
Submitted:
29 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
The reaction mechanisms of C-S borylation of aryl sulfides catalyzed by the 1,4-benzoquinone (BQ), were investigated by employing M06-2X-D3/ma-def2SVP method and basis set. In this study, the SMD model was taken to simulate the solvent effect of 1,4-dioxane. Also, TD-DFT calculations of BQ and methyl(p-tolyl)sulfane were performed in SMD solvent model. The computational results indicated that BQ and methyl(p-tolyl)sulfane, serving as a photo-catalyst, would be excited under blue LED of 450 nm, aligning well with experimental observations. Additionally, the role of 3O2 was investigated, revealing that it could be activated to 1O2 from the released energy of 1[BQ+methyl(p-tolyl)sulfane]* or 3[BQ+ methyl(p-tolyl)sulfane]*→BQ+ methyl(p-tolyl)sulfane process. Then 1O2, bis(pinacolato)diboron, and methyl(p-tolyl)sulfane would through a series of reactions to yield the final product P. The Gibbs free energy surface show that path a2-2 is optimal and this path has less steps and lower energy barrier. The electron spin density isosurface graphs were employed to analyze structures and elucidate the single electron distribution. These computational results offer valuable insights into the studied interactions and related processes and shed light on the mechanisms governing C–S borylation from aryl sulfides and b2pin2 catalyzed by BQ and methyl(p-tolyl)sulfane.
Keywords:
C-S borlation
; DFT
; 1
; 4-benzoquinone
; Bis(pinacolato)diboron
; methyl(p-tolyl)sulfane
1. Introduction
Over the past few decades, C–S bond cleavage and transformation of aryl sulfides have received considerable attention and have been rapidly developed[1,2]. Generally, the C–S bond activation of aryl sulfides could be achieved by guiding group-assisted or directing group-free transition metal-catalysis[3,4,5,6]. While the formed aromatic C–B bonds are a series of important complexes. Especially, arylboric acid has been widely used as a sensor of sugars in materials science and medical research, an inhibitor of enzymes, and a carrier of nucleosides and sugars in biology[7,8,9,10,11]. Hence, great efforts have been devoted to the formation of aromatic C–B bonds in organic chemistry[12,13,14].
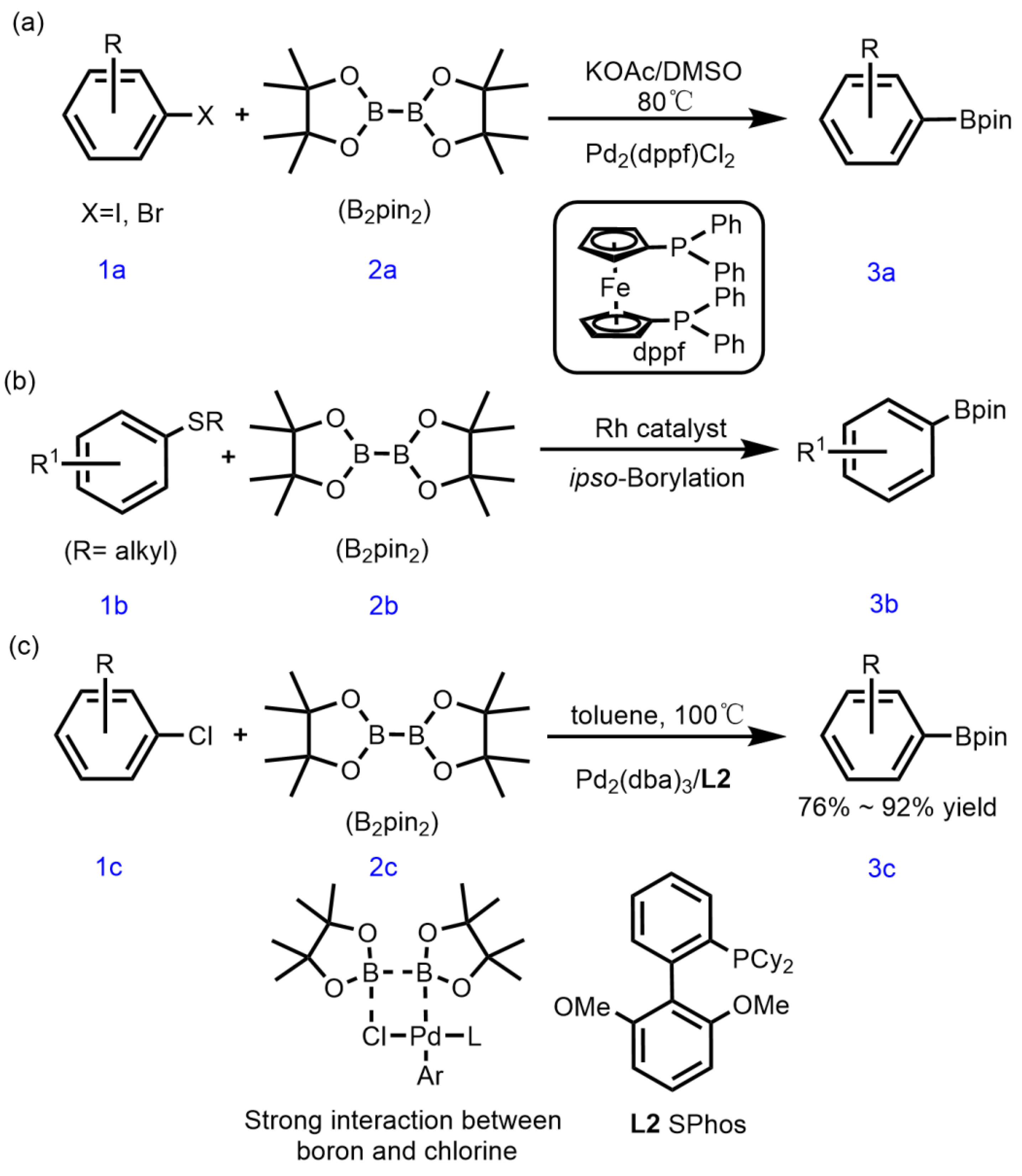
Traditionally, the Miyaura borylation reaction can be used to achieve the various C–heteroatom borylations by the corresponding aromatic electrophiles including aryl sulfides. For example, Miyaura used a palladium-catalyzed coupling reaction between halogenated aromatics and biborate pinacol ester to produce corresponding organic borates[15,16,17]; Yorimitsu[18] and Hosoya[19] finished the C–S borylation of aryl sulfides via Pd- and Rh-catalysis(Scheme 1a and 1b); Yorimitsu[20] and Gao[21] realized the C–S borylation of aryl sulfonium salts via Pd-catalysis(Scheme 1c) and UV irradiation. Unfortunately, most synthetic methods for aryl boronate esters have various disadvantages, including the need for noble metals as catalysts, complex synthetic steps, and low yields[22,23,24].
In recent years, amidst the surge in popularity of green chemistry[25,26,27], considerable attention has been directed towards identifying synthesis methods that are eco-friendly, cost-effective and highly efficient. Under the irradiation of blue LED, Xinqi Li et al. proposed a photocatalytic study that directs C–S bond activation of aryl sulfides via photoinduced aerobic borylation under transition-metal-free conditions in 2022 [28]. This synthetic methodology offers enhanced convenience, cleanliness, and efficacy, thereby bearing significant relevance in chemical research and practical applications. A thorough investigation of its reaction mechanism can undoubtedly facilitate experimenter comprehension and enable the design of similar reactions.
