
Submitted:
29 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Background: Smad4, a critical tumor suppressor gene, plays a significant role in pancreatic biology and tumorigenesis. Genetic background and sex are known to influence phenotypic outcomes, but their impact on pancreatic weight in Smad4-deficient mice remains unclear. This study investigates the impact of Smad4 deficiency on pancreatic weight in F1 mice from diverse Collaborative Cross (CC) lines, focusing on the influence of genetic background and sex. Methods: F1 mice were generated by crossbreeding female CC mice with C57BL/6J-Smad4tm1Mak males. Genotyping confirmed the presence of Smad4 knockout alleles. Mice were housed under standard conditions, euthanized at 80 weeks, and their pancreatic weights were measured, adjusted for body weight, and analyzed for effects of Smad4 deficiency, sex, and genetic background. Results: The overall population of F1 mice showed a slight but non-significant increase in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice. Sex-specific analysis revealed no significant difference in males but a significant increase in adjusted pancreatic weights in heterozygous knockout females. Genetic background analysis showed that lines CC018 and CC025 substantially increased adjusted pancreatic weights in heterozygous knockout mice. In contrast, other lines showed no significant difference or varied non-significant changes. The interplay between genetic background and sex further influenced these outcomes. Conclusion: Smad4 deficiency affects pancreatic weight in a manner significantly modulated by genetic background and sex. This study highlights the necessity of considering these factors in genetic research and therapeutic development, demonstrating the value of the Collaborative cross-mouse population in dissecting complex genetic interactions.
Keywords:
Smad4
; pancreatic weight
; genetic background
; sex differences
; Collaborative Cross
; F1 mice
; genetic modifiers
; tumor suppressor gene
1. Introduction
Pancreatic weight measurement is crucial for understanding various aspects of pancreatic health and disease progression. By quantifying pancreatic weight, researchers can assess changes in organ size over time, track growth patterns, and evaluate the impact of interventions on pancreatic morphology [1]. This information is particularly valuable in studies involving mouse models of pancreatic diseases, as it allows for the precise monitoring of disease development and treatment responses [1]. Additionally, measuring pancreatic weight can provide insights into the relationship between pancreatic size and metabolic parameters, shedding light on the physiological implications of pancreatic alterations [1]. Incorporating detailed pancreatic weight measurements in research articles enhances the validity and reliability of study findings, ultimately advancing our understanding of pancreatic biology and disease mechanisms [1].
Measuring pancreatic weights and calculating the pancreas wet weight-to-body weight ratio in mouse models is crucial for assessing pancreatic health and function [2]. The pancreas plays a vital role in digestion and glucose metabolism, and alterations in its weight can indicate underlying pathologies such as inflammation or edema [2]. In a study by Nathan JD et al., electron micrographs of the pancreas from transgenic and non-transgenic mice revealed prominent rough endoplasmic reticulum and zymogen granules in caerulein-treated animals, indicating pancreatic stress [2]. Additionally, the study demonstrated that pancreatic extracts from PSTI-I transgenic mice exhibited greater trypsin inhibitor capacity than those from non-transgenic mice, suggesting a protective role of PSTI-I in reducing trypsin activity and potentially preventing pancreatitis development [2]. By monitoring these parameters, researchers can gain valuable insights into the impact of genetic modifications or therapeutic interventions on pancreatic health and disease progression in mouse models [1,2].
Measuring pancreatic weight is crucial in understanding the development and function of the pancreas. By assessing pancreatic growth through measurements of wet weight, enzyme, and protein content, researchers can gain insights into the physiological changes in the pancreas [3]. These measurements provide valuable information about pancreatic health, such as hypertrophy or hyperplasia, and can help identify abnormalities or diseases affecting the pancreas. Understanding pancreatic growth is essential for studying conditions like diabetes, pancreatitis, and pancreatic cancer, as pancreatic weight and composition alterations can indicate underlying pathologies [3]. Therefore, the concept of measuring pancreatic weight plays a significant role in advancing our knowledge of pancreatic biology and disease mechanisms [3].
Smad4, also known as DPC4, is a crucial tumor suppressor gene involved in various cancers such as hepatocellular carcinoma, breast invasive ductal carcinoma, pancreatic cancer, colorectal cancer, and prostate cancer [4,5,6,7,8]. Loss or reduced expression of Smad4 has been associated with tumor progression, metastasis, and poor prognosis in different types of cancer [4,6,8]. Smad4 functions as a transcriptional mediator of the TGF-β signaling pathway and forms complexes with other Smad proteins to regulate gene transcription [5,7]. The location of Smad4 protein, its shuttling between the nucleus and cytoplasm, and its regulation by various signaling pathways make it a critical player in cancer development and progression [9]. Studies have shown that mutations or loss of Smad4 expression are linked to advanced disease stages, lymph node metastasis, and worse survival outcomes in cancer patients [8]. Understanding the role of Smad4 in cancer pathogenesis can provide valuable insights for developing targeted therapies and prognostic markers in oncology research.
The TGF-β/Smad4 signaling pathway is highlighted as a crucial regulator of biological processes such as epithelial-mesenchymal transition, DNA damage response, and microRNA regulation, with Smad4 acting as a tumor suppressor [10]. The interplay between the TGF-β/Smad4 pathway and other signaling cascades like MAP kinase, PI3K/Akt/mTOR, and WNT/β-catenin pathways in cancer is explored, emphasizing the importance of understanding SMAD4-mediated tumor formation and progression for the identification of potential therapeutic targets [10]. This review underscores the significance of Smad4 alterations in various cancers, including pancreatic, colorectal, gastric, and skin cancer, and the need for further research to elucidate its role in cancer progression and metastasis [10].
The role of Smad4 in pancreatic cancer, emphasizing its significance in the context of the TGF-β signaling pathway, was mentioned specifically mentioned [7]. Smad4 is a tumor suppressor in pancreatic cancer, inhibiting epithelial cell proliferation [7]. The loss of Smad4 can lead to tumor-promoting effects, highlighting the dual role of TGF-β in this disease [7]. The study underscores the importance of comprehending Smad4’s functions, structure, and regulation and its potential prognostic value in pancreatic cancer [11]. The research suggests that Smad4 protein expression profiles in pancreatic cancer cell lines can provide valuable insights into the molecular mechanisms underlying the initiation, development, and progression of pancreatic cancer [11]. Furthermore, investigations into Smad4 inactivation and its impact on the TGF-β pathway could pave the way for innovative therapeutic approaches targeting Smad4-deficient tumors, offering hope for improved outcomes in pancreatic cancer patients [11]. The study acknowledges the need for further research to elucidate the mechanisms underlying Smad4’s involvement in pancreatic cancer progression and explore novel therapeutic strategies that leverage this understanding [11].
