Submitted:
30 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Trehalose is a naturally occurring disaccharide that has recently gained significant attention for its neuroprotective properties in various models of neurodegeneration. This review provides an overview of available experimental data on the beneficial properties of trehalose for central nervous system pathological conditions. Trehalose’s impact on neuronal cell survival and function was also examined. As a result, we identified that trehalose’s neuroprotection includes autophagy modulation as well as its capability to stabilize proteins and inhibit the formation of misfolded ones. Moreover, trehalose mitigates oxidative stress-induced neuronal damage by stabilizing cellular membranes and modulating mitochondrial function. Furthermore, trehalose attenuates excitotoxicity-induced neuroinflammation by suppressing pro-inflammatory cytokine release and inhibiting inflammasome activation. A possible connection of trehalose with the gut-brain axis was also examined. These findings highlight the potential therapeutic effects of trehalose in neurodegenerative diseases. According to the conclusions drawn from this study, trehalose is a promising neuroprotective agent as a result of its distinct mechanism of action, which makes this compound a candidate for further research and development of therapeutic strategies to combat neuronal damage and promote neuroprotection in various neurological diseases.
Keywords:
trehalose
; neuroprotection
; apoptosis
; neuroinflammation
; oxidative stress
1. Introduction
Neurodegenerative diseases (NDs) pose an increasing health challenge due to their severe and prolonged clinical effects. A common pathological feature of many late-onset NDs, known as proteinopathies, is the accumulation of misfolded proteins. These diseases include Parkinson's disease (PD), Alzheimer's disease (AD), tauopathies, amyotrophic lateral sclerosis (ALS), and polyglutamine (polyQ) expansion disorders like Huntington's disease (HD) and various spinocerebellar ataxias (SCAs), such as SCA3 [1]. Misfolded proteins can arise from posttranslational modifications (e.g., hyperphosphorylated tau in AD), endoproteolytic cleavage (e.g., amyloid β peptides), as well as genetic mutations in specific proteins (such as huntingtin in HD, α-synuclein in PD, PrPC in prion disease, and superoxide dismutase 1 (SOD1) and TDP-43 in ALS) which can lead to the formation of oligomers. Normally, misfolded proteins are removed by cellular quality control systems. In addition, β-sheet-rich aggregates can interact with various cellular molecules and directly or indirectly disrupt the ubiquitin-proteasome system and chaperone-mediated autophagy. Recent evidence suggests that there is a strong link between autophagy and NDs [2]. As a result, there has been great interest in the idea that improving autophagy could alleviate neuropathology and neurodegeneration, leading to the investigation of various agents targeting the autophagy pathway as potential therapeutics in experimental studies. [3] Trehalose (Figure 1) a nonreducing disaccharide composed of two glucose molecules connected by an α, α-1,1-glycosidic bond is such a candidate.
This sugar is found in a diverse range of organisms, including bacteria, yeast, fungi, insects, and invertebrates, as well as lower and higher plants, where it can function as a source of energy and carbon. [5] Recent research has identified trehalose as a promising agent that can induce autophagy and protect against pathological changes in various neurodegenerative disorder models, which positions the compound as a potential therapeutic option. [1,6,7,8]. Research also suggests that the neuroprotective effects of this substance are not solely due to its ability to induce autophagy. Rather, these effects may be associated with trehalose's ability to reduce oxidative stress, stabilize proteins, and even affect gut-brain signaling. [9,10,11]
Therefore, this review thoroughly evaluates numerous studies and discusses, organizes, and presents the possible neuroprotective mechanisms of trehalose.
2. Materials and Methods
This systematic review was performed following the PRISMA guidelines for systematic reviews. We searched Scopus and MEDLINE/PUBMED, ResearchGate as well as ScienceDirect using the keywords “trehalose”, “neuroprotection”, “autophagy”, “biological”, “oxidative stress”, “gut-brain”, “inducing”, “Huntington’s disease”, “Parkinson’s disease”, and several others. All individual topics presented below were also searched separately. The cases published in the English literature between 1 January 1989 and 1 July 2024 were included. References of all reports were also searched manually for additional cases. In addition, we searched the articles that cited these references through PubMed. We excluded those cases where trehalose was examined for effects, not in direct correlation with the theme of this article. This comprehensive search yielded a significant number of studies. Studies that met the eligible criteria were carefully reviewed and rigorously analyzed. The insights and outcomes derived from this detailed examination are systematically presented in the subsequent sections, categorized by specific topics. During the preparation of this paper the authors used the AI model “ChatGPT-4” as a paraphraser tool.
3. Mechanisms of Neuroprotection
Five main points regarding trehalose and its neuroprotective properties were identified (Figure 2).
3.1. Autophagy Modulation
Trehalose is shown to promote autophagy through multiple different mechanisms, facilitating the cellular deterioration and recycling of damaged proteins as well as organelles. The main ways in which trehalose can influence the autophagy include:
3.1.1. mTOR Pathway Inhibition
The mechanistic target of rapamycin (mTOR) signaling pathway serves as a crucial regulator that integrates environmental stimuli with cellular responses. It exerts direct control over essential cellular processes including translation initiation, transcriptional activity, autophagy, metabolism, and organelle biogenesis. [12] These functions all together contribute to cellular homeostasis and the adaptation of organisms.
In the context of neurobiology, mTOR signaling is intricately involved in diverse facets of brain function. It orchestrates neural progenitor cell proliferation and differentiation, the formation and maintenance of synaptic connections, activity-dependent synaptic plasticity, and the regulation of complex behavioral patterns such as feeding behavior, sleep-wake cycles, and circadian rhythms.[12]
Pathological shifts in mTOR signaling are implicated in a spectrum of monogenic disorders and are contributing significantly to the pathogenesis of neurodegenerative conditions and neuropsychiatric disorders. [12] As a result of that, strategies aimed at pharmacologically modulating the mTOR pathway hold substantial promise for therapeutic approaches in neurological and psychiatric disorders, justifying further investigation and development.
