Submitted:
30 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Background Lower respiratory tract infections (LRTIs), particularly pneumonia, are a leading cause of morbidity and mortality among children worldwide, especially in low-resource settings. The World Health Organization (WHO) Integrated Management of Childhood Illness (IMCI) guidelines currently recommend antibiotic treatment for all case of with pneumonia based on clinical signs along, which can lead to over-prescription of antibiotics and contribute to antimicrobial resistance (AMR). Point of care Lung Ultrasound (LUS) has emerged as a promising diagnostic tool in this context and has demonstrated potential in distinguishing between bacterial and viral pneumonia, thus enabling more targeted antibiotic use. This study aims to explore whether integrating LUS into the IMCI-based management of clinical pneumonia will better identify those who would benefit from antibiotic treatment, reducing antibiotic overuse without compromising health outcomes. Methods An open label randomized controlled trial will enrol 3500 children aged 60 days to 12 years presenting with IMCI-defined pneumonia. Participants will be enrolled in Senegal, Tanzania, and South Africa. Participants will be randomly assigned in a ratio of 1:1, stratified by site and age group (<5years, 5-12years) to either an intervention group, where LUS will guide antibiotic prescriptions utilising a predefined interpretation framework, or a control group receiving routine IMCI-based care. The co-primary outcomes of this study are the reduction in antibiotic use on the day of enrolment (Day 1) and rate of clinical failure by Day 8, using an acceptable clinical difference of 1.5x as a non-inferiority margin (delta of 2.5%). Secondary outcomes will include adverse drug reactions, cure rates, hospital admissions including in-patient duration, and cost-effectiveness. Data will be collected at baseline (Day 1), Day 8, and Day 29 post-enrolment. Discussion This study seeks to provide evidence on the clinical and public health benefits of using LUS to improve the diagnosis and management of paediatric pneumonia in low-resource settings. If this intervention proves effective it could contribute significantly toward reducing unnecessary antibiotic prescriptions, thereby addressing AMR. The potential for LUS to be integrated into routine clinical practice could revolutionize pneumonia management, especially in areas with limited access to advanced diagnostic tools. Trial Registration This trial will be registered on ClinicalTrials.gov and The South African National Clinical Trials Register (SANCTR) prior to participant enrolment.
Keywords:
Lung Ultrasound
; IMCI
; Paediatric Pneumonia
; Antibiotic Stewardship
; Low-Resource Settings
; Antimicrobial Resistance
; Diagnostic Tools
Background
In 2019, lower respiratory tract infections (LRTI) caused 2.6 million deaths worldwide, making them the fourth leading cause of death overall [1]. While the statistic is global the effect is concentrated in low-resource settings and among children, with pneumonia being the single largest infectious cause of death in children worldwide [2].
LRTI (including pneumonia) are the most common reason for sick children to present for acute outpatient care and current best practice pneumonia management guidelines advocate for all cases to receive a course of antibiotics. This relies on the World Health Organization’s (WHO) Integrated Management of Childhood Infections (IMCI) guidelines, first developed in the 1990s - a time when providing access to antibiotics was a major goal for public health programs [3]. Today, antibiotics are readily available in most countries in sub-Saharan Africa (SSA) and antibiotic over-prescription has become a public health crisis [4]. The IMCI guidelines were re-iterated in 2014 but still rely on presumptive treatment based on clinical signs alone (cough, respiratory rate, and lower costal indrawing) [3], as evidence to include additional diagnostic tests into the IMCI approach has been lacking. However clinical features inadequately distinguish bacterial infections and complications from common, self-limiting viral infections [5]. Furthermore, recent evidence estimates the incidence of bacterial respiratory infections in SSA, using a combination of microbiology, chest x-ray (CXR), and clinical outcomes as reference standards, is as low as 2-4% in primary [6,7], and 23.3-31.6% in secondary [8], care. Therefore, approximately 9 out of 10 courses of antibiotics recommended by current guidelines for children with LRTI are estimated to be unnecessary.
Bacterial antimicrobial resistance (AMR), responsible for 1.27 million deaths in 2019 and with the highest burden in SSA [9], is thus of increasing concern; with nearly as many deaths as malaria and HIV combined. Inappropriately and excessively prescribing antibiotics represents one primary contributor of bacterial AMR [10,11,12]. In SSA, more than 50% of children who are sick receive antibiotics when visiting health facilities [13,14,15,16,17,18] - with 80–90% of such antibiotics prescribed at the outpatient level [19] and most deemed inappropriate [13,19]. Most inappropriate antibiotic use occurs with respiratory infections due to systematic overprescription as outlined above [13,19]. Antibiotic use and AMR are projected to increase over the next years, indicating urgent action [20,21,22]. Accordingly, WHO declared AMR as “one of the biggest threats to global health, food security and development today.” [4]. Effective solutions to improve antibiotic stewardship for childhood infections in SSA primary care settings remain lacking, though its well recognized improved diagnostic and management processes are essential.
Lacking more effective tools to safely identify the minority of children requiring antibiotics, frontline clinicians in low resource settings therefore prescribe antibiotics to nearly every child, driving the above-described overuse and fuelling local AMR, whilst significantly overtreating cases of viral pneumonia [5]. Besides the risk of AMR at the population level, reducing antibiotic prescriptions is also important for each individual patient. Antibiotic exposure early in life has been associated with an increased risk of health conditions, including asthma, allergic rhinitis, atopic dermatitis, autoimmune disease, obesity, and neurodevelopmental disorders [23]. Improving IMCI diagnostic criteria to better identify children with pneumonia that would benefit from antibiotic therapy is therefore a WHO research priority.
Several tools have since been proposed (currently at different stages of diagnostic development) to both improve diagnostic pathways and improve appropriateness of antimicrobial prescriptions in LMICs. These include the use of point of care C-reactive Protein (CRP), Procalcitonin (PCT), comprehensive electronic decision support algorithms [6,24,25], and more novel applications of established technologies. Two such technologies are Lung Ultrasound (LUS) and Lung Auscultation (LAusc). LUS is a well-established, near consumable-free, and non-invasive point-of-care respiratory exam. While LUS is less ubiquitous than the stethoscope, it’s new portable and affordable ultrasound-on-a-chip design, pluggable into a mobile device, has the potential to be integrated into the standard clinical exam without incurring extra costs, time, radiation, or specialist consultation. These portable ultrasound devices have reached regulatory approval and are used in medical care across SSA. This together with increasing evidence showing its ability to effectively detect lung consolidation in pneumonia have made it an increasingly attractive tool for frontline clinicians; already becoming an established practice in the outpatient case management of children with respiratory infections in many high-resource settings [26,27,28] as well as growing steadily in popularity in SSA [29].
A number of studies have compared LUS to CXR and/or Computerised tomography (CT) for pneumonia diagnosis. A multi-centre trial in Europe including 362 adult patients with pneumonia compared LUS performed by a specialist to a diagnosis of pneumonia made by CXR and in some cases CT. The authors reported a sensitivity of 93.4% (95% Confidence Interval (CI) 89.2-96.3%) and a specificity of 97.7% (95%CI 89.2-99.6%) [30]. Most studies in children (performed primarily in high-income countries and tertiary care settings) have found high diagnostic accuracy when comparing LUS to CXR, using either CXR, CT, or Magnetic Resonance Imaging (MRI) as reference standards, with a recent meta-analysis suggesting an overall sensitivity of 95% (91-97%) and specificity of 96% (90-99%) [31,32]. A smaller number of studies have investigated using LUS to assist in determining the etiological causes of the consolidations [33,34]. Although these have shown a possibility of differentiating viral from bacterial infections, the reference standards used were nasopharyngeal swab carriage, clinical syndromes, and other clinically available data. A notable limitation of this study was therefore the lack of a microbiologically confirmed diagnosis, making it hard to make definitive statements about the ability of LUS to differentiate. This lack of reliable reference standards in paediatric pneumonia is a well-documented challenge, most notably discussed in the Pneumonia Etiology Research for Child Health (PERCH) study, and thus limits the value of etiology focused diagnostic accuracy studies [8,35,36]. Although the first studies on the use of LUS to detect lung consolidation in children in LMICs are promising, as seen in Nepal, Peru, and Egypt [37,38,39,40,41], they remain limited in scope and context, with no evidence on the clinical impact of LUS assisted diagnoses on antimicrobial use or health outcomes, particularly in a primary care setting. This highlights the importance of the assessment of LUS integrated into care to assess for real changes in treatment outcomes through an interventional study, rather than observational diagnostic accuracy studies.
Studies assessing the potential of LUS in the diagnosis of pulmonary tuberculosis (TB) in children are equally limited [31]. With paediatric TB diagnosis remaining a huge challenge in low resource settings, many cases still remain undiagnosed and untreated – an issue being addressed though the development of pragmatic TB Treatment Decision Algorithms for settings with and without CXR. These Treatment Decision Algorithms are now included in the operational handbook accompanying the WHO guidelines on the management of TB in children and adolescents [42]. More evidence is however needed on the potential role of LUS as an alternative diagnostic modality in the context of these TB Treatment Decision Algorithms.
Regarding operability of LUS in SSA by novice health workers, LUS performance evidence shows it is possible to capture images/video with minimal training and achieve high diagnostic quality, as evidenced in both South Sudan and Bangladesh [43,44,45], though concerns remain regarding diagnostic quality outside research settings [44,45]. Image acquisition aside, interpretation and inter-user bias remain the largest concern, this however makes them excellent candidates for automated interpretation by objective computer-aided pattern detection. Using deep learning our collaborating team at École polytechnique fédérale de Lausanne (EPFL) have found excellent preliminary results in Swiss cohorts, matching and out-performing expert evaluation for risk stratification (Area Under the Receiver Operating characteristic Curve (AUROC) >80%) and diagnosis (AUROC >90%) of COVID-19 pneumonia (unpublished data).
As with LUS, LAsuc is another established tool being applied in a novel way. While evidence for the potential of LAusc for paediatric pneumonia is limited, some preliminary evidence on the predictive capacity of other respiratory sounds is emerging; for instance, the application of deep learning was able to predict diagnosis from breath and cough sounds collected on a mobile application with an AUC of around 80% [46]. Another group achieved above 95% sensitivity and specificity on discriminating COVID-19 coughs from other pathologies as well as healthy patients [47]. A study in South Africa included 33 culture-confirmed TB patients with healthy controls and found a diagnostic accuracy of 75% [48]. This doesn’t however speak much to the more pressing question of differentiating TB from non-TB pneumonia. While these are extremely promising results, no such trials have been performed in TB endemic areas, particularly challenging the specificity of LAusc for the detection of either pneumonia or TB. There is also as yet no regulatory approval for the use of AI powered LAusc devices in clinical practice.
This study therefore seeks to enable adoption of LUS into the routine management of IMCI clinical pneumonia through addressing key evidence gaps for translation into policy and practice, which includes generating evidence on the possible impact on health outcomes, namely: Does the integration of LUS into IMCI-based management guidelines better identify those children that would most benefit from antibiotic treatment, thus reducing antibiotic overuse without compromising health outcomes?
Study Objectives
The co-primary objectives of this study are to evaluate whether the integration of LUS into current IMCI-based management guidelines for pneumonia (intervention group) 1) Reduces antibiotic treatment at Day 1, and 2) Is non-inferior in terms of clinical failure by Day 8, among children aged 60 days - 12 years with WHO IMCI clinical pneumonia, when compared to routine care (control group).