2. Computational Details
All the calculations were performed in Gaussian 09 program package[29]. Every structure was optimized at the M06-2X-D3/ma-def2-SVP[30,31,32] level, and the frequency analysis confirmed that every transition state with only one imaginary frequency and the other structures with no imaginary frequency are correct. The SMD model[33,34]was employed to simulate the solvent effect of 1,4-dioxane, which is defined by the static dielectric constant (eps = 2.21) and the dynamic dielectric constant (epsinf = 1.90). The model has been widely used in the process of investigating the mechanisms of organic chemistry[35,36,37,38,39,40,41,42,43]. Additionally, hole-electron isosurface map, Spin density isosurface maps, frontier molecular orbital (FMO), TD-DFT[44,45] and conceptual density functional theory (CDFT) data were obtained using the Multiwfn program (version 3.3.8)[46]. FMO was drawn using VMD program 1.9.3[47]. All optimized three-dimensional (3D) structures were displayed in CYLview (version 1.0b)[48].
3. Results and Discussion
According to the reference[28], under the irradiation of blue LED, the reactants methyl(p-tolyl)sulfane (R1) and bis(pinacolato)diboron (R2) would go through C-S borylation reaction to yield the final product methyl boronic acid pinacol ester (P) in the solvent of 1,4-dioxane, in which 1,4-benzoquinone (BQ) as the photosensitizer, as depicted in Scheme 2.
3.1. Photocatalysis Process
As we all know, the circular conjugated large π-structure is the preferred site for photo-catalytic reaction. The structures of BQ and R1 have the conjugated π-bond, and BQ is usually used as the photosensitizer. Hence, there are two models designed to investigate the photo-catalytic process. When BQ was employed to explore the excitation in first model, the computational results suggest that S0→S1 of BQ in Table 1 requires to absorb 2.7539 eV energy from the wavelength of 450.22 nm, which accords with the experimental results (450 nm) within error, and the S0 →S1 transition mainly depends on HOMO→LUMO (91.62%). Moreover, the Frontier Molecular Orbital Theory (FMOT) in Figure 1b and 1c shows that this S0→S1 excitation focuses on the intramolecular transfer from σ-orbital of benzene ring to π-orbital of benzene ring, which is consistent with ρele and ρhole in Figure 2a. While the S0→S2 and S0→S3 excitations depend on the wavelengths of 405.16 nm and 286.28 nm, respectively, which mainly come from the HOMO-2→LUMO (89.04%) and HOMO-1→LUMO (99.89%) in Table 1; and they cannot be achieved with blue LED (450 nm). In the second model, BQ and R1 form a complex BQ+R1. The TD-DFT calculations suggest that 449.56 nm wavelength can finish the S0→S1 excitation of BQ+R1, which accords with the experimental results (450 nm) within error, and the S0→S1 transition mainly depends on HOMO→LUMO (57.46 %) and HOMO-3→LUMO (37.46 %). Frontier molecular orbital theory in Figure 1e-g displayed that HOMO→LUMO transition focuses on the intermolecular transfer from R1 to BQ. Meanwhile, the computational results in Table 1 indicate that S0→S2 with 436.44 nm could have the possibilities to accord with the experimental results (450 nm) within error. All above description could suggest that the photo-catalytic reaction would successfully achieve the excitation process of BQ.
3.2. R1+BQ → IM1
As highlighted in the reference[28], BQ, as an organic photocatalyst, is easy to become an excited state in the irradiation of blue LED (450 nm). The computed oscillator strengths (f=0) of BQ model in Section 3.1 indicates it has a little possibility to achieve the photoexcitation process. Hence, the second model is prior. The R1+BQ model would absorb the energy of photons to finish the S0→S1 excitation. The calculations suggest that this photo-excited process requires 51.52 kcal/mol to become excitation state 1[R1+BQ]*, which is very activate and has two possible paths, as depicted in Figure 3. In the first path, the singlet excited state 1[R1+BQ]* would become the ground state R1+BQ by releasing much energy, which results in the triplet oxygen 3O2 changing into singlet oxygen 1O2. Generally, the structure of triplet excited state 3[R1+BQ]* is much more stable than that of singlet excited state 1[R1+BQ]*. So in the second path, Intersystem Crossing (ISC) would transfer 1[R1+BQ]* into 3[R1+BQ]* and this process releases 8.63 kcal/mol of energy. Meanwhile, there is a possibility that 3[R1+BQ]* and triplet oxygen 3O2 could go through energy transfer to become R1+BQ and singlet oxygen 1O2 via releasing about 5.00 kcal/mol energy. The obtained 1O2, as IM1, is one excited state and very active, which continues to participate the following reaction.
3.3. IM1 → P
As we all know, the formed IM1(1O2) is very active, which could participate the following reactions. Hence, 1O2 reacts with R1 and R2 in paths a1 and a2, respectively. Moreover, the reaction between 3O2 and R1 (or R2) has also been investigated in path a1, elaborating in the following section.
3.3.1. Path a1
According to Figure 4, Gibbs free energy surface suggests that it is a SN2 reaction with the energy barrier of 54.87 kcal/mol, which is very high and cannot happen. Considering the large amount of 3O2, it can also have the possibility to react with R1 or R2 in the system. Figure 4 suggests that R1+3O2→TS1a1-2→IM2a1-1+IM3a1-1 (blue line) process has an energy barrier of 53.12 kcal/mol and cannot also happen in path a1-2. Finally, the reaction between 3O2 and R2 has a relative lower energy barrier than those in paths a1-1 and a1-2, 42.32 kcal/mol is still a higher barrier and it cannot continue to occur.