Genetic background is critical in shaping phenotypic variation and responses to genetic perturbations [12]. Understanding how genetic modifiers influence the phenotypic consequences of gene mutations is essential for dissecting complex traits and elucidating underlying molecular mechanisms [13]. In this study, we investigate the impact of genetic background on pancreatic phenotypes in Smad4 knockout F1 mice derived from diverse Collaborative Cross (CC) lines. To explore the role of genetic background in modulating pancreatic phenotypes associated with Smad4 deficiency, we employed a breeding strategy to generate F1 mice carrying both CC and Smad4 knockout alleles. Pancreatic weights and body weights were measured in F1 mice across multiple CC lines, allowing us to assess the impact of genetic variation on pancreatic development and overall growth in the context of Smad4 deletion.
The Collaborative Cross (CC) mouse population stands as a pioneering resource in the field of genetics, offering a sophisticated platform for investigating the complex interplay between genetic variation and phenotypic diversity [14]. Comprised of a diverse panel of recombinant inbred mouse strains derived from eight genetically distinct founder strains, the CC population encapsulates a broad spectrum of genetic diversity representative of natural mouse populations [15]. One of the primary advantages of the CC mouse population lies in its ability to capture the full breadth of genetic variation present in outbred mouse populations [12]. Unlike traditional laboratory mouse strains characterized by limited genetic diversity and fixed allelic compositions, CC lines exhibit extensive allelic diversity and genetic heterogeneity [16]. This genetic complexity mirrors the diversity found in human populations and provides a rich substrate for dissecting the genetic architecture of complex traits [17].
Moreover, the CC population facilitates the study of gene-gene interactions, or epistasis, which play a fundamental role in shaping phenotypic outcomes [18]. By crossing and intercrossing CC lines, researchers can systematically explore the combinatorial effects of allelic variants at multiple genetic loci on phenotypic traits of interest [19]. This approach allows for the identification of genetic modifiers—genes or genomic regions that modulate the phenotypic consequences of a primary genetic perturbation—thereby elucidating the underlying molecular pathways and regulatory networks governing trait variability [20].
In the context of pancreatic development and growth regulation, leveraging the genetic diversity of CC lines offers a powerful means to dissect the complex genetic determinants of pancreatic phenotypes. The pancreas is a multifunctional organ with crucial roles in metabolic homeostasis, digestion, and hormone regulation. Dysregulation of pancreatic development and function underlies various pathological conditions, including diabetes, pancreatic cancer, and exocrine insufficiency. By employing the CC mouse population, we can systematically investigate how allelic variation across different genetic backgrounds influences pancreatic morphology, function, and disease susceptibility.
Our study aims to harness the genetic diversity inherent in the CC mouse population to pinpoint genetic modifiers that shape the phenotypic responses to Smad4 knockout—a key regulator of cellular signaling pathways implicated in pancreatic development and tumorigenesis. By unraveling the molecular pathways underlying pancreatic phenotypes in the context of genetic variation, we advance our understanding of pancreatic biology and pave the way for developing targeted therapies and precision medicine approaches for pancreatic diseases.
2. Materials and Methods
Ethics and Animal Welfare Considerations
This research adhered to the national guidelines for the ethical treatment of laboratory animals. The study’s protocol received approval from the Institutional Animal Care and Use Committee (IACUC) of Tel Aviv University, under the authorization number (01-19-044). Daily health monitoring of the mice was conducted, with specific criteria set for humane euthanasia based on weight loss or observed distress, in consultation with the facility’s Veterinary staff.
Crossbreeding to Generate F1 Offspring
The Collaborative Cross (CC) mouse strains were propagated at Tel-Aviv University’s animal facility under standard conditions through about 20 generations of inbreeding, following previously described methodologies [21,22]. The C57BL/6 J-Smad4tm1Mak strain was sourced from the Jackson Laboratory (Bar Harbor, Maine, USA). Crosses between female mice from available CC strains and C57BL/6 J-Smad4tm1Mak males resulted in F1 offspring. Genotypic screening for the Smad4 gene among these produced F1 mice across lines for inclusion in subsequent experiments, detailed in Table 1.
Animal Housing and Nutritional Care
The mice were accommodated at the Sackler Faculty of Medicine’s animal facility, Tel-Aviv University, under conditions approved by the university’s Animal Use and Care Committee (01-19-044). They were kept in cages with hardwood chip bedding, separated by sex and CC lineage, under a consistent 12-hour light/dark cycle, at a room temperature of 22°C. From weaning at three weeks until the end of the study at 80 weeks, they had unrestricted access to water and a standard rodent diet (TD.2018SC, Teklad Global, Harlan Inc., Madison, WI, USA).
Extraction of Genomic DNA
For genomic DNA extraction, the NaOH method was employed, as referenced in [23]. Tail samples measuring 3-4 mm were collected into Eppendorf tubes, to which a mixture of 75µl of 25 mM NaOH and 0.2 mM EDTA was added. These samples were then heated at 98ºC for 1 hour in a thermocycler, cooled to 15°C, and neutralized with 75µl of 40mM Tris HCl (pH 5.5) post-heating. Centrifugation at 4000rpm for 3 minutes helped clarify the samples, which were then ready for PCR analysis.
F1 Mouse Genotyping Protocol
Specific primer pairs were used for PCR-based genotyping:
Primer 30403 (5′—TGT AGT TCT GTC TTT CCT TCC TG—3′)
Primer 30404 (5′—ACT GAC CTT TAT ATA CGC GCT TG—3′)
Primer oIMR2088 (5′—AGA CTG CCT TGG GAA AAG CG—3′)
Two separate PCR reactions were set up:
Reaction A targeted a 200 bp segment of the Smad4 gene’s wild-type allele using primers 30403 and 30404.
Reaction B aimed to amplify a 300 bp fragment indicative of the Smad4 knockout allele with primers 30404 and oIMR2088.