The secondary objectives are:
- a)
-
To assess the effect of the intervention (when compared to routine care) in children with WHO IMCI clinical pneumonia on:
- the proportion of children prescribed antibiotics by D8
- the proportion of adverse drug reactions related to routine antibiotic treatment by D8 (i.e., Anaphylactic reaction, severe diarrhoea, or generalised severe rash).
- the proportion of participants cured (defined as caregiver-reported recovery from illness) by D8, and by D29 (for those not cured at D8)
- the proportion and duration of inpatient admissions by D8 and D29 (for those admitted on D1 only).
- the use of diagnostic tests (in addition to LUS) on D1, and for those admitted, by D8.
- death by D29
- the proportion of participants with unscheduled health-seeking events for any cause and/or hospital admission for any cause since D8 follow-up at D29 follow-up
- b)
- To compare, where available, the aetiological diagnoses (e.g., pulmonary tuberculosis, RSV bronchiolitis, confirmed bacterial pneumonia) between the intervention and control group
- c)
-
In the subgroup of patients included in the TB substudy to evaluate:
- what proportion have confirmed, unconfirmed or unlikely intrathoracic TB according to the Classification of Intrathoracic Tuberculosis Case Definitions [49]
- whether there are any specific LUS abnormalities that may distinguish TB from non-TB lung disease.
- LUS compared to both CXR and computer aided detection (CAD) of CXR as potential alternate imaging modalities within TB Treatment Decision Algorithms
- d)
- Estimate the cost-effectiveness of integration of LUS into current IMCI-based management guidelines for pneumonia
Methods and Analysis
Study Design
This is a prospective, open label, individually randomised controlled trial (RCT) with two arms (intervention and control) investigating Lung Ultrasound (LUS) as a novel tool for the management of WHO clinical pneumonia in outpatient and emergency room settings. It is a superiority trial in terms of reduction of antibiotic prescriptions and a non-inferiority trial in terms of clinical outcomes.
Study Setting
Children eligible for this study will be those presenting to participating sites with an acute onset respiratory illness.
South African sites will include: Two hospitals (District or Regional) in the Ehlanzeni District, the largest of 3 districts that make up the Mpumalanga Province as well as Khayelitsha Hospital in the Western Cape Province in South Africa. If accrual is slower than anticipated at these sites then surrounding health care facilities (hospitals and/or clinics) in Mpumulanga and the Western Cape may be added as enrolling sites. Inclusion of these sites will be subject to local institutional approval. These hospitals will be selected based on expected paediatric case load, suitability of the infrastructure for the research question (i.e. an active emergency room and/or Paediatric OPD), and the presence of an identifiable team of clinicians allocated to paediatric care. The Ehlanzeni district includes a rural population undergoing a complex and rapid health and demographic transition underpinned by the well-described South African quadruple burden of disease - continuing high levels of HIV and TB, increasing levels of non-communicable disease, and persisting high levels of maternal and child ill-health and injury. Khayelitsha is equally known to be a densely populated urban environment with a high HIV and TB burden. Malaria, however present to a low degree in Ehlanzeni, is virtually absent in Khayelitsha barring traveller’s malaria. These districts are representative of other urban and rural areas in South Africa and many other LMIC regions in terms of socio-economic status, with a high burden of infectious diseases.
Tanzanian sites will include: 4-5 dispensary/health centres or hospitals in the Dar es Salaam Region serving primary/secondary care. Sites will be chosen to represent the urban/peri-urban area of Dar es Salaam with a low malaria risk/prevalence, low TB, and low HIV prevalence. Sites will be chosen based on number of children seen with LRTI and staffing (clinical officer/assistant medical officer or general medical doctor) in coordination with the district and regional offices.
Senegal Sites will include: Primary care clinics and District hospitals in Dakar, Senegal will be included. Sites will be chosen to represent the urban area of Dakar with a moderate malaria risk/prevalence, low TB, and low HIV prevalence. Sites will be chosen based on number of children seen with LRTI and staffing in coordination with the district and regional offices.
Eligibility Criteria:
To be eligible for inclusion, participants must be aged 60 days to 12 years and presenting for acute care to a study site with WHO IMCI clinical pneumonia, defined as: 1) Cough OR Difficulty Breathing AND, 2) One of the following: 2a. Fast breathing [> 50/minute (2–12 months), > 40/minute (1–<5 years), > 25/minute (5-12 years)] OR 2b. Lower chest wall indrawing.
Participants will be excluded if they meet any of the following criteria:
- Presenting for repeat visit/follow-up of a treated lower respiratory tract infection (index illness / non-acute) or enrolled in the study within the preceding 28 days.
- Currently taking antibiotic treatment for more than 48 hours at the time of enrolment
- WHO IMCI danger signs (inability to drink/breastfeed, vomiting everything, convulsions with this illness, lethargy/unconscious)
- Presence of jaundice
- Hypoxaemia with SpO2<88%
-
SpO2<90% (or country-specific / altitude-adjusted thresholds):
- ○ With signs of severe respiratory distress (such as nasal flaring, grunting, etc.)
- OR
- ○ In children < 6 months
- Requiring non-invasive ventilatory support (i.e., high-flow, bi-PAP, CPAP)
- Underlying disease associated with increased risk of severe pneumonia or pneumonia of unusual aetiology (e.g., WHO acute malnutrition requiring antibiotics as per local guidelines, severe immunodeficiency)
-
HIV positive participant that is either:
- ○ less than 12 months old,
- OR
- ○ requiring admission for this illness,
- OR
- ○ known to be uncontrolled on treatment (with a documented VL >1000c/ml in the previous 6 months)
- Caregiver unavailable at the time of enrolment, or unwilling, to provide informed consent
Children meeting all inclusion criteria, but meeting exclusion criteria 1-9, will be enrolled in an observational component of the study and in the TB substudy.
Study Intervention
The intervention consists of a patient management algorithm, including LUS. The trial intervention is named the “IMCI-PLUS” algorithm (“IMCI-Point-of-care Lung UltraSound algorithm”).
Intervention Group: Management Based on Lung Ultrasound Findings
The intervention will integrate LUS into current management guidelines for pneumonia (national guidelines based on IMCI). Patients will receive LUS on the day of enrolment and clinicians will be provided guidance on the interpretation of the findings and the implications on antibiotic prescription (See Table 1). Additional guidance regarding admission or site of care will not be provided and will be based on local/national guidelines based on IMCI as per routine care.
Those in the intervention group will be sent to the treating clinician with:
- Results of the LUS examination including interpretation
- Advice regarding whether the patient requires antibiotics (according to standard local guidelines) based on pre-defined thresholds.
- In the case of a normal LUS examination, advice against further investigations such as a chest x-ray or inflammatory blood markers (e.g., CRP / PCT) will be provided, although clinicians will be free to add additional tests if so desired.
LUS may be repeated during the acute course of the illness in admitted patients in the case of clinical deterioration or persistence of symptoms. Guidance on the use and interpretation of LUS in these patients will be provided. Refresher training on current local treatment guidelines will be provided prior to the start of the study as with the control group.
Lung Ultrasound
LUS will be performed using a standard point of care device with CE marking by trained study staff impartial to the management of the participant.
A standardised image acquisition and documentation protocol will be followed, adapted from that suggested by Copetti and Cattarossi et al [50]. We opt for an 8-point “Sliding protocol”, where a 10-15second video clip is recorded in sagittal orientation, sliding from the most superior region down until the diaphragm is encountered in the outlined regions. The sonographer is asked to ensure an adequate view of any pathology encountered. An additional video on each lateral region to assess for the “spine sign” is also collected – this is a static/ non-sliding video.
At selected sites, depending on the availability of an appropriate small footprint US probe, additional mediastinal views will be collected for the identification of intrathoracic lymph nodes.
Lung Ultrasound Interpretations/Classifications:
The study clinician performing the ultrasound will receive standardised training on image acquisition and interpretation. This will include adequate practice and examinations with support from a team of expert sonographers. Study clinicians, once confirmed to be at a suitable level of skill, will be responsible to interpret the images and provide classifications. Expert sonographers will provide back-up support to onsite research staff in case of uncertainty in the interpretation of certain images (either in person or remotely) as well as to conduct regular audits of interpretations.
Study clinicians, rather than hospital clinicians, will perform and interpret the LUS examination. This is done to 1) ensure standardisation of the acquisition and interpretation of images, 2) ensure the efficacy of LUS is assessed in a more ideal setting, as inter-user variability is a potential weakness of ultrasound and thus worth reducing for the aims of this study, and 3) acknowledge the current progress being made toward more advanced means of interpretation through the development of AI tools. This would enable the roll out of this intervention in regions with a paucity of ultrasound related expertise.
Interpretation risk categories for the likelihood of a pneumonia that would benefit from antibiotic treatment are outlined below:
- 1)
-
Low:
- a. Normal artefacts (e.g., A-profile),
- AND
- b. No evidence of consolidation
- AND
- c. No complications (No effusions or cavities)
- 2)
-
Intermediate:
- a. Findings compatible with a minor consolidation
- AND/OR
- b. Minor simple effusion
- 3)
-
High:
- a. Findings compatible with a moderate to large consolidation
- AND/OR
- b. Complications (e.g., significant effusions, cavities, abscess)
Exact guidance on image findings that lead to the classifications above will be based on expert consensus.
Using the above classifications, the LUS findings will be integrated into routine care guidelines as follows:
Please note, antibiotic choices for 1&2 will be site appropriate.
- South Africa: Amoxicillin/clavulanate (25mg/kg/dose of Amoxil component), IV/PO 8hrly. Switch to oral after response and cont. to complete x10days), perform blood culture. Some site adaptions will be made where appropriate. E.g., Western Cape: if HIV positive and unsuppressed, Ceftriaxone 80mg/kg (up to 2g) IV/IM + single dose cotrimoxazole
Tanzania: Severe Pneumonia: Benzyl Penicillin 50,000 units/kg IV/IM 6hourly x3days, followed by: a) Ampicillin 50mg/kg 6hrly AND Gentamicin 7.5mg/kg IV/IM OD x5days OR b) Amoxicillin 40mg/kg 8hrly x7days. Very Severe Pneumonia: Ampicillin 50mg/kg 6hrly AND Gentamicin 7.5mg/kg IV/IM OD x5days
- 2.
- South Africa: Oral Amoxicillin 45mg/kg/dose, PO 12hrly x5days
Tanzania: Oral Amoxicillin 25mg/kg/dose, PO 8hrly x5days PLUS Paracetamol/Ibuprofen
Please note, children with a known penicillin allergy should be assessed by the treating clinician and provided a suitable alternative, for example, azithromycin 10mg/kg (up to 500mg) once daily for 3days.
* Definitions as per WHO IMCI, South African/Tanzanian Standard Treatment Guidelines, except:
- HIV exposed, and HIV positive patients older than 12months controlled on treatment, will not be classified as severe/very severe (if not meeting other criteria for severe disease).
For Admitted Patients:
Those in the intervention group with clinical deterioration will have a repeat LUS performed to assess for new evidence of consolidation in patients without antibiotics and to assess for additional/severe complications in those on antibiotics. Guidance will be provided for the interpretation of the findings in line with Table 1 above. Those with “poor response” to 1st line therapy at 72hours (lack of improvement of respiratory distress, persistence of fever >38ºC) will have a repeat LUS performed and interpretations & guidance on management of complications provided in line with Table 1 above.
For Non-Admitted Patients:
All participants will be advised to re-present to the healthcare facility (i.e., the study site) for repeat review as per IMCI guidelines in the case of clinical deterioration or persistence of symptoms >3 days. If an in-person visit on D3 is standard-of-care at the site, then this visit will be conducted at the study site. Transport costs will be re-imbursed to ensure access to care for families in case of clinical deterioration. Those in the intervention group with clinical deterioration will have a repeat LUS performed to assess for new evidence of consolidation in patients without antibiotics and to assess for additional/severe complications in those on antibiotics. Guidance will be provided for the interpretation of the findings in line with Table 1 above.