3.3.2. Path a2
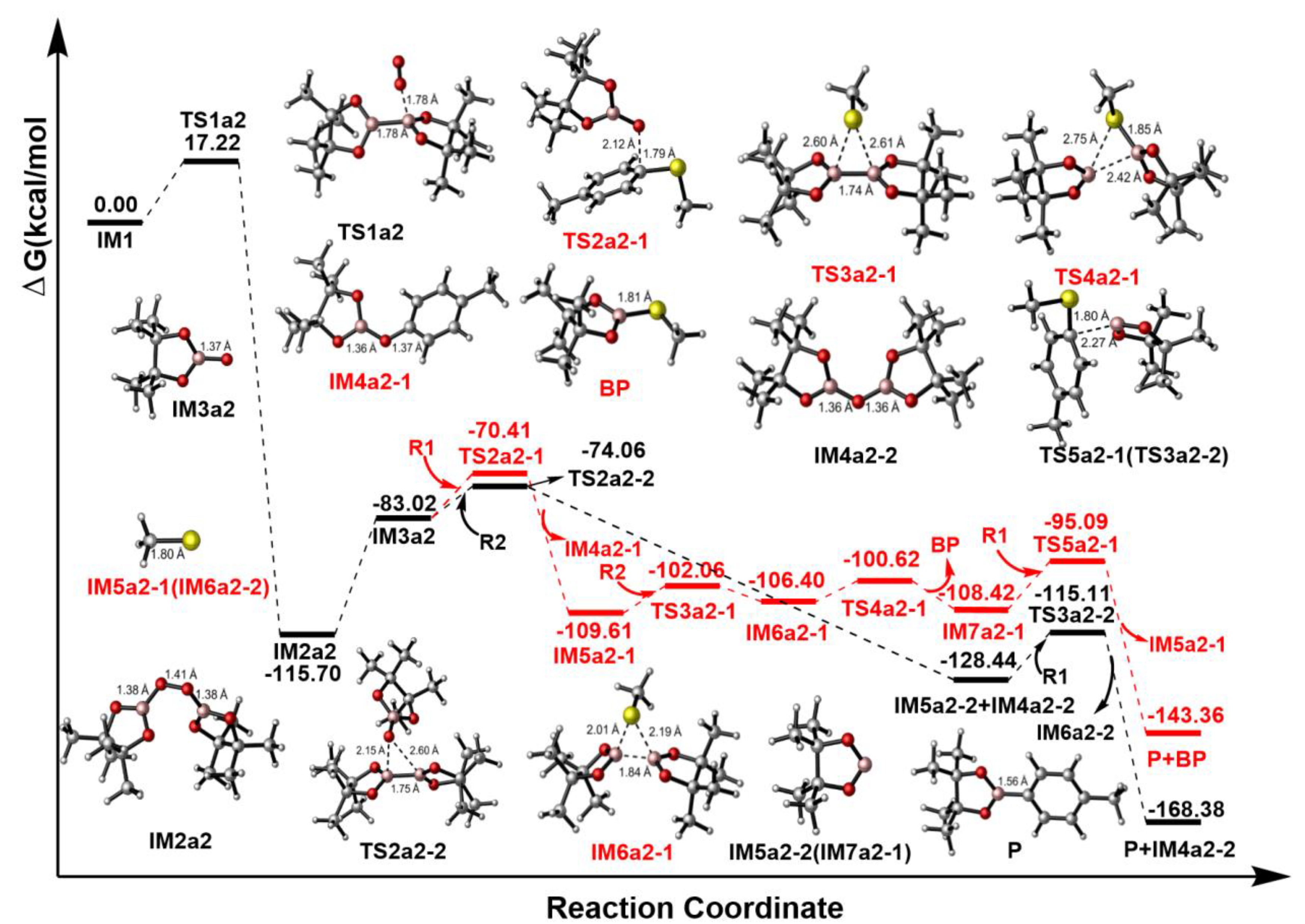
Based on the analysis of Fukui function and dual descriptor, B5(R2) atom with the positive value (=0.0746) in Table 2 is susceptible to nucleophilic attack. In Path a2 of Figure 6, B5 atom of R2 reacts with O1(IM1) atom via transition state TS1a2, which has an energy barrier of 17.22 kcal/mol and the distance of O1(IM1)•••B5(R1) in TS1a2 is 1.78 Å. Additionally, the energy of IM2a2 is 115.70 kcal/mol lower than that of IM1+R2, indicating that this step is an exothermic process in Figure 6. Then the following step would convert intermediate IM2a2 into IM3a2 via an intramolecular homolytic reaction by absorbing energy of 32.68 kcal/mol. The electron spin density isosurface graph of IM3a2 in Figure 5d show that the single electron is distributed on O1 atom, which is the reactive site to participate the following reaction. The condensed dual descriptor (CDD) values of C4 atom of R1 and B5 (or B6) atom of R2 are =0.0025 and =0.0746 in Table 2, respectively. Therefore, there are two paths (a2-1 and a2-2) can yield the final product P.
In path a2-1, IM3a2 would react with R2 at the sites O1(IM3a2) atom and C4(R2) atom via transition state TS2a2-1. The computational results displayed that it is a SN2 reaction and the distances of O1(IM3a2)•••C4(R2) and C4(R2)•••S3(R2) are 2.12 Å and 1.97 Å in TS2a2-1, respectively. Moreover, this IM3a2+R1→IM4a2-1+ IM5a2-1 process is an exothermic procedure and 26.59 kcal/mol would be released. The formed IM5a2-1 is a radical and the single electron is distributed in S3 atom in Figure 5c. The next process is an intramolecular ring-closure reaction happened between B5-B6(R2) bond and S3 atom via a three-membered(B5, B6 and S3) transition state TS3a2-1, which needs 7.55 kcal/mol energy and the distances of B5•••S3, B6•••S3 and B5•••B6 is 2.61 Å, 2.60 Å and 1.74 Å, respectively. The generated IM6a2-1 is one three-membered intermediate, and it would go through ring-opening reaction to generate byproduct BP and IM7a2-1 radical via transition state TS4a2-1. It is a fast step and 5.78 kcal/mol is the energy barrier in Figure 6. Furthermore, Figure 5 e, f and j reveal it is one single electron transfer process and the single electron is distributed in B5 atom of IM7a2-1 in Figure 5f. Finally, a SN2 reaction happened between C4(R1) and B5(IM7a2-1) atoms via TS5a2-1, which needs 13.33 kcal/mol energy. The distance of C4(R1)•••B5(IM7a2-1) decreased to 1.56 Å in P from 2.27 Å in TS5a2-1, indicating that the IM7a2-1 intermediate could successfully move to the C4(R1) atom of R1. The energy of obtained IM5a2-1+P was 34.94 kcal/mol lower than that of IM7a2-1+R1, showing that this process is an exothermic reaction in Figure 6. Meanwhile, IM5a2-1 can continue to also attack R2 reaction to realize recycling.
The B5 or B6 atom of R2 is an electron-deficient structure. So, in Path a2-2, IM3a2 with one single electron can also attack the B5 or B6 atom of R2 to complete the SN2 reaction. The calculations show that this IM3a2+R2→IM5a2-2 process with the energy barrier of 8.96 kcal/mol via TS2a2-2 is an exothermic procedure (45.42 kcal/mol) in Figure 6. Meanwhile, the B5–B6 bond of R2 is broken and an intermediate IM4a2-2 is formed. The structure of IM5a2-2 is as the same as IM7a2-1 in path a2-1, so the final reaction of IM5a2-2+R1→P+IM6a2-2 process is as the same as IM7a2-1+R1→P+IM5a2-1 procedure in path a2-1.
4. Conclusions
In summary, this paper systematically investigated the detailed formation mechanisms of methyl boronic acid pinacol ester (P) through the photoinduced aerobic borylation reaction between methyl(p-tolyl)sulfane (R1) and bis(pinacolato)diboron (R2). The computations were performed at the M06-2X-D3/ma-def2SVP level, with SMD model to simulate the solvent effect of 1,4-dioxane. Additionally, the photo-catalytic mechanisms of BQ+R1 model were explored using TDDFT method at the M06-2X-D3/ma-def2-SVP level. The computational results revealed that R1+BQ is preferentially excited into excited state 1[R1+BQ]*, which has the possibility to change into 3[R1+BQ]* via intersystem crossing (ISC). Hence, there are two possibilities existed can change 3O2 into 1O2 by energy transfer process from 1[R1+BQ]* and 3[R1+BQ]*, respectively. Subsequently, two paths (a2-1 and a2-2) were proposed to convert R2 into product P via a series of reactions. The Gibbs free energy surfaces path a2-2 was identified as the optimal route with lower energy barriers, and the electron spin density isosurface graphs can reveal that it is a free radical transfer process. The clear mechanisms offer valuable insights for the design of similar reactions and showcase the potential of computational methods in advancing catalysis research.
Funding
This work was supported by the Natural Science Foundation of Sichuan Province [grant number 2022NSFSC0629]
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supporting information
The other paths to yield the product P; the cartesian coordinates of all the transition states and the energies of all structures.