These PCR reactions included a series of steps: an initial touchdown phase, followed by 30 cycles of denaturation, annealing, and extension, concluding with a final extension and a holding step.
Tissue Harvesting
At 80 weeks, the mice were humanely euthanized using CO2, and their final body weights were recorded. Body weight changes were calculated using the formula: (final body weight—initial body weight) x100% / final body weight [14]. The pancreas was extracted, weighed, and the weight was adjusted relative to the body weight using the formula: pancreas weight × 100% body weight [24].
Data Analysis
IBM SPSS Statistics 23 was used for data analysis. An independent sample t-test was conducted to compare adjusted pancreatic weights between knockout (KO) and wild-type (WT) groups. The analysis also included assessing the impact of sex on adjusted pancreatic weights and the variation of adjusted pancreatic weights across different genetic lines to understand line effects.
3. Results
The Effect of Smad4 Kock out in the General Population of F1 Mice
Our experiment’s overall population of F1 mice showed a slight but insignificant increase in pancreatic adjusted weight in heterozygous knockout mice compared to wild-type seven-line crosses. Data supporting this finding are presented in Figure 1.
Sex Effect
Heterozygous knockout males showed a slight decrease in adjusted pancreatic weights compared to wild-type males. Meanwhile, in females, there was a significant increase in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice. Data supporting these findings are presented in Figure 2. These results suggest a sex-specific effect of heterozygous knockout on pancreatic weight regulation, highlighting the importance of further investigation into the underlying mechanisms driving this observed disparity.
Line Genetic Effect
The effect of Smad4 knockout on adjusted pancreatic weights varied significantly among the different genetic lines of F1 mice. Lines CC018 and CC025 exhibited a significant increase in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice, indicating a notable impact of Smad4 deficiency in these specific genetic backgrounds. Conversely, lines CC012, CC019, CC037, CC059, and CC084 showed no significant difference in adjusted pancreatic weights between heterozygous knockout and wild-type mice, suggesting that Smad4 deficiency does not markedly affect pancreatic weight adjustment in these genetic backgrounds. Among these, lines CC012, CC059, and CC084 showed no change in adjusted pancreatic weights, meaning that the pancreatic weight relative to body weight was consistent between heterozygous knockout and wild-type mice. Additionally, lines CC019 and CC037 presented with a non-significant decrease in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice, suggesting that the effect of Smad4 deficiency might be subtle or influenced by other genetic or environmental factors in these lines. Data supporting these findings are presented in Figure 3, which details the specific adjusted pancreatic weights for each genetic line, highlighting the variability in response to Smad4 knockout across different genetic backgrounds.
Line and Sex Effect
In this section, we compared the impact of Smad4 deficiency across seven lines (CC012, CC018, CC019, CC025, CC037, CC059, and CC084), analyzing differences in adjusted pancreatic weights between male and female mice. The results of this section are presented in Figure 4.
For female mice, a significant effect of Smad4 knockout was observed in three lines: CC018, CC025, and CC084. This indicates that Smad4 deficiency substantially increases adjusted pancreatic weights, specifically in females from these lines, suggesting a heightened sensitivity or a different regulatory mechanism in female mice in response to the knockout.
In male mice, the impact of Smad4 deficiency was less consistent across the lines. Only line CC025 significantly increased adjusted pancreatic weights, mirroring the effect seen in females of the same line. Additionally, line CC037 showed a significant decrease in adjusted pancreatic weights in male mice, contrasting with the lack of substantial change or increase in other lines and sexes. This suggests that the impact of Smad4 deficiency may not only be sex-specific but also vary widely depending on the genetic background of the mice.
Heritability
This study aimed to discover whether adjusted pancreatic weights phenotypic variance has a genetic basis in Smad4 knockout F1 populations. Table 2 summarizes the heritability (H2) values calculated to answer this question. One-way ANOVA was used to calculate the heritability of sex and genotype-specific characteristics. The traits calculated are pancreatic-adjusted weights and body weight changes for both sexes and genotypes.
4. Discussion
The Smad4 gene under scrutiny encodes a signal transduction that is essential for both embryonic development and signaling. This gene has been linked to Myhre Syndrome, Juvenile Polyposis Syndrome, Hereditary Hemorrhagic Telangiectasia Syndrome, and human pancreatic cancer [25,26]. These knock-out mice have a mutation where a NEO selection cassette has taken the place of exon 8 and a portion of exon 9. Western blot examination of homozygous immortalized embryonic fibroblasts shows no detection of any gene product (protein) [27,28]. The phenotype of homozygous null mice is embryonic fatal; they are unable to gastrulate or develop past embryonic day 7.5. At E5.5, the size of the embryo decreases, and the extraembryonic and embryonic zones become jumbled and poorly defined. In homozygous embryos, BrdU labeling at E6.5 shows reduced cell growth. The backcrossing process might not have fixed the Y chromosome to the C57BL/6 genetic background. A mouse is viable and fertile if it is heterozygous for the desired mutation [29,30].
Our study aimed to investigate the effect of Smad4 deficiency on pancreatic weight in F1 mice across different genetic backgrounds using the Collaborative Cross (CC) lines. The findings provide insights into how Smad4 knockout influences pancreatic morphology, revealing significant variability influenced by genetic background and sex. Previous reports suggested Smad4 loss alone does not affect pancreatic weight, as mice with homozygous deletion of Smad4 in the pancreas did not show any gross anatomical or physiological abnormalities, maintaining normal pancreatic cytoarchitecture. Pancreatic weight remained unaffected in this context [31].
Additionally, another report on transgenic mice expressing dnSmad4, a dominant-negative Smad4 protein, do not show significant changes in pancreatic weight, exocrine, or ductal histology compared to wild-type mice. However, these mice display an age-dependent increase in islet size. The dnSmad4 transgene expression leads to an expanded population of replicating cells expressing the transgene in the stroma between enlarged islets and pancreatic ducts. This suggests that loss of Smad4 signaling in the pancreas does not affect overall pancreatic weight but influences islet size and cell proliferation dynamics within the pancreatic tissue [32].