Control Group: Management Based on Routine Care
The control group will be managed by the treating clinician as per routine care. Routine care in South African, Tanzanian, and Senegalese District/Regional Hospitals are as per the Standard Treatment Guidelines [51] which is based on IMCI (see last column in Table 1). These guidelines recommend antibiotic prescriptions for all cases. In South Africa, despite slight local differences between provinces, recommended investigations include a chest X-ray and a blood culture for those classified as having “Very Severe Pneumonia”. No other investigations are advised for any other group. In Tanzania, recommended investigations include a full blood count, arterial blood gas, C-reactive protein, and chest X-ray, depending on availability [52]. The same follow-up instructions (return in case of clinical deterioration or persistence of symptoms >3 days) will be given to non-admitted patients. If an in-person visit on D3 is standard-of-care at the site, then this visit will be conducted at the study site. Refresher training on current local treatment guidelines will be provided prior to the start of the study. See Figure 1 for basic overview of study flow.
Study Endpoints
Co-Primary Endpoints
- (i)
- Proportion of participants prescribed antibiotic treatment in each study group on D1.
- (ii)
-
Proportion of participants with clinical failure, defined as the development of any of the following criteria during the specified time periods after enrolment.
-
Any time before or on D8:
- ■
- WHO IMCI danger sign (inability to drink/breastfeed, vomiting everything, convulsions with this illness, lethargy/unconscious)
- ■
- New or worsening severe respiratory distress (such as grunting, head nodding, severe chest indrawing)
- ■
- Secondary Hospitalization (defined as a hospitalization occurring after discharge from in-patient admission or outpatient visit) related to a deterioration of the presenting complaint on D1
- ■
- Change in level of care (e.g., admission to intensive care unit, transfer to higher level of care)
- ■
- Need for respiratory support (e.g., high flow nasal cannula, CPAP)
- ■
- Death due to any medical cause (i.e., except trauma)
-
At D8 outcome assessment:
- ■
- Report from the caregiver of non-resolution/worsening of illness
-
Antibiotic treatment is a relevant care-related outcome [53] as we expect that the integration of LUS will result in a more targeted management approach, and hence a decrease, in antibiotic treatment. The most accurate point of assessment and largest effect is expected at the time of the initial management decision. Antibiotic treatment for the acute course of the illness is clinically interesting but subject to reporting bias and additional factors not addressed by the intervention (such as adherence to treatment guidelines and seeking of antibiotic treatment by caregivers) and so we chose to therefore assess cumulative prescription by D8 as a secondary endpoint.
Secondary Endpoints
- a)
-
Evaluation of the effect of the intervention in children with clinical pneumonia on:
- The proportion of children prescribed an antibiotic by D8.
- The proportion of adverse drug reactions related to routine antibiotic treatment by D8 (i.e., Anaphylactic reaction, severe diarrhoea, or generalised severe rash).
- The proportion of patients cured (defined as caregiver reported recovery from illness) at D8 and D29.
- The proportion of patients admitted to hospital on D1
- The duration of the D1 inpatient admissions (by D29)
- The proportion of patients undergoing a non-study related diagnostic test (including CXR, blood tests, urine tests, microbiological assays) on D1, and for those admitted on D1, tests conducted up until D8.
- The proportion of deaths of any cause by D29.
- The proportion of participants with unscheduled health seeking events for any cause and/or hospital admission for any cause since D8 follow-up (at D29 follow-up)
- b)
- Proportion of different aetiological diagnoses (e.g., pulmonary tuberculosis, RSV bronchiolitis, confirmed bacterial pneumonia).
- c)
-
In children enrolled in the TB substudy:
- The proportion of children with confirmed, unconfirmed or unlikely intrathoracic TB
-
The proportion of children:
- ○ meeting exclusion criteria 1-9 and not enrolled in the RCT OR
- ○ enrolled in the RCT and classified as having clinical failure
- who ultimately had TB disease
- The ability of LUS (and mediastinal US, if done) to distinguish TB from non-TB disease
- The diagnostic accuracy of LUS compared with CXR and CAD within the context of TB Treatment Decision Algorithms
- d)
- Cost-effectiveness of integration of LUS into current IMCI-based management guidelines for pneumonia
Recruitment, Screening and Informed Consent Procedure
All paediatric patients (>60 days – 12 years) presenting to the ER or Outpatients Department (OPD) in participating study sites will be triaged as per routine care. Study staff will screen all presenting patients and consecutive patient recruitment will be done for those meeting all inclusion criteria. Those showing signs of severe disease or WHO IMCI Danger signs on triage (and hence meeting exclusion criteria) will be referred immediately to the treating clinicians as per local policy. (See Figure 1)
For those meeting inclusion criteria, sufficient time to read the informed consent form (See cover page for versions) and make an informed decision will be given to the patients/parents/caregivers before inclusion in the study. Information covered will include the nature of the study, its purpose, the procedures involved, the expected duration, the potential risks and benefits and any discomfort it may entail. Each participant and their parents/caregivers will be informed that the participation in the study is voluntary and that they may withdraw from the study at any time and that withdrawal of consent will not affect their subsequent medical assistance and treatment. The participants/parents/caregivers will be informed that the participants medical records may be examined by authorised individuals other than their treating clinician. In the event the participant’s parent/caregiver is unable to read, a member of the study team will read it to them and provide an explanation of all study procedures as described in the informed consent document. A Patient Information Sheet outlining the study and the procedures will also be provided to the participants. There will be sufficient opportunities to ask questions. After this time the study staff will obtain written informed consent from the parent/caregiver of the participant. In the event the parent/caregiver is unable to write/sign, the agreement can be signed by a witness (e.g., a nurse or attending clinician not part of the study team). Assent will be obtained from children 7 -12 years old by a similar process. If a child <7years old, or a participant deemed to be incapable of judgment, shows signs that they are unwilling to participate in the study, the participant will be excluded from participation. A copy of the signed informed consent and if applicable assent will be given to the parent/caregiver of the study participant. The consent and assent forms will be retained as part of the study records. Any refusals will be excluded from the study and participants will receive routine care.
All study documents requiring engagement with participants (e.g., informed consent forms and patient information sheets) will be translated into the local language (e.g., Swahili, Xitsonga/Shangaan, Sepedi, Swati, Afrikaans, Xhosa, French etc) and back translated to ensure accuracy.
There is no compensation or payment foreseen for the participating patients nor will there be costs incurred by the patient for any study related activities, such as consultation or investigation costs at the time of enrolment. Transportation costs for any study related in-person follow-up visits will however be compensated to ensure access to care for all children included into the study in case of clinical deterioration. In South Africa, this will depend on the site, and will be guided by the SAHPRA “Guideline for clinical trial participant time, inconvenience and expense (tie) compensation model”. A standard travel radius/catchment area will be drafted, and a standard amount set.
Study Procedures
The study will aim for a maximum recruitment period of 24months with an initial pilot phase. Each participant included will be followed up for a total of 28days after enrolment (See Figure 1). Those in the TB substudy will be followed-up for a total of 8 weeks from the point of enrolment into the substudy.
Data Collection
Clinical, diagnostic and management data, including screening, planned and unplanned study visits, will be collected and recorded in an eCRF (e.g., REDCap®). For ease of later reference and any repeat presentation to the study site, participants’ hospital folders/health cards will be marked with a study logo/marker. Study staff will receive training to standardize data collection (baseline characteristics, diagnostic test results, and outcomes).
All consented participants will have the following data collected:
Clinical Baseline Data:
This information will be collected by study staff and entered directly into the eCRF.
- Demographic and socio-economic characteristics (including age, sex, maternal education)
-
Relevant medical history such as:
- ○
-
Routine Healthcare card/booklet data
- ■
- Past medical history
- ■
- Documentation of growth pattern
- ■
- Immunisation history
- ○
- HIV status / Past TB history
- ○
- Current medications
- ○
- Visit to a traditional healer for the current illness
- ○
-
Epidemiological exposure including
- ■
- TB contact in the last 12 months Other sick contacts
- Documentation of whether Tuberculin Skin Test was done (and results, if available)
- Other notable symptoms & clinical signs relevant to current presentation (with duration, onset, and severity where applicable)
- Vital signs and anthropometric measurements: Weight, height/length, heart rate, respiratory rate, temperature, oxygen saturation, middle upper arm circumference.
- Documentation of date of CXR, if done, and digital archiving of CXR images
At the time of enrolment and as per local guidelines, the results of tests as part of routine care will be collected, such as Malaria RDT, POC Haemoglobin, and HIV screening.
IMCI-PLUS Data:
- LUS is minimally invasive, contains no ionising radiation and poses no risk to the participants. The LUS will be performed by trained study staff.
- LAusc will be performed by trained study staff.
Assessment of Outcomes:
Outcomes will be assessed through review of their available clinical records (paper record or electronic health record) and standardised patient assessment forms from the study site. For inpatients, daily record review will be conducted along with a D8 follow-up. For outpatients, treatment will be reviewed at enrolment, at unplanned follow-up visits (in person), and at D8 follow-up (see Figure 1, Table 2). The assessments will include:
- Antibiotic-related assessment - Data on antibiotic prescription, duration, type, dosage, and route of administration.
- Event-related data - Clinical failure.
Secondary Outcomes:
For assessment of secondary outcomes, key clinical and management data at enrolment, during inpatient admission, as well as D8 follow-up will be captured by research staff, including:
- Clinician derived key examination findings (including clinician interpretation of CXR or other imaging, if done and documented)
- Initial working diagnosis made by the treating clinician (e.g., IMCI classification) and/or differential diagnoses
-
Management plans at enrolment and unplanned follow-up visits
- ○
- Other investigations requested and their results (e.g., from Blood, Sputum, X-rays etc)
- ○
- Treatment (antibiotics and other)
- ○
- Discharge from OPD/ER or admission
- ○
- Deviation vs conformity to the IMCI-PLUS algorithm advice or to the expected routine care (based on local guidelines)
- ○
- Documentation of whether TB treatment or TB preventive therapy is started
-
If admitted:
- ○
- Length of admission
- ○
- Treatments received during the course of admission and on discharge
- ○
- Investigation results (e.g., Chest X-ray, Blood, Sputum, Urine etc)
- ○
- Details relating to any escalation of care
- ○
- Final hospital diagnosis
- ○
- Final hospital outcome (Referral/transfer, discharge, demise)
- All those meeting clinical failure at D8 or having recurrent/persistent symptoms at D29 will be followed until cured or chronically stable, and will be enrolled, with informed consent, into the TB substudy.
Follow-up of Participants
Details of Day 8 Follow-up Strategy:
On D8, a telephone follow-up will be conducted with the caregiver to assess for current health status, evidence of clinical failure, clinical course since enrolment, and antibiotic use. If caregivers cannot be reached by phone, other strategies will be implemented to reach these caregivers, including home visits, and establishing contact through community stakeholders (community leaders, community health workers). See Figure 2 below for a more detailed algorithm. Based on responses, participants may be flagged for in-person review and advised to return to their health facility/study site for clinical review. Participants will be compensated for transport related costs relating to this repeat visit.
Returning participants will be triaged as per routine outpatient presentation and study staff will assist in screening them for signs of clinical failure. If the participant meets WHO danger sign criteria / signs of severe disease, they will be immediately referred to a hospital clinician. A repeat LUS will be performed for all returning patients with persistent symptoms on D8 and these children will also be enrolled, with informed consent, into the TB substudy.
