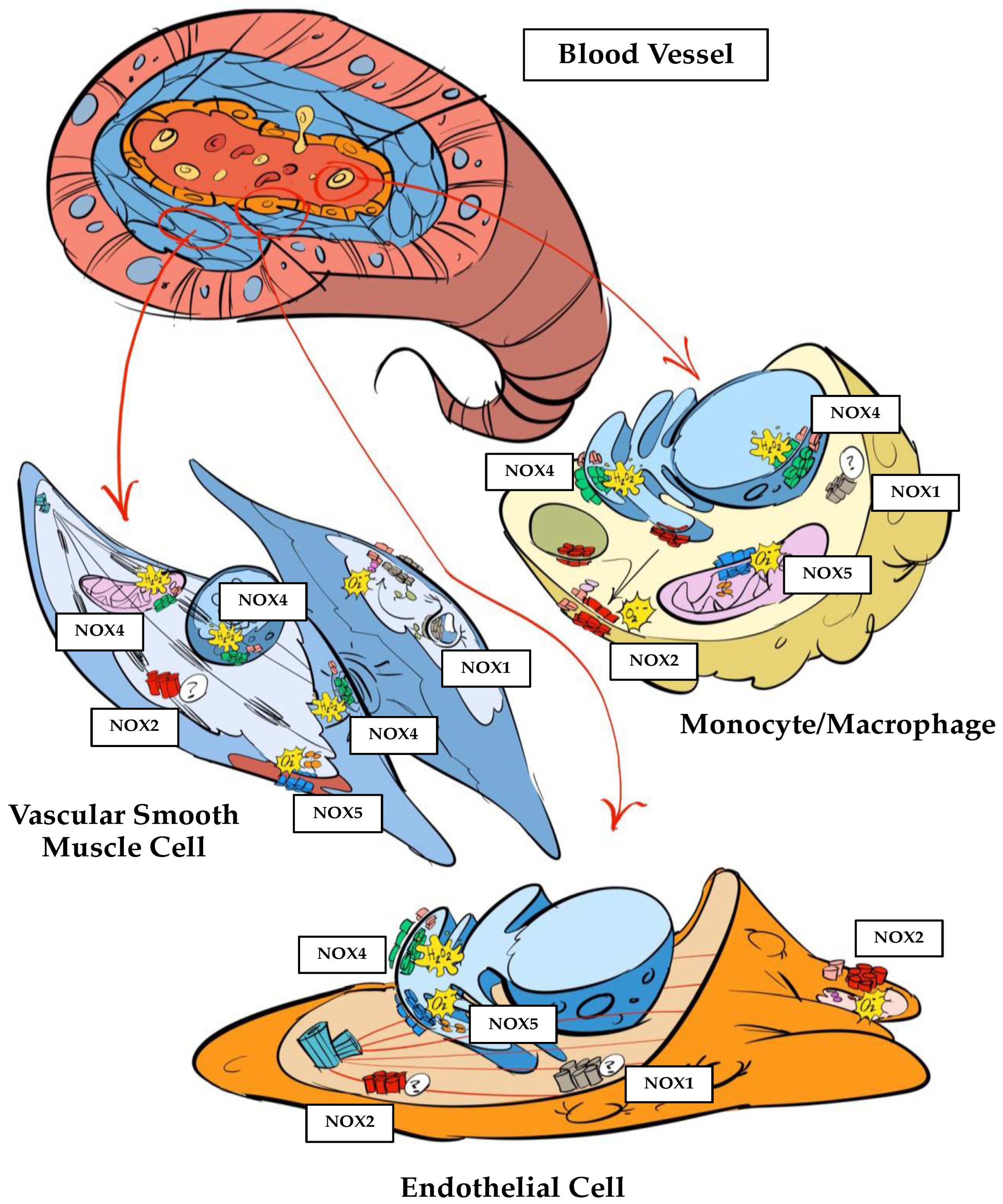
Figure 2.