Trehalose is shown to possess the ability to inhibit the mTOR pathway through various mechanisms, presented below:
- Activation of AMPK
AMP-activated protein kinase (AMPK) is an important factor in cellular energy balance and has diverse regulatory effects on several metabolic pathways. One of AMPK's primary jobs is to activate autophagy. The autophagy itself is a cellular disintegration and recycling process. This occurs through both direct and indirect processes involving Unc-51 Like Autophagy Activating Kinase 1 (ULK1) [13]. AMPK directly phosphorylates ULK1, thereby initiating the autophagy process. Additionally, AMPK has an indirect impact on autophagy. Secondly, AMPK indirectly activates ULK1 through the inhibition of mTORC1, which normally phosphorylates ULK1 and inhibits its interaction with AMPK. This coordinated regulation of ULK1 and mTORC1 facilitates the removal of damaged mitochondria and preserves mitochondrial integrity under conditions of nutrient starvation. AMPK's functions guarantee that cellular components are effectively recycled and destroyed under energy-stressed situations. [13]
AMPK also plays an important role in stimulating mitochondrial biogenesis. It controls the expression of peroxisome proliferator-activated receptor gamma coactivator gamma 1-alpha (PGC-1α), a transcription coactivator that promotes the transcription of genes involved in mitochondrial function and biogenesis. By enhancing PGC-1α activity, AMPK ensures an increase in mitochondrial number and improves mitochondrial function, which is important for maintaining cellular energy balance. [13]
In addition, AMPK also activates the cellular antioxidant defense mechanisms. These effects are reportedly achieved by upregulating the expression of antioxidant genes and enhancing the activity of antioxidant enzymes. [13] This function is vital for protecting cells against oxidative stress, which can damage cellular components and impair function.
Trehalose has been reported to activate AMPK, which as a result can inhibit the mTOR and lead to the activating of autophagy. It does so by creating a cellular environment that can mimic acute stress. [14] Kuczyńska-Wiśnik et al. (2024) in their review stated that trehalose may block the entry of glucose and fructose via the GLUT family of transporters into hepatocytes. [14] Thereby, indirectly trehalose leads to the inhibition of glycolysis and the citric acid cycle, eventually leading to ATP deficiency. AMPK is usually activated when cellular ATP levels fall and AMP/ATP or ADP/ATP rise, indicating the low energy state of the cell. This stress is sensed by the cellular machinery and AMPK and ULK1 are activated. The result is the initiation of the autophagy process.
However, the precise mechanisms by which cells absorb trehalose are not yet fully understood. SLC2A8 (GLUT8) has been identified as a transporter of trehalose in mammals, facilitating its uptake and triggering autophagy in hepatocytes. However, in neuronal cells such as N2A neuroblastoma cells, the accumulation of LC3-II, an indicator of autophagy, in response to trehalose is not dependent on SLC2A8. This discrepancy is due to the absence of SLC2A8 on the plasma membrane of neuronal cells. [10]
These findings suggest that the process of trehalose uptake may differ between neuronal cells and hepatocytes, potentially resulting in varying effects on the autophagic pathway. In contrast to observations in animal models where trehalose administration induces autophagy in the brain, experiments conducted directly on cultured cells, including neurons, indicate that trehalose may inhibit rather than stimulate autophagy. [10]
- Interference with mTORC1
It has been suggested that trehalose might interfere with the upstream regulators of mTORC1. [15] This includes Tuberous sclerosis proteins 1 and 2, also known as TSC1 (hamartin) and TSC2 (tuberin), which negatively regulate mTORC1 activity. By affecting these regulatory proteins, trehalose can suppress the activation of mTORC1 and ultimately inhibit autophagy.
Trehalose is also known to induce cellular stress responses, including endoplasmic reticulum stress.[16] In addition, trehalose is taken into lysosomes through endocytosis, causing an accumulation that raises the lysosomal pH. The heightened pH disturbs the lysosomal surroundings, leading to the deactivation of mTORC1. This deactivation disrupts mTORC1's control over cellular processes. [14] As a result these stress responses activate signaling pathways that inhibit mTORC1, such as the unfolded protein response and other stress-related kinases.
3.1.2. mTOR Independent Autophagy Induction
An in vitro study by Kuczyńska-Wiśnik et al. (2024) demonstrated that trehalose induces autophagy independently of the mTOR pathway.[14] The authors point to two possible mechanisms. The first one suggests that the two pathways may be truly independent and can also act on different components of the autophagy machinery. The second hypothesis is that trehalose may act on a middle component in the pathway between mTOR and autophagy. However, they concluded that it is impossible to test the second hypothesis without knowing what these components are, as they are not currently considered to be involved in mammalian autophagy processes.
3.1.3. TFEB Activation
Transcription factor EB or TFEB acts as a key master gene in the control of lysosomal biogenesis. [17] It encodes transcription factors that carefully regulate the expression of various lysosomal hydrolases and membrane proteins as well as genes involved in the autophagy process. [17,18] Under conditions such as nutrient deprivation or abnormal lysosomal accumulation, which often occur in lysosomal storage diseases, TFEB undergoes significant translocation from the cytoplasm to the nucleus. This translocation leads to the activation of target genes, thereby initiating a cascade of gene expression aimed at combating these cellular stressors.[17,18] Furthermore, experimental overexpression of TFEB in cultured HeLa cells demonstrated its potent ability to enhance lysosomal biogenesis. [17] This overexpression promotes not only lysosome formation but also exocytosis, the process by which cells expel waste products. In addition, it stimulates autophagy, a key cellular process for breaking down and recycling damaged cellular components. Through these mechanisms, TFEB plays a key role in maintaining cellular homeostasis and responding to metabolic stress. [17,18,19] Trehalose can activate TFEB, which then is translocated to the nucleus and promotes the expression of genes involved in autophagy and lysosomal function. [7] This increases the cells' ability to degrade and recycle cellular components. Trehalose activates TFEB through several mechanisms related to its role in cellular stress response and autophagy induction.
First, trehalose affects nutrient-sensing pathways that regulate TFEB. Under nutrient-rich conditions, TFEB is phosphorylated by mTORC1 and secreted into the cytoplasm. [14] The disaccharide promotes autophagy and alters nutrient signaling, thereby reducing mTORC1 activity and leading to TFEB dephosphorylation and nuclear translocation. [7] In the cell nucleus, TFEB activates genes involved in lysosomal biogenesis and autophagy.