Unplanned/Spontaneous in-Person Follow-up before D8:
Participants will be managed as per the D8 in-person visit outlined above and as per standard guidelines. However, as per the intervention strategy, those in the intervention group with clinical deterioration will have a repeat LUS performed to assess for new evidence of consolidation in patients without antibiotics and to assess for additional/severe complications in those on antibiotics. These children will also be enrolled, with informed consent, on the TB substudy. Guidance will be provided for the interpretation of the findings in line with Table 1 above.
Day 29-Follow-up
Will be performed telephonically to assess for repeat health seeking events for a respiratory illness, hospitalisation, history of starting TB treatment, and death. All children with persistent or recurrent respiratory illness at D29, as well as those who report starting TB treatment in routine care, will be reviewed in-person and enrolled, with informed consent, into the TB substudy.
Exploratory Outcomes and Additional Investigations
LUS Performed in the Control Group:
The control group will have LUS images collected but not interpreted at the time of enrolment and as such, findings will not affect the initial or ongoing management of the participants. This data will be collected for retrospective review to better understand and characterise the control groups outcomes and to improve risk stratification for pneumonia.
All participants enrolled in the study will meet WHO clinical pneumonia definitions and as such will require antibiotics according to local standard treatment guidelines in the routine care group (control group). As the purpose of the intervention is primarily to use LUS to reduce antibiotics, the negative LUS findings are used to motivate for restrictive use of antibiotics in cases where antibiotics are indicated. Therefore, any potentially abnormal findings in the LUS of the control group not relayed to the health care worker will not have indirect negative effects on patient management and we feel LUS is therefore justified in being collected.
The benefit of integrating LUS findings into the management of pneumonia is yet to be determined and is the focal point of this study. Hence these LUS findings will not be integrated into routine care in the control group.
Lung Auscultation (LAusc)
LAusc sounds will be recorded from all participants using a digital stethoscope (in both the intervention and control groups) pre-randomisation. These sounds will be captured by study staff and will not be interpreted at the point of enrolment. The sounds will be recorded for 10-15 seconds in each of the 8 predefined locations as illustrated in Figure 3 (4 anterior and 4 posterior quadrants = an estimated total procedure time of 2minutes)
Sample Collection and Biobanking
Subject to a separate consent from, samples for biobanking will be collected. These samples will include:
- Nasopharyngeal swab (Flock swob in UTM tube)
- Saliva Sample
- 2x Dried Blood Spot Cards
-
Venous blood sample
- ○
- 2x 0.5 mL venous tubes (SST-like) for serum collection
- ○
- 1x 1.5 mL blood mRNA tube (PAXGene tube for transcriptomics)
We will use topical anaesthetic cream and distraction techniques where possible to minimise distress. Where a participant will require blood sample collection for their routine care, and where appropriate, the collection of the study samples will coincide with this to minimize the number of blood draws required.
A biobank will be developed according to the International Society for Biological and Environmental Repositories (ISBER) best practice recommendations.
For study purposes only, and to address secondary and exploratory objectives, the biobank samples will be used for further microbiological testing (e.g., respiratory viral panel, SARS-CoV-2 genotyping, TB specific molecular and microbiological assays, HIV testing (if result not already available), and other assays for infective organisms), inflammatory biomarkers (CRP, PCT), and TB related mRNA transcriptional signatures.
No host genetic analysis will be performed. No future testing will be done on samples without approval from the relevant Human Research Ethics Committee.
In South Africa, biobank analysis will be done in cooperation with the National Institute of Communicable Diseases (NICD) in Gauteng, South Africa.
Samples from South African sites will be biobanked at an accredited biobanking facility in South Africa such as the Sydney Brenner Institute for Molecular Bioscience (SBIMB) Biobank, South Africa, 9 Jubilee Rd, Parktown, Johannesburg, 2193, South Africa (BEC20200401) (M200469), or the Biomedical Research Unit at Stellenbosch University, Francie van Zyl Drive, Parow, Cape Town, South Africa. As for biobanking in Tanzania, samples will be stored at the MUHAS biobanking facilities.
Withdrawal and Discontinuation
An individual patient may discontinue their participation in the study at any point in time (i.e., refusing further data collection or follow-up) by informing their treating clinician or the study team. At this time, they will be specifically asked whether the data/samples already collected may still be used. If consent to use the data is revoked, the records and samples will be handled as outlined in Section “Confidentiality and Coding”. Anonymity is guaranteed through the process of data coding.
For the follow-up phone calls, the participant’s primary caregiver will be asked to provide the contact of a next-of-kin in the unfortunate event of unavailability, death, or incapacitation.
Inclusion into Observational Cohort Study
Patients screened at the facility who meet all inclusion criteria but also meet any exclusion criteria numbers 1-9 (and will therefore be excluded), will instead be recruited into an observational cohort study to obtain a more detailed understanding of all children presenting with IMCI pneumonia.
These participants will be consented on a separate consent & assent form as appropriate and receive a separate patient information sheet. All above procedures will apply (including data collection, LUS and LAusc collection, biobanking request, clinical outcomes assessment on D1, and clinical data related to any admission). Participants will however not be randomised and will not receive the study intervention. Participants will also receive a D8 and D29 telephone follow-up however, will not be requested to return for an in-person visit for study purposes.
Inclusion in the TB Substudy
The following patients will be eligible for enrolment in the TB substudy:
- Children meeting all inclusion criteria but also meeting any of the exclusion criteria 1-9
- Children <2 years old, living with HIV, and/or having severe acute malnutrition
- Children with clinical failure at D8 or persistent/recurrent symptoms at or D29
These children will be specifically consented either on a separate consent form or on a separate section of the observational cohort consent form depending on their point of entry into the substudy. They will not be randomised unless they are also enrolled in the main trial but they will have all procedures outlined for the main trial and the observational cohort as well as the following additional tests:
- Collection of at least 1 respiratory sample (gastric aspirates and/or induced sputum and/or nasopharyngeal aspirates) for Xpert Ultra MTB/RIF testing and liquid culture
- Urine for lateral flow lipoarabinomannan (LAM) if living with HIV and available at the site
- CXR (both anteroposterior and lateral), if done as an investigation in the routine care pathway, for real-time interpretation by clinicians to facilitate appropriate clinical care and for archiving for retrospective interpretation with CAD software
- If not already collected as part of the biobank main study, collection of a venous blood sample 1x1.5ml blood mRNA tube for biobanking (PaxGene transcriptomics)
All children enrolled in the TB substudy will be linked to the appropriate care within the routine care setting and all will have telephonic follow-up visits with the study team on D8 and D29 as well as an additional telephonic follow-up at week 8. If caregivers cannot be reached by phone at D8 follow-up, other strategies will be implemented to reach these caregivers, including home visits, and establishing contact through community stakeholders (community leaders, community health workers). If there are persistent or recurrent symptoms an in-person visit will be undertaken. In-person visits will also take place if any sample test positive for TB or if a child is started on TB treatment in routine care (to allow the study team to evaluate for treatment response and classification according to the standard NIH TB case classifications [49]).
Statistics and Methodology
Statistical Analysis Plan and Sample Size Calculation
Statistical analysis will be performed by the trial statistician using R or Stata. Analyses will follow CONSORT guidelines [59,60] and intention-to-treat (ITT) principles. Participants missing follow-up information at D8 will be assumed not to have experienced clinical failure for the ITT analysis. A flowchart will describe the inclusion and follow-up of participants by study arm. Baseline characteristics will be described by study group with summary statistics such as median and interquartile range or number and percentage; no formal testing between groups will be performed [61]. Outcomes will be described by group using summary statistics. The first primary outcome, the proportion of children receiving antibiotics on D1, will be assessed for superiority using a logistic regression model, reporting odds ratios and a modified Poisson regression reporting risk ratios and risk differences and their respective 95% confidence intervals (CI). The second primary outcome, the proportion of children with clinical failure by D8, will be evaluated using a CI approach. A figure illustrating the CIs of the risk difference and the non-inferiority margin will be presented. Primary analyses for the non-inferiority comparison will be performed on both the ITT and per protocol sets [62]. If clinical failure in the intervention group is found to be non-inferior to the control, then we will assess for superiority using the ITT set. All models will be adjusted for the stratification factors of site and age group [63].
Binary secondary outcomes with sufficient events will be evaluated in the same way as the primary superiority outcome. Continuous secondary outcomes will be assessed using linear regression models, reporting adjusted mean differences between arms.
Further details will be provided in the statistical analysis plan.
Sample Size
With an assumed baseline proportion of clinical failure in the control group of 5% and using an acceptable clinical difference of 1.5x as a non-inferiority margin (therefore a delta of 2.5%) [54,55,56,57,58], the estimated sample size required across all sites would be 2730 (power 85%). This would equate to 1365 participants per group for the co-primary endpoints. We however aim to enrol a total of 3000 participants under the age of 5 years of age (power >85%) as this is the age group of greatest interest based on current IMCI guidelines. However due to limited data on paediatric pneumonia outcomes in the 5-12years age group we wish to include these children as well. Considering approximately 90% of children presenting for acute respiratory care in the age group 60days-12years are <5years, we would aim for a total sample size of 3500 participants, 1750 participants per group, to account for repeat visits of the same child within the enrolment period as well as potential differences in clinical failure rates between sites. We will conduct an interim analysis assessing, among other things, LTFU and clinical failure rates. This will be done approximately 25% through the RCT and the sample size adjusted accordingly if appropriate. The planned breakdown of participant enrolment per country is as follows: Tanzania – 2000 participants, Senegal – 500 participants, South Africa – 1000 participants. As the expected difference in antibiotic prescription rates is expected to be large, this sample size will provide sufficient power to test for a clinically meaningful difference of 20% on Day 1 and 15% by Day 8 (secondary outcome).
Methods of Minimising Bias
Randomisation
Participants will be randomised 1:1 to intervention and control. Participants will be randomized using block randomization with varying block sizes, stratified by site and age groups (<5, 5-12), based on randomization lists generated by an independent statistician and uploaded into the study database. If internet connectivity is of concern at the site, offline randomization will be used.
Blinding
Study site blinding is not feasible in this trial and therefore it will be an open-label study. Hospital clinicians will not be able to perform LUS or receive the LUS findings for the control group as the use of LUS is not standard practice for routine management in the site hospitals. Only study staff will be conducting LUS and clinicians will be requested not to use LUS during the study.
Standardisation of assessment and management between both groups will be achieved through the creation of standardised patient assessment forms (e.g., with triage vitals, WHO pneumonia criteria, and LUS findings, interpretation, and guidance (if appropriate)) and refresher training on local treatment guidelines provided to hospital clinicians and nurses who will be treating pneumonia cases in both groups of the study.
Due to the open-label nature of the design, there is a minor risk of cross contamination. Clinicians seeing participants with negative LUS findings doing well without antibiotics may begin to change their practice with participants in the control group – assuming in cases with minor symptoms that it may be viral and not require antibiotics (despite not having LUS findings available). This potential decrease in antibiotic prescription in the control group may decrease the overall measured impact of the intervention.
Study personnel assessing the endpoints during follow-up will be blinded to the allocation to the best extent possible.
Per Protocol Deviations:
Admitted participants may be subject to additional tests as per routine hospital practice such as inflammatory markers and CXR. Relying on these additional tests may result in a higher rate of per protocol deviations in the intervention group with clinicians overruling the “no antibiotic” recommendation. This could be mitigated through baseline training on the role and interpretation of inflammatory markers and CXRs as well as the presentation of current data to support the diagnostic potential of LUS for paediatric pneumonia.