NOX overactivation leads to endothelial cell dysfunction and inflammation. Humoral factors related with atherosclerosis (Ang II, ET-1, oxLDL) increase NOX activity. The overactivation of every NOX leads to apoptosis. The activation of NOX2, NOX4 and NOX5 by TLRs ends in cytokines secretion. NOX4 and NOX5 alter PGs signaling, inflammatory mediators that participate in atherosclerosis. Ang II: angiotensin II. ET-1: endothelin 1. GM-CSF: granulocyte-monocyte colony stimulating factor. MCP-1: monocyte chemoattractant protein 1. oxLDL: oxidized LDL. PGE2: prostaglandin E2. PGI2: prostaglandin I2. TLR: toll-like receptor.
Figure 2.
NOX overactivation leads to endothelial cell dysfunction and inflammation. Humoral factors related with atherosclerosis (Ang II, ET-1, oxLDL) increase NOX activity. The overactivation of every NOX leads to apoptosis. The activation of NOX2, NOX4 and NOX5 by TLRs ends in cytokines secretion. NOX4 and NOX5 alter PGs signaling, inflammatory mediators that participate in atherosclerosis. Ang II: angiotensin II. ET-1: endothelin 1. GM-CSF: granulocyte-monocyte colony stimulating factor. MCP-1: monocyte chemoattractant protein 1. oxLDL: oxidized LDL. PGE2: prostaglandin E2. PGI2: prostaglandin I2. TLR: toll-like receptor.
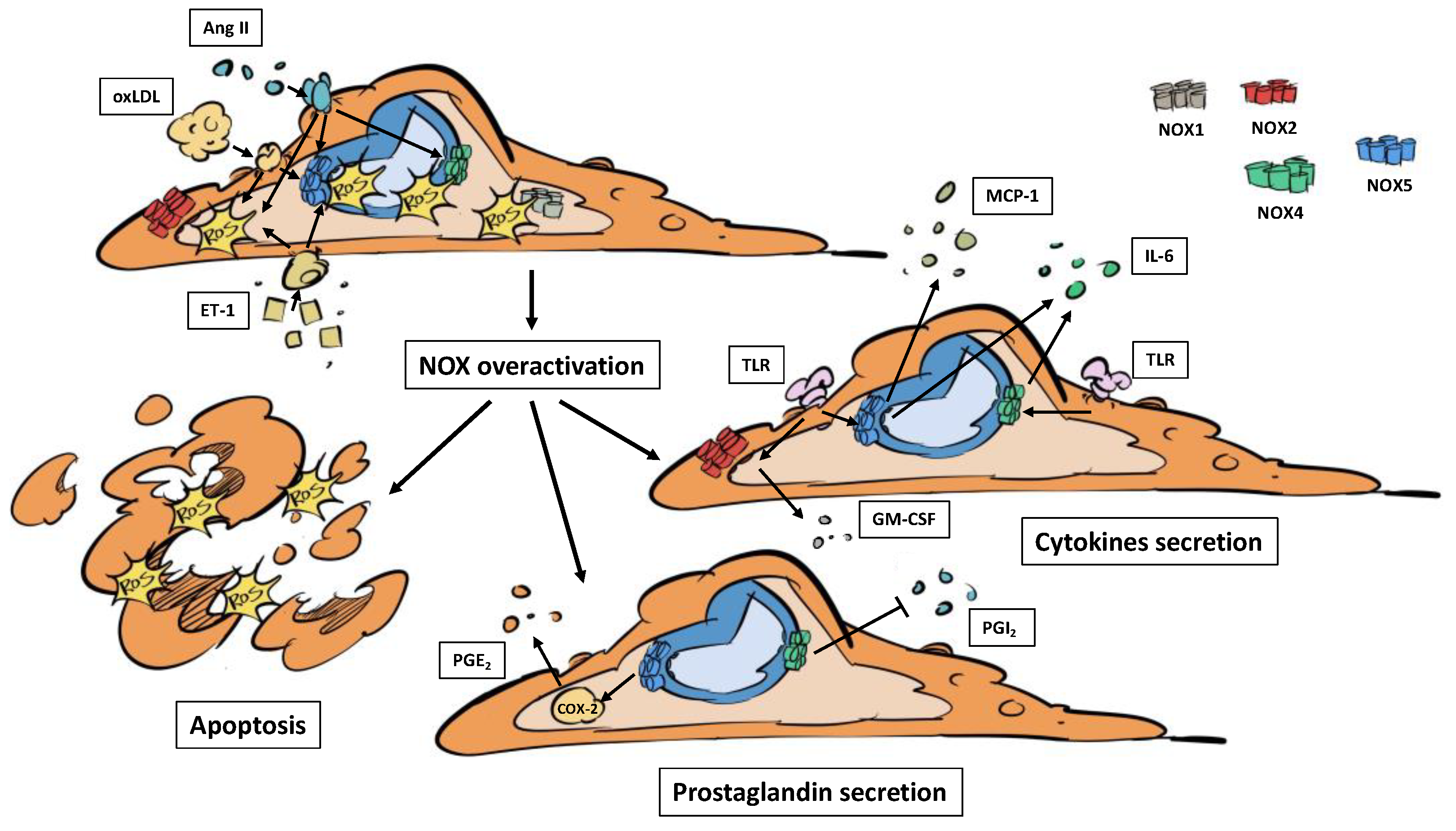
Figure 3.