Second, trehalose has been shown to affect the calcium signaling pathways, which are important for TFEB activation. [14]
A trehalose-activated pathway was identified in mouse motor neurons that is initiated by the transient expansion and subsequent permeabilization of lysosomal membranes under osmotic stress. This osmotic stress results in the release of Ca2+ ions from the destabilized lysosomes. The released Ca2+ ions then activate PPP3CB, a calcium-dependent and calmodulin-stimulated protein phosphatase. Activated PPP3CB dephosphorylates the transcription factor EB (TFEB), which is essential for its translocation to the nucleus, where TFEB can regulate gene expression involved in lysosomal biogenesis and autophagy.[20]
Third, activation of transcription factors such as TFEB promotes the expression of autophagy and lysosome-related genes. [7] Activation of TFEB leads to increased synthesis of lysosomal proteins, including those involved in chaperone-mediated autophagy. Chaperone-mediated autophagy is a selective type of autophagy in which certain proteins are transported directly across the lysosomal membrane for degradation. This is particularly important for removing misfolded or damaged proteins and is beneficial in neurodegenerative diseases.
Trehalose’s autophagy involvement is summarized in Table 1.
3.2. Inhibition of Protein Clustering
Protein aggregation is a hallmark of many neurodegenerative conditions. [15] Trehalose has been found to inhibit the aggregation of misfolded proteins. By preventing protein aggregation, trehalose can reduce cellular toxicity, including neurotoxicity.
Proteins in their native state are surrounded by a layer of water molecules that contribute to their stability. This hydration shell helps maintain the protein's three-dimensional structure and facilitates proper folding.
In their study, Carpenter and Crowe (1989), using infrared spectroscopy, showed that trehalose can replace some of these water molecules through hydrogen bonding interactions with both protein and water in bovine serum albumin. [22] The observed process leads to stabilization of the protein structure under conditions where the natural hydration shell may be disrupted. These hydrogen bonds inhibit abnormal intermolecular interactions that lead to protein aggregation. However, it remains elusive if and how this interaction results in the stabilization of proteins. [5]
Heat shock and oxidative stress can lead to protein unfolding, exposing hydrophobic regions that promote aggregation.
A comprehensive study by Singer and Lindquist from 1998 provided significant insights into the protective role of trehalose in cells exposed to heat stress. [23] Their research, which included both in vivo and in vitro experiments, highlighted the crucial function of trehalose in stabilizing proteins at elevated temperatures. By employing two different temperature-sensitive reporter proteins, they observed that enzymes retained a higher degree of activity during heat shock in cells that were able to produce trehalose.
An additional and critical finding from their study was the ability of trehalose to suppress the aggregation of proteins that had already undergone denaturation. This indicates that trehalose not only helps in maintaining protein stability but also prevents further damage to already affected proteins.
The researchers further demonstrated the importance of rapidly degrading trehalose after the end of the heat shock. They found that when the unfolded luciferase, one of the reporter proteins they used, is removed from the trehalose environment, it could effectively be refolded by molecular chaperones. However, if the concentration of trehalose remains high, it interferes with the refolding process thereby hindering the chaperones from renaturing the protein. Therefore, the study emphasizes the need for active trehalase enzyme in the environment to degrade trehalose after the heat stress is alleviated. This allows the molecular chaperones to efficiently refold denatured proteins and restore their normal functions. The findings of Singer and Lindquist (1998) provide a better understanding of the double role of trehalose in the protection and management of protein folding during and after thermal stress. [23]
Trehalose forms a crystalline structure upon drying that traps and stabilizes proteins, preventing denaturation and aggregation. [24] In this crystalline form, disaccharide molecules are arranged in an amorphous, non-crystalline matrix. Trehalose is effective in neutralizing and stabilizing biomolecules, including proteins and membranes, by forming a glass.[24] This immobilization process helps proteins maintain their functional state and not aggregate or denaturate, which often occurs under osmotic stress and dehydrating conditions.
As described above trehalose enhances autophagy, aiding in the clearance of misfolded and aggregated proteins.[6,16] This can reduce the load of misfolded proteins, decreasing the chances of further aggregation.
Tanaka et al. (2004) demonstrated that trehalose inhibits aggregation of proteins by interacting with expanded polyglutamines using a mouse model of Huntington’s disease. [25] They also indicated that trehalose has the potential to inhibit protein aggregation at the initial stage of aggregate formation. This inhibition occurs through the increased stability of polyglutamine-containing proteins, thereby preventing the early onset of aggregation.
Sarkar et al. (2007) showed that trehalose attenuates polyQ-mediated accumulation and cytotoxicity, while also facilitating the clearance of soluble mutant huntingtin. [21] In their study trehalose significantly reduced aggregation and cell death associated with EGFP-tagged huntingtin exon 1 containing 74 polyQ repeats (EGFP-HDQ74) in both COS-7 cells (non-neuronal) and SK-N-SH cells (neuronal precursor). This effect is specific to trehalose, as the authors did not observe similar results with other disaccharides such as sucrose, the trisaccharide raffinose, or the sugar alcohol sorbitol. These findings suggest that the protective properties of trehalose are unique and not shared by other structurally related sugars. Furthermore, initially, it was thought that trehalose's ability to stabilize partially unfolded mutant proteins with expanded polyQ regions accounted for its protective effects. However, the authors revealed a different underlying mechanism. [21] They found that trehalose primarily stimulates autophagy, which enhances the clearance of mutant proteins. This conclusion is supported by their observation that the effectiveness of trehalose in reducing aggregates of mutant huntingtin was completely lost when the autophagy process was inhibited.
Through the mechanisms that are mentioned above, trehalose effectively inhibits protein accumulation and promotes protein stabilization both in vivo and in vitro which is beneficial in the context of neurodegenerative diseases and other conditions associated with protein misfolding and aggregation (Table 2).