Handling of Missing Data and Drop-Outs
Missing baseline and outcome data will be summarized by study group. Participants with missing follow-up data at day 8 will not be replaced but considered to not have experienced clinical failure. In the case of extensive missing data, we may consider appropriate methods to account for missingness such as adjusting for further baseline variables which are associated with missing outcome data or multiple imputation as appropriate. Details will be provided in the statistical analysis plan.
Economic Evaluation
We will conduct a multi-national economic evaluation of integrating the Lung Ultrasound (LUS) into current IMCI based management guidelines for pneumonia for Senegal, South Africa and Tanzania. We will conduct a cost effectiveness analysis, using natural units informed by the trial outcome such as a percentage reduction in antibiotic prescribing rate and a cost utility study using Disability Adjusted Years (DALYs) as a measure of health outcome. Our study comparators are current IMCI based management for pneumonia for each country (current practice) versus current IMCI based management for pneumonia for each country plus LUS (IMCI-PLUS algorithm).
We anticipate that the heterogeneity in resource use and costs across the three countries is potentially substantial given their differing health systems therefore, study perspective will not be uniform across countries. Public primary healthcare in South Africa is largely free to the patient and almost all costs are borne by the government therefore the economic evaluation will be done from the government’s perspective. Patients in Senegal and Tanzania bear a significant portion of healthcare costs when accessing public primary care services so the analysis in these two countries will be done from a societal perspective.
The target population for the analysis are children aged 60 days-12 years with WHO IMCI clinical pneumonia. Specifications of the target population including exclusion and inclusion criteria are described elsewhere in the protocol. The time horizon for the analysis is equivalent to the treatment duration of a pneumonia episode, including a possible second-line treatment.
We will perform the economic evaluation of using an excel decision analytic model (Figure 4). We will conduct a split analysis using a multi country costing approach. We assume that clinical effects are homogenous across the three countries but not costs. We will apply unit cost estimates from individual countries to counts of resource use in each of those countries. Costs of health care and illness will be estimated at individual patient level, including costs to health services and patient participants of health care utilisation such as primary care visits, specialist referrals, emergency care visits, hospital admissions, user fees.
Identification and quantification of resources used by each participant during the trial will be extracted from patient notes, electronic medical records, patient administration systems and a trial specific form. Unit cost of resources provided by the government will be obtained from respective government documents and market prices will be used for out-of-pocket payments incurred by patients. Monetary values will be expressed in each country’s respective currencies, i.e. South African Rand, Franc of West Africa for Senegal and Tanzanian Shilling and the United States Dollar. There will be no discounting as the time horizon is less than a year.
Using the average costs and outcomes for each alternative (i.e., for each arm in the trial), the two alternatives will be compared to calculate the incremental costs and compare them to the incremental effectiveness outcomes. The results will be expressed as an Incremental cost effectiveness ratio (ICER) which is a measure of the additional cost per unit increase of benefit in the primary outcome (reduction of antibiotic prescriptions). The following formula represents the calculation of the incremental cost-effectiveness ratio (ICER):
The results of the cost-effectiveness analysis will be compared to the South African specific threshold to determine whether the new intervention is cost-effective or not. For Senegal and Tanzania, the WHO recommended threshold will be used. Deterministic and probabilistic sensitivity analysis will be carried out to assess the degree of uncertainty in the ICER. Results will be presented on tornado diagrams, scatter plots and cost-effectiveness acceptability curves.
In addition, we will carry out a cost utility analysis, Disability Adjusted Life Years (DALYs) as outcome. DALYs will be determined by calculating years of life lived with disability and years of life lost to premature mortality based on outcome data from the trial and country specific life tables. This will estimate the incremental cost per DALYs averted and the probability of cost-effectiveness at various levels of willingness to pay for a DALY.
A budget impact assessment for the South African, Tanzanian, and Senegalese governments from the healthcare payer’s perspective will be carried out for a 5-year time horizon. A model will be developed to describe the financial resources associated with the usual practice over the course of the National Department of Health’s budgeting cycle (one year). This will represent the base case or ‘reference case’. The comparative scenario will model the required changes in health service resourcing that are expected to result from the adoption of the IMCI-PLUS program, including indirect and downstream impacts on other parts of the health service. Resource use data will be sourced from the costing analysis. There will be no discounting of costs as a budget impact analysis aims to present the actual financial implications required to deliver the new intervention over each year of the analysis. Inflation rates will be applied to costs/prices of resources used in the model for subsequent years. All model assumptions and data inputs will be described in full. Justification for the inclusion or exclusion of relevant model parameters will be provided. We anticipate using a simple spreadsheet-based model for the analysis. Scenario sensitivity analyses will be conducted to assess the impact of varying model inputs on the affordability of the IMCI-PLUS program
Ethics and Dissemination
Overall Ethical Considerations
Though LUS is increasingly used in the management of clinical pneumonia in children, its additional benefit has not been rigorously studied. This is particularly relevant in IMCI settings, where additional diagnostics must demonstrate potential clinical benefit before integration into current management guidelines is considered. The inclusion criteria where chosen based on current IMCI guidelines, allowing for interpretation (including generalizability) within the IMCI framework. Randomization of participants is necessary since there is currently no diagnostic reference standard for bacterial pneumonia (e.g., a reference standard for the need for antibiotic treatment) and diagnostic accuracy studies for LUS against other reference standards (e.g., radiography or lab investigations) are of limited use.
Research Ethics Approval
Ethics approval for this study was obtained from the Cantonal Ethics Committee for Research, Bern, Switzerland (2023-01534), Muhimbili University of Health and Allied Sciences Research Ethics Committee, Dar es Salaam, Tanzania (MUHAS-REC-02-2024-2032), and the National Institute for Medical Research, Dar es Salaam, Tanzania (NIMR/HQ/R.8a/Vol.IX/4559). This protocol is also currently under review in South Africa by the Wits Health Consortium Human Research Ethics Committee and Stellenbosch University Human Research Ethics Committee. This trial is currently being registered with the South African National Clinical Trials Registry.
Risk-Benefit Assessment
The use of LUS may lead to better targeting of children in need for antibiotic treatment. This reduction in antibiotic treatment is expected to reduce side effects for patients, long-term detrimental health effects for patients from antibiotic use (including asthma, auto-immune conditions, diabetes, obesity, and neurodevelopmental conditions), and antimicrobial resistance, which is a major global health problem. The risk of the intervention is judged as comparable to standard medical care. The proposed intervention is based on previously validated tests. The risks of this study are minimal.
A potential risk is related to a participant with bacterial pneumonia not receiving an antibiotic. However, there are several layers of safety included in the study design to mitigate this risk:
- Eligibility criteria have been carefully selected to only include children that may truly benefit from the intervention. Patients with very severe symptoms requiring close inpatient monitoring are excluded. Patients with a higher baseline risk for bacterial pneumonia (immunodeficiency, severe acute malnutrition) will be excluded.
- A comprehensive risk management plan will be developed
- The establishment of an Independent Data Monitoring Committee (IDMC) for regular review of cumulative safety and study conduct data. The responsibility of the IDMC will be to safeguard the interests of trial participants, assess the safety of the interventions during the trial, and contribute to monitoring the overall conduct of the clinical trial. The IDMC is independent of, but reports to, the trial coordination group. The specific roles of the IDMC are to monitor evidence for treatment harm (e.g., Serious Adverse Events (SAEs)), recommend whether the trial should continue to recruit participants or whether recruitment should be stopped due to safety reasons. The trial statistician or a named independent delegate will produce the report to the IDMC and will participate in IDMC meetings. Further details of IDMC functioning and procedures will be provided by the Sponsor Representative in the IDMC Charter agreed and signed by all IDMC members.
Data Recording and Source Data
The electronic case report form (eCRF) will be set up by the Sponsor Representative. Individual accesses to eCRF will be given to local investigators and delegated study personnel in order to report site-specific coded study data. Data will be entered into the eCRF (e.g., REDCap®) by the study team and validated for completeness and discrepancies automatically. An audit trail system maintains a record of initial entries and changes (reasons for changes, time and date of changes, user identification of entry and changes).
Source data will be available at each site for each participant. The source data will include the original documents relating to the study, as well as the medical treatment and medical history of the participant. Each site will define the unique source data corresponding to each eCRF entry in a study-specific “Source Data Location List” (SDLL), allowing both the local investigators and monitors to access the same source. Direct entry into the eCRF will be allowed for certain data clearly identified in the SDLL. Identifying data for phone follow-up visits will be collected and kept locally at the site.
Confidentiality and Coding
Data Management and Coding
Applicable laws and regulations will be adhered to, including but not limited to the General Data Protection Regulations (GDPR) and Protection of Personal Information Act 4 of 2013 (POPIA), pertaining to the use, transfer, processing and storage of Data [70].
Trial and participant data will be handled with uttermost discretion and will only be accessible to authorised personnel who require the data to fulfil their duties within the scope of the study. On the CRFs and other study specific documents, participants are only identified by a unique participant number. Participant identifying data such as names and contact details will be captured on a single form with access restriction - accessible only to site staff for quality control, participant follow-up, and data linking purposes. Participant number coding will be done using an Electronic Data Capture (EDC) software. Biological material in this study will not be identified by participant name but by a unique participant number. Biological material will be appropriately stored in a restricted area only accessible to the authorised personnel. Data management will be defined in greater detail in a data management plan that will be put in place before initiation of the first site.
Data Security, Access and Back-up
The study team of a local site will only have access to the data of the participants and to the data entered in this center. The study team of a local site will access the data through a tablet computer or a computer. Access to the secured platform (REDCap®) will be password-protected. Back-ups will be performed on a regular basis.
Members of the site study coordination team will be authorized to enter and access data into the eCRF for the specific sections of which they are responsible (i.e., follow-up visits if applicable). They can access these data for all the patients included in the study. Only the study identifier will be entered into data storage applications of the portable ultrasound devices.
Analysis and Archiving
Completion status of each section/form of the eCRF will be predefined during database development. The system will include visual aids to inform of data entry completion.
The database will be locked after study data validation activities and monitoring review have been completed. After study completion and publication of the dataset, apart from the information directly related to the site and potentially identifying information (e.g., date of consultation), data will be made accessible in an open access data repository after the main study results are published. Data will be made available to other researchers pending approval by the Sponsor Representative.
Retention and Destruction of Study Data and Biological Material
All study data are archived for 10 years after study termination or premature termination of the study. The source data will remain in the respective hospitals and stored according to specific regulation.
The biobank, as according to best practice recommendations, ensures that participants are aware of their right to withdraw, without justification, penalty or disadvantage, the requirements for exercising that right, and the implications of and any limits to exercising that right.
The Sponsor Representative ensures that traceability of the samples and data is possible to enable participant withdrawal.
Different levels of withdrawal may apply including:
- No further contact: No further contact is made with the participant but previously obtained biospecimens and data are permitted to be retained and used, and further data can be obtained from health records.
- No further access: No further contact with the participant or access to health records is permitted but previously obtained biospecimens and data can be continued to be stored and used.
- No further use: No further contact would be made with the participant, and data and biospecimens would no longer be available for research. It would be necessary to destroy biospecimens, with archiving of data for audit purposes only.
Participant samples collected for biobanking will be frozen and kept anonymous (only with code) for a maximum of 10 years. The samples will be destroyed after 10 years, or before by decision of the study management. No future testing will be done beyond analyses specified in this protocol on samples without approval of the EC.