The overall population of F1 mice showed a slight but not statistically significant increase in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice. This suggests that Smad4 deficiency might influence pancreatic weight, but the effect is modest and could be masked by other genetic and environmental factors. Previous studies have established Smad4’s role in cellular signaling pathways and its involvement in various cancers, including pancreatic cancer. Our findings align with the idea that Smad4 has a complex role in pancreatic biology, potentially affecting growth and morphology in subtle ways that warrant further investigation.
The impact of Smad4 knockout on adjusted pancreatic weights varied significantly between male and female mice. Heterozygous knockout males showed a slight decrease in adjusted pancreatic weights compared to wild-type males. However, a significant increase in adjusted pancreatic weights was noted in females. This sex-specific response suggests that female mice might be more sensitive to Smad4 deficiency, potentially due to differences in hormonal regulation or metabolic processes that influence pancreatic development and function. The sexual dimorphism observed in our study could be crucial for understanding how genetic mutations differentially affect males and females and might help tailor sex-specific therapeutic approaches in diseases related to Smad4 deficiency.
The response to Smad4 knockout varied across different CC lines, indicating that genetic background significantly influences the phenotypic outcome as also revealed by Qahaz et al. [33]. Lines CC018 and CC025 showed a significant increase in adjusted pancreatic weights in heterozygous knockout mice compared to wild-type mice. These lines may harbor genetic modifiers interacting with Smad4, enhancing its impact on pancreatic weight. Conversely, lines CC012, CC019, CC037, CC059, and CC084 did not show significant differences, suggesting that other compensatory mechanisms might mitigate the effect of Smad4 deficiency in these genetic backgrounds. The non-significant increases and decreases observed in some lines further highlight the complexity of genetic interactions that regulate pancreatic morphology.
The analysis of sex effects within specific lines revealed additional complexity. In female mice, a significant impact of Smad4 knockout was observed in lines CC018, CC025, and CC084, whereas in male mice, significant changes were noted only in lines CC025 (increase) and CC037 (decrease). This highlights the importance of considering genetic background and sex when studying the effects of genetic mutations. The significant reduction in adjusted pancreatic weights in CC037 males, contrasting with the effects in females, underscores the intricate interplay between genetic, hormonal, and environmental factors in shaping phenotypic outcomes. These findings suggest that therapeutic strategies for conditions involving Smad4 might need to be personalized based on both genetic background and sex.
5. Conclusions
Our study underscores the importance of genetic background in modulating the phenotypic effects of gene knockouts. With its extensive genetic diversity, the CC mouse population serves as an excellent model for dissecting the complex interactions between genes and phenotypes. Future research should focus on identifying specific genetic modifiers that influence the impact of Smad4 deficiency on pancreatic weight. Understanding these modifiers could provide insights into the molecular pathways involved and identify potential targets for therapeutic intervention.
Moreover, the observed sex-specific effects warrant further exploration into the underlying mechanisms. Investigating hormonal influences and sex-specific gene expression patterns in the pancreas could explain why female mice are more affected by Smad4 deficiency. This knowledge could be pivotal in developing sex-specific treatments for pancreatic diseases and other conditions involving Smad4 mutations.
This study highlights the significant variability in the impact of Smad4 deficiency on pancreatic weight due to genetic background and sex. The findings emphasize the need to consider genetic diversity and sex differences in biomedical research, particularly in studies involving genetic modifications. By leveraging the genetic complexity of the CC mouse population, we can gain deeper insights into the biological functions of Smad4 and its role in pancreatic health and disease, ultimately contributing to the development of more effective and personalized therapeutic strategies.
Author Contributions
Conceptualization, F.A.I., A.N. and I.A.-E.; Methodology, O.Z.; Validation, F.A.I.; Investigation, O.Z. and I.M.L.; Resources, F.A.I.; Data Curation, O.Z., I.M.L., and K.M. O.Z.; Writing—Original Draft Preparation, O.Z., K.M. and I.M.L.; Writing—Review and Editing, O.Z. and F.A.I.; Supervision, F.A.I. and A.N.; Project Administration, F.A.I.; Funding Acquisition, F.A.I. and I.A.-E. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by core funding from Tel-Aviv University (TAU) for supporting staff at Iraqi’s lab and technical staff.