References
- Lou, J.; Wang, Q.; Wu, P.; Wang, H.; Zhou, Y.-G.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations: an update. Chem. Soc. Rev. 2020, 49, 4307–4359. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-L.; Zhu, Q.-L.; Dong, Z.-B.; Chen, J.-Q.; Shi, Z. Synthesis of Aryl Dithiocarbamates from Tetramethylthiuram Monosulfide (TMTM) and Aryl Boronic Acids: Copper-Catalyzed Construction of C(sp2)–S Bonds. Synthesis 2021, 54, 475–482. [Google Scholar] [CrossRef]
- Delcaillau, T.; Boehm, P.; Morandi, B. Nickel-Catalyzed Reversible Functional Group Metathesis between Aryl Nitriles and Aryl Thioethers. Journal of the American Chemical Society 2021, 143, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Boehm, P.; Müller, P.; Finkelstein, P.; Rivero-Crespo, M.A.; Ebert, M.-O.; Trapp, N.; Morandi, B. Mechanistic Investigation of the Nickel-Catalyzed Metathesis between Aryl Thioethers and Aryl Nitriles. J. Am. Chem. Soc. 2022, 144, 13096–13108. [Google Scholar] [CrossRef] [PubMed]
- Delcaillau, T.; Woenckhaus-Alvarez, A.; Morandi, B. Nickel-Catalyzed Cyanation of Aryl Thioethers. Org. Lett. 2021, 23, 7018–7022. [Google Scholar] [CrossRef] [PubMed]
- Delcaillau, T.; Morandi, B. Nickel-Catalyzed Thiolation of Aryl Nitriles. Chemistry-a European Journal 2021, 27, 11823–11826. [Google Scholar] [CrossRef]
- Yorimitsu, H. Catalytic Transformations of Sulfonium Salts via C-S Bond Activation. Chem. Rec. 2021, 21, 3356–3369. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Tan, C.; Liu, Y.; Wei, Y.; Zhao, X.; Liu, X.; Tan, J.; Yoshida, H. A leap forward in sulfonium salt and sulfur ylide chemistry. Chin. Chem. Lett. 2020, 32, 299–312. [Google Scholar] [CrossRef]
- Ma, N.-N.; Ren, J.-A.; Liu, X.; Chu, X.-Q.; Rao, W.; Shen, Z.-L. Nickel-Catalyzed Direct Cross-Coupling of Aryl Sulfonium Salt with Aryl Bromide. Org. Lett. 2022, 24, 1953–1957. [Google Scholar] [CrossRef]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2013, 57, 2832–2842. [Google Scholar] [CrossRef]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Pattison, G. Fluorination of organoboron compounds. Org. Biomol. Chem. 2019, 17, 5651–5660. [Google Scholar] [CrossRef]
- Dhital, R.N.; Sakurai, H. Oxidative Coupling of Organoboron Compounds. Asian J. Org. Chem. 2014, 3, 668–684. [Google Scholar] [CrossRef]
- Wang, J. When diazo compounds meet with organoboron compounds. Pure Appl. Chem. 2018, 90, 617–623. [Google Scholar] [CrossRef]
- Wang, M.; Shi, Z. Methodologies and Strategies for Selective Borylation of C–Het and C–C Bonds. Chem. Rev. 2020, 120, 7348–7398. [Google Scholar] [CrossRef]
- Rout, L.; Punniyamurthy, T. Recent advances in transition-metal-mediated Csp2-B and Csp2-P cross-coupling reactions. Co-ord. Chem. Rev. 2020, 431, 213675. [Google Scholar] [CrossRef]
- Gao, M.-Y.; Gosmini, C. Cobalt-Catalyzed Reductive Cross-Coupling To Construct Csp3–Csp3 Bonds via Csp3–S and Csp3–X Bonds Activation. Organic Letters 2023, 25, 7689–7693. [Google Scholar] [CrossRef]
- Bhanuchandra, M.; Baralle, A.; Otsuka, S.; Nogi, K.; Yorimitsu, H.; Osuka, A. Palladium-Catalyzed ipso-Borylation of Aryl Sulfides with Diborons. Org. Lett. 2016, 18, 2966–2969. [Google Scholar] [CrossRef]
- Uetake, Y.; Niwa, T.; Hosoya, T. Rhodium-Catalyzed ipso-Borylation of Alkylthioarenes via C–S Bond Cleavage. Organic Letters 2016, 18, 2758–2761. [Google Scholar] [CrossRef]
- Minami, H.; Otsuka, S.; Nogi, K.; Yorimitsu, H. Palladium-Catalyzed Borylation of Aryl Sulfoniums with Diborons. ACS Catal. 2017, 8, 579–583. [Google Scholar] [CrossRef]
- Huang, C.; Feng, J.; Ma, R.; Fang, S.; Lu, T.; Tang, W.; Du, D.; Gao, J. Redox-Neutral Borylation of Aryl Sulfonium Salts via C–S Activation Enabled by Light. Org. Lett. 2019, 21, 9688–9692. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, H. Research Progress towards Synthesis of Aryl Boronic Acid Compounds. Chin. J. Org. Chem. 2018, 38, 738–751. [Google Scholar] [CrossRef]
- Fang, H.-P.; Fu, C.-C.; Tai, C.-K.; Chang, K.-H.; Yang, R.-H.; Wu, M.-J.; Chen, H.-C.; Li, C.-J.; Huang, S.-Q.; Lien, W.-H.; et al. Synthesis and stability study of isocyano aryl boronate esters and their synthetic applications. RSC Adv. 2016, 6, 30362–30371. [Google Scholar] [CrossRef]