3.3. Osmoprotective Effect
Neurons are very sensitive to changes in osmotic pressure due to their unique membrane properties and rapid metabolism. Maintaining osmotic pressure near constant is important for maintaining neuronal activity and preventing cell swelling or shrinkage, which can disrupt cellular processes and lead to cell death. [26] Osmotic changes can lead to a number of complications, including brain edema. Nerve edema and inflammation can affect nerve function by increasing intracranial pressure, reducing blood flow, and causing nerve damage. Osmotic stress has been linked to a variety of neurological disorders, including stroke, traumatic brain injury, and neurodegenerative diseases. Disruption of osmotic pressure increases neuronal damage and contributes to disease progression by disrupting cellular and molecular processes required for neuronal survival.
Trehalose exerts an osmoprotective effect through several mechanisms that help cells withstand osmotic stress:
3.3.1. Membrane Stabilization
In a glassy state, trehalose molecules arrange themselves in an amorphous, non-crystalline matrix. When trehalose forms a glass, it effectively immobilizes and stabilizes biomolecules, including proteins and membranes. [24] This immobilization prevents the biomolecules from undergoing denaturation or aggregation that typically occurs under dehydrating conditions. The vitrification process helps proteins maintain their functional conformations under osmotic stress.
Trehalose interacts with phospholipid bilayers by forming hydrogen bonds between the hydroxyl groups and the phosphate groups of membrane phospholipids, as described by Crowe et al. (1992). [27] This molecular interaction strongly stabilizes the bilayer structure, especially under conditions of osmotic stress such as dehydration or exposure to hyperosmotic solutions in mammalian cells[28] and in a strain of wild-type Saccharomyces cerevisiae. [29] By integrating within the lipid bilayer, trehalose reduces membrane permeability, which is important to avoid phase transitions, and the formation of non-bilayer structures, which are usually less stable, can compromise membrane integrity and function. This stabilizing mechanism allows cell membranes to maintain their structure and function even under adverse conditions. The protective function of trehalose is important for cell strength, allowing cells to resist and adapt to various stresses, thereby protecting cell health and vitality in vitro in a dry state. [30]
3.3.2. Oxidative Stress Reduction
Trehalose has antioxidant properties that help reduce oxidative damage associated with osmotic stress. Benaroudj et al. (2001) used bacterial strains tracking their resistance to H2O2 in the presence of H2O2/FeCl3. [31] The results showed a significant resistance to reactive oxygen species. The suggested mechanism by which trehalose enhances resistance to oxidative stress was proposed to be the ability of the sugar to scavenge free radicals.
Through these mechanisms, trehalose acts as an effective osmoprotectant, helping cells survive and function under conditions of osmotic stress, such as dehydration, high salinity, or rapid changes in environmental water availability.
3.4. Anti-Inflammatory Properties
Trehalose reduces inflammation through various mechanisms that modulate immune responses and cellular signaling pathways. [32] The key mechanisms by which trehalose exerts its anti-inflammatory effects are as follows:
3.4.1. Modulation of Inflammatory Mediators: Trehalose's Role in Suppressing Pro-Inflammatory Cytokines
Studies concluded that trehalose has the ability to significantly inhibit the production of the pro-inflammatory cytokines, thereby attenuating the inflammatory response.[33,34]
Interleukin-1β (IL-1β) is crucial for orchestrating inflammatory responses through its binding to IL-1 receptors (IL-1R), which are expressed in various cell types.[35] It is synthesized in response to activation of inflammasomes, which are protein complexes that activate caspases. This activation process is pivotal in enhancing immune reactions and promoting short-term inflammation as part of the body's defense against pathogens. IL-1β also plays a significant role in shaping adaptive immunity, contributing to the overall immune response to infections and other challenges to the body's homeostasis.
Taya et al. (2009) found that stimulated mouse peritoneal macrophages, capable of producing IL-1β, showed a significant inhibition of the IL-1β production in a dose-dependent manner when treated with trehalose. [33]
Tumor necrosis factor-alpha (TNF-α) belongs to a family of inflammatory cytokines that utilize similar signaling pathways, including activation of the transcription factor nuclear factor kappa B (NF-κB) and initiation of apoptotic pathways. [36,37] These cytokines are associated with a diverse range of diseases, including cancer, arthritis, diabetes, atherosclerosis, and various inflammatory conditions.
Trehalose has been found to possess the ability to decrease TNF-a mRNA expression at 12 h in the study of Taya et al. (2009) mentioned above. [33]
Yu et al. (2023) investigated the synthesis of non-protein inflammatory mediators, specifically eicosanoid PGE2 and the arginine metabolite nitric oxide (NO), along with the transcription of their respective converting enzyme genes (Cox-2 and iNOS) in a model using the RAW 264.7 cell line, originating from murine macrophages. [34] Trehalose exhibited notable effectiveness in inhibiting the transcription of Cox-2, a key enzyme involved in inflammation. Additionally, the substance significantly down-regulated the production of PGE2, a potent inflammatory mediator. Regarding iNOS transcription, trehalose exerted pronounced suppressive effects, which corresponded to a specific reduction in the synthesis of nitric oxide (NO), highlighting trehalose's selective impact on inflammatory pathways.
As the information regarding trehalose and its effects on eicosanoids is relatively limited further studies in that direction are needed.
3.4.2. Suppression of NF-κB Signaling
The NF-κB transcription factor family plays a significant role in managing the immune response by regulating proinflammatory processes throughout the body. In its inactive state, NF-κB remains sequestered by IκB proteins found in the cytoplasm. When activated, IκB is degraded via proteasomes, leading to the liberation of NF-κB to translocate into the nucleus where its main function is as a transcription factor, followed by the expression of proinflammatory genes. Activation of NF-κB is typically initiated by a wide range of stimuli, mainly including proinflammatory cytokines and chemokines.
The downstream effects of NF-κB activation are highly specific to the cell type involved, which often culminates in the induction of proinflammatory cascades. In the central nervous system, the microglia serving as principal immune responders, prominently upregulate the NF-kB in response to many pathological stimuli. This activation primes microglia for interaction with other cell types in the central nervous system which can potentially trigger cellular death processes that can exacerbate disease pathology. [38]
Trehalose inhibits the activation of NF-κB in vitro, resulting in decreased transcription of pro-inflammatory genes. A study by Yu et al. from 2023, using the RAW 264.7 cell line, originating from murine macrophages, found that trehalose treatment did not alter the overall expression levels of NF-κB in comparison to both lipopolysaccharide (LPS)-treated and untreated (control) cells. [34] Notably, trehalose significantly reduced the expression of phosphorylated NF-κB relative to LPS-treated controls. These findings indicate that trehalose may inhibit LPS-induced inflammatory responses in vitro by modulating Toll-like receptor 4 (TLR4)-mediated NF-κB signaling.