Institutional Review Board Statement
All experimental procedures involving animals were conducted following the guidelines set forth by the Institutional Animal Care and Use Committee (IACUC) and received approval under protocol [01-19-044]. Ethical considerations regarding the care and use of animals were strictly adhered to throughout the study.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Paredes, J.L.; Orabi, A.I.; Ahmad, T.; Benbourenane, I.; Tobita, K.; Tadros, S.; Bae, K.T.; Husain, S.Z. A Non-Invasive Method of Quantifying Pancreatic Volume in Mice Using Micro-MRI. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Nathan, J.D.; Romac, J.; Peng, R.Y.; Peyton, M.; Macdonald, R.J.; Liddle, R.A. Transgenic Expression of Pancreatic Secretory Trypsin Inhibitor-I Ameliorates Secretagogue-Induced Pancreatitis in Mice. Gastroenterology 2005, 128. [Google Scholar] [CrossRef]
- Miyasaka, K.; Fujimoto, T.; Kawanami, T.; Takiguchi, S.; Jimi, A.; Funakoshi, A.; Shirasawa, S. Pancreatic Hypertrophy in Ki-Ras-Induced Actin-Interacting Protein Gene Knockout Mice. Pancreas 2011, 40. [Google Scholar] [CrossRef]
- Derynck, R.; Gelbart, W.M.; Harland, R.M.; Heldin, C.H.; Kern, S.E.; Massagué, J.; Melton, D.A.; Mlodzik, M.; Padgett, R.W.; Roberts, A.B.; et al. Nomenclature: Vertebrate Mediators of TGFbeta Family Signals. Cell 1996, 87. [Google Scholar] [CrossRef]
- Wrana, J.L. The Secret Life of Smad4. Cell 2009, 136. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Shamsul Hoque, A.T.M.; Moskaluk, C.A.; Da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, A Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science (1979) 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and Its Role in Pancreatic Cancer. Tumor Biology 2015, 36. [Google Scholar] [CrossRef]
- Liu, F.; Pouponnot, C.; Massagué, J. Dual Role of the Smad4/DPC4 Tumor Suppressor in TGF-Inducible Transcriptional Complexes. 1997. [Google Scholar]
- Miyaki, M.; Kuroki, T. Role of Smad4 (DPC4) Inactivation in Human Cancer. Biochem Biophys Res Commun 2003, 306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The Role of TGF-β/SMAD4 Signaling in Cancer. Int J Biol Sci 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srinivasan, R.; Wig, J.D. Major Molecular Markers in Pancreatic Ductal Adenocarcinoma and Their Roles in Screening, Diagnosis, Prognosis, and Treatment. Pancreas 2011, 40. [Google Scholar] [CrossRef] [PubMed]
- Zohud, O.; Lone, I.M.; Nashef, A.; Iraqi, F.A.; Fuad Iraqi, C.A. Towards System Genetics Analysis of Head and Neck Squamous Cell Carcinoma Using the Mouse Model, Cellular Platform, and Clinical Human Data. Anim Models Exp Med 2023, 6, 537–558. [Google Scholar] [CrossRef]
- Dorman, A.; Binenbaum, I.; Abu-Toamih Atamni, H.J.; Chatziioannou, A.; Tomlinson, I.; Mott, R.; Iraqi, F.A. Genetic Mapping of Novel Modifiers for Apc Min Induced Intestinal Polyps’ Development Using the Genetic Architecture Power of the Collaborative Cross Mice. BMC Genomics 2021, 22. [Google Scholar] [CrossRef]
- Zohud, O.; Midlej, K.; Lone, I.M.; Nashef, A.; Abu-Elnaaj, I.; Iraqi, F.A. Studying the Effect of the Host Genetic Background of Juvenile Polyposis Development Using Collaborative Cross and Smad4 Knock-Out Mouse Models. International Journal of Molecular Sciences 2024, 25, 5812. [Google Scholar] [CrossRef]
- Churchill, G.A.; Airey, D.C.; Allayee, H.; Angel, J.M.; Attie, A.D.; Beatty, J.; Beavis, W.D.; Belknap, J.K.; Bennett, B.; Berrettini, W.; et al. The Collaborative Cross, a Community Resource for the Genetic Analysis of Complex Traits. Nat Genet 2004, 36, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Atamni, H.J.A.T.; Botzman, M.; Mott, R.; Gat-Viks, I.; Iraqi, F.A. Mapping Liver Fat Female-Dependent Quantitative Trait Loci in Collaborative Cross Mice. Mammalian Genome 2016, 27. [Google Scholar] [CrossRef]
- Durrant, C.; Tayem, H.; Yalcin, B.; Cleak, J.; Goodstadt, L.; Pardo-Manuel De Villena, F.; Mott, R.; Iraqi, F.A. Collaborative Cross Mice and Their Power to Map Host Susceptibility to Aspergillus Fumigatus Infection. Genome Res 2011, 21, 1239–1248. [Google Scholar] [CrossRef]
- Dorman, A.; Baer, D.; Tomlinson, I.; Mott, R.; Iraqi, F.A. Genetic Analysis of Intestinal Polyp Development in Collaborative Cross Mice Carrying the Apc Min/+ Mutation. BMC Genet 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Lorè, N.I.; Sipione, B.; He, G.; Strug, L.J.; Atamni, H.J.; Dorman, A.; Mott, R.; Iraqi, F.A.; Bragonzia, A. Collaborative Cross Mice Yield Genetic Modifiers for Pseudomonas Aeruginosa Infection in Human Lung Disease. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Abu-Toamih-Atamni, H.J.; Lone, I.M.; Binenbaum, I.; Mott, R.; Pilalis, E.; Chatziioannou, A.; Iraqi, F.A. Mapping Novel QTL and Fine Mapping of Previously Identified QTL Associated with Glucose Tolerance Using the Collaborative Cross Mice. Mammalian Genome 2023, 35, 31–55. [Google Scholar] [CrossRef]