- Zhou, J.; Berthel, J.H.J.; Kuntze-Fechner, M.W.; Friedrich, A.; Marder, T.B.; Radius, U. NHC Nickel-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions of Aryl Boronate Esters with Perfluorobenzenes. J. Org. Chem. 2016, 81, 5789–5794. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.K. Green Chemistry: An Introductory Text, 3rd edition. Green Chem. Lett. Rev. 2017, 10, 30–31. [Google Scholar] [CrossRef]
- Al-Shatti, B.J.; Alsairafi, Z.; Al-Tannak, N.F. Green chemistry and its implementation in pharmaceutical analysis. Rev. Anal. Chem. 2023, 42. [Google Scholar] [CrossRef]
- Kar, S.; Sanderson, H.; Roy, K.; Benfenati, E.; Leszczynski, J. Green Chemistry in the Synthesis of Pharmaceuticals. Chem. Rev. 2021, 122, 3637–3710. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wan, Z.; Hu, X.; Zhang, H. Photoinduced aerobic C–S borylation of aryl sulfides. Org. Chem. Front. 2022, 9, 3034–3038. [Google Scholar] [CrossRef]
- M.J. Frisch, G.W.T., H.B. Schlegel, et al., , Gaussian 09, R. E. 01. Gaussian, Inc. Wallingford CT, 2013.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Saheb, V.; Bahadori, A. Theoretical studies on the kinetics of the hydrogen-abstraction reactions from 1,3,5-trioxane and 1,4-dioxane by OH radicals. Prog. React. Kinet. Mech. 2020, 45, 1–13. [Google Scholar] [CrossRef]
- Cheng, X. Computational insights into the coupling mechanism of benzoic acid, phenoxy acetylene and dihydroisoquinoline catalyzed by silver ion as polarizer and stabilizer. Appl. Organomet. Chem. 2020, 34, e5903. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Feng, T.-T.; Lin, Y.; Chen, B.; Zhou, D.-G.; Li, R. Alkali metal hydroxide-catalyzed mechanisms of Csp–H silylation of alkynes: a DFT investigation. Org. Biomol. Chem. 2024. [Google Scholar] [CrossRef]
- Zhou, D.-G.; Wang, P. Mechanisms of the reaction between benzonitrile and 4-octyne catalyzed by Ni(PMe3)2: A theoretical investigation. Journal of Physical Organic Chemistry 2019, 32, e3932. [Google Scholar] [CrossRef]
- Zhou, D.-G.; Li, Y.-Q. Mechanistic Study of 1,4-Benzodiazepine-2,5-diones from Diphenylamine and Diethyl 2-Phenylmalonate by Density Functional Theory. J. Phys. Chem. A 2019, 124, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-G.; Zhou, P.-P.; Jing, H.-W. Mechanisms of Csp 3 -H functionalization of ethyl 2-(methyl( p -tolyl)amino)acetate: A theoretical investigation. Comput. Theor. Chem. 2017, 1118, 144–152. [Google Scholar] [CrossRef]
- Zhou, D.-G. DFT investigation on the mechanism of catalytic reaction between 3-diazoindolin-2-imines and N-ethylaniline catalyzed by Rh2(Oct)4. Chem. Phys. 2019, 531, 110661. [Google Scholar] [CrossRef]
- Zheng, X.-F.; Zhou, D.-G. Mechanisms of asymmetric sulfa-Michael additions between phenylacetylene and thiolacetic acid: A DFT investigation. Comput. Theor. Chem. 2021, 1207, 113523. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Li, S.-J.; Wang, X.; Shi, Q.; Li, X.; Qu, L.-B.; Wei, D.; Lan, Y. Multiple Functional Organocatalyst-Promoted Inert C–C Activation: Mechanism and Origin of Selectivities. ACS Catal. 2021, 11, 3443–3454. [Google Scholar] [CrossRef]
- Zhou, D.-G. Mechanisms of Csp3-H functionalization of acetonitrile or acetone with coumarins: A DFT investigation. Mol. Catal. 2020, 498, 111246. [Google Scholar] [CrossRef]
- Chen, B.; Zhou, D.G.; Yang, L.J. Reaction mechanism of acetonitrile, olefins, and amines catalyzed by Ag2CO3: A DFT investigation. Journal of Physical Organic Chemistry 2023, 37. [Google Scholar] [CrossRef]
- Zheng, X.-F.; Zhou, D.-G.; Yang, L.-J. DFT investigation of the DDQ-catalytic mechanism for constructing C–O bonds. Org. Biomol. Chem. 2024, 22, 3693–3707. [Google Scholar] [CrossRef]
- Budyka, M.F. Density functional theory study of the styrylbenzoquinoline dyad and the related dibenzoquinolylcyclobutane formed in the [2 + 2] photocycloaddition reaction. Int. J. Quantum Chem. 2023, 124. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Legault, C.Y., Universite de Sherbrooke 2009.
Scheme 1.
C–S borylation of aryl sulfides.
Scheme 2.
The total reaction from R1+R2→P.
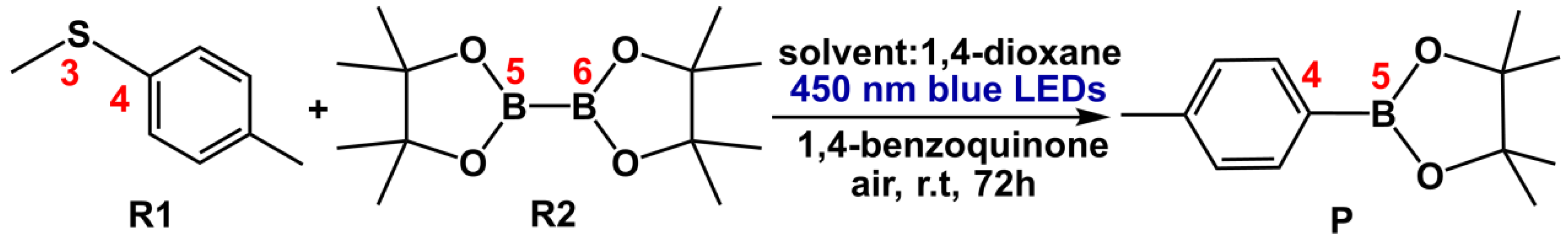
Figure 1.
Frontier molecular orbital of BQ, and R1+BQ models.
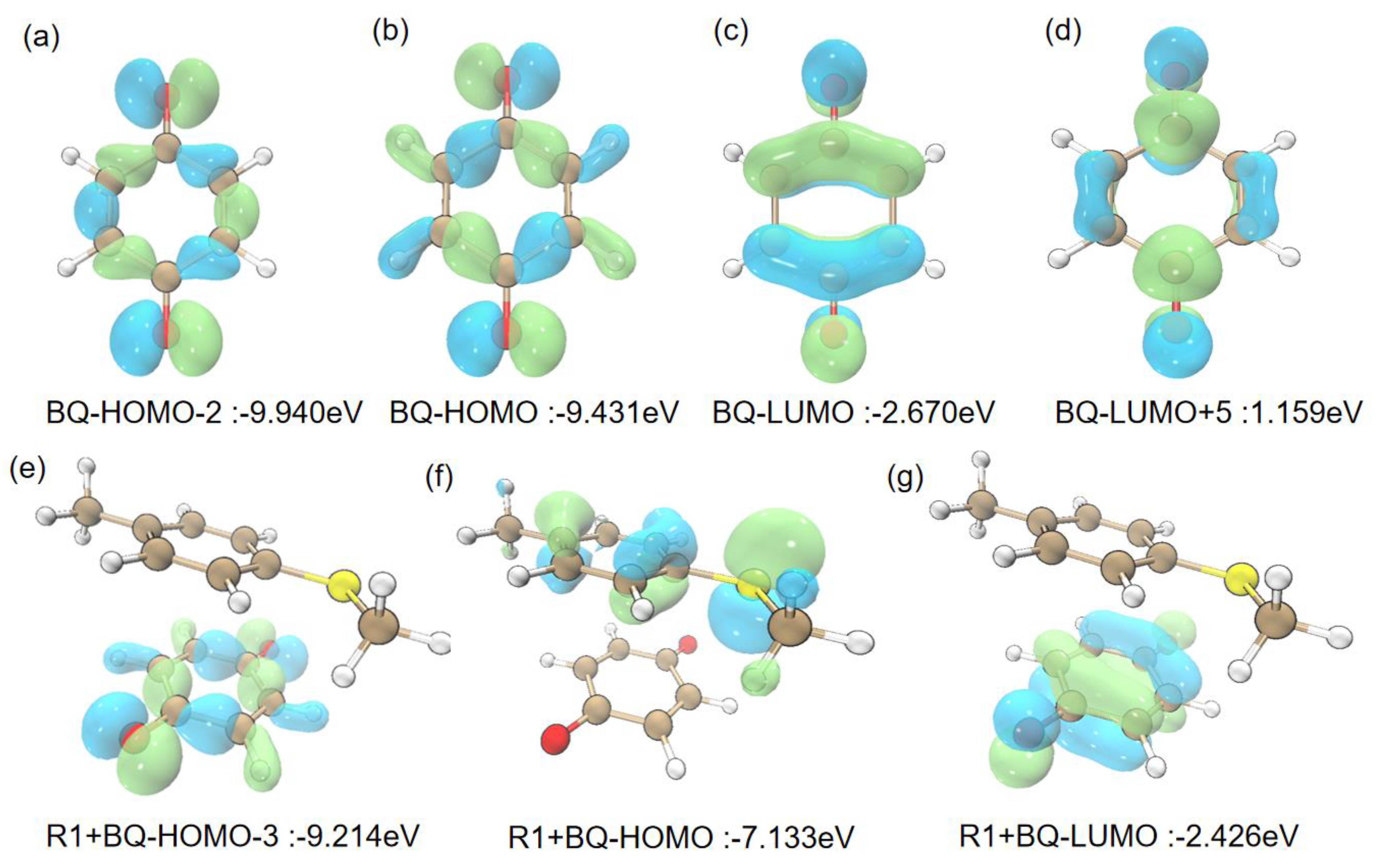
Figure 2.