Samples from the South African sites will be stored at an accredited biobanking facility, such as: Sydney Brenner Institute for Molecular Bioscience (SBIMB) Biobank, South Africa, 9 Jubilee Rd, Parktown, Johannesburg, 2193, South Africa (BEC20200401) (M200469). Should biobanking be added in Tanzania, samples will be stored at the MUHAS biobanking facilities.
Monitoring and Registration
The monitoring activities will be coordinated by the Swiss TPH Clinical Operations Unit. Local research units will be contracted by Clinical Operations Unit to perform monitoring activities at local sites.
A risk-adapted monitoring strategy will be developed in accordance with relevant regulations and the monitoring strategy (nature and extent of the monitoring activities) will be described into a joint monitoring plan. In principle, site initiation visits will be conducted on each site in order to ensure that all sites are ready to start the trial. Then interim monitoring visits will be performed remotely or on site depending on the situation and focusing mainly on safety, intervention adherence, and patient eligibility. Data from participant records (source documents) will be reviewed by local monitors against data reported in the eCRF in order to check completeness, validity, consistency, and adherence to protocol specifications. Queries regarding missing, questionable or inconsistent data will be documented. Monitoring activities will be documented in a monitoring report after each monitoring visit. A (remote) closeout monitoring visit per study site will be performed at the end of the trial to ensure all pending actions are resolved and investigator site file is ready for archival.
Funding
The study is financed by grant PZ00P3_193342 of the Swiss National Science Foundation
(Ambizione Call 2020) and by grant 101145822 of the European and Developing Countries Clinical Trials Partnership (EDCTP3). There is no other financial support.
Publication and Dissemination
The data from all centres will be analysed together and published as soon as possible in peer-reviewed journals, as well as being presented at national and/or international conferences. Individual groups and clinicians must not publish data concerning their participants that are directly relevant to questions posed by the study until the study coordination group has published its report. The study coordination group will form the basis of the Writing Committee and will advise on the nature of all publications.
Then, results will be disseminated through relevant clinical and policy groups. The results of the study will also be presented in national and international conferences relevant to the field. Several publications in high-impact factor journals are expected to be produced out of this trial. Reporting of the trial will follow CONSORT 2010 guidelines for reporting and ICMJE recommendations for authorship.
References
- World Health Organisation (WHO). WHO Fact Sheet: The top 10 causes of death (2020) [Internet]. 2020 [cited 2023 Mar 29]. Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- WHO. Pneumonia Fact Sheet [Internet]. 2022 [cited 2023 Mar 7]. Available from: https://www.who.int/news-room/fact-sheets/detail/pneumonia.
- Gera T, Shah D, Garner P, Richardson M, Sachdev HS. Integrated management of childhood illness (IMCI) strategy for children under five. Vol. 2016, Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd; 2016.
- World Health Organisation (WHO). Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022.
- Risk R, Naismith H, Burnett A, Moore SE, Cham M, Unger S. Rational prescribing in paediatrics in a resource-limited setting. Arch Dis Child. 2013;98(7):503–9.
- Keitel K, Kagoro F, Samaka J, Masimba J, Said Z, Temba H, et al. A novel electronic algorithm using host biomarker point-of-care tests for the management of febrile illnesses in Tanzanian children (e-POCT): A randomized, controlled non-inferiority trial. PLoS Med. 2017 Oct 1;14(10).
- D’Acremont V, Kilowoko M, Kyungu E, Philipina S, Sangu W, Kahama-Maro J, et al. Beyond Malaria — Causes of Fever in Outpatient Tanzanian Children. New England Journal of Medicine. 2014;370(9):809–17.
- O’Brien KL, Baggett HC, Brooks WA, Feikin DR, Hammitt LL, Higdon MM, et al. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: the PERCH multi-country case-control study. The Lancet. 2019;394(10200):757–79.
- Murray CJ, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet. 2022;399(10325):629–55.
- Laxminarayan R, Duse A, Wattal C, Zaidi AKM, Wertheim HFL, Sumpradit N, et al. Antibiotic resistance-the need for global solutions [Internet]. Vol. 13, The Lancet Infectious Diseases. Elsevier; 2013. p. 1057–98.
- Costelloe C, Metcalfe C, Lovering A, Mant D, Hay AD. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: Systematic review and meta-analysis. Vol. 340, BMJ (Online). British Medical Journal Publishing Group; 2010 [cited 2023 Jul 10]. p. 1120.
- Holmes AH, Moore LSP, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, et al. Understanding the mechanisms and drivers of antimicrobial resistance [Internet]. Vol. 387, The Lancet. Lancet Publishing Group; 2016. p. 176–87.
- van de Maat J, De Santis O, Luwanda L, Tan R, Keitel K. Primary Care Case Management of Febrile Children: Insights From the ePOCT Routine Care Cohort in Dar es Salaam, Tanzania. Front Pediatr. 2021;9:626386.
- Levine GA, Bielicki J, Fink G. Cumulative Antibiotic Exposure in the First 5 Years of Life: Estimates for 45 Low- and Middle-Income Countries From Demographic and Health Survey Data. Clin Infect Dis. 2022;75(9):1537–47.
- Fink G, D’Acremont V, Leslie HH, Cohen J. Antibiotic exposure among children younger than 5 years in low-income and middle-income countries: a cross-sectional study of nationally representative facility-based and household-based surveys. Lancet Infect Dis. 2020;20(2):179–87.
- Sulis G, Adam P, Nafade V, Gore G, Daniels B, Daftary A, et al. Antibiotic prescription practices in primary care in low- And middle-income countries: A systematic review and meta-analysis. PLoS Med. 2020;17(6):e1003139.
- UK Health Security Agency. English surveillance programme for antimicrobial utilisation and resistance (ESPAUR) report 2021 to 2022. 2022.
- Swedres-Swarm. Sales of antibiotics and occurrence of resistance in Sweden. [Internet]. Solna/Uppsala; 2021. Available from: www.folkhalsomyndigheten.sewww.sva.sewww.folkhalsomyndigheten.se/publicerat- material/oratwww.sva.se/swedres-svarm/.
- Ardillon A, Ramblière L, Kermorvant-Duchemin E, Sok T, Zo AZ, Diouf JB, et al. Inappropriate antibiotic prescribing and its determinants among outpatient children in 3 low- and middle-income countries: A multicentric community-based cohort study. PLoS Med. 2023;20(6):e1004211.
- Klein EY, Van Boeckel TP, Martinez EM, Pant S, Gandra S, Levin SA, et al. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc Natl Acad Sci U S A. 2018;115(15):E3463–70.
- O’Neill, J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. 2014.
- Allwell-Brown G, Hussain-Alkhateeb L, Kitutu FE, Strömdahl S, Mårtensson A, Johansson EW. Trends in reported antibiotic use among children under 5 years of age with fever, diarrhoea, or cough with fast or difficult breathing across low-income and middle-income countries in 2005–17: a systematic analysis of 132 national surveys from 73 countries. Lancet Glob Health. 2020;8(6):e799–807.
- Duong QA, Pittet LF, Curtis N, Zimmermann P. Antibiotic exposure and adverse long-term health outcomes in children: A systematic review and meta-analysis [Internet]. Vol. 85, Journal of Infection. W.B. Saunders Ltd; 2022. p. 213–300. Available from: https://pubmed.ncbi.nlm.nih.gov/35021114/.
- Keitel K, Samaka J, Masimba J, Temba H, Said Z, Kagoro F, et al. Safety and Efficacy of C-reactive Protein-guided Antibiotic Use to Treat Acute Respiratory Infections in Tanzanian Children: A Planned Subgroup Analysis of a Randomized Controlled Noninferiority Trial Evaluating a Novel Electronic Clinical Decision Algorithm (ePOCT). Clinical Infectious Diseases. 2019;69(11):1926–34.
- Althaus T, Greer RC, Swe MMM, Cohen J, Tun NN, Heaton J, et al. Effect of point-of-care C-reactive protein testing on antibiotic prescription in febrile patients attending primary care in Thailand and Myanmar: an open-label, randomised, controlled trial. Lancet Glob Health. 2019;7(1):e119–31.
- Cortellaro F, Colombo S, Coen D, Duca PG. Lung ultrasound is an accurate diagnostic tool for the diagnosis of pneumonia in the emergency department. Emergency Medicine Journal. 2012;29(1):19–23.
- Lichtenstein DA, Mezière GA. Relevance of lung ultrasound in the diagnosis of acute respiratory failure the BLUE protocol. Chest. 2008;134(1):117–25.
- Chavez MA, Shams N, Ellington LE, Naithani N, Gilman RH, Steinhoff MC, et al. Lung ultrasound for the diagnosis of pneumonia in adults: A systematic review and meta-analysis. Respir Res. 2014;15(1).
- Stewart KA, Navarro SM, Kambala S, Tan G, Poondla R, Lederman S, et al. Trends in Ultrasound Use in Low and Middle Income Countries: A Systematic Review. International Journal of Maternal and Child Health and AIDS (IJMA). 2020;9(1):103–20.
- Reissig A, Copetti R, Mathis G, Mempel C, Schuler A, Zechner P, et al. Lung ultrasound in the diagnosis and follow-up of community-acquired pneumonia: A prospective, multicenter, diagnostic accuracy study. Chest. 2012;142(4):965–72.
- Heuvelings CC, Bélard S, Familusi MA, Spijker R, Grobusch MP, Zar HJ. Chest ultrasound for the diagnosis of paediatric pulmonary diseases: a systematic review and meta-analysis of diagnostic test accuracy. Br Med Bull.;129(1):35–51.
- Balk DS, Lee C, Schafer J, Welwarth J, Hardin J, Novack V, et al. Lung ultrasound compared to chest X-ray for diagnosis of pediatric pneumonia: A meta-analysis. Pediatr Pulmonol. 2018 Aug 1;53(8):1130–9.
- Malla D, Rathi V, Gomber S, Upreti L. Can lung ultrasound differentiate between bacterial and viral pneumonia in children? Journal of Clinical Ultrasound. 2021 Feb 1;49(2):91–100.
- Ginsburg AS, Vitorino P, Qasim Z, Lenahan JL, Hwang J, Lamorte A, et al. Lung ultrasound patterns in paediatric pneumonia in Mozambique and Pakistan. ERJ Open Res. 2021 Jan;7(1):00518–2020.
- McIntosh, K. Community-Acquired Pneumonia in Children. New England Journal of Medicine. 2002;346(6):429–37.
- Duke, T. What the PERCH study means for future pneumonia strategies. Vol. 394, The Lancet. Lancet Publishing Group; 2019. p. 714–6.
- Amatya Y, Russell FM, Rijal S, Adhikari S, Nti B, House DR. Bedside lung ultrasound for the diagnosis of pneumonia in children presenting to an emergency department in a resource-limited setting. Int J Emerg Med. 2023;16(1).
- Ellington LE, Gilman RH, Chavez MA, Pervaiz F, Marin-Concha J, Compen-Chang P, et al. Lung ultrasound as a diagnostic tool for radiographically-confirmed pneumonia in low resource settings. Respir Med. 2017;128:57–64.
- Chavez MA, Naithani N, Gilman RH, Tielsch JM, Khatry S, Ellington LE, et al. Agreement Between the World Health Organization Algorithm and Lung Consolidation Identified Using Point-of-Care Ultrasound for the Diagnosis of Childhood Pneumonia by General Practitioners. Lung. 2015;193(4):531–8.
- Pervaiz F, Chavez MA, Ellington LE, Grigsby M, Gilman RH, Miele CH, et al. Building a Prediction Model for Radiographically Confirmed Pneumonia in Peruvian Children: From Symptoms to Imaging. Chest. 2018;154(6):1385–94.