- Iraqi, F.A.; Churchill, G.; Mott, R. The Collaborative Cross, Developing a Resource for Mammalian Systems Genetics: A Status Report of the Wellcome Trust Cohort. Mammalian Genome 2008, 19. [Google Scholar] [CrossRef]
- Iraqi, F.A.; Mahajne, M.; Salaymah, Y.; Sandovski, H.; Tayem, H.; Vered, K.; Balmer, L.; Hall, M.; Manship, G.; Morahan, G.; et al. The Genome Architecture of the Collaborative Cross Mouse Genetic Reference Population. Genetics 2012, 190. [Google Scholar] [CrossRef]
- Truett, G.E.; Heeger, P.; Mynatt, R.L.; Truett, A.A.; Walker, J.A.; Warman, M.L. Preparation of PCR-Quality Mouse Genomic Dna with Hot Sodium Hydroxide and Tris (HotSHOT). Biotechniques 2000, 29. [Google Scholar] [CrossRef] [PubMed]
- Veite-Schmahl, M.J.; Regan, D.P.; Rivers, A.C.; Nowatzke, J.F.; Kennedy, M.A. Dissection of the Mouse Pancreas for Histological Analysis and Metabolic Profiling. J. Vis. Exp 2017, 55647. [Google Scholar] [CrossRef]
- Chang, W.; Renaut, P.; Pretorius, C. SMAD4 Juvenile Polyposis Syndrome and Hereditary Haemorrhagic Telangiectasia Presenting in a Middle-Aged Man as a Large Fungating Gastric Mass, Polyposis in Both Upper and Lower GI Tract and Iron Deficiency Anaemia, with No Known Family History. BMJ Case Rep 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 Is Dispensable for Normal Pancreas Development yet Critical in Progression and Tumor Biology of Pancreas Cancer. Genes Dev 2006, 20. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Tanaka, S.; Umemori, H.; Minowa, O.; Usui, M.; Ikematsu, N.; Hosoda, E.; Imamura, T.; Kuno, J.; Yamashita, T.; et al. Negative Regulation of BMP/Smad Signaling by Tob in Osteoblasts. Cell 2000, 103. [Google Scholar] [CrossRef] [PubMed]
- Mamot, C.; Mild, G.; Reuter, J.; Laffer, U.; Metzger, U.; Terracciano, L.; Boulay, J.L.; Herrmann, R.; Rochlitz, C. Infrequent Mutation of the Tumour-Suppressor Gene Smad4 in Early-Stage Colorectal Cancer. Br J Cancer 2003, 88. [Google Scholar] [CrossRef] [PubMed]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.-G.; Boldyreff, B. Disruption of the Regulatory β Subunit of Protein Kinase CK2 in Mice Leads to a Cell-Autonomous Defect and Early Embryonic Lethality. Mol Cell Biol 2003, 23. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Yoshizawa, T.; Takahashi, Y.; Nitahara-Kasahara, Y.; Okada, T.; Nomura, Y.; Yamanaka, H.; Kosho, T.; Matsumoto, K. Backcrossing to an Appropriate Genetic Background Improves the Birth Rate of Carbohydrate Sulfotransferase 14 Gene-Deleted Mice. Exp Anim 2020, 69. [Google Scholar] [CrossRef]
- Garcia-Carracedo, D.; Yu, C.-C.; Akhavan, N.; Fine, S.A.; Schönleben, F.; Maehara, N.; Karg, D.C.; Xie, C.; Qiu, W.; Fine, R.L.; et al. Smad4 Loss Synergizes with TGFα Overexpression in Promoting Pancreatic Metaplasia, PanIN Development, and Fibrosis. 2015. [Google Scholar] [CrossRef]
- Simeone, D.M.; Zhang, L.; Treutelaar, M.K.; Zhang, L.; Graziano, K.; Logsdon, C.D.; Burant, C.F. Islet Hypertrophy Following Pancreatic Disruption of Smad4 Signaling. Am J Physiol Endocrinol Metab 2006, 291, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Qahaz, N.; Lone, I.M.; Khadija, A.; Ghnaim, A.; Zohud, O.; Nun, N.B.; Nashef, A.; Abu El-Naaj, I.; Iraqi, F.A. Host Genetic Background Effect on Body Weight Changes Influenced by Heterozygous Smad4 Knockout Using Collaborative Cross Mouse Population. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Impact of Smad4 Deficiency on Adjusted Pancreatic Weight in the General Population of F1 Mice: The graph represents the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across 11 different genetic lines of the Collaborative Cross (CC) population. The overall trend shows a slight increase in pancreatic weight in the Smad4+/- group compared to the WT group, although this increase is not statistically significant (p>0.05). Error bars denote the standard error of the mean (SEM). The lack of substantial difference suggests that Smad4 deficiency has a modest effect on pancreatic weight in the general population. The X-axis represents the genotype, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the average adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.
Figure 1.
Impact of Smad4 Deficiency on Adjusted Pancreatic Weight in the General Population of F1 Mice: The graph represents the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across 11 different genetic lines of the Collaborative Cross (CC) population. The overall trend shows a slight increase in pancreatic weight in the Smad4+/- group compared to the WT group, although this increase is not statistically significant (p>0.05). Error bars denote the standard error of the mean (SEM). The lack of substantial difference suggests that Smad4 deficiency has a modest effect on pancreatic weight in the general population. The X-axis represents the genotype, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the average adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.
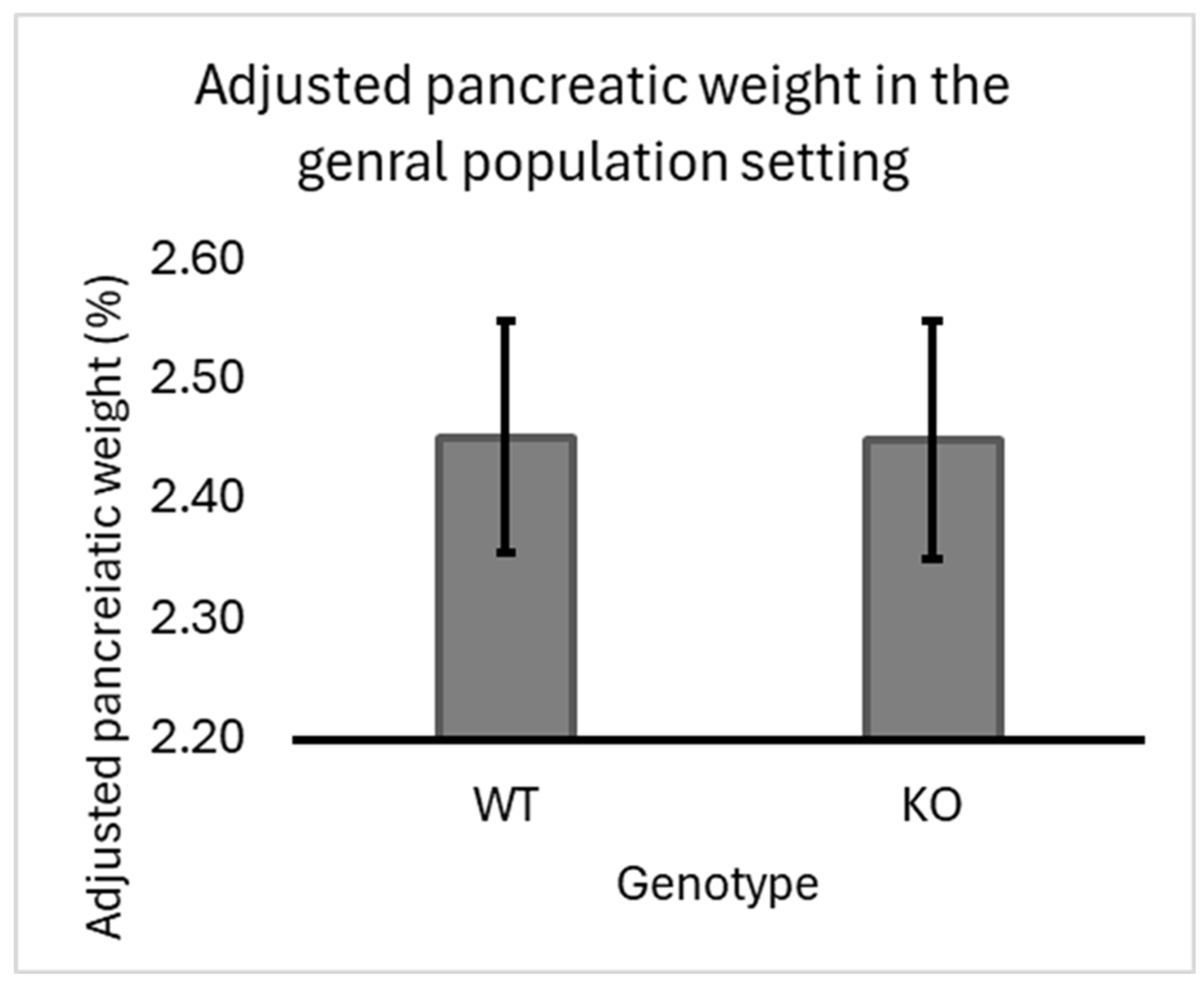
Figure 2.