The ρhole(blue) and ρele(green) of S0 → S1, S0 → S2, and S0 → S3 of R1+BQ.
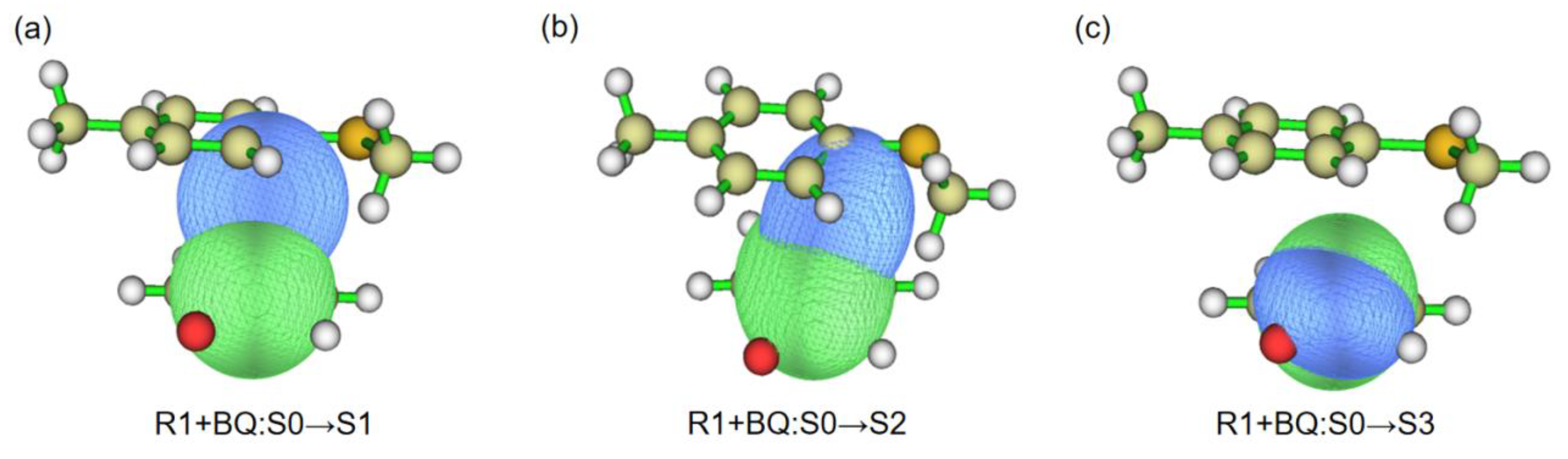
Scheme 3.
The detailed reaction photocatalysis process.
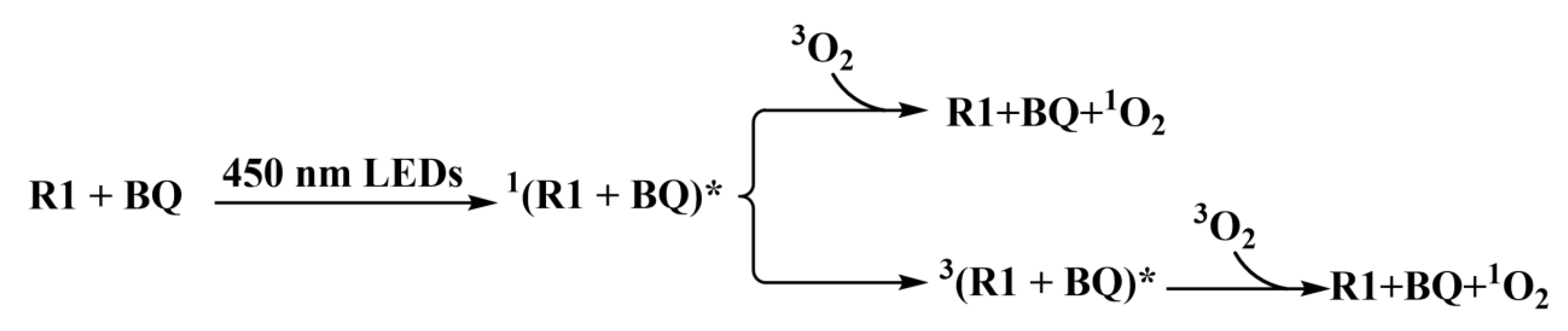
Figure 3.
The Gibbs free energy surfaces in photocatalysis process.
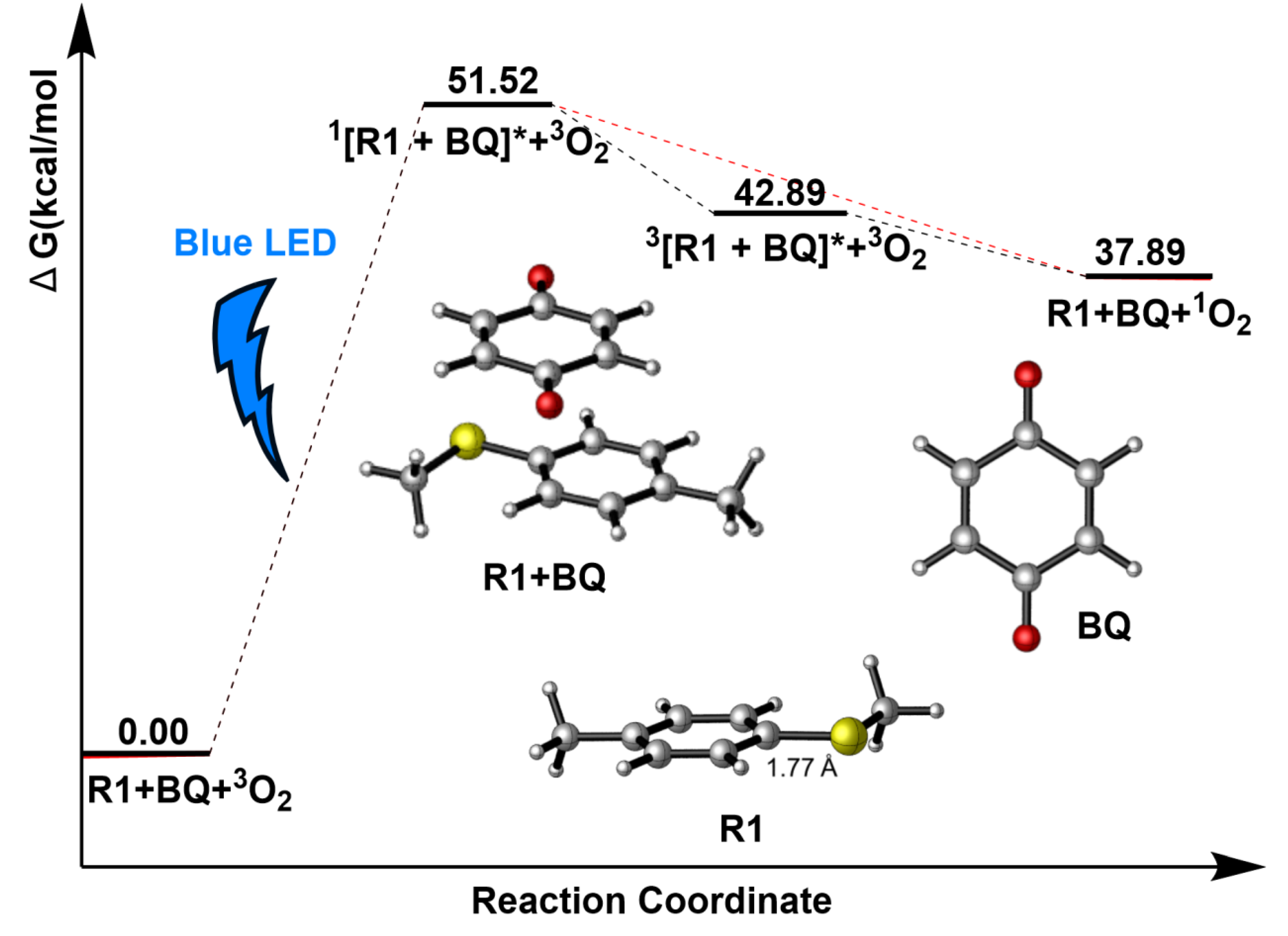
Figure 4.