The different NOX homologues play crucial roles in the immune cell infiltration into the vascular wall and the genesis of foam cells. NOX2, NOX4 and NOX5 increase ICAM-1 and VCAM-1 expression in endothelial cells, promoting monocyte adhesion. By contrast, NOX4 inhibits the infiltration of monocytes. NOX2 promotes the apoptosis of macrophages which aggravates the immune cell infiltration by inflammatory signals. NOX1 mediates the transformation of macrophages towards foam cells. NOX2 activation leads to alterations in ECM composition, cell migration and lipid accumulation in VSMC. NOX1, NOX4 and NOX5 also participate in VSMC migration. ECM: extracellular matrix. ICAM-1: intracellular adhesion molecule 1. LPS: lipopolysaccharide. TNF-α: tumoral necrosis factor α. VCAM-1: vascular cell adhesion molecule 1.
Figure 3.
The different NOX homologues play crucial roles in the immune cell infiltration into the vascular wall and the genesis of foam cells. NOX2, NOX4 and NOX5 increase ICAM-1 and VCAM-1 expression in endothelial cells, promoting monocyte adhesion. By contrast, NOX4 inhibits the infiltration of monocytes. NOX2 promotes the apoptosis of macrophages which aggravates the immune cell infiltration by inflammatory signals. NOX1 mediates the transformation of macrophages towards foam cells. NOX2 activation leads to alterations in ECM composition, cell migration and lipid accumulation in VSMC. NOX1, NOX4 and NOX5 also participate in VSMC migration. ECM: extracellular matrix. ICAM-1: intracellular adhesion molecule 1. LPS: lipopolysaccharide. TNF-α: tumoral necrosis factor α. VCAM-1: vascular cell adhesion molecule 1.