- Seif El Dien HM, Abd Ellatif DAK. The value of bedside Lung Ultrasonography in diagnosis of neonatal pneumonia. Vol. 44, Egyptian Journal of Radiology and Nuclear Medicine. Elsevier B.V.; 2013. p. 339–47.
- World Health Organisation. WHO operational handbook on tuberculosis: Module 5: Management of tuberculosis in children and adolescents. 2022.
- Fentress M, Ezibon P, Bulabek A, Schwanfelder C, Schrift D, Shah S, et al. A lung ultrasound scanning technique for children and adults in low-resource settings: Preliminary experiences in Sub-Saharan Africa. American Journal of Tropical Medicine and Hygiene. 2021;105(5):1148–51.
- Nadimpalli A, Tsung JW, Sanchez R, Shah S, Zelikova E, Umphrey L, et al. Feasibility of training clinical officers in point-of-care ultrasound for pediatric respiratory diseases in Aweil, South Sudan. American Journal of Tropical Medicine and Hygiene. 2019;101(3):689–95.
- Pervaiz F, Hossen S, Chavez MA, Miele CH, Moulton LH, McCollum ED, et al. Training and standardization of general practitioners in the use of lung ultrasound for the diagnosis of pediatric pneumonia. Pediatr Pulmonol. 2019;54(11):1753–9.
- Coppock H, Gaskell A, Tzirakis P, Baird A, Jones L, Schuller B. End-to-end convolutional neural network enables COVID-19 detection from breath and cough audio: A pilot study. BMJ Innov. 2021;7(2):356–62.
- Imran A, Posokhova I, Qureshi HN, Masood U, Riaz MS, Ali K, et al. AI4COVID-19: AI enabled preliminary diagnosis for COVID-19 from cough samples via an app. Inform Med Unlocked. 2020;20.
- Becker KW, Scheffer C, Blanckenberg MM, Diacon AH. Analysis of adventitious lung sounds originating from pulmonary tuberculosis. In: Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, EMBS. Annu Int Conf IEEE Eng Med Biol Soc; 2013. p. 4334–7.
- Graham SM, Cuevas LE, Jean-Philippe P, Browning R, Casenghi M, Detjen AK, et al. Clinical Case Definitions for Classification of Intrathoracic Tuberculosis in Children: An Update. Clinical Infectious Diseases. 2015 Oct 15;61:S179–87.
- Copetti R, Cattarossi L. Ultrasound diagnosis of Pneumonia in Children. Chest Radiology. 2008 Mar;113(2):190–8.
- South African Department of Health. Standard Treatment Guidelines & Essential Medicines List for South Africa - Hospital Level - Paediatrics. 4th Edition. 2017. Available from: http://www.health.gov.za/index.php/standard-treatment-guidelines-.
- Tanzanian Ministry of Health. Standard Treatment Guidelines & National Essential Medicines List - Tanzanian Mainland. 5th Edition. 2018. Available from: https://hssrc.tamisemi.go.tz/storage/app/uploads/public/5ab/e9b/b21/5abe9bb216267130384889.pdf.
- Schweitzer VA, van Werkhoven CH, Rodríguez Baño J, Bielicki J, Harbarth S, Hulscher M, et al. Optimizing design of research to evaluate antibiotic stewardship interventions: consensus recommendations of a multinational working group. Vol. 26, Clinical Microbiology and Infection. Elsevier B.V.; 2020. p. 41–50.
- Fox MP, Thea DM, Sadruddin S, Bari A, Bonawitz R, Hazir T, et al. Low Rates of Treatment Failure in Children Aged 2–59 Months Treated for Severe Pneumonia: A Multisite Pooled Analysis. Clinical Infectious Diseases. 2013;56(7):978–87.
- Ginsburg AS, Mvalo T, Nkwopara E, McCollum ED, Phiri M, Schmicker R, et al. Amoxicillin for 3 or 5 Days for Chest-Indrawing Pneumonia in Malawian Children. New England Journal of Medicine. 2020;383(1):13–23.
- EMPIC Study Group. Innovative, enhanced community management of non-hypoxaemic chest-indrawing pneumonia in 2-59-month-old children: A cluster-randomised trial in Africa and Asia. BMJ Glob Health. 2022;7(1):e006405.
- Jehan F, Nisar I, Kerai S, Balouch B, Brown N, Rahman N, et al. Randomized Trial of Amoxicillin for Pneumonia in Pakistan. New England Journal of Medicine. 2020 Jul 2;383(1):24–34.
- Ahmed S, Ariff S, Muhammed S, Rizvi A, Ahmed I, Soofi SB, et al. Community case management of fast-breathing pneumonia with 3 days oral amoxicillin vs 5 days cotrimoxazole in children 2-59 months of age in rural Pakistan: A cluster randomized trial. J Glob Health. 2022 Dec 29;12:04097.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 Statement: Updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol. 2010 Aug 1;63(8):834–40.
- Piaggio G, Elbourne DR, Pocock SJ, Evans SJW, Altman DG. Reporting of Noninferiority and Equivalence Randomized Trials Extension of the CONSORT 2010 Statement. JAMA. 2012;308(24):2594–604.
- Pocock SJ, Assmann SE, Enos LE, Kasten LE. Subgroup analysis, covariate adjustment and baseline comparisons in clinical trial reporting: current practice and problems. Stat Med. 2002;21:2917–30.
- Committee for Proprietary Medicinal Products (CPMP). Points to consider on switching between superiority and non-inferiority. Br J Clin Pharmacol. 2001;52(3):223.
- Kahan BC, Morris TP. Reporting and analysis of trials using stratified randomisation in leading medical journals: review and reanalysis. BMJ. 2012;345(7879).
- WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects – WMA – The World Medical Association. Available from: https://www.wma.net/policies-post/wma-declaration- of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/.
- SR 810.30 - Federal Act of 30 September 2011 on Research involving Human Beings (Human Research Act, HRA). Available from: https://www.fedlex.admin.ch/eli/cc/2013/617/en.
- ICH E6 (R2) Good clinical practice - Scientific guideline | European Medicines Agency. Available from: https://www.ema.europa.eu/en/ich-e6-r2-good-clinical-practice-scientific-guideline.
- Cancer Institute N. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0. 2009. Available from: http://www.meddramsso.com.
- SR 810.305 - Ordinance of 20 September 2013 on Clinical Trials in Human Research (Clinical Trials Ordinance; ClinO). Available from: https://www.fedlex.admin.ch/eli/cc/2013/643/en.
- ICH Topic E 2 A Clinical Safety Data Management: Definitions and Standards for Expedited Reporting Step 5 Note for guidance on clinical safety data management: definitions and standards for expedited reporting. 1995. Available from: http://www.emea.eu.int.
- SR 235.1 - Federal Act of 19 June 1992 on Data Protection (FADP). Available from: https://www.fedlex.admin.ch/eli/cc/1993/1945_1945_1945/en.
Figure 1.
Flow Diagram of Study Procedures.
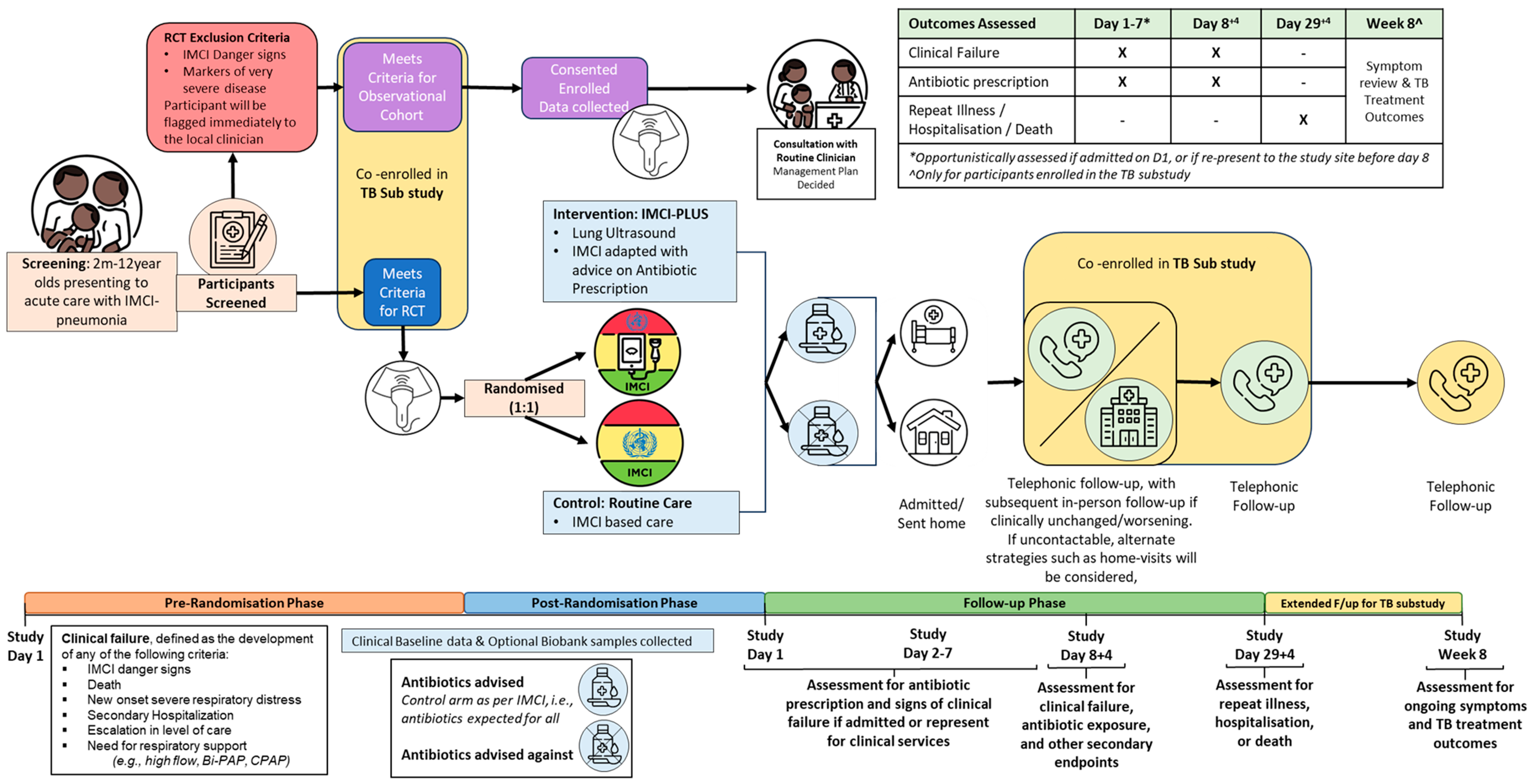
Figure 2.
Schematic Presentation of Day 8 Follow-up. *Compensation for transportation to the healthcare facility will be reimbursed accordingly. Note: If caregivers cannot be reached by phone, other strategies will be implemented (such as home visits or establishing contact through community stakeholders).
Figure 2.
Schematic Presentation of Day 8 Follow-up. *Compensation for transportation to the healthcare facility will be reimbursed accordingly. Note: If caregivers cannot be reached by phone, other strategies will be implemented (such as home visits or establishing contact through community stakeholders).