The Gibbs free energy surfaces of paths a1-1, a1-2 and a1-3.
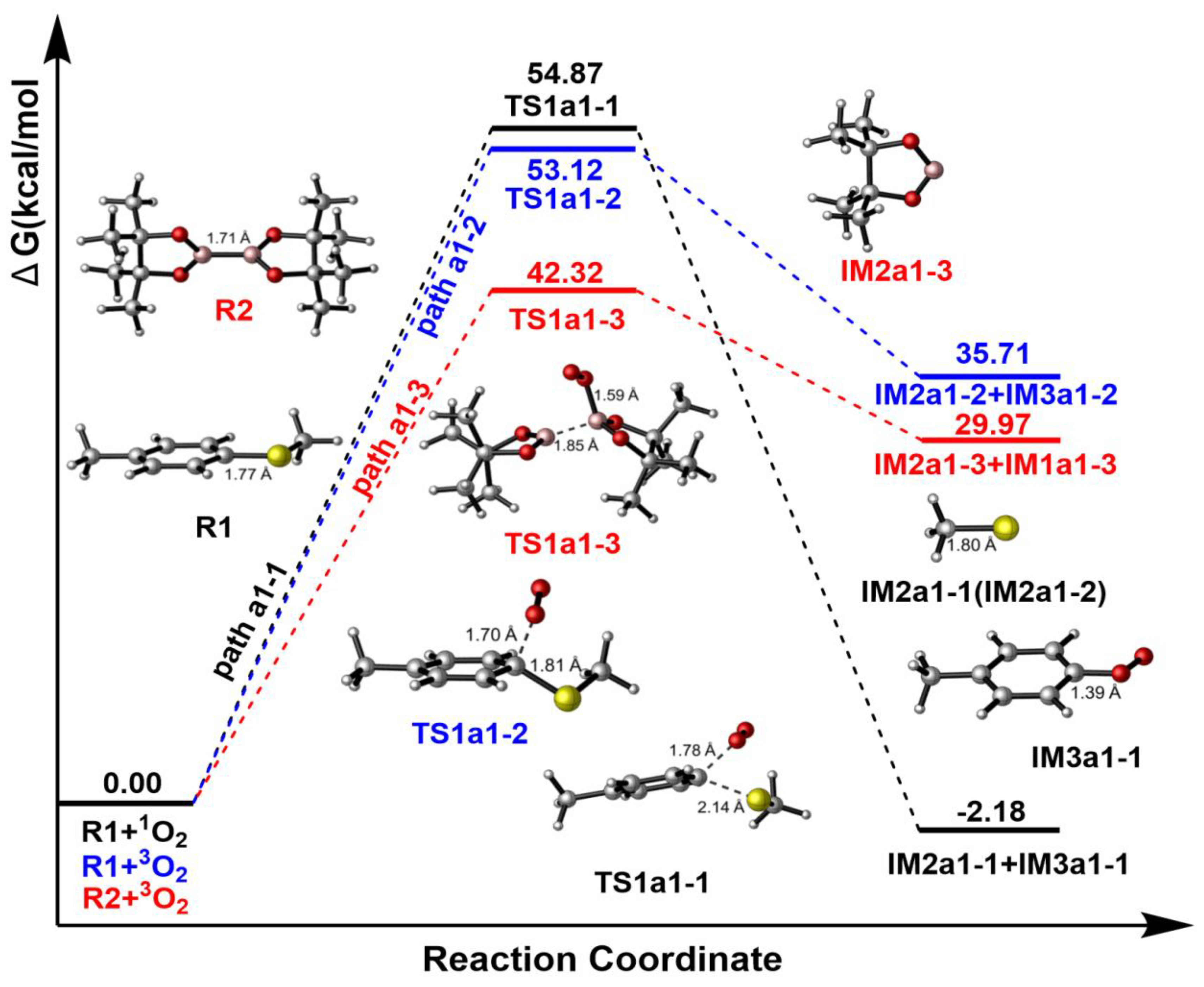
Figure 5.
The electron spin density isosurface graphs of some structures in Path a1 and a2.
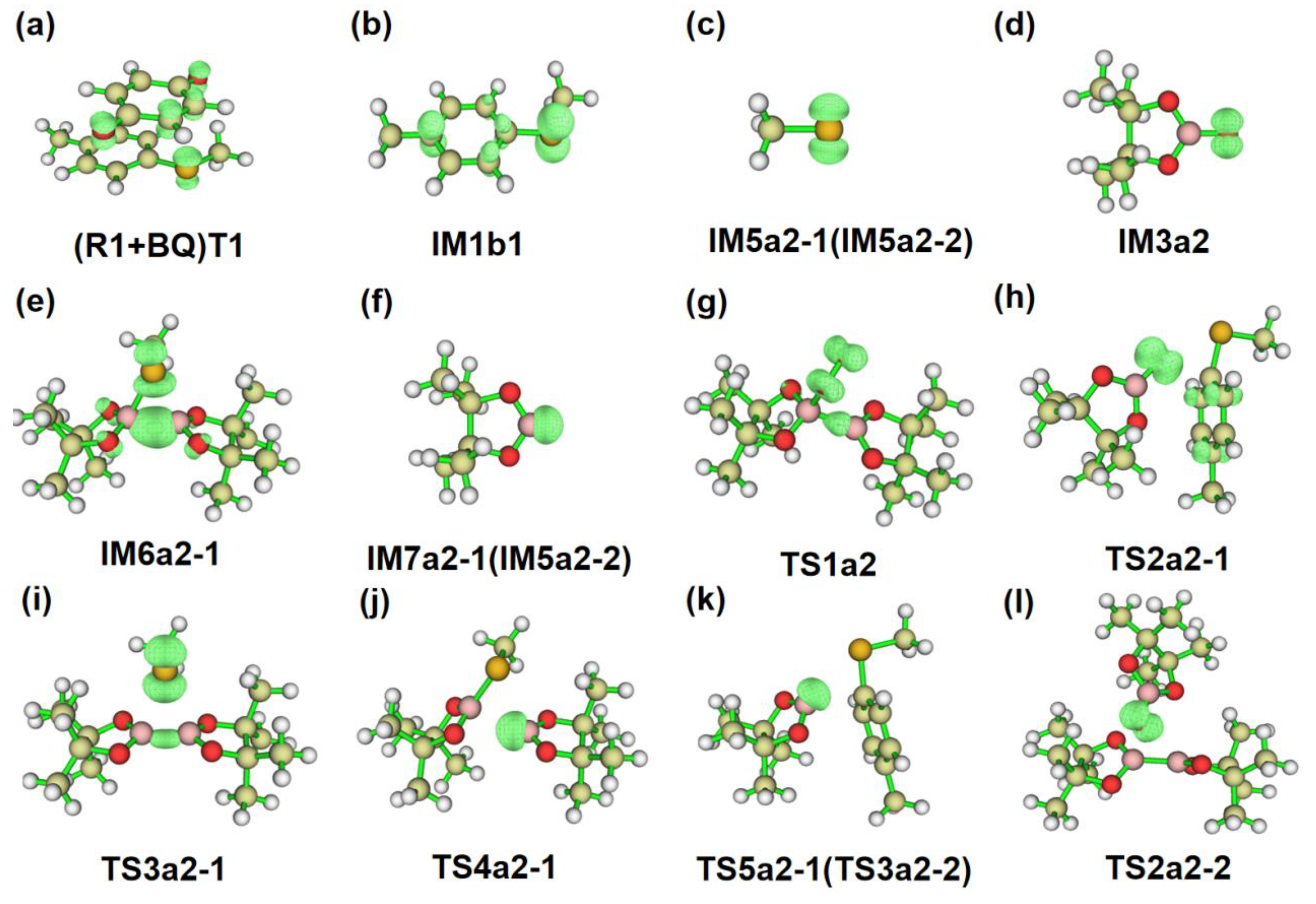
Figure 6.