3.5. Gut-Brain Signaling Modulation
The well-documented brain-gut axis refers to a bidirectional communication network between the central nervous system and the enteric nervous system. This network is mediated by neurons of the sympathetic and parasympathetic nervous systems, as well as by circulating hormones and various neuromodulatory molecules. Historically, it has been recognized as a mediator of gastrointestinal symptoms related to stress. However, interactions between the brain and gut go beyond the realms of stress, anxiety, or depression. They also include situations where both the brain and gut, along with their connection through the autonomic nervous system, are affected by the same pathological conditions, such as Parkinson's disease. The concept of this axis has now expanded to include the microbiota, forming the microbiota-gut-brain axis. Emerging evidence suggests that gut-resident bacteria can impact brain function, making the microbiome a potential target for diagnosing and treating a variety of disorders, including Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, autism, stroke, depression, and drug addiction. [39,40]
Trehalose has been investigated for its role in promoting gut health, particularly through its prebiotic effects.[41] Prebiotics like trehalose can selectively stimulate the growth and activity of beneficial gut bacteria, which in turn can influence gut-brain signaling via the gut microbiota-brain axis.
However, there are studies suggesting that trehalose can enhance the virulence of epidemic Clostridium difficile [42], though there are other studies indicating no such correlation [43]. As the results are conflicting further investigation is needed.
There is relatively limited research exploring the influence of trehalose on gut-brain signaling as compared to its effects on other aspects of health. However, some studies have suggested potential connections. [10]
When animals consume trehalose, it undergoes hydrolysis by the trehalase enzyme in the gut. Although some trehalose may enter the bloodstream, its ability to reach the brain is limited by the blood-brain barrier. Consequently, the initial effects of trehalose are likely exerted primarily within the gastrointestinal tract. For instance, trehalose could potentially impact the gut microbiota by shielding it from harmful factors and stressors, thereby enhancing its resilience and overall survival.[10] This suggests that trehalose may play a beneficial role in gut health and microbial balance, influencing broader physiological outcomes through its actions at the gut level.
Evidence has increasingly shown that the gut microbiota can have a significant impact on various physiological systems including the central nervous system which hints that trehalose might exert neuroprotective effects through modulating microbiota-gut-brain signaling. [44] In support of this, only oral intake of trehalose and not an intraperitoneal injection led to autophagy induction in mouse brains, suggesting that the neuroprotective effects of trehalose require the involvement of the gastrointestinal system. [45] But besides these findings, the possibility that the trehalose travels through the bloodstream and directly impacts neurons in the brain cannot be ruled out entirely. [10]
3.6. Additional Mechanisms Underlying the Neuroprotective Effects of Trehalose
In addition to its established function in enhancing autophagy, trehalose is implicated in influencing multiple biochemical pathways involved in secondary injury responses following traumatic brain injury (TBI), notably including the regulation of brain metal homeostasis. [46]
Essential metals such as copper, zinc, and iron are integral to fundamental biological processes like cellular metabolism, antioxidant defense mechanisms, and neurotransmission. However, dysregulation in their levels has been linked to exacerbated inflammatory responses, oxidative stress, and neuronal damage. [47,48,49,50]
Recent studies suggest that trehalose can modulate the concentrations of these essential metals within the brain following TBI, potentially preventing their depletion or augmenting their levels. The precise molecular mechanisms by which trehalose exerts these effects on metal ion homeostasis remain a subject of ongoing investigation. Current hypotheses propose that trehalose may interact with regulatory proteins involved in metal storage, chelation processes, or transport mechanisms within brain cells. By adjusting the levels of these essential elements, trehalose may contribute to maintaining an optimal biochemical environment necessary for neuronal survival and proper cognitive function.[46]
4. Discussion
Trehalose, a naturally occurring disaccharide, is known to have complex neuroprotective properties and covers several biological mechanisms essential to maintaining cell health and resilience, particularly in neurons and brains. One of the important ways that trehalose supports neuroprotection is by improving autophagy, a fundamental cell process that deals with the elimination of damaged proteins and organs. Trehalose achieves this by inhibiting the mTOR pathway, the central regulator of cell response to stress and food availability. By regulating mTOR signaling, trehalose promotes autophagy activation and facilitates cell component breakdown and recycling. This requirement is crucial to maintaining cell homeostasis and to mitigating the accumulation of toxic aggregates associated with neurodegenerative diseases.
One of the leading mechanisms by which trehalose promotes neuroprotection is by enhancing autophagy, an essential cellular process responsible for clearing damaged proteins and organelles from cells. Trehalose accomplishes this by inhibiting the mTOR pathway, a central regulator of cellular responses to stress and nutrient availability. By influencing and downregulating mTOR signaling, trehalose promotes the activation of autophagy, resulting in the breakdown and recycling of cellular components. This process is crucial for maintaining cellular homeostasis and reducing the buildup of toxic aggregates associated with neurodegenerative diseases.
Furthermore, trehalose activates AMPK, an energy-sensing enzyme that plays a main role in coordinating cellular energy metabolism and autophagy initiation. By this mechanism trehalose not only directly stimulates the ULK1 (Unc-51 Like Autophagy Activating Kinase 1) complex, a key initiator of autophagy, but also modulates other signaling pathways that contribute to autophagy flux. This dual mechanism enhances the efficiency of cellular “waste” clearance and supports cellular adaptation to stress conditions, thereby resulting in the promotion of neuronal survival and function.
In addition to its role in autophagy regulation, trehalose exerts direct effects on protein homeostasis by inhibiting the aggregation of misfolded proteins. Many neurodegenerative diseases, such as Alzheimer's and Parkinson's, are characterized by the accumulation of misfolded proteins that form toxic aggregates. Trehalose interacts with these proteins through hydrogen bonding, stabilizing their structures and preventing the aberrant interactions that lead to pathological aggregation. By maintaining protein solubility and stability, trehalose helps alleviate protein aggregation-associated toxicity, consequently preserving neuronal integrity and function.