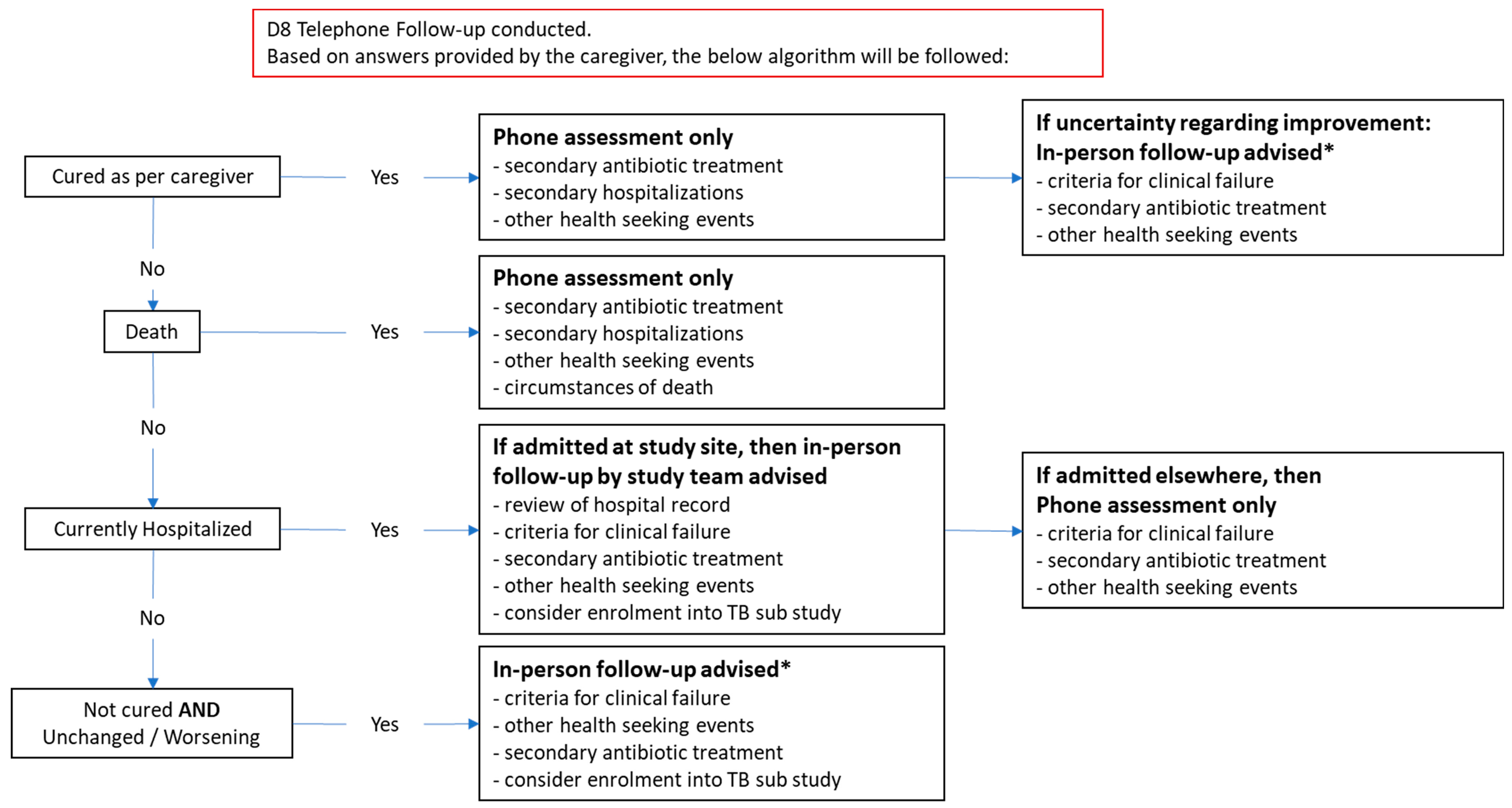
Figure 3.
Lung Auscultation Sites.
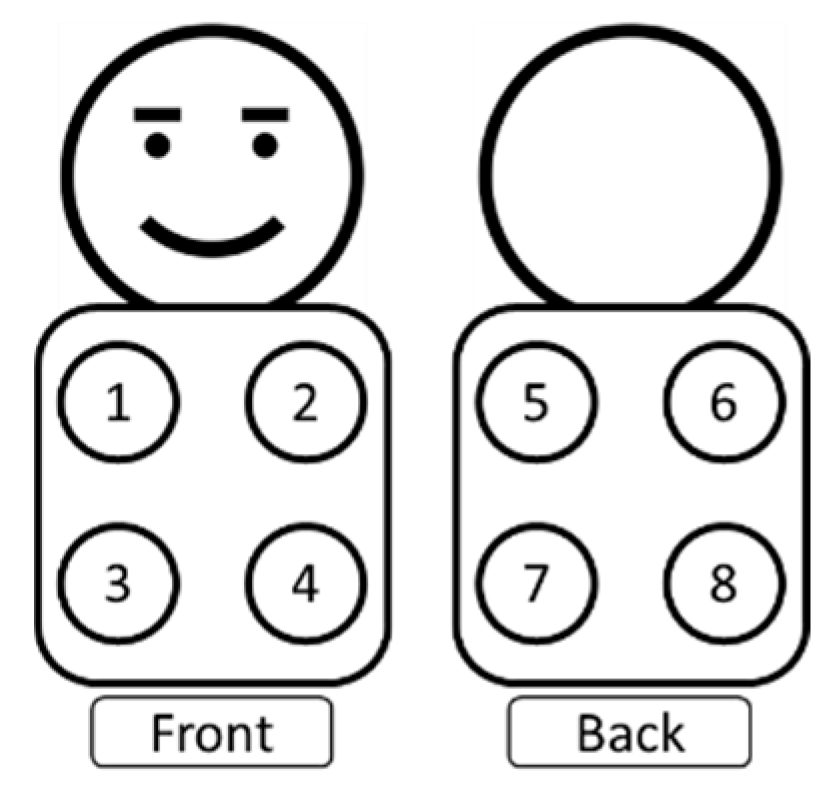
Figure 4.
Excel decision analytic model.

Table 1.
Summary of the IMCI-PLUS algorithm.
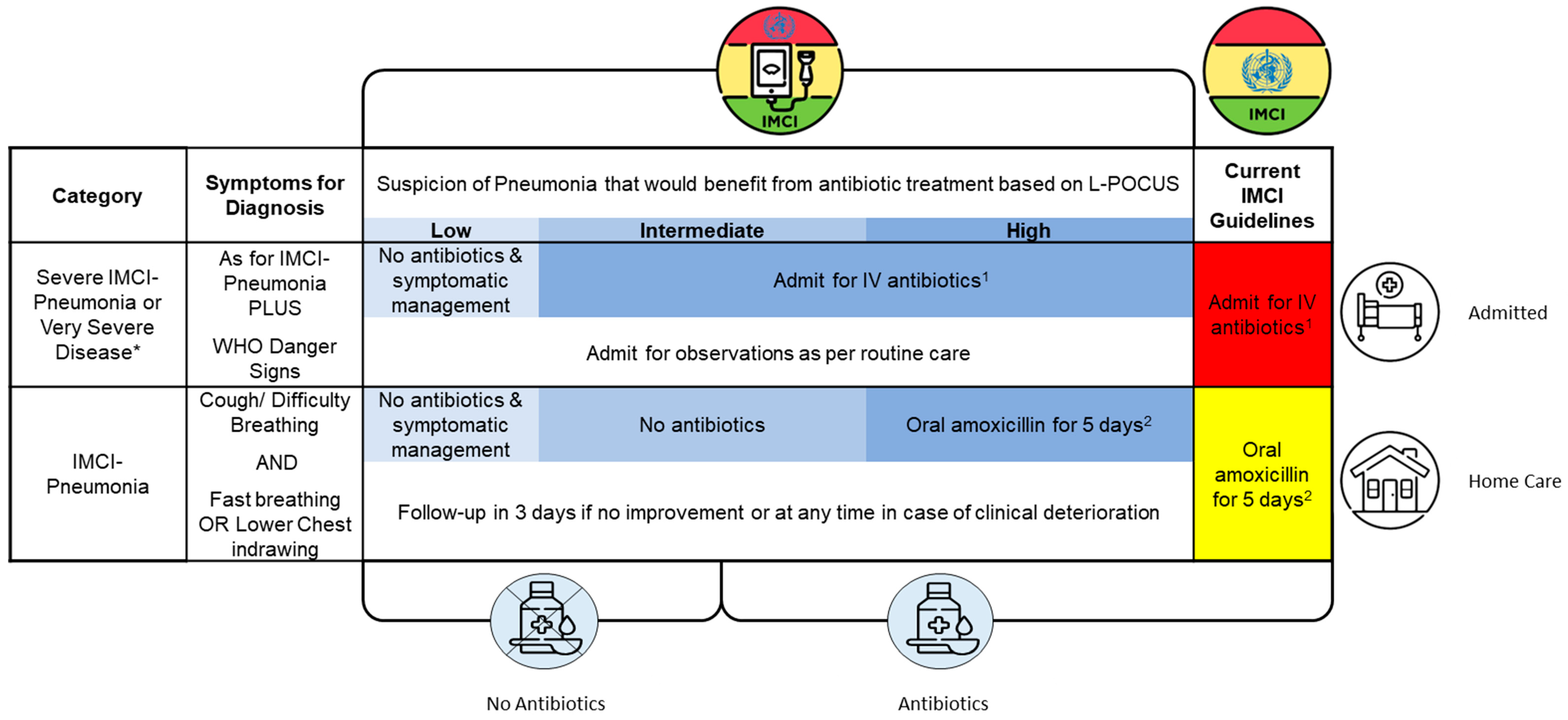
Table 2.
Overview of Study Procedure Timeline.
| Time since enrolment (days) | D1 | D1 | D2-7* | D8 | D29 | Week 8 |
|---|---|---|---|---|---|---|
| Visit | Enrolment | Clinical Visit | Spontaneous in-person Follow-up | 1st Follow-up(telephone/ in-person) | 2nd Follow-up (telephone) | 3rd Follow-up (telephone) |
| Windows | +4days | +4 days | +4days | |||
| Enrolment | ||||||
| Eligibility screening | + | |||||
| Informed consent | + | |||||
| Randomisation | + | |||||
| Demographic data collection | + | |||||
| Baseline clinical data collection | + | |||||
| LUS | + | +** | +^ | |||
| LAusc | + | |||||
| Samples collected for aetiological investigations & biobanking | + | |||||
| Interventions | ||||||
| Control group: Routine care | + | + | ||||
| Intervention group: IMCI-PLUS algorithm and guidance for clinical deterioration & persistence of symptoms | + | + | ||||
| Assessments | ||||||
| Antibiotic-related assessment | + | + | + | |||
| Event-related data (Clinical Failure) | + | + | + | |||
| Clinical signs | + | + | + | +^ | ||
| Clinical management | + | + | +^ | |||
| Survival | + | + | + | |||
| Adverse Events | + | + | + | |||
| TB substudy only – Symptoms and TB treatment history | + | |||||
* D2-7 refers to unplanned follow-up as a result of participants representing to the study site for the index illness. ** LUS performed at follow-up visits (D2-7 or D8) if participant is in the intervention group only. ^ Only collected if in-person follow-up on D8.
| 1 | WHO operational Handbook on Tuberculosis: Module 5: Cough longer than 2 weeks, Fever longer than 2 weeks, Lethargy, Weight loss, Haemoptysis, Night sweats, Swollen lymph nodes, Tachycardia, Tachypnoea |
| 2 | E.g., National TB Management Guidelines – 2014 (p.114): Cough lasting longer than two weeks (if HIV+, then
of any duration), unexplained weight loss, night sweats or a fever, and loss of appetite |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