The Gibbs free energy surfaces from IM1
to P1 via two probable Paths a2-1 and a2-2.
Scheme 4.
The detailed reaction process from IM1 to P via two probable Paths a2-1 and a2-2.
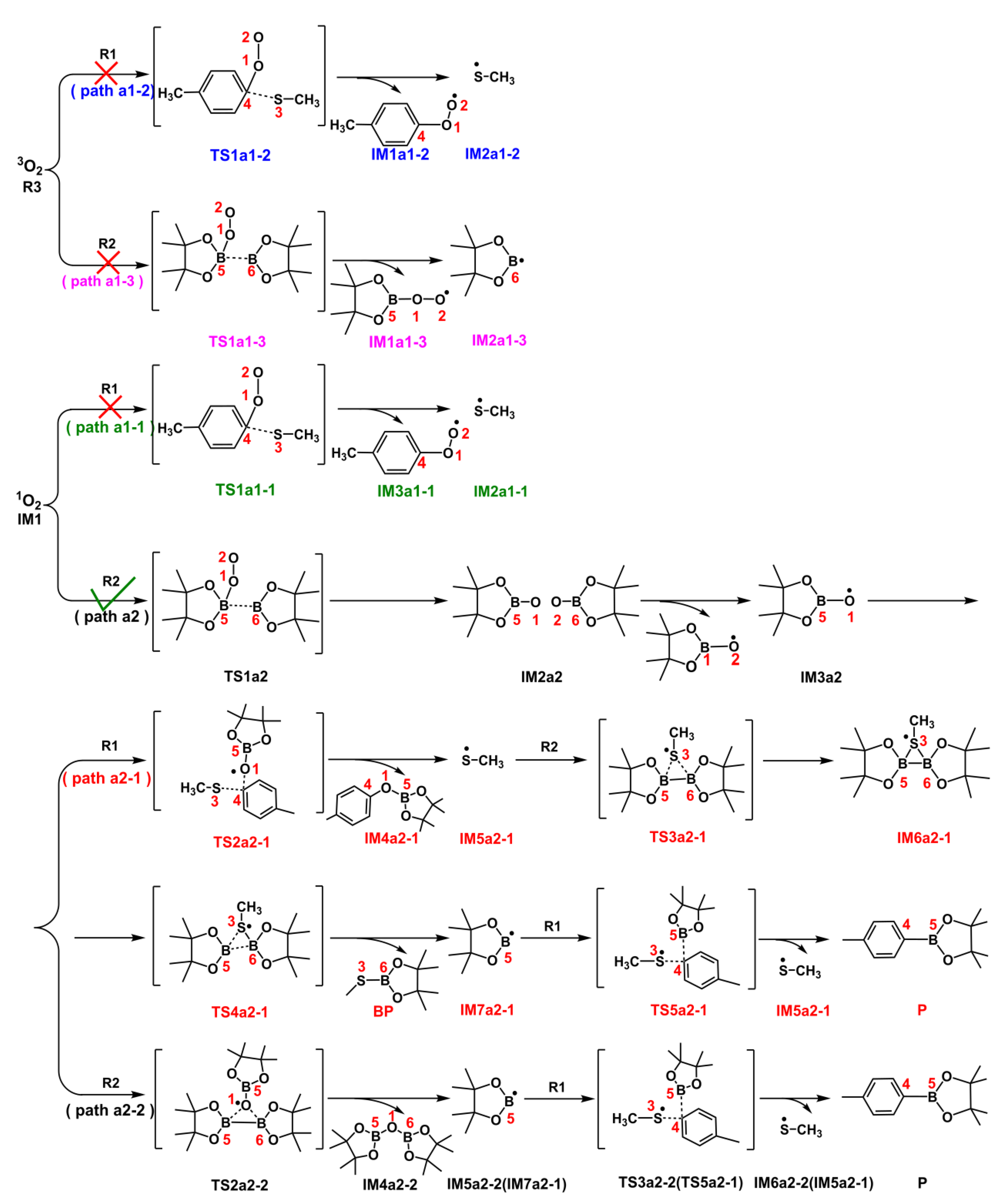

Table 1.
The excitation analysis of several models with at M06-2X-D3/ma-def2-SVP level.
| State | E(eV) | λ(nm) | ƒ | Orbital (coefficient) | |
|---|---|---|---|---|---|
| BQ | S1 | 2.7539 | 450.22 | 0.0000 | H > L (91.62%) |
| S2 | 3.0602 | 405.16 | 0.0000 | H-2 > L (89.04%) | |
| S3 | 4.3308 | 286.28 | 0.0000 | H-1 > L (99.89%) | |
| BQ+R1 | S1 | 2.7579 | 449.56 | 0.0033 | H-3 > L (37.46%) |
| H > L (57.46%) | |||||
| S2 | 2.8408 | 436.44 | 0.0029 | H-3 > L (51.47%) | |
| H > L (42.03%) | |||||
| S3 | 3.1101 | 398.65 | 0.0000 | H-5 > L (79.39%) | |
| H-4 > L (9.00%) |
Table 2.
Fukui functions and dual descriptors of R1, R2 and IM3a2 at the M06-2X-D3 /ma-def2-SVP level.
Table 2.
Fukui functions and dual descriptors of R1, R2 and IM3a2 at the M06-2X-D3 /ma-def2-SVP level.
| Atom | q(N) | q(N+1) | q(N-1) | f - | f + | CDD | |
| R1 | S3 | -0.0355 | -0.1054 | 0.2834 | 0.3189 | 0.0699 | -0.2491 |
| C4 | -0.0227 | -0.0726 | 0.0246 | 0.0473 | 0.0499 | 0.0025 | |
| R2 | B5 | 0.1650 | 0.0470 | 0.2085 | 0.0435 | 0.1181 | 0.0746 |
| B6 | 0.1650 | 0.0470 | 0.2085 | 0.0435 | 0.1181 | 0.0746 | |
| IM3a2 | O1 | -0.1258 | -0.1258 | -0.0759 | 0.0499 | 0.0176 | -0.0323 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