Moreover, trehalose acts as an osmoprotectant, safeguarding neurons against osmotic stress by stabilizing cellular membranes and protein structures. This protective function is particularly important under conditions of dehydration or exposure to high osmolarity environments. In such environments, trehalose replaces water molecules in protein hydration shells and interacts with phospholipids to maintain membrane integrity. By reducing membrane permeability and preventing cellular deformation, trehalose contributes to neuronal resilience against osmotic challenges, thereby supporting overall cellular health.
Beyond its roles in cellular maintenance and stress response, trehalose exhibits anti-inflammatory properties that are beneficial in neurodegenerative damage. It modulates immune responses by suppressing the production of pro-inflammatory cytokines such as IL-1β, and TNF-α, and inhibiting NF-κB signaling pathways. By dampening excessive inflammatory responses implicated in neurodegeneration, trehalose helps mitigate neuroinflammation and its detrimental effects on neuronal function and survival.
Furthermore, emerging research suggests that trehalose may impact neurological health indirectly through interactions with the gut microbiota and the gut-brain axis. Changes in gut microbial composition and function can influence systemic inflammation, potentially impacting brain function and neurodegenerative processes. Trehalose's ability to modulate gut microbiota and inflammatory responses highlights its broader therapeutic potential in supporting brain health and ameliorating neurodegenerative diseases.
Recent research has substantially advanced our understanding of trehalose's neuroprotective effects by proposing a novel hypothesis that highlights its involvement in essential metal homeostasis. According to this emerging hypothesis, trehalose may be instrumental in regulating and maintaining the balance of critical metals such as copper, zinc, and iron. These metals are vital for various cellular processes, including enzymatic functions, antioxidant defense, and neurotransmission. The hypothesis suggests that by modulating the levels and distribution of these essential metals, trehalose may mitigate dysregulation-related pathologies, thereby enhancing its neuroprotective effects. This perspective provides a promising new avenue for exploring how trehalose contributes to neuronal health and resilience.
In summary, trehalose stands out as a promising candidate for neuroprotective interventions due to its diverse mechanisms of action. From promoting autophagy and inhibiting protein aggregation to acting as an osmoprotectant and modulating inflammatory responses, trehalose offers comprehensive support for neuronal resilience and function. Continued research into its therapeutic applications holds promise for addressing neurodegenerative diseases and other neurological disorders where maintaining cellular health and mitigating stress responses are crucial for optimal brain function.
Author Contributions
Conceptualization, S.Dragomanova; methodology, S.Dragomanova, B.S.; software, S.Dragomanova and B.S.; formal analysis, S.Dragomanova, G.K., S.D.; investigation, B.S. and S.Dragomanova; data curation, S.Dragomanova, G.K., S.D..; writing—original draft preparation, B.S., S.Dragomanova; writing—review and editing, S.Dragomanova, G.K., S.D.; visualization, S.Dragomanova; supervision, S.Dragomanova, G.K., S.D.; project administration, S.Dragomanova. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
As the authors are not native-born English speakers they used an AI model as a paraphraser tool. The model used is ChatGPT-4, by the developer OpenAI, headquartered San Francisco, California, USA. After using this tool, the authors reviewed and edited the content as needed and they take full responsibility for the content of the publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical Inducers of Autophagy That Enhance the Clearance of Mutant Proteins in Neurodegenerative Diseases. Journal of Biological Chemistry 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of Misfolded Proteins in Neurodegenerative Diseases: Therapeutic Targets and Strategies. Exp Mol Med 2015, 47, e147–e147. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Barreto, G.; Sahebkar, A. Therapeutic Potential of Trehalose in Neurodegenerative Diseases: The Knowns and Unknowns. Neural Regen Res 2021, 16, 2026. [Google Scholar] [CrossRef] [PubMed]
- Trehalose - Structure. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Trehalose. (accessed on 12 July 2024).
- Elbein, A.D. New Insights on Trehalose: A Multifunctional Molecule. Glycobiology 2003, 13, 17R–27. [Google Scholar] [CrossRef] [PubMed]
- Casarejos, M.J.; Solano, R.M.; Gómez, A.; Perucho, J.; De Yébenes, J.G.; Mena, M.A. The Accumulation of Neurotoxic Proteins, Induced by Proteasome Inhibition, Is Reverted by Trehalose, an Enhancer of Autophagy, in Human Neuroblastoma Cells. Neurochemistry International 2011, 58, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-J.; Stitham, J.; Evans, T.D.; Zhang, X.; Rodriguez-Velez, A.; Yeh, Y.-S.; Tao, J.; Takabatake, K.; Epelman, S.; Lodhi, I.J.; et al. Trehalose Causes Low-Grade Lysosomal Stress to Activate TFEB and the Autophagy-Lysosome Biogenesis Response. Autophagy 2021, 17, 3740–3752. [Google Scholar] [CrossRef] [PubMed]
- Hui Yap, K.; Azmin, S.; Makpol, S.; Damanhuri, H.; Mustapha, M.; Hamzah, J.; Ibrahim, N. Profiling Neuroprotective Potential of Trehalose in Animal Models of Neurodegenerative Diseases: A Systematic Review. Neural Regen Res 2023, 18, 1179. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose Protects against Oxidative Stress by Regulating the Keap1–Nrf2 and Autophagy Pathways. Redox Biology 2018, 15, 115–124. [Google Scholar] [CrossRef]
- Lee, H.-J.; Yoon, Y.-S.; Lee, S.-J. Mechanism of Neuroprotection by Trehalose: Controversy Surrounding Autophagy Induction. Cell Death Dis 2018, 9, 712. [Google Scholar] [CrossRef]
- Jing, M.; Liu, K.; Liu, C.; Yan, D.; Ma, Z.; Wang, C.; Deng, Y.; Liu, W.; Xu, B. Protective Effects of Trehalose against Mn-induced A-synuclein Oligomerization in Mice: Involvement of Oxidative Stress and Autophagy. Environmental Toxicology 2020, 35, 55–65. [Google Scholar] [CrossRef]