Sex-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph illustrates the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (KO) male and female F1 mice. In male mice, there is no significant difference in adjusted pancreatic weights between WT and Smad4+/- groups. In contrast, female mice show a significant increase in adjusted pancreatic weights in the Smad4+/- group compared to the WT group (*p<0.05). The X-axis represents the genotype and Sex, while the Y-axis represents the adjusted pancreatic weights. Error bars represent the standard error of the mean (SEM). These results highlight a sex-specific response to Smad4 deficiency, with females exhibiting a more pronounced increase in pancreatic weight than males.
Figure 2.
Sex-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph illustrates the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (KO) male and female F1 mice. In male mice, there is no significant difference in adjusted pancreatic weights between WT and Smad4+/- groups. In contrast, female mice show a significant increase in adjusted pancreatic weights in the Smad4+/- group compared to the WT group (*p<0.05). The X-axis represents the genotype and Sex, while the Y-axis represents the adjusted pancreatic weights. Error bars represent the standard error of the mean (SEM). These results highlight a sex-specific response to Smad4 deficiency, with females exhibiting a more pronounced increase in pancreatic weight than males.
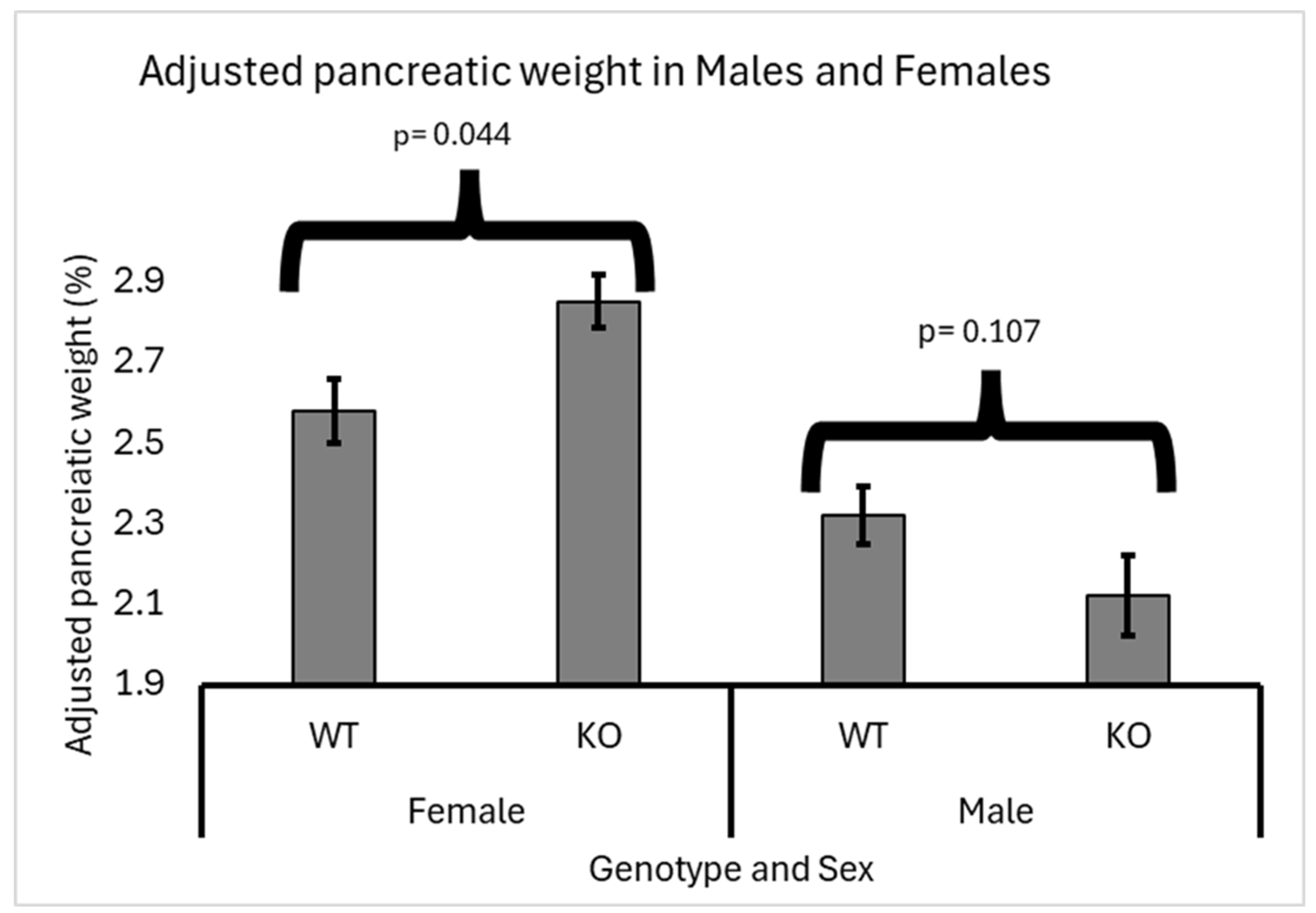
Figure 3.
Line-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph presents the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across various Collaborative Cross (CC) lines. Error bars represent the standard error of the mean (SEM). The X-axis represents the genotype and Line, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.
Figure 3.
Line-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph presents the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across various Collaborative Cross (CC) lines. Error bars represent the standard error of the mean (SEM). The X-axis represents the genotype and Line, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.