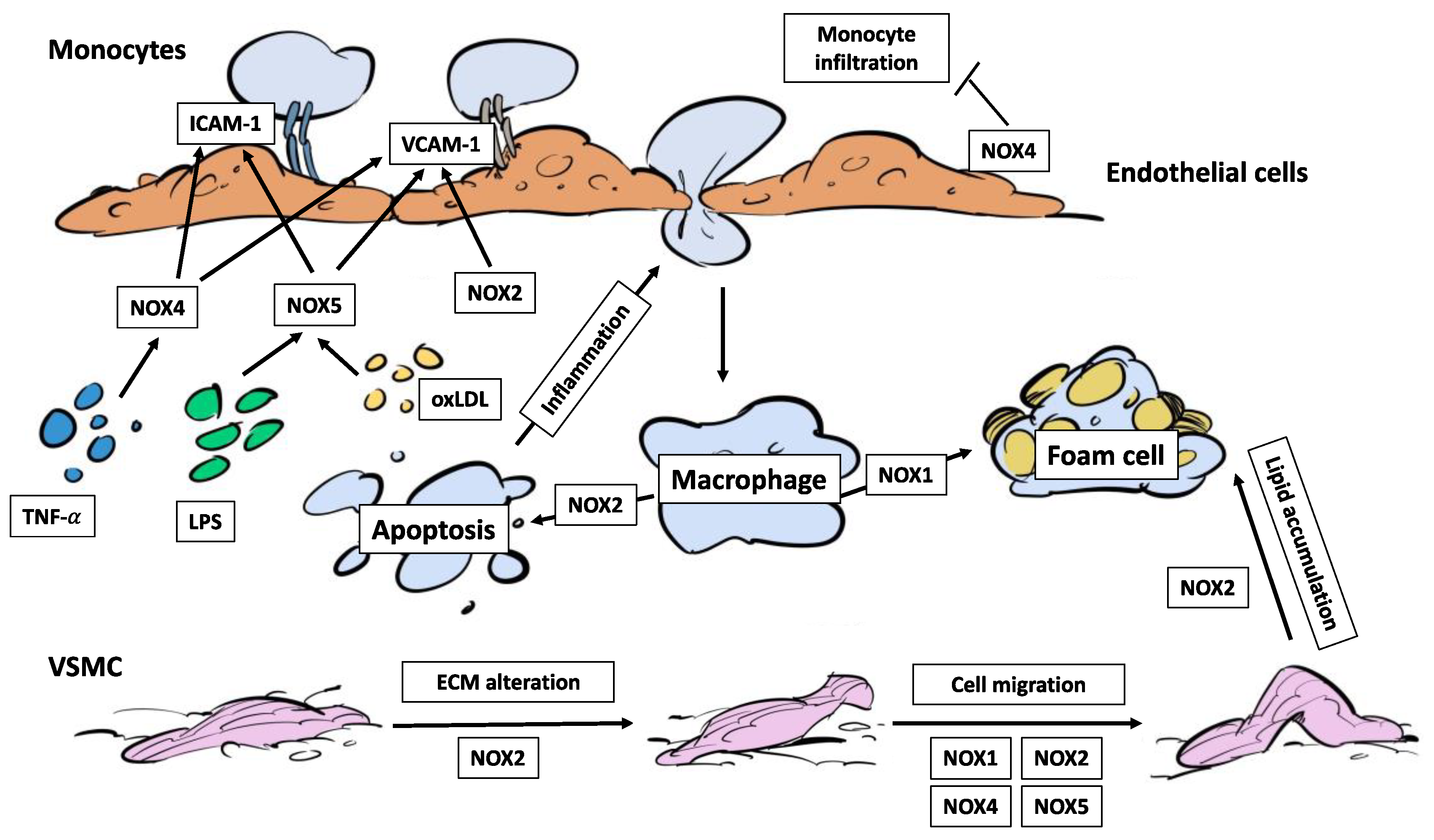
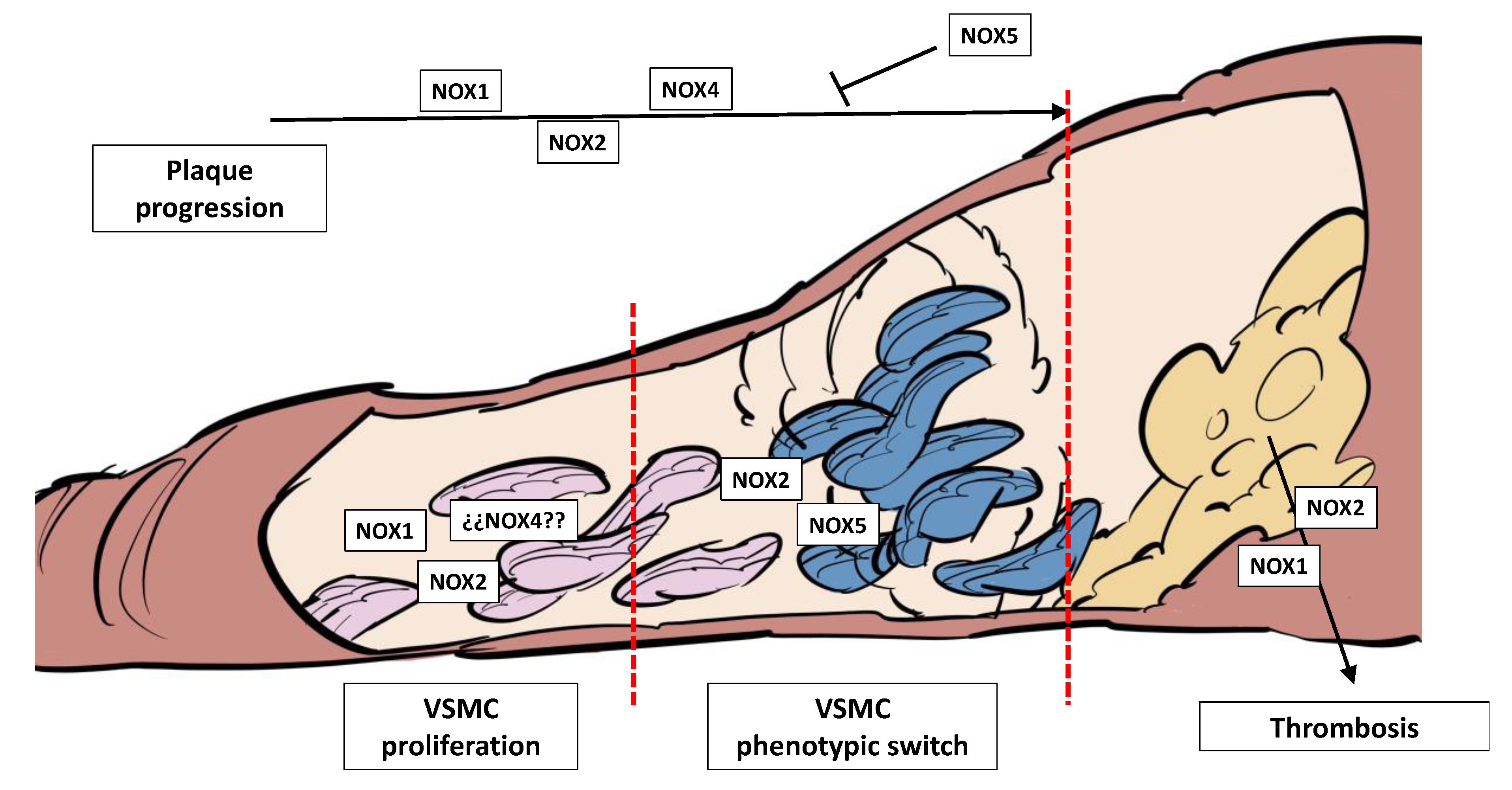
Figure 4.
The NOX homologues play crucial roles in atherosclerotic plaque growth and thrombosis. NOX1 and NOX2 increase VSMC proliferation, while the role of NOX4 in this step is controversial. NOX2 and NOX5 promote VSMC switch toward a synthetic phenotype and Ca++ accumulation. NOX1, NOX2 and NOX4 promote plaque progression in vivo, while NOX5 inhibits this process in rabbits. NOX1 and NOX2 are prothrombotic, while NOX4 has no effect in this process.
Figure 4.
The NOX homologues play crucial roles in atherosclerotic plaque growth and thrombosis. NOX1 and NOX2 increase VSMC proliferation, while the role of NOX4 in this step is controversial. NOX2 and NOX5 promote VSMC switch toward a synthetic phenotype and Ca++ accumulation. NOX1, NOX2 and NOX4 promote plaque progression in vivo, while NOX5 inhibits this process in rabbits. NOX1 and NOX2 are prothrombotic, while NOX4 has no effect in this process.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