- Lipton, J.O.; Sahin, M. The Neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M. Regulation and Function of AMPK in Physiology and Diseases. Exp Mol Med 2016, 48, e245–e245. [Google Scholar] [CrossRef] [PubMed]
- Kuczyńska-Wiśnik, D.; Stojowska-Swędrzyńska, K.; Laskowska, E. Intracellular Protective Functions and Therapeutical Potential of Trehalose. Molecules 2024, 29, 2088. [Google Scholar] [CrossRef]
- Cai, L.; Yoon, J.D.; Hwang, S.-U.; Lee, J.; Kim, E.; Kim, M.; Hyun, S.-Y.; Choi, H.; Oh, D.; Jeon, Y.; et al. Exploring the Mechanism of Trehalose: Dual Functions of PI3K/Akt and VPS34/mTOR Pathways in Porcine Oocytes and Cumulus Cells. Biology of Reproduction 2022, 107, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Developmental Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel mTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. Journal of Biological Chemistry 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.F.; Crowe, J.H. An Infrared Spectroscopic Study of the Interactions of Carbohydrates with Dried Proteins. Biochemistry 1989, 28, 3916–3922. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.A.; Lindquist, S. Multiple Effects of Trehalose on Protein Folding In Vitro and In Vivo. Molecular Cell 1998, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Roy, I. Effect of Trehalose on Protein Structure. Protein Science 2009, 18, 24–36. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose Alleviates Polyglutamine-Mediated Pathology in a Mouse Model of Huntington Disease. Nat Med 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain Oedema in Focal Ischaemia: Molecular Pathophysiology and Theoretical Implications. The Lancet Neurology 2007, 6, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.H.; Hoekstra, F.A.; Crowe, L.M. Anhydrobiosis. Annual Review of Physiology 1992, 54, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Garcı́a De Castro, A.; Tunnacliffe, A. Intracellular Trehalose Improves Osmotolerance but Not Desiccation Tolerance in Mammalian Cells. FEBS Letters 2000, 487, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Hounsa, C.-G.; Brandt, E.V.; Thevelein, J.; Hohmann, S.; Prior, B.A. Role of Trehalose in Survival of Saccharomyces Cerevisiae under Osmotic Stress. Microbiology 1998, 144, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.H.; Crowe, L.M.; Oliver, A.E.; Tsvetkova, N.; Wolkers, W.; Tablin, F. The Trehalose Myth Revisited: Introduction to a Symposium on Stabilization of Cells in the Dry State. Cryobiology 2001, 43, 89–105. [Google Scholar] [CrossRef]
- Benaroudj, N.; Lee, D.H.; Goldberg, A.L. Trehalose Accumulation during Cellular Stress Protects Cells and Cellular Proteins from Damage by Oxygen Radicals. Journal of Biological Chemistry 2001, 276, 24261–24267. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, D.; Chen, X.; Bian, F.; Qin, W.; Gao, N.; Xiao, Y.; Li, J.; Pflugfelder, S.C.; Li, D.-Q. Trehalose Induces Autophagy Against Inflammation by Activating TFEB Signaling Pathway in Human Corneal Epithelial Cells Exposed to Hyperosmotic Stress. Investigative Ophthalmology & Visual Science 2020, 61, 26–26. [Google Scholar] [CrossRef]
- Taya, K.; Hirose, K.; Hamada, S. Trehalose Inhibits Inflammatory Cytokine Production by Protecting IκB-α Reduction in Mouse Peritoneal Macrophages. Archives of Oral Biology 2009, 54, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Park, H.; Kim, W. Trehalose Inhibits Inflammatory Responses through Mitochondrial Reprogramming in RAW 264.7 Macrophages. Antioxidants 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Baraskar, K.; Thakur, P.; Shrivastava, R.; Shrivastava, V.K. Female Obesity: Association with Endocrine Disruption and Reproductive Dysfunction. Obesity Medicine 2021, 28, 100375. [Google Scholar] [CrossRef]
- Bharat, B. Aggarwal; Shishir Shishodia; Kazuhiro Ashikawa; Alok C. Bharti The Role of TNF and Its Family Members in Inflammation and Cancer: Lessons from Gene Deletion. CDTIA 2002, 1, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Mitamura, Y.; Harada, T. The Role of Cytokines and Trophic Factors in Epiretinal Membranes: Involvement of Signal Transduction in Glial Cells. Progress in Retinal and Eye Research 2006, 25, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, S.; Wright-Jin, E. NF-κB as an Inducible Regulator of Inflammation in the Central Nervous System. Cells 2024, 13, 485. [Google Scholar] [CrossRef] [PubMed]
- Yarandi, S.S.; Peterson, D.A.; Treisman, G.J.; Moran, T.H.; Pasricha, P.J. Modulatory Effects of Gut Microbiota on the Central Nervous System: How Gut Could Play a Role in Neuropsychiatric Health and Diseases. J Neurogastroenterol Motil 2016, 22, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr Neurol Neurosci Rep 2017, 17, 94. [Google Scholar] [CrossRef]
- Chen, A.; Gibney, P.A. Dietary Trehalose as a Bioactive Nutrient. Nutrients 2023, 15, 1393. [Google Scholar] [CrossRef]
- Collins, J.; Robinson, C.; Danhof, H.; Knetsch, C.W.; Van Leeuwen, H.C.; Lawley, T.D.; Auchtung, J.M.; Britton, R.A. Dietary Trehalose Enhances Virulence of Epidemic Clostridium Difficile. Nature 2018, 553, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Moura, I.B.; Wilcox, M.H. Is There a Causal Relationship between Trehalose Consumption and Clostridioides Difficile Infection? Current Opinion in Gastroenterology 2021, 37. [Google Scholar] [CrossRef] [PubMed]
- Felice, V.D.; Quigley, E.M.; Sullivan, A.M.; O’Keeffe, G.W.; O’Mahony, S.M. Microbiota-Gut-Brain Signalling in Parkinson’s Disease: Implications for Non-Motor Symptoms. Parkinsonism & Related Disorders 2016, 27, 1–8. [Google Scholar] [CrossRef]
- Tanji, K.; Miki, Y.; Maruyama, A.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; Wakabayashi, K. Trehalose Intake Induces Chaperone Molecules along with Autophagy in a Mouse Model of Lewy Body Disease. Biochemical and Biophysical Research Communications 2015, 465, 746–752. [Google Scholar] [CrossRef]
- Ghorbani, M.; Abouei Mehrizi, M.; Tajvidi, M.; Amin Habibi, M.; Mohammadi, M.; Esmaeilian, S.; Torabi, P.; Rahmanipour, E.; Daskareh, M.; Mohammadi, A. Trehalose: A Promising New Treatment for Traumatic Brain Injury? A Systematic Review of Animal Evidence. Interdisciplinary Neurosurgery 2024, 36, 101947. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and Away from Alzheimer’s Disease. 2004, 430.