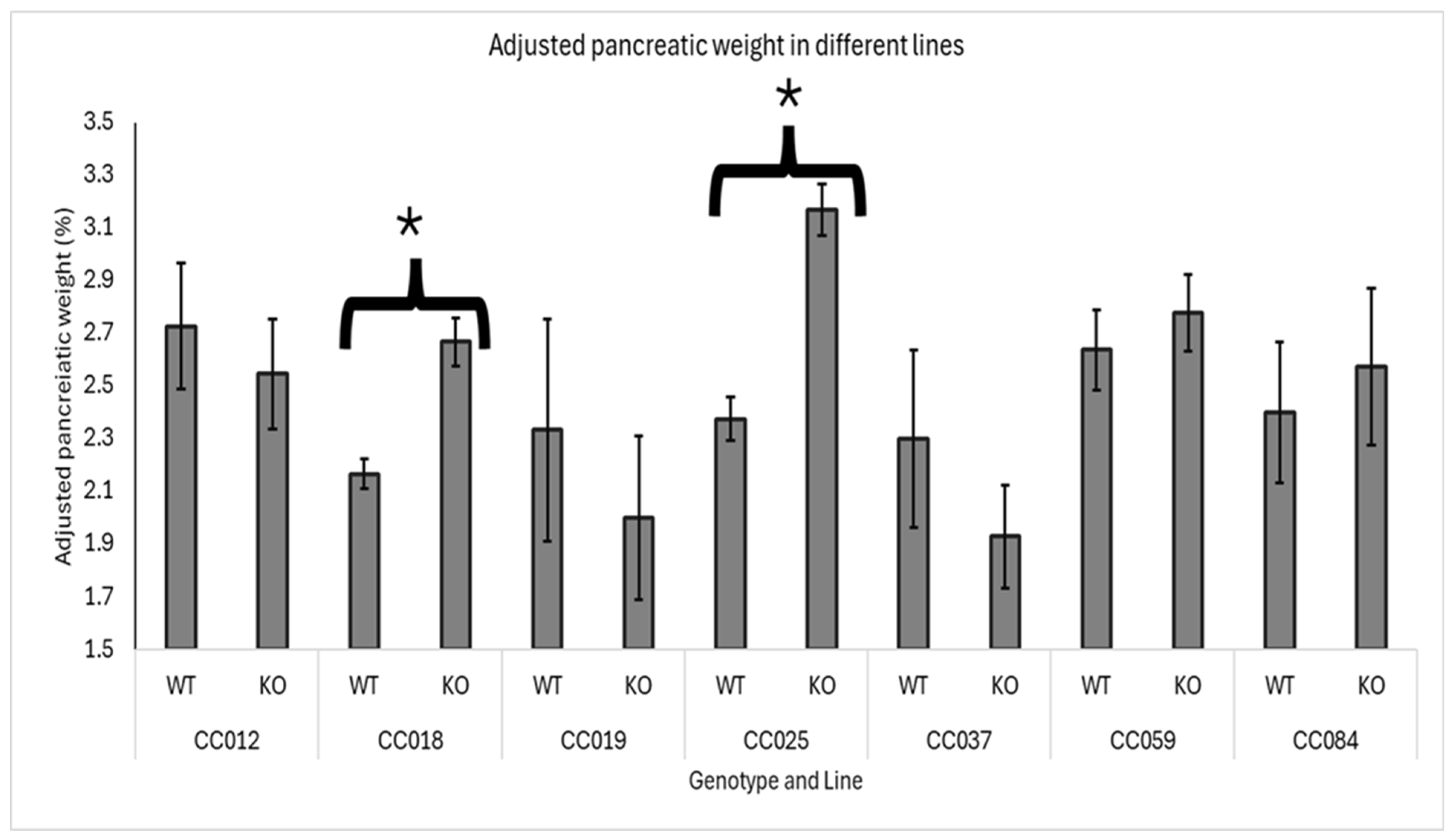
Figure 4.
Sex and Line-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph shows the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for male and female wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across seven Collaborative Cross (CC) lines. Error bars represent the standard error of the mean (SEM). The X-axis represents the genotype, Line, and Sex, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the average adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.
Figure 4.
Sex and Line-Specific Effects of Smad4 Deficiency on Adjusted Pancreatic Weight in F1 Mice: The graph shows the adjusted pancreatic weights (pancreas weight as a percentage of body weight) for male and female wild-type (WT) and heterozygous knockout (Smad4+/-) F1 mice across seven Collaborative Cross (CC) lines. Error bars represent the standard error of the mean (SEM). The X-axis represents the genotype, Line, and Sex, while the Y-axis represents the adjusted pancreatic weights. The statistical significance of differences in the average adjusted pancreatic weights between the two groups is presented as follows: (*) indicates a significant difference at p < 0.05.

Table 1.
summarizes the sample size of male and female mice from the seven Collaborative cross-lines with different genetic backgrounds.
Table 1.
summarizes the sample size of male and female mice from the seven Collaborative cross-lines with different genetic backgrounds.
| Line | ||||||||
|---|---|---|---|---|---|---|---|---|
| CC012 | CC018 | CC019 | CC025 | CC037 | CC059 | CC084 | ||
| Female | WT | 7 | 3 | 3 | 5 | 4 | 4 | 5 |
| KO | 5 | 3 | 3 | 3 | 5 | 5 | 3 | |
| Male | WT | 4 | 3 | 3 | 3 | 6 | 7 | 5 |
| KO | 6 | 3 | 4 | 3 | 9 | 4 | 4 | |
Table 2.
Results of calculating heritability (H2) and genetic variance (CVg) values. Heritability was calculated using one-way ANOVA for the traits in our study, which were calculated separately by sex and genotype.
Table 2.
Results of calculating heritability (H2) and genetic variance (CVg) values. Heritability was calculated using one-way ANOVA for the traits in our study, which were calculated separately by sex and genotype.
| Sex | Genotype | Trait | VG | H2 | CVg |
|---|---|---|---|---|---|
| Female | WT | Adjusted Pan Weight % | 0.284903 | 0.477302 | 0.214363 |
| Female | WT | Delta_BW | 14.5507 | 0.153954 | 0.062006 |
| Female | KO | Adjusted Pan Weight % | 0.257129 | 0.603407 | 0.201222 |
| Female | KO | Delta_BW | 4.166905 | 0.036585 | 0.032808 |
| Male | WT | Adjusted Pan Weight % | 0.536923 | 0.456597 | 0.319978 |
| Male | WT | Delta_BW | 6.331883 | 0.04312 | 0.04313 |
| Male | KO | Adjusted Pan Weight % | 0.243643 | 0.412505 | 0.196654 |
| Male | KO | Delta_BW | 4.073576 | 0.030492 | 0.03395 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