- Kim, B.-E.; Nevitt, T.; Thiele, D.J. Mechanisms for Copper Acquisition, Distribution and Regulation. Nat Chem Biol 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Ho, E.; Courtemanche, C.; Ames, B.N. Zinc Deficiency Induces Oxidative DNA Damage and Increases P53 Expression in Human Lung Fibroblasts. The Journal of Nutrition 2003, 133, 2543–2548. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. The Lancet Neurology 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
Figure 1.
Structure of trehalose [4].
Figure 1.
Structure of trehalose [4].
Figure 2.
Biological properties of trehalose.
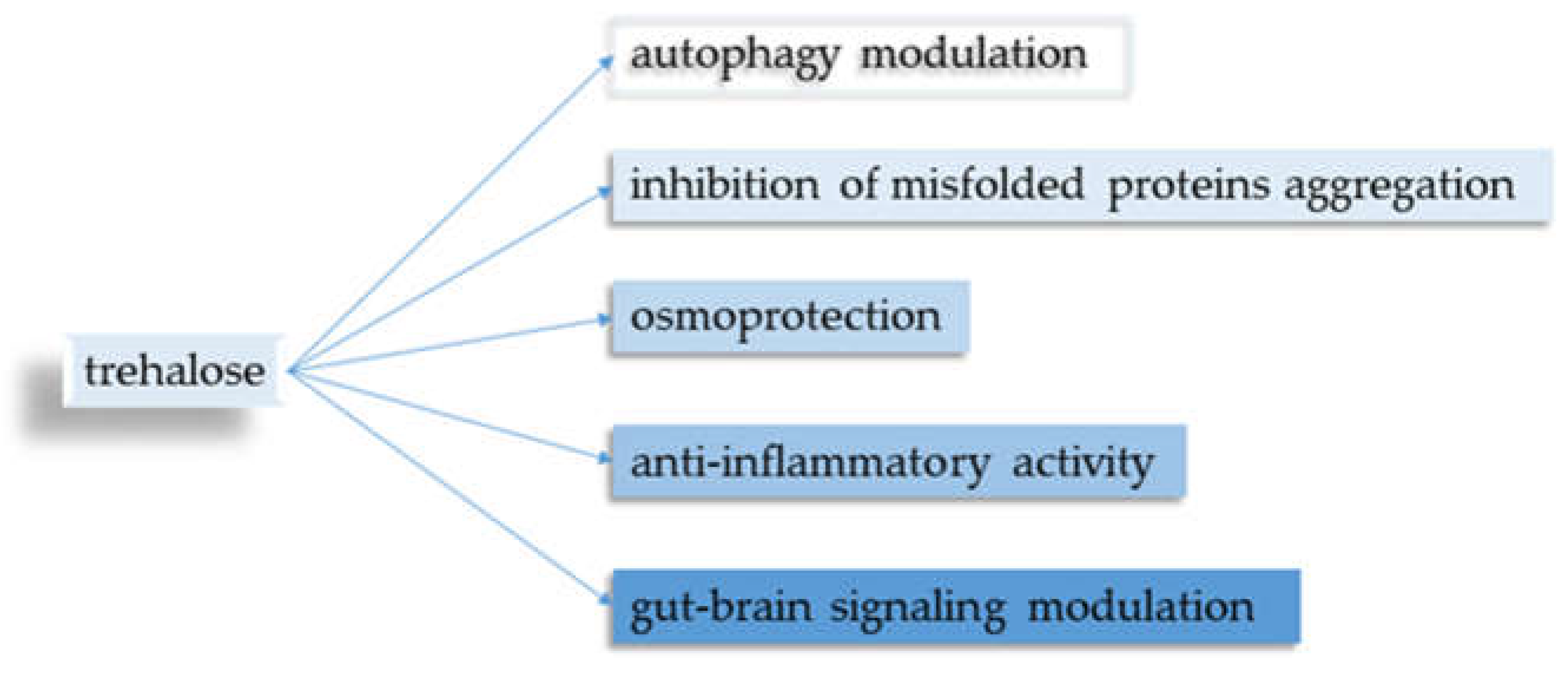
Table 1.
Trehalose’s mechanisms of autophagy involvement.
| Autophagy mechanism | Detailed mechanism of autophagy activation | Reference |
|---|---|---|
| 1mTOR Pathway involvement | direct inhibition of mTORC12 | [15] |
| stress response activation$$$AMPK3 activation | [20]$$$[14] | |
| mTOR independent | Inducing autophagy without influencing the mTOR pathway | [21] |
| TFEB4 Activation | influencing nutrient-sensing pathways that regulate TFEB $$$facilitating lysosomal calcium release | [21]$$$[20] |
| increased synthesis of lysosomal proteins | [7] |
mTOR1 – mechanistic target of rapamycin; mTORC12 – mTOR Complex 1; AMPK3 – AMP-activated protein kinase; TFEB4 – transcription factor EB.
Table 2.
Trehalose’s mechanisms of inhibition of misfolded proteins aggregation.
| Detailed mechanism | Reference |
|---|---|
| stabilizing protein molecules by forming hydrogen bonds | [5] |
| stabilization of hydration shells | [5] |
| protection against heat | [5,23] |
| water replacement | [5] |
| reduction of misfolded protein burden | [6] |
| interaction with amyloidogenic proteins | [21,25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



